

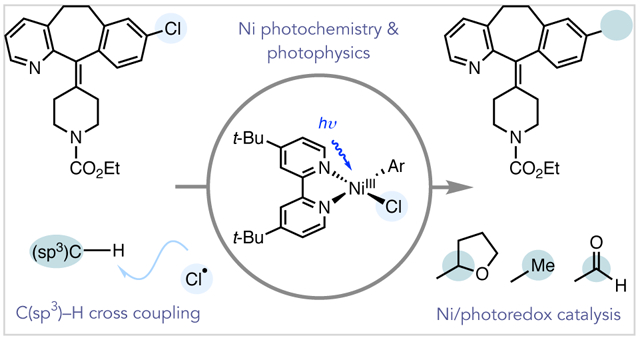
CONSPECTUS:
In recent years, the development of light-driven reactions has contributed numerous advances in synthetic organic chemistry. A particularly active research area combines photoredox catalysis with nickel catalysis to accomplish otherwise inaccessible cross-coupling reactions. In these reactions, the photoredox catalyst absorbs light to generate an electronically excited charge-transfer state that can engage in electron or energy transfer with a substrate and the nickel catalyst. Our group questioned whether photoinduced activation of the nickel catalyst itself could also contribute new approaches to cross-coupling. Over the past five years, we have sought to advance this hypothesis for the development of a suite of mild and site-selective C(sp3)–H cross-coupling reactions with chloride-containing coupling partners via photoelimination of a Ni–Cl bond.
On the basis of a report from the Nocera laboratory, we reasoned that photolysis of a Ni(III) aryl chloride species, generated by single electron oxidation of a typical Ni(II) intermediate in cross-coupling, might allow for the catalytic generation of chlorine atoms. Taken together with the ability of Ni(II) to accept alkyl radicals, we hypothesized that photocatalytically generated chlorine atoms could mediate hydrogen atom transfer (HAT) with C(sp3)–H bonds to generate a substrate-derived alkyl radical that is captured by the Ni center in cross-coupling. A photoredox catalyst was envisioned to promote the necessary single electron oxidation and reduction of the Ni catalyst to facilitate an overall redox-neutral process. Overall, this strategy would offer a visible light-driven mechanism for chlorine radical formation enabled by the sequential capture of two photons.
As an initial demonstration, we developed a Ni/photoredox-catalyzed α-oxy C(sp3)–H arylation of cyclic and acyclic ethers. This method was extended to a mild formylation of abundant and complex aryl chlorides through selective 2-functionalization of 1,3-dioxolane. Seeking to develop a suite of reactions that introduce carbon at all different oxidation states, we explored C(sp3)–H cross-coupling with trimethyl orthoformate, a common laboratory solvent. We found that trimethyl orthoformate serves as a source of methyl radical for a methylation reaction via β-scission from a tertiary radical generated upon chlorine-mediated HAT. Since chlorine radical is capable of abstracting unactivated C(sp3)–H bonds, our efforts have also been directed at cross-coupling with a range of feedstock chemicals, such as alkanes and toluenes, along with late-stage intermediates, using chloroformates as coupling partners. Taken together, this platform enables access to valuable synthetic transformations with (hetero)aryl chlorides, which, despite being the most ubiquitous and inexpensive aryl halide coupling partner, are rarely reactive in Ni/photoredox catalysis.
Little is known about the photophysics and photochemistry of organometallic Ni complexes relevant to cross-coupling. We have conducted mechanistic investigations, including computational, spectroscopic, emission quenching, and stoichiometric oxidation studies, of Ni(II) aryl halide complexes common to Ni/photoredox reactions. These studies indicate that chlorine radical generation from excited Ni(III) is operative in the described C(sp3)–H functionalization methods. More generally, the studies illustrate that the photochemistry of cross-coupling catalysts cannot be ignored in metallaphotoredox reactions. We anticipate that further mechanistic understanding should facilitate new catalyst design and lead to the development of new synthetic methods.
Graphical Abstract
INTRODUCTION
The past decade has witnessed a dramatic rise in the application of photoredox catalysis in organic synthesis.5 Integral to the success of this field is the photoredox catalyst, which becomes both a potent one-electron oxidant and reductant upon absorption of a visible light photon (photoinduced electron transfer, or PET).6 Single-electron transfer (SET) between this reactive species and a small molecule substrate affords access to radical intermediates under exceptionally mild reaction conditions.5 Moreover, the resultant reduced or oxidized catalyst retains a strong thermodynamic driving force to return to the original oxidation state, thereby facilitating access to redox-neutral transformations. For these reasons, photoredox catalysis has enabled chemists to solve challenging problems in synthetic methodology development, organometallic catalysis, and medicinal chemistry.
Over the past decade, our lab has built a program in nickel-catalyzed cross-coupling.7 Given the proclivity of nickel to both generate and accept reactive radical species, we postulated that merging nickel and photoredox catalysis could introduce a co-operative platform wherein a photoredox catalyst could supply substrate radicals as coupling partners to Ni and modulate Ni’s oxidation state in catalysis (Figure 1A). In collaboration with the MacMillan laboratory, we reported a decarboxylative C(sp3)–C(sp2) cross-coupling of amino acids, as well as α-oxy or α-phenyl-substituted carboxylic acids, with aryl halides.8 This catalyst system was also applied to the C(sp3)–H arylation of dimethylanilines, demonstrating that Ni/photoredox catalysis could serve as a mild method for the direct functionalization of C(sp3)–H bonds.8,9 Independently, the Molander laboratory reported the merger of Ni and photoredox catalysis for the cross-coupling of benzyl trifluoroborate salts as aliphatic radical precursors with aryl halides.10 Since these initial reports, many researchers have made important contributions to this area, revealing that nickel/photoredox catalysis can serve as a platform for bond formation with traditionally unreactive coupling partners.11
Figure 1.

A. Ni/photoredox cross-coupling. B. Halogen photoelimination from Ni(III) trihalide complexes (HX = hydrohalic acid). C. Photoelimination of chlorine radicals for C(sp3)–H cross-coupling.
The impact of PET on the field of Ni-catalyzed cross-coupling prompted our interest in exploring alternative mechanisms for harnessing visible light in catalysis. In this context, we were drawn to photoinduced ligand dissociation, or photoelimination, a mechanism by which light absorption by a transition metal complex leads to excited state labilization of a metal–ligand bond.12 A preeminent example is photoexcitation of metal carbonyl complexes with ultraviolet irradiation. Seminal work by Bergman, Graham, and Jones demonstrated that photolysis of Ir–and Rh–CO complexes affords coordinatively unsaturated species responsible for activation of inert hydrocarbon C–H bonds.13
Owing to its important implications for energy storage, photoelimination of halogen radicals has seen recent and growing interest. In 2015, the Nocera group reported a protocol for halogen photoelimination from mononuclear Ni(III) trihalide complexes, the first example from a first row transition metal complex.14 These Ni(III) complexes, supported by bidentate phosphine ligands, underwent photoelimination of chlorine or bromine radicals under visible light irradiation (λ > 400 nm). The liberated chlorine atom is stabilized by an arene-to-chlorine-atom charge-transfer interaction. The formation of this relatively long-lived transient species arrives from a dissociative LMCT excited state (1b), from which photoinduced cleavage of the apical Ni(III)–Cl bond produces Ni(II) species 1c and a chlorine radical (Figure 1B).
Although work by Nocera and others focused upon halogen photoelimination for energy storage applications, we questioned whether the generation of halogen radicals by photoelimination could also offer interesting synthetic opportunities. Traditionally, chlorine radical generation requires employment of reagents that are oxidizing and challenging to handle, including chlorine gas or N-chlorosuccinimide. Furthermore, oxidation of chloride to chlorine is inaccessible with most conventional photocatalysts (E = 2.03 V vs SCE in MeCN). Instead, we envisioned that access to chlorine radical could be possible from abundant chloride-containing electrophiles by combining chlorine photoelimination from Ni with metallaphotoredox cross-coupling. By design, the chloride source would serve as both the reaction coupling partner and the source of chlorine radical for hydrogen atom transfer. If successful, this approach could facilitate cross-coupling with unactivated C(sp3)–H bonds in the absence of a directing group: the thermodynamically favorable formation of H–Cl (bond dissociation energy (BDE) = 102 kcal/mol) would permit virtually any C(sp3)–H bond to undergo abstraction (Figure 1C). This Account centers upon our studies in applying the concept of photoelimination of halogen radicals toward C(sp3)–H cross-coupling and our mechanistic understanding of chlorine photoelimination in catalysis.
SYNTHETIC METHOD DEVELOPMENT
Initial Discovery of the Arylation of Ethers
In 2014 and 2015, our lab reported examples of C–H functionalization α to nitrogen atoms in anilines via an oxidation/deprotonation sequence (N-phenylpyrrolidine E1/2 = 0.70 V vs SCE in MeCN)15 (Figure 2A), wherein α-amino radicals were generated under photoredox conditions and intercepted by Ni for cross-coupling (Figure 2B).8,9 However, this approach was unsuccessful for functionalization of C–H bonds α to ethers owing to their high oxidation potential (i.e., THF E = 1.75 V vs SCE; E1/2 [*IrIII/IrII] = 1.21 V vs SCE in MeCN).16 On the other hand, the C–H bonds adjacent to oxygen atoms in ethers are significantly weakened (bond dissociation free energy (BDFE) ~ 86–89 kcal/mol) such that activation with HAT agents, including chlorine radicals, could provide a route for functionalization (Figure 2C).17 Thus we initiated our investigation of halogen photoelimination in cross-coupling with this substrate class.
Figure 2.
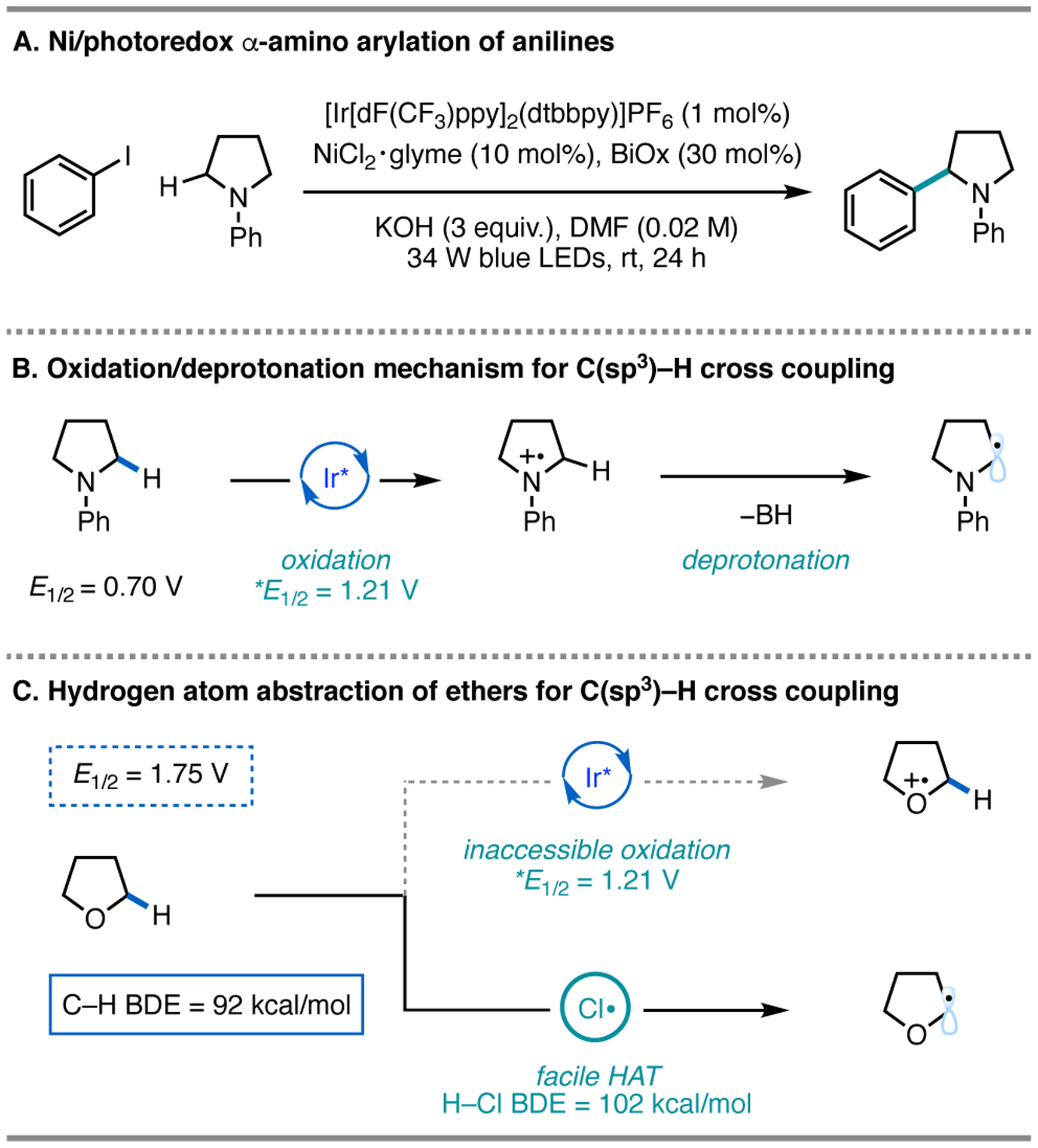
Strategies for Ni/photoredox C(sp3)–H functionalization.
We proposed that the arylation of ethers could proceed via the mechanism shown in Figure 3.1 Oxidative addition of a Ni(0) complex 2 into an aryl chloride would produce Ni(II) aryl chloride intermediate 3. Concurrently, irradiation of an iridium(III) photocatalyst 4 would afford a highly oxidizing, long-lived triplet excited *Ir(III) state (τ0 = 2.3 μs, *E1/2 = 1.21 V vs SCE in MeCN) (5) which could oxidize Ni(II) intermediate 3 to Ni(III) aryl chloride species 6. The Ni(III)–chlorine bond is then rendered sufficiently weak for a photon of visible light to homolyze the bond (BDFE = 47 kcal/mol), resulting in a Ni(II) aryl species 7 and a chlorine radical. The photocatalytically generated chlorine radical would then abstract a hydrogen atom from THF (THF BDE = 92 kcal/mol; H–Cl BDE = 102 kcal/mol), and rebound of the resulting carbon-centered radical into 7 would produce Ni(III) species 8. Subsequent reductive elimination from Ni(III) would forge the product containing a new C(sp3)–C(sp2) bond and Ni(I) species 9, which is reduced by the highly reducing Ir(II) species 10 (E1/2 = −1.37 V vs SCE in MeCN) to regenerate both the Ni(0) and Ir(III) catalysts.
Figure 3.
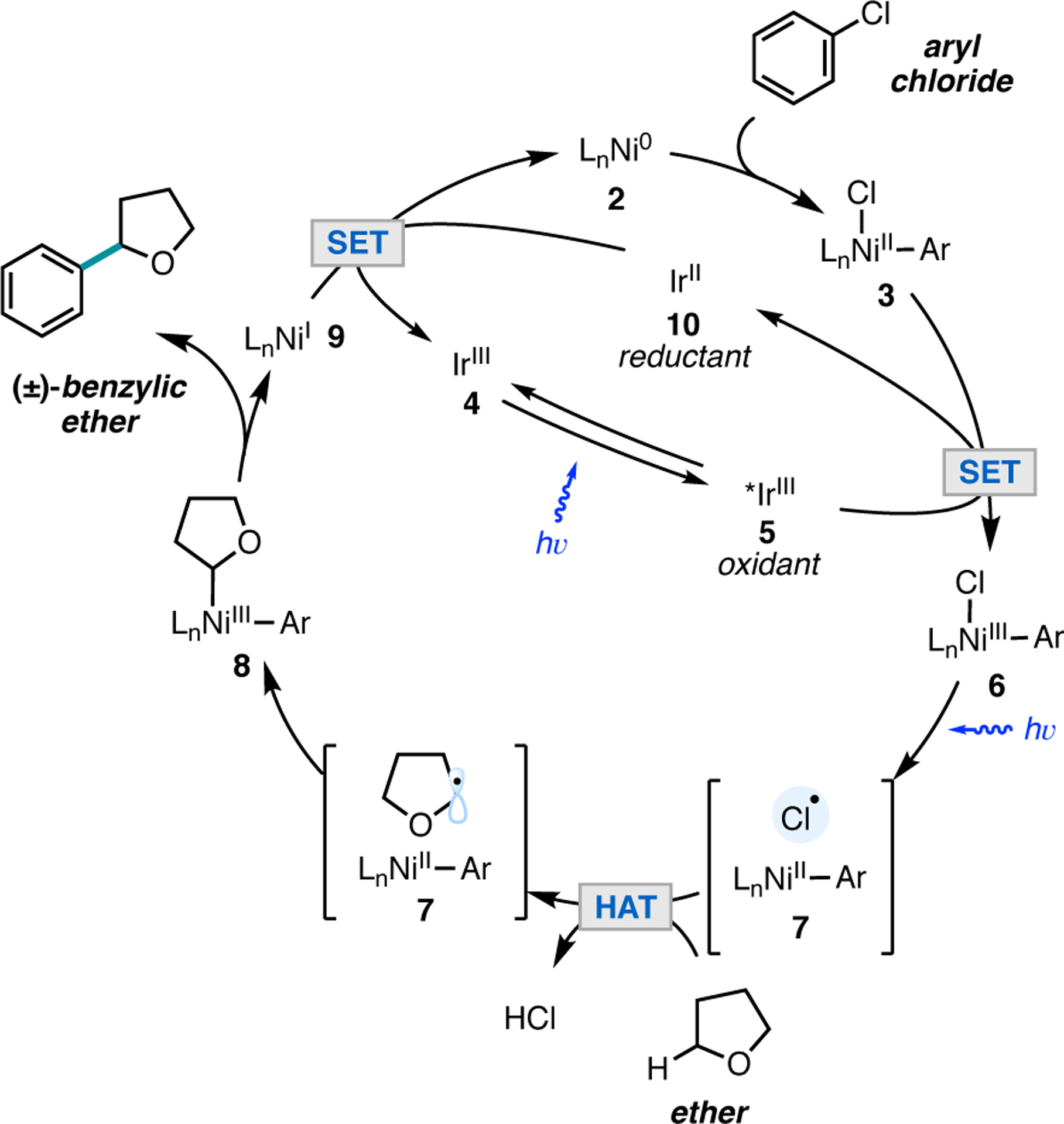
Proposed catalytic cycle for the arylation of ethers via chlorine photoelimination.
We found that employment of Ni(cod)2 (cod = 1,5-cyclooctadiene) (10 mol%), 4,4´-di-tert-butylbipyridine (dtbbpy) ligand (15 mol%), [Ir[dF(CF3)ppy]2(dtbbpy)]PF6 photocatalyst (2 mol%), and potassium phosphate (2 equiv.) under irradiation with blue LEDs enabled the cross-coupling of a variety of aryl chlorides with THF (0.04 M) (Figure 4).1 (Hetero)aryl chlorides with varying electronic and steric properties also underwent efficient coupling to give α-arylated products (Figure 4A). Additionally, other cyclic, as well as acyclic, ethers could be used in the Ni/photoredox cross-coupling reaction with 4´-chloroacetophenone, including 1,4-dioxane ((±)-11l) and anisole ((±)-11n) (Figure 4B). In reactions with 1,2-dimethoxyethane to give product (±)-11m, cross-coupling occurred at both the secondary and primary C–H bonds in a 1.35:1 ratio, indicating moderate selectivity for the weaker C–H bond. Reactions performed in the absence of Ni catalyst, photoredox catalyst, or light resulted in the formation of no cross-coupled product, indicating that all three components were necessary. At the same time that this work was disclosed, the Molander group demonstrated coupling of aryl bromides with ethers, leveraging bromine radical HAT with ethereal substrates using nickel/photoredox catalysis.18
Figure 4.

Representative (hetero)aryl chloride and ether substrate scopes.
Importantly, our platform for chlorine photoelimination allows for the use of aryl chlorides, the most ubiquitous, inexpensive, and biologically relevant aryl halide coupling partner. Generally, nickel/photoredox methodologies require aryl bromides or iodides as substrates for reactivity. In fact, at the time of the publication of this Account, more than 450 nickel/photoredox methodologies have been reported, but fewer than ten of these reports employ aryl chlorides (>5 substrates).19 In most nickel/photoredox cross-coupling methodologies that employ aryl halides, the generation of the radical species occurs independently of the nickel catalytic cycle, and so radical addition to a Ni catalyst may occur prior to oxidative addition, furnishing a Ni(I) intermediate. Indeed, computational studies by Molander, Kozlowski, and Gutierrez favor Ni(0)/Ni(I)/Ni(III) catalytic cycles that proceed via oxidative addition to Ni(I) rather than to Ni(0).20 Importantly, oxidative addition of an aryl chloride to Ni(I) is significantly more challenging than to Ni(0), which may explain why aryl chlorides are not competent coupling partners in other Ni/photoredox methodologies. In contrast, because aliphatic radical generation is dependent on Ni(III) photoelimination in the proposed catalytic cycle, which occurs only after oxidative addition, a Ni(0)/Ni(II)/Ni(III) sequence is likely favored.
Formylation of (Hetero)aryl Chlorides
The arylation of ethers served as an important proof of concept for chlorine photoelimination in a synthetic context. However, a number of other methods exist to effect α-arylation of ethers, including examples of redox-neutral methods.21 Moving forward, we sought to develop synthetic applications of the photoelimination strategy that would capitalize on its unique features: redox neutral C–H cross-coupling and employment of abundant and inexpensive chloride-containing coupling partners. One such application that we identified was a mild formylation of (hetero)aryl chlorides.
The versatility of aromatic aldehydes renders them important functional groups in the synthesis of pharmaceuticals, fragrances, fine chemicals, and natural products. One route for preparing benzaldehydes is electrophilic aromatic substitution, however both regioselectivity and reactivity are dependent on the substrate identity. Challenges associated with regioselective formylation can be circumvented by employing traditional organometallic methods, but this strategy can compromise functional group tolerance. A more general approach to aryl formylation is Pd-catalyzed reductive carbonylation of aryl halides, first reported in 1974 by Heck and coworkers.22 However, this approach presents implementation challenges on laboratory scale and tends to be limited to aryl bromides or iodides. These limitations have restricted formylation reactions to early stages in synthesis. At the same time, there are far more commercial aryl chlorides than benzaldehydes, making them attractive substrates for formylation.
As such, we envisioned that a mild formylation of aryl chlorides via chlorine photoelimination, using 1,3-dioxolane as a formyl source, would be an attractive alternative.23 A site-selective C–H functionalization at the 2-position of 1,3-dioxolane, the weakest C–H bond in the molecule, followed by radical trapping by Ni, would afford benzylidene acetal derivatives. Reaction optimization demonstrated that [Ir[dF(CF3)ppy]2(dtbbpy)]PF6 (1 mol%), NiCl2•glyme (10 mol%), dtbbpy (15 mol%), and K3PO4 (2 equiv.) under 34 W blue LED irradiation allowed for acetal intermediates to be synthesized from (hetero)aryl chlorides and dioxolane as reaction solvent (0.05 M). The resulting benzylidene acetals were not isolated, but instead directly subjected to acidic workup to provide the corresponding benzaldehyde products (Figure 5). Notably, a nickel/photoredox-catalyzed strategy similar to ours was concurrently reported by Mariano, Wang, and coworkers to enable formylation of aryl halides and triflates with diethoxyacetic acid as a formyl equivalent.19a While aryl chlorides underwent formylation in their report, only electron-deficient substrates were reactive in the method.
Figure 5.
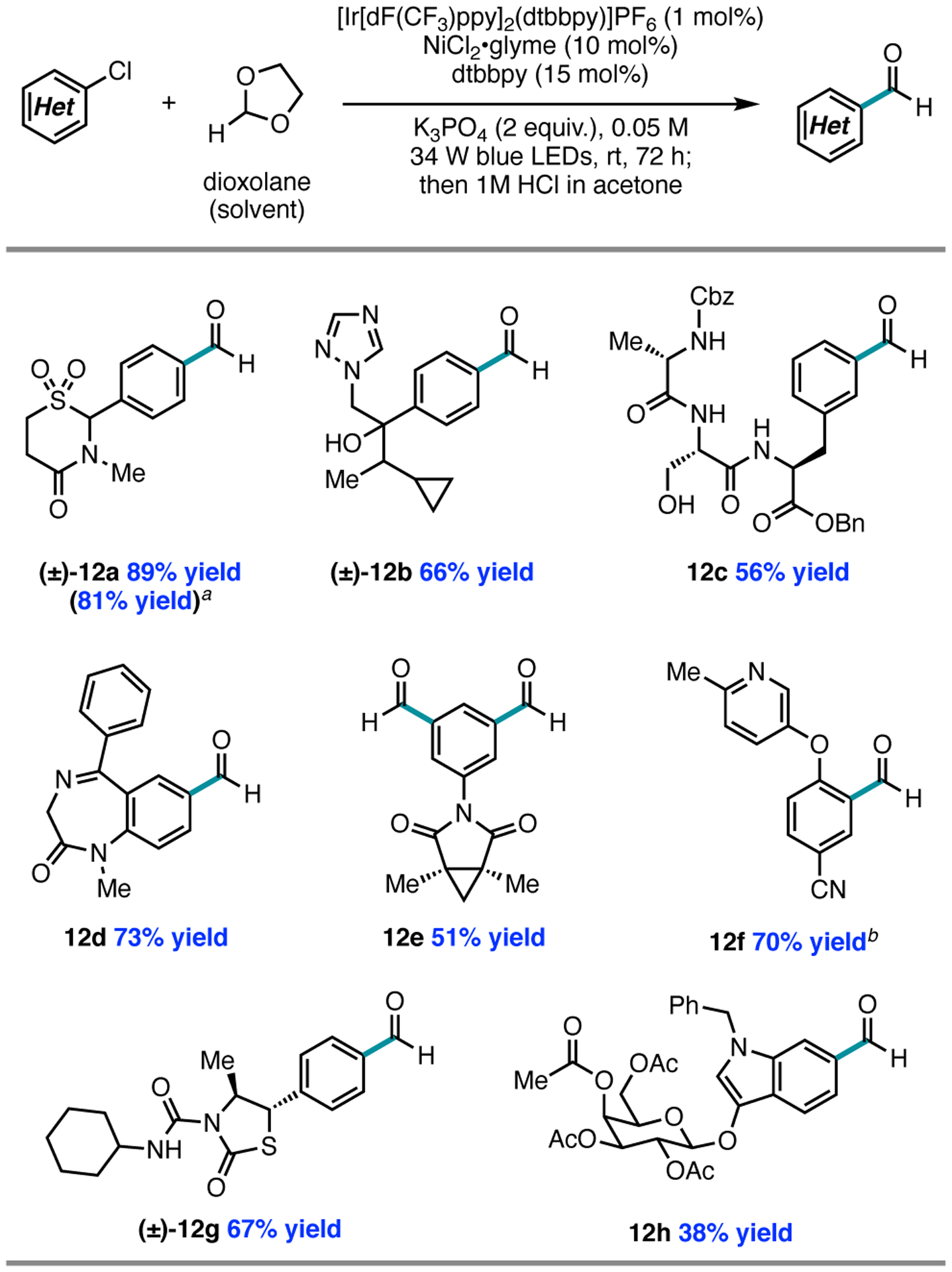
Representative substrate scope for the formylation of (hetero)aryl chlorides. a Yield using Schlenk technique. b Benchtop setup (gram scale).
We centered our scope evaluation on late-stage and biologically relevant (hetero)aryl chlorides, as well as abundant aryl chlorides that are not available as commercial aryl bromides, iodides, and aldehydes. A common limitation of photoredox reactions is their scalability, often due to issues of light penetration.24 We therefore evaluated the formylations on a gram-scale: a benchtop scale-up reaction provided 12f in 70% yield.
Methylation of (Hetero)aryl Chlorides
Benzaldehydes are valuable intermediates in large part because they enable access to esters, amines, alcohols, and toluenes via oxidation or reduction. Nevertheless, redox manipulation is not ideal from a step economy perspective and requires stoichiometric oxidants or reductants that often have an adverse impact on functional group tolerance in late-stage modification.25 Thus, we anticipated that direct installation of a single carbon substituent at different oxidation states would be attractive.
To accomplish this, we sought to extend chlorine photoelimination to a direct aryl esterification using trimethyl orthoformate as a methyl ester surrogate. Early experiments employing trimethyl orthoformate showed that the corresponding aryl methyl ester was accessible from 4´-chloroacetophenone in 8% yield (14), which we attributed to coupling with the tertiary radical of trimethyl orthoformate (Figure 6A). The benzylic ether product derived from coupling at the primary C–H bonds of trimethyl orthoformate (13) was also obtained in 27% yield. Most surprising and intriguing to us, however, was the formation of methylated product 15 in 38% yield, demonstrating that three different oxidation states were accessible by cross-coupling from a single carbon unit.2
Figure 6.
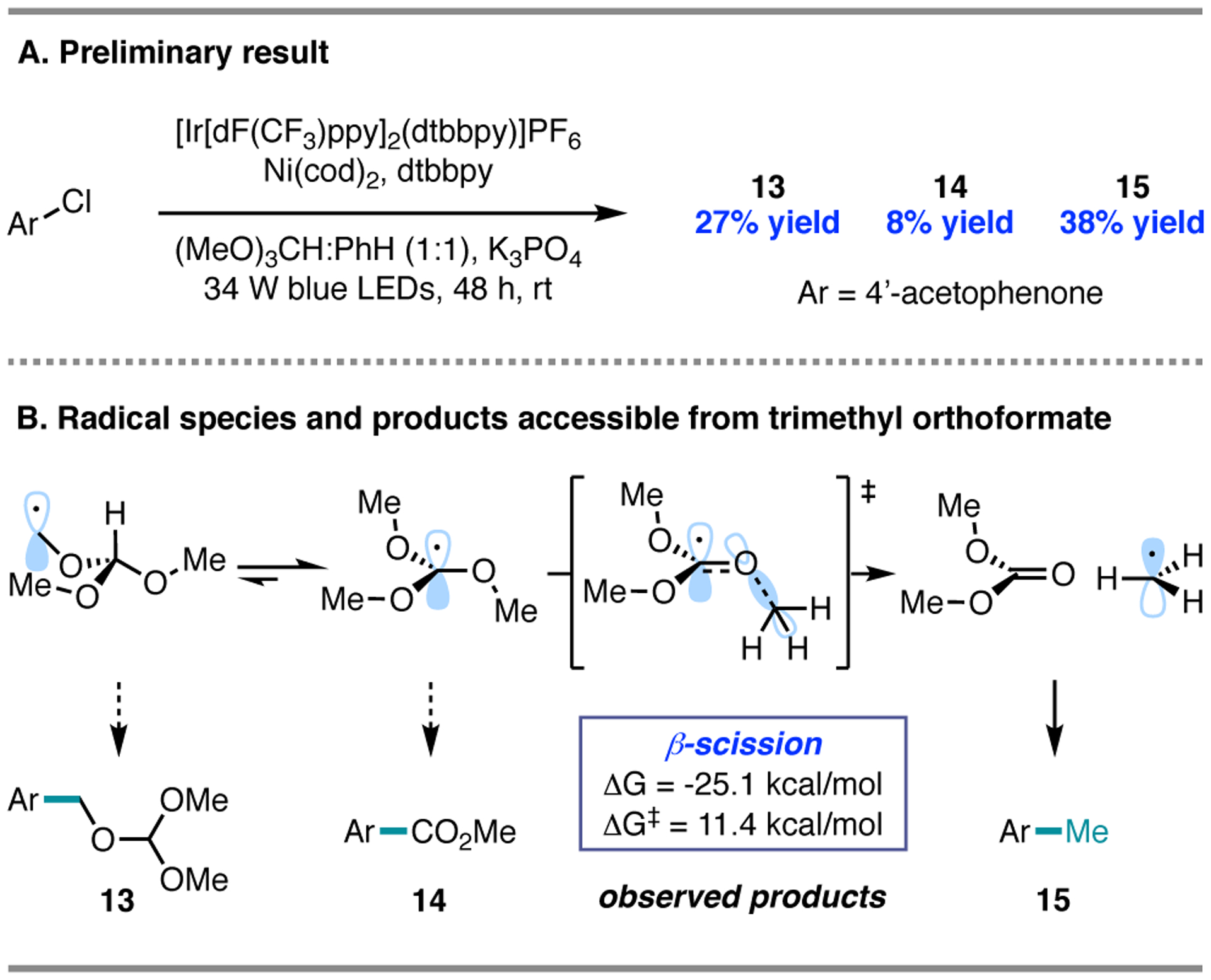
Design of a methylation reaction.
Formation of the methylated product was attributed to coupling with methyl radical, which we hypothesized was generated via β-scission of the tertiary radical of trimethyl orthoformate after C–H abstraction by chlorine radical. While β-scission is well-studied for the generation of aliphatic radicals from oxygen-centered radicals,26a–c there is significantly less precedent for β-scission from carbon-centered radicals.26d–e Our computational investigation suggested that β-scission of the tertiary radical of trimethyl orthoformate is kinetically feasible (ΔG‡ = 11.4 kcal/mol). Moreover, formation of dimethyl carbonate as a byproduct affords a thermodynamic driving force (ΔG = −25.1 kcal/mol) (Figure 6B).27 Indeed, reaction progress analysis and methyl radical trapping experiments supported the intermediacy of methyl radical and the concurrent generation of dimethyl carbonate. The computations also predicted preferential HAT at the tertiary C–H bond over the primary sites (ΔBDFE = −1.2 kcal/mol).
While our discovery of this methylation was serendipitous, we recognized that installation of a methyl substituent onto small molecules is an established and important strategy for rendering compounds with improved biological properties, a phenomenon so pervasive it has been called the “magic methyl effect.”28 However, most methylation methodologies require harsh reaction conditions or reactive electrophilic or nucleophilic methyl sources that are incompatible with late-stage functionalization of bioactive molecules.28a Recently, N-hydroxyphthalimide esters29a and methyl tosylate29b–c have been demonstrated to be competent methyl radical precursors in Ni cross-coupling with aryl iodides and bromides, respectively. Accordingly, these reactions require either preparation of the methyl radical source or employment of an electrophilic methylating reagent. In contrast, we were drawn to trimethyl orthoformate as an abundant, commercial, and functional group tolerant methyl source.
In optimizing the initial lead result for methylation, we arrived at the reaction conditions shown in Figure 7, which delivered methylation over alkoxymethylation in a ~4:1 ratio while minimizing formation of the ester product. An extensive scope of heteroaryl chlorides and late-stage compounds was established. The abbreviated scope shown in Figure 7 demonstrates the remarkable tolerance of radical methylation for functionalities that are incompatible with nucleophilic and electrophilic methyl sources. We were also able to extend this method to acyl chlorides as a mild method for the generation of ketones, esters, and tertiary amides.2
Figure 7.
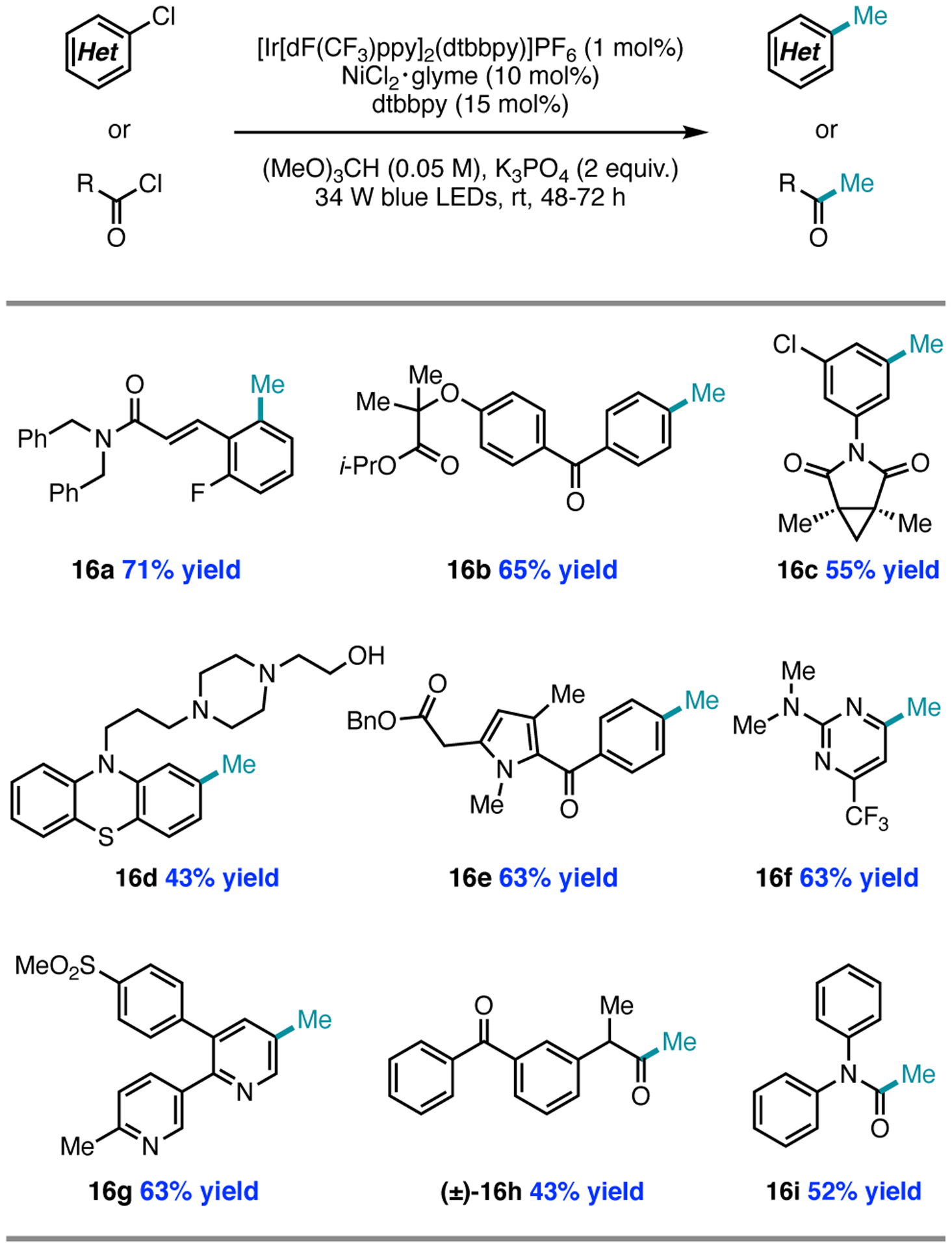
Representative substrate scope for the methylation of (hetero)aryl and acyl chlorides.
While the nickel/photoredox-catalyzed methylation reactions are high-yielding with electron-deficient (hetero)aryl chlorides, electron-rich substrates are unreactive; however, electron-rich aryl chlorides are competent reaction partners in the formylation and etherification methods. For example, para-phenoxy- and para-methoxy-substituted aryl chlorides underwent etherification or formylation respectively in 77% ((±)-11c) or 58% yield (17); in contrast, methylation of 4-chloroanisole proceeded in less than 2% yield (Figure 8A). This limitation, consistent with a more challenging oxidative addition to Ni(0), could be circumvented by employing aryl bromides (vide infra) as substrates with the addition of tetrabutylammonium chloride (TBACl) as an exogenous chloride source for halide exchange, providing 18 in 56% yield (Figure 8B). While we do not yet understand the divergence in reactivity between the methods, we found that the electronic bias between methods could be used advantageously: for example, procymidone, containing two equivalent aryl chlorides, underwent formylation in both positions to give product 12e in Figure 5. Under the methylation protocol, only one aryl chloride underwent functionalization to provide 16c in Figure 7, thereby preserving an additional reactive handle for further functionalization.
Figure 8.
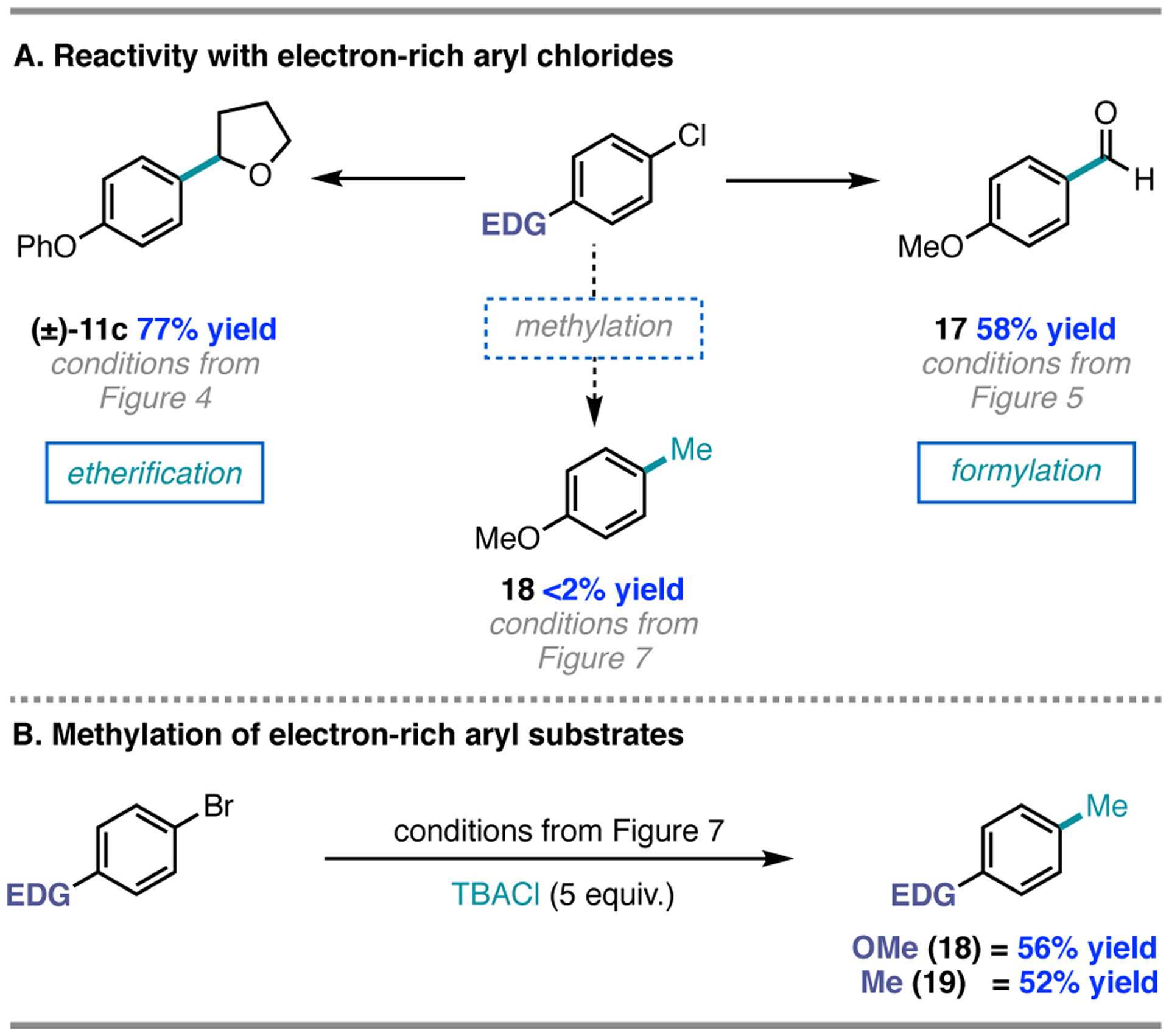
A. Comparison of electronic tolerance across photoelimination methodologies. B. Methylation of electron-rich aryl bromides via halide exchange at Ni.
Functionalization of Unactivated C–H Bonds
The use of ethereal solvents in aryl etherification, formylation, and methylation reactions provided a handle for reactivity and selectivity via the reduced bond strengths of C–H bonds adjacent to oxygen atoms. However, use of chlorine radical as a hydrogen atom abstractor should permit direct functionalization of entirely unactivated C(sp3)–H bonds, a longstanding challenge in C–H functionalization. Most examples of C(sp3)–H activation for C–C bond formation require substrates with co-ordinating directing groups or their employment in a large excess, precluding their application in synthesis at both an early and a late stage.30 Thus, the direct and modular functionalization of unactivated alkanes to form C–C bonds represents an unmet challenge in synthesis.31
To address this challenge, we aimed to apply chlorine photoelimination for the cross-coupling of 4´-chloroacetophenone with cyclohexane (BDFE = 91 kcal/mol); however, performing the reaction under the conditions shown in Figure 4 with 10 equivalents of cyclohexane delivered the desired product in only 41% yield.32 We recognized that the limited success of our initial attempts could be attributed to the C–H bond strengths and polarities of the starting materials and products. In arylation reactions, the bond strengths of the benzylic C(sp3)–H bonds in the products are considerably weaker (BDFE ~ 80 kcal/mol) than the bond strengths of the unactivated C(sp3)–H bonds of the starting materials (BDFE = 91–95 kcal/mol). This could lead to unproductive consumption of chlorine radical, particularly when C(sp3)–H substrates are employed as limiting reagents. This problem was anticipated to be particularly acute for the photoelimination strategy since it provides only one chlorine radical per substrate activation in contrast to methods where HAT catalysts are continuously regenerated.
To address this issue, we sought to exploit C–H bond polarity effects.33 We anticipated that installation of an electron-withdrawing group via C–C cross-coupling would reduce the rate of product α C–H bond abstraction by chlorine radical, an electrophilic radical, even though the product would still possess a weaker C–H bond than the starting material. Thus, we chose the introduction of an ester from chloroformates as a more promising means of achieving C–C coupling from unactivated alkanes (Figure 9).3 Such a strategy would not only deactivate the product towards overfunctionalization, but would also bestow a once unreactive alkane with the great expanse of reactivity associated with the carbonyl group.
Figure 9.

Designing a system for functionalizing unactivated C(sp3)–H substrates.
Upon optimization, we found that phenyl chloroformate underwent reaction with only a small excess of cyclohexane (3 equiv.), furnishing the esterified product in 66% yield under the conditions shown in Figure 10.34 Scope studies revealed that a variety of substrates containing unactivated C–H bonds underwent functionalization (Figure 10). For example, tetramethylsilane, which contains one of the strongest C(sp3)–H bonds (BDFE = 96 kcal/mol), underwent esterification in 51% yield (20d) (Figure 10). Acyl chlorides could also be used as substrates to provide ketone products upon coupling with aliphatic reaction partners, such as cyclooctane (20e).35 For substrates containing multiple chemically distinct C(sp3)–H bonds, preferential esterification occurred at benzylic (20f-20g) or α-oxy or α-amino positions (20h), consistent with abstraction of weaker and more hydridic C–H bonds. More complex, unactivated substrates underwent functionalization at a variety of positions. The regioselectivity observed generally was consistent with functionalization at positions with the most hydridic C–H bonds that would also result in the most stabilized radical species (tertiary > secondary > primary C–H bonds). Site-selectivity could additionally be predicted with an Evans-Polanyi plot (vide infra).33,36 In the case of product 20i, functionalization occurred only in one position, consistent with chlorine radical abstraction at the most electron-rich, sterically accessible site. Esterification of late-stage molecules, such as sclareolide (20k), proceeded to give a mixture of regioisomers, again with a preference for the most electron-rich and sterically accessible C–H bonds. Additionally, this method for direct C–C bond formation represents one of the only methods by which a quaternary carbon center can be installed with Ni/photoredox catalysis (20j).20b,29b,37
Figure 10.

Representative scope for the acylation of unactivated C–H bonds. a 6 equiv. of C–H substrate used. b Yield reported post-desilylation.
MECHANISTIC INVESTIGATIONS
Since our initial report, a variety of methods using halogen photoelimination in cross-coupling have been reported.11 However, different mechanistic proposals have been put forth to account for the generation of halogen radicals, namely between photoelimination from excited Ni(II) or excited Ni(III). Our original mechanistic proposal invoked a process similar to Nocera’s14 in the light-induced generation of chlorine radicals from Ni(III) complexes (Figure 3). Aligned with our proposal, a C(sp3)–C(sp3) cross-coupling of alkyl bromides with ethers was recently disclosed by König and coworkers wherein photoelimination of bromine radicals is proposed to proceed from a Ni(III) species.38 Wu has also invoked oxidation of Ni(II) to Ni(III) by a photocatalyst, followed by photolysis to generate chlorine radicals.39 Photoluminescence quenching studies and UV irradiation control reactions supported excitation of Ni(III) for chlorine radical elimination.
Alternatively, other researchers have proposed an energy transfer mechanism to access excited Ni(II) for halogen radical formation. Molander and coworkers leveraged bromine photoelimination in the cross-coupling of aryl bromides with ethers, which was proposed to proceed via excited state Ni(II)–Br homolysis.18 The researchers proposed energy transfer between the photocatalyst and the Ni catalyst as the operative mechanism on the basis of their observation that photocatalysts which possessed high excited state oxidation potentials but lower triplet energies failed to deliver cross-coupled product. Additionally, in the cross-coupling of allylic C(sp3)–H bonds with aryl and vinyl bromides, Rueping proposed bromine radical generation from excited Ni(II) species using an acridinium photocatalyst.40 The researchers deemed oxidation to Ni(III) prior to halogen elimination unlikely because, after oxidation, the reduced state of the photocatalyst may not be sufficiently reducing to regenerate the Ni(0) catalyst.
Thus, our mechanistic studies on chlorine photoelimination have largely focused on discerning between these two possibilities in the synthetic methods that we have reported: (1) elimination from excited Ni(II) via energy transfer or (2) elimination from excited Ni(III) via light-induced bond homolysis (Figure 11).
Figure 11.
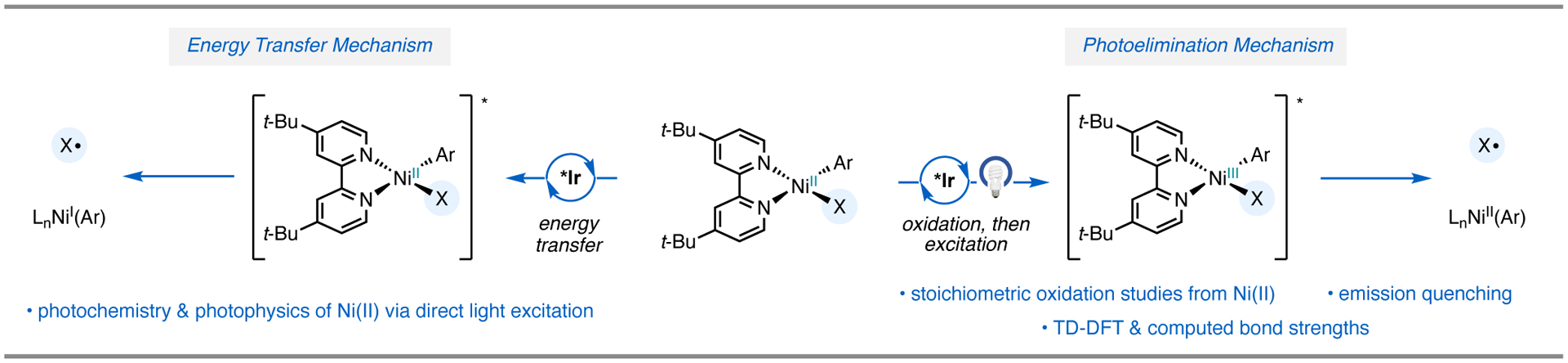
Proposed mechanisms for halogen photoelimination.
Photophysical, Photochemical, and Computational Studies of Ni(II) Complexes
Despite the tremendous advances that have been made in synthesis with Ni/photoredox catalysis, little is known about the photophysics and photochemistry of the Ni(II) complexes relevant to these processes. Thus, we prepared a series of aryl halide complexes and investigated their interaction with visible light using steady state and ultrafast spectroscopy.4 Upon excitation, these complexes were found to relax into a long-lived 3d-d excited state (Figure 12A). Although this transition features bond weakening of the Ni–ligand bonds, we found that this 3d-d state is too low in energy (12 kcal/mol) above the ground state to enable elimination of a chlorine or bromine radical at temperatures relevant to the synthetic reactions (DFT at the M06/TZVP//B3LYP/TZVP level of theory).
Figure 12.
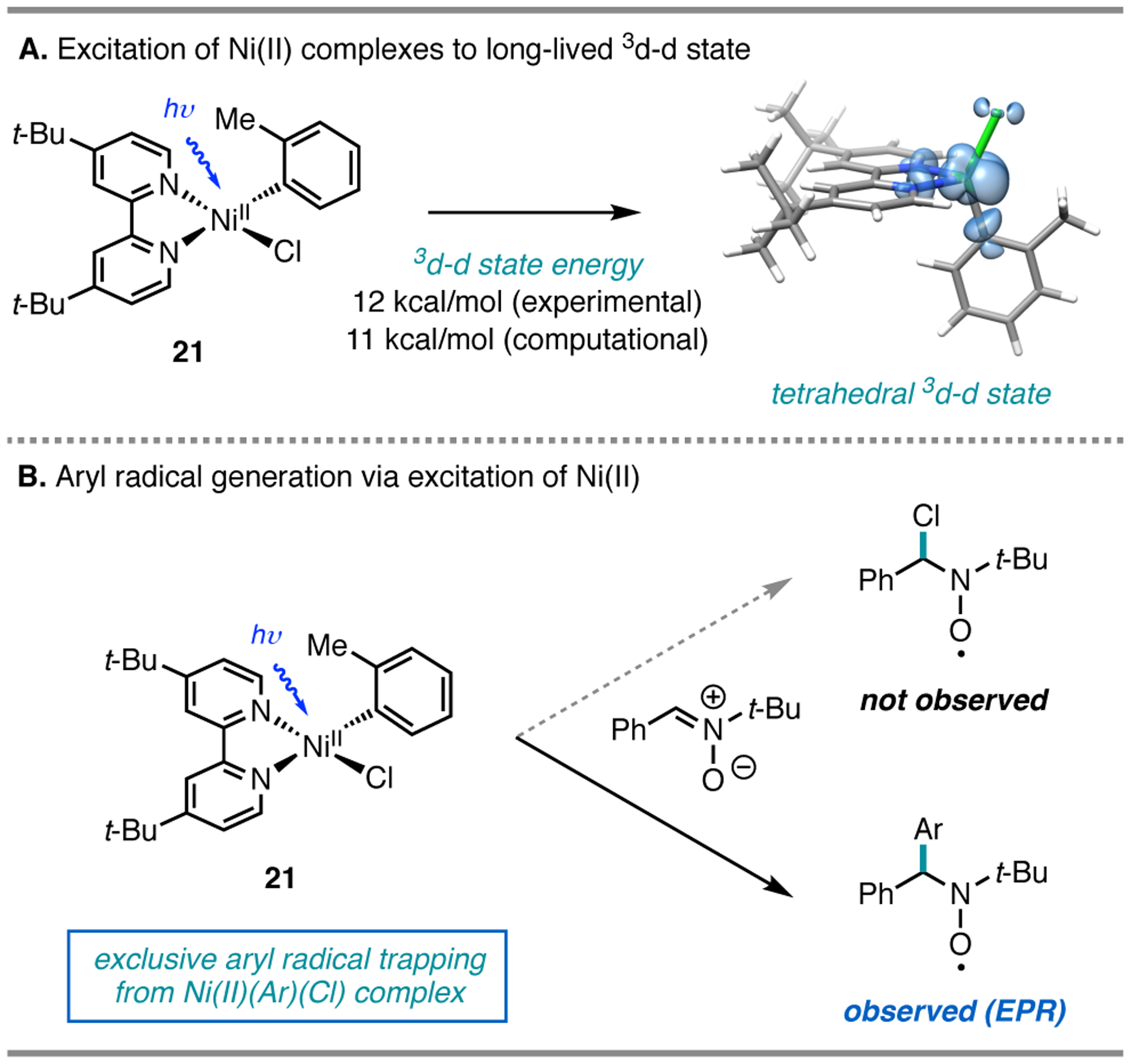
A. Irradiation of 21 gives rise to a 3d-d excited state. B. Aryl radical capture in the presence of a radical trap.
To further probe the feasibility of chlorine photoelimination at Ni(II), we turned to a computational investigation. We found that while Ni(II)–Cl has a BDFE of 77 kcal/mol, oxidation of Ni(II) induces significant bond weakening (Ni(III)–Cl BDFE = 47 kcal/mol), where now the Ni(III)–Cl bond is sufficiently weak for visible light-induced homolysis. The bond weakening is consistent across different carbon substituents: in the oxidation of (dtbbpy)NiII(CO2Ph)(Cl) to [(dtbbpy)NiIII(CO2Ph)(Cl)]+, the computed Ni–Cl BDFE is reduced from 65 to 37 kcal/mol. The significantly stronger Ni(II)–X bond strengths provide evidence that bond homolysis for halogen radical generation is unlikely, at least from the lowest energy Ni(II) excited state.
Ni(II) aryl chloride complexes can exist as stable and isolable species, providing a platform to study their photochemistry (Figure 12B). Irradiation of isolated (dtbbpy)NiII(o-Tol)(Cl) (21) in the presence of a radical trap41 provided no evidence of chlorine radical generation by EPR, although this does not necessarily reflect the lack of chlorine radical formation.42 Instead, irradiation of this isolated Ni(II) aryl chloride complex resulted in exclusive trapping of aryl radicals in low conversion (Ni(II)–Ar BDFE = 35 kcal/mol).43 As such, we expect that other pathways, specifically oxidation of Ni by the photocatalyst, could be outcompeting this step under catalytically relevant conditions.
Contrasting Photochemistry at Ni(II) with Chlorine Photoelimination from Ni(III)
Instead of chlorine radical photoelimination from excited Ni(II), we propose that the excited state of the Ir photocatalyst oxidizes the Ni(II) aryl halide species formed by oxidative addition. Because 21 was found to be competent in the cross-coupling with THF in the presence of light and photocatalyst, we chose to examine its properties to distinguish between energy transfer and oxidation pathways. Cyclic voltammetry of this complex indicated that *Ir(III) should be a suitable oxidant for accessing Ni(III) (Ep = 0.85 V vs SCE in THF). Importantly, Stern-Volmer quenching studies revealed that the Ni(II) complex is the most likely species responsible for quenching the excited state of the photocatalyst. These data, in addition to the relative stability of the isolated Ni(II) complexes under visible light irradiation, provide evidence for the generation of Ni(III) by photoinduced electron transfer from the Ir photocatalyst, consistent with the Ni(III) photoelimination proposal.
We then sought to explore the feasibility of photoelimination from excited Ni(III) complexes computationally. Using time-dependent density functional theory (TD-DFT), we found the calculated absorption spectrum for [(dtbbpy)NiIII(Ph)(Cl)]+ contains high-energy features that are characterized by significant Ni–Cl σ→σ* character. Along with the greatly weakened Ni(III)–Cl BDFE (47 kcal/mol), these states are expected to be dissociative.
To gain experimental data for photoelimination from Ni(III), we pursued stoichiometric studies. The proposed catalytic reaction requires two photons to enable chlorine radical generation: one for photocatalyst excitation to enable oxidation of Ni(II) to Ni(III), and a second photon for homolytic cleavage of the Ni(III)–Cl bond. Since the first of these steps could be accomplished using a chemical oxidant without light, the necessity of oxidation and irradiation could be interrogated separately. In a reaction employing stoichiometric (dtbbpy)NiII(Ar)(Cl) (Ar = 4-methylbiphenyl) (22) and single-electron oxidant [TBPA]SbCl6 (tris(4-bromophenyl)ammoniumyl hexachloroantimonate, E = 1.16 V vs SCE in dichloromethane) under visible light irradiation, cross-coupled product 24 was obtained in 28% yield (Figure 13). When the reaction was conducted with oxidant but without irradiation, a requirement for photoelimination from Ni(III), no cross-coupling was observed. Additionally, when reactions were performed with light but without oxidant, formation of 24 was again not observed. These experiments demonstrate that, from the isolable Ni(II) aryl chloride complex, both oxidant and light are necessary for chlorine photoelimination.
Figure 13.
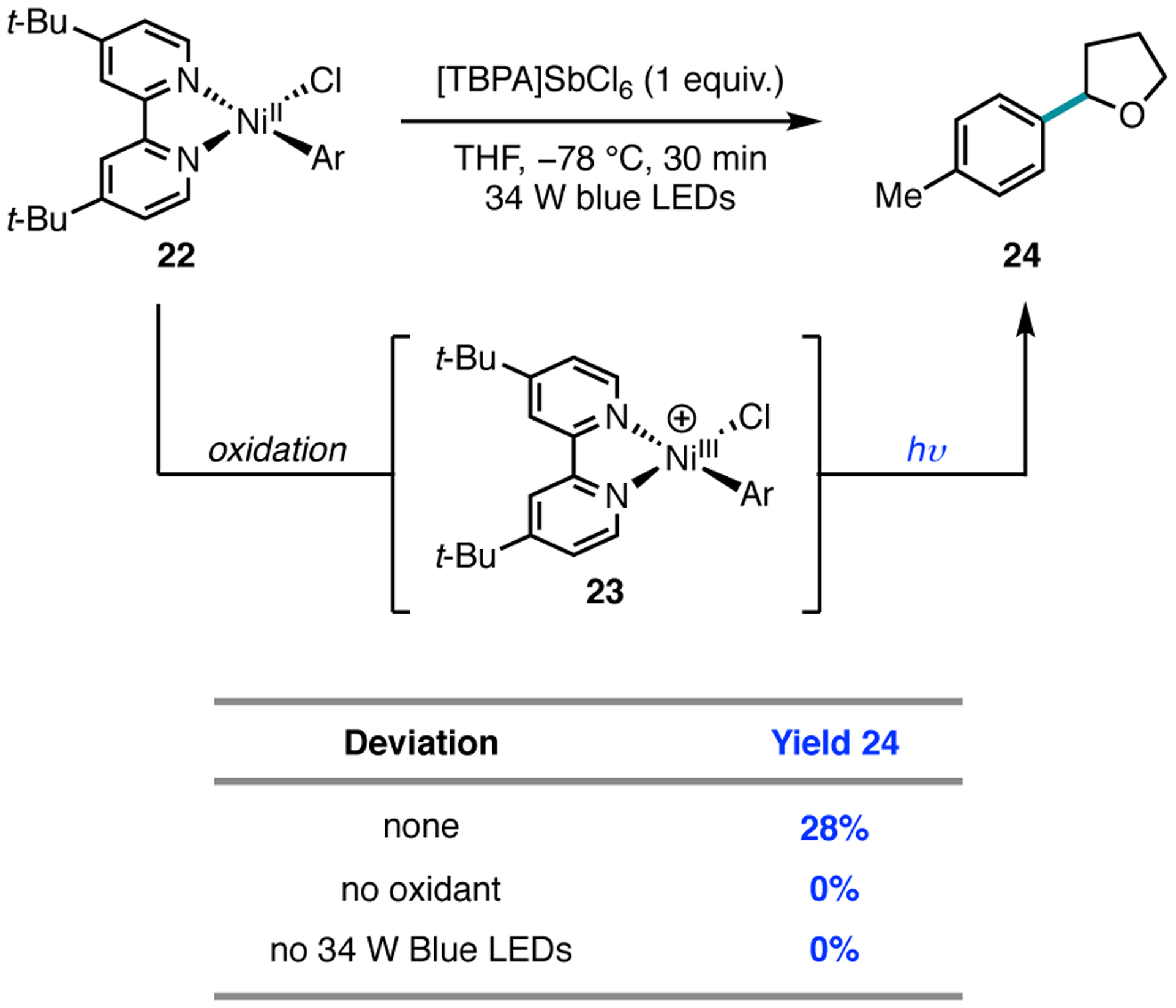
Stoichiometric studies from catalytically competent Ni(II) complex.
Evidence for Chlorine Radical in Hydrogen Atom Transfer
According to the mechanistic proposal, chlorine radicals mediate HAT with C(sp3)–H substrates. However, in light of our observation of aryl radical generation upon photolysis of Ni(II) complex 21, we also considered the possibility that aryl radicals could mediate HAT. To probe for their intermediacy in the catalytic system, we examined the coupling of aryl halides with a deuterium-labeled substrate (THF-d8). Experiments using aryl chloride 25-Cl provided strong evidence against aryl radical generation as minimal deuterodehalogenation was observed (Table 1, Entry 1). Moreover, only 50% yield of the cross-coupled product 27 could be formed if aryl radical was mediating HAT. The observation of greater than 50% yield in the cross-coupling (68% yield) with 25-Cl provides further evidence against aryl radicals acting as the operative HAT agents. Although the Ni(III)–Ar BDFE is likely sufficiently weak to be cleaved with visible light, these findings support that aryl radical generation from Ni(III) does not take place, possibly because Ni complex geometry and orbital considerations disfavor bond homolysis.
Table 1.
Deuterium labeling experiments across aryl halides. Ar = 2-(4-halophenyl)-6-methyl-pyridine.
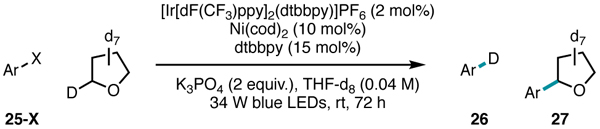
| |||
|---|---|---|---|
| Entry | 25-X | Yield 26 | Yield 27 |
| 1 | Cl | 3% | 68% |
| 2 | Br | 2% | 5% |
| 3 | I | 66% | 4% |
If aryl radicals were operative in HAT, aryl bromides and iodides should perform comparably to aryl chlorides since the former substrates undergo facile oxidative addition to Ni. However, when using aryl bromides as cross-coupling partners, only a trace amount of cross-coupled product was observed (Table 1, Entry 2). Reactions performed with aryl iodides also delivered trace yield of the cross-coupled product, but with this substrate class deuterodehalogenation proceeded in 66% yield (Table 1, Entry 3). This result suggests that, when using aryl iodides, aryl radicals are generated and mediate deuterium abstraction of THF-d8. The generation of aryl radicals with aryl iodides could be attributed to the photochemistry of Ni(II), but most likely proceeds via oxidation addition and radical cage escape as described by Kochi.44 Therefore, hydrogen atom abstraction by aryl radicals is significant only with aryl iodides and is consistent with chlorine photoelimination in the mechanistic proposal.
The fact that 25-Br and 25-I were unreactive is consistent with the role of halogen radical as the HAT agent: the weak H–I bond (BDE = 71 kcal/mol) renders an iodine radical incapable of abstracting even an activated C(sp3)–H bond (Table 2, Entry 3). However, reactivity could be restored when using aryl iodides and bromides as substrates via the addition of exogenous chloride for halide exchange (Table 2, Entry 4).45
Table 2.
Halide additive studies.
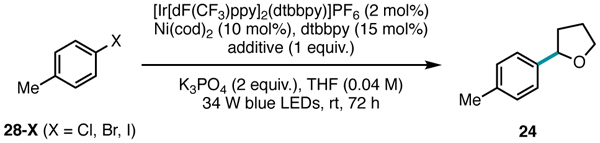
| |||
|---|---|---|---|
| Entry | 28-X | Additive | Yield 24 |
| 1 | Cl | none | 68% |
| 2 | Br | none | 10% |
| 3 | I | none | 5% |
| 4 | I | TBACl | 51% |
| 5 | I | TBABr | 37% |
| 6 | I | TBAI | 6% |
While reaction yields are lower when using aryl bromides than the analogous aryl chlorides, we observed that selectivity in C(sp3)–H abstraction is higher when employing these substrates, consistent with the Hammond postulate wherein the later transition state by bromine radical abstraction (H–Br BDE = 88 kcal/mol versus H–Cl BDE = 102 kcal/mol) results in higher selectivity for the thermodynamically favored alkyl radical product. For example, reactions conducted in 1,3-dioxolane delivered two regioisomeric products, generally in a 9:1 ratio between the desired and undesired isomers (Table 3). Selectivity in C–H abstraction was two times higher by employing aryl bromides as substrates.
Table 3.
Arylation of 1,3-dioxolane with aryl chlorides and bromides.
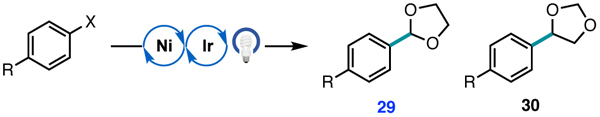
| ||||
|---|---|---|---|---|
| Entry | R | X | Yield 29 | 29 : 30 |
| 1 | CN | Cl | 73% | 7.1 : 1 |
| 2 | CN | Br | 52% | 15.8 : 1 |
| 3 | Ph | Cl | 65% | 9.0 : 1 |
| 4 | Ph | Br | 36% | 18.1 : 1 |
The involvement of chlorine radicals in hydrogen atom transfer was also indirectly examined via an Evans-Polanyi analysis (Figure 14A).33,36 While hydrogen atom abstraction can occur in a variety of positions in unactivated alkanes, functionalization of alkanes with chemically distinct C–H bonds revealed regioselectivity consistent with the influence of BDFE on the rate of C–H abstraction. For example, esterification of 2-methylbutane occurred in a 53% combined yield with preference for the tertiary C–H bond (α:β:γ:δ 1.3:9:6.3:1). An Evans-Polanyi plot for 2-methylbutane exhibited a linear correlation with an α value of 0.44, which is in agreement with tabulated α values for hydrogen atom abstraction by chlorine radical (αCl = 0.45).36
Figure 14.
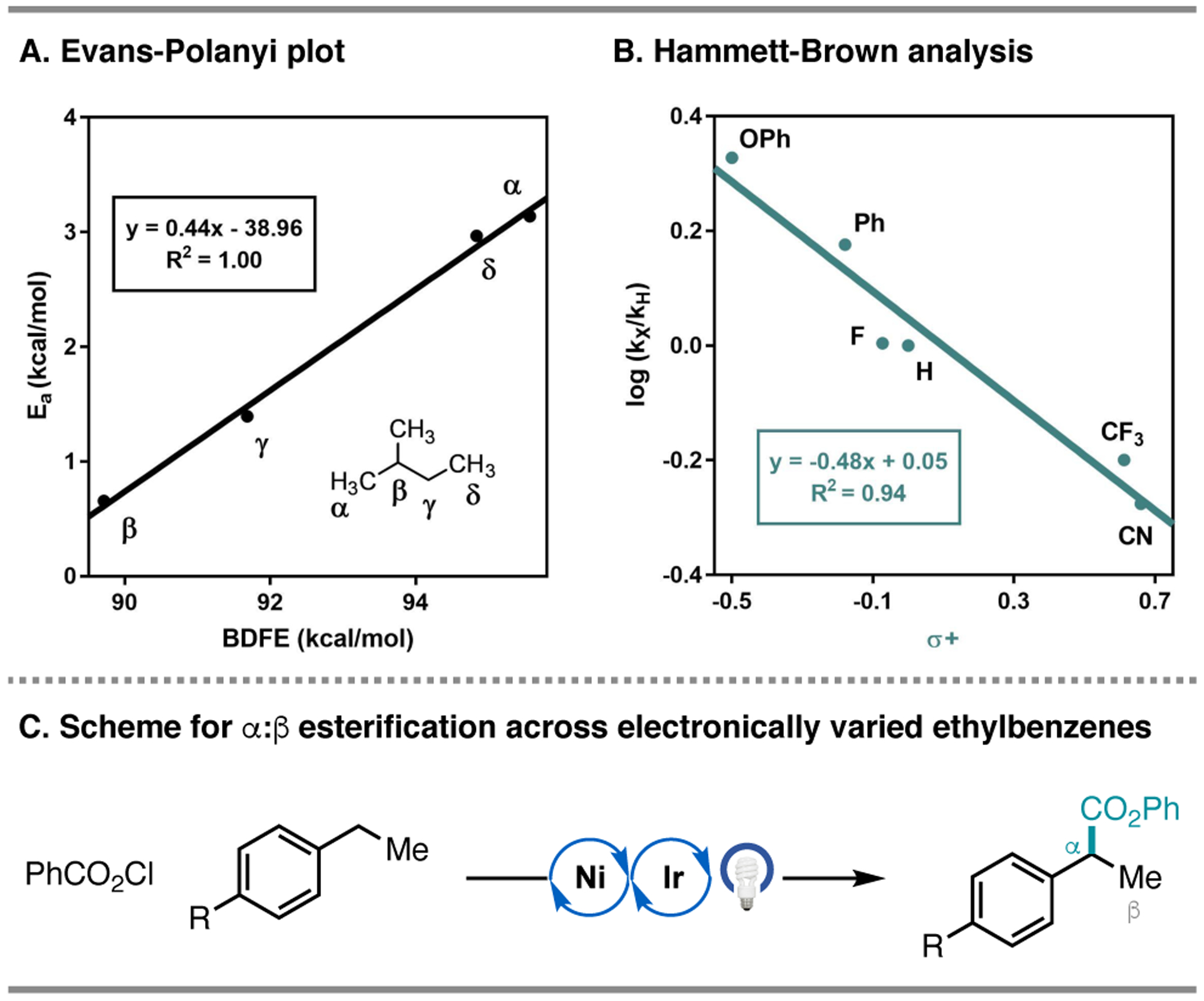
A. Evans-Polanyi for the reaction of phenyl chloroformate with 2-methylbutane. B. Hammett-Brown analysis of the relative rate of benzylic functionalization of ethylbenzenes. C. Reaction scheme for Hammett-Brown analysis.
Furthermore, selectivity in C–H abstraction by chlorine radical demonstrated a clear trend in polarity for the most electron-rich site. This influence was directly studied with a series of para-substituted ethylbenzenes, wherein the ratio of benzylic to methyl functionalization decreases with electron-withdrawing substituents (Figure 14C). Additionally, Hammett-Brown analysis revealed that electron-rich ethylbenzenes undergo benzylic C–C bond formation faster than electron-deficient substrates (Figure 14B). The small ρ value determined from this series is consistent with a highly exothermic abstraction step and in agreement with free-radical chlorination of toluenes (ρ values ranging from −0.5 to −1.0).46
The requirement for both light and oxidant in our system, in addition to trends observed via Evans-Polanyi and Hammett-Brown analyses, lend support to the generation of chlorine radicals for hydrogen atom transfer.47 Key experimental studies, namely stoichiometric oxidation from Ni(II), Stern-Volmer emission quenching experiments, studies on the photophysics of excited Ni(II) complexes, and halide identity studies, support the photoelimination of chlorine radicals from excited Ni(III). Computational studies, including TD-DFT and computed bond strengths, lend additional support for the feasibility of chlorine photoelimination. Notably, a recent report demonstrated that DFT calculations are inadequate for describing the electronic structure of certain (bpy)Ni systems, with multireference calculations being necessary for this purpose.43 These theoretical findings emphasize the need for experimental mechanistic studies, including those presented here for chlorine photoelimination, in elucidating reaction mechanisms in Ni/photoredox-catalyzed processes.
CONCLUSION AND OUTLOOK
We have shown that the photocatalytic generation of halogen radicals from Ni complexes has value in achieving otherwise challenging C(sp3)–H bond functionalization reactions. By invoking chlorine radicals in hydrogen atom transfer, virtually any C(sp3)–H bond can be activated and functionalized under exceptionally mild conditions, as has been demonstrated in etherification, formylation, methylation, and acylation reactions. Our mechanistic understanding of this process leaves several potential extensions of these methods. Ligand design could facilitate catalyst-controlled site-selective C–H cross-couplings. Stabilization of chlorine radicals by aryl groups on ligands, as demonstrated by Nocera, indicates one such promising route.14 Our studies thus far have focused on the photoelimination of chlorine radicals and their use as hydrogen atom transfer agents; the photoelimination of other reactive ligands could have broad implications for catalysis and cross-coupling. For example, the generation of N- or O-centered radicals could provide an opportunity to use alcohol- or amine-based coupling partners.
Considering the ability of a variety of first-through third-row metal complexes to interact with visible light and photoredox catalysts, it may become feasible to use other metals for photoelimination processes in cross-coupling. Very recently, Castellano has demonstrated that irradiation of phenanthroline-ligated Cu(II) species with blue light results in Cu–Cl bond homolysis to generate chlorine radicals.48 This study, in addition to our studies on Ni, highlights that the photochemistry of cross-coupling catalysts themselves must not be ignored in metallaphotoredox systems, but should serve as a design principle for the development of new catalytic reactions.
ACKNOWLEDGMENT
We are grateful to the past and present members of our research group for their contributions to this work, and to our collaborators from Celgene, the Scholes laboratory (Princeton), and the Castellano laboratory (NC State). This material is based on work supported by the National Science Foundation Graduate Research Fellowship Program under Grant Number DGE-1656466 (to S.K.K.). A.G.D. gratefully acknowledges Celgene, NIGMS (R35 GM126986), Princeton Innovation Fund, and BioLEC, an Energy Frontier Research Center (U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award no. DE-SC0019370) for funding. We thank Jesus Martinez Alvarado and Stephen Ting for helpful discussions.
Biographies
Stavros Kariofillis obtained a B.S. in Biochemistry from Lafayette College in 2013, where he worked with Professor Roxy Swails on developing new water-soluble palladium catalysts for cross-coupling. As an undergraduate student, he also worked with Professor Melanie Sanford at the University of Michigan studying the organometallic properties and reactivity of copper(I)-difluoromethyl complexes. He is currently an NSF Predoctoral Fellow in Professor Abigail Doyle’s group at Princeton University, where his research focuses on methodology development within Ni/photoredox catalysis.
Abigail Doyle is the A. Barton Hepburn Professor of Chemistry in the Chemistry Department at Princeton University. She obtained her Ph.D. in catalysis and physical organic chemistry at Harvard University in 2008 under the direction of Prof. Eric Jacobsen after receiving her A.B. and A.M. in chemistry and chemical biology from Harvard in 2002. She joined the faculty at Princeton University in 2008 and is currently a co-PI for the NSF CCI Center for Computer Assisted Synthesis (C-CAS) and the DOE EFRC Bioinspired Light-Escalated Chemistry (BioLEC). The Doyle laboratory is interested in developing new approaches to chemical synthesis and catalysis, with a focus on Ni-catalyzed cross-coupling and nucleophilic fluorination methodology.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Shields BJ; Doyle AG Direct C(sp3)–H Cross Coupling Enabled by Catalytic Generation of Chlorine Radicals. J. Am. Chem. Soc 2016, 138, 12719–12722. [DOI] [PMC free article] [PubMed] [Google Scholar]; Our first paper in the area of chlorine photoelimination demonstrated that this activation mode could be used in a synthetic context, here towards an arylation of ethers. Mechanistic studies supported that chlorine photoelimination occurs from excited Ni(III) complexes.
- (2).Kariofillis SK; Shields BJ; Tekle-Smith MA; Zacuto MJ; Doyle AG Nickel/Photoredox-Catalyzed Methylation of (Hetero)aryl Chlorides Using Trimethyl Orthoformate as a Methyl Radical Source. J. Am. Chem. Soc 2020, 142, 7683–7689. [DOI] [PMC free article] [PubMed] [Google Scholar]; We highlight the synthetic utility of chlorine photoelimination via the methylation of (hetero)aryl chlorides, illustrating reaction compatibility with sensitive functional groups and late-stage compounds by using trimethyl orthoformate as a source of methyl radical.
- (3).Ackerman LKG; Martinez Alvarado JI; Doyle AG Direct C–C Bond Formation from Alkanes Using Ni-Photoredox Catalysis. J. Am. Chem. Soc 2018, 140, 14059–14063. [DOI] [PMC free article] [PubMed] [Google Scholar]; Catalytic C–C bond formation from strong C(sp3)–H bonds is a highly desired yet outstanding synthetic challenge. Here we demonstrate that chlorine photoelimination can serve as a platform for functionalizing unactivated C(sp3)–H bonds via an acylation of hydrocarbon substrates.
- (4).Ting SI; Garakyaraghi S; Taliaferro CM; Shields BJ; Scholes GD; Castellano FN; Doyle AG 3d-d Excited States of Ni(II) Complexes Relevant to Photoredox Catalysis: Spectroscopic Identification and Mechanistic Implications. J. Am. Chem. Soc 2020, 142, 5800–5810. [DOI] [PubMed] [Google Scholar]; We studied the photophysical and photochemical properties of Ni(II) complexes, revealing that upon initial excitation, an MLCT state is generated that decays to a long-lived 3d-d state. This study provides insight into the behavior of Ni complexes in photoredox reactions.
- (5).(a) Shaw MH; Twilton J; MacMillan DWC Photoredox Catalysis in Organic Chemistry. J. Org. Chem 2016, 81, 6898–6926. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Romero NA; Nicewicz DA Organic Photoredox Catalysis. Chem. Rev 2016, 116, 10075–10166. [DOI] [PubMed] [Google Scholar]; (c) Narayanam JMR; Stephenson CRJ Visible Light Photoredox Catalysis: Applications in Organic Synthesis. Chem. Soc. Rev 2011, 40, 102–113. [DOI] [PubMed] [Google Scholar]
- (6).Energy transfer may also be operative:; Arias-Rotondo DM; McCusker JK The Photophysics of Photoredox Catalysis: A Roadmap for Catalyst Design. Chem. Soc. Rev 2016, 45, 5803–5820. [DOI] [PubMed] [Google Scholar]
- (7).(a) Huang C-Y; Doyle AG Nickel-Catalyzed Negishi Alkylations of Styrenyl Aziridines. J. Am. Chem. Soc 2012, 134, 9541–9544. [DOI] [PubMed] [Google Scholar]; (b) Arendt KM; Doyle AG Dialkyl Ether Formation by Nickel-Catalyzed Cross-Coupling of Acetals and Aryl Iodides. Angew. Chem. Int. Ed 2015, 54, 9876–9880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Zuo Z; Ahneman DT; Chu L; Terrett JA; Doyle AG; MacMillan DWC Merging Photoredox Catalysis with Nickel Catalysis: Coupling of α-Carboxyl sp3-Carbons with Aryl Halides. Science 2014, 345, 437–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Ahneman DT; Doyle AG C-H Functionalization of Amines with Aryl Halides by Nickel-Photoredox Catalysis. Chem. Sci 2016, 7, 7002–7006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Tellis JC; Primer DN; Molander GA Single-Electron Transmetalation in Organoboron Cross-Coupling by Photoredox/Nickel Dual Catalysis. Science 2014, 345, 433–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).(a) Twilton J; Le C; Zhang P; Shaw MH; Evans RW; MacMillan DWC The Merger of Transition Metal and Photocatalysis. Nat. Rev. Chem 2017, 1, 0052. [Google Scholar]; (b) Milligan JA; Phelan JP; Badir SO; Molander GA Alkyl Carbon-Carbon Bond Formation by Nickel/Photoredox Cross-Coupling. Angew. Chem. Int. Ed 2019, 58, 6152–6163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Geoffroy GL; Wrighton MS Organometallic Photochemistry. Academic Press, New York, 1979. [Google Scholar]
- (13).Arndtsen BA; Bergman RG; Mobley TA; Peterson TH Synthetic Intermolecular Carbon–Hydrogen Bond Activation by Synthetic Metal Complexes in Homogeneous Solution. Acc. Chem. Res 1995, 28, 154–162. [Google Scholar]
- (14).(a) Hwang SJ; Anderson BL; Powers DC; Maher AG; Hadt RG; Nocera DG Halogen Photoelimination from Monomeric Ni(III) Complexes Enabled by the Secondary Coordination Sphere. Organometallics 2015, 34, 4766–4774. [Google Scholar]; (b) Hwang SJ; Powers DC; Maher AG; Anderson BL; Hadt RG; Zheng S-L; Chen Y-S; Nocera DG Trap-Free Halogen Photoelimination from Mononuclear Ni(III) Complexes. J. Am. Chem. Soc 2015, 137, 6472–6475. [DOI] [PubMed] [Google Scholar]
- (15).Liu W; Ma Y; Yin Y; Zhao Y Anodic Cyanation of 1-Arylpyrrolidines. Bull. Chem. Soc. Jpn 2006, 79, 577–579. [Google Scholar]
- (16).Roth HG; Romera NA; Nicewicz DA Experimental and Calculated Electrochemical Potentials of Common Organic Molecules for Applications to Single-Electron Redox Chemistry. Synlett 2016, 27, 714–723. [Google Scholar]
- (17).Newhouse T; Baran PS If C–H Bonds Could Talk: Selective C–H Bond Oxidation. Angew. Chem. Int. Ed 2011, 50, 3362–3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Heitz DR; Tellis JC; Molander GA Photochemical Nickel-Catalyzed C–H Arylation: Synthetic Scope and Mechanistic Investigations. J. Am. Chem. Soc 2016, 138, 12715–12718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).(a) Huang H; Li X; Yu C; Zhang Y; Mariano PS; Wang W Visible-Light-Promoted Nickel- and Organic-Dye-Cocatalyzed Formylation Reaction of Aryl Halides and Triflates and Vinyl Bromides with Diethoxyacetic Acid as a Formyl Equivalent. Angew. Chem. Int. Ed 2017, 56, 1500–1505. [DOI] [PubMed] [Google Scholar]; (b) Abdiaj I; Fontana A; Gomez MV; de la Hoz A; Alcázar J Visible-Light-Induced Nickel-Catalyzed Negishi Cross-Couplings by Exogenous-Photosensitizer-Free Photocatalysis. Angew. Chem. Int. Ed 2018, 57, 8473–8477. [DOI] [PubMed] [Google Scholar]; (c) Dewanji A; Krach PE; Rueping M The Dual Role of Benzophenone in Visible-Light/Nickel Photoredox-Catalyzed C–H Arylations: Hydrogen-Atom Transfer and Energy Transfer. Angew. Chem. Int. Ed 2019, 58, 3566–3570. [DOI] [PubMed] [Google Scholar]; (d) Huang L; Zhu C; Yi L; Yue H; Kancherla R; Rueping M Cascade Cross-Coupling of Dienes: Photoredox and Nickel Dual Catalysis. Angew. Chem. Int. Ed 2019, 59, 457–464. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Park BY; Pirnot MT; Buchwald SL Visible Light-Mediated (Hetero)aryl Amination Using Ni(II) Salts and Photoredox Catalysis in Flow: A Synthesis of Tetracaine. J. Org. Chem 2020, 85, 3234–3244. [DOI] [PubMed] [Google Scholar]; (f) Sakai HA; Liu W; Le C; MacMillan DWC Cross-Electrophile Coupling of Unactivated Alkyl Chlorides. J. Am. Chem. Soc 2020, 142, 11691–11697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).(a) Gutierrez O; Tellis JC; Primer DN; Molander GA; Kozlowski MC Nickel-Catalyzed Cross-Coupling of Photoredox-Generated Radicals: Uncovering a General Manifold for Stereocon-vergence in Nickel-Catalyzed Cross-Couplings. J. Am. Chem. Soc 2015, 137, 4896–4899. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yuan M; Song Z; Badir SO; Molander GA; Gutierrez O On the Nature of C(sp3)–C(sp2) Bond Formation in Nickel-Catalyzed Tertiary Radical Cross-Couplings: A Case Study of Ni/Photoredox Catalytic Cross-Coupling of Alkyl Radicals and Aryl Halides. J. Am. Chem. Soc 2020, 142, 7225–7234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).(a) Qvortrup K; Rankic DA; MacMillan DWC A General Strategy for Organocatalytic Activation of C–H Bonds via Photoredox Catalysis: Direct Arylation of Benzylic Ethers. J. Am. Chem. Soc 2014, 136, 626–629. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jin J; MacMillan DWC Direct α-Arylation of Ethers through the Combination of Photoredox-Mediated C–H Functionalization and the Minisci Reaction. Angew. Chem. Int. Ed 2015, 54, 1565–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhang L; Si X; Yang Y; Zimmer M; Witzel S; Sekine K; Rudolph M; Hashmi ASK The Combination of Benzaldehyde and Nickel-Catalyzed Photoredox C(sp3)–H Alkylation/Arylation. Angew. Chem. Int. Ed 2019, 58, 1823–1827. [DOI] [PubMed] [Google Scholar]; (d) Twilton J; Christensen M; DiRocco DA; Ruck RT; Davies IW; MacMillan DWC Selective Hydrogen Atom Abstraction through Induced Bond Polarization: Direct α-Arylation of Alcohols through Photoredox, HAT, and Nickel Catalysis. Angew. Chem. Int. Ed 2018, 57, 5369–5373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Schoenberg A; Heck RF Palladium-Catalyzed Formylation of Aryl, Heterocyclic, and Vinylic Halides. J. Am. Chem. Soc 1974, 96, 7761–7764. [Google Scholar]
- (23).Nielsen MK; Shields BJ; Liu J; Williams MJ; Zacuto MJ; Doyle AG Mild, Redox-Neutral Formylation of Aryl Chlorides through the Photocatalytic Generation of Chlorine Radicals. Angew. Chem. Int. Ed 2017, 56, 7191–7194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).(a) Harper KC; Moschetta EG; Bordawekar SV; Wittenberger SJ A Laser Driven Flow Chemistry Platform for Scaling Photochemical Reactions with Visible Light. ACS Cent. Sci 2019, 5, 109–115. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bonfield HE; Knauber T; Lévesque F; Moschetta EG; Susanne F; Edwards LJ Photons as a 21st Century Reagent. Nat. Commun 2020, 11, 804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Newhouse T; Baran PS; Hoffmann RW The Economies of Synthesis. Chem. Soc. Rev 2009, 38, 3010–3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).(a) Kochi JK Chemistry of Alkoxy Radicals: Cleavage Reactions. J. Am. Chem. Soc 1962, 84, 1193–1197. [Google Scholar]; (b) Bacha JD; Kochi JK Polar and Solvent Effects in the Cleavage of t-Alkoxy Radicals. J. Org. Chem 1965, 30, 3272–3278. [Google Scholar]; (c) Walling C Some Aspects of the Chemistry of Alkoxy Radicals. Pure Appl. Chem 1967, 15, 69–80. [Google Scholar]; (d) Kuhn LP; Wellman C Reactions of t-Butyl Peroxide with Acetals. J. Org. Chem 1957, 22, 774–776. [Google Scholar]; (e) Hartzell GE; Huyser ES Generation of Methyl Radicals by Decomposition of Bibenzyl Compounds Containing α-Methyl Substituents. J. Org. Chem 1964, 29, 3341–3344. [Google Scholar]
- (27).Level of theory: CBS–QB3.
- (28).(a) Schönherr H; Cernak T Profound Methyl Effects in Drug Discovery and a Call for New C–H Methylation Reactions. Angew. Chem. Int. Ed 2013, 52, 12256–12267. [DOI] [PubMed] [Google Scholar]; (b) Barreiro EJ; Kümmerle AE; Fraga CAM The Methylation Effect in Medicinal Chemistry. Chem. Rev 2011, 111, 5215–5246. [DOI] [PubMed] [Google Scholar]
- (29).(a) Huihui KMM; Caputo JA; Melchor Z; Olivares AM; Spiewak AM; Johnson KA; DiBenedetto TA; Kim S; Ackerman LKG; Weix DJ Decarboxylation Cross-Electrophile Coupling of N-Hydroxphthalimide Esters with Aryl Iodides. J. Am. Chem. Soc 2016, 138, 5016–5019. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhang P; Le C; MacMillan DWC Silyl Radical Activation of Alkyl Halides in Metallaphotoredox Catalysis: A Unique Pathway for Cross-Electrophile Coupling. J. Am. Chem. Soc 2016, 138, 8084–8087. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wang J; Zhao J; Gong H Nickel-Catalyzed Methylation of Aryl Halides/Tosylates with Methyl Tosylate. Chem. Commun 2017, 53, 10180–10183. [DOI] [PubMed] [Google Scholar]
- (30).(a) Engle KM; Mei T-S Wasa M; Yu J-Q Weak Coordination as a Powerful Means of Developing Broadly Useful C–H Functionalization Reactions. Acc. Chem. Res 2012, 45, 788–802. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tang S; Wang P; Li H; Lei A Multimetallic Catalysed Radical Oxidative C(sp3)–H/C(sp)–H Cross-Coupling Between Unactivated Alkanes and Terminal Alkynes. Nat. Commun 2016, 7, 11676. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Shao B; Bagdasarian AL; Popov S; Nelson HM Arylation of Hydrocarbons Enabled by Organosilicon Reagents and Weakly Co-ordinating Anions. Science 2017, 355, 1403–1407. [DOI] [PubMed] [Google Scholar]; (d) Choi GJ; Zhu Q; Miller DC; Gu CJ; Knowles RR Catalytic Alkylation of Remote C–H Bonds Enabled by Proton-Coupled Electron Transfer. Nature 2016, 539, 268–271. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Chu JCK; Rovis T Amide-Directed Photoredox-Catalysed C–C Bond Formation at Unactivated sp3 C–H Bonds. Nature 2016, 539, 272–275. [DOI] [PMC free article] [PubMed] [Google Scholar]; A recent report described two examples of C–C bond formation using 1 equivalent of cyclooctane:; (f) Margrey KA; Czaplyski WL; Nicewicz DA; Alexanian EJ A General Strategy for Aliphatic C–H Functionalization Enabled by Organic Photoredox Catalysis. J. Am. Chem. Soc 2018, 140, 4213–4217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Rh-catalyzed C(sp3)–H insertion with donor-acceptor carbenoids represents one method for circumventing employment of excess hydrocarbon. See:; (a) Liao K; Negretti S; Musaev DG; Bacsa J; Davies HML Site-selective and Stereoselective Functionalization of Unactivated C–H Bonds. Nature 2016, 533, 230–234. [DOI] [PubMed] [Google Scholar]; (b) Liao K; Pickel TC; Boyarskikh V; Bacsa J; Musaev DG; Davies HML Site-selective and Stereoselective Functionalization of Non-activated Tertiary C–H Bonds. Nature 2017, 551, 609–613. [DOI] [PubMed] [Google Scholar]
- (32).While we were not successful developing an arylation reaction with low equivalents of C–H partner, a recent report by MacMillan and coworkers has demonstrated that, under UV irradiation, the combination of a tetrabutylammonium decatungstate HAT catalyst and a Ni cross-coupling catalyst can achieve direct arylation of aliphatic C–H bonds using five equivalents of alkane:; Perry IB; Brewer TF; Sarver PJ; Schultz DM; DiRocco DA; MacMillan DWC Direct Arylation of Strong Aliphatic C–H Bonds. Nature 2018, 560, 70–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Tedder JM The Importance of Polarity, Bond Strength and Steric Effects in Determining the Site of Attack and the Rate of Free Radical Substitution in Aliphatic Compounds. Tetrahedron 1982, 38, 313–329. [Google Scholar]
- (34).Upon omission of the tungstate salt, cross-coupling between phenyl chloroformate and cyclohexane proceeds in 62% yield after 72 hours.
- (35).Lee GS; Won J; Choi S; Baik M-H; Hong SH Synergistic Activation of Amides and Hydrocarbons for Direct C(sp3)–H Acylation Enabled by Metallaphotoredox Catalysis. Angew. Chem. Int. Ed 2020, 59, 16933–16942. [DOI] [PubMed] [Google Scholar]
- (36).Afans’ev IB Correlation Equations in Free-radical Reactions. Russ. Chem. Rev 1971, 40, 216–232. [Google Scholar]
- (37).Primer DN; Molander GA Enabling the Cross-Coupling of Tertiary Organoboron Nucleophiles through Radical-Mediated Alkyl Transfer. J. Am. Chem. Soc 2017, 139, 9847–9850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Santos MS; Corrêa AG; Paixão MW; König B C(sp3)–C(sp3) Cross-Coupling of Alkyl Bromides and Ethers Mediated by Metal and Visible Light Photoredox Catalysis. Adv. Synth. Catal 2020, 362, 2367–2372. [Google Scholar]
- (39).Deng H-P; Fan X-Z; Chen Z-H; Xu Q-H; Wu J Photoinduced Nickel-Catalyzed Chemo- and Regioselective Hydroalkylation of Internal Alkynes with Ether and Amide α-Hetero C(sp3)–H Bonds. J. Am. Chem. Soc 2017, 139, 13579–13584. [DOI] [PubMed] [Google Scholar]
- (40).Huang L; Rueping M Direct Cross-Coupling of Allylic C(sp3)–H Bonds with Aryl- and Vinylbromides by Combined Nickel and Visible-Light Catalysis. Angew. Chem. Int. Ed 2018, 57, 10333–10337. [DOI] [PubMed] [Google Scholar]
- (41).(a) Mariko K; Hirochika S; Tadashi S; Katsumi T; Masayuki Y Spin Trapping of Aryl and Arylcyclohexadienyl Radicals by N-t-Butyl-α-phenylnitrone (N-Benzylidene-t-butylamine Oxide) and α,N-Diphenylnitrone (N-Benzylideneaniline Oxide). Bull. Chem. Soc. Jpn 1977, 50, 1195–1200. [Google Scholar]; (b) Janzen EG; Knauer BR; Williams LT; Harrison WB Electron Spin Resonance of β-Chloro-alkyl Nitroxides. Angular Dependince of β-Chlorine Hyperfine Coupling. J. Phys. Chem 1970, 74, 3025–3027. [Google Scholar]
- (42).No evidence of chlorine radical generation was observed by EPR, although this does not necessarily reflect the lack of chlorine radical formation as Ni(I) may abstract chlorine radical off of the Cl spin adduct to regenerate the starting material.
- (43).A recent theoretical report calculates significantly stronger Ni–Ar bond strengths relative to DFT:; Cagan DA; Stroscio GD; Cusumano AQ; Hadt RG J. Phys. Chem. A 2020, 124, 9915–9922. [DOI] [PubMed] [Google Scholar]
- (44).Tsou TT; Kochi JK Mechanism of Oxidative Addition. Reaction of Nickel(0) Complexes with Aromatic Halides. J. Am. Chem. Soc 1979, 101, 6319–6332. [Google Scholar]
- (45).Tsou TT; Kochi JK Nickel Catalysis in Halogen Exchange with Aryl and Vinylic Halides. J. Org. Chem 1980, 45, 1930–1937. [Google Scholar]
- (46).Russell GA; Williamson RC Nature of the Polar Effect in Reactions of Atoms and Radicals. II. Reactions of Chlorine Atoms and Peroxy Radicals. J. Am. Chem. Soc 1964, 86, 2357–2364. [Google Scholar]
- (47).McDonald and coworkers demonstrated that Ni(III)–Cl and Ni(III) –F complexes can undergo hydrogen atom transfer directly with carbons without the intermediacy of halogen radicals and in the absence of visible light irradiation:; (a) Mondal P; Pirovano P; Das A; Farquhar ER; McDonald AR Hydrogen Atom Transfer by a High-Valent Nickel-Chloride Complex. J. Am. Chem. Soc 2018, 140, 1834–1841. [DOI] [PubMed] [Google Scholar]; (b) Mondal P; Lovisari M; Twamley B; McDonald AR Fast Hydrocarbon Oxidation by a High-Valent Nickel-Fluoride Complex. Angew. Chem. Int. Ed 2020, 59, 13044–13050. [DOI] [PubMed] [Google Scholar]
- (48).Fayad R; Engl S; Danilov EO; Hauke CE; Reiser O; Castellano FN Direct Evidence of Visible Light-Induced Homolysis in Chlorobis(2,9-dimethyl-1,10-phenanthroline)copper(II). J. Phys. Chem. Lett 2020, 11, 5345–5349. [DOI] [PubMed] [Google Scholar]