

SUMMARY
N6-methyladenosine (m6A) RNA modification is a fundamental determinant of mRNA metabolism, but its role in innate immunity-driven non-alcoholic fatty liver disease (NAFLD) and obesity is not known. Here, we show that myeloid lineage-restricted deletion of the m6A “writer” protein Methyltransferase Like 3 (METTL3) prevents age-related and diet-induced development of NAFLD and obesity in mice with improved inflammatory and metabolic phenotypes. Mechanistically, loss of METTL3 results in the differential expression of multiple mRNA transcripts marked with m6A, with a notable increase of DNA Damage Inducible Transcript 4 (DDIT4) mRNA level. In METTL3-deficient macrophages, there is a significant downregulation of mammalian target of rapamycin (mTOR) and nuclear factor κB (NF-κB) pathway activity in response to cellular stress and cytokine stimulation, which can be restored by knockdown of DDIT4. Taken together, our findings identify the contribution of METTL3-mediated m6A modification of Ddit4 mRNA to macrophage metabolic reprogramming in NAFLD and obesity.
Graphical abstract
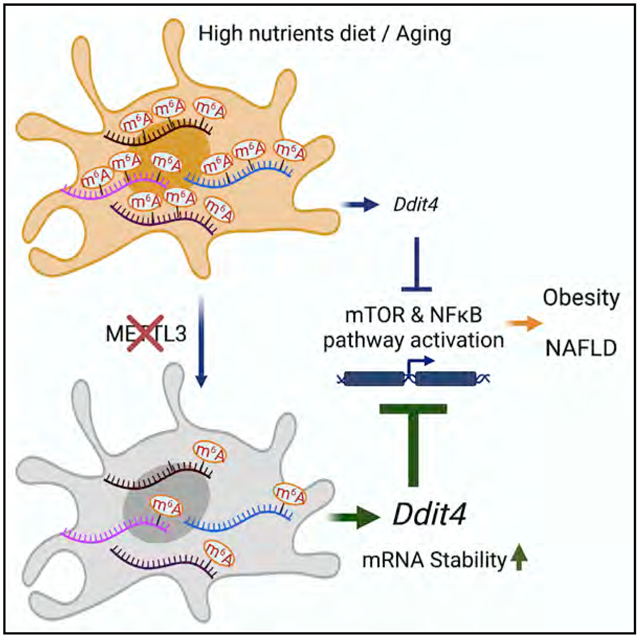
In brief
Myeloid deletion of METTL3 prevents age-related and diet-induced obesity and NAFLD in mice. Qin et al. reveal the contribution of METTL3 to maintaining macrophage homeostasis during NAFLD progression. METTL3 modulates metabolic adaptation of macrophage function in obesity and NAFLD via m6A-decorated DDIT4 mRNA degradation.
INTRODUCTION
Non-alcoholic fatty liver disease (NAFLD) due to obesity and metabolic syndrome is the leading cause of liver-related morbidity and mortality worldwide (Vernon et al., 2011). Obesity-associated chronic low-grade inflammation is critical in the progression of NAFLD toward non-alcoholic steatohepatitis (NASH), although the regulation of this important step is poorly understood (Mirrakhimov and Polotsky, 2012; Shin et al., 2012; Vernon et al., 2011). Myeloid cells such as macrophages are critical in the development of the sterile inflammation that occurs in many organs, particularly the liver and adipose tissue during the metabolic syndrome (Biswas and Mantovani, 2012; Chawla et al., 2011; McNelis and Olefsky, 2014; Odegaard and Chawla, 2011; Osborn and Olefsky, 2012). A broad range of stimuli result in the differentiation of macrophages into functionally distinct activation states that exert profound regulatory effects on tissue metabolism (Biswas and Mantovani, 2012; Chawla et al., 2011; McNelis and Olefsky, 2014; Odegaard and Chawla, 2011, 2013; Osborn and Olefsky, 2012). This is associated with significant changes in macrophage post-transcriptional mRNA modification and mRNA pool, resulting in marked changes in macrophage functional status. However, the molecular determinants that precisely control the macrophage mRNA pool and plasticity during the metabolic syndrome are still to a large extent unknown.
Control of mRNA stability is critical for managing the quantity of mRNA expression levels (Hoernes et al., 2016). N6-Methyladenosine (m6A) is the most prevalent internal modification on eukaryotic mRNAs (Desrosiers et al., 1974). Dynamic m6A modification of mRNA is post-transcriptionally installed, erased, and recognized by m6A methyltransferases (“writers”), m6A demethylases (“erasers”), and m6A-specific binding proteins (“readers”), respectively (Jia et al., 2011; Liu et al., 2014; Wang et al., 2014). m6A modification of mRNA results in lower mRNA stability and reduced mRNA levels, which affect a broad range of biological functions (Geula et al., 2015; Liu et al., 2015; Zhao et al., 2014). We have recently shown that the loss of m6A formation by the depletion of Methyltransferase Like 3 (METTL3) in T cells disrupts T cell homeostasis and the suppressive function of Treg cells, due to the loss of m6A-mediated mRNA degradation (Li et al., 2017; Tong et al., 2018). m6A modification of mRNA and m6A-modifying proteins also play major roles in regulating the innate immune response to viral infection by sensing foreign RNAs and regulating transcripts involved in innate immune signaling (Winkler et al., 2019; Zheng et al., 2017). However, whether the m6A modification participates in the regulation of innate immunity during metabolic disease and inflammation, especially in regulating macrophage activation in NAFLD/NASH pathogenesis, remains poorly understood.
RESULTS AND DISCUSSION
Mice with loss of METTL3 in myeloid cells develop less age-associated incidence of NAFLD and obesity
To examine whether expression changes of Mettl3 in macrophages accompany overnutrition-induced obesity and metabolic inflammation in the liver, we reanalyzed publicly available microarray datasets (GSE54154, GSE89632) (Arendt et al., 2015; Reddy et al., 2014) and found that Mettl3 and other m6A machinery genes were upregulated in macrophages derived from obese leptin-deficient (Lepob/ob) mice as well liver tissues from NASH patients (Figures S1A and S1B). This analysis revealed prominently increased mRNA abundance of Mettl3, Fto, Wtap, and Ythdf1, indicating that m6A machinery genes were expressed in macrophages as well as liver tissues and were upregulated by obesity and metabolic stress.
To determine whether m6A modification plays a role in macrophage during metabolic activation and function in vivo, we focused on the m6A “writer” protein METTL3 and intercrossed Mettl3 flox/flox mice (Li et al., 2017) with the Lysozyme-Cre (LysM-Cre) line to create the myeloid-specific Mettl3-knockout (KO) mice. Both the mRNA and protein expression of METTL3 were significantly reduced in the bone-marrow-derived macrophages (BMDMs) from the KO mice in comparison with the Mettl3 flox/flox (wild-type [WT]) littermates (Figures S1C and S1D), indicating successful myeloid cell-selective METTL3 deficiency. KO mice were healthy at birth, with normal body morphology and weight trajectories until 25 weeks of age when they gained significantly less weight than the WT littermates (Figure 1A). By 46 weeks of age, KO mice showed leaner body morphology (Figure 1B), significantly lower fat accumulation, and lower liver and body weight (Figure S1E). No significant differences in the organ weights of spleen, heart, kidney, and lung were observed (Figure S1E).
Figure 1. Myeloid-restricted loss of METTL3 prevents age-related incidence of obesity and NAFLD.

Eight- to 10-week-old Mettl3 KO and WT littermate mice were fed with regular chow up for 45 weeks (n = 6–9).
(A) Body weight.
(B) Body morphology.
(C) Liver sections stained by H&E, anti-CD11b, anti-Gr1, and anti-Perilipin antibody (Ab) (4 images per mouse). Circline indicates typic infiltrating area.
(D) NAFLD histological activity scores for steatosis and inflammation were determined quantitatively from (C).
(E–H) Fluorescence-activated cell sorting (FACS) analysis and quantification of liver Ly6G+/CD11b+ cells. Levels of (F) serum TGs, (G) serum cholesterol, (H) fasting blood glucose, and (I) serum ALT were measured.
(J) scWAT sections by H&E and quantification of adipose areas. Data represent mean ± SD (n = 10–15). *p < 0.05; **p < 0.01 by two-tailed Student’s t test or two-way ANOVA. Scale bars are shown as indicated.
KO mice were analyzed for the relative distribution of immune cells in the primary and secondary lymphoid organs (spleen and peripheral lymph nodes [PLNs]) and liver tissue at 46 weeks of age. Notably, aged KO mice showed fewer markers of CD11b+ (myeloid cells) and CD11b+Ly6G+ (neutrophils) populations in both spleen and PLNs (Figures S2A and S2B). There was no significant difference in the distribution of Siglec F+ (eosinophils) and CD11b+Ly6C+ (monocytes) populations in either spleen or PLNs (Figures S2A and S2B). We also characterized the phenotype of T cells and found that aged KO mice showed remarkably fewer total CD3+ and CD8+ T cells in the spleen. Consistently, mass cytometry (cytometry by time of flight [CyTOF]) revealed the reduction of infiltrating immune cells and the variable changes of T cell subpopulations in livers from aged KO mice (Figures S2F-S2H). These results demonstrated that METTL3 ablation in LysM+ cells affects general myeloid and T cell homeostasis in age.
Macrophages are known to play critical roles in obesity and metabolic syndrome that result in a sequence of pathological changes in the liver including steatosis, hepatocyte death, inflammation, and fibrogenesis (Marchesini and Marzocchi, 2007). Consistent with the reduced myeloid cell populations in spleen, PLNs, and liver tissue, aged KO mice developed fewer NAFLD/NASH features compared with WT mice as judged by decreased levels of immune cells infiltrations in liver tissue by the staining of H&E-, CD11b-, and Gr1-positive cells (Figure 1C). These are consistent with lower levels of lipid accumulation and NAFLD activity inflammation scores in KO livers (Figure 1D) and a smaller CD11+/Ly6G+ population (Figure 1E). Consistent with reduced lipid stains, aged KO mice showed lower levels of serum triglycerides (TGs) and cholesterol (Figures 1F and 1G). KO mice also displayed lower levels of fasting blood glucose, demonstrating better glucose homeostasis (Figure 1H). No obvious improvement in hepatic damage was observed, as indicated by serum ALT (Figure 1I). Compared with WT littermates, KO mice also developed fewer fatty liver and displayed fewer inflammatory phenotypes within the liver. The subcutaneous white adipose tissue (scWAT) in KO showed fewer crown-like structures and smaller adipocytes (Figure 1J). Collectively, these results indicate that METTL3 plays an important role in accelerating NAFLD and obesity development during aging.
Myeloid METTL3 deficiency prevents diet-induced NAFLD and obesity
To test whether METTL3 in macrophages accelerates overnutrition-driven NAFLD and obesity, we further examined KO mice on a high-fat diet (HFD)-induced NAFLD/NASH and obesity mouse model. In order to avoid overlapping of NAFLD and the obese phenotype observed in aged KO mice, we fed mice that were eight to ten weeks of age on an ad libitum 45 kcal% fat diet for 12 weeks. Remarkably, compared with littermate co-housed WT mice, KO mice were remarkably resistant to HFD-induced obesity, exhibiting much lower body weight (Figure 2A), much leaner body morphology (Figure 2B), and much lower liver weight (Figure 2C). In addition, KO mice exhibited significantly less hepatic steatosis by H&E and lipid staining (Figure 2D) and reduced inflammation by CD11b+ and Gr1+ staining (Figure 2E), with lower NAFLD activity inflammation scores (Figure 2F). KO mice after HFD feeding also improved aberrant morphology and function of adipose tissue reflected by lower fat weight and smaller adipocytes (Figures 2G and 2H) and displayed lower leukocyte infiltration as quantified by Gr1 staining in scWAT (Figure 2I). HFD-fed KO mice showed consistently lower CD11b+Gr1+-infiltrating cells in the liver (Figure 2J), lower hepatic damage as quantified by serum ALT (Figure 2K), and improvement of hyperlipidemia by lower serum TG and cholesterol levels. HFD-fed KO mice significantly exhibited improvement of hyperglycemia (Figure S3A) and insulin sensitivity (Figure S3B). Well-defined choline-deficient, amino-acid defined (CDAA) diets with 60% kcal of fat (HF-CDAA) provide an excellent model recapitulating key aspects of NAFLD with advanced fibrosis (Wei et al., 2020). Similarly, KO mice fed with HF-CDAA diets showed significantly improved liver histology and reduced inflammation and fibrosis compared with littermate WT controls (Figure 2N). Collectively, these data demonstrate that METTL3 in myeloid cells couples overnutrition to the development of obesity and obesity-associated metabolic deterioration such as NAFLD development, with KO mice being less prone to developing obesity and NAFLD.
Figure 2. Myeloid-restricted METTL3 deficiency protects from diet-induced obesity and NAFLD.

Eight- to 10-week-old METTL3 KO and WT littermate mice were fed with high-fat diet (HFD) for 12 weeks (n = 6–9).
(A) Body weight.
(B) Body morphology.
(C) Liver morphology and liver weight.
(D and E) Liver sections stained for H&E, anti-Perilipin, anti-CD11b, and anti-Gr1 Ab (4 images per mouse).
(F) NAFLD histological activity scores for steatosis and inflammation.
(G) scWAT morphology and quantification of scWAT weight.
(H) scWAT sections by H&E and quantification of adipocyte area.
(I) scWAT sections stained by anti-Gr1 Ab (4 images per mouse) and quantification of Gr1 stains.
(J) FACS analysis and quantification of liver Ly6G+/CD11b+ cells.
(K–M) Serum levels of ALT, TGs, and cholesterol.
(N) Eight- to 10-week-old METTL3 KO and WT littermate mice were fed with HF-CDAA diet for 12 weeks (n = 5). Liver sections stained for H&E and Sirius Red (4 images per mouse). Circline indicates typic infiltrating area. Data represent mean ± SD (n = 10–15). Two-tailed Student’s t test or two-way ANOVA: *p < 0.05; **p < 0.01. Scale bars are shown as indicated.
DDIT4 is the major m6A target gene in macrophages
To explore the molecular mechanism underlying the role of macrophages in NAFLD and obesity, we processed RNA sequencing (RNA-seq) on splenic macrophages isolated from KO and littermate WT mice and performed KEGG pathway enrichment analysis. We found significantly different mRNA expression of 896 genes in KO compared with WT macrophages. Consistent with our pathological observations, the 896 mRNAs were in pathways related to cell cycle, p53 signaling, FoxO signaling, carbon metabolism, glycolysis, and gluconeogenesis (Figure 3A). To identify the target genes of the m6A modification mediated by METTL3, we mapped the m6A RNA methylomes of macrophages from KO and WT mice by m6A epitranscriptomic microarray (m6A-array) analysis. The m6A-array analysis identified 929 mRNAs that were differentially decorated by m6A in WT and KO macrophages (Figure 3B). We further overlapped the RNA-seq with the m6A-array data and identified 37 genes that were both differentially m6A modified and differentially expressed in KO macrophages compared with WT macrophages (Figure 3B). mRNA m6A methylation is known to primarily affect RNA stability, and loss of the m6A marker by METTL3 deletion results in slower RNA decay of the m6A target mRNAs (Batista et al., 2014; Fu et al., 2014; Geula et al., 2015; Yue et al., 2015). We therefore focused on genes with decreased m6A levels and increased mRNA transcripts in the KO macrophages. Notably, DNA Damage Inducible Transcript 4 (Ddit4) was among the most significant genes with both downregulation in m6A level and upregulation in mRNA expression level (Figure 3C). By conducting DDIT4-associated gene network analysis with pre-enrichment from KEGG database and STRING functional interacting analysis, we found that 44 of 896 differentially expressed genes in KO macrophages are associated with the DDIT4 network (Figure 3D). Interestingly, these 44 DDIT4 signature genes showed a highly similar pattern of KEGG enrichment (Figure 3E) with all the differentially expressed genes (Figure 3A). In addition, the m6A-decorated DDIT4 levels showed a 50% reduction in KO macrophages from the quantification of m6A-microarray dataset (Figure 3F), which is consistent with the increased DDIT4 mRNA levels (Figure 3C). Furthermore, SRAMP browser tool (http://www.cuilab.cn/sramp) predicated three m6A modification sites with location on the RNA sequences of DDIT4, two of which exhibited very high confidence scores. The level of m6A modification sites containing identified CGACT sequence in DDIT4 was confirmed to be lost in the macrophages and the liver tissue from aged KO mice by MeRIP-m6A-PCR (Figures 3G and 3H). The RNA decay assay showed greater stability of DDIT4 mRNA levels in KO than WT macrophages after actinomycin D treatment (Figure 3I). The increase in DDIT4 transcript was further validated by qPCR from the different macrophage populations of splenic, Kupffer cells (KCs), adipose tissue macrophages (ATMs), and BMDMs (Figure 3J), indicating the induction of DDIT4 expression in METTL3 deficiency occurs systemically in macrophages. Together, these results suggest that DDIT4 mRNA is the major m6A target that could be critical to m6A-mediated macrophages activation and function during NAFLD.
Figure 3. DDIT4 represents a major m6A-decorated gene in macrophages.

Splenic macrophages (CD11b+F4/80+ cells) were sorted from 10-week-old KO and WT littermates. Total RNA was purified and applied to RNA-seq and m6A-microarray analysis.
(A) KEGG analysis of all the differentially expressed genes of KO versus WT with significance (p < 0.05).
(B) Venn diagram analysis of populations of mRNA with significant changes in expression levels or m6A decoration in KO versus WT.
(C) Heatmap of 37 genes with significant changes in expression levels and m6A decoration in KO versus WT.
(D) STRING network analysis of DDIT4-associated gene signatures with pre-enrichment from KEGG and further validated pathways from (A).
(E) Gene Ontology (GO) analysis of DDIT4-associated gene signatures from (D).
(F) Percentage of m6A methylation of mRNA in KO versus WT.
(G and H) MeRIP-m6A using mRNA from thapsigargin (thap)-treated macrophages from KO and WT control (G) and liver tissue from aged KO and WT control (H). DDIT4 transcripts quantified by qRT-PCR are shown as percentage of input RNA (n = 3).
(I) Fitted exponential decay curve of Ddit4 mRNA expression after actinomycin D (2.5 μM) treatment. The residual mRNAs were normalized to t = 0.
(J) mRNA level of DDIT4 gene expression in KO from splenic macrophages, KCs, ATMs, and BMDMs was confirmed by qRT-PCR. The gene expression level was normalized to β-actin. Data represent mean ± SD (n = 10–15). Two-tailed Student’s t test or two-way ANOVA: *p < 0.05.
DDIT4 induction improves NAFLD through downregulation of mTORC1 and NF-κB signaling pathways
DDIT4 is a key regulator of cell growth, proliferation, and survival via inhibition of the mammalian target of rapamycin C1 (mTORC1) activity (Ip et al., 2017; Ota et al., 2014). In particular, DDIT4 plays an important role in responses to cellular energy levels and cellular stress (Horak et al., 2010). DDIT4 was also recently found to be induced by interleukin-10 (IL-10) and to negatively regulate metabolic stress (Ip et al., 2017). Thus, the available evidence suggests that DDIT4 may play a more important role in energy homeostasis and metabolism than it was previously thought. Interestingly, by comparing multiple stimulatory factors including the pro-inflammatory cytokines interferon (IFN)-γ, IL-4, tumor necrosis factor alpha (TNF-α), and Toll-like receptor 3 (TLR3) agonist of Pam3CSK4 and the cellular endoplasmic reticulum (ER) stress inducer of thapsigargin, we confirmed that DDIT4 was specifically induced by ER stress at early time points of 4 h post stimulation (Whitney et al., 2009) (Figure S4), indicating ER stress response is sufficient for DDIT4 induction.
Obese individuals are at increased risk for developing insulin resistance and related comorbidities, including NASH (Hotamisligil, 2010; Sasako et al., 2019; Yang et al., 2015). Although the molecular mechanisms that underlie these associations are not completely defined, dysfunction of cellular organelles such as the ER has emerged as a key event in the alterations that follow nutrient overload (Hotamisligil, 2010). We therefore focused on mTORC1 and nuclear factor κB (NF-κB) signaling pathways that are known to be activated by ER stress and coordinate downstream metabolic and inflammatory responses and worked on the hypothesis that DDIT4 downregulates these key pathways (Figure 4A) (Brugarolas et al., 2004; Chantranupong et al., 2016; Lee et al., 2013; Peterson et al., 2009; Sancak et al., 2007). We therefore reasoned that m6A hypomethylation of DDIT4 would result in the increase of DDIT4 protein expression. Consistent with this hypothesis and our preceding data, we found much more significant induction in DDIT4 protein level in KO macrophages at baseline and after ER stress (Figure 4B) (Figure S4).
Figure 4. m6A-decorated DDIT4 signatures negatively regulate mTORC1 and NF-κB signaling activation in obesity and NAFLD.

(A) Proposed DDIT4 activity in response to ER stress and inflammatory cytokine stimulation.
(B) Western blot analysis of DDIT4, ATG5, and METTL3 protein levels and the phosphorylation of p70S6K and ATK in response to ER stress stimulation by thap (10 μM) in KO versus WT macrophages.
(C) Western blot analysis of IκBα, NF-κB, and DDIT4 protein levels and the phosphorylation of p65 in response to TNF-α (10 ng/mL) stimulation in KO versus WT macrophages.
(D) The mRNA expression levels of DDIT4 signature genes in response to ER stress stimulation in KO versus WT macrophages.
(E) Restored mRNA expression levels after Ddit4 gene ablation in KO macrophages in response to ER stress stimulation.
(F) Reduction of inflammatory gene expression by a pharmacological DDIT4 activator (DDIT4-a) in response to ER stress stimulation.
(G) Reduction of NF-κB promoter activity by DDIT4-a in the presence of forced p65 subunit expression.
(H) Eight-week-old C57B6J mice were fed with HF-CDAA diet with co-current treatment of DDI4 (1 mg/kg) and vehicle control intraperitoneally (i.p.) every other day for 6 weeks (n = 5). Liver sections stained for H&E and Sirius Red (4 images per mouse). Circline indicates typic infiltrating area. Data represent mean ± SD (n = 4). Two-tailed Student’s t test or two-way ANOVA: *p < 0.05.
mTOR acts as a key metabolic regulator and is implicated in an increasing number of pathological conditions including obesity by the control of glucose and lipid metabolism and inhibiting autophagy (Laplante and Sabatini, 2012). We further examined whether m6A ablation leads to DDIT4-dependent inhibition of mTORC1 pathway activity. In support of the idea that mTORC1 coordinates metabolic changes during macrophage activation, the ER stress inducer thapsigargin resulted in significant mTORC1 activation above the basal level in WT, while it was strongly suppressed in KO macrophages, as indicated by increased phosphorylation of the downstream targets such as p70S6K, which reached maximal levels in 4–8 h in WT, while there was no response in KO macrophages (Figure 4B). Myeloid cells lacking autophagy protein ATG5 are known to exhibit an abnormal metabolic activity (Ip et al., 2017). We therefore hypothesized that downregulation of mTORC1 activation in KO macrophages may result in increased autophagy. Indeed, consistent with downregulated p70S6K phosphorylation, the protein level of ATG5 was significantly elevated in KO macrophages, suggesting the effect of loss of METTL3 in myeloid cells on NASH is partially due to increased autophagy.
The transcriptional factor of NF-κB regulates a wide range of host genes that control prototypical pro-inflammatory and immune responses (Lawrence, 2009). We next asked whether NF-κB signaling could be reduced by m6A depletion. TNF-α is a pleiotropic inflammatory cytokine produced by many different types of cells in the body, and it induces canonical NF-κB pathway through the initiation of IκBα degradation (Parameswaran and Patial, 2010). Consistently, TNF-α stimulation resulted in a drastic reduction of IκBα in WT macrophages, but not in KO macrophages (Figure 4C). Notably, there was no difference of IκBα m6A quantity between KO and WT from m6A-array dataset, indicating an indirect role of METTL3 in the control of IκBα protein expression. Furthermore, the key downstream signal of p65 phosphorylation was significantly reduced in KO macrophages, demonstrating that m6A ablation leads to a reduction of NF-κB signaling activation (Figure 4C). In addition, TNF-α stimulation resulted in significantly higher levels of DDIT4 in the absence of METTL3, demonstrating a role for m6A methylation in this pathway as well (Figure 4C). Consistently, the phosphorylation level of p70S6K, which is the downstream activation marker of mTORC1, was elevated by TNF-α stimulation in WT macrophages, while it was significantly reduced in KO macrophages. Gene expression levels of DDIT4 as well as DDIT4 signature genes such as Il10 and Tsc2 were significantly increased and Il1b, Tnfa, and Nox2 were dramatically inhibited in response to ER stress stimulation in KO macrophages (Figures 4C and 4D).
To confirm that the various gene expression changes observed in KO macrophages were due to the upregulation of DDIT4, we knocked down DDIT4 in KO macrophages and then stimulated the cells with thapsigargin. Cells lacking DDIT4 in KO remarkably recovered Tsc2, Il10, Il1rn, Il1b, and Tnfa gene expression and resembled WT macrophages (Figure 4E). This was further confirmed by pharmacological activation of DDIT4, which significantly inhibited the gene expression of Il1b, Tnfa, and Nox2 in response to ER stress stimulation in macrophages (Figure 4F). The result was consistent with the hypothesis that upregulation of DDIT4 is responsible for changes observed in KO macrophages. The pharmacological activation of DDIT4 dramatically inhibited the forced p65-mediated NF-κB promoter activation, confirming the suppressive effect of DDIT4 activation on NF-κB signaling activity indirectly targets on p65 subunit (Figure 4G). These observations suggest that the inhibition of NF-κB, in addition to mTOR signaling by DDIT4, play an essential role in m6A depletion-mediated improvement of macrophages effector function after ER stress and cytokine stimulation. To confirm the in vivo role of DDIT4, WT mice fed with HF-CDAA diets with co-current treatment of pharmacological DDIT4 activator showed significantly improved liver histology and reduced inflammation and fibrosis (Figure 4H). Collectively, these results demonstrate that METTL3-mediated m6A decoration of DDIT4 plays a critical role in the regulation of macrophage activation and function and promotion of NAFLD pathogenesis. These results confirm that excessive nutrient load elicits metabolic ER stress leading to the increased m6A decoration of DDIT4, reduced DDIT4 levels, and increased macrophage effector function.
METTL3 controls metabolic adaptation in macrophage effector function in vivo in obesity and NAFLD mouse model
As a final step we considered whether induction of DDIT4 in KO macrophages couples to the improvement of metabolic status in NAFLD in vivo at the molecular level. To study how METTL3 in myeloid cells regulates the phenotype of NAFLD development under HFD-induced obesity in mouse model, we examined the expression profiling of metabolic genes. In both liver (Figure 5A) and scWAT tissues (Figure 5B) of HFD-fed KO mice, there were robust increases in the expression of DDIT4 signatures in cell cycle (Ccnd1) and significant decreases in the expression of oxidative stress (Nox2 and Nox4), lipogenesis (Pparg and Srebp1c), and gluconeogenesis (G6pc) and corresponding changes of inflammation and anti-inflammation markers (Il1, Ccl5, Il10, and Il1rn), with some cell-specific differences. There was consistent downregulation of pro-inflammatory Il1b and upregulation of anti-inflammation markers (Il10 and Il1rn) (Figure 5C). Serum IL-1β was also significantly lower (Figure 5D) and hepatic protein levels of DDIT4 and IκBα were consistently higher in KO after HFD-induced obesity and NASH (Figure 5E). To confirm the genetic results obtained from METTL3 KO cells, we used pharmacological activator of DDIT4, and this dramatically inhibited D-galactosamine/lipopolysaccharide (LPS)-induced acute hepatitis in vivo (Figure S5). Collectively, our results from acute liver injury and chronic overnutrition mice provide further evidence that DDIT4 acts as a negative regulator of nutrient-induced metabolic dysfunction and inflammation in obesity and NAFLD development.
Figure 5. METTL3 controls metabolic adaptation in macrophage effector function in obesity and NAFLD.

(A–C) Eight- to 10-week-old KO mice and WT littermates were fed with HFD for 16 weeks as described in Figure 2. qRT-PCR analysis of the mRNA abundance of the indicated genes in (A) liver tissue, (B) scWAT tissue, and (C) KCs isolated form HFD-fed mice.
(D) ELISA analysis of IL-1β protein secretion in serum from HFD-fed mice.
(E) Western blot of IκBα and DDIT4 protein levels in liver tissues from HFD-fed mice.
(F) Schema of myeloid METTL3 promoted NAFLD and obesity development associated at least in part with the translation of m6A-decorated DDIT4 gene transcripts. Data represent mean ± SD (n = 10–15). Two-tailed Student’s t test or two-way ANOVA: *p < 0.05; **p < 0.01.
Macrophage activation is a key event in the inflammatory response in NAFLD development. Activated macrophages undergo profound reprogramming of their cellular metabolism. Here, we provide evidence for METTL3-mediated and m6A-dependent regulation of the metabolic process in activated macrophages that is key for the development of NASH. Loss of METTL3 leads to the reduction of mTOR and NF-κB signaling pathway activity through the stabilization of m6A-decorated DDIT4 gene transcripts in response to cellular stress. The increased DDIT4 gene activation in METTL3-deficient macrophages consequently inhibits ER-triggered cellular stress response, and inflammatory cytokines including TNF-α promoted metabolic dysfunction in macrophage effector function during NASH/obesity development (Figure 5F). Our study demonstrates that m6A modification is critical in the control of macrophages-directed metabolic programming through the regulation of immune transcripts in NAFLD and obesity.
Limitations of the study
This study has potential limitations. First, the primary limitation is antibody-only-based m6A-array analysis was used in our system, and we were short of the exact m6A site at nucleotide resolution and quantification of the fraction of modification. In addition, new sequencing-based technologies will become available and could be applied. Second, a pharmacological DDIT4 activator only was used for the in vivo validation, and a genetic DDIT4 KO and/or transgene mouse study could strength the performance. Third, it is quite possible that multiple functional targets of the m6A modification mediated by METTL3 modulate macrophage function. We have used a general way to pick up DDIT4 as major candidate from omics dataset analysis. However, our overall results from comprehensive knockdown/over-activation studies both in vitro and in vivo support DDIT4 as the critical one of METTL3-decorated genes with significant functional consequences. Furthermore, the true therapeutic window for METTL3 inhibition will only be known once potent and selective inhibitors are developed.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Xinshou Ouyang (xinshou.ouyang@yale.edu)
Materials availability
All unique/stable reagents generated in this study are available from the lead contact.
Data and code availability
The RNA-Seq and m6A Epitranscriptomic microarray datasets generated in this study are deposited with Gene Expression Ommibus (GEO) under accession number GEO: GSE184318, GSE184367.
This paper does not report original code. Additional dedicated scripts developed for this work are available from the lead contact upon request.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
C57BL/6J mice were purchased from the National Cancer Institute (Frederick, MD) and were group housed (5 per cage). Mettl3 conditional knockout mice were generated by inserting two lox sites into the first and the last introns using the CRISPR/cas9 based genome-editing system as previously described (Li et al., 2017). The lysozyme-Cre (LysMcre) mice were obtained from Jackson Laboratories (Stock # 004781, Bar Harbor, ME). We then generated LysM-Cre+/− Mettl3 flox/flox (abbreviated KO) mice by intercrossing Mettl3 flox/flox and LysM-Cre mice. Mettl3 flox/flox mice without LysM-Cre gene (abbreviated WT) were used as controls for all experiments. All the KO and WT mice were littermates and co-housed for any experiments described. Animals were maintained in group-housing on a 12-h light/12-h dark cycle with free access to water and standard rodent chow. Housing rooms were maintained at 22-23°C. Both female and male mice were used in experiments. Wherever possible, preliminary experiments were performed to determine requirements for sample size, taking into account resources available and ethical, reductionist animal use. Exclusion criteria such as inadequate staining or low cell yield due to technical problems were pre-determined. Mice were randomly assigned to experimental groups. 8-10 weeks old mice were used in the study. At the conclusion of each study, mice were euthanized by general isoflurane anesthesia with additional cervical dislocation. All animal studies were approved by the Yale University Institutional Animal Care and Use Committee and were performed in accordance with all regulatory standards.
High fat diet mouse model
The 8-10 weeks old mice were fed either a high fat diet (Research Diets Inc, #D12451, New Brunswick, NJ; 45% calories from fat, 35% from carbohydrate, 20% from protein) or HF-CDAA (Research Diets Inc, #A06071302, New Brunswick, NJ; 60% kcal fat of HFD with 0.1%mehionine), ad lib for 6-12 weeks. Chow diet (Harlan Teklad #TD.2916, Madison, WI; 12% calories from fat, 48.5% from carbohydrate, 16.4% from protein) was used as control for HFD.
At the end of the protocols, the whole livers and serum were collected for histological, cytological, biochemical, and molecular analysis.
METHODS DETAILS
BMDMs, ATMs and KCs preparation and culture
Bone marrow (BM) from 6 week’s C57BL/6 mice was cultured in RPMI-1640 medium (PAA Laboratories) supplemented with 10% FBS (fetal bovine serum) (GIBCO), recombinant mouse M-CSF (10 ng/ml) (R&D). After 3 days, non-adherent cells were gently removed, and the remaining cells were further cultured with fresh medium containing M-CSF. On the sixth day, adherent cells were subjected to positive selection with magnetic beads coated with anti-mouse CD11b and were defined as BMDMs.
ATMs isolation:
White Adipose tissue was isolated from mice immediately after CO2 asphyxiation. Tissues were handled using sterile techniques and minced into fine (< 10 mg) pieces. Minced samples were placed in HEPES-buffered DMEM (Invitrogen Corp.) supplemented with 10 mg/ml fatty acid–poor BSA (FAP-BSA; Sigma-Aldrich) and centrifuged at 1,000 g for 10 minutes at room temperature to pellet erythrocytes and other blood cells. Collagenase (Sigma-Aldrich) was added to 1 mg/ml and incubated at 37°C for 20 minutes with shaking] at a concentration of 0.03 mg/ml and 50 U/ml DNase I (Sigma-Aldrich) was added to the tissue suspension and the samples were incubated at 37°C on an orbital shaker (215 Hz) for 45–60 minutes. Once digestion was complete, samples were passed through a sterile 250-μm nylon mesh. The suspension was centrifuged at 1,000 g for 10 minutes. The pelleted cells were collected as the ATMs, and the floating cells were collected as the adipocyte-enriched fraction. The adipocyte fraction was further digested for 1 hour, washed twice with DMEM, and centrifuged as above until there was no further cell/debris pellet. The ATMs were resuspended in erythrocyte lysis buffer and incubated at room temperature for 5 minutes. The erythrocyte-depleted ATMs were centrifuged at 500 g for 5 minutes, and the pellet was resuspended in FACS buffer (PBS containing 5 mM EDTA and 0.2% [wt/vol] FAP-BSA).
KCs isolation:
The mouse liver was perfused with PBS through portal vein, and liver tissue was cut into small pieces by a scissor. The single cell was made using syringe plunger to mull the tissue, and passed through a 40 μm cell strainer. The single cell suspension was centrifuged twice at low speed and supernatant was collected, in order to get non- parenchymal fraction (NPC). The cell supernatants were applied to 42%–34% Percoll gradient and centrifuged for 30 min at room temperature, in order to get KC fraction. The KC fraction was further cultured in 1h, and the cell suspension was collected. The cell suspension was further cultured to get purified KC faction.
These cells were over 90% as CD11+F4/80+ identified by FACS.
siRNA knockdown
Ddit4 siRNA and non-targeting control siRNA were purchased from Ambion® Silencer® Select siRNAs (Catalog #: 4390771, Silencer® Select Negative Control #1 siRNA )and the experiments were done by strictly following manufacturer’s manual.
Flow cytometry
Cells from spleen and peripheral lymph nodes (PLN) were obtained by gentle pressure-dissociation of spleen and PLN using FACS buffer, and passed through a 40-μm nylon cell strainer, and the cells were further centrifuged at 100 g for 5 min to pellet the cells. Red blood cells were lysed, and removed by addition of 5 mL ACK lysis buffer (Lonza, Walkersville, MD), mixed briefly to re-suspend cells and incubated for 2-3 minutes at room temperature, followed by FACS buffer wash and spin steps. Cells were re-suspended in FACS buffer and incubated with Fc block at room temperature for 15 minutes. Cells were divided into individual tubes, and incubated with 100 μL of FACS buffer containing antibody cocktails against CD45-APC (clone 30-F11), mouse Ly-6G (Gr-1) Alexa Fluor 488 (clone RB6-8C5), and/or mouse CD11b-PE (clone M1/70) (all from BD Biosciences) in FACS buffer for 30 min. Samples were washed and the data was acquired by FACS Caliber flow cytometry (BD Biosciences). The data was further analyzed using FlowJo software (FlowJo LLC, Oregon). The dead cells were excluded by propidium iodide staining.
Transfection and luciferase reporter assay
Human HeLa cells were seeded in 24 well culture plate at a density of 1.5x10^5 cells/well. The cell cultures were incubated for overnight followed by 3% FBS medium to not allow cells overgrow. The NFκB promoter luciferase reporter construct and P65 plasmid together with Renilla luciferase (Rluc) control reporter vector at the ratio of 10:10:1were mixed and diluted by Opti-MEM serum-free medium to warm to +15°C to +25°C, and vortex gently. 1 μL per well of Lipofectamine 2000 was pre-diluted in 50 μL Opti- MEM I Reduced Serum Medium. Mix gently and incubate for 5 minutes at room temperature. The DNA and lipofectamine 2000 solution were combined with gentle mix, and further incubated for 20 minutes at room temperature. The DNA containing solution was dropped down to the cell cultures, and mixed gently by rocking the plate back and forth. The cell cultures were further incubated 18-24 hr. All the luciferase activities were measured and normalized to Rluc activity. The normalized value with the relative to control group was indicated.
RNA-Seq
Splenic CD11b+ cells were isolated using CD11b MicroBeads kit (Miltenyi Biotec, Catalog #:130-049-601). The CD11b+F4/80+ cells were isolated from CD11B+ cells by FACS sorting. Total RNAs were isolated with RNeasy mini kit (QIAGEN, Cat #: 74104). The total RNA was further processed for measuring RNA quality by RNA integrity number RIN), only RIN over 9 was accepted for further processing. 1 μg of total RNA was subjected to library synthesis using the TruSeq V2 RNA-Seq kit. Total RNA was enriched for the Poly-A mRNA and reverse transcribed to double-stranded cDNA. Sequencing was performed using 12pM hybridization to a 2x100 paired end flow cell by running standard illumine HiSeq4000 sequencing, and obtained > 50 million reads for each sample by the Genomics and Bioinformatics Core of Yale Stem Cell Center.
Quantitative Real-Time RT–PCR
Total RNA was extracted using RNeasy mini kit (QIAGEN, Cat #: 74104), and cDNA was generated with an oligo (dT) primer and the Superscript II system (Invitrogen, USA) followed by analysis using LightCycler 480 system (Roche). Real-time PCR was performed using SYBR Green real-time PCR analysis (Roche). Expression of β-actin was used to standardize the samples, and the results were expressed as a ratio relative to control. Primers used for qPCR are listed below: Ddit4 forward 5′-ctcttcgtcctcgtctcg-3′, reverse 5′-ggtcaaggccctcttctc-3′; Mettl3 forward 5′- gaaacagctggactcgcttc, reverse 5′-ggcacgggactatcactacg-3′; Il10 forward 5′- catggcccagaaatcaag-3′, reverse 5′- ttcacaggggagaaatcg-3′; Nox2 forward 5′- gtgtcgaaatctgctctcct-3′, reverse 5′- gaggttcctgtccagttgtc-3′; Il1b forward 5′- caactgtgaaatgccacct-3′, reverse 5′- gaagcagcccttcatcttt-3; Tsc2 forward 5′- gctcctggtcatccttca-3′, reverse 5′-gcagcaggaagtcaaagg-3′; II1RN forward 5′- gctcattgctgggtacttaca-3′, reverse 5′-gatgcccaagaacacactatg-3′; Ccnd1 forward 5′-gatgagaacaagcagaccatc-3′, reverse 5′- tagcaggagaggaagttgttg-3′; Chop forward 5′-gaggaggaagagcaaggaa-3′, reverse 5′- cgctcgttctcttcagcta-3′; Nox4 forward 5′- gacctggatttggatttctg-3′, reverse 5′- acaggtttgttgctcctgat-3′; Pparg forward 5′-cttgctgtggggatgtct, reverse 5′- gggttcagctggtcgata-3′; Srebp1c forward 5′-gtgtactggcctttctgtgtc-3′, reverse 5′- gtagcatcagagggagtgaga-3′; G6pc forward 5′-caagggagaactcagcaagt-3′, reverse 5′- gggcttcagagagtcaaaga-3′; Ccl5 forward 5′-gctgctttgcctacctctc-3′, reverse 5′- aacacgactgcaagattgg-3′; b-actin forward 5′-aggactcctatgtgggtga-3′, reverse 5′- atgtcgtcccagttggtaac-3′.
m6A Epitranscriptomic microarray (m6A-array)
Total RNA was isolated with RNeasy mini kit (QIAGEN, Cat #: 74104) and m6A-mRNA&lncRNA Epitranscriptomic microarray analysis was performed. The purity and amount of total RNA samples were determined with NanoDrop ND-1000. Results were provided in Sample QC report. The A260/A280 ratio for all the samples were over 2.0 for pure RNA used in the further assay. Step1: m6A Immunoprecipitation. 1-3 μg total RNA and m6A spike-in control mixture were added to 300 μL 1 × IP buffer (50mM Tris-HCl, pH7.4, 150mM NaCl, 0.1% NP40, 40U/μL RNase Inhibitor) containing 2 μg anti-m6A rabbit polyclonal antibody (Synaptic Systems). The reaction was incubated with head-over-tail rotation at 4°C for 2 hours. 20 μL Dynabeads M-280 Sheep Anti-Rabbit IgG suspension per sample was blocked with freshly prepared 0.5% BSA at 4°C for 2 hours, washed three times with 300 μL 1 × IP buffer, and resuspended in the total RNA-antibody mixture prepared above. The RNA binding to the m6A-antibody beads was carried out with head-over-tail rotation at 4°C for 2 hours. The beads were then washed three times with 500 μL 1 × IP buffer and twice with 500 μL Wash buffer (50 mM Tris-HCl, pH7.4, 50 mM NaCl, 0.1% NP40, 40 U/μL RNase Inhibitor). The enriched RNA was eluted with 200 μL Elution buffer (10 mM Tris-HCl, pH7.4, 1 mM EDTA, 0.05% SDS, 40U Proteinase K) at 50°C for 1 hour. The RNA was extracted by acid phenol-chloroform and ethanol precipitated. Step2: Labeling and Hybridization. The “IP” RNA and “Sup” RNAs were added with equal amount of calibration spike-in control RNA, separately amplified and labeled with Cy3 (for “Sup”) and Cy5 (for “IP”) using RNA Labling Kit. The synthesized cRNAs were purified by RNeasy Mini Kit. The concentration and specific actitity (pmol dye/μg cRNA) were measured with NanoDrop ND-1000. 2.5 μg of Cy3 and Cy5 labeled cRNAs were mixed. The cRNA mixture was fragmented by adding 5 μL 10 × Blocking Agent and 1 μL of 25 × Fragmentation Buffer, heated at 60°C for 30 min, and combined with 25 μL 2 × Hybridization buffer. 50 μL hybridization solution was dispensed into the gasket slide and assembled to the m6A-mRNA & lncRNA Epitranscriptomic Microarray slide. The slides were incubated at 65°C for 17 hours in an Agilent Hybridization Oven. The hybridized arrays were washed, fixed and scanned using an Agilent Scanner G2505C. Step3: Data analysis. Agilent Feature Extraction software (version 11.0.1.1) was used to analyze acquired array images. Raw intensities of IP (immunoprecipitated, Cy5-labeled) and Sup (supernatant, Cy3-labeled) were normalized with average of log2-scaled Spike-in RNA intensities. After Spike-in normalization, the probe signals having Present (P) or Marginal (M) QC flags in at least 2 out of 10 samples were retained as “All Targets Value” in the Excel sheet for further “m6A methylation level” and “m6A quantity” analyses. “m6A methylation level” was calculated for the percentage of modification based on the IP (Cy5-labeled) and Sup (Cy3-labeled) normalized intensities. “m6A quantity” was calculated for the m6A methylation amount based on the IP (Cy5-labeled) normalized intensities. Differentially m6A-methylated RNAs between two comparison groups were identified by filtering with the fold change and statistical significance (p value) thresholds. Hierarchical Clustering was performed using the R software. GO analysis was performed using top GO package in R environment for statistical computing and graphics, and Pathway analysis was calculated by fisher’s exact test.
MeRIP m6A-PCR
MeRIP m6A-PCR was performed by using commercially available Magna MeRIP TM m6A kit, (Cat#: 17-10499) according to the manufactural protocol with some modifications. The intact mRNA was purified from mouse primary macrophages or liver tissues using Magnic mRNA isolation kit, (Cat#: S1550S, New England Biolabs). The mRNA integrity was checked with high quality by Bioanalyzer. The 2.5 μg of mRNA was fragmented in 1× fragmentation buffer at 94 degree for 4 minutes and immediately stopped by adding 0.5 M EDTA. The fragmented mRNA was purified by RNase MiniElute Cleanup kit (Cat#:74204, QIAGEN), and the fragment sizes were centered on ~100nt by agarose get validation. 10% fragmented mRNA was removed as input (10% of Input). 500 μL of MeRIP reaction mixture was prepared by adding 395 μL of fragmented RNA in nuclease free water and 5 μL of RNase inhibitor and 100 μL of 5× IP buffer. The MeRIP reaction mixture was added to prepared Magna protein A/G magnetic beads-m6A antibody tube (1.5 μg of anti-m6A antibody per sample), and incubated for 4 hr at 4°C by rotating head over tail and crosslinked. Each MeRIP reaction tube was briefly centrifuged to remove liquid from cap and sides of the tube and then placed on a separator for 1 minute. The supernatant was carefully aspired without disturbing the magnetic beads. The bead pellets were washed by adding 500 μL of cold 1× IP buffer in the total of 3 times. 100 μL of elution buffer was added to the beads, and mixed by gently pipetting several times to completely resuspended beads. The beads containing solution was the incubated for 2hrs with by rotating head over tail shaking at 4°C. The MeRIP reactions were centrifuged briefly to remove liquid from cap and sides of the tube, and placed on a magnetic separator for 1 minute. The supernatant containing eluted RNA fragments was transferred without aspirating the beads to a new 1.5 mL microcentrifuge tube. The eluted RNA was further purified by RNeasy mini kit to yield final 14 μL of RNA elution. The total RNA elution was applied to qRT-pCR to evaluate target gene enrichment using SYBR Green Master Mix reaction system. The quantification of qRT-pCR results was performed with Ct normalization of the anti-m6A sample, and the negative control of mouse IgG to 10% input sample. The PCR primers were used as follows: Ddit4-m6A target: Forward 5′-GGCAAGGACTGAAGGACTG-3′, reverse 5′-AGCCACCTGCATACAACCT-3.
KEGG Enrichment Analysis
The clusterProfiler package in R was utilized for the identification and visualization of enriched pathways among differentially expressed genes. The function “enrichKEGG” was used to identify enriched pathways based on the KEGG database.
Microarray Data Analysis
The microarray datasets (GSE54154 and GSE53403) were retrieved from the GEO (https://www.ncbi.nlm.nih.gov/geo/). The R package GEO query was used to download the dataset and limma package was used for normalization, calculation of gene expression, and annotation. The expression pattern of m6A genes were further analyzed. Data were visualized using ggplot2 or Complex Heatmap package.
Liver tissue processing and mass cytometry CyTOF analysis
The isolation of mouse liver nonparenchymal cell fractions was performed as previously described (Zhao et al., 2019). Briefly, mouse liver was perfused collagenase perfusion, cell suspension in HBSS was centrifuged at 50 x g for 30 minutes at room temperatures to remove hepatocytes. The supernatants containing non-parenchymal cell fraction (NPC) was pelleted at 350 x g for 6 minutes.
Red blood cells were lysed, and removed by addition of 5 mL ACK lysis buffer (Lonza, Walkersville, MD), mixed briefly to re-suspend cells and incubated for 2-3 minutes at room temperature, followed by HBSS wash and spin steps. The cell suspension was passed through a 40-μm nylon cell strainer for 2-3 times to remove debris. The NPC suspension was subjected to a density gradient centrifugation using 18% OptiPrep (Sigma) at 1200 xg for 20 minutes at room temperature. The purified NPC cells with few dead cells were used for antibody staining and mass cytometry CyTOF analyses. The purified antibodies were obtained from Fluidigm using clones as listed in Key resources table. The NPC cells were resuspended in buffer 2 (1x PBS with 0.1% BSA, 2mM EDTA and 0.05% Na Azide in MilliQ water) to achieve 1 x10^6 cells/50 μl. 5 μL of Fc block was added to the cell suspension with gentle vortex and incubated at room temperature for 10 minutes. Antibody cocktails were added to the cell suspension at concentration of 1 μL of each antibody to 1 × 10^6 cells, and incubated for 30 minutes following wash by adding 500 ul of buffer 2 at the centrifuge of 400 g x 10minutes. After second wash, cell pellets were added buffer 1 (1x PBS) containing 10 μM of Cisplatin for 5 minute sat room temperature. The cells were repeated for 2 washes with buffer 2. Cells were acquired and analyzed using a CyTOF Mass cytometer.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD11b-PE (cloneM1/70) | BD Biosciences | Cat# 553311, RRID:AB_396680 |
| CD45-APC (clone 30-F11) | BD Biosciences | Cat# 561018, RRID: AB_398672 |
| Ly-6G (Gr-1) Alexa Fluor 488 (RB6-8C5) | BD Biosciences | Cat# 553127, RRID: AB_394643 |
| CD3ε | Biolegend | 100308 (145-2c11) |
| CD4 | Biolegend | Cat# 100512, RRID:AB_312715) |
| CD8a | Biolegend | 100708(53-6.7) |
| anti-Perilipin antibody | Abcam | Abcam Cat# ab3526, RRID:AB_2167274) |
| anti-Grl antibody | Thermo Fisher Scientific | Cat#:MA1-70099 (RB6-8C5); RRID: AB_1074825 |
| Mettl3 | Proteintech | Cat#:15073-1-AP; RRID: AB_2142022 |
| DDIT4 | Proteintech | Cat#:10638-1-AP; RRID:AB_2245711 |
| IkBα phosphorylated at Ser32 (14D4 | Cell signaling technology | Cat#:2859S; RRID:B_561111 |
| IkBα | Cell signaling technology | Cat#:9234S; RRID: AB_2269803 |
| P70S6K phosphorylated at T389 | Cell signaling technology | Cat#:9234S; RRID: AB_2269803 |
| P70S6K (49D7) | Cell signaling technology | Cat#2708S; RRID: AB_390722 |
| AKT phosphorylated at T308 | Cell signaling technology | Cat#:4056S; RRID:AB_331163 |
| AKT (C67E7) | Cell signaling technology | Cat#: 4691T,AB_915783) |
| p65 phosphorylated at Ser536 | Cell signaling technology | Cat# 3031S; RRID: AB_330559 |
| NFkB P65 | Cell signaling technology | Cat#: 8242P; RRID: AB_10859369 |
| NFkB p65 (L8F6) | Cell signaling technology | Cat#: 6956S: RRID:AB_10828935 |
| anti-ATG5 antibody | Cell signaling technology | Cat#:12994S; RRID:AB_2630393 |
| anti-β-actin antibody | Cell signaling technology | Cat#: 3700S, RRID:AB_2242334 |
| Flag-HRP | Cell signaling technology | Cat#: 2044S; RRID:AB_10707327 |
| Ly6G/C (141Pr) | Fluidigm | Cat#: 3141005B, Clone: RB6-8C5 |
| CD11b (156Gd) | Fluidigm | Cat#: 3149028D, CloneEPR1344 |
| CD69 (145Nd) | Fluidigm | Cat#: 3145005C, Clone:H1.2F3 |
| CD8a(153Eu) | Fluidigm | Cat#: 3153012B, Clone:53-6.7 |
| CD11b (148Nd) | Fluidigm | Cat#: 3148003C, Clone:M1/70 |
| CD19 (149Sm) | Fluidigm | Cat#: 3149002C, Clone:6D5 |
| CD25 (151Eu) | Fluidigm | Cat#: 3151007C, Clone:3C7 |
| CD3e (152Sm) | Fluidigm | Cat#: 3152004C, Clone:145-2C11 |
| CD62L (160Gd) | Fluidigm | Cat#: 3160008C, Clone:MEL-14 |
| NK1.1 (170Er) | Fluidigm | Cat#: 3170002C, Clone:PK136 |
| F4/80 (146Nd) | Fluidigm | Cat#: 3146008B, Clone:BM8 |
| CD44 (171Yb) | Fluidigm | Cat#: 3171003C, Clone:IM7 |
| CD4 (172Yb) | Fluidigm | Cat#: 3172003C, Clone:RM4-5 |
| CD45R (176Yb) | Fluidigm | Cat#: 3176002C, Clone:RA3-6B2 |
| Chemicals, peptides, and recombinant proteins | ||
| Mouse recombinant TNFα | Peprotech | Cat#: 315-01a |
| DDIT4 activator | Sigma | Cat#: SML0561-5MG |
| Thapsigargin | TOCRIS | Cat#: 1138 |
| Critical commercial assays | ||
| LightCycler 480 SYBR Green I Master | Roche Diagnostics | Cat# 04707516001 |
| Dual-Luciferase® Reporter Assay System | Promega | Cat#: E1910 |
| Lipofectamine 2000 | Thermo Fisher Scientific | Cat#: 11668019 |
| RNeasy mini kit | QIAGEN | Cat #: 74104 |
| RNase MiniElute Cleanup kit | QIAGEN | Cat#:74204 |
| Magnic mRNA isolation kit | New Engladn Biolabs | Cat#: S1550S |
| Magna MeRIP TM m6A kit | MilliporeSigma | cat#: 17-10499 |
| Deposited data | ||
| M6A-RIP-array raw data | this manuscript | Gene Expression Omnibus (GEO:GSE184367) |
| RNA-Seq raw data | this manuscript | Gene Expression Omnibus (GEO:GSE184318) |
| Experimental models: Organisms/strains | ||
| Lysozyme-Cre mouse | Jackson Laboratories | B6.129P2-Lyz2tm1(cre)Ifo/J, Stock# 004781 |
| C57BL/6J | Jackson Laboratories | Stock No: 000664 |
| Mettl3f/f/ mice | In Lab | Li et al., 2017 |
| Software and algorithms | ||
| FlowJo, Version 10 | FlowJo | https://www.flowjo.com/solutions/flowjo/downloads |
| GraphPad Prism version 8 | Prism | https://www.graphpad.com |
| ImageJ v1.49 | NIH | https://imagej.nih.gov/ij/download.html |
| CyTOF Helios | Fluidigm | https://go.fluidigm.com/cytof |
| Other | ||
| HFD | Research Diets Inc. | Cat#: 12451 |
| Chow | Harlan Teklad | Cat#: TD.2916 |
| HF-CDAA | Research Diets Inc. | Cat#: A06071302 |
Western blot
Cells were washed twice with ice-cold PBS and ruptured with CLB buffer (Cell Signaling Technology) containing PMSF and cocktail inhibitor. Cell lysates were resolved by SDS-PAGE and transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore, Bedford, MA, USA) and then blotted. Specific antibodies used are listed below: anti-Mettl3 antibody (15073–1-AP, 1:500) was from Proteintech; anti-DDIT4 antibody (1:1000) was from Proteintech; antibody to IkBα phosphorylated at Ser32 (2859S, 1:1000), anti-IkBα antibody (9234S, 1:3000), antibody to P70S6K phosphorylated at T389 (9234S, 1:1000), antibody to AKT phosphorylated at T308 (4056S, 1:1000), antibody to p65 phosphorylated at Ser536 (3031S, 1:1000), anti-NFkB p65 antibody (6956S, 1:1000), anti-ATG5 antibody (12994S, 1:1000), anti-β-actin (3700S, 1:10000), anti-Flag-HRP (2044S, 1:20000) were from Cell Signaling Technology. All of the unprocessed scans of the blots were shown in the Source Data file.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Analysis
The sample size chosen for our animal experiments in this study was estimated based on our prior experience of performing similar sets of experiments. All animal results were included, and no method of randomization was applied. We at least independently repeated the data once, and all attempt to reproduce the results were successful. For all the bar graphs, data were expressed as mean ± SD. Statistical analyses were performed using GraphPad Prism 6. Differences were analyzed by Student’s t test, or One-way ANOVA test using GraphPad Prism 6. P values ≤ 0.05 were considered significant (*: p < 0.05; **: p < 0.001; ***: p < 0.0001); P values > 0.05; non-significant (NS). FlowJo (Treestar) was used to analyze all the flow cytometry data. The sample sizes (biological replicates), specific statistical tests used, and the main effects of our statistical analyses for each experiment were detailed in each figure legend.
RNA-Seq data analysis
Raw RNA sequencing reads were aligned to the mouse genome (mm10, GRCm38) using Partek Flow software. Gene expression levels were measured by Cufflinks and differential analysis was performed with Cuffdiff (Trapnell et al., 2010). Genes were considered significantly differentially expressed if showing ≥ 1.5 fold change and < 0.05 p value. We used CuffDiff and rMATS to analyze the possible splicing difference events, and did not find any significant difference between Mettl3 KO and WT samples (Wang et al., 2017).
CyTOF data analysis
The data were exported as a traditional flow cytometry file (.fcs) format, and cells for each barcode were deconvoluted using Boolean gating. The visualization and interpretation of CyTOF data, and One-SENSE analysis were performed as recently described (Linderman et al., 2019; Van Gassen et al., 2015)
Supplementary Material
Highlights.
Loss of METTL3 in myeloid cells prevents obesity and NAFLD in mice
METTL3 regulates Ddit4 mRNA stability via m6A RNA modification
METTL3 deficiency leads to reduced mTOR and NF-κB pathway activity upon cellular stress
Induction of DDIT4 modulates metabolic adaptation to macrophage effector function
ACKNOWLEDGMENTS
Wethank Rolando Garcia-Milian from Cushing/Whitney Medical Library of Yale University for the support of RNA-seq and IPA analysis. This study was funded by NIH UO1 grant 5U01AA026962-02 (to X.O.), NIDDK P30KD034989 Pilot Project Program (to X.O.), the National Natural Science Foundation of China (32070917 and 82030042 to H.-B.L.), NCI grant 5R01CA224023-03 (to R.A.F.), and the Howard Hughes Medical Institute (to R.A.F.). The Yale Liver Center core facilities were funded by NIH by grant DK P30-034989. The graphical abstract was created with BioRender.com.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109968.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Arendt BM, Comelli EM, Ma DW, Lou W, Teterina A, Kim T, Fung SK, Wong DK, McGilvray I, Fischer SE, and Allard JP (2015). Altered hepatic gene expression in nonalcoholic fatty liver disease is associated with lower hepatic n-3 and n-6 polyunsaturated fatty acids. Hepatology 61, 1565–1578. [DOI] [PubMed] [Google Scholar]
- Batista PJ, Molinie B, Wang J, Qu K, Zhang J, Li L, Bouley DM, Lujan E, Haddad B, Daneshvar K, et al. (2014). M(6)A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell 15, 707–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas SK, and Mantovani A (2012). Orchestration of metabolism by macrophages. Cell Metab. 15, 432–437. [DOI] [PubMed] [Google Scholar]
- Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, Witters LA, Ellisen LW, and Kaelin WG Jr. (2004). Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 18, 2893–2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chantranupong L, Scaria SM, Saxton RA, Gygi MP, Shen K, Wyant GA, Wang T, Harper JW, Gygi SP, and Sabatini DM (2016). The CASTOR Proteins Are Arginine Sensors for the mTORC1 Pathway. Cell 165, 153–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla A, Nguyen KD, and Goh YP (2011). Macrophage-mediated inflammation in metabolic disease. Nat. Rev. Immunol 11, 738–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desrosiers R, Friderici K, and Rottman F (1974). Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc. Natl. Acad. Sci. USA 71, 3971–3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Dominissini D, Rechavi G, and He C (2014). Gene expression regulation mediated through reversible m6A RNA methylation. Nat. Rev. Genet 15, 293–306. [DOI] [PubMed] [Google Scholar]
- Geula S, Moshitch-Moshkovitz S, Dominissini D, Mansour AA, Kol N, Salmon-Divon M, Hershkovitz V, Peer E, Mor N, Manor YS, et al. (2015). Stem cells. M6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science 347, 1002–1006. [DOI] [PubMed] [Google Scholar]
- Hoernes TP, Hüttenhofer A, and Erlacher MD (2016). mRNA modifications: Dynamic regulators of gene expression? RNA Biol. 13, 760–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horak P, Crawford AR, Vadysirisack DD, Nash ZM, DeYoung MP, Sgroi D, and Ellisen LW (2010). Negative feedback control of HIF-1 through REDD1-regulated ROS suppresses tumorigenesis. Proc. Natl. Acad. Sci. USA 107, 4675–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS (2010). Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 140, 900–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip WKE, Hoshi N, Shouval DS, Snapper S, and Medzhitov R (2017). Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science 356, 513–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, Yi C, Lindahl T, Pan T, Yang YG, and He C (2011). N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol 7, 885–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M, and Sabatini DM (2012). mTOR signaling in growth control and disease. Cell 140, 274–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence T (2009). The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol 1, a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Budanov AV, and Karin M (2013). Sestrins orchestrate cellular metabolism to attenuate aging. Cell Metab. 18, 792–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HB, Tong J, Zhu S, Batista PJ, Duffy EE, Zhao J, Bailis W, Cao G, Kroehling L, Chen Y, et al. (2017). M6A mRNA methylation controls T cell homeostasis by targeting the IL-7/STAT5/SOCS pathways. Nature 548, 338–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linderman GC, Rachh M, Hoskins JG, Steinerberger S, and Kluger Y (2019). Fast interpolation-based t-SNE for improved visualization of single-cell RNA-seq data. Nat. Methods 16, 243–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X, et al. (2014). A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol 10, 93–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Dai Q, Zheng G, He C, Parisien M, and Pan T (2015). N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 518, 560–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchesini G, and Marzocchi R (2007). Metabolic syndrome and NASH. Clin. Liver Dis 11, 105–117, ix. [DOI] [PubMed] [Google Scholar]
- McNelis JC, and Olefsky JM (2014). Macrophages, immunity, and metabolic disease. Immunity 41, 36–48. [DOI] [PubMed] [Google Scholar]
- Mirrakhimov AE, and Polotsky VY (2012). Obstructive sleep apnea and non-alcoholic Fatty liver disease: is the liver another target? Front. Neurol 3, 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odegaard JI, and Chawla A (2011). Alternative macrophage activation and metabolism. Annu. Rev. Pathol 6, 275–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odegaard JI, and Chawla A (2013). The immune system as a sensor of the metabolic state. Immunity 38, 644–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn O, and Olefsky JM (2012). The cellular and signaling networks linking the immune system and metabolism in disease. Nat. Med 18, 363–374. [DOI] [PubMed] [Google Scholar]
- Ota KT, Liu RJ, Voleti B, Maldonado-Aviles JG, Duric V, Iwata M, Dutheil S, Duman C, Boikess S, Lewis DA, et al. (2014). REDD1 is essential for stress-induced synaptic loss and depressive behavior. Nat. Med 20, 531–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parameswaran N, and Patial S (2010). Tumor necrosis factor-α signaling in macrophages. Crit. Rev. Eukaryot. Gene Expr 20, 87–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, Gray NS, and Sabatini DM (2009). DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 137, 873–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy MA, Chen Z, Park JT, Wang M, Lanting L, Zhang Q, Bhatt K, Leung A, Wu X, Putta S, et al. (2014). Regulation of inflammatory phenotype in macrophages by a diabetes-induced long noncoding RNA. Diabetes 63, 4249–4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, Carr SA, and Sabatini DM (2007). PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 25, 903–915. [DOI] [PubMed] [Google Scholar]
- Sasako T, Ohsugi M, Kubota N, Itoh S, Okazaki Y, Terai A, Kubota T, Yamashita S, Nakatsukasa K, Kamura T, et al. (2019). Hepatic Sdf2l1 controls feeding-induced ER stress and regulates metabolism. Nat. Commun 10, 947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin MK, Drager LF, Yao Q, Bevans-Fonti S, Yoo DY, Jun JC, Aja S, Bhanot S, and Polotsky VY (2012). Metabolic consequences of high-fat diet are attenuated by suppression of HIF-1α. PloS ONE 7, e46562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong J, Cao G, Zhang T, Sefik E, Amezcua Vesely MC, Broughton JP, Zhu S, Li H, Li B, Chen L, et al. (2018). M(6)A mRNA methylation sustains Treg suppressive functions. Cell Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, and Pachter L (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gassen S, Callebaut B, Van Helden MJ, Lambrecht BN, Demeester P, Dhaene T, and Saeys Y (2015). FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytometry A 87, 636–645. [DOI] [PubMed] [Google Scholar]
- Vernon G, Baranova A, and Younossi ZM (2011). Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther 34, 274–285. [DOI] [PubMed] [Google Scholar]
- Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, Fu Y, Parisien M, Dai Q, Jia G, et al. (2014). N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505, 117–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Pan Y, Shen S, Lin L, and Xing Y (2017). rMATS-DVR: rMATS discovery of differential variants in RNA. Bioinformatics 33, 2216–2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei G, An P, Vaid KA, Nasser I, Huang P, Tan L, Zhao S, Schuppan D, and Popov YV (2020). Comparison of murine steatohepatitis models identifies a dietary intervention with robust fibrosis, ductular reaction, and rapid progression to cirrhosis and cancer. Am. J. Physiol. Gastrointest. Liver Physiol 318,G174–G188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitney ML, Jefferson LS, and Kimball SR (2009). ATF4 is necessary and sufficient for ER stress-induced upregulation of REDD1 expression. Biochem. Biophys. Res. Commun 379, 451–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler R, Gillis E, Lasman L, Safra M, Geula S, Soyris C, Nachshon A, Tai-Schmiedel J, Friedman N, Le-Trilling VTK, et al. (2019). m6A modification controlsthe innate immune responseto infection by targeting type I interferons. Nat. Immunol 20, 173–182. [DOI] [PubMed] [Google Scholar]
- Yang L, Calay ES, Fan J, Arduini A, Kunz RC, Gygi SP, Yalcin A, Fu S, and Hotamisligil GS (2015). METABOLISM. S-Nitrosylation links obesity-associated inflammation to endoplasmic reticulum dysfunction. Science 349, 500–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Y, Liu J, and He C (2015). RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev. 29, 1343–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Yang Y, Sun BF, Shi Y, Yang X, Xiao W, Hao YJ, Ping XL, Chen YS, Wang WJ, et al. (2014). FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 24, 1403–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P, Han SN, Arumugam S, Yousaf MN, Qin Y, Jiang JX, Torok NJ, Chen Y, Mankash MS, Liu J, et al. (2019). Digoxin improves steatohepatitis with differential involvement of liver cell subsets in micethrough inhibition of PKM2 transactivation. Am. J. Physiol. Gastrointest. Liver Physiol 317, G387–G397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Q, Hou J, Zhou Y, Li Z, and Cao X (2017). The RNA helicase DDX46 inhibits innate immunity by entrapping m6A-demethylated antiviral transcripts in the nucleus. Nat. Immunol 18, 1094–1103 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-Seq and m6A Epitranscriptomic microarray datasets generated in this study are deposited with Gene Expression Ommibus (GEO) under accession number GEO: GSE184318, GSE184367.
This paper does not report original code. Additional dedicated scripts developed for this work are available from the lead contact upon request.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.