

Summary
Interactions between the microbiota and mammalian host are essential for defense against infection, but the microbial-derived cues that mediate this relationship remain unclear. Here, we find that intestinal epithelial cell (IEC)-associated commensal bacteria, Segmented Filamentous Bacteria (SFB), promote early protection against the pathogen, Citrobacter rodentium, independent of CD4+ T cells. SFB induced histone modifications in IECs at sites enriched for retinoic acid receptor motifs, suggesting SFB may enhance defense through retinoic acid (RA). Consistent with this, inhibiting RA signaling suppressed SFB-induced protection. Intestinal RA levels were elevated in SFB mice despite inhibition of mammalian RA production, indicating that SFB directly modulate RA. Interestingly, RA was produced by intestinal bacteria and loss of bacterial-intrinsic aldehyde dehydrogenase activity decreased RA levels and increased infection. These data reveal RA as an unexpected microbiota-derived metabolite that primes innate defense and suggests that pre- and probiotic approaches to elevate RA could prevent or combat infection.
Keywords: Microbiota, infection, intestinal epithelial cells, retinoic acid, Citrobacter, SFB
Graphical Abstract
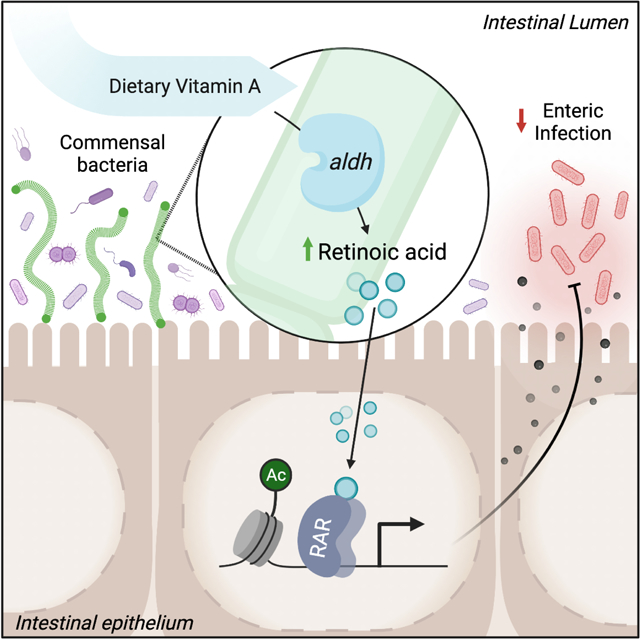
eTOC Blurb
Interactions with the microbiota alter host defense against pathogens. Woo et al. report that commensal bacteria, including segmented filamentous bacteria, can express aldehyde dehydrogenase (aldh) enzymes, producing retinoic acid in the intestine. Bacterial-derived retinoic acid primes epithelial defense and promotes innate protection against intestinal infection.
Introduction
The mammalian intestine is inhabited by trillions of commensal microbes, collectively referred to as the microbiota. In addition to innocuous commensals, the gastrointestinal tract is constantly at risk of invasion and infection by pathogenic microbes. Interactions between the intestinal microbiota and the mammalian host are essential for effective defense against pathogens, as loss of the microbiota in germ-free and antibiotic-treated animals leads to increased susceptibility to enteric and non-enteric infection (Abt and Pamer, 2014; Benson et al., 2009; Ganal et al., 2012; Ivanov et al., 2009). Intestinal epithelial cells (IECs) reside at the direct interface between the host and commensal microbes and, therefore, carry the potential to critically respond to signals from the microbiota. Besides functioning as a physical barrier, these cells actively respond to microbial challenges by secreting antimicrobial peptides, mucins, chemokines and cytokines that prime and regulate innate and adaptive immunity (Gallo and Hooper, 2012; Peterson and Artis, 2014; Ramanan and Cadwell, 2016). IECs are also equipped to sense microbial stimuli through various membrane and cytoplasmic pattern-recognition receptors (Price et al., 2018).
In addition to canonical microbial sensing pathways, epigenetic mechanisms enable environmental signals to instruct cellular responses and represent another interface by which microbiota can impact mammalian cells (Amatullah and Jeffrey, 2020; Woo and Alenghat, 2017). Consistent with this, epigenetic-modifying enzymes in IECs integrate microbiota-derived signals to regulate intestinal homeostasis and immunity (Ansari et al., 2020; Navabi et al., 2017; Takahashi et al., 2009; Wu et al., 2020). Epigenetic-modifying enzymes mediate covalent chromatin modifications that alter DNA accessibility and gene expression. Thus, epigenetic modifications that are sensitive to the microbiota may identify regulatory pathways that can enhance host defense to infection (Arrowsmith et al., 2012; Kelly et al., 2018).
Increasing evidence highlights that microbiota-derived metabolites mediate the host-microbiota relationship (Lavelle and Sokol, 2020; McCarville et al., 2020; Rooks and Garrett, 2016). Commensal bacteria generate a variety of metabolites through either direct synthesis or breakdown of dietary components that can be absorbed in the intestine and potentially travel systemically (Matsumoto et al., 2018; Wikoff et al., 2009). For example, well-characterized bacterial-derived short-chain fatty acids that are produced by bacteria in the intestine can regulate local cells as well as distant tissues (Chang et al., 2014; Dalile et al., 2019; Fellows et al., 2018; Furusawa et al., 2013; Kaiko et al., 2016; Yang et al., 2020). However, despite the appreciation that commensal bacteria prime enhanced innate defenses, the underlying pathways and microbiota-derived cues that decrease host susceptibility to pathogenic infection remain poorly defined.
Citrobacter rodentium is a well-characterized murine bacterial pathogen that causes similar pathology to human enteropathogenic Escherichia coli (Mundy et al., 2005). C. rodentium initiates intestinal infection by adhering to the apical surface of IECs in the large intestine. Defense against C. rodentium requires signals from commensal microbes, as microbiota-depleted animals exhibit higher C. rodentium levels and impaired ability to clear the infection compared to microbiota-replete counterparts (Kamada et al., 2012; Osbelt et al., 2020; Woo et al., 2019). Segmented Filamentous Bacteria (SFB) are commensal bacteria (Jonsson et al., 2020) that protect against enteric pathogens such as C. rodentium (Chung et al., 2012; Garland et al., 1982; Heczko et al., 2000; Ivanov et al., 2009; Shi et al., 2019). Unlike the majority of commensal bacteria that are spatially separated from the epithelium, SFB directly binds to IECs in the distal small intestine (Atarashi et al., 2015; Ivanov et al., 2009; Ladinsky et al., 2019). SFB protects against C. rodentium infection, despite colonizing a distinct anatomical region of the intestine. Therefore, SFB likely modulates mammalian pathways rather than directly competing with C. rodentium, as has been shown for commensal E. coli and Bacteroides thetaiotaomicron (Kamada et al., 2012).
Previous studies have described that decreased C. rodentium infection in mice colonized with SFB were associated with SFB-induced expansion of CD4+ Th17 cells that produce IL-17 and IL-22 (Goto et al., 2014; Ivanov et al., 2009). Here, we discovered that SFB also decreases initial susceptibility to C. rodentium infection prior to regulation by CD4+ T cells. ChIP-seq analyses in uninfected mice revealed that SFB colonization induced epigenetic modifications in IECs at retinoic acid receptor (RAR) motifs. Consistent with enhanced transcriptional potential, IECs from SFB-colonized mice exhibited greater induction of RAR targets relative to IECs from germ-free mice post-C. rodentium infection, suggesting that SFB may enhance innate defense through the RAR ligand, retinoic acid (RA). Interestingly, intestinal RA levels were increased in mice colonized with SFB and inhibiting RA signaling in SFB-colonized mice increased pathogen burden. However, SFB-dependent RA accumulation was not dependent on mammalian RA production. Instead, SFB and other commensal bacteria expressed dehydrogenase genes homologous to a microbial enzyme that converts vitamin A to RA. Remarkably, these enzymes are highly prevalent in the human microbiota, and their enzymatic activity alters RA levels and host regulation in the intestine.
Results
Commensal SFB protects against early infection independently of CD4+ T cells.
C. rodentium is an enteric pathogen that follows a similar pathogenesis to human enteropathogenic E. coli and establishes initial colonization within 2–3 days, reaching peak of infection around days 8–10 post-infection (Symonds et al., 2009). The presence of SFB in the intestinal microbiota protects against C. rodentium infection (Ivanov et al., 2009). Furthermore, we found that colonizing germ-free (GF) mice with SFB alone was sufficient to significantly lower pathogen burdens compared to GF mice (Figure 1A). Interestingly, C. rodentium protection in SFB-colonized mice was already evident within the early phase of infection (days 3–6), suggesting that SFB may promote innate responses that decrease C. rodentium burden. SFB-colonization has previously been shown to induce CD4+ Th17 differentiation. In mice, Th17 cells activated during the peak of C. rodentium infection (day 10–14) mediate clearance of the pathogen by producing IL-22 and IL-17 (Ivanov et al., 2009; Omenetti et al., 2019). However, it is unclear whether Th17 cells are involved in SFB-dependent defense against C. rodentium during initial stages of infection. To test the involvement of Th17 cells and other CD4+ T helper cell populations in SFB-dependent protection against early phase C. rodentium infection, anti-IL-17A or anti-CD4 depleting antibodies were administered prior and during infection (Figure 1B). Mice receiving IL-17A blocking antibodies exhibited similar pathogen levels early post-infection (Figure S1A). Anti-CD4 treatment effectively depleted CD4+ T cells systemically (Figure S1B) as well as in the colon where C. rodentium infects (Figure 1C). Interestingly, SFB colonization led to decreased C. rodentium burden even when mice lacked CD4+ T cells (Figure 1D), indicating that CD4+ T cells are not required for initial SFB-dependent protection.
Figure 1: Commensal SFB primes the intestinal epithelium at retinoic acid receptor sites.
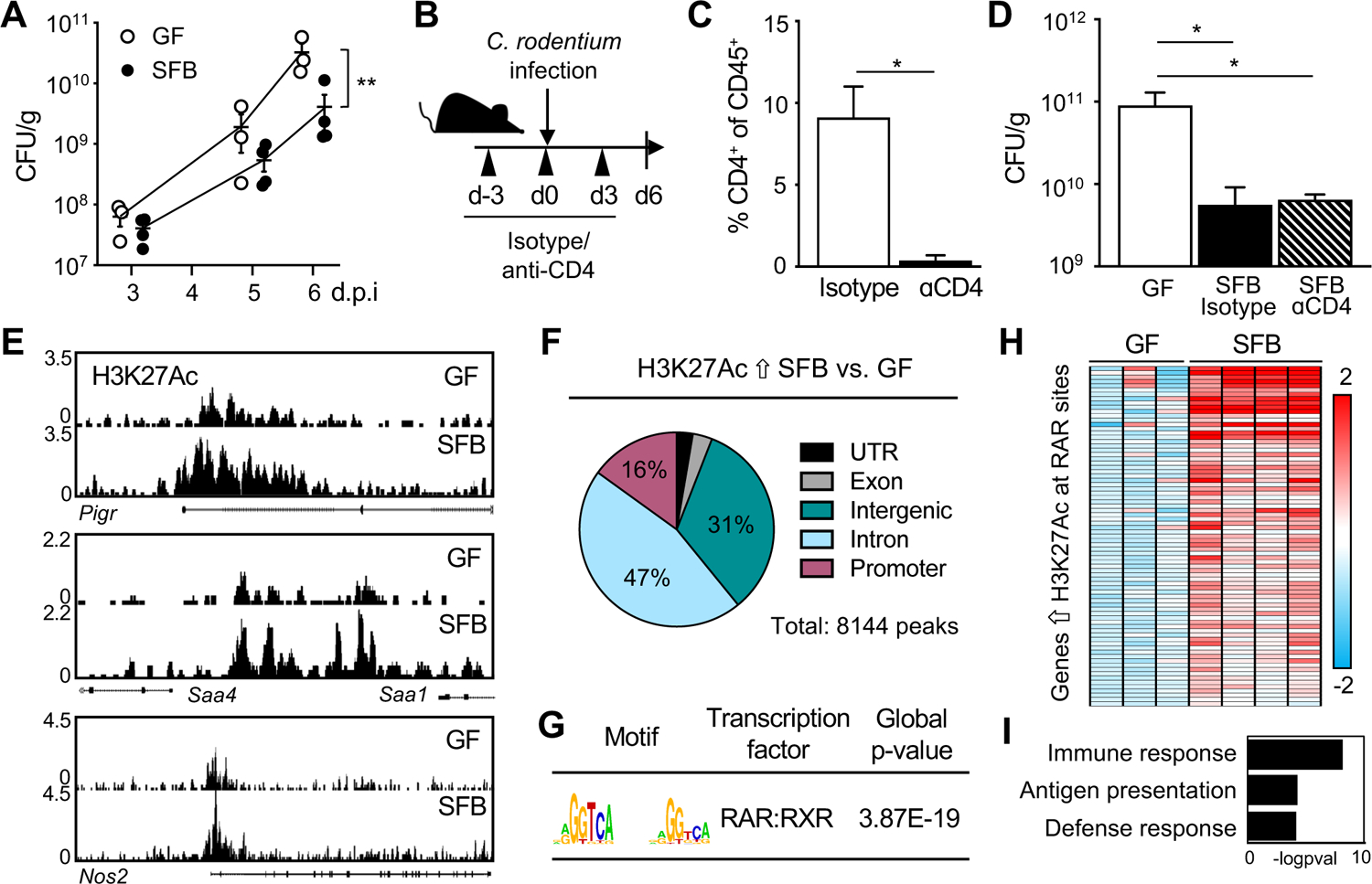
(A) Colony-forming units (CFUs) of C. rodentium in stool of infected germ-free (GF) and SFB-monoassociated mice, normalized to sample weight, days 3–6 post-infection (p.i.). (B) Experimental approach. (C) Percent CD4+ T cells in colon from isotype and anti-CD4 treated mice (n = 3). Gated on CD45+ cells. (D) C. rodentium CFUs in stool, normalized to sample weight, day 6 p.i. (n = 3). (E) Representative sequence tracks for H3K27Ac ChIP-seq from IECs isolated from ileum of GF and SFB-monoassociated mice, normalized to reads per million mapped reads. (F) Genomic distribution of H3K27Ac peaks increased in IECs of SFB versus GF mice, shown as percent of total number of differential peaks. (G) Motif enrichment of retinoic acid receptor (RAR) binding elements at SFB-induced H3K27Ac sites using JASPAR. (H) Heatmap of relative mRNA expression in ileal epithelium harvested from C. rodentium-infected GF and SFB mice at day 6 p.i., represented as relative fold change. (I) Gene ontology for RAR targets that are differentially induced in SFB-infected vs GF-infected from (H). Results are mean ± SEM. Data are representative of at least two independent experiments. *p < 0.05, **p < 0.01. See also Figure S1.
Intestinal epithelium is transcriptionally primed by SFB at retinoic acid receptor motifs.
Given that enhanced initial defense against C. rodentium was not reliant on SFB-induced CD4+ T cells (Figure 1D), we hypothesized that IECs may be important mediators of SFB-driven defense. IECs are critically poised to respond to the microbiota and pathogens and thus play an important role in innate defense. To investigate whether SFB alters the transcriptional state of IECs, we first performed chromatin-immunoprecipitation sequencing (ChIP-seq) on primary IECs isolated from ileum of GF and SFB-monoassociated mice for the histone modification H3 lysine 27 acetylation (H3K27Ac) that characterizes primed and active chromatin (Creyghton et al., 2010). These global analyses revealed many genes that exhibit increased levels of histone H3K27Ac in response to SFB colonization, as indicated by differential peaks at multiple representative genes (Figure 1E). The majority of the sites with differential H3K27Ac in IECs from SFB-colonized mice occurred in regulatory gene elements (Figure 1F), consistent with the known link between H3K27Ac and transcriptionally primed genes (Creyghton et al., 2010; Rada-Iglesias et al., 2011).
To determine whether regions of increased histone acetylation were regulated by a shared transcription factor and/or pathway, motif enrichment analyses were performed. Sites with elevated H3K27Ac in IECs of SFB-colonized mice were significantly enriched for retinoic acid receptor (RAR) motifs (Figure 1G). RARs are a family of nuclear hormone receptors that regulate chromatin accessibility and gene expression by recruiting epigenetic modifiers and cofactors. Interestingly, expression of a large proportion of RAR targets with increased H3K27Ac were significantly upregulated in IECs from SFB mice during C. rodentium infection, compared to IECs from infected GF mice (Figure 1H). Pathway analyses further revealed that the majority of the SFB-sensitive epigenetically primed RAR targets were enriched in host defense pathways (Figure 1I). These data demonstrate that commensal SFB modifies the epigenetic and transcriptional state of the intestinal epithelium and suggest that SFB-dependent regulation of RAR in IECs may prime innate defense against infection.
Retinoic acid improves C. rodentium defense through Nos2 regulation.
RARs are a family of transcription factors that bind as heterodimers with retinoid × receptors to retinoic acid response elements in the DNA. These receptors are activated by binding to the vitamin A metabolite, retinoic acid (RA). Ligand binding results in recruitment of molecular machinery that modifies local chromatin and promotes active transcription. RA and vitamin A availability can modulate C. rodentium and E.coli infection in mice and humans, respectively (Cabrera et al., 2014; McDaniel et al., 2015), provoking the hypothesis that RA may mediate the SFB-induced decrease in C. rodentium. Therefore, to first test whether RA is protective against C. rodentium in an SFB-deficient context, GF mice were treated with exogenous RA prior to and throughout the duration of infection. Administration of RA to GF mice was sufficient to protect GF mice against C. rodentium (Figure 2A), similar to protective effects described for RA-treated microbiota-sufficient mice (Snyder et al., 2019). C. rodentium growth and viability was not directly impaired by RA in vitro (Figure S2A), indicating that decreased C. rodentium infection from RA supplementation in mice required the mammalian host.
Figure 2: Retinoic acid improves C. rodentium defense through Nos2 regulation.
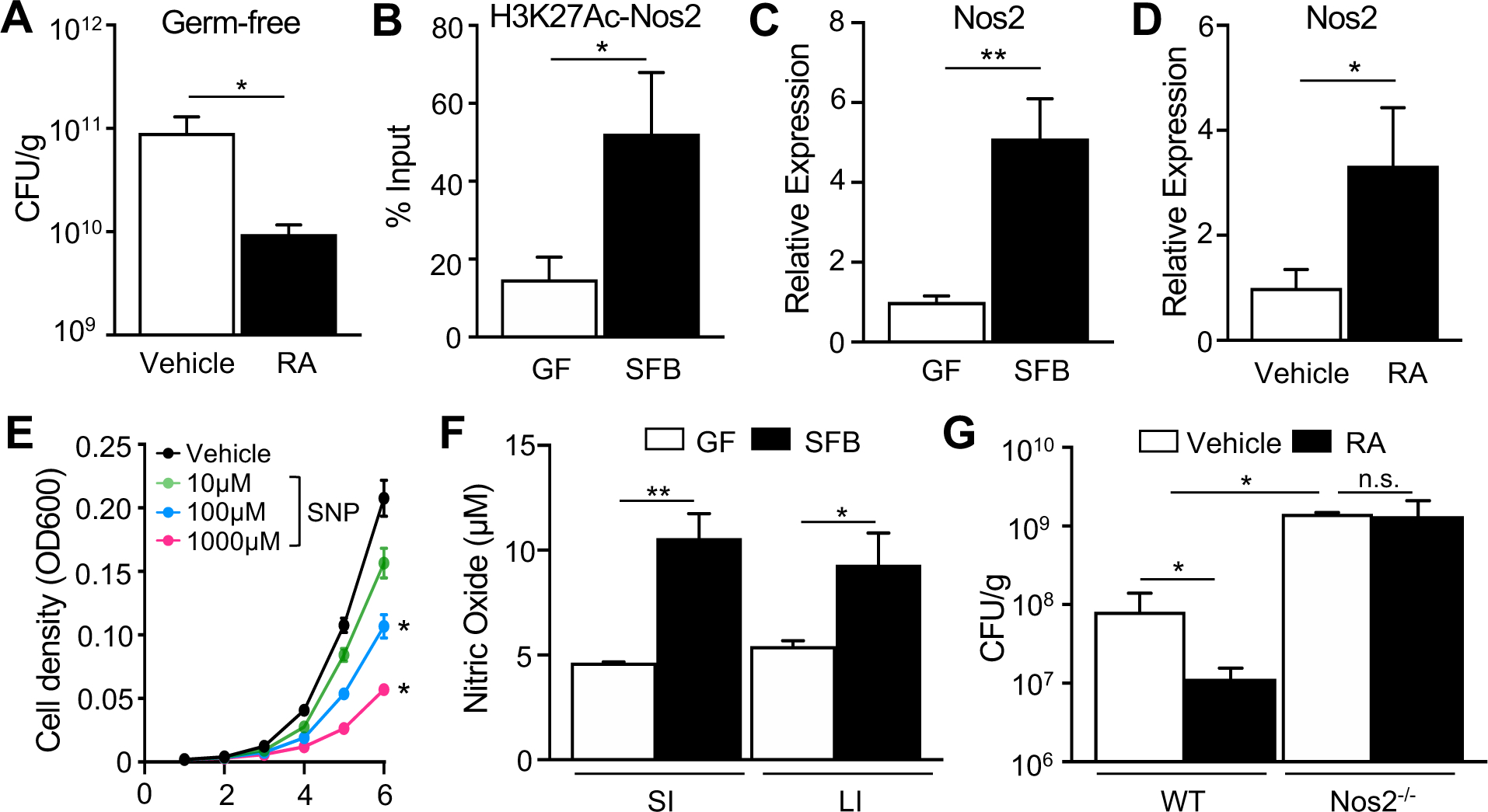
(A) C. rodentium CFUs in stool of GF mice treated with all-trans retinoic acid (RA), day 6 p.i. (n = 3–4). (B) ChIP-qPCR for H3K27Ac at Nos2 RAR site, shown as percent of input (n = 3). (C, D) mRNA expression levels in ileal epithelium, normalized to GF or vehicle-treated mice (n = 3–4). (E) Bacterial cell density of C. rodentium (n = 3). (F) Nitric oxide levels (NO3− + NO2−) in lumen of small (SI) and large (LI) intestine (n = 3). (G) C. rodentium CFUs in stool, normalized to sample weight, day 6 p.i. (n = 3). Results are mean ± SEM. Data are representative of three independent experiments. *p < 0.05, **p < 0.01. n.s., not significant. See also Figure S2.
Nitric oxide synthase 2 (Nos2, iNOS) is an enzyme that is highly expressed in the intestinal epithelium and generates nitric oxide (NO), which has potent antimicrobial activity against C. rodentium (Vallance et al., 2002). SFB significantly increased histone H3K27Ac at an RAR site within the Nos2 gene (Figure 2B). This epigenetic alteration was localized to the small intestine where SFB colonizes (Figure S2B). SFB also induced ileal epithelial Nos2 expression in relation to GF mice (Figure 2C) and similar induction of Nos2 mRNA occurred in IECs harvested from GF mice following treatment with exogenous RA (Figure 2D). Consistent with previous work (Vallance et al., 2002), C. rodentium survival in vitro was impaired with the NO-donor sodium nitroprusside (SNP) (Figure 2E). Furthermore, SFB colonization increased luminal NO levels in the small and large intestine (Figure 2F). These data collectively provoked the hypothesis that Nos2 functions downstream of RA to decrease C. rodentium levels. To test this mechanism, C. rodentium infection levels were compared in mice lacking Nos2 (Nos2−/−) and WT mice in the absence or presence of exogenous RA. As expected, Nos2−/− mice exhibited elevated infection relative to WT mice (Figure 2G). Interestingly, RA administration decreased C. rodentium infection in WT mice but did not alter pathogen levels in Nos2−/− mice (Figure 2G). Collectively, these data highlight Nos2 as an SFB-sensitive, epigenetically regulated RAR target in IECs that mediates RA-induced protection against C. rodentium.
IEC-intrinsic RAR activation enhances defense against C. rodentium.
The role of RA in infection has been extensively investigated in immune cells (Hall et al., 2011a) and recent studies demonstrated that loss of RAR expression or impaired RAR responsiveness in IECs alters intestinal development and defense (Gattu et al., 2019; Iyer et al., 2020; Jijon et al., 2018). To test whether IEC-intrinsic RA signaling specifically contributes to RA mediated protection against C. rodentium infection, we generated an IEC-specific dominant-negative RAR (dnRAR) transgenic mouse (Figure 3A) that expresses non-responsive RAR specifically in IECs (dnRARIEC) (Figure 3B). Mice expressing dnRAR did not exhibit significant differences in intestinal length (Figures S3A and S3B), histology (Figures S3C and S3D) or barrier function (Figure S3E). Furthermore, SFB and 16S DNA levels were similar in dnRARFF and dnRARIEC mice (Figures 3C and 3D). Interestingly, dnRARIEC mice exhibited significantly higher C. rodentium infection compared with floxed controls (dnRARFF) (Figures 3E and 3F), similar to the effect previously described for Salmonella Typhimurium infection (Gattu et al., 2019; Iyer et al., 2020). To next decipher whether RA-driven protection requires epithelial RAR activation, control and dnRARIEC mice were treated with RA and infected with C. rodentium. As described above, RA-treatment decreased infection in control dnRARFF mice (Figure 3G); however, this RA-induced protection was greatly reduced in dnRARIEC mice (Figure 3G), indicating that epithelial-intrinsic RAR activity significantly contributes to RA-dependent defense against C. rodentium.
Figure 3: IEC-intrinsic RAR activation enhances defense against C. rodentium.
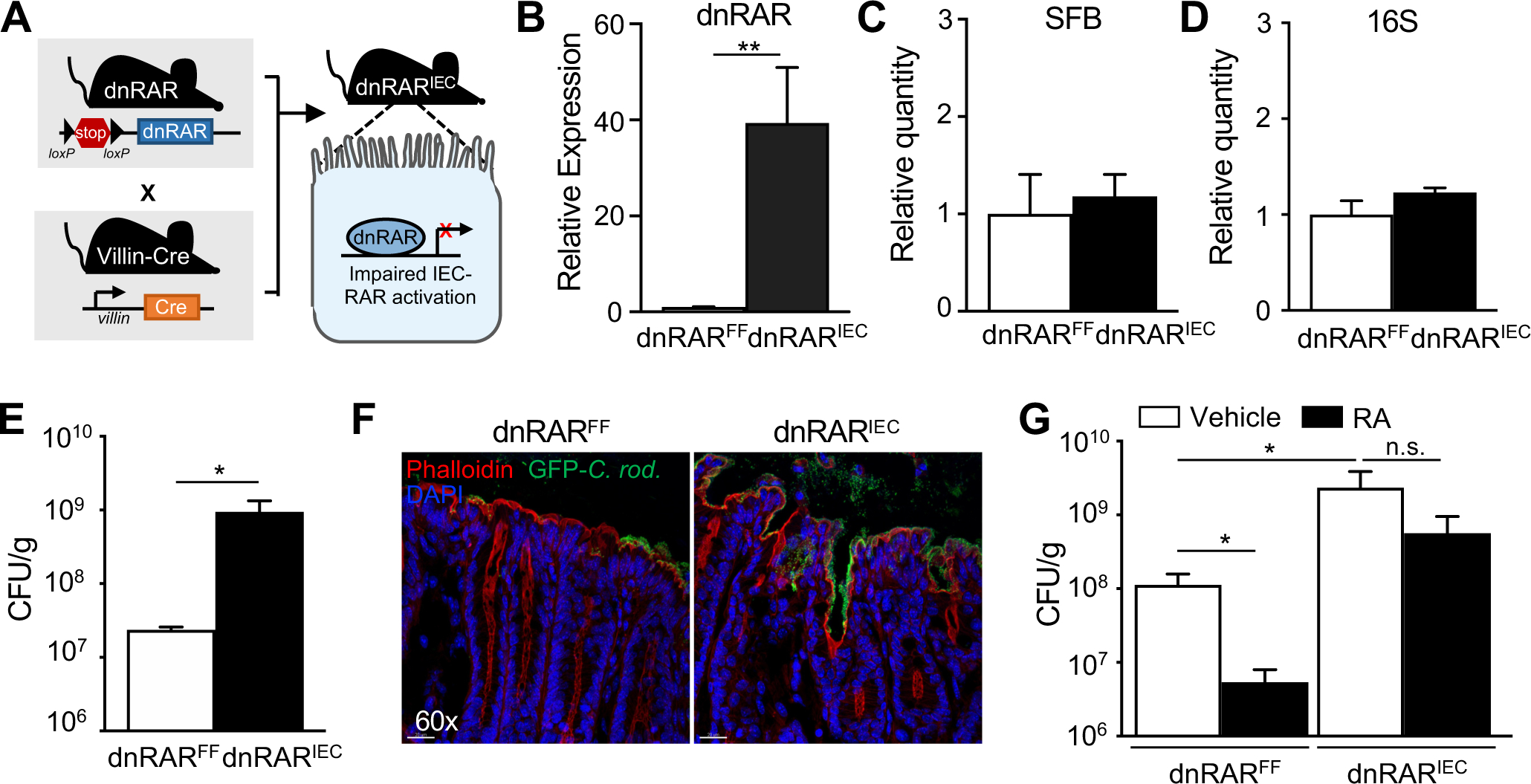
(A) Control mice with a floxed stop-codon upstream of a dominant-negative RAR (dnRARFF) were crossed to mice expressing IEC specific villin-cre recombinase to generate IEC-specific dnRAR expressing mice (dnRARIEC). (B) dnRAR mRNA expression in IECs of dnRARFF and dnRARIEC mice (n = 3–4), normalized to dnRARFF. (C, D) Relative level of (C) SFB and (D) 16S rRNA DNA by qPCR in feces from dnRARFF and dnRARIEC mice (n = 4), normalized to dnRARFF. (E) C. rodentium CFUs in stool, normalized to sample weight, day 6 p.i. (n = 3–4). (F) Immunofluorescent staining of distal large intestine from dnRARFF and dnRARIEC mice infected with GFP-expressing C. rodentium (Green: GFP-C. rodentium, red: Phalloidin, blue: DAPI). (G) C. rodentium CFUs in stool, normalized to sample weight, day 6 p.i. (n = 5–6). Results are mean ± SEM. Data are representative of three independent experiments. *p < 0.05, **p < 0.01. n.s., not significant. See also Figure S3.
SFB increases intestinal retinoic acid levels despite inhibition of host production.
Our initial findings demonstrated that SFB increased histone acetylation at RAR target genes in IECs (Figures 1E–1G) and that the timing and magnitude of RA-induced protection against C. rodentium parallels the phenotype observed with SFB colonization (Figures 1A and 2A). Thus, we hypothesized that RA may mediate SFB-dependent defense against this pathogen. In order to test whether SFB promotes protection from infection by activating RAR, GF and SFB-colonized mice were treated with an RAR-inverse agonist (RARi: BMS493) that blocks RA-RAR activation (Metzler et al., 2018). RARi treatment did not alter SFB colonization (Figure S4A). Remarkably, impaired RA-RAR activation abrogated the protective effect of SFB against C. rodentium, whereas pathogen levels in GF mice were largely unaffected (Figure 4A). Given that RARs are activated by their RA ligand, intestinal levels of RA were compared between GF and SFB-colonized mice. Interestingly, SFB mice exhibited significantly elevated RA in intestinal contents and IECs compared to GF mice (Figures 4B and 4C). Consistent with increased local RA, expression of the RA-sensitive gene Serum amyloid A-1/2 (Saa1/2) was increased in IECs from SFB-colonized mice (Figure 4D).
Figure 4: SFB increases intestinal retinoic acid levels despite inhibition of host production.
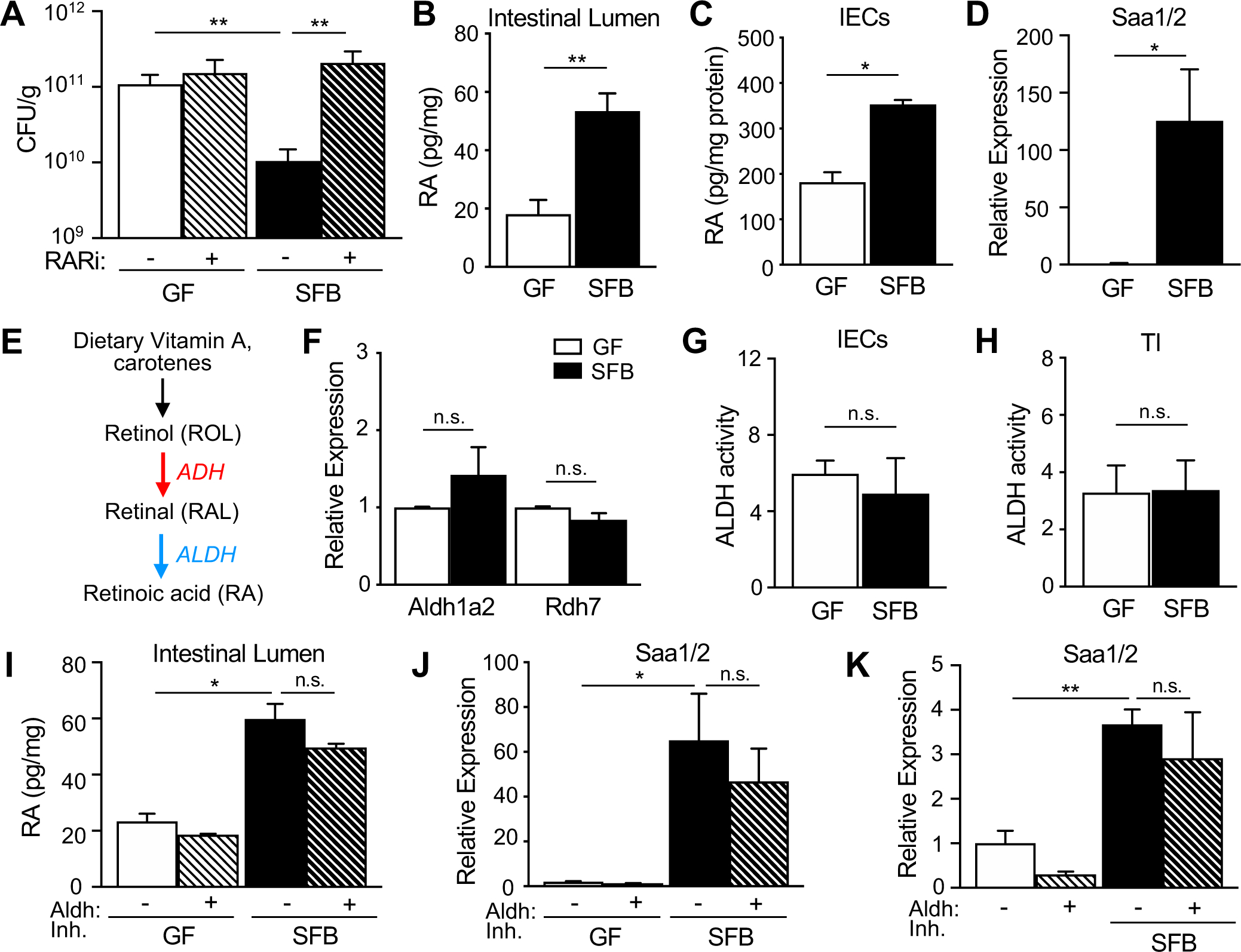
(A) C. rodentium CFUs in stool of GF and SFB mice −/+ RAR inhibitor (RARi; BMS493), normalized to sample weight, day 6 p.i. (n = 4–6). (B, C) Retinoic acid concentration in (B) fecal samples and (C) IECs collected from GF and SFB mice (n = 4), normalized to sample weight. (D) mRNA expression in IECs of GF and SFB mice (n = 4), normalized to GF. (E) Schematic of mammalian vitamin A metabolism to retinoic acid. (F) mRNA expression in IECs of GF and SFB mice (n = 4), relative to GF. (G, H) ALDH activity in (G) IECs and (H) terminal ileum (TI) tissue (n = 3). (I) Retinoic acid concentration in stool of GF and SFB mice treated with Aldh1a2 inhibitor, normalized to sample weight (n = 4). (J, K) mRNA expression in (J) IECs (n = 4) or (K) intestinal organoids (n = 4–6). Results are mean ± SEM. Data are representative of three independent experiments. *p < 0.05, **p < 0.01. n.s., not significant. See also Figure S4.
RA is generated from the vitamin A-derivative retinol in a two-step oxidation reaction involving retinol dehydrogenases (ADH, RDH) and retinaldehyde dehydrogenases (ALDH, RALDH) (Figure 4E). IECs are equipped to generate RA, and it was recently reported that microbiota modulates intestinal tissue RA levels by regulating expression of Rdh7 in IECs (Grizotte-Lake et al., 2018). However, we found that expression of key enzymes involved in the conversion of vitamin A to RA (Aldh1a2 and Rdh7) were similarly expressed in IECs of GF and SFB mice (Figure 4F). Furthermore, ALDH activity was similar in IECs and ileal tissue of GF and SFB-colonized mice (Figures 4G and 4H), suggesting that host-intrinsic enzymes required for RA production in the intestine are not primarily impacted by SFB alone. To further investigate the contribution of host RA synthesis on intestinal RA levels in SFB colonized mice, mice were treated with an Aldh1a2 inhibitor, WIN18446, that blocks mammalian RA production (Arnold et al., 2015; Paik et al., 2014; Seamons et al., 2020). As predicted, administration of WIN18446 decreased mammalian ALDH enzymatic activity (Figure S4B) and downstream gene expression (Figure S4C). However, despite host ALDH inhibition, RA levels and IEC-expression of Saa1/2 remained elevated in SFB colonized mice (Figures 4I and 4J). SFB levels in the intestine were similar following WIN18446 administration (Figure S4D). To directly interrogate the epithelial Saa1/2 response to SFB without other cellular contributions, intestinal organoids were examined in the absence or presence of SFB. As expected, inhibition of mammalian Aldh1a2 decreased basal Saa1/2 expression in intestinal organoids (Figure 4K). However, consistent with our findings in vivo, SFB-exposure induced Saa1/2 expression in intestinal organoids despite pharmacological inhibition of mammalian Aldh1a2 (Figure 4K). Taken together, these findings suggest that the SFB-dependent increase in intestinal RA and regulation of epithelial responses is largely bacterial-dependent.
Commensal bacteria provide a direct source of retinoic acid in the intestine.
Intestinal RA levels did not reflect altered mammalian RA synthesis in SFB-colonized mice (Figures 4F–4K), suggesting a distinct source of RA. Therefore, to investigate whether SFB directly generates RA by metabolizing vitamin A, explant cultures containing SFB were treated with vitamin A (retinol). Surprisingly, RA levels were significantly increased in supernatants following retinol supplementation of SFB explant cultures (Figure 5A). To further examine vitamin A metabolism by SFB, RA levels were compared in GF and SFB mice fed vitamin A-deficient diet. Interestingly, removal of vitamin A from the diet decreased intestinal RA levels in SFB mice to the amount observed in GF mice (Figure 5B). Furthermore, vitamin A-deficiency resulted in elevated C. rodentium infection burdens as compared to SFB-colonized mice fed vitamin A-sufficient diet (Figure 5C), indicating that SFB requires dietary vitamin A to increase luminal RA and protect against C. rodentium.
Figure 5: Commensal bacteria provide a direct source of retinoic acid in the intestine.
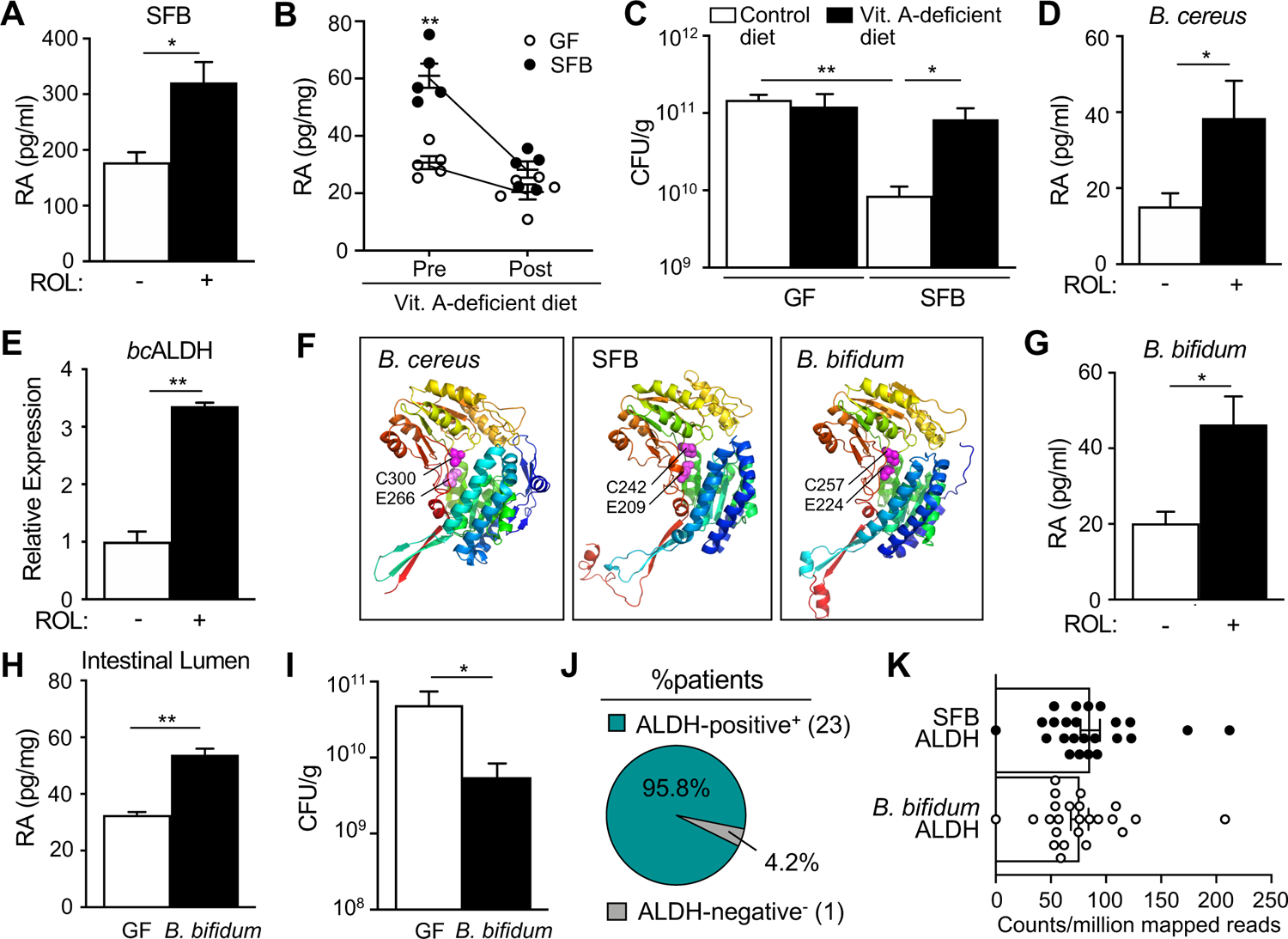
(A) Retinoic acid concentration in supernatants from intestinal explants from GF and SFB mice incubated with vehicle or all-trans retinol (ROL) (n = 3). (B) Retinoic acid levels in intestinal lumen of mice pre and post vitamin A-deficient diet. (C) C. rodentium CFUs in stool, normalized to sample weight, day 6 p.i. (n = 3) (D) Retinoic acid concentration in media from B. cereus cultured with ROL, per 108 CFU of bacteria (n = 3). (E) Expression of ALDH by B. cereus cultured −/+ ROL (n = 3). (F) Protein structure of B. cereus ALDH1A1 (KFL74159.1), SFB ALDH (WP_007440235.1) and B. bifidum ALDH (WP_015438559.1). Conserved catalytic glutamate [E] and cysteine [C] residues shown as magenta-colored spheres. Root-mean-square deviation of atomic positions (RMSD) compared to bcALDH1A1 for SFB ALDH = 2.059 Å; B. bifidum ALDH, RMSD = 1.514 Å. (G) Retinoic acid concentration following incubation with ROL, per 108 CFU of bacteria (n = 3). (H) Retinoic acid concentration in intestinal contents of GF mice monoassociated with B. bifidum, normalized to sample weight (n = 3). (I) C. rodentium CFUs in stool, normalized to sample weight, day 6 p.i. (n = 4–5) (J) Percent of patients containing intestinal microbiome reads that align to SFB or B. bifidum ALDH. (K) ALDH counts per million reads. Results are mean ± SEM. Data represent at least two independent experiments. *p < 0.05, **p < 0.01.
The bacterial strain, Bacillus cereus, has been described to express a bacterial aldehyde dehydrogenase enzyme (bcALDH1A1, KFL74159.1) that produces RA from vitamin A in vitro (Hong et al., 2016). Consistent with this work, we found that B. cereus produced RA when incubated with retinol (Figure 5D), without altered growth (Figure S5A). Furthermore, retinol increased bcALDH1A1 expression by B. cereus (Figure 5E), supporting that this bacterial RA-producing enzyme is sensitive to local retinol levels. Therefore, to investigate whether SFB and potentially other bacterial species express similar aldehyde dehydrogenases that generate RA, we compared protein sequences using bcALDH1A1 as reference. Glutamate-266 and Cysteine-300 are necessary for the catalytic activity of bcALDH1A1 (Hong et al., 2016). Interestingly, SFB, along with the well-known probiotic bacteria Bifidobacterium bifidum, encode bacterial ALDH enzymes that share these critical catalytic amino acid residues (Figure 5F). Furthermore, the predicted protein structures of ALDH enzymes from SFB and B. bifidum both exhibited marked overlap with bcALDH1A1 (Figure 5F).
Similar to SFB and B. cereus, B. bifidum cultured with retinol increased RA levels without impacting bacterial growth (Figures 5G and S5B). Therefore, to test whether B. bifidum generates RA in vivo, GF mice were colonized with B. bifidum (Figure S5C) and compared to GF controls. Mice colonized with ALDH-expressing B. bifidum demonstrated increased luminal RA relative to GF mice (Figure 5H) and enhanced protection against C. rodentium infection (Figure 5I). Thus, B. bifidum mono-colonized mice exhibited early protection against C. rodentium, similar to SFB. These results indicate that a subset of commensal bacterial populations, including SFB and B. bifidum, can provide a direct source of RA in the intestine. Given that SFB and B. bifidum both convert retinol to RA and express ALDH enzymes, the prevalence of ALDHs in the human intestinal microbiota was evaluated by aligning SFB and B. bifidum ALDH genes against microbiome sequencing from healthy human patients (Lewis et al., 2015). Interestingly, these analyses revealed that both genes were detected in nearly all samples analyzed (23/24 patients) (Figure 5J) with an average read count of approximately 80 counts per million reads (Figure 5K), indicating broad prevalence of homologous genes in the human intestinal microbiota.
SFB dehydrogenase activity produces retinoic acid and improves host defense.
While direct genetic manipulation in SFB or B. bifidum would be ideal to test the functional role of the microbial ALDH gene, this approach is technically limiting in both species. Therefore, to directly examine the contribution of bacterial ALDH enzymes in producing RA, ALDHs from SFB or B. bifidum were expressed in E. coli as the wild-type enzyme (ALDHWT) or a catalytic domain mutant variant (ALDHMUT) (Figure 6A). ALDHWT and ALDHMUT enzymes were similarly expressed for both strains (Figure 6B). Importantly, ALDHWT strains exhibit significant ALDH activity whereas ALDHMUT strains showed no enzymatic activity (Figure 6C). When cultured with vitamin A, both SFB-ALDHWT and B. bif-ALDHWT strains produced elevated RA relative to non-ALDH expressing control (Figure 6D). Importantly, this RA induction with ALDHWT bacteria was lost in ALDHMUT strains (Figure 6D), demonstrating that RA levels are indeed dependent on the bacterial enzyme. In addition, intestinal organoid cultures containing retinol exposed to ALDHWT bacteria exhibited increased Saa1/2 (Figure 6E) and Nos2 (Figure 6F) expression relative to organoids incubated with ALDHMUT bacteria, demonstrating that microbial ALDH activity increases expression of RA-sensitive targets in mammalian epithelium.
Figure 6: SFB dehydrogenase activity produces retinoic acid and improves host defense.
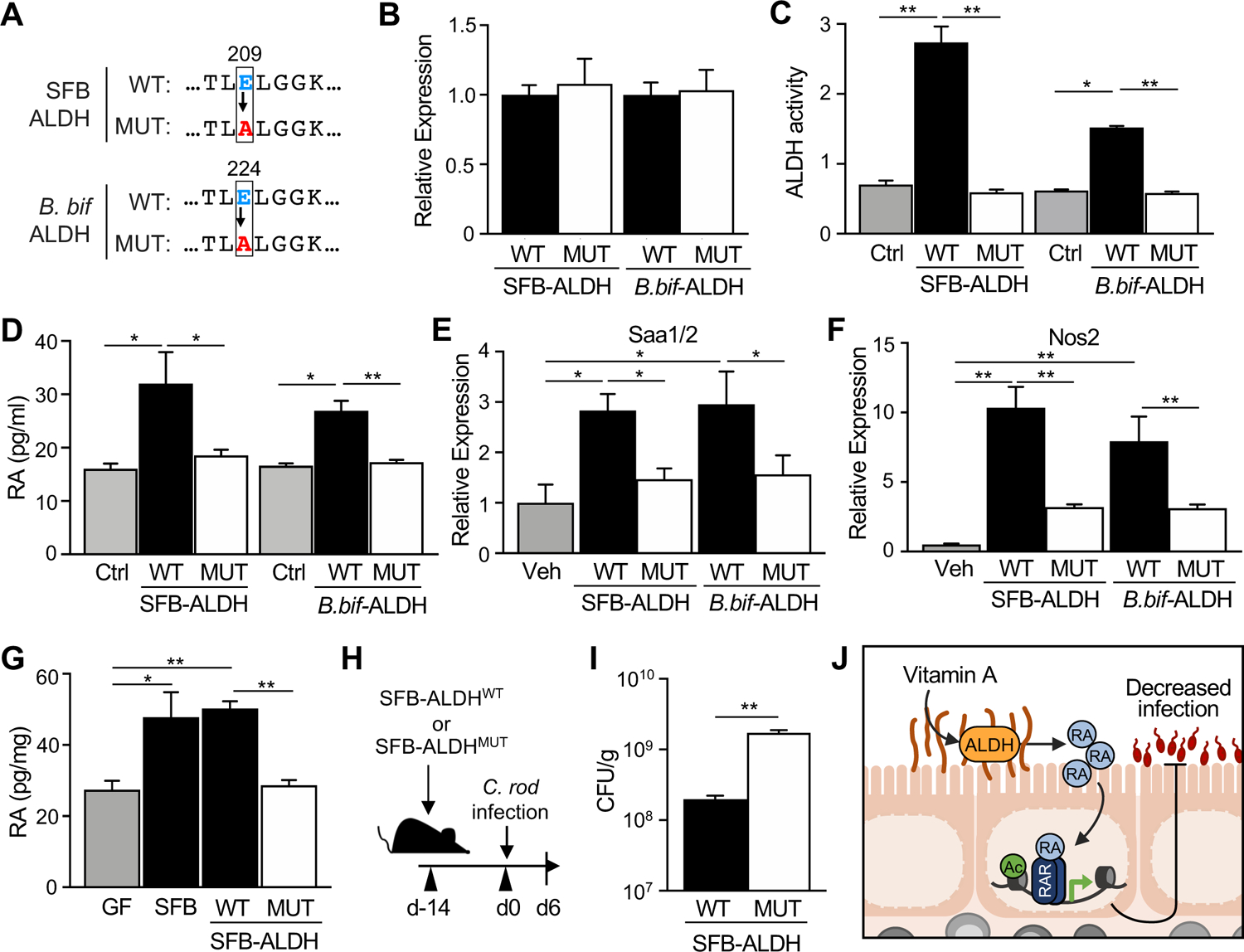
(A) Protein sequence for wildtype (WT) SFB and B. bifidum ALDH demonstrating conserved catalytic glutamate [E] that has been replaced by alanine [A] in the mutant (MUT). SFB and B. bifidum ALDH-pET-21a(+) plasmids were expressed in E.coli (ALDHWT or ALDHMUT). (B, C) ALDH (B) expression and (C) activity in ALDHWT or ALDHMUT strains (n = 3). (D) Retinoic acid concentration of ALDHWT or ALDHMUT cultures incubated with retinol (ROL) (n = 3). (E, F) mRNA expression in intestinal organoids stimulated with ALDHWT or ALDHMUT strains for 24h in 1μM retinol, normalized to vehicle control (n = 3). (G) Retinoic acid concentration in intestinal contents of GF mice monoassociated with SFB, SFB-ALDHWT or SFB-ALDHMUT (n = 3), normalized to sample weight. (H) Experimental design. (I) C. rodentium CFUs in stool, normalized to sample weight, day 6 p.i. (n = 3). (J) Microbiota-derived retinoic acid in the intestine boosts host defense against enteric infection. Results are mean ± SEM. Data are representative of at least two independent experiments. *p < 0.05, **p < 0.01. See also Figure S5.
To next test whether bacterial ALDH enzymes generate RA in vivo, GF mice were colonized with SFB-ALDH expressing strains. Interestingly, colonization of GF mice with the SFB-ALDHWT strain increased intestinal RA levels relative to GF mice, whereas RA levels in ALDHMUT-colonized mice remained relatively unchanged (Figure 6G). Furthermore, following C. rodentium infection, GF mice colonized with ALDHWT also exhibited lower pathogen levels relative to infected mice colonized with ALDH-inactive bacteria (Figures 6H and 6I). Collectively, these new data demonstrate a direct contribution of bacterial ALDH enzymes in the production of RA in the intestine, and that this microbial enzymatic activity enables improved pathogen control.
Discussion
In this study, we discovered that SFB and other beneficial commensal bacteria generate RA in the intestine and regulate epithelial RAR signaling to enhance defense against a pathogen (Figure 6J). RA is a fat-soluble metabolite derived from carotenes and vitamin A that is known for its immunomodulatory effects and role during infectious disease (Hall et al., 2011a). Vitamin A deficiency in humans is linked to increased susceptibility to numerous bacterial and viral pathogenic infections (Sommer, 2008; World Health Organization, 2009), whereas vitamin A supplementation reduces incidence and mortality of diarrheal diseases commonly caused by intestinal pathogens (Green and Mellanby, 1928; Huang et al., 2018; Semba, 1999). Although vitamin A has been used as a clinical health intervention for diarrheal disease and other health conditions, the reported outcomes have been inconsistent (Dibley et al., 1996; Long et al., 2007). This variability has been attributed to range of factors such as varying degrees of baseline vitamin A deficiencies and differences in vitamin A dosing strategies (Dibley et al., 1996; Long et al., 2007). In mice, vitamin A availability alters the severity of infection and regulates repair of infection-induced epithelial damage (McDaniel et al., 2015; Mielke et al., 2013; Snyder et al., 2019).
IECs in the small intestine express both RA-generating enzymes and RARs that are activated by RA, thereby playing a central role in vitamin A-dependent regulation. Our transcriptional analyses showed that induction of RAR targets during infection was higher with SFB-colonization and indicated RAR targets are enriched in host defense pathways. Consistent with this, recent studies have discovered that IEC-intrinsic RAR expression promotes defense by regulating antimicrobial peptide production (Gattu et al., 2019; Jijon et al., 2018). A potential increase in goblet cells has been described in the intestine of dnRARIEC mice (Iyer et al., 2020). While we did not observe significant differences in goblet cells, an increase in goblet cells would be expected to limit C. rodentium infection (Bhinder et al., 2014), as opposed to the elevated burden that occurs in dnRARIEC mice. Furthermore, the ability of IECs to sense RA is necessary for defense against Salmonella Typhimurium (Gattu et al., 2019; Iyer et al., 2020). Using a transgenic mouse model where IECs are unable to respond to RA, we observed that IEC-intrinsic activation of RAR by RA is necessary for defense against C. rodentium. Specifically, RA administration significantly lowered pathogen burdens in control mice compared to mice lacking the ability to activate RAR in IECs. Although we did not observe significant early protection against C. rodentium from RA-administration in mice with defective RAR specifically in IECs, RA also likely activates mucosal immune cells, consistent with studies showing dendritic cells, macrophages and innate-lymphoid cells also respond to RA during infection (Erkelens and Mebius, 2017; Gundra et al., 2017; Kim et al., 2015; Zeng et al., 2016). Additionally, while our studies indicate Nos2 as one downstream target in IECs that mediates RA-induced protection against C. rodentium, we cannot exclude that other factors, such as ILCs or IL-22, also contribute.
SFB-dependent defense initially occurred independently from CD4+ T cells, however, both SFB and RA are known to regulate development and function of adaptive immune cells, particularly Th17 cells which promote C. rodentium clearance (Symonds et al., 2009). Th17 cell regulation by RA appears to be largely context and dose dependent. RA supplementation at pharmacological levels has been shown to suppress Th17 differentiation and promote regulatory T cells (Benson et al., 2007; Mucida et al., 2007). In contrast, others have found that physiological concentrations of RA stimulation instead promote Th17 skewing of CD4+ T cells both in vitro and in vivo (Takahashi et al., 2012; Uematsu et al., 2008; Wang et al., 2010). Consistent with this evidence, RARα-deficient T cells are also unable to differentiate into Th17 cells in vitro under Th17-polarizing conditions (Hall et al., 2011b). Importantly, mice fed vitamin-A deficient diet are deficient in Th17 cells, further indicating that RA is required for in vivo development and/or maintenance of these cells (Cha et al., 2010; Wang et al., 2010). The presence of SFB in the intestinal microbiota of mice was shown to drive Th17 cell expansion by inducing epithelial expression of Serum Amyloid A (SAA) proteins (Ivanov et al., 2009; Sano et al., 2015). Intestinal SAA expression requires dietary vitamin A and is directly regulated by epithelial RARs (Gattu et al., 2019).
These findings, in combination with our current work, support a model in which increased RA levels and enhanced epithelial RAR activation by SFB promotes innate epithelial defense, and simultaneously drives Th17 cell differentiation, potentially through RA-dependent transcriptional regulation of SAAs. Considering that SFB directly interacts with IECs and even undergoes vesicle-mediated communication (Ladinsky et al., 2019), we suspect that SFB may provide a local dose of RA that transcriptionally primes host epithelial cells. Interestingly, SFB does not induce similar H3K27Ac levels within the Saa1/2 and Nos2 genes of colonic IECs, suggesting tissue site specificity. SFB colonizes the terminal ileum, however C. rodentium infects the colon. While we cannot exclude that a subset of epigenetic changes will overlap at distinct locations, it is unlikely to be identical given the localization of SFB to the ileum. The spatial separation of SFB and C. rodentium colonization implies that SFB does not directly block the pathogen niche. Our mechanistic analyses identified Nos2 as an epigenetically modified SFB-sensitive RAR target in the ileum that is necessary for RA-induced protection against C. rodentium. Interestingly we found that NO, the antimicrobial metabolite generated by Nos2, was increased in both ileum and colonic contents from SFB-colonized mice relative to GF mice. Thus, we suspect that NO production through microbiota-dependent regulation of Nos2 and NO may affect the pathogen as it travels in the ileum or during colonic colonization.
Despite the abundance of evidence linking host immunity and RA, relatively little is known about how the microbiota regulates RA. It was recently shown that intestinal tissue RA levels were lower in conventionally-housed mice compared to GF mice due to decreased expression of Rdh7, an enzyme that oxidizes retinol to retinal (Grizotte-Lake et al., 2018). This downregulation was found to be driven primarily by Clostridial species. Furthermore, expansion of Proteobacteria following antibiotic depletion of Clostridia correlated with induction of host Rdh7 expression, suggesting that distinct commensal bacterial species may differentially regulate RA. Our data describe a mechanism in which epithelial expression of RA-sensitive factors in mice mono-colonized with SFB, or intestinal organoids exposed to SFB, is largely bacterial-dependent. SFB induced RA-target genes Saa1/2 expression in organoids despite inhibition of mammalian Aldh1a2. Further, bacterial ALDH was necessary to produce RA, and mutation of this enzyme inhibited RA production. Taken together, these findings indicate a role for bacterial-derived RA in epithelial regulation. Thus, in addition to mammalian-produced RA, SFB also contributes RA in the intestinal environment and microbial generated RA is relevant to regulation of IECs and C. rodentium. However, the magnitude of induction in vitro was not as large as that observed in vivo, which supports that other factors not present in culture, such as IL-22, can also contribute to activation of RA-sensitive pathways in the host. Given this regulation, we anticipate that commensal bacterial species likely differentially modulate intestinal RA levels through microbe-intrinsic mechanisms and/or distinct host-dependent pathways.
Investigation of vitamin A metabolism has largely focused on mammalian enzymes, as RA production has generally been considered a mammal-specific reaction (Biesalski et al., 2007). Whether bacteria directly contribute to vitamin A metabolism has been largely unexplored. Prior studies demonstrated that E. coli were capable of generating retinal and retinyl acetate, and potentially retinoic acid, in culture (Jang et al., 2011, 2015). Genetic manipulation of putative endogenous genes in E.coli involved in converting retinol to retinal (ybbo) and RA (puuC, eutC) altered retinoid production by E. coli (Jang et al., 2011, 2015). Furthermore, a bacterial ALDH expressed in B. cereus, a gram-positive bacterium commonly found in the gastrointestinal tract of mammals, was able to directly convert retinal to RA in vitro (Hong et al., 2016). We demonstrate that commensal SFB and B. bifidum express ALDH proteins that produce RA in vitro and in vivo, and that mammalian intestinal epithelial regulation is sensitive to the catalytic activity of these bacterial enzymes. In addition, dietary vitamin A and enzymatically active bacterial ALDH are required for SFB to increase luminal RA concentrations and protect against C. rodentium. Together these findings indicate that bacteria inherently harbor retinoid metabolism pathways and revealed important dietary implications for bacterial metabolism of vitamin A in host defense.
In addition to the protective effects of bacterial RA on the host, we anticipate that there are likely bacterial-intrinsic benefits to metabolizing vitamin A. While we did not observe obvious differences in cultured bacterial growth with short-term vitamin A exposure, it is possible that metabolizing vitamin A entails a competitive advantage in the intestine. Vitamin A availability affects cellular zinc absorption, and vice versa (Christian and West, 1998; Rahman et al., 2002; Smith, 1980). Zinc is an essential micronutrient for all organisms including bacteria and is required for normal cellular physiology. However, excess zinc is toxic to bacteria and thus must be tightly controlled (Hantke, 2005; McDevitt et al., 2011). Zinc is most abundant in the intestine, so it is plausible that bacterial metabolism of retinoids improves absorption of zinc to maintain non-bactericidal levels in the intestinal environment. Alternatively, oxidation of vitamin A to retinol may provide bacteria with important reducing equivalents in the form of NADH and NADPH that are needed for energy metabolism (Spaans et al., 2015; Sporer et al., 2017).
Collectively, this work reveals a new level of regulation in which microbiota-derived RA enhances host defense to a bacterial pathogen. To our knowledge, the ability of commensal bacteria, such as SFB, to provide a direct source of RA in the intestine that physiologically regulates the mammalian host has not been described. Expansion of analyses to other human cohorts and dissecting upstream and downstream factors in this pathway will form the basis for further investigations into commensal bacterial RA production. In addition to SFB and B. cereus, we found that microbial RA regulation extended to another beneficial bacterial strain, the probiotic Bifidobacterium bifidum, and that microbial ALDH enzymes are indeed prevalent in healthy human microbiota. Thus, these findings support development of pro- and prebiotic approaches that induce elevated intestinal RA in order to decrease enteric infection.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Theresa Alenghat (Theresa.Alenghat@cchmc.org).
Materials availability
Mouse strains, bacterial strains and plasmids used in this study are available from the lead contact with a completed Materials Transfer Agreement.
Data and code availability
ChIP-seq and RNA-seq data have been deposited to the Gene Expression Omnibus (GEO) with the study accession numbers GSE182628 and GSE182630 and are publicly available as of the date of publication. This paper also analyzes existing, publicly available human stool metagenomic sequencing data (NCBI SRA: SRP057027)(Lewis et al., 2015). Accession numbers for these datasets are also listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-Histone H3 (ac-K27) | Abcam | Cat# ab4729 |
| Rat monoclonal anti-CD4 | Bio X Cell | Cat# BE0003-1; Clone: GK1.5 |
| Rat monoclonal IgG2B | Bio X Cell | Cat# BE0090; Clone: LTF-2 |
| Mouse Monoclonal anti-IL17A | Bio X Cell | Cat# BE0173; Clone: 17F3 |
| Mouse monoclonal IgG1 | Bio X Cell | Cat# BE0083; Clone: MOPC-21 |
| Mouse Anti-CD45.2 BUV395 | BD Biosciences | Cat# 564616; Clone: 104 |
| Rat Anti-CD4 APC-eFluor 780 | eBioscience | Cat# 47-0042-02; Clone: RM4-5 |
| Rat Anti-CD8a APC | eBioscience | Cat# 17-0081-82; Clone: 53-6.7 |
| Anti-GFP Alexa Fluor 488 | Invitrogen | Cat# A21311 |
| Bacterial and virus strains | ||
| Segmented filamentous bacteria (SFB) | Yakult Central Institute | N/A |
| GFP-Citrobacter rodentium | Dr. Bruce A. Vallance | N/A |
| Bacillus cereus | ATCC | ATCC 10876 |
| Bifidobacterium bifidum | ATCC | ATCC 29521 |
| Escherichia coli BL21(DE3) | Thermo Scientific | Cat# EC0114 |
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| Difco™ MacConkey Agar | BD | Cat# 212123 |
| LB Broth (Lennox) Tablet microbial | Sigma Aldrich | Cat# L7275 |
| All-trans retinoic acid (atRA) | Sigma Aldrich | Cat# R2625 |
| DMSO (Dimethyl sulfoxide) | Sigma Aldrich | Cat# D8418 |
| Corn oil | Fisher Scientific | Cat# S25271 |
| RARi (BMS 493) | Torcis Bioscience | Cat# 3509 |
| Aldh1a2 inhibitor (WIN 18,446) | Cayman Chemical | Cat# 14018 |
| All-trans retinol | Sigma Aldrich | Cat# R7632 |
| Vitamin A-deficient diet | Envigo Teklad Diets | Cat# TD.86143 |
| Vitamin A-control diet | Envigo Teklad Diets | Cat# TD.91280 |
| Brain Heart Infusion broth | Sigma Aldrich | Cat# 53286 |
| MRS broth | Sigma Aldrich | Cat# 69966 |
| L-Cysteine | Sigma Aldrich | Cat# C7352 |
| Kanamycin sulfate | Gibco | Cat# 1866779 |
| Ampicillin sodium salt | Sigma Aldrich | Cat# A0166 |
| Fluorescein isothiocyanate(FITC)–dextran | Sigma Aldrich | Cat# 46944 |
| IPTG (Isopropyl β-D-1-thiogalactopyranoside) | Thermo Scientific | Catt# 15529019 |
| Fixable Violet Dead Cell Stain Kit | Invitrogen | Cat# L34964 |
| RNAprotect Bacteria Reagent | Qiagen | Cat# 76506 |
| Power SYBR Green PCR Master Mix | Thermo Scientific | Cat# 4367659 |
| Alexa Fluor™ 594 Phalloidin | Invitrogen | Cat# A12381 |
| DAPI (4’,6-diamidino-2-phenylindole) | Life Technologies | Cat# D1306 |
| Fluoromount-G | Invitrogen | Cat# 00-4958 |
| Sodium Nitroprusside (SNP) | Sigma Aldrich | Cat# PHR1423 |
| Critical commercial assays | ||
| Mouse Retinoic Acid ELISA Kit | MyBiosource | Cat# MBS706971 |
| RNeasy Mini RNA Kit | Qiagen | Cat# 74104 |
| Pierce™ BCA Protein Assay Kit | Thermo Scientific | Cat# 23225 |
| DNase I, Amplification Grade | Invitrogen | Cat# 18068015 |
| Verso cDNA Synthesis Kit | Thermo Scientific | Cat# AB1453A |
| Nitric Oxide Assay Kit | Biovision | Cat# K262 |
| QIAamp Fast DNA Stool Mini Kit | Qiagen | Cat# 51604 |
| ALDH Activity Assay Kit | Sigma Aldrich | Cat# MAK082 |
| Deposited data | ||
| ChIP-seq raw and analyzed data | This Paper | GEO: GSE182628 |
| RNA-seq raw and analyzed data | This Paper | GEO: GSE182630 |
| Human stool metagenomic shotgun sequencing data | Lewis et al., 2015 | SRA: SRP057027 |
| Experimental models: Cell lines | ||
| Experimental models: Organisms/strains | ||
| Mouse: WT: C57BL/6J | The Jackson Laboratory | JAX: 000664 |
| Mouse: dnRARfl/fl: Gt(ROSA)26Sortm1(RARA*)Soc/HsvJ | The Jackson Laboratory | JAX: 029812 |
| Mouse: Nos2−/−: B6.129P2-Nos2tm1Lau/J | The Jackson Laboratory | JAX: 002609 |
| Mouse: Villin-Cre: B6.Cg-Tg(Vil1-cre)997Gum/J | The Jackson Laboratory | JAX: 004586 |
| Oligonucleotides | ||
| Primers for ChIP-qPCR and RT-qPCR, see Table S1 | This Paper | N/A |
| Recombinant DNA | ||
| pET-21a(+)-SFB-ALDHWT | This Paper | N/A |
| pET-21a(+)-SFB-ALDHMUT | This Paper | N/A |
| pET-21a(+)-B. bifidum-ALDHWT | This Paper | N/A |
| pET-21a(+)-B. bifidum-ALDHMUT | This Paper | N/A |
| Software and algorithms | ||
| Prism 8.0 | Graphpad | graphpad.com/scientific-software/prism/ |
| FlowJo | BD | flowjo.com/ |
| Biowardrobe | Kartashov and Barski, 2015 | biowardrobe.cchmc.org/ems/ |
| PscanChIP | Zambelli et al., 2013 | 159.149.160.88/pscan_chip_dev/ |
| DAVID Bioinformatics Resources | Huang et al., 2009 | david.ncifcrf.gov |
| Phyre2 | Kelley et al., 2015 | sbg.bio.ic.ac.uk/phyre2 |
| PyMOL Molecular Graphics System, 2.4 | Schrondinger, LLC | pymol.org/ |
| Other | ||
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Germ-free (GF) C57BL/6 mice were maintained in sterile isolators (Class Biologically Clean) or sealed positive pressure IVC racks (Allentown) in the CCHMC Gnotobiotic Mouse Facility. For mono-association studies, GF mice were colonized with singular commensal species suspended in sterile PBS via oral gavage (Bifidobacterium bifidum ATCC 29521) or by pre-colonized bedding (SFB). For SFB ALDH-expressing strains, GF mice were orally gavaged with a single dose of 109 CFU SFB-ALDHWT or SFB-ALDHMUT strains and maintained on water containing ampicillin (1g/L) and isopropylthio-β-galactoside (IPTG, 1mM) refreshed every 7 days. All GF and monoassociated mice were fed autoclaved food and water, and routinely monitored to ensure the absence of microbial contamination and/or assess level of colonization. C57BL/6 floxed dnRAR (Rajaii et al., 2008) mice were crossed to villin-Cre-recombinase expressing mice (Madison et al., 2002) to generate dnRARIEC mice. For all experiments, mice were used at 8–16 weeks old, age- and sex-matched and paired with littermates when possible. Animals were housed in ventilated cages up to 4 per cage in 12 hr light/dark cycles with unrestricted access to food and water. Nos2−/− mice (Laubach et al., 1995) were bred on site. For FITC-dextran intestinal permeability studies, mice were fasted for 4 hr and gavaged with FITC-dextran (0.6 mg/g body weight) diluted in PBS. Serum was collected 4 hr post-gavage and fluorescence intensity was measured at 485/530 nm using a micro-plate reader (Biotek Synergy 2). For vitamin A-deficiency studies, mice were fed irradiated vitamin A-deficient (Teklad, TD.86143, 0 IU vitamin A/g diet) or control (Teklad, TD.91280, 20 IU vitamin A/g diet) purified diets for at least 4 weeks. All mouse experiments were conducted according to the Institutional Animal Care and Use Committee (IACUC). Animals were cared for by a licensed veterinarian and proper steps were taken to ensure the welfare and minimize the suffering of all animals in the conducted studies.
Bacterial strains and culture
C. rodentium were cultured in vitro in 96-well round bottom plates with DMSO (Sigma), 1nM-10μM all-trans retinoic acid (RA, Sigma), 10–1000μM sodium nitroprusside (SNP, Sigma) or 40 μg/ml Kanamycin (Gibco) at 37°C shaking at medium speed in a microplate reader (Biotek Synergy 2). Bacterial density (OD600) was measured hourly over 16 hr. For bacterial retinol culture studies, bacteria were grown in liquid cultures (B. cereus: Brain-Heart Infusion broth (BHI, Sigma); B. bifidum: MRS broth (Sigma) with 0.05% cysteine (Sigma), in anaerobic chamber; ALDH-expressing E.coli BL21(DE3) strains: LB with ampicillin (Sigma, 1mg/ml)) overnight at 30°C or 37°C at 180 rpm for 16 hr. Bacterial suspensions were then washed in PBS and diluted 1:3 in fresh LB (Sigma) and incubated with 1μM all-trans retinol (Sigma) for 3 hr in a 24-well plate at 37°C with gentle shaking at 120 rpm under light-restricted conditions. To determine bacterial levels, fecal or cultured bacterial DNA was isolated using QIAamp Fast DNA Stool Mini Kit (Qiagen) following the kit protocol. Bacterial DNA was assessed by quantitative PCR (QuantStudio3; Applied Biosystems) using bacterial-specific or 16S primer pairs (Table S1).
Intestinal Organoids
Murine organoids were generated from ileal IECs isolated from male WT C57BL/6J mice as previously described (Woo et al., 2019; Wu et al., 2020). Dissected terminal ileums (12 cm) were opened longitudinally, scraped to remove intestinal contents and outer cells, washed repeatedly in ice-cold PBS, and cut into 1 cm pieces. Ileum pieces were incubated in Chelation Buffer (2 mM EDTA in PBS) for 30 min at 4°C with rotation. Tissues were subsequently transferred into new tubes containing Shaking Buffer (PBS, 43.3 mM sucrose, 54.9 mM sorbitol) and gently shaken by hand for 2–4 mins. Ileal crypts were resuspended and plated in Matrigel (Corning) overlaid with 500 μl organoid culture media (60% Advanced DMEM/F12 media supplemented with 10 mM HEPES, 2 mM L-glutamate, 40% L-WRN conditioned media, 1x N2 supplement, 1x B27 supplement, 50 ng/mL murine EGF, and 10 μM Y-27632 ROCK inhibitor). Culture media was refreshed every 3–4 days. Organoid cultures were treated with 1μM Aldh1a2 inhibitor WIN 18446 (Cayman Chemical) for 12 hours and then stimulated with 100 μl of SFB contents at 150mg/ml PBS for 24 hours. For bacterial culture treatments in organoids, 100 μl of liquid cultures were directly added to organoids containing 1μM retinol for 24 hours. After incubation, organoids were washed 3 times in PBS and lysed using the RNeasy kit (Qiagen).
METHOD DETAILS
C. rodentium infections
Mice were orally infected with 109 colony-forming units (CFUs) of C. rodentium suspended in sterile PBS. Post-infection CFUs were measured in stool homogenized in 500 μl PBS in a Tissue Lyser II at 30 Hz for 3 min. Homogenates were serially diluted 10-fold on MacConkey agar (BD) and CFUs were counted after 16 hr incubation at 37°C, normalized to fecal weight. For RA studies, mice were orally gavaged with 300μg RA or vehicle (DMSO) in 100μl corn oil q.d. 5 days prior to and during the infection. For RAR inhibitor (RARi) studies, 400μg BMS493 (Torcis Bioscience) suspended in 10% DMSO/corn oil was administered to mice via oral gavage q.o.d. over 6 days pre-infection and 6 days post-infection. For Aldh1a2 inhibition, mice were orally gavaged with 400 mg/kg of WIN 18446 (Cayman Chemical) or vehicle (DMSO) in 100 μl corn oil for 8 days q.o.d.
CD4+ T cell depletion and flow cytometry
CD4+ cells were depleted using anti-CD4 monoclonal depletion antibody (clone: GK1.5) or matching isotype control (Rat IgG2B). IL-17A neutralization was performed using anti-IL-17A monoclonal antibody (clone: 17F3) or matching isotype control (Mouse IgG1). Antibodies were administered intraperitoneally, 500μg per day every 3 days for a total of 3 doses. Efficacy of CD4+ depletion was determined in colonic lamina propria and spleen by flow cytometry. For intestinal lamina propria lymphocytes isolation, tissue pieces were washed with cold PBS and incubated in RPMI with 1 mg/ml Collagenase/Dispase for 30 min at 37°C with shaking at 200 rpm. Splenocytes were disrupted into single cell suspension by passing the organ through 70 μm filter and RBCs were lysed in ACK lysis buffer (Invitrogen) for 3 min. Cells were stained using the following monoclonal fluorescence-conjugated antibodies: BUV395 anti-CD45.2 (Clone: 104, BD Biosciences), APC-eFluor 780 anti-CD4 (Clone: RM4–5, eBioscience), and APC anti-CD8a (Clone: 53–6.7, eBioscience). All antibodies were diluted in FACS buffer (2% FBS, 0.01 Sodium Azide, PBS). Dead cells were gated out by using the Fixable Violet Dead Cell Stain Kit (Invitrogen). Samples were acquired on the BD LSRFortessa (BD Biosciences) and analyzed with FlowJo Software (Treestar).
IEC isolation and RNA analyses
IECs were isolated from distal small intestine (12 cm) or large intestine by shaking tissue in 1mM EDTA/1mM DTT 5% FBS at 37°C for 10 min as described previously (Alenghat et al., 2013). Bacteria were treated with RNAprotect Bacteria Reagent (Qiagen) for 5 min prior to RNA isolation. RNA was extracted from cells using the RNeasy Kit (Qiagen) according to manufacturer’s instructions. For RT-qPCR, RNA was treated with DNase I (Invitrogen) and reverse-transcribed with Verso reverse transcriptase (Thermo Scientific). Expression was compared using SYBR (Applied Biosystems) and analyzed in the linear range of amplification. Target gene expression was normalized to an unaffected control gene. All primers used this study are found in Table S1. For global expression analyses, 3–4 biological replicates of IECs from C. rodentium-infected GF and SFB-monoassociated mice were compared. Following removal of primers and barcodes, raw reads were processed using Kallisto, which employs pseudoalignment to assess compatibility between raw reads and genomic targets. Annotations were provided by UCSC with transcripts per million (TPM) as output, which were log2-transformed and baselined to the median of all samples. Further, transcripts were filtered to include only those with TPM >3 in 100% of samples in at least one condition. Differential expression was assessed with a moderated t-test with p<0.05 and fold-change>1.5. For gene ontology analyses, differential gene lists were submitted to DAVID bioinformatics database (david.ncifcrf.gov)(Huang et al., 2009). Pathway enrichment significance are displayed as log10-transformed p-values.
ChIP-seq
ChIP-seq on IECs was performed as described previously (Wu et al., 2020) with a few modifications. Briefly, cells were fixed for 10 min in 1% formaldehyde at room temperature, followed by quenching with 125mM glycine for 10 min. After a two-step wash with cold PBS, fixed cells were lysed, and nuclear extracts were washed in TE 0.1% SDS with protease inhibitors and sonicated using a S220 Focused-ultrasonicator (Covaris). Prior to immunoprecipitation, sheared chromatin was precleared for 20 min at 4°C using Protein G Dynabeads (Thermo Fisher Scientific). Immunoprecipitations were performed using fresh beads and anti-Histone H3 acetyl K27 (H3K27Ac) antibody (Abcam: ab4729) using a SX-8G IP-STAR automated system (Diagenode) with the following wash buffers: (1) RIPA 150mM NaCl, (2) RIPA 250mM NaCl, (3) LiCl 250mM, 0.5% sodium deoxycholate, NP40 0.5%, and (4) TE 0.2% Triton X-100. Immunoprecipitated chromatin were treated with Proteinase K (Thermo Fisher Scientific) at 42°C for 30 min, 65°C for 4 hr, and 15°C for 10 min in elution buffer (TE 250mM NaCl 0.3% SDS). Phenol:chloroform isoamyl alcohol with Tris-HCl (pH 8.0) and chloroform phase-separation were used to isolate DNA, followed by overnight ethanol precipitation. ChIP DNA was sequenced using Illumina HiSeq 2500 platform. ChIP-seq data were processed using analytic pipelines in galaxy (usegalaxy.org). Following raw read alignment to mm10, MACS2 was used for peak calling and differential peak detection. Peaks were visualized by the UCSC genome browser in Biowardrobe (Kartashov and Barski, 2015). Transcription factor-binding site motifs were identified within 150 bp of the center of the differential peaks using PscanChIP (JASPAR 2018 database)(Zambelli et al., 2013), displayed as the global p-value.
Nitric Oxide Quantification
Dissected mouse ileum and colon tissues were opened longitudinally and scraped using a clean microscope slide to collect mucosal scrape. Samples were homogenized in Nitric Oxide (NO) Assay Buffer (Biovision, K262) and treated with perchloric acid (PCA) and potassium hydroxide (KOH) to precipitate interfering proteins. Deproteinized samples were run on a Nitric Oxide Assay kit (Biovision, K262) according to manufacturer instructions. Briefly, samples were added to a 96-well plate and incubated with Nitrate Reductase and enzyme cofactor for 1 hr at room temperature and incubated with enhancer for an additional 10 min. 50 μl of Griess Reagent R1 and R2 were sequentially added to each well. Color was developed for 10 min at room temperature and absorbance was read at 540 nm using a micro-plate reader (Biotek Synergy 2).
Histological tissue analyses
Sections of intestine were fixed in 4% paraformaldehyde overnight at 4°C, paraffin embedded, sectioned, and stained with hematoxylin and eosin or periodic acid-Schiff/Alcian blue. For immunofluorescence, distal large intestine was fixed in 4% paraformaldehyde overnight at 4°C and then placed in 30% sucrose for 24 hr. Tissues were embedded in OCT compound and cut as frozen sections (10 μm). Frozen sections were thawed and blocked with 1% BSA for 1 hr at room temperature. The following antibodies were diluted in 0.5% BSA and incubated with the tissue for 1.5 hr at room temperature: Alexa Fluor 488-anti-GFP (5.0 μg/ml, Invitrogen) and Alexa Fluor 594-Phalloidin (1:200, Invitrogen). Nuclei were stained with DAPI (4’,6-Diamidino-2-Phenylindole, Dihydrochloride, 0.5 ug/ml, Life Technologies). Slides were washed and then mounted using Fluoromount-G (Invitrogen) and imaged on a Nikon A1R LUN-V inverted confocal microscope.
SFB and B. bifidum ALDH constructs
SFB ALDHWT (WP_007440235.1) and B. bifidum ALDHWT (WP_015438559.1) genes were codon-optimized for expression in E. coli and cloned into the NdeI/BamHI site of the pET-21a(+) plasmid (GenScript). ALDHMUT variants were generated by substituting the catalytic glutamate [E] with an alanine [A] residue at amino acid position 209 and 244 of SFB ALDH and B. bifidum ALDH, respectively. Plasmids were transformed into BL21(DE3) E. coli Competent Cells (Thermo Scientific) and positive transformants were selected using ampicillin (Sigma, 1mg/ml) and screened by PCR. Successfully transformed clones were grown in LB broth (Sigma) containing Ampicillin (Sigma, 1mg/ml) at 37°C 180 rpm until optical density at 600 (OD600) reached 0.4–0.6, and further cultured in presence of 0.5mM isopropylthio-β-galactoside (IPTG, Invitrogen) for 4 hours to induce protein expression.
Retinoic acid quantification
Intestinal contents and IECs pellets were collected under dark conditions and homogenized in PBS. Extracts were run on a retinoic acid ELISA kit (MyBiosource, MBS706971) according to manufacturer instructions. Briefly, samples were incubated with 50 μl HRP-conjugated antibody for 40 min at 37°C, washed 5 times with wash buffer, and incubated with TMB substrate for 20 min at 37°C. The reaction was quenched, and absorbance was measured of each well using a micro-plate reader (Biotek Synergy 2) set to 450 nm. For explant experiments, equal sections of terminal ileum were taken from GF and SFB-monoassociated mice and cultured in a 24-well plate with 1μM all-trans retinol (Sigma) for 3 hr at 37°C without light. RA was measured in culture supernatant or bacterial culture media after incubation. For RA quantification of ALDHWT and ALDHMUT strains, cultures were treated with 1μM all-trans retinol (Sigma) and IPTG for 4 hours.
Aldehyde dehydrogenase activity
Samples were homogenized in ice-cold ALDH assay buffer (Sigma, MAK082) for 3 min at 30 Hz and spun down at 13,000 × g for 10 min. ALDH activity was measured according to manufacturer’s instructions (Sigma, MAK082). Briefly, 50 μl of supernatants with equal protein concentrations determined using Pierce BCA Protein Assay Kit (Thermo Scientific) were combined with ALDH substrate and acetaldehyde in 96-well plates. Background controls were performed in parallel by omitting acetaldehyde from the reaction. Reactions were incubated at room temperature for 5 min and absorbance at 450 nm was measured in 5 min intervals using a micro-plate reader (Biotek Synergy 2). Enzymatic activity was calculated as a function of amount NADH generated over time.
Protein modeling and sequence alignment
To predict 3D structures, protein sequences were submitted to the Phyre2 server (http://www.sbg.bio.ic.ac.uk/phyre2) (Kelley et al., 2015) and modeled after existing Protein Data Bank templates (bcALDH1A1: PDB c4pt3C; SFB ALDH: PDB c6k0zA; B. bifidum ALDH: PDB c4f9iA). Figures were generated using the PyMOL Molecular Graphics System, Version 2.4 Schrodinger, LLC (https://pymol.org/). The superimpose function was used to determine structural similarity to bcALDH1A1, reported as the overall root-mean-square deviation (RMSD) value. Microbiome shotgun sequencing data obtained from the stool of 24 healthy patients that had not received antibiotics within 6 months prior to the study (Lewis et al., 2015) were aligned against the SFB ALDH (WP_007440235.1) and B. bifidum ALDH (WP_015438559.1) sequences using Bowtie2. Prevalence of these genes are expressed as ALDH counts per million mapped bacterial reads.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were performed using GraphPad Prism 8.0. Statistical significance was determined by Student’s t-test or ANOVA. All data meet the assumptions of the statistical tests used. Results are shown as mean ± SEM and considered significant at p<0.05 (*); p<0.01 (**); p<0.001 (***). Additional quantification and statistical information (including exact value of n, and what n represents) are included in the figures or figure legends where appropriate
DATA AND CODE AVAILABILITY
Datasets from this study have been deposited in the NCBI Gene Expression Omnibus (GEO) database under the following accession identifiers: RNA-seq (GSE182630), ChIP-seq (GSE182628).
Supplementary Material
LIFE SCIENCE TABLE WITH EXAMPLES FOR AUTHOR REFERENCE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-Snail | Cell Signaling Technology | Cat#3879S; RRID: AB_2255011 |
| Mouse monoclonal anti-Tubulin (clone DM1A) | Sigma-Aldrich | Cat#T9026; RRID: AB_477593 |
| Rabbit polyclonal anti-BMAL1 | This paper | N/A |
| Bacterial and virus strains | ||
| pAAV-hSyn-DIO-hM3D(Gq)-mCherry | Krashes et al., 2011 | Addgene AAV5; 44361-AAV5 |
| AAV5-EF1a-DIO-hChR2(H134R)-EYFP | Hope Center Viral Vectors Core | N/A |
| Cowpox virus Brighton Red | BEI Resources | NR-88 |
| Zika-SMGC-1, GENBANK: KX266255 | Isolated from patient (Wang et al., 2016) | N/A |
| Staphylococcus aureus | ATCC | ATCC 29213 |
| Streptococcus pyogenes: M1 serotype strain: strain SF370; M1 GAS | ATCC | ATCC 700294 |
| Biological samples | ||
| Healthy adult BA9 brain tissue | University of Maryland Brain & Tissue Bank; http://medschool.umaryland.edu/btbank/ | Cat#UMB1455 |
| Human hippocampal brain blocks | New York Brain Bank | http://nybb.hs.columbia.edu/ |
| Patient-derived xenografts (PDX) | Children’s Oncology Group Cell Culture and Xenograft Repository | http://cogcell.org/ |
| Chemicals, peptides, and recombinant proteins | ||
| MK-2206 AKT inhibitor | Selleck Chemicals | S1078; CAS: 1032350-13-2 |
| SB-505124 | Sigma-Aldrich | S4696; CAS: 694433-59-5 (free base) |
| Picrotoxin | Sigma-Aldrich | P1675; CAS: 124-87-8 |
| Human TGF-β | R&D | 240-B; GenPept: P01137 |
| Activated S6K1 | Millipore | Cat#14-486 |
| GST-BMAL1 | Novus | Cat#H00000406-P01 |
| Critical commercial assays | ||
| EasyTag EXPRESS 35S Protein Labeling Kit | PerkinElmer | NEG772014MC |
| CaspaseGlo 3/7 | Promega | G8090 |
| TruSeq ChIP Sample Prep Kit | Illumina | IP-202-1012 |
| Deposited data | ||
| Raw and analyzed data | This paper | GEO: GSE63473 |
| B-RAF RBD (apo) structure | This paper | PDB: 5J17 |
| Human reference genome NCBI build 37, GRCh37 | Genome Reference Consortium | http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/ |
| Nanog STILT inference | This paper; Mendeley Data | http://dx.doi.org/10.17632/wx6s4mj7s8.2 |
| Affinity-based mass spectrometry performed with 57 genes | This paper; Mendeley Data | Table S8; http://dx.doi.org/10.17632/5hvpvspw82.1 |
| Experimental models: Cell lines | ||
| Hamster: CHO cells | ATCC | CRL-11268 |
| D. melanogaster: Cell line S2: S2-DRSC | Laboratory of Norbert Perrimon | FlyBase: FBtc0000181 |
| Human: Passage 40 H9 ES cells | MSKCC stem cell core facility | N/A |
| Human: HUES 8 hESC line (NIH approval number NIHhESC-09-0021) | HSCI iPS Core | hES Cell Line: HUES-8 |
| Experimental models: Organisms/strains | ||
| C. elegans: Strain BC4011: srl-1(s2500) II; dpy-18(e364) III; unc-46(e177)rol-3(s1040) V. | Caenorhabditis Genetics Center | WB Strain: BC4011; WormBase: WBVar00241916 |
| D. melanogaster: RNAi of Sxl. y[1] sc[*] v[1]; P{TRiP.HMS00609}attP2 | Bloomington Drosophila Stock Center | BDSC:34393; FlyBase: FBtp0064874 |
| S. cerevisiae: Strain background: W303 | ATCC | ATTC: 208353 |
| Mouse: R6/2: B6CBA-Tg(HDexon1)62Gpb/3J | The Jackson Laboratory | JAX: 006494 |
| Mouse: OXTRfl/fl: B6.129(SJL)-Oxtrtm1.1Wsy/J | The Jackson Laboratory | RRID: IMSR_JAX:008471 |
| Zebrafish: Tg(Shha:GFP)t10: t10Tg | Neumann and Nuesslein-Volhard, 2000 | ZFIN: ZDB-GENO-060207-1 |
| Arabidopsis: 35S::PIF4-YFP, BZR1-CFP | Wang et al., 2012 | N/A |
| Arabidopsis: JYB1021.2: pS24(AT5G58010)::cS24:GFP(-G):NOS #1 | NASC | NASC ID: N70450 |
| Oligonucleotides | ||
| siRNA targeting sequence: PIP5K I alpha #1: ACACAGUACUCAGUUGAUA | This paper | N/A |
| Primers for XX, see Table SX | This paper | N/A |
| Primer: GFP/YFP/CFP Forward: GCACGACTTCTTCAAGTCCGCCATGCC | This paper | N/A |
| Morpholino: MO-pax2a GGTCTGCTTTGCAGTGAATATCCAT | Gene Tools | ZFIN: ZDB-MRPHLNO-061106-5 |
| ACTB (hs01060665_g1) | Life Technologies | Cat#4331182 |
| RNA sequence: hnRNPA1_ligand: UAGGGACUUAGGGUUCUCUCUAGGGACUUAGGGUUCUCUCUAGGGA | This paper | N/A |
| Recombinant DNA | ||
| pLVX-Tight-Puro (TetOn) | Clonetech | Cat#632162 |
| Plasmid: GFP-Nito | This paper | N/A |
| cDNA GH111110 | Drosophila Genomics Resource Center | DGRC:5666; FlyBase:FBcl0130415 |
| AAV2/1-hsyn-GCaMP6- WPRE | Chen et al., 2013 | N/A |
| Mouse raptor: pLKO mouse shRNA 1 raptor | Thoreen et al., 2009 | Addgene Plasmid #21339 |
| Software and algorithms | ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Samtools | Li et al., 2009 | http://samtools.sourceforge.net/ |
| Weighted Maximal Information Component Analysis v0.9 | Rau et al., 2013 | https://github.com/ChristophRau/wMICA |
| ICS algorithm | This paper; Mendeley Data | http://dx.doi.org/10.17632/5hvpvspw82.1 |
| Other | ||
| Sequence data, analyses, and resources related to the ultra-deep sequencing of the AML31 tumor, relapse, and matched normal | This paper | http://aml31.genome.wustl.edu |
| Resource website for the AML31 publication | This paper | https://github.com/chrisamiller/aml31SuppSite |
PHYSICAL SCIENCE TABLE WITH EXAMPLES FOR AUTHOR REFERENCE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| QD605 streptavidin conjugated quantum dot | Thermo Fisher Scientific | Cat#Q10101MP |
| Platinum black | Sigma-Aldrich | Cat#205915 |
| Sodium formate BioUltra, ≥99.0% (NT) | Sigma-Aldrich | Cat#71359 |
| Chloramphenicol | Sigma-Aldrich | Cat#C0378 |
| Carbon dioxide (13C, 99%) (<2% 18O) | Cambridge Isotope Laboratories | CLM-185-5 |
| Poly(vinylidene fluoride-co-hexafluoropropylene) | Sigma-Aldrich | 427179 |
| PTFE Hydrophilic Membrane Filters, 0.22 μm, 90 mm | Scientificfilters.com/TischScientific | SF13842 |
| Critical commercial assays | ||
| Folic Acid (FA) ELISA kit | Alpha Diagnostic International | Cat# 0365-0B9 |
| TMT10plex Isobaric Label Reagent Set | Thermo Fisher | A37725 |
| Surface Plasmon Resonance CM5 kit | GE Healthcare | Cat#29104988 |
| NanoBRET Target Engagement K-5 kit | Promega | Cat#N2500 |
| Deposited data | ||
| B-RAF RBD (apo) structure | This paper | PDB: 5J17 |
| Structure of compound 5 | This paper; Cambridge Crystallographic Data Center | CCDC: 2016466 |
| Code for constraints-based modeling and analysis of autotrophic E. coli | This paper | https://gitlab.com/elad.noor/sloppy/tree/master/rubisco |
| Software and algorithms | ||
| Gaussian09 | Frish et al., 2013 | https://gaussian.com |
| Python version 2.7 | Python Software Foundation | https://www.python.org |
| ChemDraw Professional 18.0 | PerkinElmer | https://www.perkinelmer.com/category/chemdraw |
| Weighted Maximal Information Component Analysis v0.9 | Rau et al., 2013 | https://github.com/ChristophRau/wMICA |
| Other | ||
| DASGIP MX4/4 Gas Mixing Module for 4 Vessels with a Mass Flow Controller | Eppendorf | Cat#76DGMX44 |
| Agilent 1200 series HPLC | Agilent Technologies | https://www.agilent.com/en/products/liquid-chromatography |
| PHI Quantera II XPS | ULVAC-PHI, Inc. | https://www.ulvac-phi.com/en/products/xps/phi-quantera-ii/ |
Highlights.
Commensal Segmented filamentous bacteria protect against C. rodentium infection
Commensal bacteria express enzymes that generate retinoic acid in the intestine
Commensal bacterial-derived retinoic acid promotes epithelial defense in the host
Acknowledgements
We thank A. Herr, and the Way, Qualls, Haslam, and Deshmukh labs for useful discussions and members of the Alenghat lab for critical reading of the manuscript. We thank CCHMC Veterinary Services, Research Flow Cytometry Core, Pathology Research Core, and S. Jagannathan, and A. Barski for services and technical assistance. We thank the Yakult Central Institute for providing SFB. This research is supported by the National Institutes of Health (DK114123, DK116868 to T.A., and F32AI147591 to E.M.E.), Cardell Fellowship to V.W., Pew Charitable Trust, and a Kenneth Rainin Foundation award to T.A. T.A. holds an Investigator in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund. This project is supported in part by PHS grant P30 DK078392 and the CCHMC Trustee Award and Procter Scholar’s Program.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abt MC, and Pamer EG (2014). Commensal bacteria mediated defenses against pathogens. Curr. Opin. Immunol. 29, 16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alenghat T, Osborne LC, Saenz SA, Kobuley D, Ziegler CGK, Mullican SE, Choi I, Grunberg S, Sinha R, Wynosky-Dolfi M, et al. (2013). Histone deacetylase 3 coordinates commensal-bacteria-dependent intestinal homeostasis. Nature 504, 153–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amatullah H, and Jeffrey KL (2020). Epigenome-metabolome-microbiome axis in health and IBD. Curr. Opin. Microbiol. 56, 97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari I, Raddatz G, Gutekunst J, Ridnik M, Cohen D, Abu-Remaileh M, Tuganbaev T, Shapiro H, Pikarsky E, Elinav E, et al. (2020). The microbiota programs DNA methylation to control intestinal homeostasis and inflammation. Nat. Microbiol. 5, 610–619. [DOI] [PubMed] [Google Scholar]
- Arnold SLM, Kent T, Hogarth CA, Griswold MD, Amory JK, and Isoherranen N (2015). Pharmacological inhibition of ALDH1A in mice decreases all-trans retinoic acid concentrations in a tissue specific manner. Biochem. Pharmacol. 95, 177–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrowsmith CH, Bountra C, Fish PV, Lee K, and Schapira M (2012). Epigenetic protein families: A new frontier for drug discovery. Nat. Rev. Drug Discov. 11, 384–400. [DOI] [PubMed] [Google Scholar]
- Atarashi K, Tanoue T, Ando M, Kamada N, Nagano Y, Narushima S, Suda W, Imaoka A, Setoyama H, Nagamori T, et al. (2015). Th17 Cell Induction by Adhesion of Microbes to Intestinal Epithelial Cells. Cell 163, 367–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson A, Pifer R, Behrendt CL, Hooper LV, and Yarovinsky F (2009). Gut Commensal Bacteria Direct a Protective Immune Response against Toxoplasma gondii. Cell Host Microbe 6, 187–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson MJ, Pino-Lagos K, Rosemblatt M, and Noelle RJ (2007). All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. J. Exp. Med. 204, 1765–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhinder G, Stahl M, Sham HP, Crowley SM, Morampudi V, Dalwadi U, Ma C, Jacobson K, and Vallance BA (2014). Intestinal epithelium-specific MyD88 signaling impacts host susceptibility to infectious colitis by promoting protective goblet cell and antimicrobial responses. Infect. Immun. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biesalski HK, Chichili GR, Frank J, von Lintig J, and Nohr D (2007). Conversion of β-Carotene to Retinal Pigment. Vitam. Horm. 75, 117–130. [DOI] [PubMed] [Google Scholar]
- Cabrera G, Fernández-Brando RJ, Abrey-Recalde MJ, Baschkier A, Pinto A, Goldstein J, Zotta E, Meiss R, Rivas M, and Palermo MS (2014). Retinoid levels influence enterohemorrhagic Escherichia coli infection and shiga toxin 2 susceptibility in mice. Infect. Immun. 82, 3948–3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha H-R, Chang S-Y, Chang J-H, Kim J-O, Yang J-Y, Kim C-H, and Kweon M-N (2010). Downregulation of Th17 Cells in the Small Intestine by Disruption of Gut Flora in the Absence of Retinoic Acid. J. Immunol. 184, 6799–6806. [DOI] [PubMed] [Google Scholar]
- Chang PV, Hao L, Offermanns S, and Medzhitov R (2014). The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl. Acad. Sci. U. S. A. 111, 2247–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian P, and West KP (1998). Interactions between zinc and vitamin A: An update. Am. J. Clin. Nutr. 68, 435S–441S. [DOI] [PubMed] [Google Scholar]
- Chung H, Pamp SJ, Hill JA, Surana NK, Edelman SM, Troy EB, Reading NC, Villablanca EJ, Wang S, Mora JR, et al. (2012). Gut immune maturation depends on colonization with a host-specific microbiota. Cell 149, 1578–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. (2010). Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. U. S. A. 107, 21936–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalile B, Van Oudenhove L, Vervliet B, and Verbeke K (2019). The role of short-chain fatty acids in microbiota–gut–brain communication. Nat. Rev. Gastroenterol. Hepatol. 16, 461–473. [DOI] [PubMed] [Google Scholar]
- Dibley MJ, Sadjimin T, Kjolhede CL, and Moulton LH (1996). Vitamin A supplementation fails to reduce incidence of acute respiratory illness and diarrhea in preschool-age Indonesian children. J. Nutr. 126, 434–442. [DOI] [PubMed] [Google Scholar]
- Erkelens MN, and Mebius RE (2017). Retinoic Acid and Immune Homeostasis: A Balancing Act. Trends Immunol. 38, 168–180. [DOI] [PubMed] [Google Scholar]
- Fellows R, Denizot J, Stellato C, Cuomo A, Jain P, Stoyanova E, Balázsi S, Hajnády Z, Liebert A, Kazakevych J, et al. (2018). Microbiota derived short chain fatty acids promote histone crotonylation in the colon through histone deacetylases. Nat. Commun. 9, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, Nakanishi Y, Uetake C, Kato K, Kato T, et al. (2013). Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504, 446–450. [DOI] [PubMed] [Google Scholar]
- Gallo RL, and Hooper LV (2012). Epithelial antimicrobial defence of the skin and intestine. Nat. Rev. Immunol. 12, 503–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganal SC, Sanos SL, Kallfass C, Oberle K, Johner C, Kirschning C, Lienenklaus S, Weiss S, Staeheli P, Aichele P, et al. (2012). Priming of Natural Killer Cells by Nonmucosal Mononuclear Phagocytes Requires Instructive Signals from Commensal Microbiota. Immunity 37, 171–186. [DOI] [PubMed] [Google Scholar]
- Garland CD, Lee A, and Dickson MR (1982). Segmented filamentous bacteria in the rodent small intestine: Their colonization of growing animals and possible role in host resistance to Salmonella. Microb. Ecol. 8, 181–190. [DOI] [PubMed] [Google Scholar]
- Gattu S, Bang Y-J, Pendse M, Dende C, Chara AL, Harris TA, Wang Y, Ruhn KA, Kuang Z, Sockanathan S, et al. (2019). Epithelial retinoic acid receptor β regulates serum amyloid A expression and vitamin A-dependent intestinal immunity. Proc. Natl. Acad. Sci. 116, 2018–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y, Panea C, Nakato G, Cebula A, Lee C, Diez MG, Laufer TM, Ignatowicz L, and Ivanov II (2014). Segmented filamentous bacteria antigens presented by intestinal dendritic cells drive mucosal Th17 cell differentiation. Immunity 40, 594–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green HN, and Mellanby E (1928). Vitamin a as an anti-infective agent. Br. Med. J. 2, 691–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grizotte-Lake M, Zhong G, Duncan K, Kirkwood J, Iyer N, Smolenski I, Isoherranen N, and Vaishnava S (2018). Commensals Suppress Intestinal Epithelial Cell Retinoic Acid Synthesis to Regulate Interleukin-22 Activity and Prevent Microbial Dysbiosis. Immunity 49, 1103–1115.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundra UM, Girgis NM, Gonzalez MA, Tang MS, Van Der Zande HJP, Lin J. Da, Ouimet M, Ma LJ, Poles J, Vozhilla N, et al. (2017). Vitamin A mediates conversion of monocyte-derived macrophages into tissue-resident macrophages during alternative activation. Nat. Immunol. 18, 642–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JA, Grainger JR, Spencer SP, and Belkaid Y (2011a). The role of retinoic acid in tolerance and immunity. Immunity 35, 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JA, Cannons JL, Grainger JR, Dos Santos LM, Hand TW, Naik S, Wohlfert EA, Chou DB, Oldenhove G, Robinson M, et al. (2011b). Essential role for retinoic acid in the promotion of CD4+ T cell effector responses via retinoic acid receptor alpha. Immunity 34, 435–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hantke K (2005). Bacterial zinc uptake and regulators. Curr. Opin. Microbiol. 8, 196–202. [DOI] [PubMed] [Google Scholar]
- Heczko U, Abe A, and Finlay BB (2000). Segmented filamentous bacteria prevent colonization of enteropathogenic Escherichia coli O103 in rabbits. J. Infect. Dis. 181, 1027–1033. [DOI] [PubMed] [Google Scholar]
- Hong SH, Ngo HPT, Nam HK, Kim KR, Kang LW, and Oh DK (2016). Alternative biotransformation of retinal to retinoic acid or retinol by an aldehyde dehydrogenase from Bacillus cereus. Appl. Environ. Microbiol. 82, 3940–3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4. [DOI] [PubMed] [Google Scholar]
- Huang Z, Liu Y, Qi G, Brand D, and Zheng S (2018). Role of Vitamin A in the Immune System. J. Clin. Med. 7, 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, et al. (2009). Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer N, Grizotte-Lake M, Duncan K, Gordon SR, Palmer ACS, Calvin C, Zhong G, Isoherranen N, and Vaishnava S (2020). Epithelium intrinsic Vitamin A signaling coordinates pathogen clearance in the gut via IL-18. PLoS Pathog. 16, e1008360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang HJ, Yoon SH, Ryu HK, Kim JH, Wang CL, Kim JY, Oh DK, and Kim SW (2011). Retinoid production using metabolically engineered Escherichia coli with a two-phase culture system. Microb. Cell Fact. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang HJ, Ha BK, Zhou J, Ahn J, Yoon SH, and Kim SW (2015). Selective retinol production by modulating the composition of retinoids from metabolically engineered E. coli. Biotechnol. Bioeng. 112, 1604–1612. [DOI] [PubMed] [Google Scholar]
- Jijon HB, Suarez-Lopez L, Diaz OE, Das S, De Calisto J, Yaffe MB, Pittet MJ, Mora JR, Belkaid Y, Xavier RJ, et al. (2018). Intestinal epithelial cell-specific RARα depletion results in aberrant epithelial cell homeostasis and underdeveloped immune system. Mucosal Immunol. 11, 703–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson H, Hugerth LW, Sundh J, Lundin E, and Andersson AF (2020). Genome sequence of segmented filamentous bacteria present in the human intestine. Commun. Biol. 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiko GE, Ryu SH, Koues OI, Collins PL, Solnica-Krezel L, Pearce EJ, Pearce EL, Oltz EM, and Stappenbeck TS (2016). The Colonic Crypt Protects Stem Cells from Microbiota-Derived Metabolites. Cell 165, 1708–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada N, Kim YG, Sham HP, Vallance BA, Puente JL, Martens EC, and Núñez G (2012). Regulated virulence controls the ability of a pathogen to compete with the gut microbiota. Science (80-.). 336, 1325–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kartashov AV, and Barski A (2015). BioWardrobe: An integrated platform for analysis of epigenomics and transcriptomics data. Genome Biol. 16, 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN, and Sternberg MJE (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly D, Kotliar M, Woo V, Jagannathan S, Whitt J, Moncivaiz J, Aronow BJ, Dubinsky MC, Hyams JS, Markowitz JF, et al. (2018). Microbiota-sensitive epigenetic signature predicts inflammation in Crohn’s disease. JCI Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MH, Taparowsky EJ, and Kim CH (2015). Retinoic Acid Differentially Regulates The Migration Of Innate Lymphoid Cell Subsets To The Gut. Immunity 43, 107–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladinsky MS, Araujo LP, Zhang X, Veltri J, Galan-Diez M, Soualhi S, Lee C, Irie K, Pinker EY, Narushima S, et al. (2019). Endocytosis of commensal antigens by intestinal epithelial cells regulates mucosal T cell homeostasis. Science (80-.). 363, eaat4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laubach VE, Shesely EG, Smithies O, and Sherman PA (1995). Mice lacking inducible nitric oxide synthase are not resistant to lipopolysaccharide-induced death. Proc. Natl. Acad. Sci. U. S. A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavelle A, and Sokol H (2020). Gut microbiota-derived metabolites as key actors in inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 17, 223–237. [DOI] [PubMed] [Google Scholar]
- Lewis JD, Chen EZ, Baldassano RN, Otley AR, Griffiths AM, Lee D, Bittinger K, Bailey A, Friedman ES, Hoffmann C, et al. (2015). Inflammation, Antibiotics, and Diet as Environmental Stressors of the Gut Microbiome in Pediatric Crohn’s Disease. Cell Host Microbe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long KZ, García C, Santos JI, Rosado JL, Hertzmark E, DuPont HL, and Ko GP (2007). Vitamin A supplementation has divergent effects on norovirus infections and clinical symptoms among Mexican children. J. Infect. Dis. 196, 978–985. [DOI] [PubMed] [Google Scholar]
- Madison BB, Dunbar L, Qiao XT, Braunstein K, Braunstein E, and Gumucio DL (2002). Cis elements of the villin gene control expression in restricted domains of the vertical (crypt) and horizontal (duodenum, cecum) axes of the intestine. J. Biol. Chem. 277, 33275–33283. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Kunisawa A, Hattori T, Kawana S, Kitada Y, Tamada H, Kawano S, Hayakawa Y, Iida J, and Fukusaki E (2018). Free D-amino acids produced by commensal bacteria in the colonic lumen. Sci. Rep. 8, 17915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarville JL, Chen GY, Cuevas VD, Troha K, and Ayres JS (2020). Microbiota Metabolites in Health and Disease. Annu. Rev. Immunol. 38, 147–170. [DOI] [PubMed] [Google Scholar]
- McDaniel KL, Restori KH, Dodds JW, Kennett MJ, Ross AC, and Cantornaa MT (2015). Vitamin A-deficient hosts become nonsymptomatic reservoirs of Escherichia coli-like enteric infections. Infect. Immun. 83, 2984–2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDevitt CA, Ogunniyi AD, Valkov E, Lawrence MC, Kobe B, McEwan AG, and Paton JC (2011). A molecular mechanism for bacterial susceptibility to Zinc. PLoS Pathog. 7, e1002357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzler MA, Raja S, Elliott KH, Friedl RM, Tran NQH, Brugmann SA, Larsen M, and Sandell LL (2018). RDH10-mediated retinol metabolism and RARα-mediated retinoic acid signaling are required for submandibular salivary gland initiation. Dev. 145, dev164822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mielke LA, Jones SA, Raverdeau M, Higgs R, Stefanska A, Groom JR, Misiak A, Dungan LS, Sutton CE, Streubel G, et al. (2013). Retinoic acid expression associates with enhanced IL-22 production by γδ T cells and innate lymphoid cells and attenuation of intestinal inflammation. J. Exp. Med. 210, 1117–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, and Cheroutre H (2007). Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science (80-.). 317, 256–260. [DOI] [PubMed] [Google Scholar]
- Mundy R, MacDonald TT, Dougan G, Frankel G, and Wiles S (2005). Citrobacter rodentium of mice and man. Cell. Microbiol. 7, 1697–1706. [DOI] [PubMed] [Google Scholar]
- Navabi N, Whitt J, Wu S. en, Woo V, Moncivaiz J, Jordan MB, Vallance BA, Way SS, and Alenghat T (2017). Epithelial Histone Deacetylase 3 Instructs Intestinal Immunity by Coordinating Local Lymphocyte Activation. Cell Rep. 19, 1165–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omenetti S, Bussi C, Metidji A, Iseppon A, Lee S, Tolaini M, Li Y, Kelly G, Chakravarty P, Shoaie S, et al. (2019). The Intestine Harbors Functionally Distinct Homeostatic Tissue-Resident and Inflammatory Th17 Cells. Immunity 51, 77–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osbelt L, Thiemann S, Smit N, Lesker TR, Schröter M, Gálvez EJC, Schmidt-Hohagen K, Pils MC, Mühlen S, Dersch P, et al. (2020). Variations in microbiota composition of laboratory mice influence Citrobacter rodentium infection via variable short-chain fatty acid production. PLoS Pathog. 16, e1008448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik J, Haenisch M, Muller CH, Goldstein AS, Arnold S, Isoherranen N, Brabb T, Treuting PM, and Amory JK (2014). Inhibition of retinoic acid biosynthesis by the bisdichloroacetyldiamine WIN 18,446 markedly suppresses spermatogenesis and alters retinoid metabolism in mice. J. Biol. Chem. 289, 15104–15117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson LW, and Artis D (2014). Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 14, 141–153. [DOI] [PubMed] [Google Scholar]
- Price AE, Shamardani K, Lugo KA, Deguine J, Roberts AW, Lee BL, and Barton GM (2018). A Map of Toll-like Receptor Expression in the Intestinal Epithelium Reveals Distinct Spatial, Cell Type-Specific, and Temporal Patterns. Immunity 49, 560–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, and Wysocka J (2011). A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470, 279–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman MM, Wahed MA, Fuchs GJ, Baqui AH, and Alvarez JO (2002). Synergistic effect of zinc and vitamin A on the biochemical indexes of vitamin A nutrition in children. Am. J. Clin. Nutr. 75, 92–98. [DOI] [PubMed] [Google Scholar]
- Rajaii F, Bitzer ZT, Xu Q, and Sockanathan S (2008). Expression of the dominant negative retinoid receptor, RAR403, alters telencephalic progenitor proliferation, survival, and cell fate specification. Dev. Biol. 316, 371–382. [DOI] [PubMed] [Google Scholar]
- Ramanan D, and Cadwell K (2016). Intrinsic Defense Mechanisms of the Intestinal Epithelium. Cell Host Microbe 19, 434–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooks MG, and Garrett WS (2016). Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 16, 341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano T, Huang W, Hall JA, Yang Y, Chen A, Gavzy SJ, Lee JY, Ziel JW, Miraldi ER, Domingos AI, et al. (2015). An IL-23R/IL-22 Circuit Regulates Epithelial Serum Amyloid A to Promote Local Effector Th17 Responses. Cell 163, 381–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seamons A, Haenisch M, Meeker S, Pershutkina O, Brabb T, Treuting PM, and Paik J (2020). Protective effects of aldh1a enzyme inhibition on helicobacter-induced colitis in smad3−/− mice are associated with altered α4ß7 integrin expression on activated t cells. Nutrients 12, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semba RD (1999). Vitamin A and immunity to viral, bacterial and protozoan infections. Proc. Nutr. Soc. 58, 719–727. [DOI] [PubMed] [Google Scholar]
- Shi Z, Zou J, Zhang Z, Zhao X, Noriega J, Zhang B, Zhao C, Ingle H, Bittinger K, Mattei LM, et al. (2019). Segmented Filamentous Bacteria Prevent and Cure Rotavirus Infection. Cell 179, 644–658.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JC (1980). THE VITAMIN A- ZINC CONNECTION: A REVIEW. Ann. N. Y. Acad. Sci. 355, 62–75. [DOI] [PubMed] [Google Scholar]
- Snyder LM, McDaniel KL, Tian Y, Wei CH, Kennett MJ, Patterson AD, Catharine Ross A, and Cantorna MT (2019). Retinoic acid mediated clearance of Citrobacter rodentium in vitamin A deficient mice requires CD11b+ and T cells. Front. Immunol. 9, 3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer A (2008). Vitamin A deficiency and clinical disease: An historical overview. J. Nutr. 138, 1835–1839. [DOI] [PubMed] [Google Scholar]
- Spaans SK, Weusthuis RA, van der Oost J, and Kengen SWM (2015). NADPH-generating systems in bacteria and archaea. Front. Microbiol. 6, 742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sporer AJ, Kahl LJ, Price-Whelan A, and Dietrich LEP (2017). Redox-based regulation of bacterial development and behavior. Annu. Rev. Biochem. 86, 777–797. [DOI] [PubMed] [Google Scholar]
- Symonds EL, Riedel CU, O’Mahony D, Lapthorne S, O’Mahony L, and Shanahan F (2009). Involvement of T helper type 17 and regulatory T cell activity in Citrobacter rodentium invasion and inflammatory damage. Clin. Exp. Immunol. 157, 148–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H, Kanno T, Nakayamada S, Hirahara K, Sciumè G, Muljo SA, Kuchen S, Casellas R, Wei L, Kanno Y, et al. (2012). TGF-β and retinoic acid induce the microRNA miR-10a, which targets Bcl-6 and constrains the plasticity of helper T cells. Nat. Immunol. 29, 587–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Sugi Y, Hosono A, and Kaminogawa S (2009). Epigenetic Regulation of TLR4 Gene Expression in Intestinal Epithelial Cells for the Maintenance of Intestinal Homeostasis. J. Immunol. 183, 6522–6529. [DOI] [PubMed] [Google Scholar]
- Uematsu S, Fujimoto K, Jang MH, Yang BG, Jung YJ, Nishiyama M, Sato S, Tsujimura T, Yamamoto M, Yokota Y, et al. (2008). Regulation of humoral and cellular gut immunity by lamina propria dendritic cells expressing Toll-like receptor 5. Nat. Immunol. 9, 769–776. [DOI] [PubMed] [Google Scholar]
- Vallance BA, Deng W, De Grado M, Chan C, Jacobson K, and Finlay BB (2002). Modulation of inducible nitric oxide synthase expression by the attaching and effacing bacterial pathogen Citrobacter rodentium in infected mice. Infect. Immun. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Kang SG, HogenEsch H, Love PE, and Kim CH (2010). Retinoic Acid Determines the Precise Tissue Tropism of Inflammatory Th17 Cells in the Intestine. J. Immunol. 184, 5519–5526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikoff WR, Anfora AT, Liu J, Schultz PG, Lesley SA, Peters EC, and Siuzdak G (2009). Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. U. S. A. 106, 3698–3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo V, and Alenghat T (2017). Host–microbiota interactions: epigenomic regulation. Curr. Opin. Immunol. 44, 52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo V, Eshleman EM, Rice T, Whitt J, Vallance BA, and Alenghat T (2019). Microbiota Inhibit Epithelial Pathogen Adherence by Epigenetically Regulating C-Type Lectin Expression. Front. Immunol. 10, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization (2009). Global prevalence of vitamin A deficiency in populations at risk 1995–2005 : WHO global database on vitamin A deficiency. WHO; Iris 55. [Google Scholar]
- Wu S. en, Hashimoto-Hill S, Woo V, Eshleman EM, Whitt J, Engleman L, Karns R, Denson LA, Haslam DB, and Alenghat T (2020). Microbiota-derived metabolite promotes HDAC3 activity in the gut. Nature 586, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Yu T, Huang X, Bilotta AJ, Xu L, Lu Y, Sun J, Pan F, Zhou J, Zhang W, et al. (2020). Intestinal microbiota-derived short-chain fatty acids regulation of immune cell IL-22 production and gut immunity. Nat. Commun. 11, 4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambelli F, Pesole G, and Pavesi G (2013). PscanChIP: Finding over-represented transcription factor-binding site motifs and their correlations in sequences from ChIP-Seq experiments. Nucleic Acids Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng R, Bscheider M, Lahl K, Lee M, and Butcher EC (2016). Generation and transcriptional programming of intestinal dendritic cells: Essential role of retinoic acid. Mucosal Immunol. 9, 183–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
ChIP-seq and RNA-seq data have been deposited to the Gene Expression Omnibus (GEO) with the study accession numbers GSE182628 and GSE182630 and are publicly available as of the date of publication. This paper also analyzes existing, publicly available human stool metagenomic sequencing data (NCBI SRA: SRP057027)(Lewis et al., 2015). Accession numbers for these datasets are also listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-Histone H3 (ac-K27) | Abcam | Cat# ab4729 |
| Rat monoclonal anti-CD4 | Bio X Cell | Cat# BE0003-1; Clone: GK1.5 |
| Rat monoclonal IgG2B | Bio X Cell | Cat# BE0090; Clone: LTF-2 |
| Mouse Monoclonal anti-IL17A | Bio X Cell | Cat# BE0173; Clone: 17F3 |
| Mouse monoclonal IgG1 | Bio X Cell | Cat# BE0083; Clone: MOPC-21 |
| Mouse Anti-CD45.2 BUV395 | BD Biosciences | Cat# 564616; Clone: 104 |
| Rat Anti-CD4 APC-eFluor 780 | eBioscience | Cat# 47-0042-02; Clone: RM4-5 |
| Rat Anti-CD8a APC | eBioscience | Cat# 17-0081-82; Clone: 53-6.7 |
| Anti-GFP Alexa Fluor 488 | Invitrogen | Cat# A21311 |
| Bacterial and virus strains | ||
| Segmented filamentous bacteria (SFB) | Yakult Central Institute | N/A |
| GFP-Citrobacter rodentium | Dr. Bruce A. Vallance | N/A |
| Bacillus cereus | ATCC | ATCC 10876 |
| Bifidobacterium bifidum | ATCC | ATCC 29521 |
| Escherichia coli BL21(DE3) | Thermo Scientific | Cat# EC0114 |
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| Difco™ MacConkey Agar | BD | Cat# 212123 |
| LB Broth (Lennox) Tablet microbial | Sigma Aldrich | Cat# L7275 |
| All-trans retinoic acid (atRA) | Sigma Aldrich | Cat# R2625 |
| DMSO (Dimethyl sulfoxide) | Sigma Aldrich | Cat# D8418 |
| Corn oil | Fisher Scientific | Cat# S25271 |
| RARi (BMS 493) | Torcis Bioscience | Cat# 3509 |
| Aldh1a2 inhibitor (WIN 18,446) | Cayman Chemical | Cat# 14018 |
| All-trans retinol | Sigma Aldrich | Cat# R7632 |
| Vitamin A-deficient diet | Envigo Teklad Diets | Cat# TD.86143 |
| Vitamin A-control diet | Envigo Teklad Diets | Cat# TD.91280 |
| Brain Heart Infusion broth | Sigma Aldrich | Cat# 53286 |
| MRS broth | Sigma Aldrich | Cat# 69966 |
| L-Cysteine | Sigma Aldrich | Cat# C7352 |
| Kanamycin sulfate | Gibco | Cat# 1866779 |
| Ampicillin sodium salt | Sigma Aldrich | Cat# A0166 |
| Fluorescein isothiocyanate(FITC)–dextran | Sigma Aldrich | Cat# 46944 |
| IPTG (Isopropyl β-D-1-thiogalactopyranoside) | Thermo Scientific | Catt# 15529019 |
| Fixable Violet Dead Cell Stain Kit | Invitrogen | Cat# L34964 |
| RNAprotect Bacteria Reagent | Qiagen | Cat# 76506 |
| Power SYBR Green PCR Master Mix | Thermo Scientific | Cat# 4367659 |
| Alexa Fluor™ 594 Phalloidin | Invitrogen | Cat# A12381 |
| DAPI (4’,6-diamidino-2-phenylindole) | Life Technologies | Cat# D1306 |
| Fluoromount-G | Invitrogen | Cat# 00-4958 |
| Sodium Nitroprusside (SNP) | Sigma Aldrich | Cat# PHR1423 |
| Critical commercial assays | ||
| Mouse Retinoic Acid ELISA Kit | MyBiosource | Cat# MBS706971 |
| RNeasy Mini RNA Kit | Qiagen | Cat# 74104 |
| Pierce™ BCA Protein Assay Kit | Thermo Scientific | Cat# 23225 |
| DNase I, Amplification Grade | Invitrogen | Cat# 18068015 |
| Verso cDNA Synthesis Kit | Thermo Scientific | Cat# AB1453A |
| Nitric Oxide Assay Kit | Biovision | Cat# K262 |
| QIAamp Fast DNA Stool Mini Kit | Qiagen | Cat# 51604 |
| ALDH Activity Assay Kit | Sigma Aldrich | Cat# MAK082 |
| Deposited data | ||
| ChIP-seq raw and analyzed data | This Paper | GEO: GSE182628 |
| RNA-seq raw and analyzed data | This Paper | GEO: GSE182630 |
| Human stool metagenomic shotgun sequencing data | Lewis et al., 2015 | SRA: SRP057027 |
| Experimental models: Cell lines | ||
| Experimental models: Organisms/strains | ||
| Mouse: WT: C57BL/6J | The Jackson Laboratory | JAX: 000664 |
| Mouse: dnRARfl/fl: Gt(ROSA)26Sortm1(RARA*)Soc/HsvJ | The Jackson Laboratory | JAX: 029812 |
| Mouse: Nos2−/−: B6.129P2-Nos2tm1Lau/J | The Jackson Laboratory | JAX: 002609 |
| Mouse: Villin-Cre: B6.Cg-Tg(Vil1-cre)997Gum/J | The Jackson Laboratory | JAX: 004586 |
| Oligonucleotides | ||
| Primers for ChIP-qPCR and RT-qPCR, see Table S1 | This Paper | N/A |
| Recombinant DNA | ||
| pET-21a(+)-SFB-ALDHWT | This Paper | N/A |
| pET-21a(+)-SFB-ALDHMUT | This Paper | N/A |
| pET-21a(+)-B. bifidum-ALDHWT | This Paper | N/A |
| pET-21a(+)-B. bifidum-ALDHMUT | This Paper | N/A |
| Software and algorithms | ||
| Prism 8.0 | Graphpad | graphpad.com/scientific-software/prism/ |
| FlowJo | BD | flowjo.com/ |
| Biowardrobe | Kartashov and Barski, 2015 | biowardrobe.cchmc.org/ems/ |
| PscanChIP | Zambelli et al., 2013 | 159.149.160.88/pscan_chip_dev/ |
| DAVID Bioinformatics Resources | Huang et al., 2009 | david.ncifcrf.gov |
| Phyre2 | Kelley et al., 2015 | sbg.bio.ic.ac.uk/phyre2 |
| PyMOL Molecular Graphics System, 2.4 | Schrondinger, LLC | pymol.org/ |
| Other | ||
Datasets from this study have been deposited in the NCBI Gene Expression Omnibus (GEO) database under the following accession identifiers: RNA-seq (GSE182630), ChIP-seq (GSE182628).