

Abstract
Following on from the devastating spread of COVID‐19, a major global priority has been the production, procurement, and distribution of effective vaccines to ensure that the global pandemic reaches an end. However, concerns were raised about worrying side effects, particularly the occurrence of thrombosis and thrombocytopenia after administration of the Oxford/AstraZeneca and Johnson & Johnson's Janssen COVID‐19 vaccine, in a phenomenon being termed vaccine‐induced thrombotic thrombocytopenia (VITT). Similar to heparin‐induced thrombocytopenia (HIT), this condition has been associated with the development of anti‐platelet factor 4 antibodies, purportedly leading to neutrophil‐platelet aggregate formation. Although thrombosis has also been a common association with COVID‐19, the precise molecular mechanisms governing its occurrence are yet to be established. Recently, increasing evidence highlights the NLRP3 (NOD‐like, leucine‐rich repeat domains, and pyrin domain‐containing protein) inflammasome complex along with IL‐1β and effete neutrophils producing neutrophil extracellular traps (NETs) through NETosis. Herein, we propose and discuss that perhaps the incidence of VITT may be due to inflammatory reactions mediated via IL‐1β/NLRP3 inflammasome activation and consequent overproduction of NETs, where similar autoimmune mechanisms are observed in HIT. We also discuss avenues by which such modalities could be treated to prevent the occurrence of adverse events and ensure vaccine rollouts remain safe and on target to end the current pandemic.
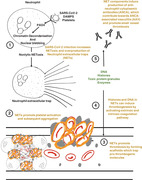
Graphical Abstract
Review on the scientific premise for incidence of vaccine‐induced thrombotic thrombocytopenia as a result of inflammatory reactions mediated via IL‐1β/NLRP3 inflammasome activation and consequent overproduction of NETs.
1. INTRODUCTION
Ever since COVID‐19, caused by SARS‐CoV‐2, was declared by the World Health Organization (WHO) as a global pandemic, the leading global strategy against this disease has been the effective development, procurement, and distribution of vaccines. Indeed, many vaccines have been authorized by global regulatory authorities since December 2020. 1 , 2 Coronaviruses are large single‐stranded positive‐sense RNA viruses with a helical nucleocapsid (N) and an envelope composed of matrix protein (M), an envelope protein (E), and spike protein (S). The spike protein is the receptor‐binding site for angiotensin‐converting enzyme 2 for viral entry into the cell. The developmental stage of the COVID‐19 vaccine saw various approaches, all of which utilize S protein as the immunogen: recombinant vaccines using viral vectors; nucleic acid vaccines or mRNA vaccines; inactivated vaccines; nanoparticle or virus‐like particle vaccines; protein subunit vaccines; and live attenuated vaccines. 3 , 4
However, as one would expect from vaccines developed at such an unprecedented pace, several side effects have been reported termed adverse events of special interests (AESI). Variable AESIs observed include acute myocardial infarction, hemorrhagic/nonhemorrhagic stroke, deep vein thrombosis, pulmonary embolism, Bell's palsy, transverse myelitis, Guillain‐Barre syndrome, immune thrombocytopenia, disseminated intravascular coagulation (DIC), anaphylaxis, encephalomyelitis, narcolepsy, pericarditis, cerebral venous sinus thrombosis (CVST), splanchnic vein thrombosis (SVT), and appendicitis. 5 , 6 , 7 There is an immediate need to elucidate whether these AESIs and vaccines share a causal relationship to further optimize vaccine protocols, provide therapeutic strategies, and reduce public fear and hesitancy over the vaccines. 8 It should, however, be emphasized beforehand that the vaccines are largely safe, as AESIs are exceptional findings, and the benefits of receiving the vaccine far outweigh any potential risks.
Reports of thrombosis in atypical locations, particularly CVST and SVT, with concomitant thrombocytopenia following immunization with the adenoviral ChAdOx1 nCoV‐19 AstraZeneca and Ad26.COV2.S Janssen vaccines resulted in these vaccines being temporarily withdrawn in Europe and the United States, respectively. Upon further testing and being declared efficacious and safe, these vaccines were re‐authorized for use. Elucidating the mechanisms and investigating potential therapeutic strategies have been the subject of intense study ever since. The temporal coincidence of thrombosis and thrombocytopenia following vaccine administration suggested a mechanism resembling that of heparin‐induced thrombocytopenia (HIT), with the term vaccine‐induced thrombotic thrombocytopenia (VITT) being coined to refer to this clinical entity. This was further strengthened by the demonstration of anti‐platelet factor 4 (PF4) antibodies in the sera of patients afflicted with VITT. 9 , 10 , 11
Recently, neutrophil extracellular traps (NETs) have gained traction as being key in mediating thrombotic events characterizing severe COVID‐19 and various auto‐immune conditions, including HIT. 12 , 13 , 14 , 15 Accordingly, in this article, we detail the similarities and differences in the pathogenesis of HIT and VITT, discuss how the different components of NETs facilitate thrombosis, elucidate how NET production could occur in the setting of COVID‐19 vaccine administration to subsequently mediate thrombosis and thrombocytopenia, and provide questions that should be addressed by future research to scrutinize the validity of this hypothesis. Last, we briefly review validated therapeutic strategies reported thus far and suggest drugs that could prove to be efficacious by inhibiting NETs. Elucidating whether NETs play a major or minor role in VITT should be the focus of future studies as this could have significant therapeutic implications far beyond the treatment of VITT; further, NET inhibitors could represent viable options to mitigate VITT as well as numerous other prothrombotic disorders.
2. COVID‐19 VACCINATION AND THROMBOCYTOPENIA
The Oxford/AstraZeneca vaccine gained rapid emergency approval and clearance for use following confirmation of efficiency (76–80%), and despite exceptional cases of thrombosis being reported, the vaccine was considered safe by both the European Medicines Agency and the WHO. The efficacy of other vaccines in comparison is variable, with the Johnson & Johnson's (J&J) Janssen vaccine (66%) showing lower efficacy, and Moderna and Pfizer exhibiting efficacies higher than 90%. 16 By March 18, 2021, close to 20 million doses had been administered in Europe and the United Kingdom. Soon, however, an increasing number of cases reporting “embolic and thrombotic events” after taking the AstraZeneca vaccine began to be reported, predominantly in females aged between 20 and 50 years. A total of 269 thrombotic events (45 fatalities) were reported, with CVST and SVT being disproportionately prevalent. 17
An article reported 11 patients, aged 22–49 years and 9 of whom were female, who experienced thrombotic events beginning 5–16 d postvaccination with the ChAdOx1 vector vaccine. 7 All patients studied developed thrombosis, with the most common site being CVST, and tested positive for anti‐PF4 antibodies and in a PF4‐enhanced platelet activation test independent of heparin, demonstrating that thrombosis was mediated by activation of platelets via auto‐antibodies against PF4. 18 Accordingly, the clinical criteria for the diagnosis of VITT include 1 exposure to the AstraZeneca or Janssen COVID‐19 vaccines 4–30 d before presentation, 2 thrombosis, 3 thrombocytopenia, and 4 positive for anti‐PF4 antibodies on a standard ELISA. Besides positive anti‐PF4 antibodies, other important lab findings reveal an elevated D‐dimer, normal or low fibrinogen levels, and DIC leading to a consumption coagulopathy with consequent hemorrhage. 7 , 9 Because the pathophysiology seemed to mimic that of HIT, the term VITT was coined to refer to this clinical entity.
3. INFLAMMASOMES, NETS, AND VITT
Neutrophils are the most abundant type of circulating cell in the body, and are critical components of the innate immune response against infectious and noninfectious disease pathogenesis. 18 Recently, the expulsion of chromatin by neutrophils to form specialized structures known as NETs has gained attention in describing the function of neutrophils in health and disease. 19 These unique, web‐like structures, are composed of DNA, histones, and enzymes, which are released from neutrophils as they undergo a specialized type of cell death called “NETosis.” 20 Although the primary function of NETs is to trap microbes and associated debris, an uncontrolled proliferation of NETs from neutrophils culminates in alveolar damage, endothelial injury and coagulopathy (Fig. 1). Importantly, NETs are now also increasingly associated with central roles in COVID‐19 infection and pathogenesis. 20
FIGURE 1.
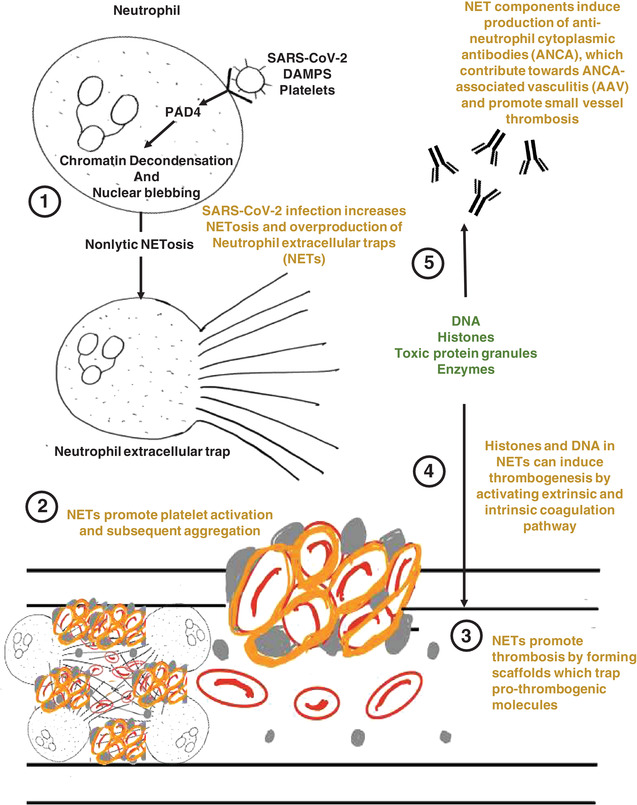
Schematic summary indicating the proposed pathophysiology underlying the role of NETosis in thrombosis. damage‐associated molecular patterns (DAMPs), platelets, and complement receptor engagement trigger nonlytic neutrophil extracellular trap (NET) formation, allowing NETing neutrophils to retain basic phagocytic and chemotactic functions. Nonlytic NETosis directly activates PAD‐4, which converts arginine to citrulline on histones, causing loss of positive charges of histones and disrupting electrostatic attractions between histones and DNA. The result is decondensation of chromatin. Subsequently, chromatin and associated proteins are lost via blebbing of the nuclear envelope, which is resealed afterward. NETs provide a structural basis for thrombosis and promote platelet aggregation and coagulation, manifesting as a pro‐thrombogenic state. Additionally, NETs can lead to the production of ANCA, leading to ANCA‐associated vasculitis which has been noted in severe cases of COVID‐19
Although the role of neutrophils in thrombosis is currently poorly understood, neutrophils are among the first cells that are recruited at inflammation or infection sites, 21 and promote coagulation via fibrin deposition to prevent microbial spread. 22 Excessive activation or dysregulation of neutrophils in blood vessels results in coagulopathy, 23 suggesting the potential importance of NET‐mediated thrombosis regulation. 19 Indeed, NETs form scaffolds, which trap platelets, RBCs, and platelet adhesion molecules such as fibrinogen, vWF, and fibronectin, 24 which may not only provide the structural basis for thrombosis, but also can potentially induce coagulation through its various constituents by activation of platelets. 25 Indeed, NET‐specific markers are significantly elevated in COVID‐19 patient sera, whereas Interestingly, COVID‐19 patient sera triggered NET production in healthy control‐derived neutrophils, 13 , 26 , 27 , 28 , 29 and COVID‐19 neutrophils exhibited significantly higher levels of NETs at baseline. 29 Although NET release during infections is predominantly considered to be physiologically beneficial, perhaps excessive NET production is harmful, resulting in tissue injury and thrombosis. 21 , 30
Although NETosis is physiologically beneficial, when excessive, NETs are key in propagating thrombosis and thrombocytopenia characterizing various disorders, as well as being implicated in the pathogenesis of auto‐immune diseases such as systemic lupus erythematosus, rheumatoid arthritis, and HIT. 31 , 32 Moreover, early data on COVID‐19 suggested that neutrophils were the primary immune cell type recruited during SARS‐CoV2 infection, mediating direct damage to resident cells through the production of reactive oxygen species and NETs. 33 , 34
NETs, as a consequence of their fibrous structure, constitute a platform onto which platelets, fibrin, and RBCs are deposited, thereby providing a structural basis for thrombosis. 24 , 35 , 36 Furthermore, certain NET components, including cell‐free DNA, histones, and tissue factor are well‐established inducers of coagulation. Histones cause platelet and endothelial cell activation via TLR 2 and 4, the latter of which leads to cell vacuolization and subsequent cell death, which precipitates a prothrombotic state. 24 , 35 , 37 , 38 Activated platelets interacting with neutrophils can also induce NETosis through binding via P‐selectin and high mobility group box 1. NET expulsion in turn facilitates the adhesion and activation of more platelets (Table 1). Therefore, neutrophil‐platelet interactions constitute a feed‐forward loop that exacerbates thrombosis. 39 , 40
TABLE 1.
Causes of acquired thrombocytopenia with shortened platelet survival
| Acquired thrombocytopenia with shortened platelet survival | |
| Associated with bleeding | Associated with thrombosis |
| Acute Immune thrombocytopenic purpura (ITP) | Thrombotic thrombocytopenic purpura |
| Drug‐induced thrombocytopenia | Disseminated intravascular coagulation (DIC) |
| Chronic ITP | Heparin‐ induced thrombocytopenia (HIT) |
| Most other causes | Vaccine‐induced prothrombotic immune thrombocytopenia |
Furthermore, the release of neutrophil elastase (NE) and myeloperoxidase (MPO) leads to endothelial damage, and exposure of vWF, which independently activates platelets to cause thrombosis. Extracellular DNA binds various coagulation factors, thereby promoting thrombin‐dependent fibrin formation. NET components such as serine proteases inhibit tissue factor pathway inhibitor (TFPI), whereas histones (another NET component) bind thrombomodulin preventing thrombomodulin‐dependent protein C activation. 22 , 41 Furthermore, NETs also augment activation of the coagulation cascade by inhibiting natural anticoagulants.
The general immunologic sequence culminating in NETosis involves the initial activation of macrophages secondary to binding of pathogen‐associated molecular patterns or damage‐associated molecular patterns (DAMPs) to pattern recognition receptors, most commonly NLPR3 monomers, which form an inflammasome complex through oligomerization. 42 The NLPR3 inflammasome promotes secretion of IL‐1β, which positively reinforces inflammasome formation and perpetuates neutrophil recruitment and activation. These neutrophils undergo NETosis, which, in turn, further drive inflammasome formation through DAMPs. This loop circuitry of activation has been implicated in acute lung injury, acute respiratory distress syndrome, and organ dysfunction in COVID‐19. 13 , 27 Accordingly, in cases of excessive inflammasome activity, seen in conditions such as COVID‐19 and sepsis, consequent NETosis and elevated serum NET markers, including MPO, elastase, cell‐free DNA, and citrullinated histone H3, are also observed. Indeed, viable SARS‐CoV‐2 directly induced NET release, in a manner dependent on levels of angiotensin‐converting enzyme 2, serine protease, virus replication, and Peptidyl Arginine Deiminase 4 (PAD‐4) in COVID‐19 patients. 43 Illness severity was directly correlated with plasma MPO‐DNA complexes in COVID‐19 patients, with soluble and cellular factors triggering NETs observed at significantly higher levels alongside NET‐containing microthrombi with neutrophil‐platelet infiltration in pulmonary autopsies. 29
NETs provably contribute to vaccine immunogenicity and thereby efficacy. This is due to the use of vaccine adjuvants, which bolster host defense against the immunogen the vaccine delivers. The common mechanism of all adjuvants is the recruitment of innate immune cells, such as neutrophils, dendritic cells, and macrophages, which activate B‐ and T‐cells to confer long‐term immunity. Adjuvants are generally considered safer than live‐attenuated vaccines and, by increasing vaccine efficacy, they lead to dosage sparing, which increases global vaccine supply. Examples of common adjuvants include aluminum salts, saponins, and emulsions, which activate inflammasomes and proinflammatory cytokines, especially IL‐1β. 44 , 45 The inflammasome targeted by adjuvants is NLRP3, which is induced through NF‐κB signaling.
4. HIT AND VITT: THE ROLE OF NETS
The temporal association of thrombosis and thrombocytopenia following administration of the Oxford/AstraZeneca vaccine suggested a phenomenon similar to the pathogenesis of HIT. The pathophysiology of HIT involves binding of heparin to PF4, prompting a conformational change which renders the complex immunogenic that then elicits the production of IgG heparin‐PF4 antibodies (HIT antibodies). 46 This heparin‐PF4‐IgG immune complex (HIT‐IC) then activates platelets via FcγRIIa receptors, triggering their activation and releasing prothrombotic platelet‐derived microparticles, including PF4, thereby precipitating a hypercoagulable state. The result is platelet consumption and eventual thrombocytopenia. 46 , 47 Importantly, however, individuals have been known to present with a clinical entity resembling HIT without prior exposure to heparin. In this context, antibodies were directed against PF4‐polyanion complexes, as was the case with VITT. 48 , 49 It is essential to determine which component of the ChAdOx1 nCoV‐19 and AD26.COV2.S binds PF4 to further optimize vaccine protocols.
Recent research has suggested that the pathogenesis of HIT extends beyond that of simply involving platelets and implicates NETs, as evidenced by elevated NET markers in HIT. The HIT‐IC has been shown to activate neutrophils directly through engaging the FcγRIIa receptor and indirectly through activation of platelets by FcγRIIa, which subsequently induce NETosis via P‐selectin binding to P‐selectin glycoprotein ligand‐1 on the surface neutrophils. 14 , 15 The thrombocytopenia is believed to be secondary to enhanced phagocytosis of HIT‐IC‐covered platelets by macrophages. 39 , 50
As stated earlier, NETs could play roles in enhancing vaccine immunogenicity through adjuvants that activate the NLRP3 inflammasome. 42 , 51 Additionally, NET components such as nucleic acids and various proteins are negatively charged and, therefore, are capable of binding PF4 on the platelet surface to subsequently elicit the generation of autoantibodies against PF4‐anionic complexes. Therefore, in the context of VITT, many of the clinically relevant vaccine adjuvants, such as aluminum salts, saponins, and emulsions, activate the NLRP3 inflammasome and trigger NETosis. 44 , 45 , 52 Subsequently, NET components bind PF4 and elicit the production of autoantibodies against PF4‐polyanionic complexes. A question that arises in response is why only a few individuals are affected despite widespread vaccinations? Perhaps genetic factors predisposing individuals to develop VITT should be considered, although their role in HIT is considered minimal. The 131RR genotype of the FCG2RA gene encoding the FcγRIIa receptor, polymorphism of the IL‐10 microsatellite sequence, and PIA2 variant of GPIIIa of platelets, have all been reported to increase the risk of thrombosis in HIT. 53 , 54 , 55 Therefore, a potential genetic predisposition merits further investigation.
Nevertheless, regardless of whether NETs and VITT share a causal relationship, it is highly probable that NETs do contribute to the propagation of thrombosis and thrombocytopenia in VITT as established by extensive research into the role of NETs in thrombosis (Fig. 2). To elucidate whether NETs play a major role in VITT, first, measuring serum NET markers, Cf‐DNA, MPO‐DNA, and citrullinated histone H3 (Cit‐H3) in VITT would show the activity of NETosis. Subsequently, analyzing whether the degree of NETosis activity correlates with the clinical course of VITT could, at least to a degree, implicate NETs as important mediators of VITT.
FIGURE 2.
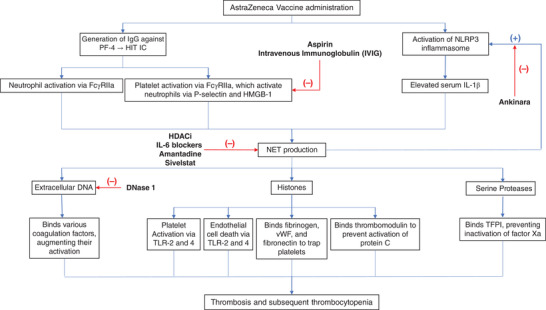
Schematic summary of the proposed mechanism of the role played by neutrophil extracellular traps (NETs) in development of thrombosis and thrombocytopenia in individuals receiving the Oxford/AstraZeneca vaccine, with proposed treatments indicated at relevant stages. Many clinically relevant vaccine adjuvants (aluminum, emulsions, saponins, among others) activate the NLRP3 inflammasome, releasing IL1β, inducing NETosis. Elevated levels of antibodies platelet factor 4 (PF4)‐related immune complexes (IC) in individuals receiving the Oxford/AstraZeneca vaccine indicates a mechanism similar to heparin‐induced thrombocytopenia (HIT), whereby NET production occurs directly via the FcγRIIa receptor on neutrophils, and indirectly via FcγRIIa‐mediated activation of platelets which in turn stimulate NETosis. Various components of NETs, including extracellular DNA, histones, and serine proteases, render them procoagulant. Extracellular DNA is known to bind and augment activation of proteases of the coagulation cascade. Histones induce endothelial cell damage via TLR2 and 4, precipitating hypercoagulation. Histones activate platelets via TLR2 and 4, which in turn enhance NET production, establishing a feed‐forward loop. Histones and extracellular DNA bind fibrinogen, vWF, and fibronectin to enhance platelet trapping. Last, histones bind thrombomodulin, preventing thrombomodulin‐dependent activation of protein C. Serine proteases bind tissue factor pathway inhibitor (TFPI), inhibiting inactivation of factor Xa. All these factors culminate in rendering NETs prothrombotic
An intriguing feature of VITT not seen in HIT is the predilection for causing CVST and/or SVT, the latter encompassing mesenteric and portal vein thrombosis and Budd‐Chiari syndrome. 56 , 57 Although the mechanism behind this remains undetermined, NETs likely form important constituents of thrombi in both locations. A recent review article exploring the role of NETs in different forms of liver disease showed that liver damage secondary to portal vein thrombi and intravascular sinusoidal microthrombi in various diseases, including portal hypertension (PHTN) and sepsis, could be mitigated through inhibition of NETs via sivelstat, an NE inhibitor. 31 Likewise, a study analyzing 68 ischemic stroke patients demonstrated the presence of NETs within these thrombi through immunostaining NE and by measuring serum NET markers, showing that NET components were a prominent feature in almost all thrombi. Additionally, ex vivo degradation of retrieved thrombi was enhanced with the addition of DNase to standard tissue plasminogen activator (tPA). 56 Thus, unraveling the role of NETs in the setting of VITT could yield novel therapeutic strategies that have implications far beyond VITT, including the treatment of stroke patients and patients suffering from PHTN. For a start, a similar study aiming to analyze whether NETs constitute a major portion of thrombi in VITT by immunostaining for NET components and evaluating the efficacy of thrombolysis via DNase against that of conventional thrombolytic drugs alone would indicate how involved NETs are in VITT thrombosis.
5. THE ROLE OF VACCINE COMPONENTS IN NET‐INDUCED VITT
With the caveat that detailed vaccine component specifications are not current available, multiple hypotheses also suggest that perhaps either specific factors, or even combinations of such factors, present as vaccine components such as adjuvants may play some kind of role. 58 Indeed, Greinacher et al., 59 recently suggested the identification of multiple cell‐culture derived human proteins in the ChAdOx1 nCov‐19 vaccine, potentially exposing predisposed individuals to strong alloimmune reactions against antigens. The authors also further suggested that ChAdOx1 nCoV‐19 vaccine constituents were able to form antigenic complexes with PF4, whereas levels of EDTA present in the vaccine increased microvascular permeability, collectively leading to acute inflammatory reactions. 59 However, other vaccines also contain a milieu of human proteins, although these are not always specified. Furthermore, the J&J Janssen vaccine is not reported to contain EDTA, suggesting that perhaps more multifactorial roles are in play than expected.
Cases of severe allergic reactions (anaphylaxis) reported following the first dose of the Pfizer‐BioNTech or Moderna SARS‐CoV‐2 mRNA vaccines 60 are perhaps triggered by the PEG or lipid components of these vaccines, 61 leading to mast cell activation in an IgE‐independent manner as in the complement mediated pseudo‐allergy reaction that has been described in the case of liposomal carriers, 61 , 62 or perhaps via complement activation initiated by IgG or IgM raised against the PEGylated lipids of these vaccine formulations. 61
The active component in the ChAdOx1 nCoV‐19 vaccine is of course an adenovirus, which in itself can activate platelets. 58 , 63 , 64 To this degree, perhaps the replication‐deficient adenoviral vector in the ChAdOx1 nCoV‐19 vaccine contributes toward the release of platelet‐derived PF4, leading to VITT via damage of endothelial cells (easily transduced following intramuscular injection 65 ) by the synthesized spike protein. 58 , 65 , 66 , 67 Recruited platelets could then be activated by the spike protein bound to endothelial cells, 58 , 68 resulting in the formation of immunogenic PF4. 58 Indeed, PF4‐immune complexes can be recognized by C1q that binds to the Fc portion of IgG molecules, leading to downstream generation of potent proinflammatory mediators and effectors that can potentiate thromboinflammation similar to those seen in COVID19 patients. 61 , 69 , 70
COX enzymes (induced via the spike proteins of coronaviruses) can catalyze thromboxane A2 (TxA2) production, 71 perhaps underlying the thromboinflammation observed in VITT. 72 Indeed, in mice infected with SARS‐CoV, TxA2 production was markedly increased in younger mice compared to middle aged mice, 72 in line with the higher risk for thrombosis in adults aged <60 yr. 7 , 72 Intriguingly, platelets from female mice seem significantly more reactive than from male mice, 73 whereas TxA2 generation, TxA2‐platelet interaction, and platelet activation also seem increased in women compared to men. 74 , 75 All such observations seem consistent with the apparent increased risk of thrombosis in women following the AstraZeneca and J&J vaccines. 72 However, it is important to note that based on the current evidence, specific risk factors have yet to be confirmed.
6. PREVENTION AND TREATMENT OF NET‐INDUCED VITT
Similar to the management approach in the setting of HIT, a high dose of i.v. immunoglobulin (IVIG) of 1 g/kg for 2 d may also be administered. 1 , 9 , 10 , 76 Initiating a high‐dose steroid therapy with or without high‐dose immunoglobulins has shown to rapidly increase the platelet count in VITT patients in whom IVIG is delayed or platelet count is <50 × 109. 77 Low molecular weight heparin should not be used in these patients as strong reactivity with antibodies could worsen the condition. Warfarin is also contraindicated as it can potentially worsen thrombosis. Fondaparinux, danaproid, lepirudin, and argotraban can be used to treat thrombotic events in these patients. 78 , 79
Many avenues can be considered for the treatment of thrombocytopenia associated with thrombosis, all of which can be associated with varying modes of pathogenesis (Table 2). Considering the body of evidence supporting the role of NETs in VITT, it is perhaps prudent to suggest prophylaxis of low‐dose aspirin in populations susceptible to VITT, which predominantly seems to be females aged 20–50 yr, administered for 2 wk to prevent this potentially dangerous adverse effect. The basis of this recommendation is the prevention of platelet activations and platelet aggregation by aspirin. As discussed earlier, a complex interplay exists between platelets and neutrophils that induces NETosis and exacerbates thrombosis. Therefore, by abrogating platelet activation and aggregation, NETosis can potentially be prevented. Sivelstat, a neutrophil elastase inhibitor, ameliorates liver damage due to portal vein thrombi and intravascular sinusoidal microthrombi in PHTN and sepsis, and probably numerous other disorders characterized by thrombosis. 31 A more comprehensive review of various drugs shown to inhibit NETs can be found here. 80 Further work is required to fully elucidate the therapeutic potential of these drugs in conjunction with unraveling the role of NETs in various prothrombotic disorders including VITT.
TABLE 2.
Summary of causes of thrombocytopenia associated with thrombosis, alongside recommended treatment pathways
| Disorder | Pathogenesis | Recommended treatments |
|---|---|---|
| Thrombotic thrombocytopenic purpura | Defective regulation of VWF activity by a circulating metalloprotease ADAMTS13 64 | Treat the cause and Plasma therapy either Fresh frozen plasma or plasmapheresis |
| Disseminated Intravascular coagulation (DIC) | Increased thrombin generation, suppression of the physiologic anticoagulant pathways, impaired fibrinolysis and activation of the inflammatory pathways 65 |
1. Treatment of cause 2. Replacement therapy (platelets, cryoprecipitate, fresh frozen plasma) 3. Sometimes heparin |
| Heparin‐induced thrombocytopenia | Formation of IgG antibodies against the platelet factor 4 (PF4)/heparin complex leading to the creation of an immunocomplex which will activate platelets and monocytes. 66 | Immediate cessation of heparin and initiation of alternative anticoagulation treatment, for example, danaproid, lepirudin and argotraban. |
| Vaccine‐induced thrombotic thrombocytopenia | Not known; postulated as associated with PF4 antibodies/neutrophil extracellular traps (NETs). 10 , 14 , 15 , 18 , 36 | I.v. immunoglobulin (IVIG) and high dose steroids. |
7. CONCLUSION
We propose that NETs could play major roles in VITT, as earlier research has firmly established NETs as key in precipitating a prothrombotic state characterizing various disorders, including HIT. To this end, we detailed the various components of NETs propagating thrombosis, and how NET formation in the context of ChAdOx1 nCoV‐19 and Ad26.COV2.S vaccine administration could perpetuate VITT. We also highlighted currently unaddressed research questions that, if answered, would not only further our understanding of VITT but also of a plethora of other thrombotic disorders. Determining if NETs play a major or minor role in VITT will significantly impact pharmacologic approaches to treating this disorder as various drugs are known to inhibit NET formation, or in the early detection of susceptibility to NET‐VITT. Specific biomarkers for NETs in body fluids include cell free DNA, MPO‐DNA, and Cit‐H3, which are easily and rapidly detectable using ELISA‐based or strip assays. 13 , 27 , 81
Therapeutic options that could be investigated include recombinant DNase‐1 (dornase alfa) in combination with tPA for resolution of thrombi caused by excessive NET production, whereas NET production was also blocked using histone deacteylase inhibitors and IL6 blockers. 13 , 20 , 27 , 82 Amantadine can block viroporins and calcium influx, which is crucial for PAD‐4 activation in NET pathogenesis, 13 , 83 , 84 whereas NET‐induced inflammatory tissue damage, coagulopathy, and vasculitis can be controlled by exogenous administration of resolvins. 13 , 27 , 85 , 86 , 87 Ankinara is another potential target that potentially disrupts the IL1‐β‐NET feedback loop. 13 , 20 Another alternative, although no changes in survival rates were observed for treatment of ARDS, includes the neutrophil elastase inhibitor Sivelestat could also be a potential target to tackle NET‐induced VITT. 13 , 88
Finally, considering the inflammatory‐like conditions presented by COVID‐19, IVIG administration has been examined as a possible route to reduce proinflammatory markers, with numerous studies now showing that deployment of IVIG within specific dosages would reduce mortality and length of hospitalization of severe COVID‐19 patients (refer to Yaqinuddin et al. 1 for a more detailed review). Furthermore, a strong correlation was observed between anti‐NET IgG and IgM with high levels of circulating NETs, impaired oxygenation efficiency, and high circulating D‐dimer, potentially impairing NET clearance and exacerbating SARS‐CoV‐2‐mediated thromboinflammation. 89 To this degree, perhaps a therapeutic path in treatment of VITT would be high‐dose IVIG administration, which was able to reduce antibody‐induced platelet activation in serum in three patients exhibiting heterogeneous symptoms of VITT. 90
AUTHORSHIP
The manuscript was conceived and context led by A.Y. and J.K. with input and support from A.R.A., A.S., M.R.S., and K.A.K. All authors contributed toward writing the manuscript, which was submitted following the approval of all authors.
DISCLOSURES
The authors declare no conflicts of interest.
ACKNOWLEDGMENT
This work was supported by a COVID‐19 project grant (#C20323) awarded by Alfaisal University to M.R.S.
Kashir J, Ambia AR, Shafqat A, Sajid MR, AlKattan K, Yaqinuddin A. Scientific premise for the involvement of neutrophil extracellular traps (NETs) in vaccine‐induced thrombotic thrombocytopenia (VITT). J Leukoc Biol. 2022;111:725–734. 10.1002/JLB.5COVR0621-320RR
REFERENCES
- 1. Yaqinuddin A, Ambia AR, Elgazzar TA, MbM AlSaud, Kashir J. Application of intravenous immunoglobulin (IVIG) to modulate inflammation in critical COVID‐19—a theoretical perspective. Med Hypotheses. 2021;151:110592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yaqinuddin ARA, Kazkaz H, MishariAlSaud M, Khaled A, Kashir J. Advantageous non‐specific effects of live‐attenuated vaccines in COVID‐19 treatment. African J Respir Med. 2021;16. [Google Scholar]
- 3. Koirala A, Joo YJ, Khatami A, Chiu C, Britton PN. Vaccines for COVID‐19: the current state of play. Paediatr Respir Rev. 2020;35:43‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dai L, Gao GF. Viral targets for vaccines against COVID‐19. Nat Rev Immunol. 2020;21:73‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li X, Ostropolets A, Makadia R, et al. Characterising the background incidence rates of adverse events of special interest for covid‐19 vaccines in eight countries: multinational network cohort study. BMJ. 2021;n1435. 10.1136/bmj.n1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Connors JM, Levy JH. COVID‐19 and its implications for thrombosis and anticoagulation. Blood. 2020;135:2033‐2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Greinacher A, Thiele T, Warkentin TE, Weisser K, Kyrle PA, Eichinger S. Thrombotic thrombocytopenia after ChAdOx1 nCov‐19 vaccination. N Eng J Med. 2021.;384(22):2092‐2101. 10.1056/nejmoa2104840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rzymski P, Perek B, Flisiak R. Thrombotic Thrombocytopenia after COVID‐19 Vaccination: In Search of the Underlying Mechanism. Vaccines. 2021;9(6):559. 10.3390/vaccines9060559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Oldenburg J, Klamroth R, Langer F, et al. Diagnosis and management of vaccine‐related thrombosis following AstraZeneca COVID‐19 vaccination: guidance statement from the GTH. Hamostaseologie. 2021;41:184‐189. [DOI] [PubMed] [Google Scholar]
- 10. Scully M, Singh D, Lown R, et al. Pathologic antibodies to platelet factor 4 after ChAdOx1 nCoV‐19 vaccination. N Engl J Med. 2021;384:2202‐2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cines DB, Bussel JB. SARS‐CoV‐2 vaccine‐induced immune thrombotic thrombocytopenia. N Engl J Med. 2021;384:2254‐2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yaqinuddin A, Kvietys P, Kashir J. COVID‐19: role of neutrophil extracellular traps in acute lung injury. Respir Investig. 2020;58:419‐420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yaqinuddin A, Kashir J. Novel therapeutic targets for SARS‐CoV‐2‐induced acute lung injury: targeting a potential IL‐1β/neutrophil extracellular traps feedback loop. Med Hypotheses. 2020;143:109906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lisman T. Platelet–neutrophil interactions as drivers of inflammatory and thrombotic disease. Cell Tissue Res. 2017;371:567‐576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Perdomo J, Leung HHL, Ahmadi Z, et al. Neutrophil activation and NETosis are the major drivers of thrombosis in heparin‐induced thrombocytopenia. Nat Commun. 2019;10(1). 10.1038/s41467-019-09160-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mallapaty S, Callaway E. What scientists do and don't know about the Oxford–AstraZeneca COVID vaccine. Nature. 2021;592:15‐17. [DOI] [PubMed] [Google Scholar]
- 17. (PRAC) PRAC. Signal Assessment Report on Embolic and Thrombotic Events (SMQ) with COVID‐19 Vaccine (ChAdOx1‐S [Recombinant])—COVID‐19 Vaccine AstraZeneca (Other Viral Vaccines). Belgium: European Medicines Agency; 2021. [Google Scholar]
- 18. Christoffersson G, Phillipson M. The neutrophil: one cell on many missions or many cells with different agendas?. Cell Tissue Res. 2018;371:415‐423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Thålin C, Hisada Y, Lundström S, Mackman N, Wallén H. Neutrophil extracellular traps: villains and targets in arterial, venous, and cancer‐associated thrombosis. Arterioscler Thromb Vasc Biol. 2019;39:1724‐1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Barnes BJ, Adrover JM, Baxter‐Stoltzfus A, et al. Targeting potential drivers of COVID‐19: neutrophil extracellular traps. J Exp Med. 2020;217(6). 10.1084/jem.20200652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Laridan E, Martinod K, De Meyer SF. Neutrophil extracellular traps in arterial and venous thrombosis. Semin Thromb Hemost. 2019;45:86‐93. [DOI] [PubMed] [Google Scholar]
- 22. Massberg S, Grahl L, von Bruehl ML, et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med. 2010;16:887‐896. [DOI] [PubMed] [Google Scholar]
- 23. Pfeiler S, Stark K, Massberg S, Engelmann B. Propagation of thrombosis by neutrophils and extracellular nucleosome networks. Haematologica. 2017;102:206‐213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fuchs TA, Bhandari AA, Wagner DD. Histones induce rapid and profound thrombocytopenia in mice. Blood. 2011;118:3708‐3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xu J, Zhang X, Pelayo R, et al. Extracellular histones are major mediators of death in sepsis. Nat Med. 2009;15:1318‐1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zuo Y, Yalavarthi S, Shi H, et al. Neutrophil extracellular traps in COVID‐19. JCI Insight. 2020; 10.1172/jci.insight.138999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yaqinuddin A, Kvietys P, Kashir J. COVID‐19: role of neutrophil extracellular traps in acute lung injury. Respir Investig. 2020;58:419‐420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yaqinuddin A, Kashir J. Novel therapeutic targets for SARS‐CoV‐2‐induced acute lung injury: targeting a potential IL‐1β/neutrophil extracellular traps feedback loop. Med Hypotheses. 2020;143:109906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Middleton EA, He XY, Denorme F, et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID‐19 acute respiratory distress syndrome. Blood. 2020;136:1169‐1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Boeltz S, Amini P, Anders HJ, et al. To NET or not to NET:current opinions and state of the science regarding the formation of neutrophil extracellular traps. Cell Death Differ. 2019;26:395‐408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hilscher MB, Shah VH. Neutrophil extracellular traps and liver disease. Semin Liver Dis. 2020;40:171‐179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Klopf J, Brostjan C, Eilenberg W, Neumayer C. Neutrophil extracellular traps and their implications in cardiovascular and inflammatory disease. Int J Mol Sci. 2021;22:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tomar B, Anders HJ, Desai J, Mulay SR. Neutrophils and neutrophil extracellular traps drive necroinflammation in COVID‐19. Cells. 2020;9(6):1383. 10.3390/cells9061383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Maxwell AJ, Ding J, You Y, et al. Identification of key signaling pathways induced by SARS‐CoV2 that underlie thrombosis and vascular injury in COVID‐19 patients. J Leukoc Biol. 2020;109:35‐47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fuchs TA, Brill A, Duerschmied D, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A. 2010;107:15880‐15885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gould TJ, Lysov Z, Liaw PC. Extracellular DNA and histones: double‐edged swords in immunothrombosis. J Thromb Haemost. 2015;13:S82‐S91. [DOI] [PubMed] [Google Scholar]
- 37. Martinod K, Wagner DD. Thrombosis: tangled up in NETs. Blood. 2014;123:2768‐2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Saffarzadeh M, Juenemann C, Queisser MA, et al. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS One. 2012;7:e32366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Maugeri N, Campana L, Gavina M, et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J Thromb Haemost. 2014;12:2074‐2088. [DOI] [PubMed] [Google Scholar]
- 40. Etulain J, Martinod K, Wong SL, Cifuni SM, Schattner M, Wagner DD. P‐selectin promotes neutrophil extracellular trap formation in mice. Blood. 2015;126:242‐246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ammollo CT, Semeraro F, Xu J, Esmon NL, Esmon CT. Extracellular histones increase plasma thrombin generation by impairing thrombomodulin‐dependent protein C activation. J Thromb Haemost. 2011;9:1795‐1803. [DOI] [PubMed] [Google Scholar]
- 42. Kahlenberg JM, Carmona‐Rivera C, Smith CK, Kaplan MJ. Neutrophil extracellular trap–associated protein activation of the NLRP3 inflammasome is enhanced in lupus macrophages. J Immunol. 2012;190:1217‐1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Veras FP, Pontelli MC, Silva CM, et al. SARS‐CoV‐2‐triggered neutrophil extracellular traps mediate COVID‐19 pathology. J Exp Med. 2020;217(12): 10.1084/jem.20201129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Demento SL, Eisenbarth SC, Foellmer HG, et al. Inflammasome‐activating nanoparticles as modular systems for optimizing vaccine efficacy. Vaccine. 2009;27:3013‐3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Reinke S, Thakur A, Gartlan C, Bezbradica JS, Milicic A. Inflammasome‐mediated immunogenicity of clinical and experimental vaccine adjuvants. Vaccines. 2020;8:554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chong BH. Evolving concepts of pathogenesis of heparin‐induced thrombocytopenia: diagnostic and therapeutic implications. Int J Lab Hematol. 2020;42:25‐32. [DOI] [PubMed] [Google Scholar]
- 47. Gollomp K, Kim M, Johnston I, et al. Neutrophil accumulation and NET release contribute to thrombosis in HIT. JCI Insight. 2018;3(18):e99445. 10.1172/jci.insight.99445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Miyata S. Spontaneous heparin‐induced thrombocytopenia (HIT) syndrome: hIT without any heparin exposure]. Rinsho Ketsueki. 2016;57:2124‐2135. [DOI] [PubMed] [Google Scholar]
- 49. Andres E, Dali‐Youcef N, Serraj K, Zimmer J. Recognition and management of drug‐induced cytopenias: the example of idiosyncratic drug‐induced thrombocytopenia. Expert Opin Drug Saf. 2009;8:183‐190. [DOI] [PubMed] [Google Scholar]
- 50. Maugeri N, Malato S, Femia EA, et al. Clearance of circulating activated platelets in polycythemia vera and essential thrombocythemia. Blood. 2011;118:3359‐3366. [DOI] [PubMed] [Google Scholar]
- 51. Mitroulis I, Kambas K, Chrysanthopoulou A, et al. Neutrophil extracellular trap formation is associated with IL‐1β and autophagy‐related signaling in gout. PLoS One. 2011;6:e29318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Neumann S, Burkert K, Kemp R, Rades T, Rod Dunbar P, Hook S. Activation of the NLRP3 inflammasome is not a feature of all particulate vaccine adjuvants. Immunol Cell Biol. 2014;92:535‐542. [DOI] [PubMed] [Google Scholar]
- 53. Arepally G, McKenzie SE, Jiang XM, Poncz M, Cines DB. Fc gamma RIIA H/R 131 polymorphism, subclass‐specific IgG anti‐heparin/platelet factor 4 antibodies and clinical course in patients with heparin‐induced thrombocytopenia and thrombosis. Blood. 1997;89:370‐375. [PubMed] [Google Scholar]
- 54. Pouplard C, Cornillet‐Lefebvre P, Attaoua R, et al. Interleukin‐10 promoter microsatellite polymorphisms influence the immune response to heparin and the risk of heparin‐induced thrombocytopenia. Thromb Res. 2012;129:465‐469. [DOI] [PubMed] [Google Scholar]
- 55. Harris K, Nguyen P, Van Cott EM. Platelet PlA2 polymorphism and the risk for thrombosis in heparin‐induced thrombocytopenia. Am J Clin Pathol. 2008;129:282‐286. [DOI] [PubMed] [Google Scholar]
- 56. Laridan E, Martinod K, De Meyer S. Neutrophil extracellular traps in arterial and venous thrombosis. Semin Thromb Hemost. 2019;45:086‐093. [DOI] [PubMed] [Google Scholar]
- 57. Singh B, Kaur P, Maroules M. Splanchnic vein thrombosis in COVID‐19: a review of literature. Dig Liver Dis. 2020;52:1407‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Goldman M, Hermans C. Thrombotic thrombocytopenia associated with COVID‐19 infection or vaccination: possible paths to platelet factor 4 autoimmunity. PLoS Med. 2021;18:e1003648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Andreas G, Kathleen S, Jan W, et al. Towards Understanding ChAdOx1 nCov‐19 Vaccine‐induced Immune Thrombotic Thrombocytopenia (VITT). PrePrint, Research Square
- 60. Castells MC, Phillips EJ. Maintaining safety with SARS‐CoV‐2 vaccines. N Engl J Med. 2021;384:643‐649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mastellos DC, Skendros P, Lambris JD. Is complement the culprit behind COVID‐19 vaccine‐related adverse reactions?. J Clin Invest. 2021;131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Klimek L, Novak N, Cabanillas B, Jutel M, Bousquet J, Akdis CA. Allergenic components of the mRNA‐1273 vaccine for COVID‐19: possible involvement of polyethylene glycol and IgG‐mediated complement activation. Allergy. 2021;00:1‐7. 10.1111/all.14794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Othman M, Labelle A, Mazzetti I, Elbatarny HS, Lillicrap D. Adenovirus‐induced thrombocytopenia: the role of von Willebrand factor and P‐selectin in mediating accelerated platelet clearance. Blood. 2006;109:2832‐2839. [DOI] [PubMed] [Google Scholar]
- 64. Coughlan L. Factors which contribute to the immunogenicity of non‐replicating adenoviral vectored vaccines. Front Immunol. 2020;11(909):1‐20. 10.3389/fimmu.2020.00909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mercier S, Gahéry‐Segard H, Monteil M, et al. Distinct roles of adenovirus vector‐transduced dendritic cells, myoblasts, and endothelial cells in mediating an immune response against a transgene product. J Virol. 2002;76:2899‐2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lei Y, Zhang J, Schiavon CR, et al. SARS‐CoV‐2 spike protein impairs endothelial function via downregulation of ACE 2. Circ Res. 2021;128:1323‐1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Nuovo GJ, Magro C, Shaffer T, et al. Endothelial cell damage is the central part of COVID‐19 and a mouse model induced by injection of the S1 subunit of the spike protein. Ann Diagn Pathol. 2021;51:151682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhang S, Liu Y, Wang X, et al. SARS‐CoV‐2 binds platelet ACE2 to enhance thrombosis in COVID‐19. J Hematol Oncol. 2020;13:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Skendros P, Mitsios A, Chrysanthopoulou A, et al. Complement and tissue factor‐enriched neutrophil extracellular traps are key drivers in COVID‐19 immunothrombosis. J Clin Invest. 2020;130:6151‐6157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Risitano AM, Mastellos DC, Huber‐Lang M, et al. Complement as a target in COVID‐19?. Nat Rev Immunol. 2020;20:343‐344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Liu M, Gu C, Wu J, Zhu Y. Amino acids 1 to 422 of the spike protein of SARS associated coronavirus are required for induction of cyclooxygenase‐2. Virus Genes. 2006;33:309‐317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Vijay R, Hua X, Meyerholz DK, et al. Critical role of phospholipase A2 group IID in age‐related susceptibility to severe acute respiratory syndrome‐CoV infection. J Exp Med. 2015;212:1851‐1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Leng XH, Hong SY, Larrucea S, et al. Platelets of female mice are intrinsically more sensitive to agonists than are platelets of males. Arterioscler Thromb Vasc Biol. 2004;24:376‐381. [DOI] [PubMed] [Google Scholar]
- 74. Kim BS, Auerbach DA, Sadhra H, et al. A sex‐specific switch in platelet receptor signaling following myocardial infarction. bioRxiv. 2019:580415. [Google Scholar]
- 75. Eikelboom JW, Hirsh J, Weitz JI, Johnston M, Yi Q, Yusuf S. Aspirin‐resistant thromboxane biosynthesis and the risk of myocardial infarction, stroke, or cardiovascular death in patients at high risk for cardiovascular events. Circulation. 2002;105:1650‐1605. [DOI] [PubMed] [Google Scholar]
- 76. Kelton JG, Arnold DM, Bates SM. Nonheparin anticoagulants for heparin‐induced thrombocytopenia. N Eng J Med. 2013;368:737‐744. [DOI] [PubMed] [Google Scholar]
- 77. Long B, Bridwell R, Gottlieb M. Thrombosis with thrombocytopenia syndrome associated with COVID‐19 vaccines. Am J Emerg Med. 2021;49:58‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ahmed I, Majeed A, Powell R. Heparin induced thrombocytopenia: diagnosis and management update. Postgrad Med J. 2007;83:575‐582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Linkins L‐A, Dans AL, Moores LK, et al. Treatment and prevention of heparin‐induced thrombocytopenia. Chest. 2012;141:e495S‐e530S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Mutua V, Gershwin LJ. A review of neutrophil extracellular traps (NETs) in disease: potential anti‐NETs therapeutics. Clin Rev Allergy Immunol. 2021;61(2):194‐211. 10.1007/s12016-020-08804-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Zuo Y, Yalavarthi S, Shi H, et al. Neutrophil extracellular traps in COVID‐19. JCI Insight. 2020;5(11):e138999. 10.1172/jci.insight.138999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Hamam HJ, Palaniyar N. Post‐translational modifications in NETosis and NETs‐mediated diseases. Biomolecules. 2019;9(8):369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Jing X, Ma C, Ohigashi Y, et al. Functional studies indicate amantadine binds to the pore of the influenza A virus M2 proton‐selective ion channel. Proc Natl Acad Sci U S A. 2008;105:10967‐10972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Dey D, Siddiqui SI, Mamidi P, et al. The effect of amantadine on an ion channel protein from Chikungunya virus. PLoS Negl Trop Dis. 2019;13:e0007548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Cherpokova D, Jouvene CC, Libreros S, et al. Resolvin D4 attenuates the severity of pathological thrombosis in mice. Blood. 2019;134:1458‐1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Conte MS, Desai TA, Wu B, Schaller M, Werlin E. Pro‐resolving lipid mediators in vascular disease. J Clin Invest. 2018;128:3727‐3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Serhan CN, Levy BD. Resolvins in inflammation: emergence of the pro‐resolving superfamily of mediators. J Clin Invest. 2018;128:2657‐2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Tagami T, Tosa R, Omura M, et al. Effect of a selective neutrophil elastase inhibitor on mortality and ventilator‐free days in patients with increased extravascular lung water: a post hoc analysis of the PiCCO Pulmonary Edema Study. J Intensive Care. 2014;2:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Zuo Y, Yalavarthi S, Navaz SA, et al. Autoantibodies stabilize neutrophil extracellular traps in COVID‐19. JCI Insight. 2021;6(15):150111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Bourguignon A, Arnold DM, Warkentin TE, et al. Adjunct immune globulin for vaccine‐induced thrombotic thrombocytopenia. N Eng J Med. 2021;385(8):720‐728. 10.1056/nejmoa2107051 [DOI] [PMC free article] [PubMed] [Google Scholar]