

Abstract
Microtubules play an important role in the process of cell mitosis and can form a spindle in the mitotic prophase of the cell, which can pull chromosomes to the ends of the cell and then divide into two daughter cells to complete the process of mitosis. Tubulin inhibitors suppress cell proliferation by inhibiting microtubule dynamics and disrupting microtubule homeostasis. Thereby inducing a cell cycle arrest at the G2/M phase and interfering with the mitotic process. It has been found that a variety of chalcone derivatives can bind to microtubule proteins and disrupt the dynamic balance of microtubules, inhibit the proliferation of tumour cells, and exert anti-tumour effects. Consequently, a great number of studies have been conducted on chalcone derivatives targeting microtubule proteins. In this review, synthetic or natural chalcone microtubule inhibitors in recent years are described, along with their structure-activity relationship (SAR) for anticancer activity.
Keywords: Chalcones, anticancer, microtubule, tubulin polymerisation inhibitors
1. Introduction
Tubulin is a globular protein mainly in kinetoplastid protozoa, which consisting of the alpha (α), beta (β), gamma (γ), delta (δ), epsilon (ε), and zeta (ζ) tubulin families1. Meanwhile, α-tubulin and β-tubulin form a heterodimer, which in turn forms long-chain fibres2. Then 13 parallel long-chain fibres are interlinked to form a hollow tube, which is called microtubule1,3. Microtubules are key components of the cytoskeleton and are involved in a variety of cellular functions such as cell signalling, intracellular transport, secretion, formation and maintenance of cell shape, motor regulation, and cell division4,5. The heterodimers are bound to each other by non-covalent bonds, so the heterodimers can be continuously bound and separated. And the two heterodimers at the two ends of the microtubule can be continuously added and released so that the microtubule can be continuously polymerised and discrete, so there is a dynamic balance state in this process6. When the microtubule is in dynamic balance, one end of the microtubule will release a set of heterodimers, and the other end will bind a set of heterodimers, which can not only keep the length of the microtubule constant but also enable the microtubule to move from one side to the other, thus completing the necessary physiological function.
In mammalian cells, microtubules are closely related to cell division. Tubulin inhibitors can disturb the dynamic balance of microtubules by inhibiting the kinetic properties of microtubules. Thus, leading to the induction of cell cycle arrest at the G2/M phase and interfering with the mitotic process to inhibit cell proliferation7. Four targets of action have been identified in the study of microtubule proteins, which can be divided into two classes: microtubule-stabilizing binder including taxanes binding site and laurimalide binding sites; and microtubule destabilising binder including vinca alkaloids binding site and colchicine binding site. The former can promote the polymerisation of microtubule proteins, while the latter inhibits the polymerisation of tubulin, both of which can arrest the process of tumour cell growth in the cell division phase8. Many anti-mitotic drugs, such as vinblastine alkaloids, taxanes, and nocodazole, have been used clinically for many years9, while problems such as drug resistance and toxic side effects remain to be solved. Thus, it is of great significance to discover and develop new tubulin polymerisation inhibitors10,11.
Chalcone is a natural product with the structure of 1,3-diphenyl-2-propene-1-one, which can act on a variety of targets and has antioxidant, antifungal, antitumor, antibacterial, anti-inflammatory, analgesia, immunoregulation, and other biological activities12–15. As early as two decades ago, Edwards et al. discovered that chalcone can bind with tubulin and disrupts the formation of blood vessels around the tumour, lead to the tumour tissue with inadequate oxygen and nutrient supply, eventually lead to tumour cell death16. With the development of research on chalcone in recent years, it has been found that chalcone has a simple structure, easy to synthesise, and potent antitumor activity17. However, there are no clinical antitumor drugs with chalcone structure, so chalcone derivatives have received wide attention and are expected to advance the progress of antitumor drug research8. The core structure of chalcone is composed of two aromatic rings where are connected by three carbon α, β-unsaturated carbonyl bridges (Figure 1). Due to its simple chemical synthesis and ease of modification, most of the current studies on the structural modification of chalcone are focussed on the replacement of the two aromatic rings or the α, β-unsaturated carbonyl bridges. In this review, we focus on the biological activities of chalcone derivatives as tubulin polymerisation inhibitors reported in recent years, classifies them according to the structure type of modified substituents, and summarise the effect of substituent groups on anticancer activity.
Figure 1.
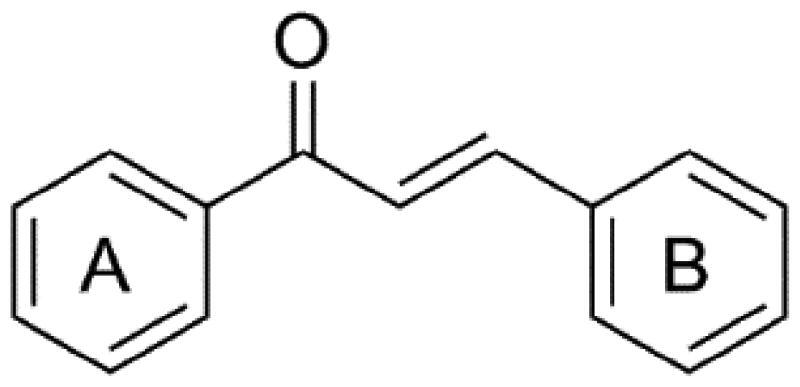
Chemical structure of chalcone.
2. Simple chalcones
Chalcones are widely distributed in natural medicinal plants and generally have hydroxyl or methoxy substituents on their aromatic rings. By the Claisen-Schmidt aldol condensation of an aldehyde with a ketone, a range of chalcones and (E)-4-(4′-hydroxyphenyl)but-3-en-2-one have been designed and synthesised by Lawrence et al.18. In this study, all tested compounds exhibited superior cytotoxicity (IC50 < 3 µM) in human leukaemia cancer cells (K562), human breast cancer cells (MCF-7), human breast cancer cells (BT20), and various multidrug-resistant cell lines than the lead butenone l (Figure 2). Especially, compounds 2b–c (Figure 2) displayed high toxicity against tested cancer cell lines. The structure-activity relationship studies (SARs) showed that the methyl group of 1 is replaced with a substituted aryl group and an additional substitution alternative pattern of the B ring could improve the antitumor activity. Mechanism studies showed these compounds can block cells in the G2/M phase of the cell cycle. In addition, the microtubule network in untreated cells was destroyed by incubation with some target compounds, and 2a (Figure 2) was the most effective inhibitor of tubulin assembly (IC50 = 10 µM) in this series of compounds.
Figure 2.

Simple chalcone compounds of 1 and 2.
Tu et al.19 synthesised a range of novel 2′,5′-dimethoxylchalcone derivatives 3 (Figure 3) and tested their cytotoxicities against human bladder cancer cell lines (NTUB1) and human prostate cancer cell line (PC3). Most of them had strong cytotoxic effects on NTUB1 and PC3 cell lines, with the best effects of 3a and 3b. Interestingly, Western blot results showed that compound 3b increased α-tubulin levels in a dose-dependent manner, while 500 nM paclitaxel showed similar effects in NTUB1 cells. These results demonstrated that the substituted carbamoyl group on C-4 position of B-ring may be an important structural feature of the binding to tubulin. In addition, 3b has the specificity to inhibit the growth of urinary cancer cells and minor toxicity for normal cells.
Figure 3.

Simple chalcone compounds of 3.
Kong et al.20 compared the bioactivity of novel boronic acid analogs 4 (Figure 4) of chalcones with tubulin polymerisation inhibitor combretastatin A-4 (CA-4). The results showed that compound 4b was the most potent compound in this series, and its inhibitory activity was similar to that of CA-4 in the tubulin assay. However, 4b was 3–4 times less active against the growth of MCF-7 cells than CA-4 and was even less active in destroying the microtubules in A-10 cells. In contrast, the inhibitory activity of 4a in colchicine binding and tubulin polymerisation was weak, whereas showed effective cytotoxic activity against MCF-7 cancer cells (IC50, 0.9 µM). Besides, compound 4a has a 10–200 nM inhibitory effect on 16 kinds of human cancer cell lines (e.g. colon cancer HCT-15, CNS cancer SNB-19, and ovarian cancer SK-OV-3) with GI50 values below 10 nM. It also has a significant anti-angiogenesis effect on the human umbilical vein endothelial cells HUVEC. The SAR study showed that the carbonyl adjacent to the A-ring is more active than B-ring.
Figure 4.

Simple chalcone compounds of 4.
Based on the classical models of colchicine ligand and colchicine binding sites to β-tubulin, two series of chalcones were designed and synthesised, and their effects on the inhibition of tubulin assembly and toxicity in human cancer cell lines were investigated21. Among them, compounds 5a and 6a (Figure 5) inhibited tubulin assembly comparable to colchicine in vitro and were only one order of magnitude weaker than colchicine in human cancer cell toxicity tests. Docking studies showed that the direction of binding of the chalcone scaffold to the colchicine site of tubulin was similar to that of natural products. Particularly, the 3,4,5-trimethoxyphenyl moiety closed to the carbonyl group seems to favour the ligand's interaction with tubulin, occupying the same position as the colchicine corresponding part. Furthermore, 5a and 6a inhibited the growth of human leukaemia cell lines at nanomolar concentration, caused microtubule instability and mitosis arrest in human cervical cancer cells, and inhibited the migration of human breast cancer cells in scratch wounds and Boyden chamber assays.
Figure 5.

Simple chalcone compounds of 5–9.
Two series of chalcones (7 and 8) were evaluated for their antiproliferative inhibition and promotion of tubulin polymerisation activities22. Thereinto, compounds in series 8 (Figure 5) showed strong inhibitory effect on human colon cancer stage II (HT-29) cells, with IC50 value of 19.26–36.95 μM, which was much lower than that of the control compound (153.96 µM). Especially, compounds 8a and 8b showed activities as inhibitors and promoters of microtubule dynamics, respectively. Meanwhile, Tajuddin and co-workers have found that compounds containing dimethoxybenzene on ring B and furan on ring A inhibited tubulin polymerisation and HT-29 cell growth.
Aryapour and co-workers prepared a series of chloro-substituted-2′-hydroxychalcones 9 (Figure 5) and tested their cytotoxic effects on K562 and human neuroepithelioma cell lines (SK-N-MC), as well as their role as inhibitors of tubulin polymerisation23. Compared with curcumin, they had better tubulin inhibition and cytotoxic activity against K562 and SK-N-MC. Meanwhile, compound 9a inhibited the assembly of the protofilaments by 89% and could bind to tubulin with a dissociation constant of 3.7 µM and change the far-ultraviolet circular dichroism spectra of tubulin. By comparing the unsubstituted analogs and chlorine-substituted analogs, it was found that the addition of chlorine-substituted analogs to the A/B ring increased the hydrophobic interaction and bioactivity of the compounds. While, the additional chlorine substitution (2-chlorine) reduces the bioactivity, which may be due to the steric hindrance of the substitution.
Several novel chalcones 10 (Figure 6) containing fluoro atom at B ring were prepared and evaluated for their antiproliferative activity against a panel of human tumour cell lines24. Some of them showed potent antiproliferative effects to certain human tumour cells, and compound 10a displayed the most promising anticancer activity against most cell lines. However, none of them showed any cytotoxicity to normal human embryonic kidney cell lines (HEK 293). Acridine orange staining data indicated that these compounds could induce cytotoxicity and anti-proliferation of tumour cells by inducing apoptosis, and further studies showed that compounds 10a and 10b exhibited a concentration-dependent antiproliferative activity on human hepatocellular carcinoma cell lines. Meanwhile, the silico molecular docking study of the compounds on tubulin suggested that they could interfere with cell division. SAR analysis showed that the antiproliferative activity of chalcone structure can be improved by removing the ketone or double bond on the two benzene ring linking groups, and the presence of fluoro substituents in the B ring increased the antitumor potency, but at least two fluoro substituents were in the 2,5 position of the B ring.
Figure 6.

Simple chalcone compounds of 10.
Abosalim et al.25 synthesised a class of new chalcone analogs 11 (Figure 7) and evaluated their anticancer activity against breast cancer and liver cancer. Several of them were revealed a broad superlative inhibitory activity against both HepG2 and MCF-7 cell lines. Particularly, 11a was the best compound in both MCF-7 and HepG2 cell lines (GI50: 5.43 ± 0.170 μM, 1.80 ± 0.50 μM, respectively), besides the tubulin polymerisation inhibition with IC50 of 4.51 ± 0.13 μM. Staining analysis and DNA flow cytometry indicated that 11a arrested the cell cycle arrest in G2/M phase and stimulated apoptosis by effectively inhibiting microtubule polymerisation. In addition, molecular docking results showed that 11a interacts with various amino acid residues at tubulin binding sites, indicating that 11a has a good binding pattern.
Figure 7.

Simple chalcone compounds of 11.
3. Aromatic ring modifications
3.1. Benzopyran-chalcones
Millepachine (12) is a bioactive natural product with the structure of benzopyran (Figure 8), which exhibits significant potential for cancer treatment26,27, and its derivatives have the potential to become new anticancer agents. In 2012, Wang et al.28 designed and synthesised a novel series of millepachine derivatives 13, and evaluated for their in vitro and in vivo antiproliferative activity (Figure 8). Among them, compound 13a with 4-diethylamino substitute on the B ring was found to be superior antiproliferative activity than millepachine against HepG2, K562, human ovarian cancer cells (SKOV3), HCT116, HT29, and SW620 tumour cells (mean IC50 = 0.64 vs. 2.86 µM, respectively). In addition, 13a could significantly suppress tubulin aggregation in HepG2 cells and arrest the HepG2 cell cycle in the G2/M phase in a concentration-dependent manner. In vivo studies demonstrated that 13a could significantly inhibit the growth of tumour, and its anticancer effect was stronger than millepachine and cisplatin. SAR analysis showed that the introduction of EWG into the right phenyl ring was detrimental to anti-tumour activity, while the introduction of dialkylamine groups into the para-position of the right ring remarkably improved the anti-tumour activity. Besides, the introduction of electron-donating groups (EDG) at para-position was more beneficial to inhibit the activity than that of EWGs.
Figure 8.

Benzopyran-chalcone compounds of millepachine and 13.
Duo to the poor solubility and low bioavailability (7.08%) of compound 13a possibly limited the druggability. To obtain new tubulin inhibitors with better antiproliferative activity and oral bioavailability, a range of novel millepachine derivatives 14 (Figure 9) were designed and synthesised based on the SAR of compound 13a29. All newly synthesised compounds showed strong tubulin polymerisation inhibition and anti-proliferation activities. Especially, compound 14a exhibited the most potent inhibition activity which inhibited the growth of HepG2, A549, human malignant melanoma (A375), human hepatocellular carcinoma (SMMC-7221), and K562 cancer cell lines with IC50 values in the range of 0.15–0.52 µM. In addition, compound 14a inhibited the G2/M phase of the cell cycle and tubulin aggregation. A molecular docking study suggested that compound 14a binds into the colchicine binding site of tubulin. Together, these findings suggested that 14a was a promising new anti-mitotic compound that may be used in the treatment of cancer. Importantly, oral efficacy results show that 14a displays modest to excellent oral bioavailability (oral bioavailability of 26.25% at a dose of 12 mg/kg) as compared to 13a (oral bioavailability of 7.08% at a dose of 12 mg/kg).
Figure 9.

Benzopyran-chalcone compounds of 14.
Wang and colleagues also synthesised a series of novel pyrano chalcone derivatives containing indole moiety 15 (Figure 10) and evaluated their antiproliferative activities30. Most of them displayed moderate to strong cytotoxic activity against HepG2 cancer cell lines. Thereinto, compound 15a with a propionyloxy group at the 4-position of the left phenyl ring and N-methyl-5-indoly on the right ring displayed the most potent cytotoxic activity against SMMC-7221, HepG2, PC-3, A549, K562, HCT116, SKOV3, MCF-7, vincristine resistant HCT-8 (HCT-8/V) and taxol resistant HCT-8 (HCT-8/T) with IC50 values ranging from 0.22 to 1.80 µM and low cytotoxicity on the normal human cell line Lo2. In addition, 15a was found to significantly induce cell cycle arrest in the G2/M phase and suppressed the polymerisation of tubulin. Docking studies showed that the interaction of 15a at the colchicine binding site of tubulin. SAR analysis indicated that N-methyl-5-indoly on the right ring and propionyloxy group at the 4-position of the left phenyl ring could increase the cytotoxicity.
Figure 10.

Benzopyran-chalcone compounds of 15.
In the same year, Yang et al.31 synthesised a series of novel millepachine derivatives and their anti-proliferation activities in vitro were evaluated. Among them, the IC50 value of compound 16 (Figure 11) on three cancer cells (HepG2, A375, and K562) was 163–253 times higher than that of millepachine, and the IC50 value was between 8 nM and 26 nM. Preliminary studies on the mode of action showed that 16 induced cell accumulation in the G2/M phase of the cell cycle and induced apoptosis of HepG2 cells, and it was not a substrate for the glycoprotein drug pump, nor was it affected by the β-tubulin III gene. Compound 16 also exhibited anti-vascular activity. Microtubule dynamics confirmed 16 could bind to the colchicine site. Moreover, the hydrochloride salt of 16 (16·HCl) effectively increased the bioavailability up to 47% and maintained the antiproliferative activity. Importantly, 16·HCl was a promising oral anticancer drug because 16·HCl significantly inhibited tumour growth in four xenograft tumour models (including drug-resistant tumour-bearing mice) without causing significant weight loss in mice.
Figure 11.

Benzopyran-chalcone compounds of 16–18.
Through modification of millepachine, a new class of chalcone tubulin inhibitors 17 (Figure 11) were designed and synthesised32. Compounds 16 and 17 showed potent and similar growth inhibition against both sensitive and multidrug drug-resistance (MDR)1 or multidrug resistance-associate protein (MRP)1 (A2780/T, HCT-8/T and HCT-8V) cell lines, with a resistance factor ranging from 1.5 to 2.5. By biotinylation of millepachine and pull-down experiments, Yang et al. finally identified β-tubulins as cellular targets for millepachine and its derivatives and showed that these compounds bind to colchicine sites in an irreversible manner. Besides, since chalcone occupies colchicine in the trans-conformation (s-trans), naturally occurring chalcone with the s-trans conformation was more active.
Many novel millepachine derivatives induce apoptosis in the double-digit nanomolar range of multiple tumour cell lines by attacking microtubules33. Among them, compound 18 (Figure 11) showed strong antitumor activity against 21 tumour cell lines including multi-drug resistant phenotypes, with IC50 values in the range of 0.09 to 1.30 μM. Moreover, 18 could induce G2/M phase cell cycle arrest, promote tubulin aggregation into microtubules, and stabilise the microtubule. Besides during in vivo study, compound 18 exhibited significantly inhibitory activity in human HepG2 xenograft tumour models. Thus, as a new microtubule stabiliser, 18 has good antitumor activity.
3.2. Naphthalene-chalcones
The naphthalene ring system is found widely in natural products and has a variety of biological activities, including antitumor34, anti-inflammatory35, antibacterial36, antifungal37, and antioxidant38. Notably, a large number of naphthalene-substituted chalcones derivatives have also been reported in recent years due to their good biological activity, which prompted researchers to study naphthalene-substituted chalcones in the hope of obtaining compounds with strong anti-tumour activities.
In 2014, Lee et al.39 synthesised a group of methoxylchalcones and discussed the mechanisms by which these compounds inhibit tumour growth. Among them, the newly synthesised methoxychalcone HMNC-74 19 (Figure 12) showed the best clonogenicity inhibitory effect on SW620 colon cancer cells. Further study showed that 19 can inhibit tubulin polymerisation and trigger mitotic arrest, in turn causing both p53-dependent and caspase-2-mediated apoptosis.
Figure 12.

Naphthalene-chalcone compounds of 19 and 20.
To study the antitumor mechanism of benzochalcone derivatives, Shin et al.40 synthesised a range of benzochalone derivatives. In this study, 2-hydroxy-4-methoxy-2′,3′-benzochalcone (HymnPro) 20 (Figure 12) showed the best activities against the clonogenicity of Capan-1 human pancreatic cancer cells, and also suppressed cell proliferation in some human solid tumour cell lines as well as inhibited tumour growth in nude mice. The molecular mechanisms showed that compound 20 exerts antitumor activity by disrupting microtubule assembly, which leads to mitotic arrest and sequential activation of the caspase pathway, resulting in apoptosis. In 2016, Lee and colleagues observed that treatment CaPan-1 pancreatic cancer cells with 20 resulted in a dose-dependent accumulation of ROS due to intracellular glutathione depletion41. Meanwhile, N-acetylcysteine, a ROS scavenging agent, can inhibit Hymnpro-induced caspase activation and cell death, but G2/M cell cycle arrest and microtubule assembly were not significantly affected.
Based on the lead compound HMNC-74 (19), Wang et al.42 prepared a new class of naphthalene-chalcone derivatives 21 (Figure 13) and evaluated their inhibitory activities against breast cancer MCF-7 cell line. Most of them show strong antiproliferative activity. Particularly, compound 21a was the most active with an IC50 value of 1.42 ± 0.15 µM, as superior to cisplatin (IC50 = 15.24 ± 1.27 µM). In addition, 21a showed lower cytotoxicity to normal cells (HEK 293) than tumour cells. Besides, 21a could significantly induce cell cycle arrest at the G2/M phase and cell apoptosis. Compound 21a showed a strong tubulin polymerisation inhibition activity with an IC50 value of 8.4 µM, which was slightly higher than that of colchicine (IC50 = 10.6 µM). Docking studies indicated that 21a interact and bind at the colchicine binding site of the tubulin.
Figure 13.

Naphthalene-chalcone compounds of 21.
To discover novel tubulin polymerisation inhibitors, Wang et al.43 designed and synthesised a hybrid scaffold by incorporating millepachine with naphthalene moiety in a single molecule 22 (Figure 14). These compounds were evaluated for their in vitro anticancer activity on selected human cancer cell lines. Compared with the lead compound millepachine, compound 22a showed the most effective anti-tumour activity against human colon cancer cells (HCT116) and human hepatocellular liver carcinoma cells (HepG2) with IC50 values of 1.20 and 1.02 μM, respectively. Furthermore, tubulin polymerisation experiments showed that 22a could effectively inhibit tubulin polymerisation, and flow cytometry indicated that 22a could inhibit the activity of HepG2 cells in the G2/M phase in a dose-dependent manner. SAR analysis summarised that the change of substituent affects anticancer activity. First, introducing the electron-withdrawing group (EWG) into the meta position of the benzene ring significantly increased its anticancer activity, while moving the EWG to the ortho or para-position destroyed its anticancer activity. Additionally, the introduction of methoxy in the phenyl ring can improve the biological activity, but too much is not good for the antitumor activity. Furthermore, molecular docking studies suggested that 22a binds to the colchicine binding site of tubulin.
Figure 14.

Naphthalene-chalcone compounds of 22.
Many previous studies have indicated that the 3-amino-4-methoxy phenyl moiety is an essential pharmacophoric group for binding to the colchicine site of tubulin and enhance antiproliferative activity44–47. Based on the structure of compound 22a, Wang et al. design and synthesis a novel series of aminochalcones by the introduction of 3-amino-4-methoxyphenyl moiety into the molecules (Figure 15)48. The antiproliferative activity of this series of amino-chalcone derivatives 23 against HepG2 and HCT116 human cancer cell lines was evaluated. Most of the newly synthesised compounds showed moderate to strong antiproliferative activity against tested cancer cell lines. Among the derivatives, compound 23a had the strongest inhibitory activity against HepG2 and HCT116 with IC50 in the range of 0.15–0.34 μM. And this compound showed low cytotoxicity on normal human cell lines (Lo2). Compared with colchicine (IC50 = 9.0 µM), compound 23a exhibited concentration-dependent tubulin assembly with IC50 value of 7.1 μM in vitro. The further biological evaluation showed that compound 23a could arrest the cell cycle in the G2/M phase. SAR analysis indicated that the antiproliferative activity of the amino group was significantly reduced when the alkyl substituents were introduced into the amino group and the normal or branched-chain alkyl substituents were introduced into the phenyl ring amino group. Docking study demonstrated the interaction of compound 23a at the colchicine binding site of tubulin.
Figure 15.

Naphthalene-chalcone compounds of 23.
3.3. Benzofuran-chalcones
Furan structures are widely found in bioactive compounds and possess a wide range of biological activities such as antitumor49, anti-inflammatory50, antifungal51, anti-tuberculosis52, histamine antagonism53, anti-bacterial54–56. Because of such a wide range of pharmacological activities of furans, researchers introduced them into the chalcone molecule to modify the structure to obtain derivatives with better antitumor activity.
To investigated the antiproliferative activities against MCF-7 cell line and the potential to induce apoptosis and also to inhibit tubulin polymerisation and/or epidermal growth factor receptor-tyrosine kinase (EGFR-TK) phosphorylation, Mphahlele et al. synthesised a range of 2-arylbenzo[c]furan-chalcones 24 (Figure 16)57. Compared with control actinomycin D, several of them showed moderate to significant antigrowth effects on MCF-7 cell lines in vitro. Thereinto compounds 24a and 24b displayed the strongest activities (IC50 = 3.55 × 10−4– 0.79 µM). Docking studies showed that the compounds have the potential to inhibit tubulin polymerisation and EGFR-TK phosphorylation. Besides, the model structures of 24a and 24b suggested hydrophobic interactions and the bonding of hydrogen and/or halogens to protein residues, which may contribute to the increased binding affinity of both receptors and their significant antigrowth effects on MCF-7 cell lines.
Figure 16.

Benzofuran-chalcone compounds of 24.
3.4. Indole-chalcones
Indole ring is a very important heterocyclic, widely exists in active natural products and synthetic compounds, such as strychnine and Vinblastine58,59. Moreover, indole is often used as a component of many natural products and drugs and attracted wide attention for its anti-inflammatory60, antifungal61, antibacterial62, antitumor, and other biological activities63.
Mirzaei et al. have demonstrated that a series of indole-derived methoxylated chalcones 25 (Figure 17) as anti-dermatophyte agents64. Most of the compounds showed strong antifungal activity in the antifungal susceptibility tests of different dermatophytes in vitro. Particularly, the 4-ethoxy analog 25a with minimum inhibitory concentration (MIC) values of 0.25–2 μg/mL was the most active against Trichophyton interdigitale, Trichophyton veruccosum and Microsporum fulvum. Furthermore, the 4-butoxy compound 25b showed the most potent activities against Trichophyton mentagrophytes, Microsporum canis, and Arthroderma benhamiae (MIC values of 1 − 16 μg/mL). Model docking assay showed that compound 25a could bind with tubulin, and by molecular dynamics (MD) simulation, the interaction between compound 25b and the active site of the target protein was stable.
Figure 17.

Indole-chalcone compounds of 25.
Boumendjel et al. reported that compound 26 (Figure 18) had the potential to treat the tumours of the central nervous system (CNS)65. In the four human cell lines (U118, U138, U373, and LN229) and one mouse glioblastoma cell line (GL26), compound 26 suppressed proliferation and arrested cells in the M phase of the cell cycle at the concentration of 10 μM. The mechanism of action shows that this compound is a microtubule depolymerising agent, which binds to the colchicine binding site of tubulin with an association constant of 2 × 105 M−1. Compound 26 also suppressed the activity of the P-glycoprotein (P-gp/ABCB1) and the breast cancer resistance protein (BCRP/ABCG2), without being a substrate of these efflux pumps. Moreover, 26 was found to induce the delay of tumour onset and inhibition of tumour growth in vivo studies and was detected in tumours of treated animals by the high-performance liquid chromatography method (HPLC). Besides, 26 is also able to cross the blood-brain barrier (BBB).
Figure 18.

Indole-chalcone compounds of 26 and 27.
Chalcone derivative 27 (Figure 18) is considered a potential anticancer drug because of its ability to kill bladder cancer cells through caspase-dependent apoptosis following cell cycle arrest at the prometaphase stage. Studies have shown that 27 can inhibit the proliferation of bladder and cervical cancer cells without toxicity to normal urothelial and pulmonary cells, which prompted the authors to further investigate the characteristics of 27 and to identify its cell targets66. They demonstrated that 27 selectively inhibits tumour-derived cell proliferation and interferes with spindle formation and mitotic chromosomal alignment. In cells, 27 induced an increase in soluble tubulin and showed anti-angiogenic activity. The results of docking studies displayed that the indole group of 27 could be adapted to the colchicine binding site of tubulin, and it could compete with colchicine for soluble tubulin. Furthermore, in a human bladder xenograft mouse model, 27 inhibited tumour growth and was non-toxic to normal cells. In summary, 27 was a novel microtubule-targeting drug with potential chemotherapeutic value.
Because of the importance of 3-bromo-3,5-dimethoxyphenyl as a fragment of tubulin inhibitor, Hassan and co-workers prepared new indole-derived chalcones 28 (Figure 19) as tubulin-targeting antiproliferative agents67. Cytotoxicity tests on tumour cell lines showed that several analogs were superior to or as effective as the control drug etoposide. Particularly, the IC50 value of compound 28a was 4.3 µg/mL, which was stronger than etoposide in inhibiting A549 cell line with adenocarcinoma. The further biological evaluation showed that compound 28a induced apoptosis of cancer cells by inhibiting tubulin aggregation and reducing mitochondrial thiol content. The docking model with tubulin showed that 28a could bind to the colchicine binding site. SAR analysis showed that the unsubstituted indole substances were superior to N-methyl indole congeners in MCF-7 cells, but the pattern of functionalities on the phenyl ring would affect the cytotoxicity for A549 N-Substitution and SKOV3.
Figure 19.

Indole-chalcone compounds of 28.
A class of novel chalcone derivatives containing 3′,4′,5′- trimethylyl and indole fragments 29 (Figure 20) have been obtained, and their antiproliferative activity against a panel of HeLa, human colon carcinoma (HT29), MCF-7, and human lymphoma (HL-60) cells (3–5 × 104 cells) lines also tested68. The majority of the synthesised compounds showed superior activities to the reference drug. Especially, compound 29a showed the most potent antiproliferative activity against HeLa, HT29, and MCF-7 cancer cell lines, with IC50 values of 0.37, 0.16, and 0.17 µM, respectively, and with considerably lower activity against HL-60 cells (IC50:18 µM). In the human myeloid leukaemia U-937 cell line overexpressing human Bcl-2 (U-937/Bcl-2), the cytotoxicity (IC50 ∼1 μM) of 29a was observed by inhibiting G2-M cell cycle progression and inducing cell apoptosis. Furthermore, the antiproliferative activity of this molecule was found to be associated with the inhibition of tubulin polymerisation. SAR analysis indicated that the introduction of methoxy at the C5- or C6-position of the indole nucleus and the absence of substituents at the N-1-indole site favoured the best activity of indole-propenone-3′,4′,5′-trimethoxyphenyl scaffold.
Figure 20.

Indole-chalcone compounds of 29.
3.5. Benzoxazole-chalcones
Benzoxazoles exhibit a variety of effects, and these compounds are widely used in medicine and optical materials69. Previous studies have indicated that benzoxazoles have a variety of biological activities such as antitumor70,71, antiviral72, antimicrobial73, antifungal74, and anti-inflammatory properties75. Current clinical drugs that contain this structure fragment include chlorzoxazone, benoxaprofen, and flurbiprofen70,75.
Konieczny et al.76 prepared a range of benzoxazole-chalcone derivatives 30 (Figure 21) and tested the antitumor activity of these compounds. Some of them demonstrated cytotoxic activity at sub-micromolar concentrations, and compound 30a showed potent activity against three tumour cell lines (A549, HCT116, and HeLa) with IC50 values in the range of 0.0035 ± 0.015 μM. The study on the mechanism of activity showed that these compounds interfered with tubulin polymerisation by binding to the colchicine binding site. Moreover, molecular modelling showed that compounds 30 could use two distinct poses in the colchicine binding domain of tubulins according to the substitution pattern of the ring B and alkoxy chain in position 6 of the ring A. SAR studies revealed that, in α, β-unsaturated ketones structures, the activity of the compounds with the carbonyl group attached to the C-5 position of the benzoxazole ring is generally higher than that of the compounds attached to the C-4 position of the compounds.
Figure 21.

Benzoxazole-chalcone compounds of 30.
Based on previous studies on benzoxazole-chalcone, Konieczny et al.77 oxidised the sulphur atoms in the oxazole ring to investigate in vitro cytotoxic activity and interaction with tubulin of these compounds (31 and 32) (Figure 22). The majority of them displayed cytotoxic activity at submicromolar concentrations, particularly, 31a (IC50 = 0.006 μM) and 32a (IC50 = 0.003 μM) showed the strongest cytotoxic activity in A549 cells. In this study, they found that the oxidation and location of sulphur atoms in the oxathiole-fused chalcones greatly influenced the activity of these compounds. Moreover, oxidation of isomers with a sulphur atom at the para-position of chalcone carbonyl increased the activity, while oxidation of isomers with a sulphur atom at the meta-position of carbonyl decreased the activity. Meanwhile, they described that the cytotoxic activity produced by these compounds derives in part from their interaction with the colchicine binding sites of tubulin, but that the profound effect of sulphur atomic oxidation on cytotoxic activity is related only to the interaction of the compounds with tubulin.
Figure 22.

Benzoxazole-chalcone compounds of 31 and 32.
3.6. Quinoline-chalcones
Quinoline is a structural fragment with strong pharmacological activity, and some compounds exhibit antibacterial, anti-inflammatory and analgesic, anticonvulsant, hypoglycaemic, antihypertensive, antitumor, and antiviral activities when introduced into quinoline78–81. And many structurally simple agents can also be modified by quinoline to increase their physiological activity and efficacy. Thus, the researchers obtained a suggestion that the introduction of quinoline into the chalcone molecule may increase the antitumor activity of chalcone. Mirzaei and co-workers synthesised a series of quinoline-chalcone hybrids 33 (Figure 23) and tested the in vitro anticancer activity of those compounds against four human cancer cell lines including human ovarian cancer cell (A-2780), cisplatin-resistant human ovarian cancer cell (A-2780/RCIS), MCF-7, mitoxantrone-resistant human breast cancer cells (MCF-7/MX), and normal cells (HUVEC) using MTT assay82. Benzoyl-containing quinolines 33a–33c displayed potent activity against both resistant cancer cells and their parents. In particular, analogs 33b–33c showed the strongest antiproliferative activity with IC50 values range from 2.32 to 22.4 µM. They have also been evaluated as tubulin inhibitors that arrested the cell cycle in the G2/M phase and induced apoptosis. SAR analysis indicated that the groups on the quinoline ring increase cytotoxicity and antiproliferative activity in the following order: 8-benzoyl > 6-benzoyl > 5,6,7-trimethoxy > 6,7-dimethoxy > phenyl (fused to 7,8 position of quinoline), and which with (3,4,5-trimethoxy phenyl) propy-2-en-1-one group is generally more cytotoxic at 3-position than the corresponding quinoline with (4-methoxy phenyl) propy-2-en-1-one group. In addition, molecular dynamics simulation and molecular docking studies on the binding site of 33c with colchicine indicated that the compound might interact with the active site of tubulin.
Figure 23.

Quinoline-chalcone compounds of 33.
Lindamulage and colleagues synthesised and tested a small chemical library of novel quinolone chalcones based on the postulation that certain chalcones could effectively overcome multidrug resistance problems with minimal side effects83. They found that these compounds were generally quite effective in killing cancer cells, and they chose to study 34a and 34b (Figure 24) in more detail as they significantly and preferentially killed a wide range of cancer over non-cancer cells. Thereinto, both 34a and 34b were resistively bound to colchicine binding pouches on β-tubulin, and 34b had strong inhibitory activity against 65 different tumour cell lines from 12 different tissues mainly in a cancer-specific manner. Interestingly, these two compounds significantly inhibited multidrug-resistant cancer cells compared with colchicine, paclitaxel, and vinblastine. Furthermore, 34b overcame multidrug resistance by inhibiting the function of MRP1 rather than maintaining strong anti-microtubule activity. Data obtained from mice engrafted with the human breast cancer cells (MDA-MB-231) triple-negative breast cancer cells exhibited that 34a and 34b exhibited potent antitumor activity and not causing any significant side effects, either alone or in combination with paclitaxel. As a result, these two compounds are potent and safe in the treatment of various kinds of cancers, especially multidrug-resistant tumours.
Figure 24.

Quinoline-chalcone compounds of 34a-35a.
Tseng et al.84 evaluated the antiproliferative activity of several 3-phenylquinolinyl chalcone analogs in vitro. Thereinto, 35a (Figure 24) showed inhibitory activity against the growth of MCF-7, MDA-MB-231, and SKBR-3 with IC50 values of 1.05, 0.75, and 0.78 μM, respectively, and no obvious cytotoxicity to normal cells. The mechanism of action of compound 35a was further studied. It was found that 35a induced G2/M cell cycle arrest by regulating cyclins B1, CDK1, and CDC25, inhibiting tubulin aggregation. Compound 35a finally induced apoptosis by increasing the apoptotic protein Bax and decreasing the anti-apoptotic protein Bcl-2. Besides, compound 35a was found to induce cell cycle arrest at the G2/M phase via cleavage of PARP, induces caspase-3 and -8 activities, and consequently cause cell death.
3.7. Other heterocyclic chalcones
Besides the above widely reported hybrid chalcone derivatives, researchers have also introduced pyridine, benzothiazole imidazole, and other structures into the molecular structure of chalcone, designed and synthesised many novel chalcone derivatives. A series of novel imidazole-chalcone derivatives 36 (Figure 25) were prepared as tubulin polymerisation inhibitors and anticancer agents by Sara et al.85 In this series, all derivatives exhibited more cytotoxicity on A549 cancer cells in comparison to the other three cell lines (MCF-7, MCF/7-MX, and HEPG2). Particularly, 36a–b displayed potent cytotoxicity with IC50 values ranging from 7.05 to 63.43 μM against all the four human cancer cells. By flow cytometry, they found that low concentrations of 36a–b induced G2/M arrest in A549 cells, while high concentrations of them increased the number of apoptotic cells (cells in subG1 phase). Further experiments showed that 36b induced apoptosis of A549 cells in a dose-dependent manner, and 36a–b, inhibited tubulin polymerisation similar to CA-4. The molecular docking study of 36b binding sites with tubulin colchicine indicated that this compound might interact with tubulin.
Figure 25.

imidazole-chalcone compounds of 36.
A range of benzo[d]imidazo[2,1-b]thiazole–chalcone derivatives 37 (Figure 26) was obtained and tested the anticancer potential86. Most conjugates showed effective activities against the tested cancer cell lines. Thereinto, 37a and 37b displayed potent antiproliferative effects against MDA MB-231 with IC50 values of 1.3 and 1.2 µM respectively. SAR analysis revealed that the order of enhancement of different substituents on the A ring could be expressed as H > OMe > OEt > F > Me, while the order was expressed as 4-OMe > 3-OH-4-OMe > 3, 4-diOMe > 3, 4, 5-triOMe > 2-Br-3, 4, 5-triOMe > 3, 5-diOMe on ring D. The flow cytometric analysis results indicated that 37 could induce cell-cycle arrest in the G2/M phase, and they could effectively inhibit microtubule assembly in the tubulin polymerisation assay. Furthermore, morphological changes, reactive oxygen species (ROS) detection by 2′,7′–dichlorofluorescein diacetate (DCFDA), and annexin V-FITC/PI assays as well as revealed that 37a and 37b induces apoptosis.
Figure 26.

Benzo[d]imidazo[2,1-b]thiazole-chalcone compounds of 37.
A range of Combretastatin A-4-based chalones 38 (Figure 27) were synthesised and their antiproliferation of human lung (A549), breast (MCF-7), melanoma (A375) and colon (HT-29) carcinoma cells were examined87. Some of them showed significant antiproliferative activity with IC50 < 2 µM and the compound 38a, the best one, which showed the strongest inhibitory activities against MCF-7 (IC50: 10 nM), A375 (IC50: 12 nM), and A549 (IC50: 65 nM) cell lines, and was 18 times that of CA-4. Moreover, all the tested compounds have shown stronger binding affinity than CA-4 in the colchicine binding site of tubulin dimer.
Figure 27.

Combretastatin A-4-based chalone compounds of 38.
4. Modification of the α, β-unsaturated carbonyl bridges of chalcone
4.1. Double bond modification
The relative position of the two aryl rings in chalcone plays a crucial role in the activity of the binding tubulin, which allows the ketene bridge to be modified. It has been reported that hydrogenation or bromination significantly reduces chalcone activity through carbon-carbon double bonds or halogenation or conversion to corresponding epoxides. Therefore, the double bond in the ketene structure may be the key site affecting the bioactivity of the compound. However, when the double bond of chalcone remains unchanged, it is unstable in vivo due to its ability to undergo Michael addition reactions with biological nucleophiles. Thus, the researchers substituted the ketene-bridge double bond with a heterocyclic ring, which not only maintained the relative tertiary stability of the two aryl rings but also avoided nucleophilic reactions in vivo, thus improving the stability of chalcones88.
Based on the crystal structure of the newly discovered tubulin inhibitor VERU-111 and its analog DJ101, Wang et al.89 synthesised several new VERU-111 analogs 39 (Figure 28) and investigated their SAR evaluation by focussing on the 3,4,5-trimethoxyphenyl (TMP) moiety modification. In this study, the antiproliferative activity of compound 39a with 3-methoxybenzo[4,5]dioxyclic ring in melanoma cell lines was better than that of VERU-111 with IC50 of 1.1–3.3 μM, and without observable toxicity to normal cells. In addition, the resistance index of 39a was significantly increased in parental and taxane resistance prostate cancer cell lines compared to paclitaxel, and the isosteric (conformationally restricted) replacement of TMP could significantly increase the antiproliferative activity of the scaffolds. Compound 39a inhibits tubulin polymerisation by the same mechanism as VERU-111. Overall, they demonstrated that modifying the TMP component of CBSIs in this scaffold could improve antiproliferative activity without affecting the mechanism or safety.
Figure 28.

VERU-111 analogs of 39.
In 2010, Han and colleagues have been found that chalcone epoxides (A, B-epoxone) 40 (Figure 29) were the growth inhibitors of two pancreatic cancer cell lines, BXPC-3 and MIA PACA-290. Three of them were active, with GI50 values of 5.6 to 15.8 µM. The average GI50 of compound 40a on the NCI 60-cell-line panel was 14.1 μM, and it interfered with the enhancement effect of paclitaxel on tubulin polymerisation, suggesting that the microtubules were its active site. Furthermore, 40a was found to block the cell cycle at the G2/M checkpoint or during mitosis leading to apoptosis. Therefore, chalcone epoxides are worthy of further study as a potential drug for the treatment of cancer.
Figure 29.

Chemical structure of compounds 40 and 41.
A range of chalcone-like derivatives 41 (Figure 29) have been obtained and tested for their biological activity and mechanism88. All of the synthesised compounds inhibited cancer cell growth at nanomolar to low micromolar concentrations. Among them, compound 41a had the strongest anti-proliferation activity, which inhibited the assembly and binding of colchicine and tubulin. Generally, all of them with potent antiproliferative activity inhibited tubulin polymerisation with IC50 < 2 μM. Some of them could arrest the G2/M phase of the cell cycle of K562 cells. SAR studies revealed that using a thiophene nucleus between the aryl and 3′,4′,5′-trimethoxybenzoyl moieties of chalcones replace the double bond of the enone system will enhance the anti-proliferation activity of these compounds.
4.2. Carbonyl modification
Guanidine analogs were widely reported as their significant antiviral and anticancer activities91,92. Methylglyoxal-bisguanylhydrazone (MAGA) has been used as a single drug in clinical therapy for malignant lymphoma, carcinoma of the head, oesophageal cancer, and non-small cell lung cancer93. A series of novel chalcone guanidine analogs 42 (Figure 30) were studied as part of the study of novel anti-tubulin polymerisation inhibitors94. Some of them exhibited remarked effects on antiproliferative activities. Compared with the positive control, compound 42a showed the strongest inhibitory activity against the growth of MCF-7 cells with IC50 of 0.09 ± 0.01 µM and the polymerisation of tubulin with IC50 of 8.4 ± 0.6 µM. On the A ring, the SAR analysis indicated that the para-substituted compounds are more active than the ortho-substituted and meta-substituted compounds. Meanwhile, the halogen-substituted compounds on B-ring showed good antiproliferative activity, and p-Br substituted 42a showed the strongest antiproliferative activity. Besides, docking simulation revealed that 42a could bind to the colchicine site of tubulin.
Figure 30.

Chalcone guanidine compounds of 42.
Wang et al. reported a range of novel chalcone oxime derivatives 43 (Figure 31) have the activity of inhibiting tubulin polymerisation95. Most of them showed effective antiproliferative activity and tubulin polymerisation inhibition activity. Thereinto, substance 43a exhibited the most potent inhibitory activity (tubulin IC50 = 1.6 µM) and showed significant antiproliferative activity against A549, Hela, and MCF-7 with GI50 values of 2.1, 3.5, and 3.6 µM, respectively in the antiproliferative assay. SARs showed that compounds with the p-substituted group showed slightly more potent activities than those of o-substituted, and the more substituted groups in the A ring, the better antiproliferative activity it was. However, the antiproliferative activity of the compounds with meta-substituted in the B ring is slightly better than that of the compounds with para-substituted, and the ERG of the B ring may have better antiproliferative activity. Studies on the mechanism of action have confirmed that chalcone oxime derivatives could still inhibit tubulin aggregation at colchicine binding sites, block cells in the G2/M phase, and induce cell apoptosis.
Figure 31.

Chalcone oxime compounds of 43.
4.3. Rigidified chalcone derivatives
Chalcone has an α, β-unsaturated ketones with a single bond that can be rotated when a group is introduced into the α position of the carbonyl group making it easier for the chalcone to maintain the trans configuration. The α position of the compound MDL-27048 is methyl, which is a typical alpha-substituted chalcone compound. Earlier years, Peyrot et al.96,97 demonstrated that MDL-27048 effectively and reversibly binds to the colchicine binding site of tubulin and has antitumor activity. In addition, Lawrence et al.98 revealed the effects of different α-substitutions, in particular α-fluorination, on the structure and biological activity of several chalcones 44–46 (Figure 32). In this study, x-ray analysis showed that α-position fluorine chalcone has s-trans conformation, which displayed strong cytotoxicity and tubulin inhibition properties. Moreover, the 3-hydroxy-4-methoxy B-ring was important for improving biological activity, and no alternative model (including 3-fluoro-4-methoxy) could match its effect.
Figure 32.

Chemical structure of compounds 44–46.
Kerr et al.99 prepared a series of aryl, hydroxyl substitutions in the α position of chalcone, all of which showed potent inhibitory activities of tubulin polymerisation. But only compound 47 (Figure 33) had activity similar to that of CA-4 at low concentrations but did not exhibit anti-mitotic or cytotoxic effects.
Figure 33.

Chemical structure of compounds 47–49.
Ducki et al.100 synthesised a series of chalcone analogs and tested the inhibitory activity against the K562 human chronic myelogenous leukaemia cell line. Among the tested compounds, the most cytotoxic analogs were those most similar to the CA-4 itself, while 48 (Figure 33) with both the A- and B-ring substitutions of CA-4 was the strongest. Interestingly, compound 49 (Figure 33), in the presence of an alkyl group at the α-position, increased its activity to 10 times that of 49. Cell cycle analysis by flow cytometry showed that these drugs had anti-mitotic effects, and they also were found to inhibit tubulin assembly at low concentrations, and competitively binding tubulin with [3H] colchicine.
In the same year, another series of novel α-aryl chalcones 50 and 51 (Figure 34) was be reported as mimics of podophyllotoxin by Ducki et al.101. These substances showed effective bioactivity, and compound 51a exhibited the strongest activity against K562 cancer cells with IC50 value of 12 nM, and the inhibition of tubulin was higher than that of the control. However, molecular studies have revealed that the B rings of chalcone and CA-4 do not occupy the same pockets of tubulin, which belonged to different pharmacophore groups. Interestingly, the binding mode of chalcone is very similar to that of podophyllotoxin, and planar hydrophobic groups such as benzene ring are added to the alpha position of chalcone skeleton, which well simulates the B ring of podophyllotoxin.
Figure 34.

Chemical structure of compounds 50 and 51.
The antiproliferative activity of some novel indole-chalcone derivatives 52 (Figure 35) against different human cancer cell lines was evaluated using the MTT assay102. In this series, analog 52a showed the strongest bioactivity against six cancer cells (A549, HeLa, Bel-7402, MCF-7, A2780, HCT-8) with IC50 values in the range of 3 − 9 nM. SAR analysis indicated that the position of the methoxy group on indole was very important for its antiproliferative activity, and the order of potency was 6-OCH3 > 5-OCH3 > 4-OCH3. Interestingly, the methyl group at the N-1 position has the opposite effect on the acetophenone and propiophenone analogs. Mechanism studies revealed that 52a could arrest the cell cycle at the G2/M phase and induce apoptosis along with the decrease of mitochondrial membrane potential. In addition, 52a was found to be a novel tubulin polymerisation inhibitor that could bind colchicine sites. Importantly, 52a (including its phosphate) showed better metabolic stability in mouse liver microsomes, as well as exhibited good inhibitory activity against the tumour growth in xenograft models in vivo without apparent toxicity, which was superior to the control compound.
Figure 35.

Indole-chalcone compounds of 52.
Novel pyridine-chalcone conjugates 53 and 54 (Figure 36) were evaluated as microtubule polymerisation inhibitors by Li et al.103. In vitro antiproliferative activity screening showed that most of the compounds exhibited potent antiproliferative activities in a nanomolar range. Among them, compound 53a with 3-amino-4-methoxyphenyl moiety exhibited the most potent activity with IC50 values ranging from 0.009 to 0.016 μM in a panel of cancer cell lines. SAR analysis indicated that Quinoline N is a critical for activity and the methyl substituent at the α-position of α, β-unsaturated ketone improved the antiproliferative activity. The order of potency of substitute in the C2-position of quinoline moiety was CH3 > H > NHCH3 > OCH3 > NHC2H5 ≈ N(CH3)2 ≈ Cl ≈ CH2OH > cyclopropylamine > CHO > COOC2H5 > morpholine > pyrrolidin > 4-methoxybenzylamine. Mechanism studies demonstrated that 53a bound to the colchicine site of tubulin, arrested the cell cycle at the G2/M phase, induced apoptosis, depolarised mitochondria, and induced reactive oxidative stress generation in K562 cells. Molecular docking study shown that compound 53a adopted a location very similar to that of CA-4.
Figure 36.

Pyridine-chalcone compounds of 53.
A range of novel pyridine-chalcone derivatives 54 (Figure 37) was found to have potential as anti-tubulin agents, and all of them were evaluated for their antiproliferative activities104. In this series, compound 54a exhibited significantly activities (IC50 = 23–45 µM) against five cancer cell lines, HepG2, epidermoid carcinoma of the nasopharynx (KB), HCT-8, MDA-MB-231, and hepatoma 22 cells (H22), which was 3 times superior to the parent compound. SAR analysis showed that the introduction of the methyl at the α-position of unsaturated carbonyl group was beneficial to the antiproliferative activity, and the exposed hydroxyl on ring B was the key to maintaining the activity. Additionally, the substituents of the EDGs on pyridine are fitted better than that of the EWGs. Studies of mechanism demonstrated that compound 54a could bind to the colchicine site of tubulin. Besides, cellular mechanism results indicated that 54a arrested the G2/M phase, induced cell apoptosis, and destroyed the intracellular microtubule network.
Figure 37.

Chemical structure of compounds 54.
Cong and colleagues prepared an αmethyl-substituted indolecardol 55a (Figure 38) and found that it exhibited potent inhibitory activity against NCI-60 cell lines (average concentration resulted in 50% growth inhibition = 6 nM)105. Interestingly, some multidrug-resistant cancer cell lines were not resistant to 55a, which also showed significant selective toxicity to cancer cells and normal CD34+ blood progenitor cells. The mechanism studies showed 55a inhibited the cells associated with tubulin binding and inhibited microtubule dynamics. Moreover, the National Cancer Institute COMPARE analysis and molecular modelling suggested that 55a acted in a similar way to colchicine.
Figure 38.

Chemical structure of compounds 55 and 56.
Canela et al.106 have studied the tubulin-binding, vascular targeting, anti-tumour, and anti-metastatic activities of such conjugates of chalcone. Compound 56a (Figure 38) showed the most potent activity against the proliferation of endothelial cells and tumour cells, and the inhibition concentration of 50% was 1–4 nM, which was slightly better than the control substances colchicine and combretastatin A4-phosphate (CA-4P). In addition, they showed that these conjugates destabilise microtubules by binding colchicine sites to tubulins, and 56a arrested tumours and endothelial cells in the G2/M phase and induced apoptosis of these cells at nanomolar concentrations. Meanwhile, In the chicken allantoic membrane (CAM) experiment, 56a could inhibit endothelial cell migration and angiogenesis. A water-soluble L-lys-L-Pro conjugate of 56a could result in rapid vascular closure and massive tumour necrosis in melanoma and breast cancer xenograft tumour models. 56a as well as showed anti-metastatic activity comparable to CA-4P. These data suggested that this novel range of derivatives was an important lead molecule for the design of vascular-destroying agents (VDAs).
5. Chalcone hybrids
5.1. Platinum-chalcone hybrids
Multi-target drugs have attracted more and more attention. To obtain compounds with high cytotoxicity, the pharmacophore binding to a single compound molecule has become the design method of new anti-tumour drugs107–109. Platinum compounds are widely used in clinical practice but have serious toxic and side effects, such as nephrotoxicity, neurotoxicity, and myelosuppression110. To overcome these shortcomings, researchers have designed novel platinum-based complexes that combine platinum with other pharmacophore groups to reduce toxic side effects. Prompted by this idea, the researcher's combination of the active groups of microtubule inhibitors with cytotoxic DNA-damaging platinum compounds to obtain complexes can also be considered as an effective strategy to target tubulin and DNA, at least theoretically, to enhance the antitumor activity of platinum drugs and overcome their side effects.
Schobert et al.111 synthesised a class of chalcone conjugates 57 (Figure 39) by introducing platinum (II) into chalcone. In this study, the activity level of the compound 57 was the same as that of parent product 48 (Figure 39), showing good activity against all kinds of tumour cells, while 57 could effectively inhibit the reproliferation of tumour cells. In addition, compound 57 could interact with various forms of DNA, but not as tightly as cisplatin. At the same time, cancer cells treated with 48 progressed more slowly from caspase-9 to caspase-3 than those treated with 57. In 2010, Zoldakova et al.112 further investigated the mechanism of 57 and found that the compound 57 has strong anti-tumour activity against a variety of cancer cells. Cellular uptake of compound 57 was similar to oxaliplatin and was mainly dependent on the organic cationic transporter (OCT-1/2) and copper transport-associated protein (CTR1), while compound 48 was mainly dependent on endocytosis. Compound 57 has strong anti-proliferative activity against tumour cells with higher P-gp expression but has relatively low activity against corresponding cancer cell lines as the substrate of BCRP, MRP3, and MRP1 ABC-transporter. In conclusion, unlike compound 48, its platinum complex 57 has a high cell line specificity and induces apoptosis through multiple targets.
Figure 39.

Chemical structure of compounds 48 and 57.
The antiproliferative activity of Pt(IV) complexes 58–60 (Figure 40) comprising chalcone compounds was evaluated by MTT assay113. All of the synthetic complexes exhibited better and stronger activity against three types of human cancer cells, including cisplatin (CDDP) resistant cells than their parent PT (II) species. Among all of the tested compounds, substances 58a–b were found to possess superior targeting of cancer cells than CDDP. Compounds 58a–b also significantly inhibited HUVEC cells migration in vitro. Docking study showed that complexes 58a–b binding to the colchicine site of tubulin. The studies of molecular mechanism have shown that 58a–b could induce the production of reactive oxygen species (ROS) and cell cycle arrest at the G2/M phase and mediate mitochondrial apoptosis by regulating the expression of Bcl-2 family members.
Figure 40.

Platinum-chalcone hybrids of 58–63.
In 2018, novel Pt(IV) species and millepachine derivatives 61–63 (Figure 40) were tested against human cancer cell lines by Huang et al.114 The results of in vitro biological experiments showed that all the tested substances had better antitumor activity against human tumour cells (including cisplatin sensitive and resistant ones) than Pt(II), and lower cytotoxicity against two kinds of normal cells than Pt(II). Among the evaluated substances, 61a was the most active, it could arrest the cell cycle at the G2/M phases and finally induce cell apoptosis. Besides, 61a showed no obvious toxicity to the two normal cells but was sensitive to the two cisplatin-resistant cells. Importantly, 61a showed good antitumor activity in SK-OV-3 xenograft tumour model, which was superior to cisplatin and its corresponding millepachine analogs.
To overcome the shortcomings of cisplatin, Huang et al.115 successfully synthesised four tubulin-targeting platinum (IV) prodrugs and tested them for anti-proliferative activity using MTT assay. Compared to the positive drugs cisplatin and 48, compounds 64 (Figure 41) exhibited stronger antiproliferative activity in human cancer cells tested including multidrug-resistant cancer cell lines, while the cytotoxicity of human normal liver HL-7702 was also lower than that of positive drugs (cisplatin and 48). Particularly, compound 64a, with IC50 values of 0.19–0.37 μM, was the best compound against tested human cancer cell lines. Additionally, 64a could induce G2/M phase arrest and apoptosis of A549 cells, the mechanism of which is related to mitochondrial membrane potential (MMP) collapse, the expression of some apoptosis-related proteins, and the increase of intracellular reactive oxygen species (ROS). More importantly, compound 64a significantly inhibited tumour growth in the A549 xenograft tumour model, but with no significant toxicity hints (Figure 41).
Figure 41.

Simple chalcone compounds of 48 and 64.
5.2. Nitrogen mustard-chalcone hybrids
Nitrogen mustard is one of the earliest antitumor drugs used clinically. As a representative of bioalkylation agents, nitrogen mustard can stably bind to affinity groups after entering the body, affect cell metabolism and promote cell apoptosis116,117. Based on molecular hybridisation strategy118, Sabina and co-workers synthesised a novel range of p-[N, N-bis(2-chloroethyl)amino]benzaldehyde substituted chalcone conjugates 65 (Figure 42) and evaluated for their inhibitory activity against A549 and HepG2 cancer cells119. Compound 65a displayed more activity in both the cancer cell lines and tubulin than the other substances (IC50 in the range of 0.089 to 0.200 µM). Docking results showed that the synergistic effect of nitrogen mustard made compound 65a bind tightly to tubulin. Besides, SAR analysis showed that the compounds with methyl or nitro groups in the A ring were more active. Interestingly, dimethoxy substituents at the 2 and 4 sites of the A ring showed the highest activity against A549, while the lowest activity against HepG2.
Figure 42.

Nitrogen mustard-chalcone hybrids of 65.
5.3. Benzothiazole-chalcone hybrids
Benzothiazole is an important structure for drug development due to its favourable biological activity120–122. Benzothiazole compounds have biological activities such as antitumor123, anti-inflammatory124, antioxidation125, antibacterial126, and anti-diabetic127 et al. Benzothiazole-chalcones retain the basic structure of chalcones and replace the A or B rings with benzothiazoles to obtain chalcones with high antitumor activity and low toxicity.
A novel series of chalcone-amidobenzothiazole derivatives 66 and 67 (Figure 43) were synthesised and evaluated as potential antitumor drugs by Kamal et al.128. All of them showed potent inhibitory activity against a panel of five human cancer cell lines (A549, A375, MCF-7, HT-29, and ACHN), and the active compounds 66a–b inhibited the growth of various cancer cell lines with IC50 ranging from 0.85 to 3.3 µM. Mechanism studies have shown that the synthesised substances arrested the A549 cell cycle in the G2/M phase, and resulting in the death of caspase-3-dependent apoptotic cells. The tubulin polymerisation assay (IC50: 66a = 3.5 µM; 66b = 5.2 µM) and immunofluorescence analysis indicated the synthesised conjugates could significantly inhibit the microtubule assembly at both molecular and cellular levels in A549 cells. Moreover, docking studies demonstrated that they interacted with tubulin and bonded effectively.
Figure 43.

Benzothiazole-chalcone hybrids of 66 and 67.
5.4. Triazole-chalcone hybrids
Triazole compounds have five-membered heterocycles, which are easy to bind to targets in vivo and have a wide range of biological activities129,130. Triazole compounds were originally mainly used in pesticides, but there are also triazole antibiotics in clinical use, such as triazole, terconazole, itraconazole, and so on. Later studies have found that triazole drugs can be used not only for antibacterial and insecticidal purposes but also for anticonvulsant, anti-anxiety, anti-inflammatory, and anti-tumour purposes131–134. Therefore, they were introduced into chalcones to obtain novel tubulin inhibitors.
A new library of chalcone derivatives 68 (Figure 44) with triazoloquinoxaline-linked moiety was prepared and their anticancer activity also has been tested135. Most of them displayed potent activities against EGFR TK and tubulin at micromolar or submicromolar concentrations. Thereinto compounds 68b–c, and 68e–g showed superior antiproliferative activities against the majority of the cell lines, with selective or non-selective behaviour (IC50 = 1.65–34.28 µM), and compounds 68a–c, 71e, and 68g could effectively inhibit the EGFR TK with IC50 ranging from 0.093 to 0.661 µM. Additionally, docking study results revealed that derivative 68g embedded colchicine binding pocket at the interface of tubulin α and β with lowest binding energy. SAR analysis indicated that quinoxaline rings play a key role in the inhibition of EGFR, and improved the stability of the structure in the colchicine bind site. The compounds which containing -OH or -OCH3 groups showed anti-EGFR and tubulin polymerisation, and the 3rd and 4th substituents, such as OCH3, had potent effects on the activity.
Figure 44.

Triazole-chalcone hybrids of 68.
5.5. Other chalcone hybrids
The nitro group is a unique functional group. Its strong electron attraction ability produces local or local electron defects in the molecule, which enables the part with a strong electrophilic ability to bind closely to amino acids, proteins, and enzymes. The introduction of the nitro group enables compounds to have a variety of chemical and biological effects. For example, Zhang and co-workers obtained a series of compounds 69 (Figure 45) by introducing o-nitroaromatic rings into chalcone and evaluated for their biological activities as anti-tubulin agents136. All of them showed significant activities against tubulin polymerisation and the growth of MCF-7 and A549 cell lines. Thereinto, compound 69a had the strongest inhibitory activity against MCF-7 and A549 cells (IC50 = 0.03 and 0.95 µg/mL) and was the best compound in the anti-tubulin polymerisation assay with IC50 of 1.42 µg/mL. SAR analysis indicated that the antiproliferative activity of the electron substituents introduced into the A ring was stronger than that of the electron-withdrawing substituents, and the potency order was CH3 > OCH3. However, compounds of salicylaldehyde have two halogen atoms at position-3 and position-5, their anticancer activity was lower than that of introducing only one halogen atom at position-5. In the molecular docking study, one hydrogen bond and one π-cation of 69a could interact with the colchicine binding site protein residues, which might play a key role in its anti-tubulin polymerisation and anti-proliferative activities.
Figure 45.

Chemical structure of compounds 69.
The diaryl ether scaffold is frequently found in many natural products and biologically important molecules137,138, and some molecules with a diaryl ether nucleus possess cytotoxic activity by inhibiting tubulin polymerisation139–141. Based on these finding, Wang et al. synthesised several novel chalcone analogs with diaryl ether moiety 70 (Figure 46) and evaluated as inhibitors of tubulin polymerisation and human cancer cell lines142. Most of the tested compounds displayed moderate to good antiproliferative activity of three human cancer cell lines of MCF-1, HepG2, and HCT116 (IC50 of 3.44 ± 0.19–8.89 ± 0.42 μM), and compound 70a displayed the most potent activities against three human cancer cell lines with IC50 values of 3.44 ± 0.19, 4.64 ± 0.23, and 6.31 ± 0.27 μM, respectively. In addition, 70a could significantly inhibit tubulin polymerisation in vitro, and the mechanism results showed that 70a could induce G2/M phase arrest and apoptosis. Besides, 70a was found to interact and bind at the colchicine binding site of the tubulin by the molecular docking study.
Figure 46.

Chemical structure of compounds 70.
A series of novel chalcone-containing shikonin conjugates 71 (Figure 47) have been obtained and evaluated as potential inhibitors of tubulin polymerisation by Qiu et al.143 Majority of them displayed significant anti-proliferative activity, and 71a was the best compound with IC50 values of 2.98 ± 0.53 μM and MCF-7 cells with IC50 values of 2.36 ± 0.32 μM. Mechanism studies showed that 71a induced MCF-7 cell apoptosis, decreased mitochondrial membrane potential, led to cell accumulation in the G2/M phase of the cell cycle, and seriously damaged the microtubule system, which is equivalent to the colchicine. Meanwhile, three hydrogen bonds of 71a were found in the protein residues of the colchicine binding site, which may be the reason for its resistance to tubulin polymerisation.
Figure 47.

Chemical structure of compounds 71.
Shankaraiah and co-workers have been obtained a range of new heterocycles-linked chalcone conjugates 72 (Figure 48) by varying different alkane spacers, and the in vitro cytotoxic potential against a panel of selected human cancer cell lines of them were be tested144. Several of them showed a wide range of inhibitory activities on the tested human cancer cell lines (IC50 = 1.48 ± 0.19 µM-10.98 ± 0.81 µM), and 72a showed the strongest activity against the NCI-H460 (lung cancer) cells (IC50 of 1.48 ± 0.19 µM). Meanwhile, 72a was found to effectively inhibit the tubulin polymerisation and destroy microtubule formation with IC50 of 9.66 ± 0.06 µM. SARs of the tested compounds have been exhibited that piperidine and morpholine on the B ring and the introduction of EDGs on the A ring enhanced the activity. In addition, the cytotoxicity of the 2 - or 4-link conjugates was lower than that of the 3-link conjugates. Mechanism studies showed that compound 72a could block NCI-H460 cells in the G2/M phase of the cell cycle and induce cell apoptosis. Besides, docking studies have shown that these novel conjugates may dock with colchicine sites of tubulin.
Figure 48.

Chemical structure of compounds 72.
A class of novel chalcone derivatives 73 (Figure 49) were considered as potential antitumor agents by Huang et al.145. These newly synthesised chalcones showed good anticancer activity, which the IC50 values are mainly at the micromolar level. Thereinto, derivative 73a showed the most effective activity against tested tumour cell lines, including multidrug-resistant human cancer lines (IC50 of 3.75–8.42 µM). SAR analysis indicated that the introduction of EWG in the para-position of the lateral benzene ring may beneficial to the activity of the selected cancer cell lines. While the 4-position of the lateral benzene ring is modified with a halogen or hydroxyl group to render them ineffective. Additionally, the mechanism results showed that 73a induced apoptosis of NCI-H460 cells through the mitochondrial pathway, which also could effectively induce cell cycle arrest in the G2/M phase. Besides, the novel analogs could inhibit tubulin polymerisation through the tubulin polymerisation assay, and docking studies revealed that 73a could bind to the colchicine site of tubulin.
Figure 49.

Chemical structure of compounds 73.
Kumar et al.146 prepared a range of new resveratrol-chalcone conjugates and tested the anticancer activity against 60 human cancer cell lines. Thereinto, conjugate 74a (Figure 50) displayed the strongest activity and high selectivity towards certain ovarian cancer, non-small cell lung cancer, and breast cancer cell lines (GI50 = 1.28–34.1 µM). Docking studies revealed 74a could bind to the colchicine site of tubulin, and observe a potent H-bonding interaction (1.852 Å) with Cys-241 amino acid present in the binding pocket. Therefore, compound 74a might be a precursor for the development of new anticancer drugs.
Figure 50.

Chemical structure of compounds 74–76.
A series of phenstatin/isocombretastatin-chalcones 75 and 76 (Figure 50) have been prepared and evaluated the anticancer activity against various human cancer cell lines147. Several targets chalcones showed potent antiproliferative activity against 60 human cancer cell lines of the NCI (GI50 = 0.11–19.0 µM). The three compounds 75a–c exhibited a broad spectrum of anti-proliferation activities in the submicromolar range for most cell lines, and all of them showed moderate to excellent cytotoxicity against breast cancer cells such as MCF-7 and MDA-MB-231 (IC50 of 0.5 to 19.9 µM). By the tubulin polymerisation assay and immunofluorescence analysis, compound 75b–c effectively inhibited tubulin assembly with IC50 values of 0.8 μM and 0.6 μM, respectively. The competitive binding assay indicated that these chalcones bind at the colchicine-binding site of tubulin, and which is blocked in the G2/M phase and leads to apoptosis.
A number of novel phenstatin based indole linked chalcone analogs 77 (Figure 51) were prepared and evaluated for their antiproliferative activity on few cancer cell lines by Kode et al.148 In this series, analog 77a showed potent anti-proliferative activity against oral cancer squamous cancer cells as well as oral cancer stem-like spheroids, and the tumour volume of experimental mice was reduced significantly, and there was no toxic effect. Interestingly, 77a also reduced angiogenesis in mice xenografts, reduced collagen levels, and led to a significant reduction in cell process, cell integrity, and cytoskeletal organisation. In addition, 77a successfully eliminated glucose uptake in tumour xenografts. Further studies showed that 77a could directly interact with tubulin and inhibit tubulin polymerisation, resulting in the formation of tubulin ring intermediates, profilament bending, and destabilisation. Besides, molecular docking results indicated that 77a showed better binding affinity to target binding sites, thus inhibiting microtubule polymerisation and glucose metabolism.
Figure 51.

Chemical structure of compounds 77.
Ahmed and co-workers have been synthesised a new range of α-phthalimido-substituted chalcones-based hybrids 78 (Figure 52) as dual histone deacetylase (HDAC)/tubulin inhibitors, and the in vitro anticancer activity was tested by MTT assay using CA-4 as reference compound149. All of them displayed significant antitumor activity with IC50 at micromolar concentrations against MCF-7 and HepG2 cell lines. In particular, the antitumor activity of 78a was 2.57 and 4.51 times that of CA-4, respectively, and could effectively inhibit the activity of β-tubulin polymerase and HDAC 1 and 2. Moreover, the mechanism studies showed that 78a could successfully arrest the cell cycle in G2/M phase and induce the apoptosis of preG1 cells. Besides, molecular docking studies indicated that these compounds could efficiently bind to the colchicine binding site of both tubulin polymerase enzyme and HDAC2 active sites with energy scores higher than the reference ligands.
Figure 52.

Chemical structure of compounds 78.
6. Conclusion
Chalcone is an important natural product with a variety of biological activities. Some compounds with a chalcone structure exhibit potent antitumor effects, which have attracted a lot of attention. Further studies revealed that the mechanism of antitumor action of chalcone compounds is that they can reversibly or irreversibly bind tubulin, thereby inhibiting or promoting tubulin polymerisation, disturbing the dynamic balance of microtubules, inhibiting cell cycle progression, inducing apoptosis, or inhibiting the formation of capillaries around tumour cells, and thus inhibiting tumour cell proliferation. The structure of chalcone consists of two aryl groups and an α, β-unsaturated alkenone. Therefore, modification of the two aryl rings and α, β-unsaturated alkenone structure is a major research direction in recent years. In previous studies on CA-4, hydroxyl and methoxy substitutions were critical for the compounds to exhibit a favourable activity, and a variety of methoxy substitution patterns were effective in increasing the antitumor activity of chalcone compounds and their derivatives, but excessive methoxy substitutions may conversely reduce the antitumor activity of the compounds. From some natural products, researchers began to replace the two aromatic rings of chalcone with heterocycles. Many attempts have been made to design heterocyclic substitution of chalcone compounds. It has been found that the substitution of the A or B ring of chalcone with the heterocycle of naphthalene ring, benzopyran, benzofuran, indole, benzothiazole, benzoxazole, quinoline, etc. exhibits excellent cytotoxicity and inhibitory effects against tubulin polymerisation. Moreover, the introduction of methoxy into the heterocyclic ring could improve the antitumor activity of heterocyclic-chalcone derivatives. Chalcone has an α, β-unsaturated ketone structure with a single bond that is rotatable, but the α-position priming group in the carbonyl group makes it easier for the compound to maintain the trans configuration and better bind to the colchicine binding site of tubulin. Hence, the introduction of thiophene, thiazole, imidazole, and benzothiophene into the alkenone system likewise forces the double bonds in alkenones to maintain a trans conformation, increasing the affinity of the compounds to tubulin. Overall, the inhibitory effects of chalcone derivatives against tubulin polymerisation and tumour cells could be enhanced by structurally modifying.
Disclosure statement
The authors confirm that this article content has no conflict of interest.
References
- 1.Kaur R, Kaur G, Gill RK, et al. Recent developments in tubulin polymerization inhibitors: an overview. Eur J Med Chem 2014;87:89–124. [DOI] [PubMed] [Google Scholar]
- 2.Peerzada MN, Hamel E, Bai R, et al. Deciphering the key heterocyclic scaffolds in targeting microtubules, kinases and carbonic anhydrases for cancer drug development. Pharmacol Ther 2021;225:107860. [DOI] [PubMed] [Google Scholar]
- 3.Jordan A, Hadfield J, Lawrence J, et al. Tubulin as a target for anticancer drugs: agents which interact with the mitotic spindle. MRR 1998;18:259–96. [DOI] [PubMed] [Google Scholar]
- 4.Sorger PK, Dobles M, Tournebize R, Hyman AA.. Coupling cell division and cell death to microtubule dynamics. Curr Opin Cell Biol 1997;9:807–14. [DOI] [PubMed] [Google Scholar]
- 5.Jordan MA, Wilson L.. Microtubules as a target for anticancer drugs. Nat Rev Cancer 2004;4:253–65. [DOI] [PubMed] [Google Scholar]
- 6.Menezes C. Arylidene indanone scaffold: medicinal chemistry and structure–activity relationship view. RSC Adv 2017;7:9357–72. [Google Scholar]
- 7.Risinger AL, Giles FJ, Mooberry SL.. Microtubule dynamics as a target in oncology. Cancer Treat Rev 2009;35:255–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li L, Jiang S, Li X, et al. Recent advances in trimethoxyphenyl (tmp) based tubulin inhibitors targeting the colchicine binding site. Euro J Med Chem 2018;151:482–94. [DOI] [PubMed] [Google Scholar]
- 9.Pellegrini F, Budman DR.. Review: tubulin function, action of antitubulin drugs, and new drug development. Cancer Invest 2005;23:264–73. [DOI] [PubMed] [Google Scholar]
- 10.Haider K, Rahaman S, Yar MS, Kamal A.. Tubulin inhibitors as novel anticancer agents: an overview on patents (2013-2018). Expert Opin Ther Pat 2019;29:623–41. [DOI] [PubMed] [Google Scholar]
- 11.Cheng ZQ, Lu X, Feng BM.. A review of research progress of antitumor drugs based on tubulin targets. Transl Cancer Res 2020;9:4020–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jandial DD, Blair CA, Zhang S, et al. Molecular targeted approaches to cancer therapy and prevention using chalcones. Curr Cancer Drug Targets 2014;14:181–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu M, Wu PY, Shen F, et al. Chalcone derivatives and their antibacterial activities: current development. Bioorg Chem 2019;91:103133. [DOI] [PubMed] [Google Scholar]
- 14.Qin HL, Zhang ZW, Lekkala R, et al. Chalcone hybrids as privileged scaffolds in antimalarial drug discovery: a key review. Eur J Med Chem 2020;193:112215. [DOI] [PubMed] [Google Scholar]
- 15.Thapa P, Upadhyay SP, Suo WLZ, et al. Chalcone and its analogs: therapeutic and diagnostic applications in alzheimer's disease. Bioorg Chem 2021;108:104681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Edwards ML, Stemerick DM, Sunkara PS.. Chalcones: a new class of antimitotic agents. J Med Chem 1990;33:1948–54. [DOI] [PubMed] [Google Scholar]
- 17.Xiao JQ, Gao MX, Diao Q, et al. Chalcone derivatives and their activities against drug-resistant cancers: an overview. Curr Top Med Chem 2021;21:348–62. [DOI] [PubMed] [Google Scholar]
- 18.Lawrence NJ, McGown AT, Ducki S, et al. The interaction of chalcones with tubulin. Anti Cancer Drug Design 2000;15:135–41. [PubMed] [Google Scholar]
- 19.Tu HY, Huang AM, Hour TC, et al. Synthesis and biological evaluation of 2',5'-dimethoxychalcone derivatives as microtubule-targeted anticancer agents. Bioorg Med Chem 2010;18:2089–98. [DOI] [PubMed] [Google Scholar]
- 20.Kong Y, Wang K, Edler MC, et al. A boronic acid chalcone analog of combretastatin a-4 as a potent anti-proliferation agent. Bioorg Med Chem 2010;18:971–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Salum LB, Altei WF, Chiaradia LD, et al. Cytotoxic 3,4,5-trimethoxychalcones as mitotic arresters and cell migration inhibitors. Euro J Med Chem 2013;63:501–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tajuddin Y, Harun Z, Ghazali AR, et al. Synthesis of chalcone derivatives and their effects on proliferation and tubulin dynamics instability of ht-29 cells. Letters in Drug Design 2016;13:662–7. [Google Scholar]
- 23.Aryapour H, Riazi GH, Foroumadi A, et al. Biological evaluation of synthetic analogues of curcumin: chloro-substituted-2′-hydroxychalcones as potential inhibitors of tubulin polymerization and cell proliferation. Med Chem Res 2011;20:503–10. [Google Scholar]
- 24.Burmaoglu S, Aktas Anil D, Gobek A, et al. Design, synthesis and antiproliferative activity evaluation of fluorine-containing chalcone derivatives. J Biomol Struct Dyn 2020. DOI: 10.1080/07391102.2020.1848627 [DOI] [PubMed] [Google Scholar]
- 25.Abosalim HM, Nael MA, El‐Moselhy TF.. Design, synthesis and molecular docking of chalcone derivatives as potential anticancer agents. Chem Select 2021;6:888–95. [Google Scholar]
- 26.Ye H, Fu A, Wu W, et al. Cytotoxic and apoptotic effects of constituents from millettia pachycarpa benth. Fitoterapia 2012;83:1402–8. [DOI] [PubMed] [Google Scholar]
- 27.Wu W, Ye H, Wan L, et al. Millepachine, a novel chalcone, induces g2/m arrest by inhibiting cdk1 activity and causing apoptosis via ros-mitochondrial apoptotic pathway in human hepatocarcinoma cells in vitro and in vivo. Carcinogenesis 2013;34:1636–43. [DOI] [PubMed] [Google Scholar]
- 28.Wang G, Wu W, Peng F, et al. Design, synthesis, and structure-activity relationship studies of novel millepachine derivatives as potent antiproliferative agents. Eur J Med Chem 2012;54:793–803. [DOI] [PubMed] [Google Scholar]
- 29.Wang G, Peng F, Cao D, et al. Design, synthesis and biological evaluation of millepachine derivatives as a new class of tubulin polymerization inhibitors. Bioorg Med Chem 2013;21:6844–54. [DOI] [PubMed] [Google Scholar]
- 30.Wang G, Li C, He L, et al. Design, synthesis and biological evaluation of a series of pyrano chalcone derivatives containing indole moiety as novel anti-tubulin agents. Bioorg Med Chem 2014;22:2060–79. [DOI] [PubMed] [Google Scholar]
- 31.Yang Z, Wu W, Wang J, et al. Synthesis and biological evaluation of novel millepachine derivatives as a new class of tubulin polymerization inhibitors. J Med Chem 2014;57:7977–89. [DOI] [PubMed] [Google Scholar]
- 32.Yang J, Yan W, Yu Y, et al. The compound millepachine and its derivatives inhibit tubulin polymerization by irreversibly binding to the colchicine-binding site in β-tubulin. J Biol Chem 2018;293:9461–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cao D, Han X, Wang G, et al. Synthesis and biological evaluation of novel pyranochalcone derivatives as a new class of microtubule stabilizing agents. Euro J Med Chem 2013;62:579–89. [DOI] [PubMed] [Google Scholar]
- 34.Milelli A, Tumiatti V, Micco M, et al. Structure-activity relationships of novel substituted naphthalene diimides as anticancer agents. Euro J Med Chem 2012;57:417–28. [DOI] [PubMed] [Google Scholar]
- 35.Sharma S, Singh T, Mittal R, et al. A study of anti-inflammatory activity of some novel alpha-amino naphthalene and beta-amino naphthalene derivatives. Archiv Der Pharmazie 2006;339:145–52. [DOI] [PubMed] [Google Scholar]
- 36.Ateş-Alagöz Z, Kuş C, Coban T.. Synthesis and antioxidant properties of novel benzimidazoles containing substituted indole or 1,1,4,4-tetramethyl-1,2,3,4-tetrahydro-naphthalene fragments. Journal of Enzyme Inhibition and Medicinal Chemistry 2005;20:325–31. [DOI] [PubMed] [Google Scholar]
- 37.Altintop MD, Atli O, Ilgin S.. Synthesis and biological evaluation of new naphthalene substituted thiosemicarbazone derivatives as potent antifungal and anticancer agents. Euro J Med Chem 2016;108:406–14. [DOI] [PubMed] [Google Scholar]
- 38.Voskiene A, Sapijanskaite B, Mickevicius V, et al. Synthesis and microbiological evaluation of new 2- and 2,3-diphenoxysubstituted naphthalene-1,4-diones with 5-oxopyrrolidine moieties. Molecules 2012;17:14434–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee JM, Lee MS, Koh D, et al. A new synthetic 2'-hydroxy-2,4,6-trimethoxy-5',6'-naphthochalcone induces g2/m cell cycle arrest and apoptosis by disrupting the microtubular network of human colon cancer cells. Cancer Lett 2014;354:348–54. [DOI] [PubMed] [Google Scholar]
- 40.Shin SY, Kim JH, Yoon H, et al. Novel antimitotic activity of 2-hydroxy-4-methoxy-2',3'-benzochalcone (hymnpro) through the inhibition of tubulin polymerization. J Agric Food Chem 2013;61:12588–97. [DOI] [PubMed] [Google Scholar]
- 41.Lee K, Lee DH, Kim J-H, et al. The chalcone derivative hymnpro generates reactive oxygen species through depletion of intracellular glutathione. Appl Biol Chem 2016;59:391–6. [Google Scholar]
- 42.Wang G, Liu W, Gong Z, et al. Synthesis, biological evaluation, and molecular modelling of new naphthalene-chalcone derivatives as potential anticancer agents on mcf-7 breast cancer cells by targeting tubulin colchicine binding site. J Enzyme Inhibition Med Chem 2020;35:139–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang G, Qiu J, Xiao X, et al. Synthesis, biological evaluation and molecular docking studies of a new series of chalcones containing naphthalene moiety as anticancer agents. Bioorg Chem 2018;76:249–57. [DOI] [PubMed] [Google Scholar]
- 44.dos Santos EA, Hamel E, Bai R, et al. Synthesis and evaluation of diaryl sulfides and diaryl selenide compounds for antitubulin and cytotoxic activity. Bioorg Med Chem Lett 2013;23:4669–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu Q, Sun M, Bai Z, et al. Design, synthesis and bioevaluation of antitubulin agents carrying diaryl-5,5-fused-heterocycle scaffold. Eur J Med Chem 2017;139:242–9. [DOI] [PubMed] [Google Scholar]
- 46.Wu-Wong JR, Alder JD, Alder L, et al. Identification and characterization of a-105972, an antineoplastic agent. Cancer Res 2001;61:1486–92. [PubMed] [Google Scholar]
- 47.Cao D, Liu Y, Yan W, et al. Design, synthesis, and evaluation of in vitro and in vivo anticancer activity of 4-substituted coumarins: a novel class of potent tubulin polymerization inhibitors. J Med Chem 2016;59:5721–39. [DOI] [PubMed] [Google Scholar]
- 48.Wang G, Peng Z, Zhang J, et al. Synthesis, biological evaluation and molecular docking studies of aminochalcone derivatives as potential anticancer agents by targeting tubulin colchicine binding site. Bioorg Chem 2018;78:332–40. [DOI] [PubMed] [Google Scholar]
- 49.Kossakowski J, Ostrowska K, Hejchman E, et al. Synthesis and structural characterization of derivatives of 2- and 3-benzo[b]furan carboxylic acids with potential cytotoxic activity. Farmaco 2005;60:519–27. [DOI] [PubMed] [Google Scholar]
- 50.Hwang JW, Choi DH, Jeon JH, et al. Facile preparation of 2-arylbenzo[b]furan molecules and their anti-inflammatory effects. Bull Korean Chem Soc 2010;31:965–70. [Google Scholar]
- 51.Wahab Khan M, Jahangir Alam M, Rashid MA, et al. A new structural alternative in benzo[b]furans for antimicrobial activity. Bioorg Med Chem 2005;13:4796–805. [DOI] [PubMed] [Google Scholar]
- 52.Manna K, Agrawal YK.. Design, synthesis, and antitubercular evaluation of novel series of 3-benzofuran-5-aryl-1-pyrazolyl-pyridylmethanone and 3-benzofuran-5-aryl-1-pyrazolylcarbonyl-4-oxo-naphthyridin analogs. Euro J Med Chem 2010;45:3831–9. [DOI] [PubMed] [Google Scholar]
- 53.Peschke B, Bak S, Hohlweg R, et al. Benzo[b]thiophene-2-carboxamides and benzo[b]furan-2-carboxamides are potent antagonists of the human h3-receptor. Bioorg Med Chem Lett 2006;16:3162–5. [DOI] [PubMed] [Google Scholar]
- 54.Wang XD, Deng RC, Dong JJ, et al. 3-aryl-4-acyloxyethoxyfuran-2(5h)-ones as inhibitors of tyrosyl-trna synthetase: synthesis, molecular docking and antibacterial evaluation. Bioorg Med Chem 2013;21:4914–22. [DOI] [PubMed] [Google Scholar]
- 55.Wang XD, Wei W, Wang PF, et al. Novel 3-arylfuran-2(5h)-one-fluoroquinolone hybrid: design, synthesis and evaluation as antibacterial agent. Bioorg Med Chem 2014;22:3620–8. [DOI] [PubMed] [Google Scholar]
- 56.Wei W, Shi WK, Wang PF, et al. Adenosine analogs as inhibitors of tyrosyl-trna synthetase: design, synthesis and antibacterial evaluation. Bioorg Med Chem 2015;23:6602–11. [DOI] [PubMed] [Google Scholar]
- 57.Mphahlele MJ, Maluleka MM, Parbhoo N, et al. Synthesis, evaluation for cytotoxicity and molecular docking studies of benzo[c]furan-chalcones for potential to inhibit tubulin polymerization and/or egfr-tyrosine kinase phosphorylation. Inter J Mol Sci 2018;19:2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sravanthi TV, Manju SL.. Indoles – a promising scaffold for drug development. Eur J Pharm Sci 2016;91:1–10. [DOI] [PubMed] [Google Scholar]
- 59.Singh TP, Singh OM.. Recent progress in biological activities of indole and indole alkaloids. Mini Rev Med Chem 2018;18:9–25. [DOI] [PubMed] [Google Scholar]
- 60.Gil-Turnes MS, Hay ME, Fenical W.. Symbiotic marine bacteria chemically defend crustacean embryos from a pathogenic fungus. Science 1989;246:116–8. [DOI] [PubMed] [Google Scholar]
- 61.Sivakumar PM, Kumar TM, Doble M.. Antifungal activity, mechanism and qsar studies on chalcones. Chem Biol Drug Des 2009;74:68–79. [DOI] [PubMed] [Google Scholar]
- 62.Hashemi SM, Badali H, Faramarzi MA, et al. Novel triazole alcohol antifungals derived from fluconazole: design, synthesis, and biological activity. Mol Divers 2015;19:15–27. [DOI] [PubMed] [Google Scholar]
- 63.Aryapour H, Riazi GH, Ahmadian S, et al. Induction of apoptosis through tubulin inhibition in human cancer cells by new chromene-based chalcones. Pharm Biol 2012;50:1551–60. [DOI] [PubMed] [Google Scholar]
- 64.Mirzaei H, Abastabar M, Emami S.. Indole-derived chalcones as anti-dermatophyte agents: in vitro evaluation and in silico study. Comput Biol Chem 2020;84:107189. [DOI] [PubMed] [Google Scholar]
- 65.Boumendjel A, McLeer-Florin A, Champelovier P, et al. A novel chalcone derivative which acts as a microtubule depolymerising agent and an inhibitor of p-gp and bcrp in in-vitro and in-vivo glioblastoma models. BMC Cancer 2009;9:242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Martel-Frachet V, Keramidas M, Nurisso A, et al. Ipp51, a chalcone acting as a microtubule inhibitor with in vivo antitumor activity against bladder carcinoma. Oncotarget 2015;6:14669–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mirzaei H, Shokrzadeh M, Modanloo M, et al. New indole-based chalconoids as tubulin-targeting antiproliferative agents. Bioorg Chem 2017;75:86–98. [DOI] [PubMed] [Google Scholar]
- 68.Preti D, Romagnoli R, Rondanin R, et al. Design, synthesis, in vitro antiproliferative activity and apoptosis-inducing studies of 1-(3',4',5'-trimethoxyphenyl)-3-(2'-alkoxycarbonylindolyl)-2-propen-1-one derivatives obtained by a molecular hybridisation approach. J Enzyme Inhib Med Chem 2018;33:1225–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lokwani P, Nagori B, Batra N, et al. Benzoxazole: the molecule of diverse biological activities. J Chem Pharma Res 2011;3:302–11. [Google Scholar]
- 70.Paramashivappa R, Phani Kumar P, Subba Rao PV, et al. Design, synthesis and biological evaluation of benzimidazole/benzothiazole and benzoxazole derivatives as cyclooxygenase inhibitors. Bioorg Med Chem Letters 2003;13:657–60. [DOI] [PubMed] [Google Scholar]
- 71.Hall IH, Peaty NJ, Henry JR, et al. Investigations on the mechanism of action of the novel antitumor agents 2-benzothiazolyl, 2-benzoxazolyl, and 2-benzimidazolyl hydrazones derived from 2-acetylpyridine. Arch Pharm 1999;332:115–23. [DOI] [PubMed] [Google Scholar]
- 72.Saari WS, Wai JS, Fisher TE, et al. Synthesis and evaluation of 2-pyridinone derivatives as hiv-1-specific reverse transcriptase inhibitors. 2. Analogues of 3-aminopyridin-2(1h)-one. J Med Chem 1992;35:3792–802. [DOI] [PubMed] [Google Scholar]
- 73.Temiz-Arpaci O, Aki-Sener E, Yalcin I, et al. Cheminform abstract: synthesis and antimicrobial activity of some 2-[p-substituted-phenyl]benzoxazol-5-yl-arylcarboxyamides. Cheminform 2010;33:49. [DOI] [PubMed] [Google Scholar]
- 74.Modiya PR, Patel CN.. Synthesis and screening of antibacterial and antifungal activity of 5-chloro-1,3-benzoxazol-2(3 h)-one derivatives. Org Med Chem Lett 2012;2:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nargund LV, Reddy GR, Hariprasad V.. Anti-inflammatory activity of substituted 1,3,4-oxadiazoles. J Pharm Sci 1994;83:246–8. [DOI] [PubMed] [Google Scholar]
- 76.Konieczny MT, Buɬakowska A, Pirska D, et al. Structural factors affecting affinity of cytotoxic oxathiole-fused chalcones toward tubulin. Euro J Med Chem 2015;89:733–42. [DOI] [PubMed] [Google Scholar]
- 77.Konieczny MT, Buɬakowska A, Pirska D, et al. Influence of s-oxidation on cytotoxic activity of oxathiole-fused chalcones. Chem Biol Drug Design 2016;88:519–33. [DOI] [PubMed] [Google Scholar]
- 78.Kumar S, Bawa S, Gupta H.. Biological activities of quinoline derivatives. Mini Rev Med Chem 2009;9:1648–54. [DOI] [PubMed] [Google Scholar]
- 79.Xiao ZP, Wang XD, Wang PF, et al. Design, synthesis, and evaluation of novel fluoroquinolone-flavonoid hybrids as potent antibiotics against drug-resistant microorganisms. Eur J Med Chem 2014;80:92–100. [DOI] [PubMed] [Google Scholar]
- 80.Solomon VR, Lee H.. Quinoline as a privileged scaffold in cancer drug discovery. Curr Med Chem 2011;18:1488–508. [DOI] [PubMed] [Google Scholar]
- 81.Chokkar N, Kalra S, Chauhan M, et al. A review on quinoline derived scaffolds as anti-hiv agents. Mini Rev Med Chem 2019;19:510–26. [DOI] [PubMed] [Google Scholar]
- 82.Mirzaei S, Hadizadeh F, Eisvand F, et al. Synthesis, structure-activity relationship and molecular docking studies of novel quinoline-chalcone hybrids as potential anticancer agents and tubulin inhibitors. J Mol Struc 2020;1202:127310. [Google Scholar]
- 83.Lindamulage IK, Vu HY, Karthikeyan C, et al. Novel quinolone chalcones targeting colchicine-binding pocket kill multidrug-resistant cancer cells by inhibiting tubulin activity and mrp1 function. Sci Rep 2017;7:10298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tseng CH, Tzeng CC, Hsu CY, et al. Discovery of 3-phenylquinolinylchalcone derivatives as potent and selective anticancer agents against breast cancers. Euro J Med Chem 2015;97:306–19. [DOI] [PubMed] [Google Scholar]
- 85.Rahimzadeh Oskuei S, Mirzaei S, Reza Jafari-Nik M, et al. Design, synthesis and biological evaluation of novel imidazole-chalcone derivatives as potential anticancer agents and tubulin polymerization inhibitors. Bioorg Chem 2021;112:104904. [DOI] [PubMed] [Google Scholar]
- 86.Sultana F, Reddy Bonam S, Reddy VG, et al. Synthesis of benzo[d]imidazo[2,1-b]thiazole-chalcone conjugates as microtubule targeting and apoptosis inducing agents. Bioorg Chem 2018;76:1–12. [DOI] [PubMed] [Google Scholar]
- 87.Rathnakar B, Kumar GS, Mahammad SP, et al. Design, synthesis, and evaluation of novel combretastatin a‐4 based chalcone derivatives as anticancer agents. J Heterocyclic Chem 2021;58:488–501. [Google Scholar]
- 88.Romagnoli R, Baraldi PG, Carrion MD, et al. Design, synthesis, and biological evaluation of thiophene analogues of chalcones. Bioorg Med Chem 2008;16:5367–76. [DOI] [PubMed] [Google Scholar]
- 89.Wang Q, Arnst KE, Wang Y, et al. Structural modification of the 3,4,5-trimethoxyphenyl moiety in the tubulin inhibitor veru-111 leads to improved antiproliferative activities. J Med Chem 2018;61:7877–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Han H, Zhao Y, Cuthbertson T, et al. Cell cycle arrest and apoptosis induction by an anticancer chalcone epoxide. Arch Pharm 2010;343:429–39. [DOI] [PubMed] [Google Scholar]
- 91.Berlinck RGS, Trindade-Silva AE, Santos MFC.. The chemistry and biology of organic guanidine derivatives. Nat Prod Rep 2012;29:1382–406. [DOI] [PubMed] [Google Scholar]
- 92.Kim SH, Semenya D, Castagnolo D.. Antimicrobial drugs bearing guanidine moieties: a review. Eur J Med Chem 2021;216:113293. [DOI] [PubMed] [Google Scholar]
- 93.Kim JS, Lee J, Chung HW, et al. Methylglyoxal-bis(guanylhydrazone), a polyamine analogue, sensitized γ-radiation-induced cell death in HL-60 leukemia cells Sensitizing effect of MGBG on γ-radiation-induced cell death. Environ Toxicol Pharmacol 2006;22:160–6. [DOI] [PubMed] [Google Scholar]
- 94.Qian Y, Zhang HJ, Lv PC, et al. Synthesis, molecular modeling and biological evaluation of guanidine derivatives as novel antitubulin agents. Bioorg Med Chem 2010;18:8218–25. [DOI] [PubMed] [Google Scholar]
- 95.Wang Y-T, Qin Y-J, Zhang Y-L, et al. Synthesis, biological evaluation, and molecular docking studies of novel chalcone oxime derivatives as potential tubulin polymerization inhibitors. RSC Adv 2014;4:32263–75. [Google Scholar]
- 96.Peyrot V, Leynadier D, Sarrazin M, et al. Interaction of tubulin and cellular microtubules with the new antitumor drug mdl 27048. J Biol Chem 1989;264:21296–301. [PubMed] [Google Scholar]
- 97.Peyrot V, Leynadier D, Sarrazin M, et al. Mechanism of binding of the new antimitotic drug mdl 27048 to the colchicine site of tubulin: equilibrium studies. Biochemistry 1992;31:11125–32. [DOI] [PubMed] [Google Scholar]
- 98.Lawrence NJ, Patterson RP, Ooi LL, et al. Effects of alpha-substitutions on structure and biological activity of anticancer chalcones. Bioorg Med Chem Lett 2006;16:5844–8. [DOI] [PubMed] [Google Scholar]
- 99.Kerr DJ, Hamel E, Jung MK, et al. The concise synthesis of chalcone, indanone and indenone analogues of combretastatin a4. Bioorg Med Chem 2007;15:3290–8. [DOI] [PubMed] [Google Scholar]
- 100.Ducki S, Rennison D, Woo M, et al. Combretastatin-like chalcones as inhibitors of microtubule polymerization. Part 1: synthesis and biological evaluation of antivascular activity. Bioorg Med Chem 2009;17:7698–710. [DOI] [PubMed] [Google Scholar]
- 101.Ducki S, Mackenzie G, Greedy B, et al. Combretastatin-like chalcones as inhibitors of microtubule polymerisation. Part 2: structure-based discovery of alpha-aryl chalcones. Bioorg Med Chem 2009;17:7711–22. [DOI] [PubMed] [Google Scholar]
- 102.Yan J, Chen J, Zhang S, et al. Synthesis, evaluation, and mechanism study of novel indole-chalcone derivatives exerting effective antitumor activity through microtubule destabilization in vitro and in vivo. J Med Chem 2016;59:5264–83. [DOI] [PubMed] [Google Scholar]
- 103.Li W, Xu F, Shuai W, et al. Discovery of novel quinoline-chalcone derivatives as potent antitumor agents with microtubule polymerization inhibitory activity. J Med Chem 2019;62:993–1013. [DOI] [PubMed] [Google Scholar]
- 104.Xu F, Li W, Shuai W, et al. Design, synthesis and biological evaluation of pyridine-chalcone derivatives as novel microtubule-destabilizing agents. Euro J Med Chem 2019;173:1–14. [DOI] [PubMed] [Google Scholar]
- 105.Cong H, Zhao X, Castle BT, et al. An indole-chalcone inhibits multidrug-resistant cancer cell growth by targeting microtubules. Mol Pharm 2018;15:3892–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Canela M-D, Noppen S, Bueno O, et al. Antivascular and antitumor properties of the tubulin-binding chalcone tub091. Oncotarget 2017;8:14325–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Berube G. An overview of molecular hybrids in drug discovery. Exp Opin Drug Disc 2016;11:281–305. [DOI] [PubMed] [Google Scholar]
- 108.Shaveta Mishra S, Singh P.. Hybrid molecules: the privileged scaffolds for various pharmaceuticals. Eur J Med Chem 2016;124:500–36. [DOI] [PubMed] [Google Scholar]
- 109.Ivasiv V, Albertini C, Goncalves AE, et al. Molecular hybridization as a tool for designing multitarget drug candidates for complex diseases. Curr Top Med Chem 2019;19:1694–711. [DOI] [PubMed] [Google Scholar]
- 110.Gibson D. Platinum(iv) anticancer agents; are we en route to the holy grail or to a dead end? J Inorg Biochem 2021;217:111353. [DOI] [PubMed] [Google Scholar]
- 111.Schobert R, Biersack B, Dietrich A, et al. Pt(ii) complexes of a combretastatin a-4 analogous chalcone: effects of conjugation on cytotoxicity, tumor specificity, and long-term tumor growth suppression. J Med Chem 2009;52:241–6. [DOI] [PubMed] [Google Scholar]
- 112.Zoldakova M, Kornyei Z, Brown A, et al. Effects of a combretastatin a4 analogous chalcone and its pt-complex on cancer cells: a comparative study of uptake, cell cycle and damage to cellular compartments. Biochem Pharmacol 2010;80:1487–96. [DOI] [PubMed] [Google Scholar]
- 113.Huang X, Huang R, Wang Z, et al. Pt(iv) complexes conjugating with chalcone analogue as inhibitors of microtubule polymerization exhibited selective inhibition in human cancer cells. Euro J Med Chem 2018;146:435–50. [DOI] [PubMed] [Google Scholar]
- 114.Huang X, Hua S, Huang R, et al. Dual-targeting antitumor hybrids derived from pt(iv) species and millepachine analogues. Euro J Med Chem 2018;148:1–25. [DOI] [PubMed] [Google Scholar]
- 115.Huang X, Liu Z, Wang M, et al. Platinum(iv) complexes conjugated with chalcone analogs as dual targeting anticancer agents: in vitro and in vivo studies. Bioorg Chem 2020;105:104430. [DOI] [PubMed] [Google Scholar]
- 116.Diethelm-Varela B, Ai Y, Liang DD, et al. Nitrogen mustards as anticancer chemotherapies: HISTORIC perspective, current developments and future trends. Curr Top Med Chem 2019;19:691–712. [DOI] [PubMed] [Google Scholar]
- 117.More GS, Thomas AB, Chitlange SS, et al. Nitrogen mustards as alkylating agents: a review on chemistry, mechanism of action and current usfda status of drugs. Anticancer Agents Med Chem 2019;19:1080–102. [DOI] [PubMed] [Google Scholar]
- 118.Chen YM, Jia YP, Song WG, et al. Therapeutic potential of nitrogen mustard based hybrid molecules. Front Pharmacol 2018;9:1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sabina XJ, Karthikeyan J, Velmurugan G, et al. Design and in vitro biological evaluation of substituted chalcones synthesized from nitrogen mustards as potent microtubule targeted anticancer agents. New Journal of Chemistry 2017;41:4096–109. [Google Scholar]
- 120.Sharma PC, Sinhmar A, Sharma A, et al. Medicinal significance of benzothiazole scaffold: an insight view. J Enzyme Inhib Med Chem 2013;28:240–66. [DOI] [PubMed] [Google Scholar]
- 121.Keri RS, Patil MR, Patil SA, et al. A comprehensive review in current developments of benzothiazole-based molecules in medicinal chemistry. Eur J Med Chem 2015;89:207–51. [DOI] [PubMed] [Google Scholar]
- 122.Rouf A, Tanyeli C.. Bioactive thiazole and benzothiazole derivatives. Eur J Med Chem 2015;97:911–27. [DOI] [PubMed] [Google Scholar]
- 123.Mohamed LW, Taher AT, Rady GS, et al. Synthesis and cytotoxic activity of certain benzothiazole derivatives against human mcf-7 cancer cell line. Chem Biol Drug Design 2017;89:566–76. [DOI] [PubMed] [Google Scholar]
- 124.Kharbanda C, Alam MS, Hamid H, et al. Synthesis and evaluation of pyrazolines bearing benzothiazole as anti-inflammatory agents. Bioorg Med Chem 2014;22:5804–12. [DOI] [PubMed] [Google Scholar]
- 125.Uremis N, Uremis MM, Tolun FI, et al. Synthesis of 2-substituted benzothiazole derivatives and their in vitro anticancer effects and antioxidant activities against pancreatic cancer cells. Anticancer Res 2017;37:6381–9. [DOI] [PubMed] [Google Scholar]
- 126.Soni B, Ranawat MS, Sharma R, et al. Synthesis and evaluation of some new benzothiazole derivatives as potential antimicrobial agents. Euro J Med Chem 2010;45:2938–42. [DOI] [PubMed] [Google Scholar]
- 127.De SK, Chen LH, Stebbins JL, et al. Discovery of 2-(5-nitrothiazol-2-ylthio)benzo[d]thiazoles as novel c-jun n-terminal kinase inhibitors. Bioorg Med Chem 2009;17:2712–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kamal A, Mallareddy A, Suresh P, et al. Synthesis of chalcone-amidobenzothiazole conjugates as antimitotic and apoptotic inducing agents. Bioorg Med Chem 2012;20:3480–92. [DOI] [PubMed] [Google Scholar]
- 129.Maddila S, Pagadala R, Jonnalagadda SB.. 1,2,4-triazoles: a review of synthetic approaches and the biological activity. Lett Org Chem 2013;10:693–714. [Google Scholar]
- 130.Sathyanarayana R, Poojary B.. Exploring recent developments on 1,2,4-triazole: synthesis and biological applications. J Chin Chem Soc 2020;67:459–77. [Google Scholar]
- 131.Shibasaki M, Fujimori A, Kusayama T, et al. Antihypertensive activity of a nonpeptide angiotensin ii receptor antagonist, ym358, in rats and dogs. Eur J Pharmacol 1997;335:175–84. [DOI] [PubMed] [Google Scholar]
- 132.Mizuno M, Inagaki A, Yamashita M, et al. Process development of a disease-modifying antirheumatic drug, tak-603, based on optimization of friedel–crafts reaction and selective substitution of a triazole ring. Tetrahedron 2006;62:4065–70. [Google Scholar]
- 133.Aiken SP, Brown WM.. Treatment of epilepsy: existing therapies and future developments. Front Biosci 2000;5:E124–52. [DOI] [PubMed] [Google Scholar]
- 134.Grana G. Adjuvant aromatase inhibitor therapy for early breast cancer: a review of the most recent data. J Surg Oncol 2006;93:585–92. [DOI] [PubMed] [Google Scholar]
- 135.Alswah M, Bayoumi AH, Elgamal K, et al. Design, synthesis and cytotoxic evaluation of novel chalcone derivatives bearing triazolo[4,3-a]-quinoxaline moieties as potent anticancer agents with dual egfr kinase and tubulin polymerization inhibitory effects. Molecules 2017;23:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zhang H, Liu JJ, Sun J, et al. Design, synthesis and biological evaluation of novel chalcone derivatives as antitubulin agents. Bioorg Med Chem 2012;20:3212–8. [DOI] [PubMed] [Google Scholar]
- 137.Pitsinos EN, Vidali VP, Couladouros EA.. Diaryl ether formation in the synthesis of natural products. Euro J Organic Chem 2011;2011:1207–22. [Google Scholar]
- 138.Bedos-Belval F, Rouch A, Vanucci-Bacque C, et al. Diaryl ether derivatives as anticancer agents – a review. Med Chem Comm 2012;3:1356–72. [Google Scholar]
- 139.Zhao T-T, Lu X, Yang X-H, et al. Synthesis, biological evaluation, and molecular docking studies of 2,6-dinitro-4-(trifluoromethyl)phenoxysalicylaldoxime derivatives as novel antitubulin agents. Bioorg Med Chem 2012;20:3233–41. [DOI] [PubMed] [Google Scholar]
- 140.Wang W, Kong D, Cheng H, et al. New benzimidazole-2-urea derivates as tubulin inhibitors. Bioorg Med Chem Lett 2014;24:4250–3. [DOI] [PubMed] [Google Scholar]
- 141.Bai L-F, Qian H-H, Jiang D-W, et al. Design, synthesis and anti-tubulin activity of novel dinitro diphenyl ether derivatives as potent anticancer agent. Med Chem 2017;7:393–7. [Google Scholar]
- 142.Wang G, Liu W, Gong Z, et al. Design, synthesis, biological evaluation and molecular docking studies of new chalcone derivatives containing diaryl ether moiety as potential anticancer agents and tubulin polymerization inhibitors. Bioorg Chem 2020;95:103565. [DOI] [PubMed] [Google Scholar]
- 143.Qiu HY, Wang F, Wang X, et al. Design, synthesis, and biological evaluation of chalcone-containing shikonin derivatives as inhibitors of tubulin polymerization. Chem Med Chem 2017;12:399–406. [DOI] [PubMed] [Google Scholar]
- 144.Shankaraiah N, Nekkanti S, Brahma UR, et al. Synthesis of different heterocycles-linked chalcone conjugates as cytotoxic agents and tubulin polymerization inhibitors. Bioorg Med Chem 2017;25:4805–16. [DOI] [PubMed] [Google Scholar]
- 145.Huang X, Huang R, Li L, et al. Synthesis and biological evaluation of novel chalcone derivatives as a new class of microtubule destabilizing agents. Euro J Med Chem 2017;132:11–25. [DOI] [PubMed] [Google Scholar]
- 146.Kumar D, Raj KK, Malhotra SV, et al. Synthesis and anticancer activity evaluation of resveratrol–chalcone conjugates. Med Chem Comm 2014;5:528–35. [Google Scholar]
- 147.Kamal A, Kumar GB, Vishnuvardhan MV, et al. Synthesis of phenstatin/isocombretastatin-chalcone conjugates as potent tubulin polymerization inhibitors and mitochondrial apoptotic inducers. Org Biomol Chem 2015;13:3963–81. [DOI] [PubMed] [Google Scholar]
- 148.Kode J, Kovvuri J, Nagaraju B, et al. Synthesis, biological evaluation, and molecular docking analysis of phenstatin based indole linked chalcones as anticancer agents and tubulin polymerization inhibitors. Bioorg Chem 2020;105:104447. [DOI] [PubMed] [Google Scholar]
- 149.Mourad AAE, Mourad MAE, Jones PG.. Novel hdac/tubulin dual inhibitor: design, synthesis and docking studies of α-Phthalimido-Chalcone Hybrids as Potential Anticancer Agents with Apoptosis-Inducing Activity. Drug Des Devel Ther 2020;14:3111–30. [DOI] [PMC free article] [PubMed] [Google Scholar]