

Abstract
Antibiotic resistance poses an immediate and growing threat to human health. Multidrug efflux pumps are promising targets for overcoming antibiotic resistance with small-molecule therapeutics. Previously, we identified a diaminoquinoline acrylamide, NSC-33353, as a potent inhibitor of the AcrAB−TolC efflux pump in Escherichia coli. This inhibitor potentiates the antibacterial activities of novobiocin and erythromycin upon binding to the membrane fusion protein AcrA. It is also a substrate for efflux and lacks appreciable intrinsic antibacterial activity of its own in wild-type cells. Here, we have modified the substituents of the cinnamoyl group of NSC-33353, giving rise to analogs that retain the ability to inhibit efflux, lost the features of the efflux substrates, and gained antibacterial activity in wild-type cells. The replacement of the cinnamoyl group with naphthyl isosteres generated compounds that lack antibacterial activity but are both excellent efflux pump inhibitors and substrates. Surprisingly, these inhibitors potentiate the antibacterial activity of novobiocin but not erythromycin. Surface plasmon resonance experiments and molecular docking suggest that the replacement of the cinnamoyl group with naphthyl shifts the affinity of the compounds away from AcrA to the AcrB transporter, making them better efflux substrates and changing their mechanism of inhibition. These results provide new insights into the duality of efflux substrate/inhibitor features in chemical scaffolds that will facilitate the development of new efflux pump inhibitors.
Keywords: efflux pump inhibitors, AcrAB−TolC, Escherichia coli, antibiotic permeation, antibiotic potentiation
Graphical Abstract
Nosocomial infections caused by Gram-negative (GN) pathogens continue to rise, constituting a serious threat to public health and safety as well as having a tremendous economic impact.1–3 The ability of many of these pathogens to develop additional drug resistance against frontline antibiotics further increases the threat.3,4 In 2013, the Centers for Disease Control (CDC) reported approximately 51,000 infections from Pseudomonas aeruginosa with roughly 6,000 of those attributed to drug-resistant strains.5 By 2019, the CDC reported that the number of drug-resistant infections had increased to an estimated 32,600, resulting in approximately 2,700 deaths.2 As a result, there is an urgent need to not only discover new antibiotics but also develop new strategies to combat drug-resistant GN bacteria.6,7
The search for new antibiotics against GN bacteria has proven extremely challenging.8,9 The difficulty in finding new lead matter for GN bacteria is borne out by the poor hit rates typically seen in high-throughput screening campaigns performed by Big Pharma,10,11 which can be traced to the rarity of chemical scaffolds that accumulate in GN bacteria. This is a consequence of compounds having poor permeation across the outer membrane (OM)12 or being extruded from the cell by efflux pumps after crossing the OM or a combination of both.13 The OM permeation barrier and active efflux often work together synergistically, severely limiting the chemical space able to target GN pathogens.14–16
The OM of GN bacteria is made up of an asymmetric bilayer comprising a highly charged lipopolysaccharide (LPS)-rich outer leaflet and a phospholipid-based inner leaflet.17 As a result of the distinctly different chemical makeup of the outer and inner leaflets, it is very challenging to identify molecules with chemical properties suitable for passive permeation through both leaflets.18,19 Some compounds can cross the OM by diffusing slowly through porins, which are trimeric β-barrel proteins that control the uptake of small molecules.20 However, compounds able to use porins to cross the OM tend to be highly polar and therefore may be less likely to permeate the more lipophilic inner membrane (IM) to reach their cytoplasmic targets.8,18
The other major challenge associated with GN bacteria is the extrusion of xenobiotics via efflux pumps. GN pathogens express multidrug efflux pumps that are able to extrude a wide range of antibiotics. Across different GN bacterial strains, the type and number of pumps expressed as well as their drug specificity can vary significantly, though some homology also exists.21,22
In GN bacteria, the major efflux pumps belong to the resistance nodulation cell division (RND) superfamily.23 The RND transporter associates with a specific OM channel and a membrane fusion protein (MFP) to form a tripartite complex that exploits proton gradients across the IM to shuttle substrates out of the cell. In E. coli, the primary tripartite efflux pump system is the AcrAB−TolC complex in which AcrB is the RND transporter, AcrA is the MFP, and TolC is the OM channel.23,24 The substrate binds to the AcrB trimer located in the IM while the TolC trimer forms a channel in the OM allowing molecules to be extruded out of the cell. The MFP, AcrA, assembles as a trimer of dimers and serves to span the gap between AcrB and TolC, forming the pump complex. The structural asymmetry of the AcrB protomers revealed a “functional rotation” mechanism in which each of the three monomers constituting the IM transporter cyclically assumes three conformations, referred to as loose, tight, and open (or access, binding, and extrusion, respectively). Only the fully assembled AcrAB−TolC system is able to extrude substrates.25–27
Because so many different classes of antibiotics are affected by efflux, one strategy that has emerged to combat drug resistance is to target bacterial efflux pumps with small-molecule efflux pump inhibitors (EPIs). Inhibition of these complex molecular machines should restore the effectiveness of antibiotics that are susceptible to efflux activity. In general, this EPI strategy has focused mainly on identifying compounds that bind to and inhibit RND transporters such as AcrB in E. coli or the homologous MexB in Pseudomonas aeruginosa.28,29 Although several novel EPIs have been reported over the years, only a few of these compounds have reached clinical trials and none have reached commercial development.30–35
At least two classes of EPIs, pyridopyrimidines and pyranopyridines, have been shown to bind in a particular hydrophobic pocket (often referred to as the “hydrophobic trap”), which is located within the distal substrate-binding site of AcrB (hereafter referred to as the deep binding pocket, DBP).28,29,36 The hydrophobic trap is lined with phenylalanine residues that can stabilize inhibitors mainly through π-stacking interactions.37 The mechanism of inhibition, as suggested by structural analyses of inhibitor-bound AcrB and MexB structures, involves the stabilization of the transporter in the ligand-bound conformation and the failure to “functionally rotate” out of this state. The binding sites and mechanisms of other EPIs that are proposed to target RND pumps remain to be identified.
Recently, we disclosed a new strategy for identifying EPIs that focuses on targeting the MFP (AcrA) component of the tripartite efflux pumps in E. coli as opposed to the RND transporter, AcrB.38 Screening the NCI Diversity Set V, a collection of freely available compounds (1593) from the NCI and the Developmental Therapeutics Program (DTP), led to the identification of seven apparent AcrA binders that improved the potency of the antibiotics novobiocin (NOV) and erythromycin (ERY) in E. coli. Two compounds, NSC-60339 (1) and NSC-33353 (2), had attractive profiles as potential EPIs (Figure 1). They exhibited only weak antibacterial activity and were shown to bind to AcrA using surface plasmon resonance (SPR) experiments. These compounds inhibited efflux in a biochemical assay that measures intracellular accumulation of the fluorescent bisbenzimide probe, Hoechst 33342.38 This probe is a good substrate of the AcrAB−TolC pump and is unable to accumulate in the cytoplasm of E. coli in the absence of an EPI.
Figure 1.
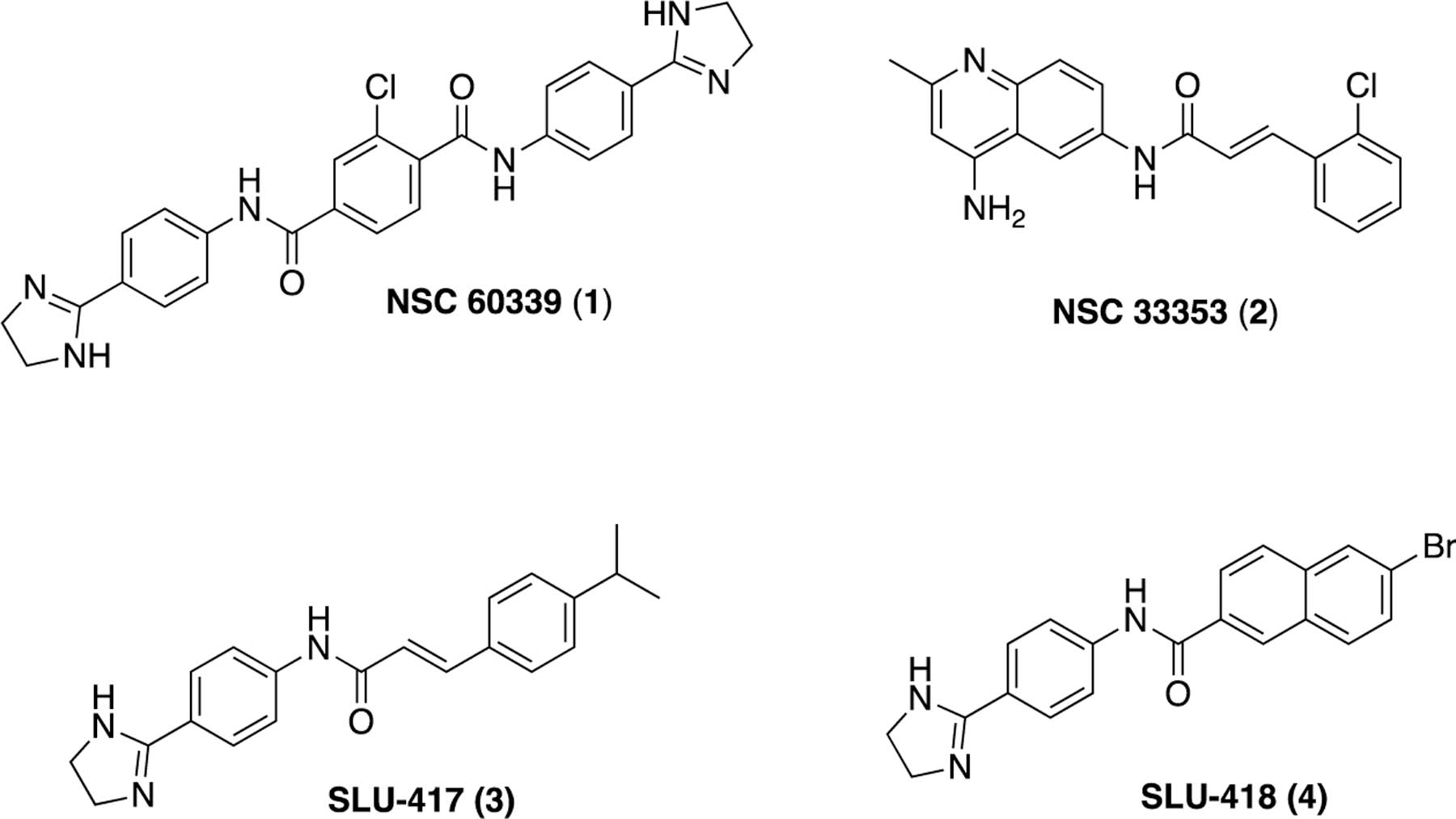
Previously identified compounds that bind to the membrane fusion protein AcrA of E. coli and inhibit efflux.38,39
Medicinal chemistry efforts to elaborate NSC-60339 (1) led to the identification of compounds such as 3 and 4 (Figure 1),39 which show modest improvements in terms of potentiation, exhibit stronger binding to AcrA, and feature increased permeation in wild-type (WT) E. coli. Interestingly, 1 and 2 differ in their mechanism of action, as they inhibit Hoechst 33342 efflux with different kinetics and only 1 alters the proteolytic profile of intracellular AcrA.
NSC-33353 (2) is a 4,6-diaminoquinoline substituted with a cinnamoyl amide attached through the C6 amine (Table 1). It was originally studied as a potential chemokine receptor modulator40 and later found to be an inhibitor of condensins.41 It was shown to potentiate the antibacterial activities of NOV and ERY with minimum potentiation concentration (MPC4) values of 1.56 and 3.1 μM, respectively (Table 1). The MPC4 value is the lowest concentration of test compound, in this case 2, that potentiates (lowers) the MIC of the corresponding antibiotic by 4-fold. These antibiotics were chosen because they operate through different antibacterial mechanisms (NOV targets DNA replication and transcription while ERY targets the translation machinery) and are unable to effectively accumulate in E. coli due to a combination of efflux by the AcrAB−TolC pump and/or permeation across the OM.38 NOV effectively permeates the OM but is an excellent substrate of AcrAB−TolC and is efficiently pumped out (Table 1). In contrast, ERY is a poor substrate of AcrAB−TolC and its activity is mostly diminished due to slow permeation across the OM (Table 1).42
Table 1.
Antibacterial and Potentiation Activity for Substituted Cinnamoyl Analogsi
| |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| MIC (μM)a |
potentiationb (μM) |
||||||||||
| # | R | WT | WT-pore | ΔtolC | ΔtolC-pore | MPC4 (NOV) | MPC4 (ERY) | CC50 (μM)c | WT/WTpored | WT-pore/ΔtolC-poree | efflux inhibition,f SS ratio 32 μM/0 μM |
| Ng | 128 | 32−64 | 0.5 | 0.125 | NA | NA | NA | NA | NA | NA | |
| Eh | 64 | 4−8 | 2 | 0.125 | NA | NA | NA | NA | NA | NA | |
| 2 | 2-Cl | 200 | 100 | 12.5 | 12.5 | 1.56 | 3.1 | 10.0 | 2 | 8 | 6.5 |
| 6 | 2-Br | 25 | 25 | 12.5 | 12.5 | 12.5 | 3.1 | 11.7 | 1 | 2 | 7.2 |
| 7 | 2-F | 100 | 100 | 50 | 50 | 12.5 | 12.5 | 28.3 | 1 | 2 | 3.9 |
| 8 | 2-Me | 50 | 25 | 25 | 50 | 6.25 | 12.5 | ND | 2 | 0.5 | 5.9 |
| 9 | 2-NO2 | >400 | >400 | >400 | >400 | >200 | >200 | ND | 1 | 1 | 1 |
| 10 | 3-Cl | 25 | 25 | 12.5 | 6.25 | 6.25 | 6.25 | 10.1 | 1 | 4 | 8.3 |
| 11 | 4-Cl | 25 | 12.5 | 12.5 | 6.25 | 6.25 | 6.25 | 11.2 | 2 | 2 | 11.2 |
| 12 | 4-Br | >200 | >200 | 25 | 12.5 | 200 | 200 | ND | 1 | >16 | 12.3 |
| 13 | 4-iPr | 50 | 25 | 25 | 25 | 12.5 | 12.5 | 13.3 | 2 | 1 | 5.5 |
| 14 | 2,3-diCl | >200 | 400 | 12.5 | 6.25 | 400 | 200 | ND | 1 | 64 | 1 |
MIC values, determined as the average of two replicates.
Minimum concentration of test compound required to reduce the MIC of novobiocin (NOV) or erythromycin (ERY) by 4-fold measured in WT-pore cells.
Measured in A549 cells; values are the average of two replicates.
The ratio of MIC values for compounds in WT cells vs permeated WT-pore cells.
The ratio of MIC values for compounds in WT-pore cells vs ΔtolC-pore cells.
Accumulation of Nile Red fluorescent probe in WT-pore cells in the presence (32 μM) and absence (0 μM) of each compound.
Values for novobiocin.
Values for erythromycin.
MIC = minimum inhibitory concentration; WT = wild-type; ND = not determined; SS = steady state; NA = not applicable.
Compound 2 was also tested for its intrinsic antibacterial activity in a set of four E. coli strains: WT and ΔtolC where the AcrAB−TolC pump has been inactivated and their hyperporinated derivatives, WT-pore and ΔtolC-pore cells.42 The MICs for 2 were 200 and 100 μM in the WT and WT-pore cells, respectively. However, in both efflux-deficient cell types, 2 displayed strong antibacterial activity with an MIC of 12.5 μM. These findings suggest that 2 is likely a substrate of the efflux pump, rendering it ineffective as an antibacterial agent. It also displayed cytotoxicity in A549 cells with a CC50 of 10 μM.
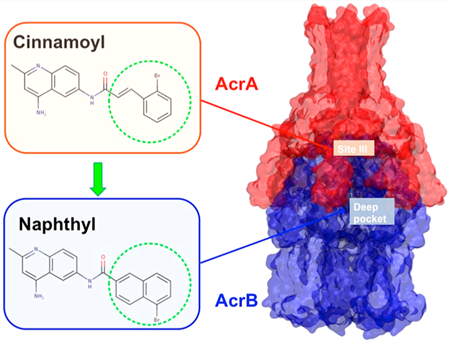
Herein, we describe follow-up medicinal chemistry efforts as well as biochemical and in silico-based mechanistic evaluation on the second screening hit, NSC-33353 (2), as a potential EPI of AcrAB−TolC. Modifications of the cinnamoyl group in 2 led to analogs with both potent antibacterial and EPI properties. The replacement of the cinnamoyl group with naphthyl isosteres gave compounds with very different EPI profiles, likely due to changes in how they bind to and inhibit AcrAB−TolC.
RESULTS
In our previous efforts to optimize 1, we found that the cinnamoyl analog, 3 (Figure 1), displayed a strong affinity for AcrA.39 The presence of the same cinnamoyl group in both 2 and 3 suggested that the quinolone ring of 2 and the phenyl dihydroimidazoline ring of 3 might bind to AcrA in a similar manner. This observation further suggested that we might follow similar structure−activity relationships (SAR) developed with 1 to explore modifications of 2. We therefore sought to investigate additional substitutions in the phenyl ring of the cinnamoyl group to further probe the SAR in this region and assess the cinnamoyl group as a possible contributor to the observed cytotoxicity of 2. To mitigate this potential liability, the replacement of the cinnamoyl moiety with substituted naphthyl groups was also explored. In the earlier SAR study of 1, naphthyl analog 4 (Figure 1) was found to greatly potentiate NOV and bind strongly to AcrA.
Chemistry.
The synthesis of all new compounds was carried out according to Scheme 1. Selective acylation of the 6-amino group in 2-methylquinoline-4,6-diamine (5) was achieved via the procedure described by Hagmann and Springer.40 Diamine 5 was dissolved in glacial acetic acid, and the resulting solution was added to the requisite acid chloride, which was dissolved in acetic acid to exclusively give the desired 6-substituted amides. The acid chlorides were typically prepared in situ from the corresponding carboxylic acids with either oxalyl chloride or thionyl chloride. Further elaboration of bromo-substituted naphthyl analogs (15 and 16) via a standard Suzuki coupling reaction with various arylboronic acids led to the aryl-substituted naphthyl analogs (24−36).
Scheme 1.
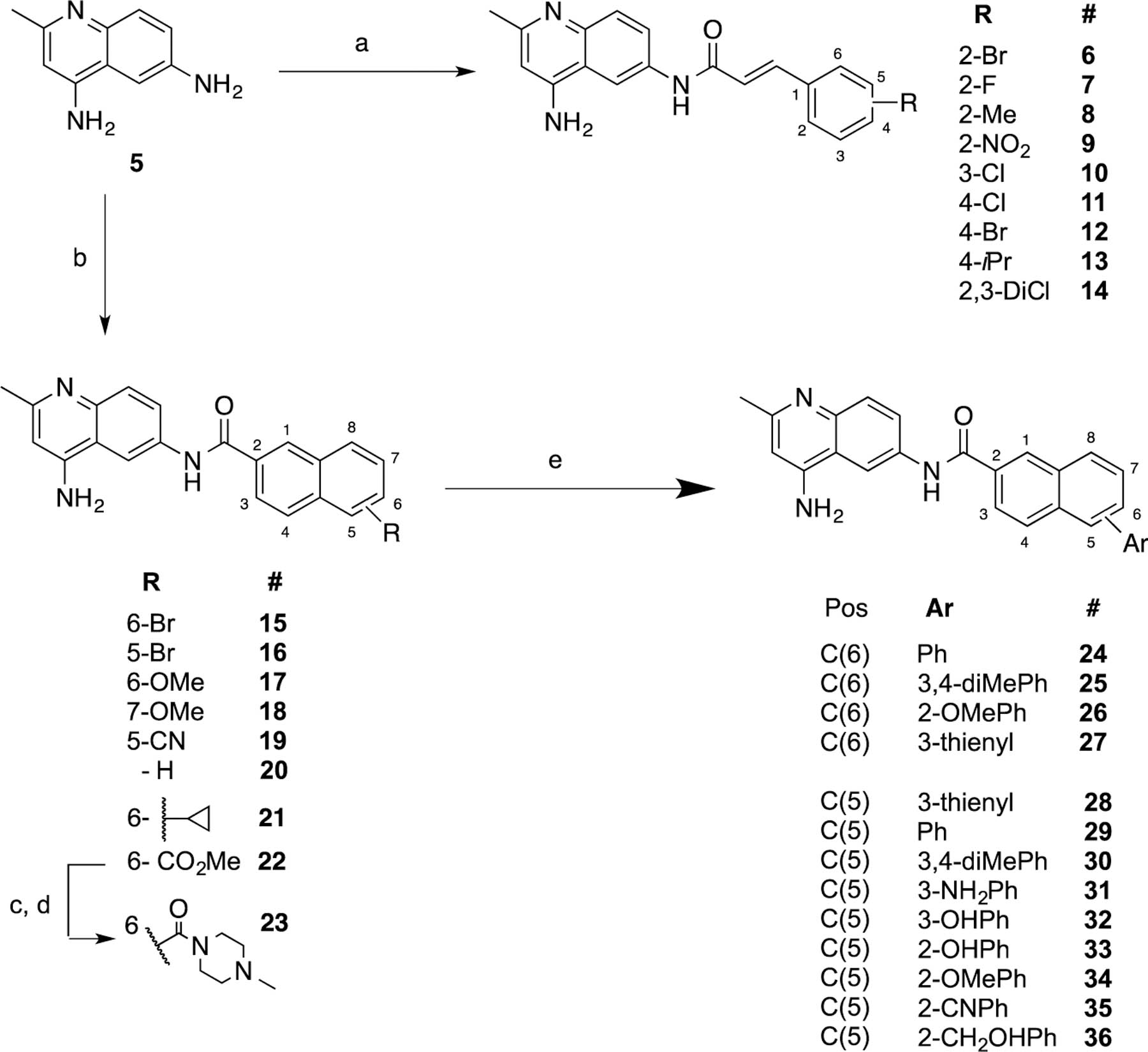
Synthetic Routes and Conditions for the Preparation of New Analogsa
a(a) HOAc (gl), then add cinnamoyl chloride, rt, 12 h. (b) HOAc (gl), then add naphthyl chloride, rt, 12 h. (c) LiOH, THF-H2O (9:1). (d) TBTU, CH2Cl2, 4-Me piperazine. (e) 15 or 16, Pd(PPh3)4, K2CO3, microwave, ArB(OH)2.
Antibacterial and Potentiation Activities of the Cinnamoyl Derivatives.
Substitution on the phenyl ring of the cinnamoyl group was initially explored (Table 1). The replacement of the chlorine with bromine (6) at the 2-position yielded an analog with antibacterial activity (MIC = 25 μM) in WT cells, representing a 8-fold decrease in the MIC compared to 2. The MIC was unchanged in the hyperporinated cells and only slightly lower in the efflux deficiesnt cells (MIC = 12.5 μM). Several other analogs in this series displayed antibacterial activity (≤50 μM) in E. coli and appeared to be only weak substrates for efflux (Table 1). However, substitution with the electron-withdrawing 2-NO2 group (9) led to a complete loss of antibacterial activity, while replacing Cl with Br (12) at the 4-position or the dichloro-substituted phenyl (14) led to a loss of antibacterial activity in WT and WT-pore cells. These compounds did show activity in the efflux-deficient ΔtolC cells with MICs ≤ 25 μM, suggesting that these substitutions promote efflux by AcrAB−TolC (Table 1).
Potentiation for NOV and ERY was also evaluated. Several compounds (6, 7, 8, 10, 11, 13) were able to potentiate NOV with MPC4 values in the range of 6.25−12.5 μM, which is slightly higher than what was observed with 2 (1.56 μM, Table 1). These compounds also potentiated ERY with MPC4 values similar to their NOV potentiation. One exception was bromo analog, 6, which potentiated ERY (MPC4 = 3.1 μM) about 4× better than it potentiated NOV (MPC4 = 12.5 μM). Three analogs (9, 12, and 14) failed to potentiate the activities of either NOV or ERY. These were the same analogs that also lacked antibacterial activity in the WT cells. In general, analogs displayed similar cytotoxicity to that observed with 2, except for fluoro analog, 7, which gave a CC50 (28.3 μM) about 3-fold higher than the original lead, 2.
Because most of these analogs possess intrinsic fluorescence that increases when compounds are bound to lipids or nucleic acids,43 we also analyzed their intracellular accumulation and the effect of efflux and permeation across the OM using an activity-independent fluorescence assay. In this assay, increasing concentrations of compounds were incubated with WT and WT-pore cells and the efflux-deficient derivatives ΔtolC and ΔtolC-pore. The change in fluorescence was then monitored as a function of time (Figure 2). In agreement with their respective antibacterial activities, the cinnamoyl analogs 7, 8, 10, and 11 accumulated with similar kinetics in the efflux-proficient and efflux-deficient cells, suggesting that these compounds avoid efflux by AcrAB−TolC (Figure S1). However, these compounds slowly permeated the OM as observed from the increased rates of their intracellular accumulation in the hyperporinated strains.
Figure 2.
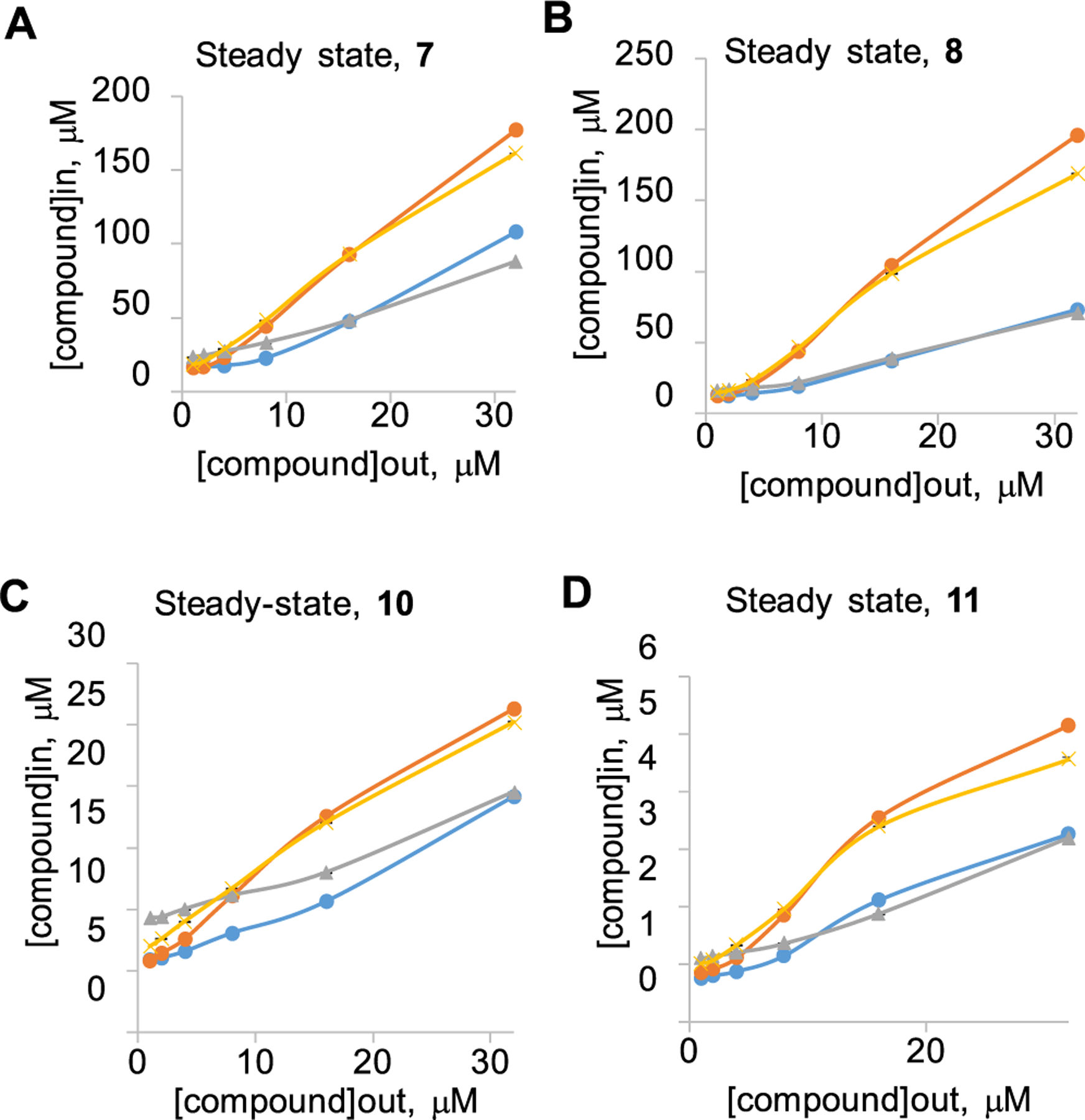
Intracellular accumulation of compounds (A) 7, (B) 8, (C) 10, and (D) 11 in E. coli cells with different genetic backgrounds. Doubling concentrations of compounds from 1 to 32 μM were added to WT (blue), WT-pore (yellow), ΔtolC (gray), and ΔtolC-pore (orange); cells were incubated at room temperature for indicated periods of time, and fluorescence was measured in real time. Data from at least two independent experiments were fitted to a two-exponential equation to extract the steady-state intracellular concentrations. Error bars are SD (n = 2).
Antibacterial and Potentiation Activities of the Naphthyl Derivatives.
Attention was next shifted to analogs where the cinnamoyl group was replaced with various substituted naphthyl groups, as in previous efforts going from 1 to 4 (Table 2). Bromo (15 and 16) and methoxy-substituted (17 and 18) analogs displayed antibacterial activity in ΔtolC and ΔtolC-pore cells with only 15 giving any appreciable activity in the WT-pore cells (MIC = 50 μM). Thus, all compounds are substrates for efflux. Analog 15 potentiated the activities of both NOV and ERY with identical MPC4 values (12.5 μM), while 16−18 potentiated NOV (MPC4 = 12.5−25 μM) but surprisingly did not potentiate ERY (MPC4 ≥ 200 μM). These naphthyl analogs gave only slightly higher CC50’s (∼20 μM) than the corresponding cinnamoyl compounds.
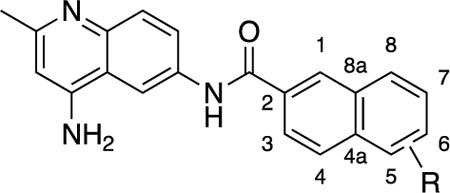
Table 2.
Antibacterial and Potentiation Activity for Substituted Naphthyl Analogsg
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| # | R | Position | MIC (μM)a | Potentiation (μM)b | CC50 (μM)c | WT/WT-pored | WT-pore/ΔtolC- poree |
Efflux inhibitionf, SS ratio 32 μM/0 μM | ||||
| WT | WT-pore | ΔtolC | ΔtolC-pore | MPC4 (NOV) | MPC4 (ERY) | |||||||
| 15 | -Br | C(6) | 100 | 50 | 25 | 12.5 | 12.5 | 12.5 | 21.3 | 2 | 4 | 1 |
| 16 | -Br | C(5) | >200 | >400 | 25 | 50 | 12.5 | >200 | 18.8 | 0.5 | >8 | 1.6 |
| 17 | -OMe | C(6) | >200 | >400 | 25 | 25 | 25 | >200 | 20.9 | 0.5 | 16 | 6.9 |
| 18 | -OMe | C(7) | >200 | 400 | 25 | 50 | 25 | 100 | ND | 1 | 16 | 2.8 |
| 19 | -CN | C(5) | >200 | >200 | 100 | 400 | 200 | 200 | ND | 1 | 1 | ND |
| 20 | -H | - | 200 | 100 | 50 | 200 | 25 | 50 | ND | 2 | 2 | 4.9 |
| 21 |
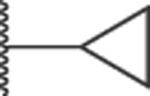
|
C(6) | 200 | 50 | 100 | 100 | 25 | 100 | 31.4 | 4 | 0.5 | 1 |
| 22 | -CO2Me | C(6) | >200 | >200 | >200 | >200 | 100 | 100 | ND | 1 | 1 | ND |
| 23 |
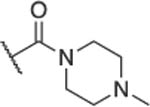
|
C(6) | >200 | >200 | >200 | 200 | >200 | >200 | ND | 1 | 1 | 1 |
MIC values, determined as the average of two replicates.
Minimum concentration of test compound required to reduce the MIC of novobiocin (NOV) or erythromycin (ERY) by 4-fold measured in WT-pore cells.
Measured in A549 cells; values are the average of two replicates.
The ratio of MIC values for compounds in WT cells vs permeated WT-pore cells.
The ratio of MIC values for compounds in WT-pore cells vs ΔtolC-pore cells.
Accumulation of Nile Red fluorescent probe in WT-pore cells in the presence (32 μM) and absence (0 μM) of each compound.
MIC = minimum inhibitory concentration; WT = wild-type; ND = not determined; SS = steady state.
Substitution with electron-withdrawing groups such as cyano (19), carbomethoxy (22), or an amide moiety (23) led to a complete loss of antibacterial and potentiation activity. The unsubstituted 20 displayed some antibacterial activity (MIC = 50 μM) in ΔtolC cells and potentiated both NOV (MPC4 = 25 μM) and ERY (MPC4 = 50 μM). The cyclopropyl analog, 21, lacked antibacterial activity in WT cells but was active in hyperporinated WT-pore cells (MIC = 50 μM), making it one of the few analogs affected by permeation. It also had a CC50 of 31 μM, which was the lowest observed in this series.
Previous docking calculations suggested that adding aromatic rings at either the C5- or C6-positions might increase the affinity for AcrA (vide infra).44 As a result, a set of aromatic substituted naphthyl groups was prepared. The corresponding C6-substituted analogs (24−27) failed to show any antibacterial activity, and only phenyl-substituted analog 24 exhibited any potentiation (Table S1). Following the trend observed with other naphthyl compounds, 24 only potentiated NOV (MPC4 = 25 μM) and not ERY.
The C5-aryl-substituted analogs are shown in Table 3. Most of these analogs (28, 29, 31, 34, and 36) had similar profiles. They showed no antibacterial activity in either WT or WT-pore cells but did exhibit similar antibacterial activity in the efflux-deficient ΔtolC and ΔtolC-pore cells. They potentiated NOV (MPC4 = 6.25−12.5 μM) but not ERY. Two analogs, the dimethylphenyl, 30, and 3-hydroxyphenyl analogs, 32, had no antibacterial activity in any of the cell types. Compound 30 potentiated NOV (MPC4 = 25 μM) but not ERY, while 32 failed to potentiate either antibiotic. Three analogs (31, 33, and 36) were able to potentiate ERY in addition to NOV. The 3-aminosubstituted analog, 31, was the weakest of the three in terms of potentiation values while the 2-hydroxyphenyl analog, 33, was the best, potentiating both NOV and ERY with MPC4 = 12.5 μM. Compound 33 was the only naphthyl analog that possessed antibacterial activity even in the WT cells (MIC = 50 μM). The CC50 value for 33 was 27.3 μM. The replacement of hydroxy or methoxy with an electron-withdrawing cyano group (35) led to a loss of both antibacterial and potentiation activity.
Table 3.
Antibacterial and Potentiation Activity for C(5)-Aryl Substituted Naphthyl Analogsg
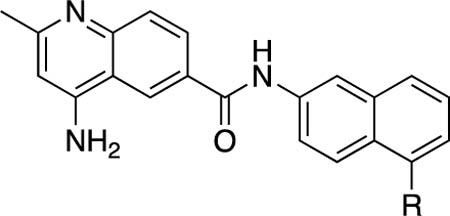
| |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| # | R | MIC (μM)a | Potentiation (μM)b | CC50 (μM)c | WT/WT-pored | WT-pore/ΔtolC-poree | Efflux inhibitionf, SS ratio 32 μM/0 μM | ||||
| WT | WT-pore | ΔtolC | ΔtolC-pore | MPC4 (NOV) | MPC4 (ERY) | ||||||
| 28 |
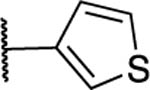
|
>200 | 100 | 25 | 50 | 12.5 | >200 | 10.5 | 2 | 4 | 4.4 |
| 29 |
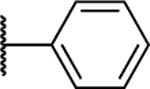
|
>200 | 100 | 25 | 25 | 12.5 | >200 | ND | 2 | 4 | 1.7 |
| 30 |
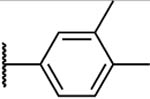
|
>200 | >400 | >200 | 200 | 25 | >200 | 7.2 | - 1 | 2 | ND |
| 31 |
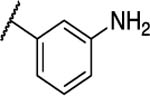
|
>200 | >400 | 12.5 | 50 | 25 | 50 | ND | 0.5 | 8 | 4.2 |
| 32 |
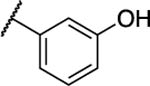
|
>200 | >200 | >200 | >200 | >200 | >200 | ND | 1 | 1 | 4.5 |
| 33 |
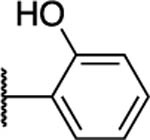
|
50 | 25 | 6.25 | 12.5 | 12.5 | 12.5 | 27.3 | 2 | 2 | 4.6 |
| 34 |
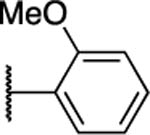
|
>200 | >400 | 6.25 | 12.5 | 12.5 | >200 | 10.2 | 1 | >64 | 5.4 |
| 35 |
|
>200 | >400 | 50 | 100 | >400 | >200 | ND | 1 | >4 | 1 |
| 36 |
|
>200 | >200 | 25 | 12.5 | 25 | 25 | ND | 1 | 8 | 9.8 |
MIC values, determined as the average of two replicates.
Minimum concentration of test compound required to reduce the MIC of novobiocin (NOV) or erythromycin (ERY) by 4-fold measured in WT-pore cells.
Measured in A549 cells; values are the average of two replicates.
The ratio of MIC values for compounds in WT cells vs permeated WT-pore cells.
The ratio of MIC values for compounds in WT-pore cells vs ΔtolC-pore cells.
Accumulation of Nile Red fluorescent probe in WT-pore cells in the presence (32 μM) and absence (0 μM) of each compound.
MIC = minimum inhibitory concentration; WT = wild-type; ND = not determined; SS = steady state.
Taken together, these results show that, unlike the cinnamoyl-derived analogs, the majority of the naphthyl analogs lacked antibacterial activity in the WT or WT-pore cells. Several compounds exhibited antibacterial activity in ΔtolC cells, suggesting that the naphthyl analogs were more susceptible to efflux than the cinnamoyl analogs. Another difference between the two sets of compounds was the finding that, with only a few exceptions (15, 20, 31, 33, and 36), the naphthyl analogs could potentiate only NOV and not ERY. Aryl substitution at the C5 position of the naphthyl ring yielded the best results in terms of NOV potentiation.
Efflux Inhibition in Bacterial Growth-Independent Assays.
Intrinsic antibacterial activities of compounds can contribute to the observed MPC values, and in the case of the cinnamoyl derivatives, the MICs are very close to the observed MPCs (Table 1). To establish whether the potentiation of antibiotic activities is indeed due to the inhibition of efflux, we first analyzed MPCs in efflux-deficient ΔtolC-pore cells. We found no potentiation of NOV or ERY activities in these cells (Table S2).
We next analyzed the ability of compounds to inhibit efflux in a bacterial growth-independent assay using fluorescent probes that are substrates of AcrAB−TolC. Since these compounds are intrinsically fluorescent (Figure 2), we needed a probe with fluorescence properties distinct from those of the compounds. We chose the Nile Red probe, which is not fluorescent in water solutions but emits red light in hydrophobic environments such as lipid membranes. Cells were incubated with increasing concentrations of compounds, and the kinetics of the change in fluorescence was analyzed (Tables 1–3 and Figure S2). Most of the tested cinnamoyl derivatives inhibited efflux of Nile Red with efficiency similar to that of 2. Compounds such as 6 and 10−12 were more effective than 2 at the highest concentration (32 μM), whereas 9 and 14 failed to inhibit Nile Red efflux (Table 1).
Similarly, naphthyl analogs were comparable to 2 as inhibitors of Nile Red efflux. Among this series, 17 and 36 were more effective and 16, 18, and 20 were all less effective than 2 (Figure S2 and Tables 2 and 3). Thus, compounds inhibited efflux in both bacterial growth-dependent assays (i.e., by potentiating the activities of the antibiotics) and in growth-independent assays. The inhibition of Nile Red efflux and potentiation of antibiotic activities were observed in WT-pore cells but not in efflux-deficient ΔtolC-pore cells, further confirming the dependence of these activities of compounds on efflux.
Interactions with AcrA.
To gain insight into the mechanism of antibiotic potentiation by 2 and its analogs, we next analyzed how these compounds interact with AcrA. In SPR-based assays with purified AcrA protein, we found that all cinnamoyl analogs bound AcrA with equilibrium dissociation constants (KD) ranging from 0.02 to 0.3 mM (Table 4). In contrast, naphthyl substituents that bound AcrA varied considerably in their affinities. Among all tested compounds, 17 and 36 were found to bind AcrA with the highest affinity (KD = 1−2 μM).
Table 4.
Interactions with AcrA and AcrB Determined by SPR and Molecular Dockingd
|
KD (mM)a |
AcrA docking score (kcal mol−1)b |
||||||
|---|---|---|---|---|---|---|---|
| # | AcrA | AcrB | site I | site II | site III | site IV | AcrB docking score (kcal mol−1)b |
| NOVc | 0.04 | 0.53 | NA | NA | NA | NA | NA |
| MCc | 0.28 | 0.11 | NA | NA | NA | NA | NA |
| 2 | + | ND | −7.3 | −7.3 | −8.1 | −7.5 | −11.9 |
| 6 | 0.02 | 0.02 | −7.0 | −7.4 | −7.9 | −7.3 | −12.1 |
| 7 | 0.28 | 0.03 | −7.6 | −7.7 | −8.1 | −7.5 | −12.1 |
| 8 | 0.26 | ND | −7.3 | −7.5 | −8.0 | −7.6 | −12.5 |
| 9 | 0.05 | 0.04 | −6.9 | −7.1 | −7.9 | −7.7 | −11.5 |
| 10 | 0.02 | 0.06 | −8.3 | −8.8 | −9.5 | −8.4 | −12.0 |
| 11 | 0.01 | 0.17 | −6.8 | −7.3 | −7.9 | −7.4 | −11.2 |
| 12 | 0.08 | 0.01 | −7.0 | −7.5 | −7.9 | −7.6 | −11.6 |
| 13 | 0.05 | 0.06 | −7.2 | −7.5 | −8.1 | −7.9 | −12.6 |
| 14 | 0.02 | 0.05 | −7.3 | −7.4 | −8.1 | −7.5 | −12.3 |
| 15 | ND | 0.01 | −7.6 | −8.2 | −8.7 | −8.3 | −13.3 |
| 16 | 0.25 | 0.25 | −7.8 | −8.2 | −8.8 | −8.1 | −13.5 |
| 17 | 0.001 | 0.02 | −7.6 | −8.4 | −8.6 | −8.0 | −12.9 |
| 18 | ND | ND | −7.6 | −7.7 | −8.5 | −7.9 | −12.8 |
| 19 | ND | ND | −8.6 | −8.7 | −9.0 | −8.4 | ND |
| 20 | ND | 0.07 | −7.8 | −8.1 | −8.7 | −8.1 | −13.1 |
| 21 | ND | ND | −7.8 | −8.2 | −8.7 | −8.3 | ND |
| 22 | ND | ND | −7.8 | −8.5 | −8.5 | −8.4 | ND |
| 23 | NB | NB | −8.5 | −8.9 | −9.3 | −9.2 | −14.6 |
| 24 | ND | ND | −8.3 | −9.0 | −9.2 | −9.2 | ND |
| 25 | ND | ND | −8.6 | −9.3 | −9.6 | −9.6 | ND |
| 26 | 0.02 | 0.05 | −8.3 | −9.0 | −8.9 | −8.9 | −14.2 |
| 27 | ND | ND | −7.8 | −8.4 | −9.1 | −8.9 | ND |
| 28 | ND | 4.63 | −8.3 | −8.1 | −9.0 | −8.1 | −14.5 |
| 29 | 1.18 | 0.05 | −8.3 | −8.8 | −9.5 | −8.4 | −15.6 |
| 30 | ND | ND | −8.6 | −9.2 | −9.6 | −8.6 | ND |
| 31 | 0.06 | 0.02 | −9.1 | −8.9 | −9.4 | −8.4 | −15.3 |
| 32 | ND | ND | −8.8 | −8.7 | −9.2 | −8.2 | −15.3 |
| 33 | ND | 4.92 | −8.6 | −8.9 | −9.4 | −8.3 | −15.3 |
| 34 | ND | ND | −8.0 | −8.8 | −8.9 | −8.1 | −14.2 |
| 35 | ND | 0.07 | −8.6 | −9.1 | −9.6 | −8.6 | −16.3 |
| 36 | 0.002 | 0.04 | −8.2 | −9.2 | −9.0 | −7.9 | −15.1 |
For compounds with EPI activities, we also analyzed their interactions with AcrA in vivo using a proteolysis assay.38 In this assay, AcrA assembled into the AcrAB−TolC complex is cleaved into two major proteolytic fragments comprising amino acid residues K46-K396 and K46-T296, respectively. The proteolytic profiles of the untreated cells are dominated by these two AcrA fragments (Figure 3). In contrast, an additional K46-K346 fragment is accumulated when AcrA fails to assemble into a complex. Although 2 and most of its derivatives generated in this study did not affect the proteolytic profiles of AcrA in the periplasm of WT-pore cells, 12 and 17 and, to a lesser extent, 31 and 36 altered the AcrA profiles, as seen from the accumulation of the characteristic K46-K346 fragment. This result suggests that treatment of cells with these compounds interfered with the assembly of the AcrAB−TolC complex (Figure 3). All four compounds bound the purified AcrA with relatively high affinities, with 17 and 36 being the strongest binders (Table 4). This result further demonstrated that, although many compounds bind the purified AcrA, their specific interactions can lead to different outcomes and certain interactions may affect the assembly of the AcrAB−TolC pump.
Figure 3.
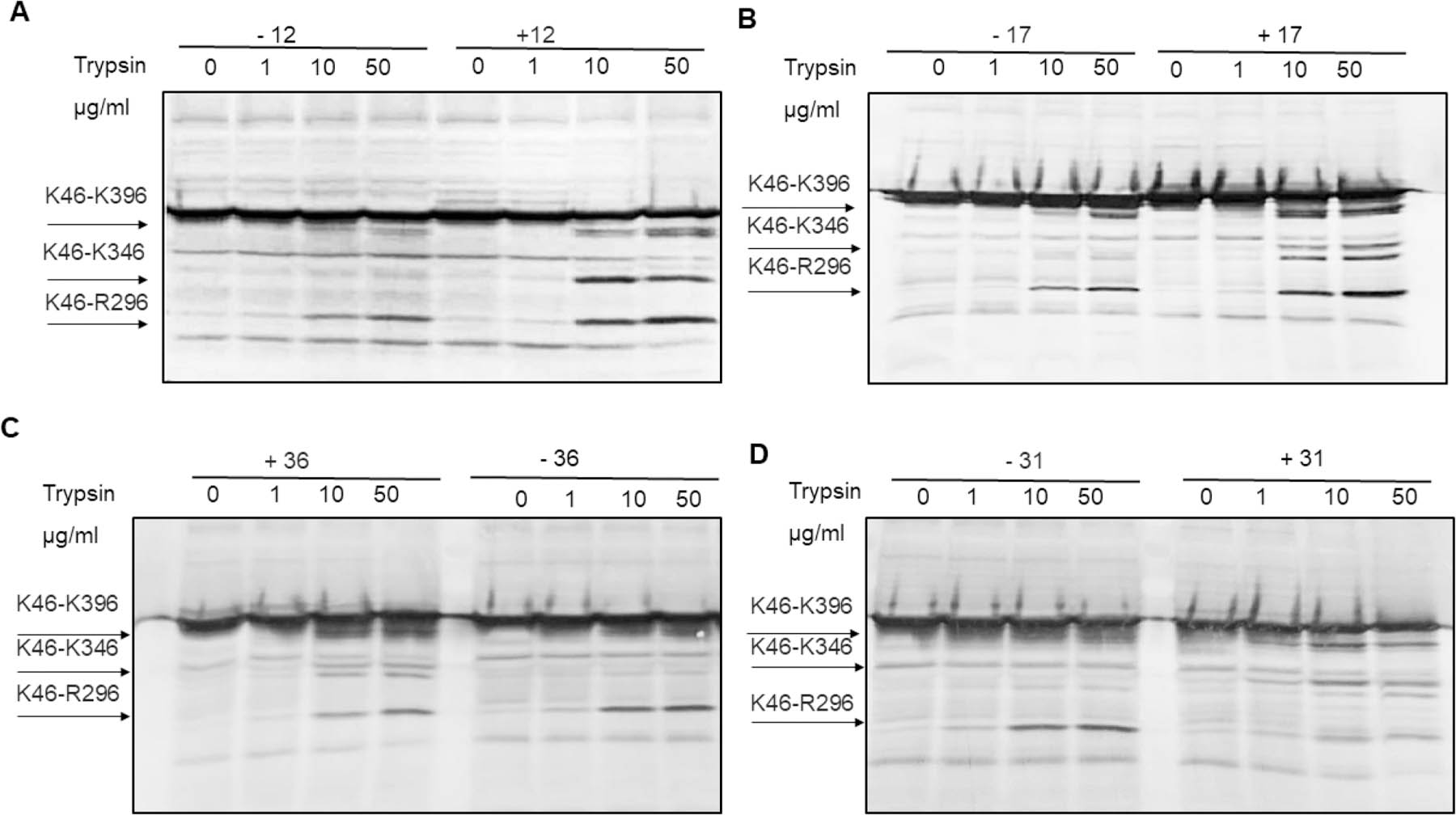
Proteolytic profiles of periplasmic AcrA in the presence and absence of (A) 12, (B) 17, (C) 36, and (D) 31. Cells were osmotically shocked with sucrose and treated with the indicated concentrations of trypsin for 30 min at 37 °C. Reactions were terminated by SDS sample buffer and boiling the samples for 5 min. Proteins and proteolytic fragments were separated by 12% SDS-PAGE, and AcrA fragments were visualized using polyclonal anti-AcrA antibody. The characteristic proteolytic fragments of AcrA as identified in ref 70 are indicated.
We next used ensemble docking to gain insight into the molecular mechanism underlying the interactions of compounds with AcrA. The unbound AcrA monomer is quite flexible and samples a wide range of conformations among its four structural domains: α-hairpin, lipoyl, β-barrel, and membrane proximal (MP).45 The two-predominant cis and trans conformations of AcrA differ in their inter-residue contacts between the β-barrel and MP domains. In the complex with AcrB and TolC, AcrA adopts a trans conformation. Previous studies have shown that 1 binds predominantly at site IV, which is located between the lipoyl and β-barrel domains of AcrA (Figure 4A).44
Figure 4.
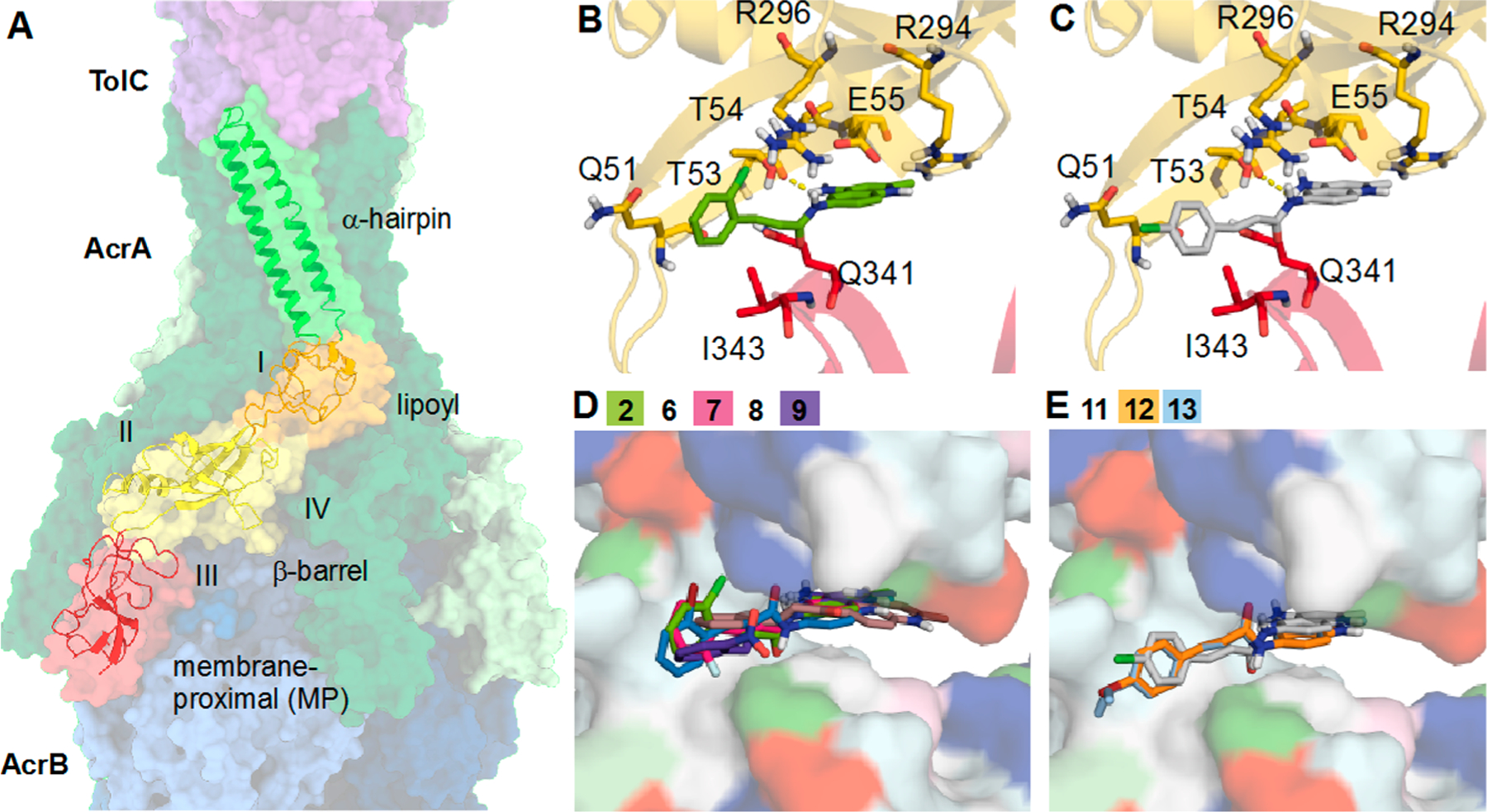
(A) Cryo-EM structure of AcrAB−TolC (PDB ID: 5NG5).71 Individual subunits of TolC (purple), AcrA (green), and AcrB (blue) are shown in a surface representation. A single monomer of AcrA is shown as a cartoon and colored by domain (α-hairpin = green, lipoyl = orange, β-barrel = yellow, membrane proximal (MP) = red). Sites I−IV used for docking are labeled. Top binding poses of (B) o-substituted cinnamoyl compound 2 (green) and (C) p-substituted cinnamoyl derivative 11 (gray) from AcrA docking. AcrA is shown as a cartoon and colored by domain with nearby residues shown as sticks and polar interactions represented by dashed yellow lines. Top site III docking poses of (D) o-substituted and (E) p-substituted cinnamoyl derivatives. Analogs are shown as sticks, and AcrA is shown as a surface and colored by residue type (hydrophobic = white, polar = cyan, positive = blue, negative = red, aromatic = magenta, proline = light green, glycine = dark green).
To analyze AcrA interactions with 2 and its analogs, we used an ensemble of 29 conformations of full-length monomeric AcrA structures obtained from molecular dynamics simulations. Compounds were docked to four previously identified sites on AcrA (Figure 4A), and the top pose was selected on the basis of the Vina docking score (Table 4).44
For most of the cinnamoyl and naphthyl analogs, the top docking score corresponded to binding at site III, which is located at the flexible interface of the β-barrel and MP domains. The overall most favorable docking score for the cinnamoyl analogs was −9.5 kcal mol−1, which was obtained for 10, and −9.6 kcal mol−1, obtained for naphthyl analogs 25, 30, and 35 (Table 4). For site III, the top docking scores were all within 1.6 kcal mol−1 of each other, which is within the expected error range for docking calculations of this type.46 Thus, we cannot reliably distinguish between the predicted binding affinities of the cinnamoyl and naphthyl analogs.
Regardless of the substituent position, the R groups do not participate in any polar interactions with the protein, and these analogs have similar docking poses at this site (Figure 4). For example, both the ortho-substituted compound 2 (Figure 4B) and the para-substituted analog 11 (Figure 4C) form polar contacts with T53 and are near Q51, T54, E55, R294, Q341, and I343. Importantly, these residues are known to be critical for the cis-to-trans conformational transitions in AcrA and for the functional activity of the efflux pump.45 All C(6)-substituted analogs as well as the nonaryl-substituted C(5)-analogs were also predicted to bind in a similar position regardless of substitution (Figure 5E−G) with the exception of 23, which was predicted to bind elsewhere in site III. For the C5-substituted aryl compounds, the naphthyl ring is flipped ∼180° in its predicted binding pose (Figure 5H).
Figure 5.
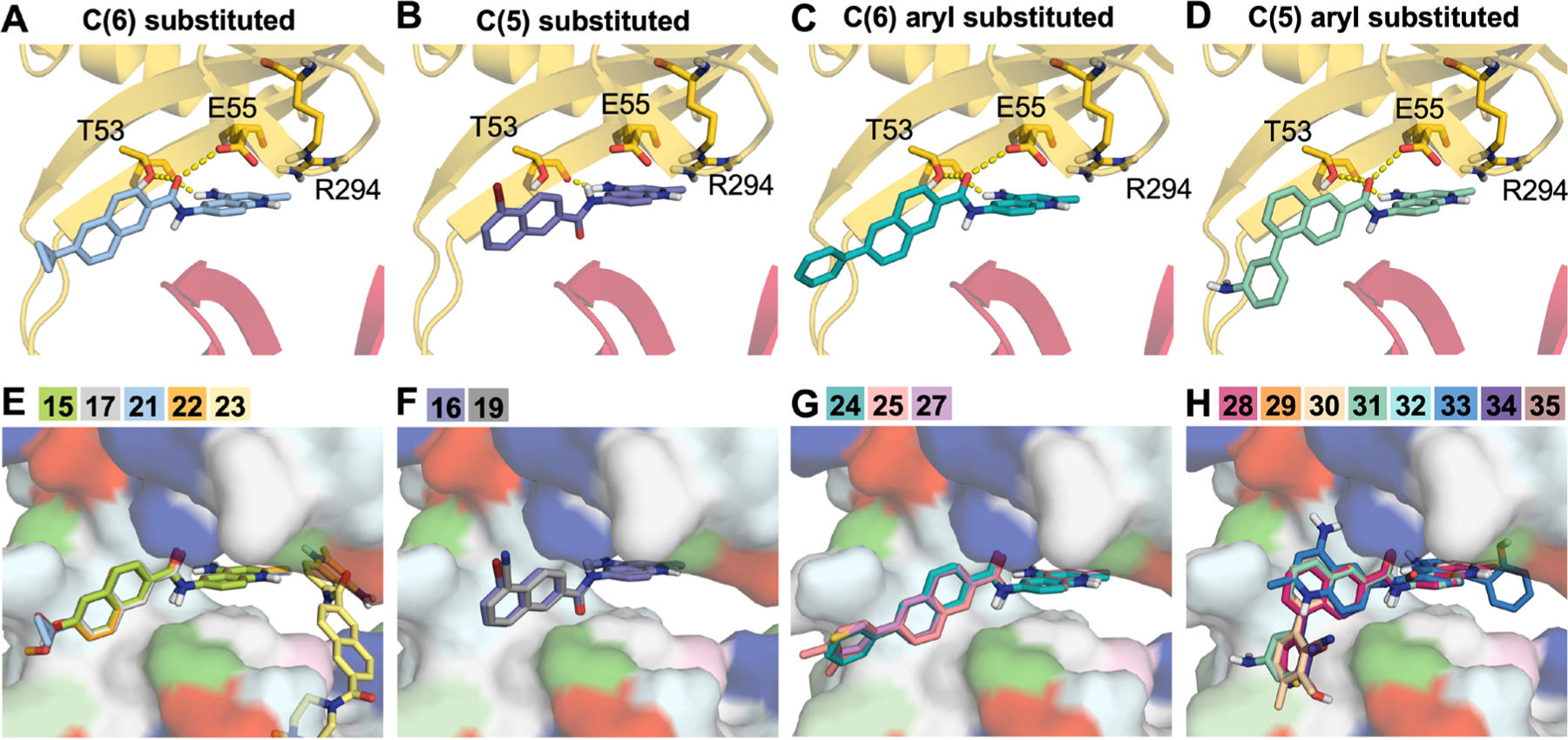
Top poses from docking naphthyl analogs to AcrA. Example top binding poses for (A) C6, (B) C5, (C) C6 aryl, and (D) C5 aryl-substituted analogs are shown as sticks. Polar interactions are represented by dashed yellow lines, and residues that form polar interactions are also shown as sticks and colored by domain (β-barrel = yellow, membrane proximal (MP) = red). (E−H) Top-scoring poses of naphthyl analogs grouped on the basis of substitution. Analogs are shown as sticks, and AcrA is shown as a surface and colored by residue type (hydrophobic = white, polar = cyan, positive = blue, negative = red, aromatic = magenta, proline = light green, glycine = dark green).
Therefore, SPR experiments suggest that compounds from both the cinnamoyl and naphthyl series interact with AcrA. These interactions apparently are not sensitive to chemical modifications and cannot explain the observed differences in the activities of the compounds. Compounds 12, 17, and 36, which display high affinities for purified AcrA, affect its proteolytic profiles and possibly the assembly of the AcrAB−TolC complex. However, we note that these ligands share similar docking scores and poses with other compounds in the AcrA model. Thus, additional factors besides interactions with AcrA alone likely contribute to the observed activities of these compounds.
Interactions with AcrB.
The analogs with cinnamoyl and naphthyl substitutions were notably different in both their growth-inhibitory and EPI activities. In particular, the naphthyl group increased the propensity of the compounds to be expelled by the AcrAB−TolC pump and reduced their potency in combination with ERY but not with NOV. These results suggested either changes in how the compounds interact with the efflux pump components, or in their mechanism of action, or both. Using SPR-based assays with purified AcrB protein, we found that the binding affinities of all cinnamoyl and a few naphthyl substituents toward AcrB varied from low micromolar range to low millimolar range and were comparable to those toward AcrA (Table 4). These affinities were also similar to the previously reported affinities of AcrB to antibiotics NOV, doxorubicin, and minocycline and such EPIs as MC207,110.47,48 However, for several naphthyl-containing compounds such as 20, 28, 33, and 35, a binding signal was detected for the immobilized AcrB but not for AcrA, suggesting that these naphthyl substituents have higher affinity for the AcrB transporter.
No crystal structure of NOV bound to AcrB is available, but the recent cryo-EM study of the MexAB−OprM tripartite pump from P. aeruginosa tentatively assigns a density to NOV in the DBP of MexB.49 Additionally, small molecule substrates such as minocycline and some previously characterized EPIs have been crystallized within the DBP of AcrB.28,29,50 Specifically, EPIs were found to bind AcrB in the hydrophobic trap. We therefore carried out molecular docking runs of 2 and its analogs to the DBP of AcrB (Figure 6). The best docking pose of compound 2 (Vina docking score of −11.9 kcal mol−1) is more favorable than the one calculated for AcrA. The ligand binds in the DBP with the cinnamoyl scaffold in the hydrophobic trap and the quinolinium heterocycle in the most hydrophilic portion of the pocket (Figure 6A). Two parallel displaced π−π-stacking interactions were observed, one involving the o-chloro-cinnamoyl moiety engaged by F178 and the other being the peptidyl quinolinium π-system with F628. No hydrogen bonds were observed, pointing to hydrophobic contacts as the main source of affinity. Indeed, the main residues contributing to binding are F610, V612, V139, V571, Y327, P326, and I277. The role of the chloro substituent at the ortho position of the phenyl ring is unclear upon first inspection, but it could be important for affecting directionality and bonding with F178.
Figure 6.
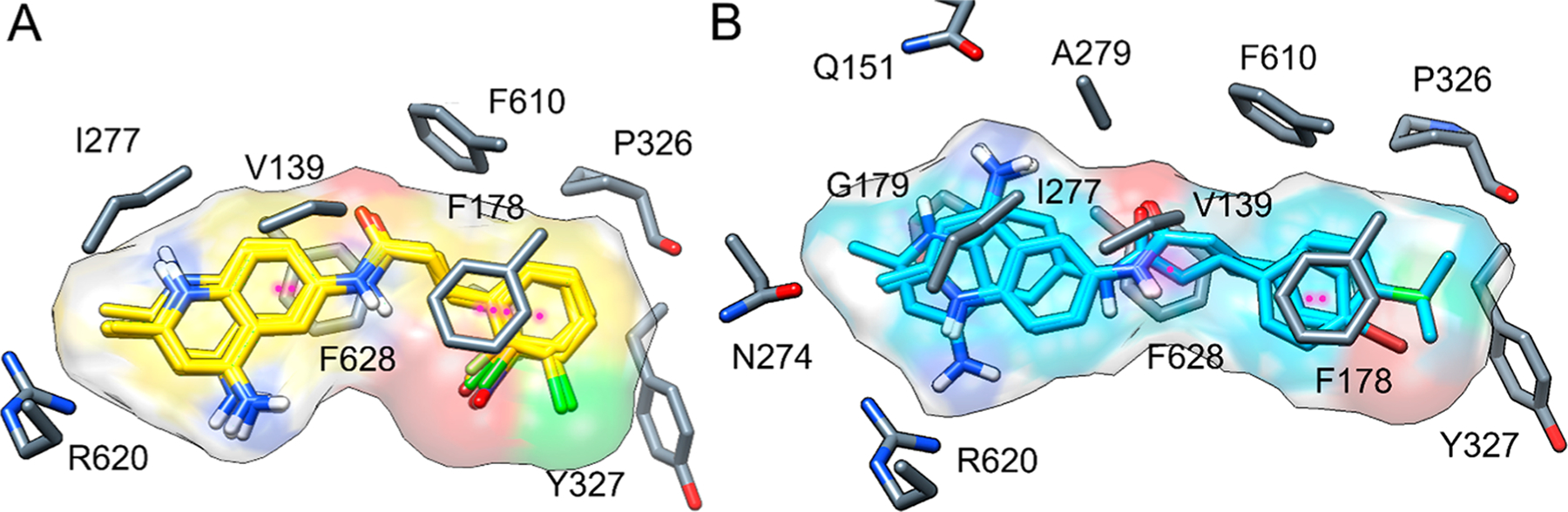
(A) Top docking pose and ligand surface of o-substituted cinnamoyl derivatives 6−9 and 14 at the DBP of AcrB; π−π-stacking interactions are shown as magenta dotted lines (ligand, yellow sticks; protein, gray sticks). (B) Top docking pose and ligand surface of p-substituted cinnamoyl derivatives 11−13 at the same site (ligand, cyan sticks; protein, gray sticks).
Cinnamoyl Derivatives.
The cinnamoyl derivatives display the same chemical interaction pattern (Figure 6A) with similar average docking scores (−12.1 ± 0.4 kcal mol−1, Table 4) than their parent compound 2. Consistent with the experimental data, these results indicate that all tested ligands can be effectively accommodated within the DBP of AcrB by exploiting two important chemical features: aromaticity and amphiphilicity. Both features contribute to the optimal fit within the protein cavity, dictating ligand orientation and defining the interaction pattern.
Upon visual inspection, p-substituted ligand poses differ subtly from o/m-analogues: Specifically, they assume more extended conformations than the latter, which instead are more bent and are all superposable (Figure 6B). Compound 14, characterized by a dichlorobenzene moiety, is a substrate of AcrB (Table 1), but in contrast to monosubstituted inhibitors 10 and 2, it lacks EPI activity in both bacterial growth-dependent and -independent assays. Both computational and experimental data suggest that the 2,3-dichloro substitution negatively affects the interaction with F178 by altering electrostatic and/or local steric effects. F178, in fact, is one of the most important phenylalanine residuess in the hydrophobic trap for binding both inhibitors and substrates, on the basis of X-ray experiments and previous computational studies.36,51,52
The o-nitro compound 9 constitutes a special case. Although retaining the above-mentioned interaction pattern, the nitro group provides polarity to the most hydrophobic portion of the ligand, such that amphiphilicity is reduced and the interaction with the hydrophobic trap is less favorable (−11.5 kcal mol−1). In agreement, 9 lacks EPI activity.
The preferred conformation of the three p-substituted cinnamoyl analogues within the DBP is linear and extended with the main substituent oriented toward Y327 and P326 (Figure 6B). These ligands share the same interaction pattern reported for the o-substituted analogues with some distinctive exceptions represented by Q151, G179, A279, and N274, and the latter interaction observed only in the docking pose of 12. The binding modes of 11 and 13 are superimposable with each other but not with 12. In 11 and 13, the anilinic amine of the quinolinium is oriented toward Q151, while in 12, it is oriented toward N274 on the opposite side. The brominated ligand binds less deeply in the DBP than the chlorinated and isopropyl derivatives, probably due to the greater size of the bromo substituent, which affects ligand directionality to some extent. Due to its positioning, the quinolinium group of 12 is too far from F628 to establish substantial parallel-displaced stacking. The interaction with F628 is preserved by the peptide bond π-system, but it is likely weaker in strength than when it is made with the quinolinium group. Consistent with this observation, the o-substituted derivatives show on average better docking scores than the corresponding p-substituted ones (Table 4).
Naphthyl Derivatives.
The docking scores for selected naphthyl derivatives are generally more favorable than cinnamoyl derivatives (Table 4). The introduction of the naphthyl ring isostere in place of the cinnamoyl moiety leads to a more favorable interaction within the DBP of AcrB. This difference is especially evident in the interaction pattern of 20 with the protein, in which the naphthalene is unsubstituted and therefore represents a good reference molecule for this series (Figure 7). Even in this case, π−π bonds appear to determine the interaction pattern observed, while amphiphilicity sets the molecular orientation. Differing from the cinnamoyl analogues, F628 further stabilizes the ligand in the pocket by means of two aromatic interactions (one parallel-displaced stacking and one sandwich stacking). The stacking interaction with F178 is preserved with the quinolinium ring interacting with F615 as well. However, F615 does not seem relevant for other compounds. The amino acids V139, G179, I277, A279, P326, Y327, V571, F610, and V612 are the most contacted residues, contributing primarily through hydrophobic interactions, even if, in some cases, Q151 and N274 are also involved.
Figure 7.
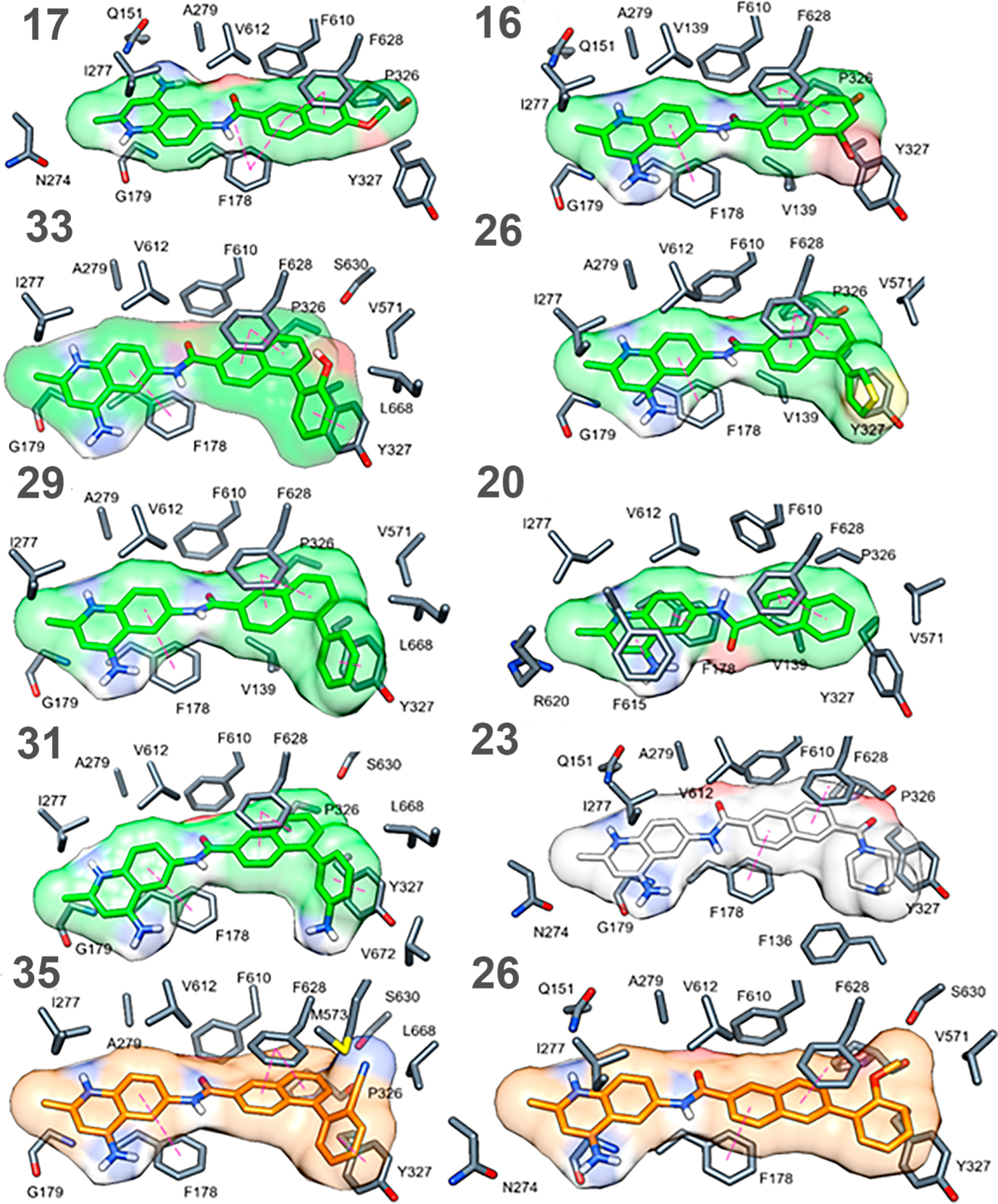
Docking poses and ligand surfaces of naphthyl AcrB binder inhibitors (green), nonbinders (light gray) and substrates (orange) in the DBP of AcrB; π−π-stacking interactions are reported as magenta dotted lines, and protein residues are depicted as gray sticks.
As illustrated in Figure 6, both inhibitors and substrates follow the binding trend described so far, but peculiarities can be detected, depending on functionalization of the naphthalene ring at position 5 or 6. Specifically, C5-substituted ligands feature an overall higher affinity than the unsubstituted compound 20 or the related C6-substituted complexes (−15.1 vs −13.2 kcal mol−1 according to the docking scores).
The computational data are consistent with the experimental results. Specifically, functionalization with a phenyl moiety at the 5-position in 29 allows for the formation of an additional π−π sandwich stacking with the Y327 phenolic side chain, preserved for all the related C5-substituted compounds but absent in the C6-subsituted ligand top poses. For the latter, in fact, the ligand is hindered and forced into its own extended orientation so that it cannot interact by the same mode. Moreover, large substituents could lead to unfavorable contacts compared to the less hindered ligand 17. Compound 35 is characterized by the presence of an acetonitrile group, which might give rise to unfavorable electrostatic interactions with M573. For the docking pose of substrate 26, stacking between the quinolinium heterocycle and F178 is lacking, which could partially explain its activity profile. Compound 23, which has a saturated piperazine amide substituent, is unable to engage in π-stacking interactions with Y327 and suffers from additional unfavorable steric clashes supporting the observed experimental data that the ligand does not bind to AcrB.
Potentiation of Antibacterial Activities in Other Gram-Negative Bacteria.
We previously showed that 2 can potentiate the activities of NOV and ERY in other enterobacteria such as Enterobacter cloacae and Klebsiella pneumoniae but not Pseudomonas aeruginosa.38 In agreement with these findings, most of the cinnamoyl derivatives with EPI activities in E. coli also potentiated the activities of NOV, ERY, or both in E. cloacea and K. pneumoniae (Table 5). Compounds 6, 7, and 8 also potentiated the activity of ciprofloxacin (CIP) but only against K. pneumoniae. In contrast, the naphthyl derivatives were notably less potent in these bacteria, likely due to their low permeation resulting from the synergistic action of active efflux and the OM barrier. Only 10 and 13 showed EPI activity in P. aeruginosa but only in combination with NOV and not with ERY or CIP (Table S3). The reasons for species- and antibiotic-specific differences in EPI activities are not immediately clear, but these could be the result of species-specific differences in the intracellular permeation of EPIs and antibiotics.
Table 5.
MIC and Potentiation Data for Selected EPIs in E. cloacae and K. pneumoniaed
|
E. cloacae ATCC13047 |
K. pneumoniae ATCC13883 |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| # | MICa | MPC4b (NOV) | Fold MICc (NOV) | MPC4b (ERY) | fold MICc (ERY) | MPC4b (CIP) | fold MICc (CIP) | MICa | MPC4b (NOV) | fold MICc (NOV) | MPC4b (CIP) | fold MICc (CIP) | MPC4b (CIP) | fold MICc (CIP) |
| NOV | 400 | NA | NA | NA | NA | NA | NA | 50 | NA | NA | NA | NA | NA | NA |
| ERY | 400 | NA | NA | NA | NA | NA | NA | 400 | NA | NA | NA | NA | NA | NA |
| CIP | 0.031 | NA | NA | NA | NA | NA | NA | 0.031 | NA | NA | NA | NA | NA | NA |
| 2 | >100 | 12.5 | 64 | 12.5 | 8 | >100 | 1 | >100 | 12.5 | 64 | >100 | 2 | >100 | 1 |
| 6 | >100 | 25 | 64 | 50 | 2 | >100 | 1 | >100 | >100 | 1 | >100 | 1 | 25 | 4 |
| 7 | 50 | 25 | 4 | >100 | 1 | >100 | 1 | 100 | 25 | 8 | 100 | 1 | 25 | 4 |
| 8 | >100 | 25 | 32 | 25 | 8 | >100 | 1 | >100 | 25 | 64 | 100 | 1 | 25 | 4 |
| 10 | >100 | 12.5 | 32 | 6.25 | 8 | >100 | 1 | 25 | 6.25 | NA | 50 | 1 | 25 | NA |
| 11 | >100 | 12.5 | 32 | >100 | 1 | >100 | 1 | 50 | 12.5 | 64 | 50 | 1 | 12.5 | NA |
| 13 | >100 | 50 | 1 | 50 | 1 | >100 | 1 | 100 | 25 | 16 | 100 | 1 | 25 | 8 |
| 15 | >100 | 100 | 2 | 100 | 1 | ND | ND | >100 | 100 | 1 | >100 | 1 | ND | ND |
| 16 | >100 | >100 | 1 | >100 | 1 | ND | ND | >100 | >100 | 1 | >100 | 1 | ND | ND |
| 21 | >100 | 25 | 1 | >100 | 1 | ND | ND | >100 | >100 | 1 | >100 | 1 | ND | ND |
| 28 | >100 | >100 | 1 | >100 | 1 | ND | ND | >100 | >100 | 1 | >100 | 1 | ND | ND |
| 33 | >100 | >100 | 1 | >100 | 1 | ND | ND | 100 | 12.5 | 64 | 50 | 1 | ND | ND |
| 34 | >100 | >100 | 1 | 25 | 64 | ND | ND | >100 | >100 | 1 | >100 | 1 | ND | ND |
| 36 | >100 | >100 | 1 | >100 | 1 | ND | ND | >100 | 50 | 1 | >100 | 1 | ND | ND |
MIC values (μM for test compounds, mg/L for antibiotics), determined as the average of two replicates.
Minimum concentration of test compounds required to reduce the MIC of novobiocin (NOV), erythromycin (ERY), or ciprofloxacin (CIP) by 4-fold measured in the indicated strains.
The ratio of MIC values for novobiocin, erythromycin, and ciprofloxacin in the absence and presence of 25 μM of test compounds.
ND = no data; NA = not applicable.
DISCUSSION
We have prepared a series of cinnamoyl- and naphthyl-derived amides of the 4,6-diamino-2-methylquinoline scaffold as potential EPIs. The cinnamoyl analogs profiled very similar to the original hit. Many of them were shown to bind strongly to AcrA, potentiate NOV and ERY in E. coli, and inhibit efflux in a fluorescent probe accumulation assay. A number of these analogs also exhibited strong antibacterial activity in addition to efflux inhibition and avoided TolC-dependent efflux. Unfortunately, they also demonstrated cytotoxicity that limits their utility. The replacement of the cinnamoyl group with a set of corresponding naphthyl-based analogs led to compounds with different profiles. In general, these analogs did not bind to AcrA but did potentiate NOV. However, most of these analogs did not potentiate ERY in E. coli. They appear to permeate the OM of E. coli and show antibacterial activity but are likely substrates of the pump, which limits their antibiotic effects. The naphthyl analogs did show lower cytotoxicity, typically 2- to 3-fold relative to the cinnamoyl compounds.
NOV and ERY differ in their interactions with AcrA and AcrB and in their propensity to be expelled by the AcrAB−TolC pump. The antibacterial activity of ERY is strongly enhanced by hyperporination of the OM, despite fully active efflux (Table 1).42 In contrast, the activity of NOV is almost exclusively affected by AcrAB−TolC. Our previous studies also showed that NOV but not ERY binds to both AcrA and AcrB in the SPR assays.38,48 The results of this study further demonstrate how specific chemical modifications govern the effect of EPIs on antibacterial activities of these antibiotics. Most of the analyzed compounds are likely to interact with both AcrA and AcrB, although the naphthyl derivatives have higher affinities in docking to the DBP of AcrB. In the crystal structure of the ERY-bound AcrB, this large antibiotic is located in a proximal multisite drug-binding pocket of AcrB (known as the proximal binding pocket, PBP) that extends farther into the interface with DBP.53 Hence, the PBP is believed to be the primary recognition and binding site of high-molecular mass compounds such as ERY (∼735 Da). Interestingly, in the assembled AcrAB−TolC complex (Figure 4A), site III of AcrA, which is the preferred binding site of 2 and its analogs, is adjacent to and faces the PBP of AcrB. Hence, the potentiation of ERY by cinnamoyl derivatives could be due to the direct interference of AcrA-bound compounds with the access of ERY into the PBP. Note that, according to a previous docking study,54 NOV appeared to bind preferentially to the region of the DBP, including some of the phenylalanine residues lining the hydrophobic trap (F178, F610, F615). This binding mode is very similar to that tentatively assigned to NOV in MexB where it interacts with residues F178, F615, and F617.49 Our results therefore suggest that the observed antibacterial potentiation of NOV and Nile Red efflux inhibition activities are enabled by the interactions of the compounds with the hydrophobic trap within the DBP of AcrB.
Previous mutagenesis studies on AcrB showed that Y327A substitution decreased the MICs (i.e., reduced efflux activity) of NOV but not ERY, whereas F178A and F628A substitutions in AcrB decreased the MICs of both antibiotics.55–57 In agreement, our molecular docking analyses further support the role of F178 and F628 for the binding of both substrate and inhibitor compounds to AcrB because these residues are involved in well-ordered π-stacking interactions of differing stability. We also identified Y327 as a crucial residue for both recognition and activity of the ligands, especially of naphthyl derivatives, and other contact residues (polar and apolar) that contribute to the general interaction pattern observed.
The results presented here expand on our previous efforts to develop novel EPIs targeting the MFP AcrA of the AcrAB−TolC efflux pump of E. coli. By following up on a novel hit from the NCI collection, we have identified structure−activity relationships for compounds that bind to AcrA and prevent efflux of both NOV and ERY. Additional modifications led to compounds with stronger affinities to AcrB and thus changed the mechanism of inhibition relative to our previously reported AcrA inhibitors. Future efforts will be directed toward further exploring the mechanism of potentiation of NOV for the naphthyl compounds and expanding the SAR around this series to optimize binding to AcrA.
METHODS
MTS Cytotoxicity Assay (CC50).
A549 cells (human alveolar basal epithelial cells, 1.0 × 104 cells/well) were seeded in 96-well plates and incubated in Dulbecco’s Modified Eagle Medium with 10% fetal bovine serum plus 1% penicillin/streptomycin solution, 1% nonessential amino acids, and 1% glutamine. The compounds were diluted in the medium to the indicated concentrations to a final concentration of 1% DMSO and added to the cells 48 h after plating, with each concentration tested in triplicate. Soluble MTS reagent [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, Promega] was added 72 h after incubation; the cultures were incubated for 90 min, and absorbance was read at 490 nm. The CC50 was calculated as the concentration of the inhibitor required to reduce cell viability 50% relative to untreated cells. The data are plotted as log[inhibitor] versus response and fit to a variable slope model using Graph Pad Prism (v6; GraphPad Software, Inc.).58
Microbiological Assay.
E. coli BW25113 (WT, Δ(araD-araB)567 Δ(rhaD-rhaB)568 ΔlacZ4787 (::rrnB3) hsdR514 rph-1), and ΔtolC (BW25113 ΔtolC ygiBC) strains of E. coli and their hyperporinated WT-pore (BW25113 attTn7::mini-Tn7T Kmr araC ParaBAD fhuAΔC/Δ4L) and ΔtolC-pore (ΔtolC ygiBC attTn7::mini-Tn7T Kmr araC ParaBAD fhuAΔC/Δ4L) variants used in this study are derivatives of BW 25113 and GD102, respectively.42 The E. coli BL21(DE3) strain was used for overexpression and purification of AcrA,59 and the AG100AX strain was used for overexpression and purification of AcrB.48 MICs were analyzed using a 2-fold broth dilution method.60 For the checkerboard assay, an antibiotic and a test compound were serially diluted into 96-well plates as described previously.61
Surface Plasmon Resonance.
For the amino coupling of AcrA and AcrB, CM5 chip surfaces were activated with 0.05 M N-hydroxysuccinimide and 0 . 2 M N-ethyl-N-(3-diethylaminopropyl)carbodiimide (BIAcore). AcrA and AcrB were injected over surfaces immediately after activation. After immobilization, the excess of reactive groups was blocked by injecting 0.5 M ethanolamine HCl (pH 8.0). The immobilization and subsequent binding experiments were conducted in running buffer containing 20 mM HEPES-KOH (pH 7.0), 150 mM NaCl, and 0.03% DDM supplemented with 5% DMSO. The CM5 chip contains four chambers, whereas the first (control surface) was activated and processed in the same way but the protein was omitted during the immobilization step. The second and third chambers contained the immobilized AcrA and AcrB (ligand). The immobilized densities of both proteins (ligand) were 7167 and 7072 response units (RU), respectively. For kinetic modeling, we considered only the simplest models that would be compatible with one or two distinct events during both inhibitor binding and dissociation. These four models are (i) a simple 1:1 binding model; (ii) a heterogeneous ligand (HL), in which different protein populations on the on-chip surface have different kinetic properties; (iii) two-state reaction or ligand-induced conformational change, wherein conformational change occurs on the same time scale as ligand binding; (iv) bivalent analyte, where multiple analytes bind independently at nonidentical sites.62,63
Fluorescence Accumulation Assay.
An accumulation assay was performed in a temperature-controlled microplate reader (Tecan Spark 10M multimode microplate reader equipped with a sample injector) in fluorescence mode.14 Cells from frozen stocks were inoculated into LB medium and incubated for 16 h at 37 °C. Cells were then subcultured into a fresh 30 mL volume of LB medium and grown at 37 °C to an optical density at 600 nm (OD600) of 0.3. The cells were then induced with 0.1% arabinose and grown to an OD600 of 1.0, collected by centrifugation at 4000 rpm for 20 min at room temperature, and washed in 25 mL of HEPES-KOH buffer (50 mM; pH 7.0) containing 1 mM magnesium sulfate and 0.4 mM glucose (HMG buffer). The cells in HMG buffer were adjusted to an OD600 of ∼1.0 and kept at room temperature during the experiment. Fluorescence intensities of Nile Red accumulated in the cells were plotted as a function of time and normalized to the emission of the dye before the cells were added. The data were imported into MatLab (MathWorks, Inc.) to be fitted to a simple exponential equation in the form of F = A1 + A2 [1 − exp(kt)], where A1 represents the amplitude of the initial fast uptake of a dye, and A2 and kt are the amplitude and rate, respectively, that are associated with the subsequent slower uptake.14
Ensemble Docking to AcrA and AcrB.
Molecular docking calculations targeting both AcrA and AcrB were performed using AutoDock Vina,46 which implements a stochastic global optimization approach. The program was used with default settings but for the exhaustiveness parameter (giving a measure of the exhaustiveness of the local search), which was set to 1024 (default 8). Ligand protonation states were calculated using Marvin calculator plugins,64 and compounds were considered flexible during docking (number of rotatable bonds in the range of 2 to 5). Protein and ligand input files in PDBQT format were prepared with AutoDock Tools.65 Protein flexibility was considered indirectly using the ensemble of conformations.66 For AcrA, we used 29 previously identified conformations extracted from a 50 ns molecular dynamics simulation of a model of the full-length AcrA monomer.44 In the case of AcrB, we employed 10 available X-ray conformations (PDB_IDs: 2DHH, 2GIF, 2J8S, 3W9H, 4DX5, 4DX7, 4U8V, 4U8Y, 4U95, 4U96).67 Guided docking runs were performed in both cases. For AcrA, we adopted a 25 Å × 25 Å × 25 Å box centered at four different sites defined on the basis of the center of mass of selected residues: site I (E67), site II (K241), site III (I343, I252), and site IV (F81, F254).68 For AcrB, we used a 40 Å × 40 Å × 40 Å box centered in the center of mass of the DBP. The top 10 docking poses were retained for each protein−ligand pair, and the top ones were selected for further analysis. Molecular graphics were rendered using UCSF Chimera69 or PyMOL 2.4 (https://pymol.org).
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the National Institute of Health, AI052293 (H.I.Z.) and AI136799 (H.I.Z., G.M., E.M., A.V.V., P.R., V.V.R., and J.K.W.), who supported this work. G.M., E.M., A.V.V., and P.R. acknowledge technical support by Giovanni Serra and Andrea Bosin (University of Cagliari). C.J.C. was supported by a National Science Foundation Graduate Research Fellowship under Grant No. 2017219379. This research used resources of the Compute and Data Environment for Science (CADES) at Oak Ridge National Laboratory, which is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC05–00OR22725. We also gratefully acknowledge Dr. Fahu He and the St. Louis University NMR facility for acquiring 13C samples on the 700 MHz NMR.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsinfecdis.1c00100.
C(6)-Aryl substituted naphthyl analogs, MIC and potentiation data, fluorescence vs time plots, activities of compounds, abbreviation list, and chemistry procedures with analytical data for all new synthetic compounds including 1H NMR, 13C NMR, and mass spectra data (PDF)
The authors declare no competing financial interest.
Contributor Information
Napoleon D’Cunha, Department of Pharmacology and Physiology, Saint Louis University School of Medicine, St. Louis, Missouri 63110, United States.
Mohammad Moniruzzaman, Department of Chemistry and Biochemistry, University of Oklahoma, Norman, Oklahoma 73072, United States.
Keith Haynes, Department of Pharmacology and Physiology, Saint Louis University School of Medicine, St. Louis, Missouri 63110, United States.
Giuliano Malloci, Department of Physics, University of Cagliari, Cagliari 09042, Italy.
Connor J. Cooper, Graduate School of Genome Science and Technology, University of Tennessee, Knoxville, Tennessee 37996, United States; Biosciences Division, Oak Ridge National Laboratory, Oak Ridge, Tennessee 37831, United States.
Enrico Margiotta, Department of Physics, University of Cagliari, Cagliari 09042, Italy.
Attilio V. Vargiu, Department of Physics, University of Cagliari, Cagliari 09042, Italy.
Muhammad R. Uddin, Department of Chemistry and Biochemistry, University of Oklahoma, Norman, Oklahoma 73072, United States
Inga V. Leus, Department of Chemistry and Biochemistry, University of Oklahoma, Norman, Oklahoma 73072, United States
Feng Cao, John Cochran Division, Department of Veteran Affairs Medical Center, St. Louis, Missouri 63106, United States.
Jerry M. Parks, Biosciences Division, Oak Ridge National Laboratory, Oak Ridge, Tennessee 37831, United States.
Valentin V. Rybenkov, Department of Chemistry and Biochemistry, University of Oklahoma, Norman, Oklahoma 73072, United States.
Paolo Ruggerone, Department of Physics, University of Cagliari, Cagliari 09042, Italy.
Helen I. Zgurskaya, Department of Chemistry and Biochemistry, University of Oklahoma, Norman, Oklahoma 73072, United States.
John K. Walker, Department of Pharmacology and Physiology, Saint Louis University School of Medicine, St. Louis, Missouri 63110, United States.
REFERENCES
- (1).Serra-Burriel M; Keys M; Campillo-Artero C; Agodi A; Barchitta M; Gikas A; Palos C; Lopez-Casasnovas G Impact of multi-drug resistant bacteria on economic and clinical outcomes of healthcare-associated infections in adults: Systematic review and meta-analysis. PLoS One 2020, 15 (1), No. e0227139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).CDC Antibiotic Resistance Threats in the United States, 2019; U.S. Department of Health and Human Services, CDC: Atlanta, GA, 2019. [Google Scholar]
- (3).Walsh TR; Toleman MA The emergence of pan-resistant Gram-negative pathogens merits a rapid global political response. J. Antimicrob. Chemother 2012, 67, 1–3. [DOI] [PubMed] [Google Scholar]
- (4).Blazquez J; Oliver A; Gomez-Gomez J-M Mutation and evolution of antibiotic resistance: Antibiotics as promoters of antibiotic resistance? Curr. Drug Targets 2002, 3, 345–349. [DOI] [PubMed] [Google Scholar]
- (5).CDC Antibiotic Resistance Threats in the United States, 2013; U.S. Department of Health and Human Services, CDC: Atlanta, GA, 2013. [Google Scholar]
- (6).Demain AL; Spizek J The antibiotic crisis. In Antimicrobial drug discovery: emerging strategies; CABI, 2012; pp 26–43. [Google Scholar]
- (7).Livermore DM The need for new antibiotics. Clin. Microbiol. Infect 2004, 10 (Suppl 4), 1–9. [DOI] [PubMed] [Google Scholar]
- (8).Reck F; Jansen JM; Moser HE Challenges of antibacterial drug discovery. ARKIVOC (Gainesville, FL, U. S.) 2020, 2019, 227–244. [Google Scholar]
- (9).McKenna M The antibiotic paradox: why companies can’t afford to create life-saving drugs. Nature 2020, 584, 338–341. [DOI] [PubMed] [Google Scholar]
- (10).Isabella VM; Campbell AJ; Manchester J; Sylvester M; Nayar AS; Ferguson KE; Tommasi R; Miller AA Toward the Rational Design of Carbapenem Uptake in Pseudomonas aeruginosa. Chem. Biol 2015, 22, 535–547. [DOI] [PubMed] [Google Scholar]
- (11).Payne DJ; Gwynn MN; Holmes DJ; Pompliano DL Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discovery 2007, 6, 29–40. [DOI] [PubMed] [Google Scholar]
- (12).Huwaitat R; McCloskey AP; Gilmore BF; Laverty G Potential strategies for the eradication of multidrug-resistant Gram-negative bacterial infections. Future Microbiol 2016, 11, 955–972. [DOI] [PubMed] [Google Scholar]
- (13).De Kievit TR; Parkins MD; Gillis RJ; Srikumar R; Ceri H; Poole K; Iglewski BH; Storey DG Multidrug efflux pumps: expression patterns and contribution to antibiotic resistance in Pseudomonas aeruginosa biofilms. Antimicrob. Agents Chemother 2001, 45, 1761–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Westfall DA; Krishnamoorthy G; Wolloscheck D; Sarkar R; Zgurskaya HI; Rybenkov VV Bifurcation kinetics of drug uptake by Gram-negative bacteria. PLoS One 2017, 12, No. e0184671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Saha P; Sikdar S; Krishnamoorthy G; Zgurskaya HI; Rybenkov VV Drug Permeation against Efflux by Two Transporters. ACS Infect. Dis 2020, 6, 747–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Krishnamoorthy G; Leus IV; Weeks JW; Wolloscheck D; Rybenkov VV; Zgurskaya HI Synergy between active efflux and outer membrane diffusion defines rules of antibiotic permeation into Gram-negative bacteria. mBio 2017, 8, e01172171–e011721716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Bader MW; Navarre WW; Shiau W; Nikaido H; Frye JG; McClelland M; Fang FC; Miller SI Regulation of Salmonella typhimurium virulence gene expression by cationic antimicrobial peptides. Mol. Microbiol 2003, 50, 219–230. [DOI] [PubMed] [Google Scholar]
- (18).Brown DG; May-Dracka TL; Gagnon MM; Tommasi R Trends and Exceptions of Physical Properties on Antibacterial Activity for Gram-Positive and Gram-Negative Pathogens. J. Med. Chem 2014, 57, 10144–10161. [DOI] [PubMed] [Google Scholar]
- (19).O’Shea R; Moser HE Physicochemical Properties of Antibacterial Compounds: Implications for Drug Discovery. J. Med. Chem 2008, 51, 2871–2879. [DOI] [PubMed] [Google Scholar]
- (20).Acosta-Gutierrez S; Ferrara L; Pathania M; Masi M; Wang J; Bodrenko I; Zahn M; Winterhalter M; Stavenger RA; Pages J-M; Naismith JH; van den Berg B; Page MGP; Ceccarelli M Getting Drugs into Gram-Negative Bacteria: Rational Rules for Permeation through General Porins. ACS Infect. Dis 2018, 4, 1487–1498. [DOI] [PubMed] [Google Scholar]
- (21).Levy SB; Marshall B Antibacterial resistance worldwide: causes, challenges and responses. Nat. Med 2004, 10, S122–S129. [DOI] [PubMed] [Google Scholar]
- (22).Li X-Z; Plésiat P; Nikaido H The Challenge of Efflux-Mediated Antibiotic Resistance in Gram-Negative Bacteria. Clin. Microbiol. Rev 2015, 28, 337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Ruggerone P; Murakami S; Pos KM; Vargiu AV RND Efflux Pumps: Structural Information Translated into Function and Inhibition Mechanisms. Curr. Top. Med. Chem 2013, 13, 3079–3100. [DOI] [PubMed] [Google Scholar]
- (24).Krishnamoorthy G; Tikhonova EB; Dhamdhere G; Zgurskaya HI On the role of TolC in multidrug efflux: the function and assembly of AcrAB–TolC tolerate significant depletion of intracellular TolC protein. Mol. Microbiol 2013, 87, 982–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Jeong H; Kim J-S; Song S; Shigematsu H; Yokoyama T; Hyun J; Ha N-C Pseudoatomic Structure of the Tripartite Multidrug Efflux Pump AcrAB–TolC Reveals the Intermeshing Cogwheel-like Interaction between AcrA and TolC. Structure 2016, 24, 272–276. [DOI] [PubMed] [Google Scholar]
- (26).Murakami S; Nakashima R; Yamashita E; Matsumoto T; Yamaguchi A Crystal structures of a multidrug transporter reveal a functionally rotating mechanism. Nature 2006, 443, 173–179. [DOI] [PubMed] [Google Scholar]
- (27).Seeger MA; Schiefner A; Eicher T; Verrey F; Diederichs K; Pos KM Structural Asymmetry of AcrB Trimer Suggests a Peristaltic Pump Mechanism. Science 2006, 313, 1295–1298. [DOI] [PubMed] [Google Scholar]
- (28).Nakashima R; Sakurai K; Yamasaki S; Hayashi K; Nagata C; Hoshino K; Onodera Y; Nishino K; Yamaguchi A Structural basis for the inhibition of bacterial multidrug exporters. Nature 2013, 500, 102–106. [DOI] [PubMed] [Google Scholar]
- (29).Sjuts H; Vargiu AV; Kwasny SM; Nguyen ST; Kim H-S; Ding X; Ornik AR; Ruggerone P; Bowlin TL; Nikaido H; Pos KM; Opperman TJ Molecular basis for inhibition of AcrB multidrug efflux pump by novel and powerful pyranopyridine derivitives. Proc. Natl. Acad. Sci. U. S. A 2016, 113, 3509–3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Thorarensen A; Presley-Bodnar AL; Marotti KR; Boyle TP; Heckaman CL; Bohanon MJ; Tomich PK; Zurenko GE; Sweeney MT; Yagi B. H 3-Arylpiperidine as Potentiators of Existing Antibacterial Agents. Bioorg. Med. Chem. Lett 2001, 11, 1903–1906. [DOI] [PubMed] [Google Scholar]
- (31).Bohnert JA; Kern WV Selected Arylpiperazines Are Capable of Reversing Multidrug Resistance in Escherichia coli Overexpressing RND Efflux Pumps. Antimicrob. Agents Chemother 2005, 49, 849–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Kinana AD; Vargiu AV; May T; Nikaido H Aminoacyl β-naphthylamides as substrates and modulators of AcrB multidrug efflux pump. Proc. Natl. Acad. Sci. U. S. A 2016, 113, 1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Nguyen ST; Kwasny SM; Ding X; Cardinale SC; McCarthy CT; Kim H-S; Nikaido H; Peet NP; Williams JD; Bowlin TL; Opperman TJ Structure–activity relationships of a novel pyranopyridine series of Gram-negative bacterial efflux pump inhibitors. Bioorg. Med. Chem 2015, 23, 2024–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Renau TE; Léger R; Filonova L; Flamme EM; Wang M; Yen R; Madsen D; Griffith D; Chamberland S; Dudley MN; Lee VJ; Lomovskaya O; Watkins WJ; Ohta T; Nakayama K; Ishida Y Conformationally-restricted analogues of efflux pump inhibitors that potentiate the activity of levofloxacin in Pseudomonas aeruginosa. Bioorg. Med. Chem. Lett 2003, 13, 2755–2758. [DOI] [PubMed] [Google Scholar]
- (35).Yoshida K.-i.; Nakayama K; Ohtsuka M; Kuru N; Yokomizo Y; Sakamoto A; Takemura M; Hoshino K; Kanda H; Nitanai H; Namba K; Yoshida K; Imamura Y; Zhang JZ; Lee VJ; Watkins WJ MexAB-OprM specific efflux pump inhibitors in Pseudomonas aeruginosa. Part 7: Highly soluble and in vivo active quaternary ammonium analogue D13–9001, a potential preclinical candidate. Bioorg. Med. Chem 2007, 15, 7087–7097. [DOI] [PubMed] [Google Scholar]
- (36).Aron Z; Opperman TJ The hydrophobic trap—the Achilles heel of RND efflux pumps. Res. Microbiol 2018, 169, 393–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Blair JM; Bavro VN; Ricci V; Modi N; Cacciotto P; Kleinekathfer U; Ruggerone P; Vargiu AV; Baylay AJ; Smith HE; Brandon Y; Galloway D; Piddock LJ AcrB drug-binding pocket substitution confers clinically relevant resistance and altered substrate specificity. Proc. Natl. Acad. Sci. U. S. A 2015, 112, 3511–3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Abdali N; Parks JM; Haynes KM; Chaney JL; Green AT; Wolloscheck D; Walker JK; Rybenkov VV; Baudry J; Smith JC; Zgurskaya HI Reviving Antibiotics: Efflux Pump Inhibitors That Interact with AcrA, a Membrane Fusion Protein of the AcrAB––TolC Multidrug Efflux Pump. ACS Infect. Dis 2017, 3, 89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Haynes KM; Abdali N; Jhawar V; Zgurskaya HI; Parks JM; Green AT; Baudry J; Rybenkov VV; Smith JC; Walker JK Identification and Structure–Activity Relationships of Novel Compounds that Potentiate the Activities of Antibiotics in Escherichia coli. J. Med. Chem 2017, 60, 6205–6219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Hagmann WK; Springer MS Substituted Aminoquinolines as Modulators of Chemokine Receptor Activity. WO 9827815, February 7, 1998. [Google Scholar]
- (41).Zhao H; Petrushenko ZM; Walker JK; Baudry J; Zgurskaya HI; Rybenkov VV Small Molecule Condensin Inhibitors. ACS Infect. Dis 2018, 4, 1737–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Krishnamoorthy G; Wolloscheck D; Weeks JW; Croft C; Rybenkov VV; Zgurskaya HI Breaking the permeability barrier of Escherichia coli by controlled hyperporination of the outer membrane. Antimicrob. Agents Chemother 2016, 60, 7372–7381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Walker JK; Haynes K; Abdali N; Rybenkov VV; Zgurskaya HI Fluorescent probes for drug permeability in Gram-negative bacteria. US 20200018705 A1, 2020. [Google Scholar]
- (44).Darzynkiewicz ZM; Green AT; Abdali N; Hazel A; Fulton RL; Kimball J; Gryczynski Z; Gumbart JC; Parks JM; Smith JC; Zgurskaya HI Identification of Binding Sites for Efflux Pump Inhibitors of the AcrAB–TolC Component AcrA. Biophys. J 2019, 116, 648–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Hazel AJ; Abdali N; Leus IV; Parks JM; Smith JC; Zgurskaya HI; Gumbart JC Conformational Dynamics of AcrA Govern Multidrug Efflux Pump Assembly. ACS Infect. Dis 2019, 5, 1926–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Trott O; Olson AJ AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem 2010, 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Mowla R; Wang Y; Ma S; Venter H Kinetic analysis of the inhibition of the drug efflux protein AcrB using surface plasmon resonance. Biochim. Biophys. Acta, Biomembr 2018, 1860, 878–886. [DOI] [PubMed] [Google Scholar]
- (48).Tikhonova EB; Yamada Y; Zgurskaya HI Sequential mechanism of assembly of multidrug efflux pump AcrAB–TolC. Chem. Biol 2011, 18, 454–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Tsutsumi K; Yonehara R; Ishizaka-Ikeda E; Miyazaki N; Maeda S; Iwasaki K; Nakagawa A; Yamashita E Structures of the wild-type MexAB-OprM tripartite pump reveal its complex formation and drug efflux mechanism. Nat. Commun 2019, 10, 1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Zwama M; Yamasaki S; Nakashima R; Sakurai K; Nishino K; Yamaguchi A Multiple entry pathways within the efflux transporter AcrB contribute to multidrug recognition. Nat. Commun 2018, 9, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Vargiu AV; Nikaido H Multidrug binding properties of the AcrB efflux pump characterized by molecular dynamics simulations. Proc. Natl. Acad. Sci. U. S. A 2012, 109, 20637–20642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Vargiu AV; Ramaswamy VK; Malloci G; Malvacio I; Atzori A; Ruggerone P Computer simulations of the activity of RND efflux pumps. Res. Microbiol 2018, 169, 384–392. [DOI] [PubMed] [Google Scholar]
- (53).Ababou A; Koronakis V Structures of gate loop variants of the acrB drug efflux pump bound by erythromycin substrate. PLoS One 2016, 11, No. e0159154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Takatsuka Y; Chen C; Nikaido H Mechanism of recognition of compounds of diverse structures by the multidrug efflux pump AcrB of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A 2010, 107 (15), 6559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Yao X-Q; Kimura N; Murakami S; Takada S Drug Uptake Pathways of Multidrug Transporter AcrB Studied by Molecular Simulations and Site-Directed Mutagenesis Experiments. J. Am. Chem. Soc 2013, 135, 7474–7485. [DOI] [PubMed] [Google Scholar]
- (56).Nakashima R; Sakurai K; Yamasaki S; Nishino K; Yamaguchi A Structures of the multidrug exporter AcrB reveal a proximal multisite drug-binding pocket. Nature 2011, 480, 565–569. [DOI] [PubMed] [Google Scholar]
- (57).Bohnert JA; Schuster S; Seeger MA; Faehnrich E; Pos KM; Kern WV Site-directed mutagenesis reveals putative substrate binding residues in the Escherichia coli RND efflux pump AcrB. J. Bacteriol 2008, 190, 8225–8229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Donlin MJ; Zunica A; Lipnicky A; Garimallaprabhakaran AK; Berkowitz AJ; Grigoryan A; Meyers MJ; Tavis JE; Murelli RP Troponoids can inhibit growth of the human fungal pathogen Cryptococcus neoformans. Antimicrob. Agents Chemother 2017, 61, e02574–16/1–e02574–16/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Zgurskaya HI; Nikaido H AcrA is a highly asymmetric protein capable of spanning the periplasm1. J. Mol. Biol 1999, 285, 409–420. [DOI] [PubMed] [Google Scholar]
- (60).Tikhonova EB; Wang Q; Zgurskaya HI Chimeric analysis of the multicomponent multidrug efflux transporters from Gram-negative bacteria. J. Bacteriol 2002, 184, 6499–6507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Lomovskaya O; Warren MS; Lee A; Galazzo J; Fronko R; Lee M; Blais J; Cho D; Chamberland S; Renau T; Leger R; Hecker S; Watkins W; Hoshino K; Ishida H; Lee VJ Identification and characterization of inhibitors of multidrug resistance efflux pumps in Pseudomonas aeruginosa: novel agents for combination therapy. Antimicrob. Agents Chemother 2001, 45, 105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Li W; Stevens CM; Pandya AN; Darzynkiewicz Z; Bhattarai P; Tong W; Gonzalez-Juarrero M; North EJ; Zgurskaya HI; Jackson M Direct inhibition of MmpL3 by novel antitubercular compounds. ACS Infect. Dis 2019, 5, 1001–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Tikhonova EB; Dastidar V; Rybenkov VV; Zgurskaya HI Kinetic control of TolC recruitment by multidrug efflux complexes. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 16416–16421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Marvin 17.9.0; ChemAxon: Cambridge, MA, 2017. [Google Scholar]
- (65).Morris GM; Huey R; Lindstrom W; Sanner MF; Belew RK; Goodsell DS; Olson AJ AutoDock and AutoDockTools: Automated docking with selective receptor flexibility. J. Comput. Chem 2009, 30, 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Ellingson SR; Miao Y; Baudry J; Smith JC Multi-Conformer Ensemble Docking to Difficult Protein Targets. J. Phys. Chem. B 2015, 119, 1026–1034. [DOI] [PubMed] [Google Scholar]
- (67).Atzori A; Malloci G; Prajapati JD; Basciu A; Bosin A; Kleinekathofer U; Dreier J; Vargiu AV; Ruggerone P Molecular Interactions of Cephalosporins with the Deep Binding Pocket of the RND Transporter AcrB. J. Phys. Chem. B 2019, 123, 4625–4635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Green AT; Moniruzzaman M; Cooper CJ; Walker JK; Smith JC; Parks JM; Zgurskaya HI Discovery of multidrug efflux pump inhibitors with a novel chemical scaffold. Biochim. Biophys. Acta, Gen. Subj 2020, 1864, 129546–129554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Pettersen EF; Goddard TD; Huang CC; Couch GS; Greenblatt DM; Meng EC; Ferrin TE UCSF Chimera-A visualization system for exploratory research and analysis. J. Comput. Chem 2004, 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- (70).Ge Q; Yamada Y; Zgurskaya H The C-terminal domain of AcrA is essential for the assembly and function of the multidrug efflux pump AcrAB–TolC. J. Bacteriol 2009, 191, 4365–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Wang Z; Fan G; Hryc CF; Blaza JN; Serysheva; Schmid MF; Chiu W; Luisi BF; Du D An allosteric transport mechanism for the AcrAB–TolC multidrug efflux pump. eLife 2017, 6, No. e24905. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.