

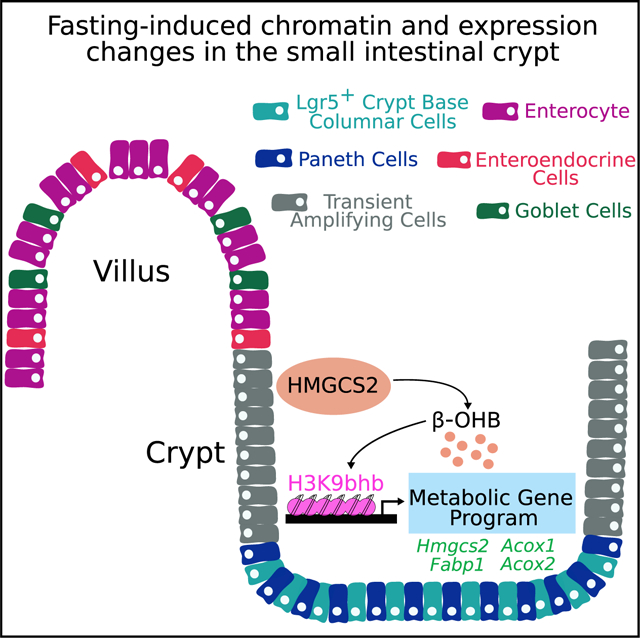
SUMMARY
β-hydroxybutyrate (β-OHB) is an essential metabolic energy source during fasting and functions as a chromatin regulator by lysine β-hydroxybutyrylation (Kbhb) modification of the core histones H3 and H4. We report that Kbhb on histone H3 (H3K9bhb) is enriched at proximal promoters of critical gene subsets associated with lipolytic and ketogenic metabolic pathways in small intestine (SI) crypts during fasting. Similar Kbhb enrichment is observed in Lgr5+ stem cell-enriched epithelial spheroids treated with β-OHB in vitro. Combinatorial chromatin state analysis reveals that H3K9bhb is associated with active chromatin states and that fasting enriches for an H3K9bhb-H3K27ac signature at active metabolic gene promoters and distal enhancer elements. Intestinal knockout of Hmgcs2 results in marked loss of H3K9bhb-associated loci, suggesting that local production of β-OHB is responsible for chromatin reprogramming within the SI crypt. We conclude that modulation of H3K9bhb in SI crypts is a key gene regulatory event in response to fasting.
Graphical Abstract
In brief
Terranova et al. demonstrate that fasting induces production of HMGCS2 and β-hydroxybutyrate in small intestine (SI) crypt cells. This causes enrichment of H3K9bhb within regulatory regions of critical metabolic genes in crypt epithelial cells. Loss of intestinal Hmgcs2 impairs H3K9bhb enrichment and affects expression of H3K9bhb-associated metabolic gene programs.
INTRODUCTION
In addition to providing energy to tissues when glucose is scarce, β-hydroxybutyrate (β-OHB) alters the epigenome by signature-specific, post-translational modification of histones and by inhibiting class I histone deacetylases (Shimazu et al., 2013; Xie et al., 2016). The epigenome is defined by chromatin modifications, including DNA methylation and histone post-translational modifications (PTMs), which collectively form a dynamic code that correlates with cell-dependent and temporal changes in gene expression in response to stimuli. The combinatorial patterns of these modifications are represented as “chromatin states” (Jenuwein and Allis, 2001) and can be used for functional annotation of the epigenome. Specific histone or DNA modifications are associated with and regulate transcriptional activation or repression of associated promoters and enhancers to influence cellular behavior (Ernst and Kellis, 2010, 2012; Lee et al., 2013; Strahl and Allis, 2000). For example, the presence of histone H3 lysine trimethylation (H3K27me3) is associated with a repressive environment, whereas H3K4me3 is generally associated with transcriptionally active promoters (Roh et al., 2006). H3K4me1- and H3K27ac-modified nucleosomes are highly enriched at enhancer elements (Birney et al., 2007; Creyghton et al., 2010; Heintzman et al., 2009), whereas H3K79me2 or H3K36me3 coincides with transcribed regions (Guenther et al., 2007). Finally, poised promoters and enhancers are frequently marked by co-occupancy of H3K27me3 with H3K4me3 or H3K4me1, respectively (Bernstein et al., 2006; Rada-Iglesias et al., 2011). Recently, lysine β-hydroxybutyrylation (Kbhb) has been identified as a new type of histone mark that is dramatically induced in the livers of fasted mice (Xie et al., 2016). The liver is the major source of β-OHB during fasting, and marked upregulation of genes involved in starvation-responsive pathways in the liver are associated with enhanced Kbhb on histone H3 (H3K9bhb).
We previously reported that fasting induces gene expression changes in small intestine (SI) stem cells and that 3-Hydroxy-3-Methylglutaryl-CoA Synthase 2 (Hmgcs2), which catalyzes the first reaction of ketogenesis to generate β-OHB, is encoded by the second most highly induced gene in SI stem cells of fasted mice (Tinkum et al., 2015). The SI epithelium is preserved by stem cells within the bottom third of the crypts, known as crypt base columnar intestine stem cells (ISCs) and identified by high expression of LGR5 (Barker et al., 2007). ISCs generate a pool of progenitor transient amplifying (TA) daughter cells in the mid third of crypts. TA cells, in turn, expand rapidly and then undergo terminal differentiation as they migrate to the top third of the crypt to differentiate and exit to the villi (Kozar et al., 2013; Ritsma et al., 2014; Sato et al., 2009; Simons and Clevers, 2011). Through dynamic and adaptive cell turnover, the SI can respond to various stimuli, like oxidative stress (Al-Asmari et al., 2016; Basivireddy et al., 2002; Moorefield et al., 2017), DNA damage (Lee et al., 2011; Miyoshi et al., 2012; Tinkum et al., 2015), and dietary restriction or nutrient overabundance (Beyaz et al., 2016; Mihaylova et al., 2018). Furthermore, the SI epithelium is a metabolic organ critical for nutrient absorption that can transcriptionally respond to nutrient fluctuations (Hakvoort et al., 2011; Mihaylova et al., 2018; Tognini et al., 2017). How promoters and enhancers become modified as a result of fasting and the effect on transcriptional changes in the SI crypt are unknown.
To determine whether fasting induces epigenomic changes in SI crypt cells, we defined the regulatory landscape of the SI crypt by analyzing 6 histone marks (H3K4me1, H3K4me3, H3K9me3, H3K27ac, H3K27me3, and H3K79me2). Further, we determined the contribution made by two distinct histone Kbhb modifications (H3K9bhb and Kbhb on histone H4 [H4K12bhb]) to chromatin states concordant with SI crypt response during fasting. Histone Kbhbs were found to associate with active and transcribed chromatin states during fasting, and these chromatin domains corresponded with fasting-induced gene expression patterns within ISC and TA cell populations of the crypts. We further showed that fasting enriched for an H3K9bhb-H3K27ac signature at active metabolic gene promoters and distal DNA enhancer elements and that β-OHB was sufficient to induce H3K9bhb enrichment at fasting-responsive genes in cultured SI stem cell-enriched epithelial spheroids. Further analysis of SI crypts conditionally deleted for Hmgcs2 revealed a marked decrease in H3K9bhb-associated loci and an altered gene expression profile in the fasted condition. These results provide important insights into how the transcriptomes of SI crypt epithelial cells (Lgr5+ stem cells and TA cells) and the epigenome of SI crypt cells are directly modified in response to changes in energy sources under conditions of metabolic stress.
RESULTS
Fasting triggers a ketogenic program in ISCs and TA cells
To investigate how fasting affects Lgr5+ ISCs and TA cell populations, we isolated SI crypts and purified matched ISCs or TA cells from replicate Lgr5EGFP-IRES-CreER/+ reporter mice using flow cytometry and performed transcriptional profiling by ultra-low mRNA sequencing (Figures 1A and S1A). A 24-h fasting regimen led to approximately 20% whole-body weight loss (Figure S1B), and, as expected, principal-component analysis (PCA) showed cells segregated first by cell type (PC1, 69% of total variance) and then by diet (PC2, 11% of total variance) (Figure S1C). Fasting altered expression of 304 genes in ISCs and 401 genes in TA cells across multiple animals (log fold change (FC) > log2(1.5-fold) or logFC < log2(1/1.5), adjusted p < 0.05) (Figure 1B; Table S1). Importantly, gene set enrichment analysis (GSEA) utilizing the Molecular Signatures Database (MSigDB)(Subramanian et al., 2005) identified pathways associated with fatty acid, lipid, and ketone body metabolism among the top 5 pathways upregulated in ISCs and TA cells with fasting, using HALLMARK, REACTOME, and KEGG gene collections (Figures 1C, 1D, and S1D; Table S1). Intersection of ISC and TA cell gene sets with top GSEA pathways identified that Hmgcs2, the gene encoding the rate-limiting enzyme for β-OHB synthesis (Cotter et al., 2013), was highly differentially expressed in ISCs and TA cells (Figure 1E). Furthermore, we confirmed that fasting evoked a fatty acid oxidation transcriptional program within ISCs (Mihaylova et al., 2018) and that pathways involved in the cell cycle and DNA replication were suppressed (Figure 1E) (Tinkum et al., 2015). We validated the expression of key genes (Hmgcs2 and Bdh1) associated with fatty acid/ketogenic metabolism in ISCs and TA cells by qPCR (Figures 1F and S1E) and observed a marked accumulation of HMGCS2 in SI crypts of fasted mice and in purified populations of Lgr5+ stem and TA cells (Figures 1G and 1H). Studies have demonstrated that Hmgcs2 expression is not specific to stem and TA cells but also present in other cell types (such as SI Paneth cells), albeit at much lower levels, and that fasting induces expression of Hmgcs2 in stem, TA, and Paneth cells (Cheng et al., 2019).
Figure 1. Fasting triggers a ketogenic program in ISC and TA cells.
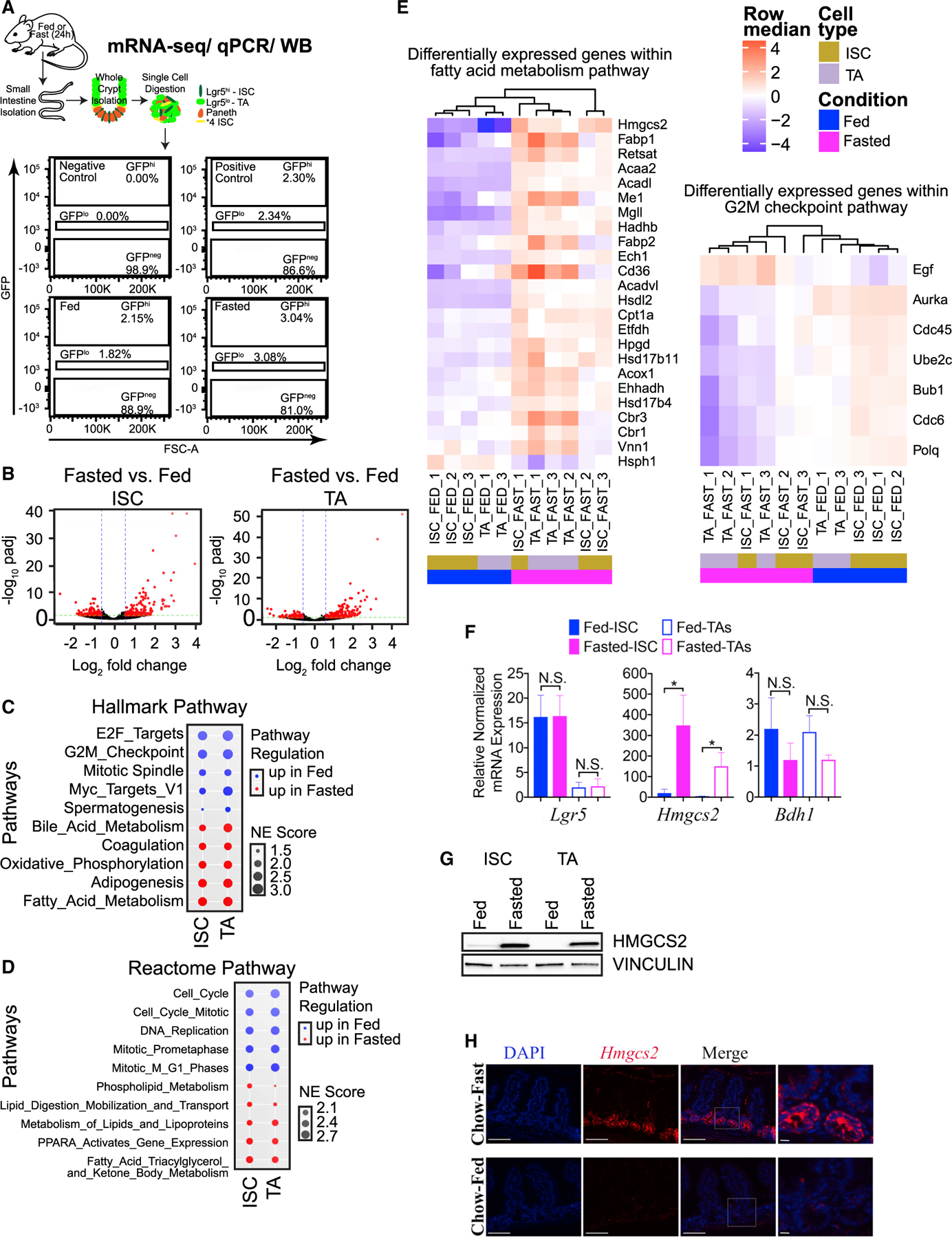
(A) Schematic of Lgr5+ cell collection (ISCs or TA cells) from SI crypts for ultra-low mRNA-seq (20,000 total cells per sample), qPCR (5,000 total cells per sample), or western blot (20,000 total cells per sample) analyses. Representative flow cytometry plots are included. FSC, forward scatter parameter.
(B) Volcano plots identify (304) differentially enriched genes by fasting (up- and downregulated) in ISCs (Lgr5−GFPhi) and (401) differentially enriched genes from matched TA progenitor cells (Lgr5−GFPlo). n = 3 mice per group.
(C) Top 5 up- or downregulated HALLMARK pathways enriched by fasting in ISCs (Lgr5−GFPhi) and TA cells (Lgr5−GFPlo), ranked by normalized enrichment (NE) score prevalence in ISCs.
(D) Top 5 up- or downregulated Reactome pathways enriched by fasting in ISCs (Lgr5−GFPhi) and TA cells (Lgr5−GFPlo), ranked by NE score prevalence in ISCs.
(E) Heatmap of RNA-seq analysis, showing relative expression of “fatty acid metabolism” and “G2M checkpoint” HALLMARK gene collections in ISCs and TA cells from fed and fasted replicate mice. All genes displayed are differentially expressed (false discovery rate [FDR] ≤ 0.05, log2FC > = 1). n = 3 mice per group.
(F) qPCR analysis confirming that Lgr5+ ISCs and TA cells were isolated by Lgr5 expression and fasting responsive gene expression of Hmgcs2 in ISCs and TA cells and Bdh1 in TA cells. Gapdh was multiplexed and used as a housekeeping gene per sample. n = 3 experiments, n = 3 mice per group. Data are mean ± SEM, analyzed by Student’s t test, unpaired. N.S., not significant.
(G) Representative western blot of HMGCS2 in ISCs and matched TAs of fed and fasted mice. VINCULIN was used as a loading control for 20,000 cells per sample. n = 4 experiments (n = 6–8 total mice per group).
(H) Immunofluorescence analysis of HMGCS2 in SI tissues (jejunum) of fed and fasted mice. Representative images from all groups are shown. Scale bars, 100 μm. Magnification, 40×. Magnified images, 40 μm.
See also Figure S1.
H3K9bhb is enriched with fasting in SI crypts
Fasting has been reported to increase circulating levels of the ketone body β-OHB within 24 h in mice (Shimazu et al., 2013; Xie et al., 2016). We confirmed high circulating serum levels of β-OHB and decreased serum glucose in mice fasted for 24 h (Figures S2A and S2B). β-OHB is the precursor for PTM of histone H3 by Kbhb, and high serum levels of β-OHB are linked to increased H3K9bhb deposition in the fasted liver (Xie et al., 2016). To investigate the contribution of fasting to Kbhb levels in SI crypt cells, we performed chromatin immunoprecipitation sequencing (ChIP-seq) with antibodies recognizing Kbhb on histone H3 (H3K9bhb) or H4 lysine residues (H4K12bhb) (Figure 2A). To identify which histone Kbhb modification is enriched with fasting in the SI crypt, we performed unsupervised clustering and differential binding affinity analysis between fed and fasted conditions, which revealed that H3K9bhb was markedly enriched upon fasting (Figures 2B and 2C). In contrast, H4K12bhb did not display clear separation between conditions, and a low number of differentially enriched loci were identified upon fasting (Figures 2B and 2C). Moreover, analysis of individual replicates and co-localization of consensus peak sites (Figures S2C–S2E) for each Kbhb mark revealed a marked increase in H3K9bhb enrichment under the fasted condition compared with the fed condition (Figure 2D). Based on consensus peaks, we identified over 21,000 H3K9bhb-enriched, genome-wide locations in SI crypt cells that were unique to fasting (Figures 2D, S2D, and S2E; Table S2). Intriguingly, compared with H3K9bhb, H4K12bhb only showed a modest enrichment under fasted conditions (Figure 2D, S2D–S2F; Table S2). These results demonstrate that fasting induced a marked enrichment of H3K9bhb compared with H4K12bhb at a large number of loci across distinct regions of the genome.
Figure 2. Fasting differentially enriches H3K9bhb at proximal promoters of metabolic programs in small intestine crypts.
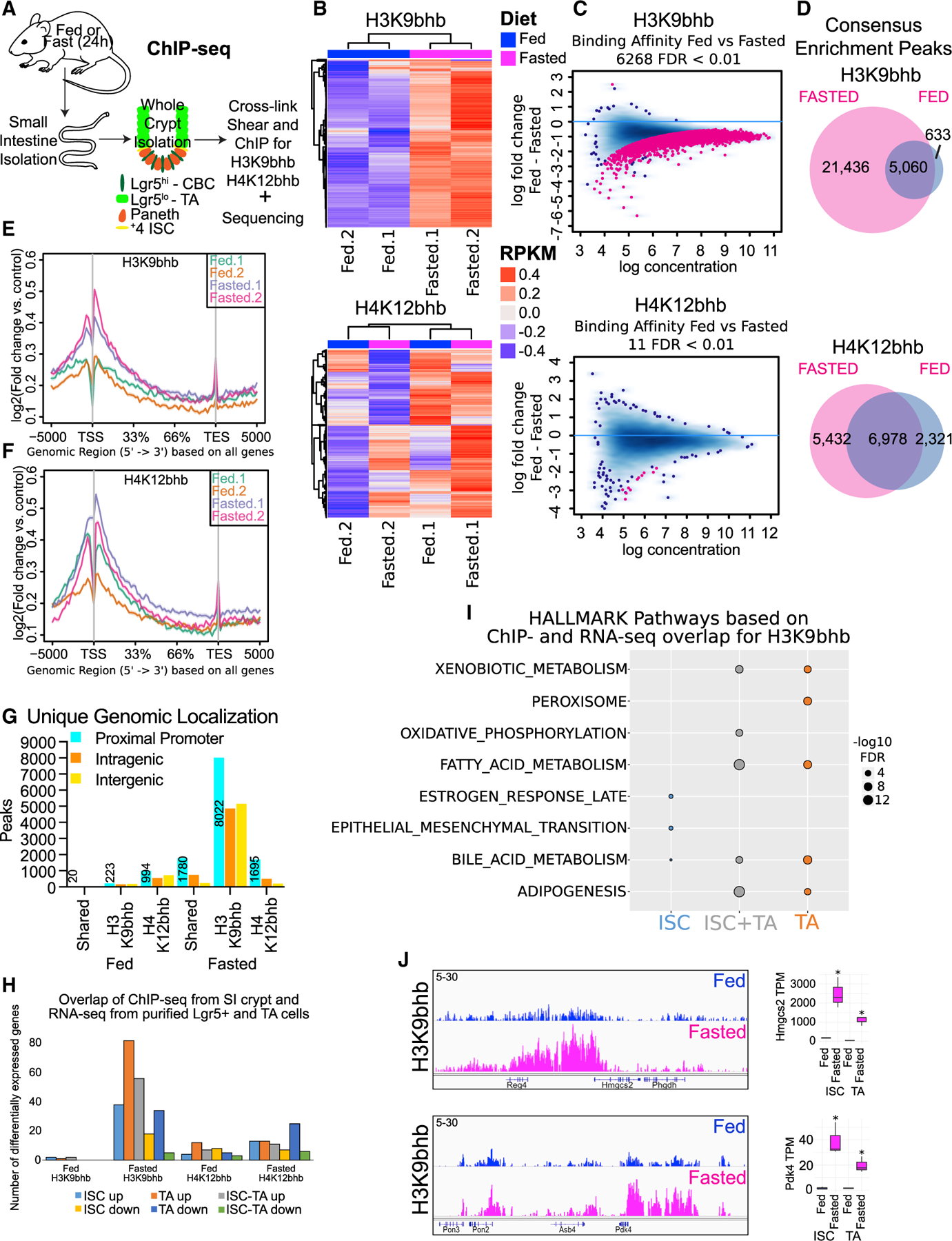
(A) Schematic of whole-crypt isolation for ChIP-seq analyses with Kbhb marks.
(B) Unsupervised clustering of the top 5,000 most variable genomic regions for H3K9bhb and H4K12bhb in SI crypt replicates under fed and fasted conditions, based on 2-sample minimal overlap for Model-based analysis of ChIP-seq (MACS) peaks using Diffbind. n = 4 mice per pool per replicate per group. RPKM, reads per kilobase per million mapped reads.
(C) Differential binding affinity for H3K9bhb and H4K12bhb, based on 2-sample minimal overlap for MACS peaks using Diffbind between fed and fasted treatments; FDR < 0.01.
(D) Co-localization analysis of H3K9bhb and H4K12bhb consensus enrichment peaks in fed and fasted SI crypts.
(E and F) Signal intensity gene body plots of H3K9bhb and H4K12bhb in individual replicates. Ensembl genes are arranged 5′ to 3′ from −5 kb to the TSS, TSS, 33%, 66%, transcription end site (TES), and +5 kb from the TES. An arrow denotes the TSS at the 5′ region of the gene.
(G) Genomic distribution for unique H3K9bhb and H4K12bhb. The proximal promoter encompasses ±5 kb of the TSS, and peak numbers of proximal promoters are listed. Intragenic encompasses regions +5 kb to the TES. Intergenic encompasses regions outside of ±5 kb of the TSS to TES.
(H) Barplot displaying H3K9bhb and H4K12bhb proximal promoter peaks from whole SI crypt ChIP-seq overlapping with differentially expressed genes from isolated ISCs and TA cells.
(I) Top 5 MSigDB GSEA pathways based on H3K9bhb or H4K12bhb proximal promoter peaks overlapping differentially expressed genes in ISCs, ISCs and TA cells, and TA cells under the fasted condition. Size of dot represents −log10 FDR value.
(J) Genome browser view of ChIP-seq tracks for H3K9bhb-enriched regions in SI crypts at the fasting-responsive genes Hmgcs2 and Pdk4 under fed and fasted conditions. All tracks are set at 5–30 data range. RNA counts (means with 95% confidence interval [CI]) in ISCs and TA cells of genes within the displayed H3K9bhb regions. TPM, transcripts per million. *q < 3.18e–18.
See also Figure S2.
Fasting enriches H3K9bhb at proximal promoters
To assess the location of specific Kbhbs at distinct regions of the genome, we performed average density of individual replicates and genomic localization analysis of consensus peaks for proximal promoter, intragenic, and intergenic regions, which revealed that H3K9bhb is enriched at the proximal promoter (within ± 5 kb of the transcription start site [TSS]) region upon fasting (Figures 2E–2G and S2F; Table S2). Enrichment of H3K9bhb at proximal promoters during fasting was identified using the genomic region enrichment of annotations tool (GREAT) with the MSigDB Gene Ontology (McLean et al., 2010). Significantly enriched pathways were associated with fatty acid, lipolysis, and ketogenic metabolism (Figures S2G and S2H), many of which were also identified by differential gene expression analysis (Figures 1C and 1D). Importantly, integration of ChIP-seq and RNA sequencing (RNA-seq) datasets confirmed that H3K9bhb was enriched on genes that were significantly upregulated in ISCs and TA cells upon fasting (Figure 2H), suggesting that chromatin reprogramming in the SI crypt might be associated with a transcriptional response in ISCs and TA regulatory cells. H3K9bhb-enriched differentially expressed genes were present in ketogenic pathways, including fatty acid and xenobiotic metabolism (Figure 2I; Table S3). Indeed, H3K9bhb was enriched within the proximal regions of critical fasting-induced genes such as Hmgcs2 and Pdk4 and absent from unexpressed genes such as Myod1 (Figures 2J and S2I).
In contrast to H3K9bhb, we found that H4K12bhb did not display concurrent enrichment across all promoters between replicates and that consensus peaks were present on a lower number of genes upon fasting (Figures 2F–2H), suggesting that H4K12bhb may not be affected by fasting in the same manner as H3K9bhb. More importantly, under fasting conditions, H4K12bhb did not display enrichment of pathways associated with fasting. Instead, fed or fasted enriched H4K12bhb displayed an association with pathways related to cellular homeostasis, including those related to the immune system, adaptive immune system, and metabolism of RNA, mRNA, or proteins (Figure S2H). Overall these results demonstrate a fasting-induced role of H3K9bhb in the SI crypts, similar to the fasted liver (Xie et al., 2016), which suggests a conserved function of this histone mark in coordinating fasting response in diverse metabolic tissues.
Histone Kbhb marks active chromatin states
Fasting has been shown to increase additional active histone marks in the liver in addition to H3K9bhb (Xie et al., 2016). To determine whether this also occurred in the SI crypt, we further assessed the global levels of H4K12bhb, H3K9ac, H3K14ac, and H3K27ac under fed and fasted conditions. With the exception of H3K9bhb, we did not detect global fasting-induced enhancement of modified histones (Figure 3A). ChIP-seq analysis further confirmed that the majority of H3K9ac enriched chromatin loci were shared between fed and fasted conditions (Figures 3B and S3A–S3C). Interestingly however, unlike with H3K9ac, we observed a site-specific increase in H3K27ac on a genome-wide and meta-gene level in response to fasting (Figures 3C, S3A, and S3D). Based on consensus peaks, we observed marked enrichment of H3K27ac around the TSS as well as a higher number of loci within promoter regions (n = 2,612) that harbor H3K9bhb and H3K27ac (Figures 3D, S3E, and S3F; Table S4). In contrast, this was not observed within promoters (n = 51) that harbor H3K9bhb and H3K9ac (n = 83) or H3K9bhb + H3K9ac + H3K27ac (Figures 3D, S3E, and S3F), suggesting that H3K9bhb displays concurrent enrichment with H3K27ac but not with H3K9ac during the fasting response.
Figure 3. Histone Kbhb is associated with an active chromatin landscape.
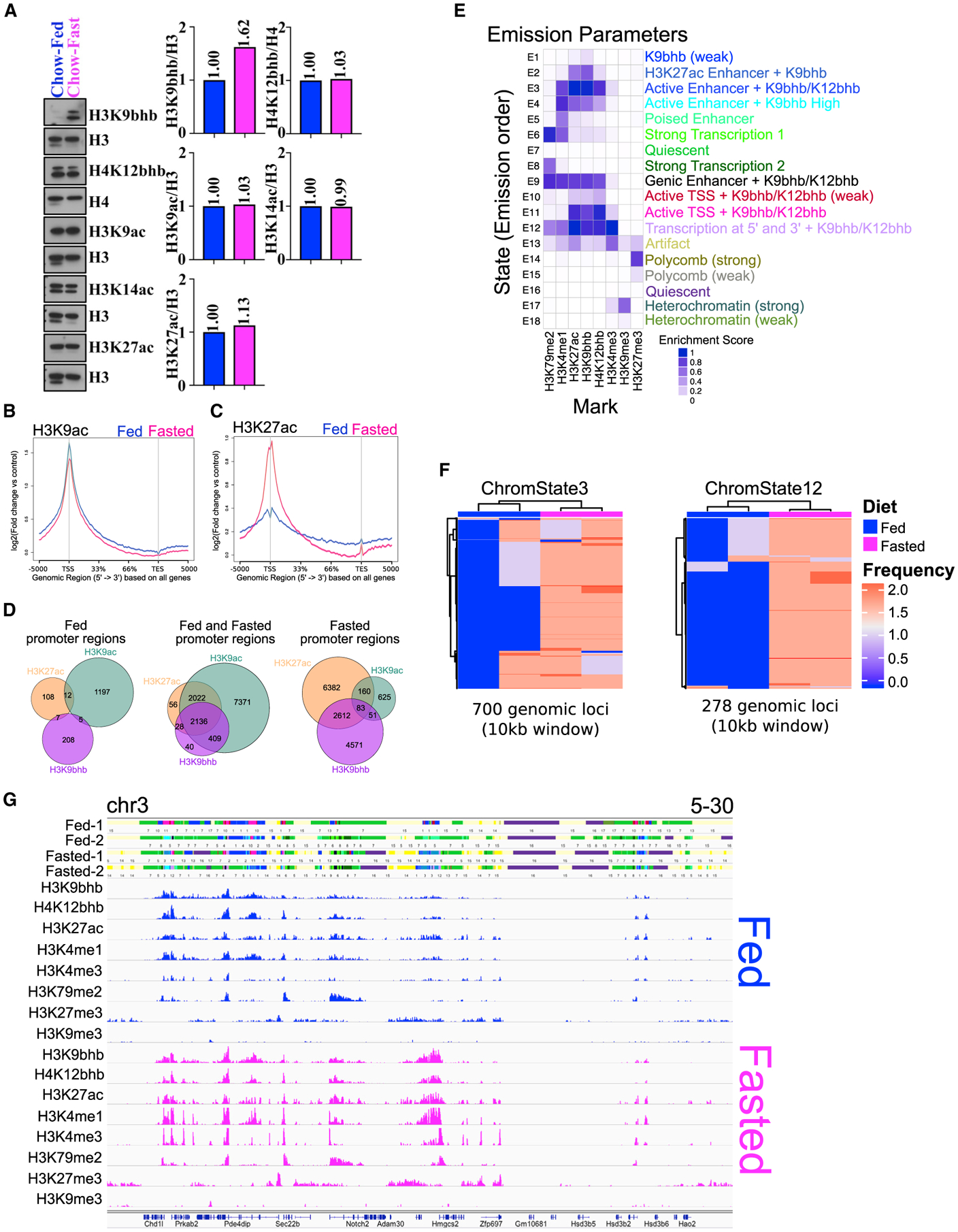
(A) Western blot analysis of histones isolated from small intestinal crypts probed with histone antibodies for H3K9bhb, H4K12bhb, H3K9ac, H3K14ac, and H3K27ac under fed and fasted conditions. Antibodies against H3 and H4 were used as loading controls. The relative quantification for all antibodies was normalized to total levels of H3 or H4.
(B and C) Signal intensity gene body plots of H3K9ac and H3K27ac based on all Ensembl genes under fed and fasted conditions. Ensembl genes are arranged 5′ to 3′ from −5 kb to the TSS, TSS, 33%, 66%, TES, and +5 kb from the TES. An arrow denotes the TSS at the 5′ region of the gene.
(D) Co-localization analysis of H3K9bhb + H3K9ac + H3K27ac consensus peaks within proximal promoter regions (±5 kb of the TSS) of SI crypt cells under the fed and fasted conditions.
(E) ChromHMM chromatin state profile of SI crypts from 1-kb interval genome bin size. User assigned functionalization of emission parameters is included.
(F) Unsupervised clustering analysis of the most variable regions for active chromatin state E3_active enhancers (marked by high levels of H3K27ac and H3K4me1 outside of the TSS) and E12_transcription at 5′ and 3′ (marked by high levels of H3K4me3, H3K27ac, H3K4me1, and H3K79me2) under fed and fasted conditions (two biological replicates in each). Frequency is the occurrence of the specified chromatin state within a 10-kb genomic window.
(G) 18-state ChromHMM model annotations in individual fed and fasted replicates around the Hmgcs2 locus, with each emission state marked by a separate color within the track. For each 1-kb interval, the corresponding input consensus ChIP-seq peak profiles of each histone mark utilized for ChromHMM binary response calls are shown for fed (blue) and fasted (magenta) sequence tag mark profiles.
See also Figures S3–S5.
To gain a better understanding of how H3K9bhb (and H4K12bhb) may work in concert with other active or repressive histone marks during the fasting response, we used ChIP-seq to define additional key components of the chromatin landscape. These modifications included PTMs generally associated with enhancers (H3K27ac and H3K4me1), promoters (H3K4me3), active transcription (H3K79me2), and polycomb (H3K27me3) or heterochromatin (H3K9me3) repression (Lee et al., 2013). Using the ChromHMM algorithm, we first assigned an 18-combinatorial chromatin state model to characterize the SI crypt (Figure 3E). In accordance with the Roadmap Epigenomics Project (Kundaje et al., 2015), our analysis revealed canonical chromatin state patterns representing active and repressive domains (Figure S3G). Such states included E3_active enhancers (marked by high levels of H3K27ac and H3K4me1 outside of the TSS), E11_active TSSs (marked by high levels of H3K4me3 and H3K27ac within the TSS), E12_transcription at 5′ and 3′ (marked by high levels of H3K4me3, H3K27ac, H3K4me1, and H3K79me2), and E14_polycomb (marked by H3K27me3) or E17_heterochromatic (marked by H3K9me3) repressive states (Figures 3E and S3G). Multidimensional scaling (MDS) and unsupervised clustering analysis of each chromatin state revealed Kbhb enrichment specific to active chromatin domains upon fasting, most notably within E3_active enhancers and E12_transcription at 5′ and 3′ (Figures 3F and S4A). Although E14_polycomb and E17_heterochromatic repression states were also enriched in fasting (Figures S4A and S4B), these were mutually exclusive with Kbhb marks (Figures 3E, 3G, S4C, and S4D). Average intensity plots of all profiled histone modification marks surrounding genes showed co-enrichment of H3K9bhb with active marks and depletion of repressive marks (Figure S5A). k-means clustering of all eight chromatin marks under fed and fasted conditions identified two active clusters that were highly enriched in H3K9bhb and depleted in H3K27me3 or H3K9me3 (Figure S5B). Specifically, we observed enrichment of H3K9bhb with H3K27ac and H3K4me3 in promoters and gene bodies in active clusters 1 and 2 (Figure S5C). These data demonstrate that H3K9bhb associates with other active histone modifications during the chromatin reprogramming that occurs in SI crypt cells during fasting.
Fasting enriches H3K9bhb and H3K27ac co-localization at metabolic enhancers
Enhancers are long-range regulatory elements that activate transcription by delivering important accessory factors to the promoter region to regulate target gene expression. With the concurrent enrichment of H3K9bhb and H3K27ac in promoter regions (Figure 3), we next examined the effect of H3K9bhb alterations during fasting on potential enhancer elements marked by H3K27ac. Spearman correlation analysis further revealed co-enrichment of H3K9bhb and H3K27ac at a genome-wide level (Figures 4A and 4B). Average density of individual replicates and co-localization analysis of consensus peaks for H3K9bhb and H3K27ac within enhancer regions (excluding ± 5 kb of the TSS) identified a marked increase in H3K9bhb-H3K27ac-associated loci upon fasting (Figures 4C and 4D; Table S4). Intriguingly, this co-enrichment profile predominates the same MSigDB pathways as H3K9bhb alone (such as lipid, fatty acid, ketone, and triacylglycerol metabolism), including additional target genes such as Cebpb, Ptpn1, Ppargc1b, and Cdc42 (Figure 4E–H), suggesting that, in addition to its association in promoters, H3K9bhb may also mediate enhancer function in concert with H3K27ac for specific metabolic genes in SI crypt cells during fasting.
Figure 4. Histone Kbhb is associated with fasting coupled enhancers.
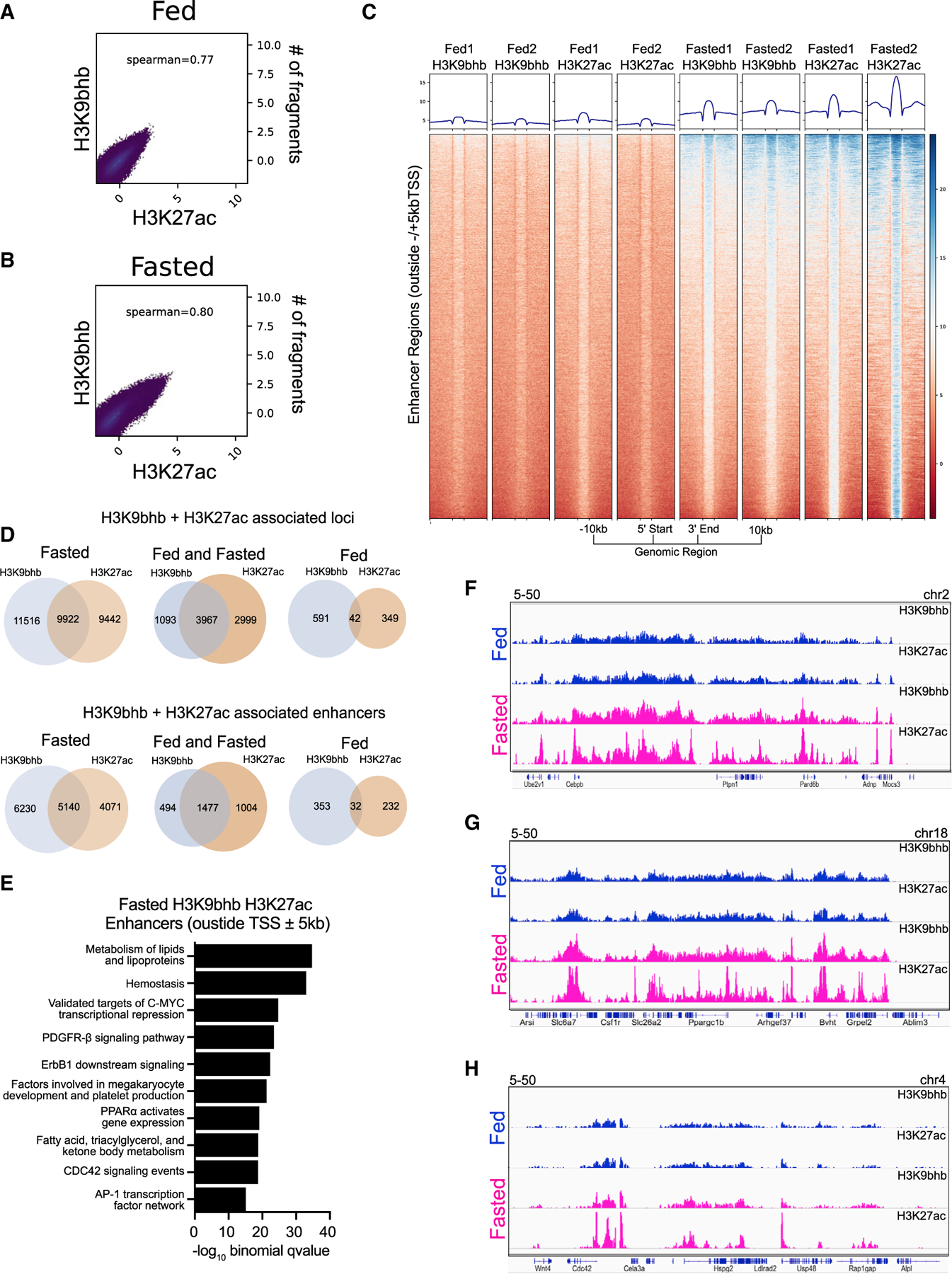
(A and B) Spearman correlation analysis for H3K9bhb and H3K27ac under fed and fasted conditions based on 500-bp genomic bins across the entire genome.
(C) Individual replicate signal intensity plots of H3K9bhb and H3K27ac for enhancer regions (excluding ±5 kb of the TSS) under the fed and fasted conditions. Regions are displayed from −10 kb to the 5′ start and +10 kb from the 3′ end of enhancer peaks.
(D) Co-localization analysis for H3K27ac- and H3K9bhb-associated loci genome wide and within enhancer regions (excluding ±5 kb of the TSS) between fed and fasted crypts.
(E) Top 10 MSigDB pathways enriched by −log10 binomial q value from GREAT annotation predictions of fasting enhancer (excluding ±5 kb of the TSS) cis-regulatory regions.
(F) Genome browser view of ChIP-seq tracks for H3K27ac + H3K9bhb enhancer-enriched regions at the Cebpb and Ptpn1 loci. Fed (blue) and fasted (magenta) tracks are shown, with all tracks set at the indicated data range.
(G) Genome browser view of ChIP-seq tracks for H3K27ac + H3K9bhb enhancer-enriched regions at the Ppargc1b locus. Fed (blue) and fasted (magenta) tracks are shown, with all tracks set at the indicated data range.
(H) Genome browser view of ChIP-seq tracks for H3K27ac + H3K9bhb enhancer-enriched regions at the Cdc42 locus. Fed (blue) and fasted (magenta) tracks are shown, with all tracks set at the indicated data range.
See also Figure S3.
Ketone bodies induce fasting-like H3K9bhb enrichment
To determine whether β-OHB alone was capable of inducing H3K9bhb peak enrichment at fasting-regulated proximal promoters, we incubated Lgr5+ stem cell-enriched epithelial spheroids (de la Cruz Bonilla et al., 2018; Miyoshi and Stappenbeck, 2013) in vitro with the bioactive β-OHB derivative (R)-(–)-3-hydroxybutyric acid (R-βHB). This resulted in a dose-dependent increase in global H3K9bhb and H4K12bhb (Figure 5A) but not H3K9ac, H3K14ac, or H3K27ac (Figure S6A). Importantly, H3K9bhb chromatin enrichment was observed at selected proximal promoter elements identified in the SI crypt cells of fasted animals (Figures 5B and S6B). Interestingly, this enrichment did not lead to similar gene expression changes (Figure 5C) as observed in ISCs and TA cells upon fasting. These data demonstrate that exogenous β-OHB can mimic fasting-induced enrichment of H3K9bhb at selected proximal promoters of critical fasting genes (i.e., Hmgcs2), but H3K9bhb enrichment alone was not sufficient to induce gene expression changes in vitro similar to those induced by fasting in vivo.
Figure 5. Exogenous ketone bodies induce fasting-like H3K9bhb enrichment without altering gene expression in stem-cell-enriched epithelial spheroids.
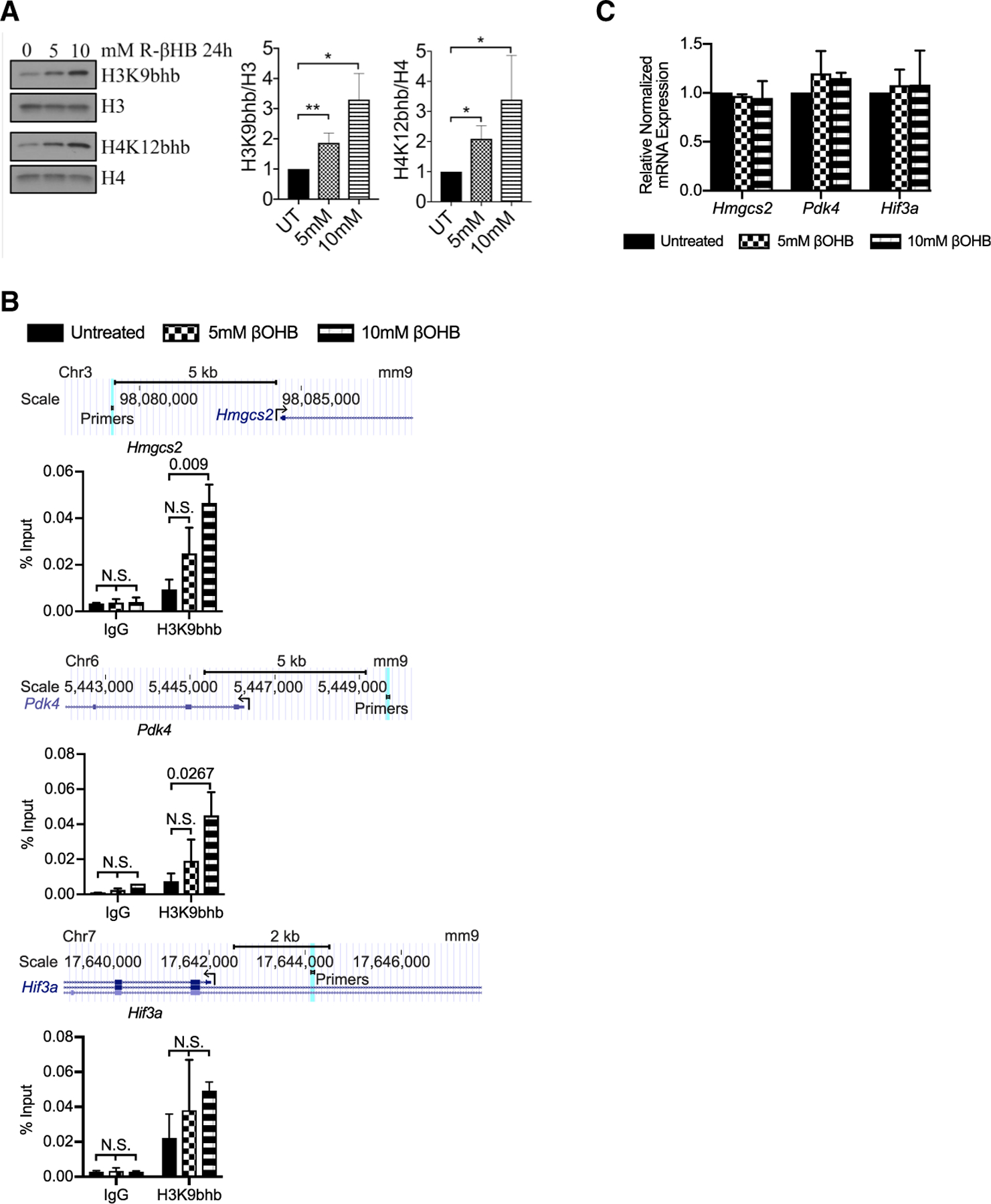
(A) Representative western blot of H3K9bhb and H4K12bhb of acid-extracted histones from stem-cell-enriched epithelial spheroids untreated (UT) or treated with 5 mM or 10 mM (R)-(–)-3-hydroxybutyric acid (R-βHB) for 24 h in rich conditioned medium. 2 μg of extracted histones loaded per well. Histone H3 and H4 are included as loading controls. Intensities were normalized to total histone protein and UT controls. n = 3 experiments. Data are mean ± SEM. *p < 0.05, **p < 0.01. Source data are provided as a source data file.
(B) ChIP-qPCR analysis of H3K9bhb distribution on Hmgcs2, Pdk4, and Hif3a gene regions from stem-cell-enriched epithelial spheroids left UT or treated with 5 mM or 10 mM R-βHB for 24 h in rich conditioned medium. The scale denotes genomic position separated by the indicated distance in kilobases. A light blue line denotes the location of primers used in ChIP-qPCR. Data are mean ± SEM, analyzed by 2-way ANOVA, n = 2 replicates per 2 experiments.
(C) qPCR analysis of Hmgcs2, Pdk4, and Hif3a gene expression of stem-cell-enriched epithelial spheroids left UT or treated with 5 mM or 10 mM R-βHB for 24 h in rich conditioned medium. Gapdh was multiplexed and used as a housekeeping gene per sample. n = 2 replicates per group. Data are mean ± SEM, analyzed by Student’s t test, unpaired.
See also Figure S6.
Loss of Hmgcs2 decreases H3K9bhb at a genome-wide level during fasting
Mice conditionally deleted for Hmgcs2 in intestinal epithelial cells (inducible knock-out [iKO mice])(Cheng et al., 2019) were used to determine whether local production of HMGCS2 contributed to the fasting-induced Kbhb modification of histones in SI crypt cells. Average density of individual replicates and differential binding analysis revealed a marked loss of H3K9bhb-associated loci in SI crypt cells of iKO mice compared with those of wild-type (WT) mice under the fasted condition (Figures 6A–6D). A similar loss for consensus and concatenated H3K9bhb peaks was observed in SI crypt cells of fasted iKO mice, and pathways associated with this loss included “fatty acid metabolism” (Figures 6E, 6F, and S6C–S6J; Table S5). These results suggest that local production of β-OHB by intestinal HMGCS2 contributes to the fasting-specific H3K9bhb changes observed in SI crypt cells. Interestingly, H3K9bhb was maintained in Hmgcs2 iKO mice that were allowed to feed but lost in mice that were fasted (Figures 6A, 6B, S6D, and S6E), suggesting that food was able to compensate for loss of HMGCS2 in fed iKO mice.
Figure 6. Loss of Hmgcs2 affects H3K9bhb enrichment on fasting-specific genes in the SI.
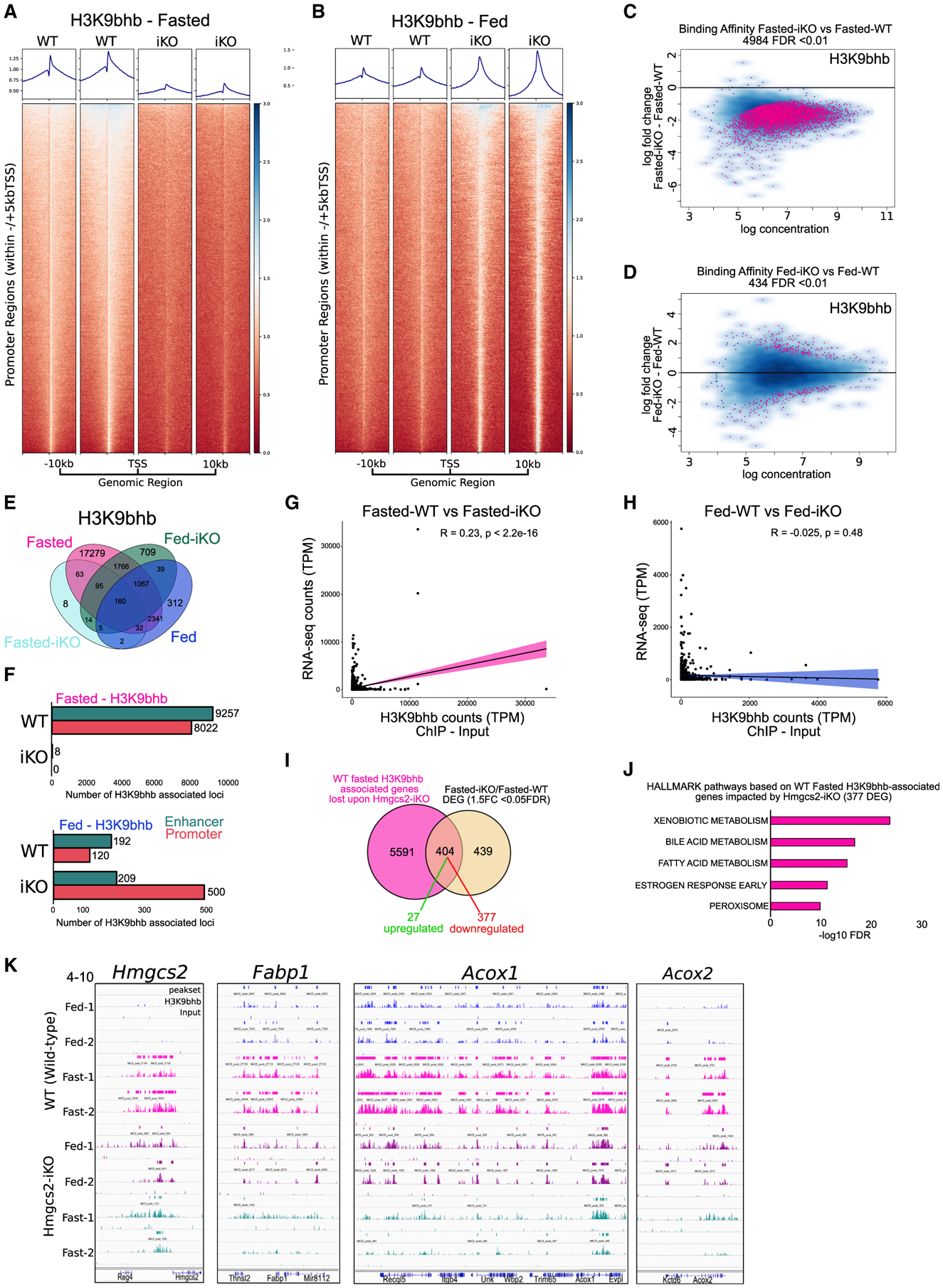
(A and B) Individual replicate signal intensity plots of H3K9bhb in Ensembl gene promoter regions (within ±5 kb of the TSS) from WT and iKO SI crypt cells under fasted and fed conditions. Ensembl genes are arranged 5′ to 3′ from −10 kb to the TSS and +10 kb from the TES.
(C and D) Differential binding affinity for H3K9bhb, based on 4-sample minimal overlap for MACS peaks using Diffbind between fasted WT and iKO and fed WT and iKO mice. FDR < 0.01.
(E) Co-localization analysis of H3K9bhb consensus peaks from WT and iKO SI crypt cells under fasted and fed conditions.
(F) Genomic distribution of H3K9bhb consensus peaks within promoter and enhancer regions from WT and iKO SI crypt cells under fasted and fed conditions. Proximal promoters encompass ±5 kb of the TSS, and enhancers encompass regions excluding ±5 kb of the TSS.
(G and H) Pearson correlation plots for H3K9bhb and RNA-seq data based on all H3K9bhb-associated promoter regions under the fasted and fed conditions.
(I) Overlaps of H3K9bhb peaks (±5 kb of the TSS) from fasted WT samples in (F) with differentially expressed genes from fasted Hmgcs2-iKO versus WT SI crypts.
(J) Top 5 MSigDB GSEA pathways enriched by −log10 FDR based on fasted H3K9bhb-associated genes significantly affected by loss of Hmgcs2.
(K) Genome browser view of ChIP-seq tracks for the H3K9bhb enriched region at the Hmgcs2, Fabp1, Acox1, and Acox2 loci. Fed (blue) and fasted (magenta) tracks are shown in WT and fed (purple) and fasted (green) in iKO SI crypt cells, with all tracks set at the indicated data range.
See also Figure S6.
To determine how Hmgcs2 loss influences gene expression patterns, RNA-seq was performed on SI crypt cells isolated from fed and fasted WT and iKO mice. As expected, Hmgcs2 was downregulated in crypt cells isolated from iKO mice, and PCA showed crypts segregated by iKO under the fasted and fed conditions (Figures S6K–S6N; Table S6). Overall, Hmgcs2 loss significantly altered the expression of 843 genes in fasted mice (fasted-WT/fasted-iKO) and 184 genes in fed mice (fed-WT/fed-iKO) (Figures S6O–S6Q; Table S6). At a genome-wide level, we observed a moderate correlation (R = 0.23) between H3K9bhb ChIP and RNA-seq data under the fasted condition (Figure 6G) and no correlation under the fed condition (Figure 6H), suggesting that the increase of H3K9bhb is not associated with global changes in gene expression levels. Importantly however, overlaps of H3K9bhb with genes significantly affected by intestinal knockout of Hmgcs2 displayed a loss of H3K9bhb on 47.9% of all differentially expressed genes (404 of 843) under the fasted condition, and 93% of these genes (377 of 404) were downregulated in fasted Hmgcs2-iKO mice relative to fasted WT mice (Figure 6I; Table S7). Although we observed a marked loss of H3K9bhb across thousands of promoters upon Hmgcs2-iKO, the genes significantly affected by this loss were primarily associated with metabolic gene programs, including “xenobiotic metabolism,” “fatty acid metabolism,” and “bile acid metabolism,” encompassing critical fasting-related genes such as Hmgcs2, Fabp1, Acox1, and Acox2 (Figures 6J and 6K; Table S7). In contrast, this was not observed for H3K9bhb-associated genes that were not affected by Hmgcs2-iKO in which we did not observe any fasting-related pathways (Figure S6R). Moreover, fed-specific enrichment of H3K9bhb was associated with only 2 differentially expressed genes in WT samples (Figure S6S), further suggesting that Hmgcs2 activation of H3K9bhb is specific to the fasting response. These findings demonstrate that H3K9bhb loss occurs on a subset of critical fasting-responsive genes significantly affected by Hmgcs2-iKO.
DISCUSSION
Here we demonstrate that fasting induced production of β-OHB, which resulted in marked enrichment of H3K9bhb on regulatory elements near genes of lipolytic and ketogenic transcriptional programs in SI crypt cells. Notably, H3K9bhb enrichment at proximal promoters (±5 kb of the TSS) in SI crypt cells in fasted mice was associated with transcriptional upregulation of critical metabolic genes such as Hmgcs2 and Pdk4. We also identified co-enrichment of H3K9bhb with H3K27ac at proximal and distal enhancers (excluding ± 5 kb of the TSS) of genes regulating the same metabolic programs. In contrast, H4K12bhb did not show similar fasting-induced enrichment, suggesting that not all histone Kbhb modifications serve similar regulatory roles. However, additional experiments are needed to confirm the functional role of different Kbhbs during fasting and ketogenesis. Although ChIP-seq experiments from SI crypts contain cell populations in addition to SI stem and TA cells, the same lipolytic and ketogenic gene programs were upregulated in SI stem and TA cells, and K9bhb was specifically enriched on the same target genes. This was consistent with enrichment of H3K9bhb at the TSS of Hmgcs2 and Pdk4 when Lgr5+ stem-cell-enriched epithelial spheroids were treated with a bioactive β-OHB derivative in vitro (Figure 5). These results suggest that H3K9bhb enrichment is a key response during fasting-induced activation of metabolic gene programs in SI crypt cells, including in Lgr5+ stem and TA cells.
Epigenetic landscape analysis with ChromHMM and k-means clustering showed that fasting-induced enrichment of H3K9bhb was associated with active/permissive chromatin states, as defined by co-enrichment with H3K4me3 and H3K27ac. Although fasting also induced specific repressive chromatin states, as defined by H3K27me3 or H3K9me3, Kbhb was not associated with these. Interestingly, experiments performed ex vivo with Lgr5+ stem-cell-enriched epithelial spheroids demonstrated that β-OHB was sufficient to enrich H3K9bhb marks at the TSS of selected genes, such as Hmgcs2 and Pdk4, but it was insufficient to activate transcription of these genes. Thus, Kbhb marks may cooperate with other chromatin modifications, such as those associated with active/permissive chromatin states, to induce gene expression programs during conditions of limited glucose in vivo. Indeed, alterations in other epigenetic markers, such as 5-methylcytosine, are known to occur during calorie restriction (Maegawa et al., 2017).
We demonstrated previously that Hmgcs2, which catalyzes the first reaction of ketogenesis to generate β-OHB, is the second most highly induced gene in SI stem cells of fasted mice (Tinkum et al., 2015), and here we show a corresponding increase in HMGCS2 protein in SI stem and TA cells of fasted animals (Figures 1G and 1H). Although ketogenesis occurs mostly in the liver, HMGCS2 in the SI crypt may contribute to the local pool of β-OHB for use not only as an energy source but also as a substrate for PTM of histones to drive metabolic expression programs in these cells. In support of this, we found that loss of intestinal Hmgcs2 impaired the ability of fasting to induce H3K9bhb and altered the expression of critical fasting-related genes within SI crypts, such as Hmgcs2, Fabp1, Acox1, and Acox2. A marked loss of H3K9bhb within promoter regions of genes and pathways necessary for the fasting response (such as fatty acid metabolism) suggests that, during fasting, HMGCS2 may indirectly control gene expression patterns by altering the distribution of specific histone Kbhb’s within regulatory regions. In support of this, although we observed a marked loss of H3K9bhb across thousands of promoters upon Hmgcs2-iKO, the genes significantly affected by this loss were associated with metabolic gene programs, further suggesting a role of H3K9bhb in the fasting response.
In contrast to fasting, H3K9bhb was maintained in fed Hmgcs2 iKO mice but lost in fasted Hmgcs2-iKO mice (Figures 6A, 6B, and S6C–S6E). Interestingly, dietary fiber ingested by fed mice is converted into short-chain fatty acids, such as butyrate, by intestinal microbiota. Butyrate can then be converted to β-OHB by an alternate pathway that does not utilize HMGCS2 (https://www.genome.jp/kegg-bin/show_pathway?map00650; Kanehisa et al., 2017; Puchalska and Crawford, 2017). Thus, we speculate that an alternate pathway maintains β-OHB levels (and, therefore, Kbhb marks) in fed Hmgcs2-iKO mice. Another notable observation under the fed condition was the lower reproducibility of H3K9bhb peak replicates relative to the fasted condition. A plausible explanation for this finding is the low levels of H3Kbhb for capture by ChIP-seq in fed mice because of low levels of HMGCS2 and β-OHB. We did not synchronize time of feeding, and therefore levels of H3Kbhb were not only low but also potentially variable between experiments. In contrast, all mice were fasted for an equivalent period of time (24 h) to generate high levels of HMGCS2 and β-OHB, enabling higher levels of H3K9bhb to be captured from a larger number of cells. These conditions favor reproducibility from experiment to experiment.
Limitations of the study
One limitation of this study was our inability to identify distinct epithelial cell populations within SI crypts that harbor fasting-induced H3K9bhb. To obtain enough cells for performing ChIP seq, epithelial cells were purified from SI crypts and consisted of a mixture primarily of stem, TA, and Paneth cells. Technologies such as CUT&TAG and single-cell CUT&TAG, which enable ChIP-seq from a very low number of cells or at the single-cell level, respectively, should enable identification of specific cell populations that become enriched for H3K9bhb in response to fasting and how this affects their transcriptomes.
Another limitation of this study is our inability to determine whether the presence of H3K9bhb in the promoter of critical fasting genes is a casual or correlative event in transcriptional regulation. It is plausible that specific promoters enriched with H3K9bhb are active, permissive, or poised for activation, and further classification of additional histones, co-activators, and transcription factors regulating this process is needed. Experiments to establish causality between Kbhb marks and fasting-induced transcriptional responses in the SI crypts, such as identification of specific readers, writers, and erasers, are critical goals in the field. Studies have identified SIRT3 as a class-selective histone de-β-hydroxybutyrylase capable of removing bhb sites from H3K9bhb (Zhang et al., 2019). A recent study has demonstrated that HDAC1 and HDAC can also serve as Kbhb erasers, whereas p300, a well-known acetyltransferase, can catalyze enzymatic addition of β-hydroxybutyrate to lysine (Huang et al., 2021). Although these studies are important advances in identification of regulatory proteins for H3K9bhb, their ability to modify histone acetylation in addition to H3K9bhb makes it difficult to perform the requisite functional experiments to investigate the causality of H3K9bhb gains during fasting (Huang et al., 2021).
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Helen Piwnica-Worms (hpiwnica-worms@mdanderson.org).
Materials availability
This study did not generate any new unique reagents.
Data and code availability
The ChIP and mRNA sequencing data have been deposited in the NCBI GEO BioProject database with the following accession number PRJNA:506838.
Code used for unsupervised clustering of chromatin states is available on https://rpubs.com/cjt5 and https://github.com/sccallahan/ChromXploreR.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Study Approval
This study was carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Animal care and use was approved by the MD Anderson Institutional Animal Care and Use Committee (IACUC). The protocol was approved by the IACUC at MD Anderson Cancer Center (1101RN02). Animals were euthanized as dictated by the Association for Assessment and Accreditation of Laboratory Animal Care International and IACUC euthanasia endpoints.
Animals and feeding
Male and female mice (strains: Lgr5EGFP-IRES-CreERT2/+ Strain name: B6.129P2-Lgr5tm1(cre/ERT2)Cle/J, JAX, 008875) and C57BL/6J (JAX, 000664)) were purchased, bred in house, and maintained at 72°F ± 2°F on a 12h light/dark cycle. Six- to twelve-week-old aged matched males and females were randomly assigned to either fed or fasted groups. Fed animals were provided ad libitium access to food (PicoLab 5053, #0007688) and water for 24h, fasted animals were deprived of food for 24 h with ad libitium access to water. Experimental mice were singly housed on aspen bedding for the duration of experiments. Animal numbers used per experiment are reported in Figure legends.
METHOD DETAILS
Glucose and serum ketone body quantification
Whole blood from tail nicks were quantified for glucose and ketone bodies using a Precision Xtra glucose monitor (Abbott) with either blood glucose or β-ketone test strips.
β-Hydroxyl-butyrate quantitation
Whole blood from fed or fasted mice at endpoint was centrifuged at 8,000rpm for 10min in serum separator tubes. β-Hydroxyl-butyrate levels were quantified using β-Hydroxyl-butyrate LiquiColor Test (StanBio CAT# 2440–058) according to the manufacturer’s instructions.
Small intestine crypt isolation
Crypt isolation followed previously published protocols with minor modifications (Guo et al., 2009; Tinkum et al., 2015). Briefly fed or fasted mice were euthanized by CO2, the entire SI was collected, flushed with PBS (Ca2+- and Mg2+-free, 2mM EDTA, 100 nM TSA) to remove feces, and the mesentery was removed. The SI sample was cut longitudinally then cut transversely into 4 equal pieces. Each sample was placed on ice in PBS (Ca2+- and Mg2+-free, 100nM TSA) while the remaining samples were collected. After collection of all samples, SI were incubated in PBS (Ca2+- and Mg2+-free, 2 mM EDTA, 100 nM TSA) for 10 min then transferred to HBSS (Ca2+- and Mg2+-free, 2 mM EDTA, 100 nM TSA). Crypts were released through a series of vortex washes at 1,600rpm in HBSS (Ca2+- and Mg2+-free, 2 mM EDTA, 100 nM TSA) at 4°C. Supernatants from all vortex washes were filtered through 70-μm mesh and pooled into 50ml conical tubes to remove villus material and tissue fragments. Isolated crypts were pelleted at 1000 rpm at 4°C. After this step, whole crypts were utilized for flow cytometry (detailed below), establishment of spheroid cultures (de la Cruz Bonilla et al., 2018) and treatment (detailed below), or ChIP-seq (detailed below). For ChIP-seq, crypts were first suspended in ADMEM/F12 (D6421, Millipore Sigma) supplemented with 10 U/mL penicillin, 10 μg/mL streptomycin, 2 mM L-glutamine, 10 mM HEPES, 10μM TGF-β RI Kinase Inhibitor VI (SB431542, Calbiochem), 10 μM Y-27632 dihydrochloride (Millipore Sigma), 0.5 mM N-acetylcysteine amide (Millipore Sigma), and 100 nM TSA.
Intestinal Hmgcs2 deletion and crypt sample preparation for ChIP-seq analysis
Intestinal-specific Hmgcs2 knockout was achieved as previously described (Cheng et al., 2019). Briefly, adult Hmgcs2loxp/loxp;Villin-CreERT2 mice received tamoxifen injections (10 mg/ml, 250ul per 25 g of body weight) every other day, for up to ten days for a total of five doses. Two weeks after the last tamoxifen injection, mice were assigned to ad libitum (AL) and fasting groups (Fast) for 24hrs. Intestinal crypts were isolated as described in the “Small intestine crypt isolation.” Isolated crypt samples were then immediately processed for cross-linking (1% formaldehyde, 10 min, 37°C), quenching (125mM Glycine, 5 min, 37°C) and washing (ice-cold PBS, 934 × g for 5 min at 4°C) and stored at −80°C before “ChIP-sequencing analysis.”
Generation of stem cell-enriched epithelial spheroid cultures
Whole crypt isolates were suspended in Matrigel and plated in a 24-well tissue culture dish (30 μL per well) as described (de la Cruz Bonilla et al., 2018). Cultures were grown in 50% L-WRN conditioned media supplemented with 10 μM TGF-β RI Kinase Inhibitor VI (SB431542, Calbiochem) and 10 μM Y-27632 dihydrochloride (Millipore Sigma). Media was changed every second day and spheroids were subcultured every third day. For experimental treatments, spheroids were cultured in 50% L-WRN conditioned media supplemented with either 5 or 10mM (R)-(–)-3-Hydroxybutyric acid (298360, Millipore Sigma), 10 μM TGF-β RI Kinase Inhibitor VI and 10 μM Y-27632 dihydrochloride. Standard media changes occurred for untreated control culture wells. All treatments were 24h prior to spheroid isolation from Matrigel for ChIP-qPCR or immunoblotting analyses.
Chromatin immunoprecipitation (ChIP)-qPCR of spheroids
Spheroids were isolated from Matrigel, cross-linked with 1% formaldehyde for 10 min at 37°C, and quenched in 125 mM glycine for 5 min at 37°C. The spheroids were then collected, lysed, and processed as previously described (Xi et al., 2018). Enriched genomic DNA was extracted with phenol:chloroform:isoamyl alcohol followed by ethanol precipitation. The ChIPed DNA was dissolved in water and analyzed by qPCR using the genomic location-specific primers (Table S8).
Flow cytometry
All FACS experiments were carried out using mice that were heterozygous for GFP. For Lgr5-EGFP+ cell purification, isolated crypts were dissociated to individual cells with TrypLE Express (Invitrogen) supplemented with 10 μM Y-27632 dihydrochloride and 0.5 mM N-acetylcysteine amide. Media (ADMEM/F12, 10 U/mL penicillin, 10 μg/mL streptomycin, 2 mM L-glutamine, 10 μM TGF-β RI Kinase Inhibitor VI, 10 μM Y-27632 dihydrochloride, and 100 nM TSA) was added to quench the reaction, then passed through 40-μm mesh to generate a single cell suspension. Lgr5-EGFP samples were enumerated and labeled with SYTOX Red (S34859, Thermo Fisher) to remove dead cells from the analysis. Lgr5-ISCs were isolated as the top 1%–3% of EGFPhi cells and matched Lgr5-progenitor/transient amplifying cells were isolated as the low to middle 1%–3% of EGFPlo cells with a BD influx sorter, BD FACS Aria III cell sorter or BD FACS Aria Fusion cell sorter (BD, Franklin Lakes, NJ, USA).. Gates were set such that no cells appeared in the GFP+ gate in negative controls. Isolated cells were utilized for immunoblotting (detailed below), ultra-low mRNA-sequencing (detailed below), or qPCR (detailed below). Analyses were performed with SoftWare.
Immunoblotting of FACS purified cells
Twenty thousand Lgr5-EGFPhi ISCs, Lgr5-EGFPlo progenitors, or Lgr5-EGFPnegative crypt cells were sorted directly into Laemmli sample buffer and boiled for 5 min as in (Beyaz et al., 2016). Samples were resolved by SDS–PAGE, transferred to 0.22 um PVDF membranes, analyzed by immunoblotting with horseradish peroxidase (HRP)-conjugated IgG secondary antibodies, and visualized using enhanced chemiluminesence (ECL) (34080, Thermo Fisher Scientific) on a G-Box imager (Syngene, Frederick, MD, USA). See Key resources table for antibodies used.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-HMGCS2 antibody [EPR8642] | Abcam | Cat#ab137043: RRID:N/A |
| Anti-Vinculin antibody [EPR8185] | Abcam | Cat#ab129002: RRID:AB_11144129 |
| H3K4me3 | Abcam | Cat#ab8580: RRID:AB_306649 |
| H3K4me1 | Abcam | Cat#ab8895: RRID:AB_306847 |
| H3K9me3 | Abcam | Cat#ab8898: RRID:AB_306848 |
| H3K27ac | Abcam | Cat#ab4729: RRID:AB_2118291 |
| H3K27me3 | Abcam | Cat#ab6002: RRID:AB_305237 |
| H3K79me2 | Abcam | Cat#ab3594: RRID:AB_303937 |
| H3K9ac | Abcam | Cat#ab4441: RRID:AB_2118292 |
| H3K14ac | Millipore | Cat#07-353: RRID:AB_310545 |
| H3 | Cell Signaling | Cat#4499: RRID:AB_10544537 |
| H4 | Abcam | Cat#ab31830: RRID:AB_1209246 |
| H3K9-β-hydroxybutyryllysine | PTM Bio | Cat#PTM-1250: RRID:N/A |
| H4K12-β-hydroxybutyryllysine | PTM Bio | Cat#PTM-1206: RRID:N/A |
| Anti-rabbit IgG, horseradish peroxidase (HRP)-linked | Cell Signaling | CAT#7074S: RRID:AB_2099233 |
| Goat Anti-Rabbit IgG (H+L) Antibody, Alexa Fluor 594 Conjugated | ThermoFisher | Cat#A-11012: RRID:AB_141359 |
| Chemicals, peptides, and recombinant proteins | ||
| TGF-β RI Kinase Inhibitor VI | Calbiochem | CAT#SB431542 |
| Y-27632 dihydrochloride | Millipore Sigma | CAT#Y0503 |
| N-acetylcysteine amide | Millipore Sigma | CAT#A0737 |
| Trichostatin A (TSA), Ready Made Solution | Millipore Sigma | CAT#T1952-200UL |
| (R)-(–)-3-Hydroxybutyric acid | Millipore Sigma | CAT#298360 |
| Corning® Matrigel® Basement Membrane Matrix, *LDEV-Free, 10 mL | Corning | CAT#356234 |
| SYTOX Red | Thermo Fisher | CAT# S34859 |
| enhanced chemiluminesence (ECL) | Thermo Fisher | CAT#34080 |
| Protease Inhibitor Cocktail Set I - Calbiochem | Millipore Millipore Sigma | CAT#539131 |
| Dynabeads Protein G for Immunoprecipitation | Thermo Fisher | CAT#10004D |
| ProLong Gold Antifade Mountant with DAPI | Thermo Fisher | CAT#P36935 |
| Borg Decloaker | Biocare Medical | CAT#BD1000 |
| Solid-Phase Reversible Immobilization (SPRI) beads; Reagent, SPRIselect, 450mL | Beckman-Coulter | CAT#B23319 |
| ADMEM/F12 | Millipore Sigma | CAT#D6421 |
| Dulbecco’s Phosphate Buffered Saline (DPBS) (Ca2+- and Mg2+-free) | Millipore Sigma | CAT#D8537-500ML |
| HBSS (Ca2+- and Mg2+-free) | Thermo Fisher | CAT#88284 |
| Invitrogen UltraPure 0.5M EDTA, pH 8.0 | Thermo Fisher | CAT#15575020 |
| L-Glutamine (200 mM) | Millipore Sigma | CAT#25030081 |
| HEPES solution | Millipore Sigma | CAT#H0887-100ML |
| 50% L-WRN conditioned media | Guo et al., 2009 | N/A |
| Pierce 16% Formaldehyde (w/v), Methanol-free | Thermo Fisher | CAT#28908 |
| TrypLE Express Enzyme (1X), phenol red | Thermo Fisher | CAT#12605028 |
| Fetal Bovine Serum (FBS) – Premium Select | Atlanta Biologicals | CAT# |
| HyClone Penicillin Streptomycin 100X Solution | Fisher Scientific | CAT#SV30010 |
| Critical commercial assays | ||
| β-Hydroxyl-butyrate LiquiColor Test | StanBio | CAT#2440-058 |
| PicoPure RNA Isolation Kit | Thermo Fisher | CAT#KIT0204 |
| SMART-Seq® v4 Ultra® Low Input RNA Kit for Sequencing | Clontech | CAT#634894 |
| Invitrogen PureLink RNA Mini Kit | Thermo Fisher | CAT#12183018A |
| High Sensitivity D1000 Reagents | Agilent Technologies | CAT#5067-5585 |
| Invitrogen Qubit dsDNA HS Assay Kit | Thermo Fisher | CAT#Q32851 |
| RNA Clean & Concentrator-25 | Zymo Research | CAT#R1017 |
| Invitrogen SuperScript III First-Strand Synthesis System | Thermo Fisher | CAT#18080051 |
| NEBNext® Ultra II DNA Library Prep Kit for Illumina® | New England Biolabs | CAT#E7645S |
| TruSeq DNA LT Sample Prep Kit | Illumina | CAT# FC-121-2001 and FC-121-2002 |
| Deposited data | ||
| Raw and analyzed data | This paper | PRJNA:506838 |
| Mouse reference genome (UCSC mm9) | Illumina iGenome | http://bowtie-bio.sourceforge.net/index.shtml |
| Ensemble annotated mouse genome features | Ensembl | https://genome.ucsc.edu/cgi-bin/hgTables |
| Experimental models: Cell lines | ||
| Primary small intestine spheroids of C57BL/6 mice | This paper | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: B6.129P2-Lgr5tm1(cre/ERT2)Cle/J | The Jackson Laboratory | JAX:008875 |
| Mouse: C57BL/6J | The Jackson Laboratory | JAX:000664 |
| Oligonucleotides | ||
| Gapdh (Mm99999915_g1) | Life Technologies | CAT#4448484 |
| Lgr5 (Mm00438890_m1) | Life Technologies | CAT#4331182 |
| Olfm4 (Mm01320260_m1) | Life Technologies | CAT#4331182 |
| Hmgcs2 (Mm00550050_m1) | Life Technologies | CAT#4331182 |
| Bdh1 (Mm00558330_m1) | Life Technologies | CAT#4331182 |
| Ascl2 (Mm01268891_g1) | Life Technologies | CAT#4331182 |
| Primers for ChIP qRT-PCR, see Table S8 | This paper | See Table S8 |
| Software and algorithms | ||
| Paired-end RNaseq mapping: Bowtie2 (v2.2.2) | Langmead and Salzberg, 2012; Trapnell et al., 2009 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Paired-end RNaseq mapping: Tophat2 (v2.0.14) | Kim et al., 2013 | https://ccb.jhu.edu/software/tophat/index.shtml |
| Gene-level read counts: HTseq | Anders et al., 2015 | https://htseq.readthedocs.io/en/release_0.11.1/ |
| Gene-level FPKM: Cufflinks | Trapnell et al., 2012 | http://cole-trapnell-lab.github.io/cufflinks/ |
| DEG analysis: DESeq2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Rawfastq ChIP-seq reads processing: snakemake based pipeline | https://github.com/crazyhottommy/pyflow-ChIPseq | |
| Raw ChIP read processing: FastQC | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ | |
| Genome alignment: Bowtie (v1.1.2) | Langmead et al., 2009 | http://bowtie-bio.sourceforge.net/index.shtml |
| Duplicate read removal: SAMBLASTER | Faust and Hall, 2014 | https://github.com/GregoryFaust/samblaster |
| Downsampling: samtools (v1.5) | Li et al., 2009 | http://samtools.sourceforge.net/ |
| ChIP-seq library visualization: deepTools (v2.4.0) | Ramírez et al., 2016 | https://deeptools.readthedocs.io/en/develop/ |
| ChIP-seq peak calling: Model-based analysis of ChIP-seq (MACS) (v1.4.2) | Zhang et al., 2008 | http://liulab.dfci.harvard.edu/MACS/00README.html |
| Kbhb binding: DiffBind R package | Stark, 2011 | https://bioconductor.org/packages/release/bioc/html/DiffBind.html |
| Histone PTM site identification: Galaxy/Cistrome | Liu et al., 2011 | http://cistrome.org/ap/root |
| Gene body heatmaps and average density plots: ngs.plot | Shen et al., 2014 | https://github.com/shenlab-sinai/ngsplot |
| Chromatin state patterns: ChromHMM | Ernst and Kellis, 2012 | http://compbio.mit.edu/ChromHMM/ |
| Gene enrichment analysis: genomic regions enrichment of annotations tool (GREAT) | McLean et al., 2010 | http://great.stanford.edu/public/html/ |
| GraphPad Prism 8.0 | GraphPad Software Inc. | https://www.graphpad.com:443/scientific-software/prism/ |
Immunofluorescence
As previously described (de la Cruz Bonilla et al., 2019), tissues were fixed in 10% neutral buffered formalin, paraffin embedded and sectioned. Antigen retrieval was performed with Borg Decloaker RTU solution (Biocare Medical). Sections were blocked using protein block (Dako). Following blocking, sections were incubated at 4°C overnight with primary antibody Hmgcs2 1:100 (ab137043, Abcam) diluted in antibody diluent (Dako). Sections were washed and then incubated with secondary antibody Alexa Fluor 594 (A-11012, ThermoFisher) for 60 min at room temperature in the same antibody diluent (Dako). Sections were washed with PBS, mounted with ProLong Gold Antifade Mountant with DAPI (Thermo Fisher. Cat. P36935). Fluorescent images were acquired using a Nikon Eclipse Ni-E microscope, with Nikon Plan Fluor 40x/1.30 objective, Andos Zyla sCMOS camera, and NISElements Advanced Research software.
Histone Extraction
Histones were extracted and purified as described earlier in Schechter et al., 2007. Briefly, small intestine crypts were homogenized in 1 mL lysis buffer (10 mM Tris-Cl pH 8, 1 mM KCl, 1.5 mM MgCl2, 1 mM DTT and 1 mM PMSF) supplemented with protease and phosphatase inhibitor cocktail. Nuclei pellet obtained by centrifugation at 10,000 g for 10 min at 4°C was resuspended in 0.2 M H2SO4 and incubated for 1 hour at 4°C. This mixture was then centrifuged at 16,000 g for 10 min at 4°C. Supernatant containing histones were precipitated by TCA (33%) for overnight at 4°C. The pellet obtained post-centrifugation at 16,000 g for 5 min at 4°C was washed twice in ice-cold acetone. The histone pellet was air-dried and suspended in 100 μL of milliQ water and stored at −20°C or −80°C.
Resolution and analysis of histones
The purified histones from small intestine crypts were resolved on 4%–12% NuPAGE Bis-Tris protein gels (Invitrogen), then transferred to a polyvinylidene difluoride (PVDF) membrane and probed with histone antibodies, H3K9bhb (PTM BioLabs#1250), H4H12bhb (PTM BioLabs#1206), H3K27ac (Abcam#4729), H3K9ac (Abcam#4441) and H3K14ac (Millipore#07–353). Histone H3(CST# D1H2) and Histone H4 (Abcam#197517) antibodies were used as loading control.
Ultra-low mRNA-sequencing
Total RNA was isolated from twenty thousand purified Lgr5-EGFPhi ISC and Lgr5-EGFPlo TA cell populations with the PicoPure RNA Isolation Kit (KIT0204, ThermoFisher) following the manufacturer’s instructions including the on column DNase I treatment step. Illumina compatible low input cDNA libraries were prepared using the Smart-Seq V4 Ultra Low Input RNA (Clontech) and KAPA Hyper-Plus Library Preparation kits according to the manufacturer’s instructions. Briefly, full length, double stranded cDNA was generated from 3 ng of Total RNA using Clontech’s SMART (Switching Mechanism at 5′ End of RNA Template) technology. The double stranded cDNA was amplified by ten cycles of LD-PCR, then purified using Ampure Beads (Agencort). Following bead elution, the cDNA was evaluated for size distribution and quantity using the TapeStation 4200 High Sensitivity DNA Kit (Agilent Technologies) and the Qubit dsDNA HS Assay Kit (ThermoFisher) respectively. The cDNA was enzymatically fragmented and 20 ng of the fragmented cDNA was used to generate Illumina compatible libraries utilizing the KAPA HyperPlus Library Preparation kit. The KAPA libraries were purified and enriched with 2 cycles of PCR to create the final cDNA library. The libraries were quantified using the Qubit dsDNA HS Assay (ThermoFisher), then multiplexed 7–8 libraries per pool. The pooled libraries were quantified by qPCR using the KAPA Library Quantification Kit (KAPA Biosystems), and assessed for size distribution using the TapeStation 4200 (Agilent Technologies). The libraries were then sequenced, one pool per lane, on the Illumina HiSeq4000 sequencer using the 76 bp paired end format. All conditions were performed in biological triplicate.
Quantitative PCR
Five thousand purified Lgr5-EGFPhi ISC, Lgr5-EGFPlo TA, and Lgr5-EGFPnegative crypt cell populations were sorted directly into RNA lysis buffer (PureLink RNA Mini Kit, Ambion) and RNA was isolated according to the manufacturer’s instructions. Total RNA was treated with TURBO DNase (Life Technologies) followed by column purification (RNA Clean & Concentrator, Zymo Research). A dynamic range test (standard curve) was performed using reverse transcription conditions to determine the amount of RNA to be used in all subsequent reactions. Undiluted total RNA from 5,000 cells was therefore converted to cDNA using the Superscript III system (Invitrogen). qPCR was performed on undiluted cDNA in duplicate for each primer and probe set using the TaqMan Gene Expression Assays (Applied Biosystems), and data were normalized to a multiplexed endogenous control, Gapdh, and further normalized to Lgr5-EGFPnegative crypt cell gene expression. No-template controls were run for each probe set and amplification was not observed for any samples. qPCR was performed on the ViiA 7 Real-Time PCR System (Life Technologies). All assays were performed in biological duplicate.
Chromatin immunoprecipitation (ChIP)
Chromatin immunoprecipitations were performed using a previously published protocol with modifications for SI crypts (Blecher-Gonen et al., 2013; Garber et al., 2012; Terranova et al., 2018). Briefly, whole isolated crypts from 4 animals were pooled per treatment per replicate and cross linked for 10 min at 37°C with 1% formaldehyde in supplemented media and quenched with 0.125M glycine for 5 min at 37°C. Crypts were washed with PBS with 1 × protease inhibitor cocktail (Millipore Sigma-Aldrich) and stored at −80°C. Pellets were thawed and then lysed for 30 min on ice with I-ChIP buffer (12mM Tris–HCl pH 8.0, 6mM EDTA pH 8.0, 0.1x PBS, 0.5% SDS) plus cOmplete mini protease inhibitors. Sonication conditions were optimized (60 cycles; 30 s on/off) for SI crypt cells using a Bioruptor (Diagnode) to achieve shear length of 250–500bp. 10% total chromatin was reserved as an input control. Chromatin was diluted 5 fold and immunoprecipitation was performed overnight by incubation of the sonicated cell lysate with 30 ul of protein G magnetic dynabeads (Invitrogen) previously coupled to target antibody for a minimum of 1h at 4°C. Immune complexes were then washed five times with cold RIPA buffer (10 mM Tris–HCl, pH 8.0, 1mM EDTA, pH 8.0, 140 mM NaCl, 1% Triton X-100, 0.1% SDS, 0.1% DOC), twice with cold high-salt RIPA buffer (10 mM Tris–HCl, pH 8.0, 1 mM EDTA, pH 8.0, 500 mM NaCl, 1% Triton X-100, 0.1% SDS, 0.1% DOC), and twice with cold LiCl buffer (10 mM Tris–HCl, pH 8.0, 1 mM EDTA, pH 8.0, 250 mM LiCl, 0.5% NP-40, 0.5% DOC). Elution and reverse cross linking was performed in 50 ul direct elution buffer (10 mM Tris–HCl, pH 8.0, 5 mM EDTA, pH 8.0, 300 mM NaCl and 0.5% SDS) with Proteinase K and RNaseA at 65°C overnight. Eluted DNA was cleaned up with solid-phase reversible immobilization (SPRI) beads (Beckman-Coulter). Antibody details are listed in the Key resources table.
ChIP-sequencing library preparation
Library preparation was performed as described in (Blecher-Gonen et al., 2013; Garber et al., 2012; Terranova et al., 2018). Briefly, enzymes from New England Biolabs were used for the following library construction processes: DNA end-repair, A-base addition, adaptor ligation, U Excision, and PCR enrichment. ChIP libraries were barcoded using TruSeq DNA LT Adapters, multiplexed together, and sequencing was performed on HiSeq 2000 (Illumina) or NextSeq 500 (Illumina).
QUANTIFICATION AND STATISTICAL ANALYSIS
Ultra-low mRNA-sequencing analysis
Paired-end RNaseq reads were mapped to the mouse reference genome (UCSC mm9) using Tophat2 (v2.0.14) (Kim et al., 2013) and Bowtie2 (v2.2.2) (Langmead and Salzberg, 2012; Trapnell et al., 2009), allowing a maximum of two mismatches per 76 bp read end. An average of 79.9 million total reads were generated per sample with a mean mapping rate of 83.1%.
Gene-level read counts were quantified using HTseq (Anders et al., 2015). For the generation of heatmaps, gene-level FPKM (Fragments Per Kilobase of transcript per Million mapped reads) values were quantified using Cufflinks (Trapnell et al., 2012). Normalization and differential expression analyses for gene/isoform expression were performed using DESeq2 (Love et al., 2014). Read counts from technical replicates were merged using the “collapseReplicates” function of the DESeq2 package. Wald test defined in the DESeq function of the package was used for differential expression analysis and shrunken log fold-changes (i.e., obtaining reliable variance estimates by pooling information across all the genes) were used for further analysis. Genes with absolute log2 fold change greater or equal to 1.5 and adjusted p value (or padj) less than 0.05 were identified as differentially expressed genes. Pathway analysis was performed using GSEA pre-ranked gene list (Mootha et al., 2003; Subramanian et al., 2005).
ChIP-seq data processing
Raw fastq reads for all ChIP-seq experiments were processed using a snakemake pipeline https://github.com/crazyhottommy/pyflow-ChIPseq as described in (Terranova et al., 2018). Briefly, raw reads were first processed using FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and uniquely mapped reads were aligned to the mm9 reference genome using Bowtie version 1.1.2 (Langmead et al., 2009). Duplicate reads were removed using SAMBLASTER (Faust and Hall, 2014) before compression to bam files. To directly compare fed and fasted ChIP-seq samples uniquely mapped reads for each mark were down-sampled per condition to 20 million, sorted and indexed using samtools version 1.5 (Li et al., 2009). To visualize ChIP-seq libraries on the IGV genome browser, we used deepTools version 2.4.0 (Ramírez et al., 2016) to generate bigWig files by scaling the bam files to reads per kilobase per million (RPKM). Super ChIP-seq tracks were generated by merging, sorting and indexing replicate bam files using samtools and scaled to RPKM using deepTools.
Identification and visualization of ChIP-seq peaks
We used Model-based analysis of ChIP-seq (MACS) version 1.4.2 (Zhang et al., 2008) peak calling algorithm with a p value threshold of 1e-5 to identify H3K9bhb, H4K12bhb and H3K27ac enrichment over “input” background. Count data for H3K9bhb and H4K12hbhb unsupervised clustering analyses were obtained using the functions dba.object and dba.peakset in the DiffBind R package version2 (Stark, 2011). Heatmaps for these sites were generated using ComplexHeatmap R package. Differential binding affinity analyses were performed using dba.analyze function in DiffBind version2.
Consensus replicate sites, unique fed and fasted sites and unique H3K9bhb, H4K12bhb or H3K27ac sites were identified using the concatenate, cluster and subtract tools from the Galaxy/Cistrome web based platform (Liu et al., 2011). Briefly, a consensus peak set was first generated by clustering intervals of replicate peaks that directly overlapped by a minimum of 1bp. Next, a shared peak set was generated by clustering intervals of fed consensus peaks that directly overlapped fasted consensus peaks by a minimum of 1bp. Unique peaks were then identified by subtracting the total number of peaks in each condition by the shared peak set. The same method was used to identify unique H3K9bhb and H4K12bhb fed and fasted sites. For the comparison of WT to iKO SI crypt cells all samples were randomly sampled to 15 million uniquely mapped reads using the same processing methods. To identify unique peaks, we identified consensus peaks in WT samples and Hmgcs2-iKO samples (fed and fasted) and compared the four datasets using intervene (Khan and Mathelier, 2017). Unique peaks between consensus WT samples and concatenated Hmgcs2-iKO samples in the fed and fasted conditions were identified using intervene (Khan and Mathelier, 2017).
Assigning ChIP-seq peaks to genes
A list of Ensembl genes was obtained from the UCSC Table browser (http://genome.ucsc.edu/). Proximal promoters were defined as ± 5kb from the transcription start site (TSS) and the gene body was defined as all genic regions outside of the +5kb promoter region. Intergenic regions were defined as all regions outside of both the proximal promoter and gene body. Kbhb peaks were assigned to genes if they overlapped the promoter by a minimum of 1bp. H3K9bhb and H3K27ac enhancers were defined as all sites outside of the proximal promoter. Gene body heatmaps and average density plots were generated using ngs.plot (Shen et al., 2014) based on all Ensembl promoters. Heatmaps for Figure 4B and Figure 6A were generated using deepTools ComputeMatrix and plotHeatmap commands based on input-subtracted bigWig files.
Chromatin state calls and differences
ChromHMM (Ernst and Kellis, 2012) was used to identify combinatorial chromatin state patterns based on the histone modifications studied. Normalized bam files were converted to bed files and binarized at a 1000bp resolution using the BinarizeBed command. We specified that ChromHMM should learn a model based on 18 chromatin states. As we considered models between 8 and 30 chromatin states, we chose an 18-state model because it is large enough to identify important functional elements while still being small enough to interpret easily. We developed an approach for characterizing epigenomic differences between the sample types. Our approach for calculating the chromatin state differences is motivated by ChromDiff (Yen and Kellis, 2015). ChromDiff is applied to gene bodies, while our approach focuses on differences in genomic regions. The method is a two-step process. First, using the segmentation calls from the ChromHMM output the entire genome is divided into non-overlapping windows of 10 Kb. We next count the number of times a chromatin state is observed in each of the 10 Kb windows and obtain a frequency matrix for each state in the ChromHMM model (E1–E18). In the second step, low variable genomic loci are removed from the frequency matrix and significant differences between two groups of samples types are calculated by using a nonparametric Mann Whitney Wilcoxon test with a P value < 0.05 for each state separately. ChromHMM labels are based on ENCODE annotations and the genomic location encompassing distinct combinations of different chromatin marks.
ChIP-seq pathway analysis
Gene enrichment analysis was performed using MSigDB pathways from genomic regions enrichment of annotations tool (GREAT) (McLean et al., 2010). To associate H3K9bhb and H4K12bhb genomic regions with genes we used the basal plus extension setting for both 5kb upstream and 5kb downstream of the TSS with a 50kb distal extension. For H3K27ac and H3K27ac-H3K9bhb genomic regions we used the same promoter setting with a 1000kb distal extension. All pathways are significant based on a binomial FDR q-value. Figure 2I and Figure 6I were performed using GSEA/MSigDB (Subramanian et al., 2005) HALLMARK pathways based on ensemble gene lists from peaks within ± 5kb of the TSS and differentially expressed genes from RNA-seq.
Other statistical analysis
Parametric data are expressed at mean ± SEM. Statistical analyses included in Figure legends.
Supplementary Material
Highlights.
Small intestine (SI) crypt cells produce HMGCS2 and β-OHB in response to fasting
β-OHB enriches H3K9bhb at regulatory elements of metabolic genes in SI crypt cells
H3K9bhb enrichment correlates with activation of metabolic pathways in SI crypt cells
Intestinal Hmgcs2 loss decreases H3K9bhb and expression of fasting-responsive genes
ACKNOWLEDGMENTS
The authors thank all members of the Piwnica-Worms and Rai laboratories for constructive criticism throughout the study. We also thank Erika Thompson for assistance with ultra-low mRNA sequencing, Deepavali Chakravarti for technical assistance, Karen Clise-Dwyer for flow cytometry insight, and Ming Tang for programming source codes. Mark Bedford is thanked for assistance in running histone acetylation reader arrays. Experiments performed in this study utilized the MDACC Flow Cytometry and Cellular Imaging Core Facility, funded by National Cancer Institute Cancer Center Support Grant CA016672. Additional funding sources that supported this work include the National Cancer Institute of the National Institutes of Health under award R01CA207236 (to H.P.W.). K.R. and C.J.T. were supported by R00160578 and R01CA245395 from the NCI, RP170407 from CPRIT, a research scholar award from the American Cancer Society, and the Center for Cancer Epigenetics at MDACC. Support for M.C.B. came from T32CA186892 and F31CA210631.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.110044.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Al-Asmari AK, Khan AQ, Al-Asmari SA, Al-Rawi A, and Al-Omani S (2016). Alleviation of 5-fluorouracil-induced intestinal mucositis in rats by vitamin E via targeting oxidative stress and inflammatory markers. J. Complement. Integr. Med 13, 377–385. [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, and Clevers H (2007). Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449, 1003–1007. [DOI] [PubMed] [Google Scholar]
- Basivireddy J, Vasudevan A, Jacob M, and Balasubramanian KA (2002). Indomethacin-induced mitochondrial dysfunction and oxidative stress in villus enterocytes. Biochem. Pharmacol 64, 339–349. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. (2006). A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125, 315–326. [DOI] [PubMed] [Google Scholar]
- Beyaz S, Mana MD, Roper J, Kedrin D, Saadatpour A, Hong SJ, Bauer-Rowe KE, Xifaras ME, Akkad A, Arias E, et al. (2016). High-fat diet enhances stemness and tumorigenicity of intestinal progenitors. Nature 531, 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birney E, Stamatoyannopoulos JA, Dutta A, Guigó R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE, et al. ; ENCODE Project Consortium; NISC Comparative Sequencing Program; Baylor College of Medicine Human Genome Sequencing Center; Washington University Genome Sequencing Center; Broad Institute; Children’s Hospital Oakland Research Institute (2007). Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447, 799–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blecher-Gonen R, Barnett-Itzhaki Z, Jaitin D, Amann-Zalcenstein D, Lara-Astiaso D, and Amit I (2013). High-throughput chromatin immunoprecipitation for genome-wide mapping of in vivo protein-DNA interactions and epigenomic states. Nat. Protoc 8, 539–554. [DOI] [PubMed] [Google Scholar]
- Cheng CW, Biton M, Haber AL, Gunduz N, Eng G, Gaynor LT, Tripathi S, Calibasi-Kocal G, Rickelt S, Butty VL, et al. (2019). Ketone Body Signaling Mediates Intestinal Stem Cell Homeostasis and Adaptation to Diet. Cell 178, 1115–1131.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotter DG, Schugar RC, Wentz AE, d’Avignon DA, and Crawford PA (2013). Successful adaptation to ketosis by mice with tissue-specific deficiency of ketone body oxidation. Am. J. Physiol. Endocrinol. Metab 304, E363–E374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. (2010). Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 107, 21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Cruz Bonilla M, Stemler KM, Taniguchi CM, and Piwnica-Worms H (2018). Stem cell enriched-epithelial spheroid cultures for rapidly assaying small intestinal radioprotectors and radiosensitizers in vitro. Sci. Rep 8, 15410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Cruz Bonilla M, Stemler KM, Jeter-Jones S, Fujimoto TN, Molkentine J, Asencio Torres GM, Zhang X, Broaddus RR, Taniguchi CM, and Piwnica-Worms H (2019). Fasting Reduces Intestinal Radiotoxicity, Enabling Dose-Escalated Radiation Therapy for Pancreatic Cancer. Int. J. Radiat. Oncol. Biol. Phys 105, 537–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, and Kellis M (2010). Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat. Biotechnol 28, 817–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, and Kellis M (2012). ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods 9, 215–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust GG, and Hall IM (2014). SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics 30, 2503–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber M, Yosef N, Goren A, Raychowdhury R, Thielke A, Guttman M, Robinson J, Minie B, Chevrier N, Itzhaki Z, et al. (2012). A high-throughput chromatin immunoprecipitation approach reveals principles of dynamic gene regulation in mammals. Mol. Cell 47, 810–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther MG, Levine SS, Boyer LA, Jaenisch R, and Young RA (2007). A chromatin landmark and transcription initiation at most promoters in human cells. Cell 130, 77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Longshore S, Nair R, and Warner BW (2009). Retinoblastoma protein (pRb), but not p107 or p130, is required for maintenance of enterocyte quiescence and differentiation in small intestine. J. Biol. Chem 284, 134–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakvoort TB, Moerland PD, Frijters R, Sokolović A, Labruyère WT, Vermeulen JL, Ver Loren van Themaat E, Breit TM, Wittink FR, van Kampen AH, et al. (2011). Interorgan coordination of the murine adaptive response to fasting. J. Biol. Chem 286, 16332–16343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al. (2009). Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Zhang D, Weng Y, Delaney K, Tang Z, Yan C, Qi S, Peng C, Cole PA, Roeder RG, and Zhao Y (2021). The regulatory enzymes and protein substrates for the lysine β-hydroxybutyrylation pathway. Sci. Adv 7, eabe2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenuwein T, and Allis CD (2001). Translating the histone code. Science 293, 1074–1080. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Furumichi M, Tanabe M, Sato Y, and Morishima K (2017). KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45 (D1), D353–D361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A, and Mathelier A (2017). Intervene: a tool for intersection and visualization of multiple gene or genomic region sets. BMC Bioinformatics 18, 287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozar S, Morrissey E, Nicholson AM, van der Heijden M, Zecchini HI, Kemp R, Tavaré S, Vermeulen L, and Winton DJ (2013). Continuous clonal labeling reveals small numbers of functional stem cells in intestinal crypts and adenomas. Cell Stem Cell 13, 626–633. [DOI] [PubMed] [Google Scholar]
- Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, et al. ; Roadmap Epigenomics Consortium (2015). Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G, Origanti S, White LS, Sun J, Stappenbeck TS, and Piwnica-Worms H (2011). Contributions made by CDC25 phosphatases to proliferation of intestinal epithelial stem and progenitor cells. PLoS ONE 6, e15561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Chong MF, Liu JC, Libedinsky C, Gooley JJ, Chen S, Wu T, Tan V, Zhou M, Meaney MJ, et al. (2013). Dietary disinhibition modulates neural valuation of food in the fed and fasted states. Am. J. Clin. Nutr 97, 919–925. [DOI] [PubMed] [Google Scholar]
- Li J, Ng EK, Ng YP, Wong CY, Yu J, Jin H, Cheng VY, Go MY, Cheung PK, Ebert MP, et al. (2009). Identification of retinoic acid-regulated nuclear matrix-associated protein as a novel regulator of gastric cancer. Br. J. Cancer 101, 691–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Ortiz JA, Taing L, Meyer CA, Lee B, Zhang Y, Shin H, Wong SS, Ma J, Lei Y, et al. (2011). Cistrome: an integrative platform for transcriptional regulation studies. Genome Biol. 12, R83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maegawa S, Lu Y, Tahara T, Lee JT, Madzo J, Liang S, Jelinek J, Colman RJ, and Issa JJ (2017). Caloric restriction delays age-related methylation drift. Nat. Commun 8, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, and Bejerano G (2010). GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihaylova MM, Cheng CW, Cao AQ, Tripathi S, Mana MD, Bauer-Rowe KE, Abu-Remaileh M, Clavain L, Erdemir A, Lewis CA, et al. (2018). Fasting Activates Fatty Acid Oxidation to Enhance Intestinal Stem Cell Function during Homeostasis and Aging. Cell Stem Cell 22, 769–778.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi H, and Stappenbeck TS (2013). In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat. Protoc 8, 2471–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi H, Ajima R, Luo CT, Yamaguchi TP, and Stappenbeck TS (2012). Wnt5a potentiates TGF-β signaling to promote colonic crypt regeneration after tissue injury. Science 338, 108–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorefield EC, Andres SF, Blue RE, Van Landeghem L, Mah AT, Santoro MA, and Ding S (2017). Aging effects on intestinal homeostasis associated with expansion and dysfunction of intestinal epithelial stem cells. Aging (Albany NY) 9, 1898–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Bunkenborg J, Olsen JV, Hjerrild M, Wisniewski JR, Stahl E, Bolouri MS, Ray HN, Sihag S, Kamal M, et al. (2003). Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell 115, 629–640. [DOI] [PubMed] [Google Scholar]
- Puchalska P, and Crawford PA (2017). Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 25, 262–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, and Wysocka J (2011). A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470, 279–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44 (W1), W160–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritsma L, Ellenbroek SIJ, Zomer A, Snippert HJ, de Sauvage FJ, Simons BD, Clevers H, and van Rheenen J (2014). Intestinal crypt homeostasis revealed at single-stem-cell level by in vivo live imaging. Nature 507, 362–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh TY, Cuddapah S, Cui K, and Zhao K (2006). The genomic landscape of histone modifications in human T cells. Proc. Natl. Acad. Sci. USA 103, 15782–15787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, and Clevers H (2009). Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459, 262–265. [DOI] [PubMed] [Google Scholar]
- Shen L, Shao N, Liu X, and Nestler E (2014). ngs.plot: Quick mining and visualization of next-generation sequencing data by integrating genomic databases. BMC Genomics 15, 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, Grueter CA, Lim H, Saunders LR, Stevens RD, et al. (2013). Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 339, 211–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons BD, and Clevers H (2011). Stem cell self-renewal in intestinal crypt. Exp. Cell Res 317, 2719–2724. [DOI] [PubMed] [Google Scholar]
- Stark RBG (2011). DiffBind: differential binding analysis of ChIP-Seq peak data (Bioconductor).
- Strahl BD, and Allis CD (2000). The language of covalent histone modifications. Nature 403, 41–45. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terranova C, Tang M, Orouji E, Maitituoheti M, Raman A, Amin S, Liu Z, and Rai K (2018). An Integrated Platform for Genome-wide Mapping of Chromatin States Using High-throughput ChIP-sequencing in Tumor Tissues. J. Vis. Exp (134) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinkum KL, Stemler KM, White LS, Loza AJ, Jeter-Jones S, Michalski BM, Kuzmicki C, Pless R, Stappenbeck TS, Piwnica-Worms D, and Piwnica-Worms H (2015). Fasting protects mice from lethal DNA damage by promoting small intestinal epithelial stem cell survival. Proc. Natl. Acad. Sci. USA 112, E7148–E7154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tognini P, Murakami M, Liu Y, Eckel-Mahan KL, Newman JC, Verdin E, Baldi P, and Sassone-Corsi P (2017). Distinct Circadian Signatures in Liver and Gut Clocks Revealed by Ketogenic Diet. Cell Metab. 26, 523–538.e5. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, and Salzberg SL (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, and Pachter L (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Y, Shi J, Li W, Tanaka K, Allton KL, Richardson D, Li J, Franco HL, Nagari A, Malladi VS, et al. (2018). Histone modification profiling in breast cancer cell lines highlights commonalities and differences among sub-types. BMC Genomics 19, 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Zhang D, Chung D, Tang Z, Huang H, Dai L, Qi S, Li J, Colak G, Chen Y, et al. (2016). Metabolic Regulation of Gene Expression by Histone Lysine β-Hydroxybutyrylation. Mol. Cell 62, 194–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen A, and Kellis M (2015). Systematic chromatin state comparison of epigenomes associated with diverse properties including sex and tissue type. Nat. Commun 6, 7973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Behbod F, Atkinson RL, Landis MD, Kittrell F, Edwards D, Medina D, Tsimelzon A, Hilsenbeck S, Green JE, et al. (2008). Identification of tumor-initiating cells in a p53-null mouse model of breast cancer. Cancer Res. 68, 4674–4682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Cao R, Niu J, Yang S, Ma H, Zhao S, and Li H (2019). Molecular basis for hierarchical histone de-β-hydroxybutyrylation by SIRT3. Cell Discov. 5, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The ChIP and mRNA sequencing data have been deposited in the NCBI GEO BioProject database with the following accession number PRJNA:506838.
Code used for unsupervised clustering of chromatin states is available on https://rpubs.com/cjt5 and https://github.com/sccallahan/ChromXploreR.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.