

Abstract
We investigated the mechanisms associated with E22K mutation in myosin regulatory light chain (RLC), found to cause hypertrophic cardiomyopathy (HCM) in humans and mice. Specifically, we characterized the mechanical profiles of papillary muscle fibers from transgenic mice expressing human ventricular RLC wild-type (Tg-WT) or E22K mutation (Tg-E22K). Because the two mouse models expressed different amounts of transgene, the B6SJL mouse line (NTg) was used as an additional control. Mechanical experiments were carried out on Ca2+- and ATP-activated fibers and in rigor. Sinusoidal analysis was performed to elucidate the effect of E22K on tension and stiffness during activation/rigor, tension-pCa, and myosin cross-bridge (CB) kinetics. We found significant reductions in active tension (by 54%) and stiffness (active by 40% and rigor by 54%). A decrease in the Ca2+ sensitivity of tension (by ΔpCa ~ 0.1) was observed in Tg-E22K compared with Tg-WT fibers. The apparent (=measured) rate constant of exponential process B (2πb: force generation step) was not affected by E22K, but the apparentrate constant of exponential process C (2πc: CB detachment step) was faster in Tg-E22K compared with Tg-WT fibers. Both 2πb and 2πc were smaller in NTg than in Tg-WT fibers, suggesting a kinetic difference between the human and mouse RLC. Our results of E22K-induced reduction in myofilament stiffness and tension suggest that the main effect of this mutation was to disturb the interaction of RLC with the myosin heavy chain and impose structural abnormalities in the lever arm of myosin CB. When placed in vivo, the E22K mutation is expected to result in reduced contractility and decreased cardiac output whereby leading to HCM.
Keywords: Ca2+ sensitivity, papillary muscle, skinned fiber, stiffness, tension
Introduction
Hypertrophic cardiomyopathy (HCM) is a disease of the myocardium characterized by asymmetric hypertrophy of the left ventricle (LV), myocardial fibrosis, and impaired diastolic function. Recent evidence suggests the prevalence of HCM is about 1 : 200 of the general population, which is higher than previously estimated (1 in 500 [1,2]). Familial HCM is caused by mutations in genes encoding major sarcomeric proteins that include the β-myosin heavy chain (β-MHC), cardiac myosin binding protein C, tropomyosin (Tm), three subunits of troponin (TnT, TnI, and TnC), actin, and titin [3]. HCM-associated mutations in myosin light chains (MLCs) are rarer, causing varying degrees of HCM, but they often result in malignant outcomes indicating important functional roles that MLCs play in cardiac muscle contraction and heart performance [4]. One HCM variant, which is the focus of this investigation, is caused by an E22K mutation in the ventricular myosin regulatory light chain (RLC, MYL2 gene), and was characterized by mid-left ventricular obstruction phenotype due to massive papillary muscle hypertrophy [5]. In a later study by Kabaeva et al., this E22K mutation was associated with moderate septal hypertrophy, late onset of clinical manifestation, and benign disease course, and prognosis [6]. Furthermore, Claes et al. reported that in addition to E22K expression, copresence of additional risk factors for hypertrophy such as hypertension, obesity, or other sarcomeric gene mutation in MYL2-E22K-positive patients was seen to increase disease penetrance and observe a full manifestation of HCM phenotype [7].
Initial attempts to use the E22K-RLC mutation in mice were disappointing, as the animals did not exhibit any of the HCM symptoms observed in humans [8]. This shortfall was later overcome by the generation of ‘humanized’ transgenic (Tg) mice overexpressing the E22K mutant of the human ventricular RLC [9]. Histologically, the hearts of Tg-E22K animals resembled those of human patients although the mice had normal echocardiograms compared with Tg-WT-RLC and NTg animals. The close morphological resemblance of HCM between E22K-positive patients and Tg-E22K mice has made this mouse model highly suitable to study the molecular mechanisms of MYL2-E22K-associated human disease [9–11], and the subject of the current investigation. The 2005 investigation [9], as well as 2007 study [11], performed on the hearts of humanized Tg-E22K mice, suggested that perturbations in the Ca2+-binding properties to RLC and reduced [Ca2+] and force transients were responsible for altered intracellular calcium homeostasis, myofilament dysfunction, and abnormal heart function in Tg-E22K mice that ultimately resulted in HCM. Solution studies on human recombinant E22K-RLC mutant indeed demonstrated that apparent calcium association constant, KCa, for E22K mutant was ~ 17-fold lower compared with human recombinant WT-RLC protein [12]. No changes in the KCa value were observed when human recombinant E22K mutant was reconstituted in skinned porcine cardiac muscle fibers [13].
In this report, we focused on the mechanical properties of humanized Tg-E22K myocardium and studied the isometric tension, Ca2+ sensitivity of force, active and rigor stiffness, and cross-bridge (CB) kinetics in skinned papillary muscle fibers from LVs of Tg-E22K hearts compared with Tg-WT. Because both mouse models contained a different expression of human RLC, with 20% reported for Tg-WT and 80% for Tg-E22K [9], all experiments also included the B6SJL mouse line (NTg) as an additional control. Rate constants of a CB cycle were deduced from tension transients induced by sinusoidal length perturbations during maximal Ca2+ activation, as described previously [14–16]. We found a 54% decrease in active tension and slightly decreased Ca2+ sensitivity of force (by ΔpCa ~ 0.1). We also observed reductions in the active (~ 40%) and rigor (~ 54%) stiffness in Tg-E22K compared with Tg-WT mice. The data are discussed in light of the previously observed physiological and biophysical properties of the E22K myocardium and the potential structural destabilization of the myosin lever arm domain that might be triggered by E22K-induced changes in the biochemical properties of RLC.
Results
Comparison of the amino acid sequences of human (NCBI #P10916) vs mouse (NCBI #P51667) ventricular RLC (MYL2 gene) shows 96.4% of sequence identity in 166 residues overlap (Fig. 1A). Despite the small difference between both sequences, depicted by the four amino acids located at the N-terminal region of RLC (Fig. 1A, amino acids labeled in black), both isoforms present with similar molecular weights (huRLC = 18 789 Da vs mseRLC = 18 864 Da). Therefore, both transgenic mouse models expressed the myc-tagged human ventricular RLC causing its slower migration on SDS/PAGE and allowing for identification of human vs mouse RLC [9] (Fig. 1B). Similar to what we reported earlier [9], the expression of human RLC in myofibrils purified from Tg-WT and Tg-E22K lines was ~ 26% and ~ 84%, respectively (Fig. 1B,C). Because of these differences in transgenic protein expression between the models, all biochemical and muscle mechanics experiments also included the B6SJL mouse line (NTg).
Fig. 1.
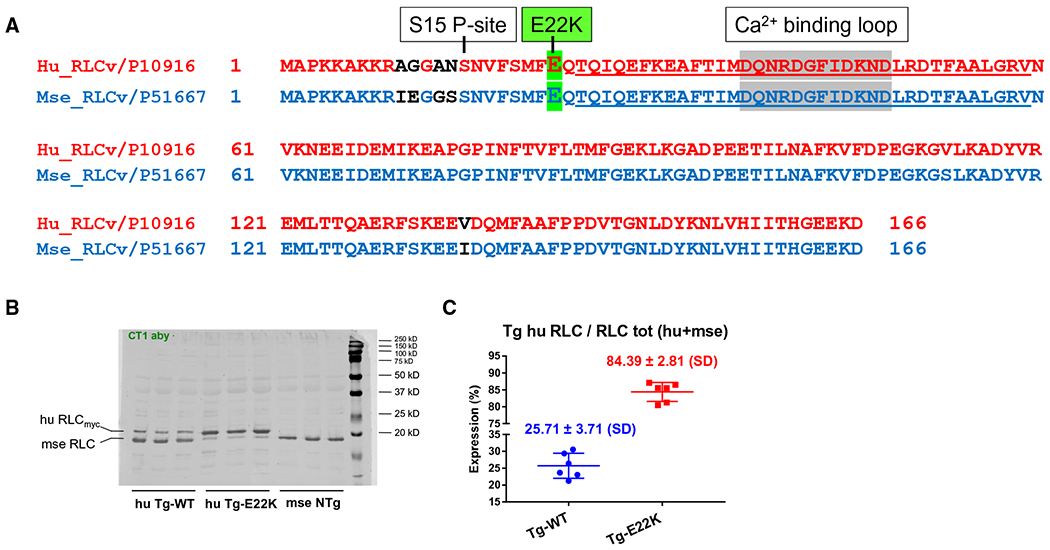
(A) Alignment of amino acid sequences of human (NCBI# P10916) vs mouse (NCBI #P51667) ventricular RLC (MYL2 gene). Note 96.4% identity in 166 residues overlap; score: 828.0; gap frequency: 0.0%. Highlighted in green is the site of E22K mutation. Underlined is the EF-hand helix–loop–helix Ca2+ binding site of RLC. Amino acid residues that are different in the sequence of human vs mouse RLC are indicated in black. The alignment and analysis of sequences were made with the EMBL-EBI Programme. (B) SDS/PAGE and western blotting against myosin RLC protein of myofibrillar preparation purified from the hearts (two hearts per group) of humanized Tg-WT and Tg-E22K mice compared with NTg mice. The blot presents samples from n = 3 different animals that were run independently on two different SDS/PAGE. Note, a slower migration of the human RLC was due to a ’myc’ tag attached to the N terminus of RLC. CT1 antibody, specific for myosin RLC protein was used. (C) Quantification of human RLC expression in myofibrils from the hearts of Tg-WT and Tg-E22K mice. Three hearts per group were used.
As shown in Fig. 1A, the E22K mutation in the myosin RLC is flanked by the N-terminal Ser15 phosphorylation site (S15 P-site) and the EF-hand helix–loop–helix Ca2+-binding site of RLC (Fig. 1A, underlined sequence: T24 to V59). Considering that the E22K mutation is located just one residue away from the incoming EF-hand helix of the Ca2+-binding site of the RLC, it is not surprising that a significant alteration (~ 17-fold decrease) in the Ca2+-binding (KCa) to the isolated human ventricular recombinant E22K-RLC protein was observed previously [12]. However, the apparent Ca2+ dissociation constants (Kapp) of E22K-RLC-reconstituted porcine cardiac myofibrils and fibers were unchanged compared with WT-RLC-reconstituted preparations [13]. Studies with humanized transgenic Tg-E22K mice carried out previously [9,11] also reported the different effect of the E22K mutation on the Ca2+ sensitivity of force (pCa50) that either increased [9] or was unchanged [11] in skinned papillary muscle fibers from Tg-E22K compared with Tg-WT hearts.
E22K mutation decreases Ca2+ sensitivity of force development and reduces Ca2+-activated tension
To address these discrepancies and elucidate the effect of E22K on the tension–pCa relationship, the development of steady-state force was recorded in skinned papillary muscle fibers from Tg-E22K, Tg-WT, and NTg hearts (Fig. 2). The experimental protocol is schematically presented in Fig. 2A. Fibers were first relaxed (R), and then, they were immersed in standard activating (A) solution followed by a brief relaxation and the force–pCa measurements in a solution of increasing Ca2+ concentrations (see Materials and methods for details on the composition of solutions). To test the reproducibility, the standard activation was repeated twice, before and after the force–pCa dependence, ensuring a steady level of tension (Fig. 2A). Subsequently, the fibers were washed off the ATP and the ‘Rigor’ was induced. To ensure complete removal of ATP, the rigor solution (free of ATP and phosphocreatine) was applied three times, as marked by solution change artifacts (Fig. 2A). As seen in Fig. 2A, rigor tension initially increased, peaked, and then declined to reach the steady-state level, which was followed by the 2nd and 3rd solution changes. The decline in rigor tension is thought to be due to the rigor CBs dissociating from actin because of the high force that is applied to the fiber in rigor. But as soon as the CB dissociates from actin, it attaches to the next available actin site because of the high affinity of the nucleotide-free myosin to actin [17,18].
Fig. 2.
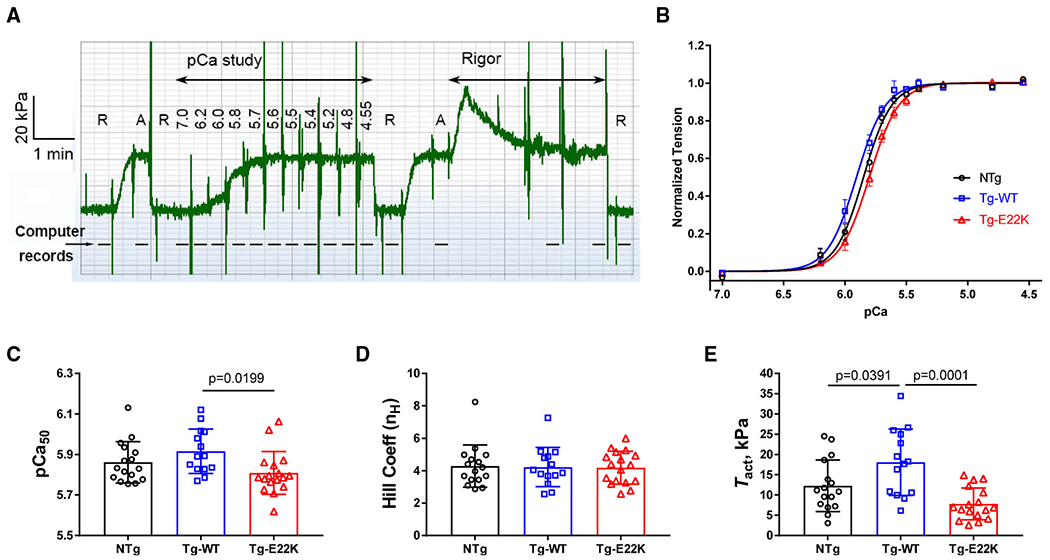
Tension–pCa study in skinned papillary muscle fibers from Tg-E22K (N = 16), Tg-WT (N = 14), and NTg (N = 17) hearts. (A) Experimental protocol in a slow time course record of force for Tg-WT (18LW119X2). R = relaxing solution, A = standard activating solution, and Rigor = rigor solution. Numbers under ’pCa study’ indicate pCa values. Most solutions were administered once (as seen by solution change artifacts). The rigor solution was applied three times. This time course was recorded every 0.1 s without filter; thus, large solution change artifacts are visible. The dashed line includes the records of sinusoidal analyses. (B) The tension–pCa relationship in papillary muscle fibers from the hearts of Tg-E22K, Tg-WT, and NTg mice. Discrete points (mean ± SEM are shown at each pCa) are from experimental observations, and continuous curves represent best-fit results to Eq. 1. (C, D) Ca2+ sensitivity (pCa50) and the Hill coefficient (nH) of tension–pCa relationship. (E) Ca2+-activated tension (Tact) in kPa in a standard activating solution. Bar graphs with individual fiber data are shown in C-E. Data are the mean ± SD. Significance was calculated by one-way ANOVA.
The plot of tension–pCa for NTg, Tg-WT, and Tg-E22K mice is presented in Fig. 2B. Experimental data were individually fitted to a 4-parameter Hill equation (Eq. 1):
| (1) |
where TLC = low [Ca2+] tension, Tact = Ca2+ activatable tension, Ca50 = [Ca2+] at half Tact, and nH = cooperativity (Hill coefficient). Ca50 is the ‘apparent Ca2+ dissociation constant’, and pCa50 = −log10Ca50 is called ‘Ca2+ sensitivity’. TLC defines baseline tension or ‘low Ca2+ tension’ (tension at pCa ≥ 7). Due to the noise associated with tension measurements, basal tension is represented by TLC depicting an averaged value of tension under the relaxing condition.
The best-fit curves are represented by smooth lines in Fig. 2B, while the fitted parameters are shown in Fig. 2C–E. The difference between two controls, NTg and Tg-WT, was found to be statistically insignificant, but the curve for Tg-E22K was right-shifted compared with Tg-WT indicating a decreased myofilament Ca2+ sensitivity of force in Tg-E22K hearts (Fig. 2B,C). Average pCa50 values ± SEM were 5.862 ± 0.102 (N = 16) for NTg, 5.916 ± 0.110 (N = 14) for Tg-WT, and 5.809 ± 0.106 (N = 17) for Tg-E22K fibers (Fig. 2C). While pCa50 was not significantly different between NTg and Tg-WT, it was significantly lower in Tg-E22K compared with Tg-WT, with a difference of 0.107 pCa units (Fig. 2C). The latter result is in accord with our solution study showing a mutation-induced decrease in the Ca2+ binding affinity to recombinant human cardiac RLC [12]. The Hill coefficient (nH), depicting the myofilament cooperativity, was not different between the three genotypes (Fig. 2D). The Ca2+-activated tension (Tact) was highest in Tg-WT fibers: Tact = 18.11 ± 8.22 (SD) kPa, which was statistically different from NTg: Tact = 12.29 ± 6.39 (SD) kPa, and Tg-E22K: Tact = 7.82 ± 3.93 (SD) kPa fibers (Fig. 2E). The similarity between the plots represents good reproducibility of the data obtained for each of the three genotypes. Both isometric tension and stiffness (Y∞) were significantly less in Tg-E22K than in Tg-WT, and so did their ratio (Tension/Y∞).
Tg-E22K fibers show decreased stiffness and altered complex modulus in standard activation study
As presented in Fig. 2A, fiber preparations from the three groups of mice were initially tested in the standard activating solution, pCa = 4.55. Then, measurements of isometric tension during Ca2+ activation, pCa-tension, rigor, and sinusoidal analysis (range: 0.35–70 Hz in 15 frequencies at 0.25% amplitude) were performed as described previously [14,16]. The plot of isometric tension is shown in Fig. 3A, the associated fiber stiffness in Fig. 3B, and their ratio in Fig. 3C. The isometric tension plot is similar in appearance to Tact (Fig. 2E) because the plots represent the same property of the fibers, except that the measurements were done separately, and many more preparations were used to plot Fig. 3A than Fig. 2E. The similarity between the plots represents a good reproducibility of the data obtained for each of the three genotypes.
Fig. 3.
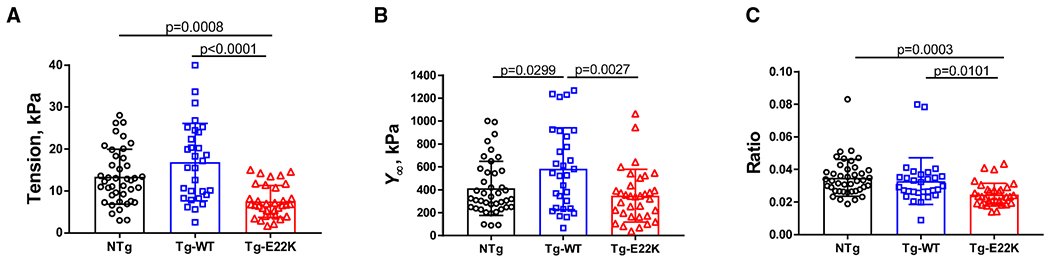
Standard activation study in Tg-E22K, Tg-WT, and NTg mice. (A) Isometric tension; (B) stiffness (Y∞); and (C) tension/stiffness (Y∞) ratio. The number of experimental fibers was as follows: N = 41 for NTg, N = 30 for Tg-WT, and N = 34 for Tg-E22K. Data are the mean ± SD, and the statistics were analyzed by one-way ANOVA.
The E22K mutation was observed to significantly decrease (56%) the level of tension compared with NTg and Tg-WT controls (Fig. 3A). We also observed a 40% reduction in fiber stiffness (Y∞) in Tg-E22K compared with Tg-WT muscles (Fig. 3B). The stiffness was also 30% less in NTg muscles than in Tg-WT, suggesting that some amino acid sequence differences between the mouse RLC of NTg myocardium and human RLC of Tg-WT (Fig. 1A) are responsible for altered interaction of the RLC with its immediate binding partner, the MHC. Figure 3C demonstrates the ratio of tension/stiffness, which approximates the amount of force generated by attached CBs to the total number of CBs. Tg-E22K fibers demonstrated the lowest ratio of force/CB compared with NTg and Tg-WT fibers indicating that the mutation leads to a reduced force that an E22K CB can develop (Fig. 3C).
During standard activation, the sinusoidal analysis was performed, and complex modulus data Y(f) were collected at 15 frequencies between 0.35 and 70 Hz. The frequency range was limited to ≤ 70 Hz, due to noise observed at 100 Hz. The complex modulus data for NTg, Tg-WT, and Tg-E22K fibers are presented in Fig. 4. The data are shown in the Nyquist plots (Fig. 4A), viscous modulus (VM) vs frequency (Fig. 4B), and elastic modulus (EM) vs frequency (Fig. 4C). Discrete points are from experimental observations, in which filled symbols are used to indicate 1 and 11 Hz frequency points. The Nyquist plots (Fig. 4A) are convenient to identify exponential processes because each process consists of a semicircle with its center on the x-axis. Fig. 4A indicates that there are at least two exponential processes B and C. The VM (Fig. 4B) indicates the frequency-dependent viscosity; it has a major peak at 25–50 Hz, which approximates frequency c (Eq. 2). The EM (Fig. 4C) indicates the frequency-dependent elasticity; it has a minimum at 7–11 Hz, which approximates frequency b (Eq. 2). The complex modulus data were fitted to Eq. 2:
| (2) |
Fig. 4.
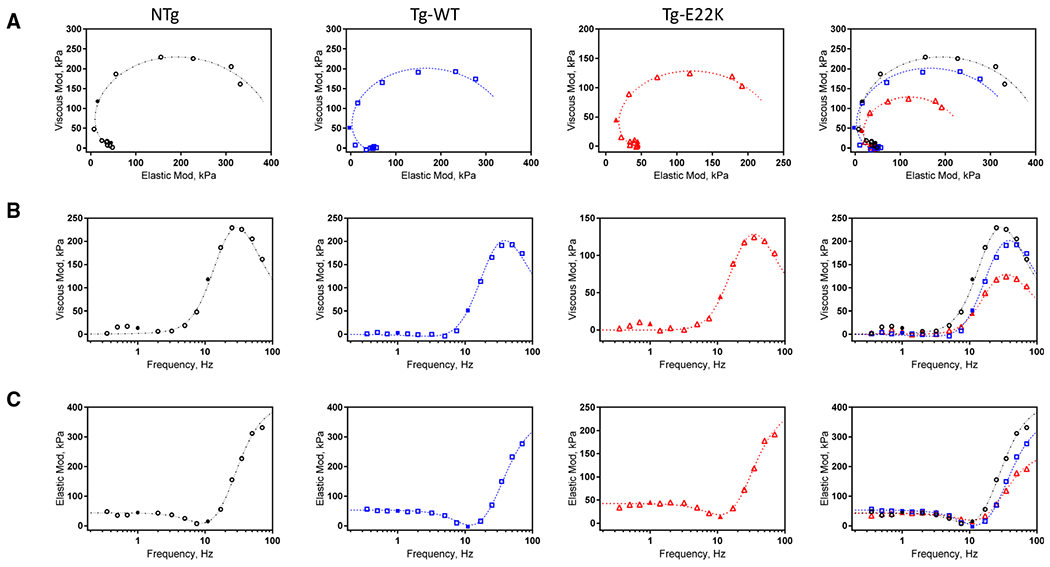
Complex modulus (Y(f) of the standard activation for NTg, Tg-WT, and Tg-E22K fibers. The data are shown in the Nyquist plots (A), VM vs frequency plots (B), and EM vs frequency plots (C). Discrete points are from experimental observations, in which filled symbols are used to indicate 1Hz and 11Hz frequency points. Continuous curves represent best-fit results to Eq. 2. Data are shown as averages from standard activation experiments performed in 10–12 fibers.
Continuous curves in Fig. 4 represent the best-fit results to Eq. 2, where H is a constant and , b and c are characteristic frequencies of exponential processes B and C, respectively, as previously described [19]. 2πb and 2πc are apparent (=measured) rate constants of processes B and C, respectively, with 2πb depicting the CB recruitment (force generation) rate and 2πc depicting the CB detachment rate [19–21].
The complex modulus extrapolated to the infinite frequency (f →∞) is called ‘stiffness’ and is defined in Eq. 3. This property is the EM of active fibers.
| (3) |
As presented in Fig. 4, in the frequency range 1–25 Hz, the E22K mutation was observed to exhibit the lowest values of VM (Fig. 4B, b ~ 3.2 Hz) and EM (Fig. 4C, c ~ 11 Hz).
E22K mutation increases CB detachment rate 2πc with standard activation
The kinetic parameters of the standard activation for Tg-E22K, Tg-WT and NTg are presented in Fig. 5. The apparent rate constant 2πb was slightly higher in Tg-WT compared with NTg fibers, but no changes were observed in Tg-E22K mice compared with NTg and Tg-WT controls (Fig. 5A). However, the apparent rate constant 2πc was significantly faster in Tg-E22K fibers compared with Tg-WT and NTg preparations (Fig. 5B). As 2πc is influenced by myosin detachment rate from actin, a faster 2πc would mean that the myosin heads stay bound to actin for less time, which would contribute to the observed decrease in force and stiffness in Tg-E22K fibers (Fig. 3). Both magnitude parameters B and C were significantly larger in Tg-WT compared with NTg and Tg-E22K fibers (Fig. 5C,D). The B and C processes characterize work-producing (CB recruitment or force generation) and work-absorbing (CB detachment) responses, respectively [19,22], and the fact that the E22K mutant was observed to have the lowest B (Fig. 5C) and C (Fig. 5D) among all three genotypes indicates the poor mechanical performance of E22K fibers.
Fig. 5.
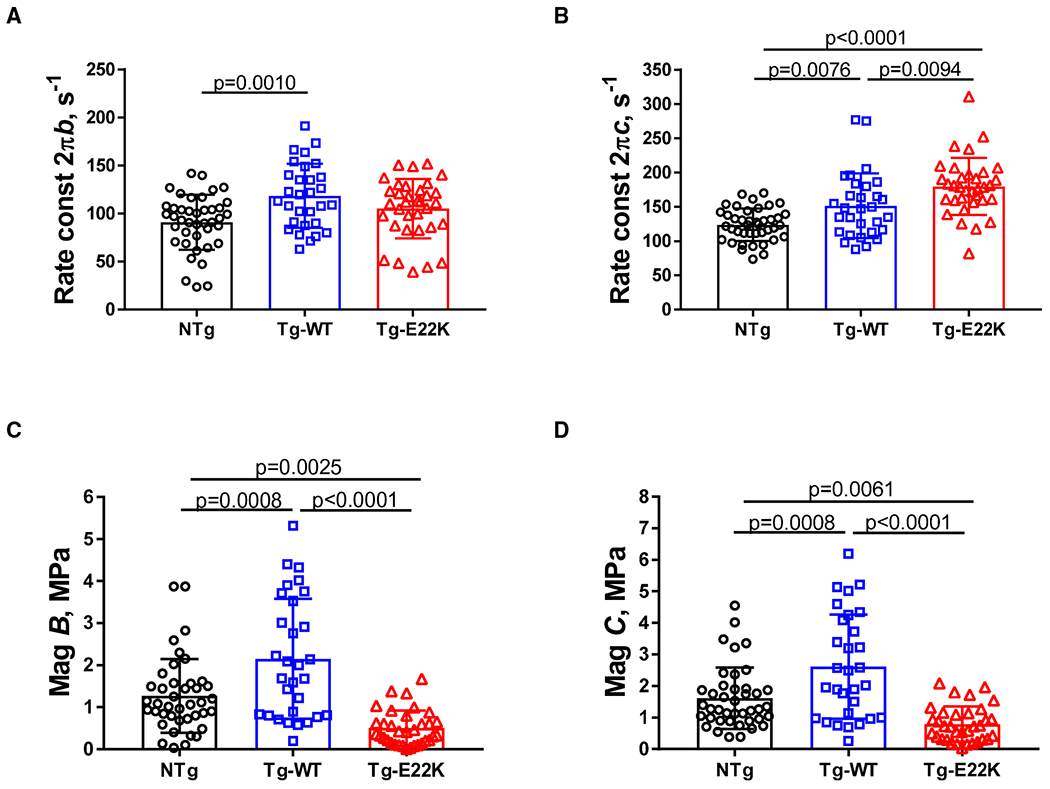
Kinetic parameters of the standard activation for Tg-E22K, Tg-WT, and NTg. (A) Apparent rate constant 2πb. (B) Apparent rate constant 2πc. (C) Magnitude B, and (D) Magnitude C. N = 41 fibers were used for NTg, N = 30 for Tg-WT, and N = 34 for Tg-E22K. Data are the mean ± SD, and the significance was analyzed by one-way ANOVA.
E22K mutation decreases fiber stiffness during rigor
Rigor was induced from full activation, and the stiffness of fibers was measured at 70 Hz (Fig. 2A, ‘Rigor’) in NTg, Tg-WT, and Tg-E22K fibers. The early decline in rigor tension, as observed in Fig. 2A, is a result of the finite dissociation of myosin CBs from actin due to the stress applied to fibers in rigor. After dissociation, however, nucleotide-free CB reattaches to the next available actin site reaching a steady level of tension [17,18]. Rigor tension results are plotted in the EM units (kPa) in Fig. 6A. As shown, the E22K mutation decreased the rigor stiffness of Tg-E22K fibers by ~ 47% compared with Tg-WT (Fig. 6A). A similar effect of E22K mutation vs WT was observed during the standard activation experiments (Fig. 3B). Interestingly, rigor stiffness was also 39% less in NTg fibers compared with Tg-WT (Fig. 6A), resembling the result from standard activation, where a 40% reduction in CB stiffness in Tg-E22K vs Tg-WT and a 30% lower stiffness in NTg vs Tg-WT muscles were compared (Fig. 3B).
Fig. 6.
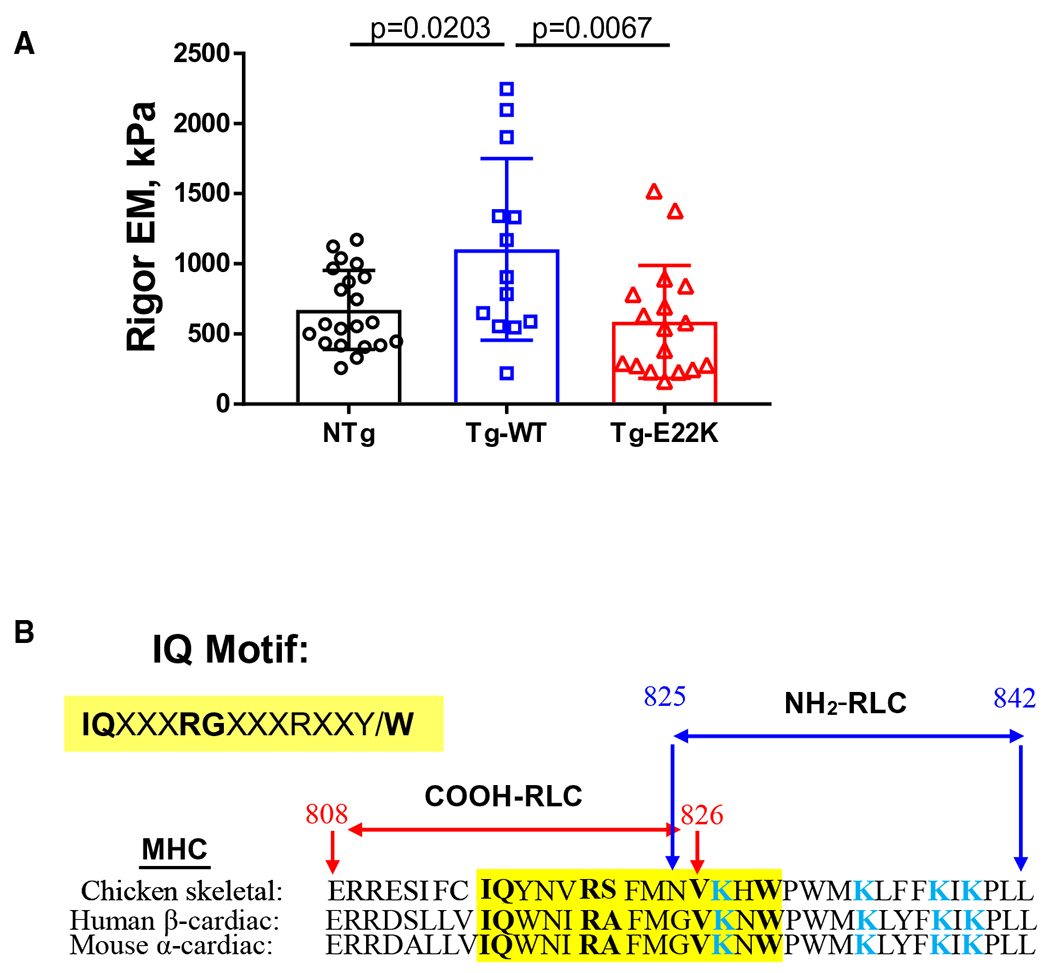
(A) Rigor was induced following standard activation (shown in Fig. 2A), and EM (in kPa) was measured at 70 Hz. N = 21 fibers were used for NTg, N = 12 for Tg-WT, and N = 17 for Tg-E22K. Data are the mean ± SD, and the data were analyzed by one-way ANOVA. (B) Overlapped sequences of MHC containing the IQ motif of chicken skeletal MHC (PDB: 2MYS), human β-MHC (MYH7 gene), and mouse α-MHC (MYH6 gene). Note that the N terminus of RLC containing the E22K mutation binds to the region of α-MHC in mice containing several Lys (K) residues (labeled in blue). The alignment and analysis of sequences was made with the EMBL-EBI Programme.
The fact that we observe reduced stiffness in NTg compared with Tg-WT fibers during activation (Fig. 3B) and rigor (Fig. 6A) suggests that the interaction between the mouse RLC in NTg fibers with the IQ motif of α-MHC on the lever arm is weaker than the interaction of human RLC in Tg-WT and α-MHC. One can speculate that this difference may result from the N-terminal amino acid sequence differences between the human vs mouse RLC (Fig. 1A) that makes their interaction with this region of the lever arm different. On the other hand, a comparison of stiffness during activation and in rigor between Tg-E22K and Tg-WT fibers (Figs 3B and 6A) reveals a lower stiffness in the mutant, while no differences were seen between NTg and E22K fibers. This reduced stiffness caused by E22K vs WT is most likely due to the impaired interaction between E22K-RLC with the α-MHC compared with WT-RLC interacting with α-MHC. As presented in Fig. 6B, the binding site for the RLC on α-MHC contains several lysine residues (Fig. 6B, K residues labeled in blue) that may exert some repulsive forces toward Lys22 on the E22K-RLC mutant weakening its interaction with the IQ motif. These Lys-Lys repulsive forces are not present in Tg-WT fibers containing E22-RLC, and thus, the interaction of WT-RLC with the IQ motif of α-MHC is unchanged. Since the myosin RLC structurally supports the lever arm of the myosin head [23], our combined results indicate that the stiffness of this region of myosin is most likely reduced due to the E22 to K22 charge change in the RLC E22K variant.
E22K mutation does not obstruct myosin RLC and troponin-I phosphorylation in situ
The N-terminal domain of the human ventricular RLC contains a MLCK-specific S15 P-site (Fig. 1A). The mouse ventricular RLC can additionally be phosphorylated (+P) at Ser-14 that is replaced by asparagine (Asn-14) in the sequence of human ventricular RLC (Fig. 1A). We previously reported that the bacterially expressed E22K mutant of human cardiac RLC had dramatically altered phosphorylation properties compared with RLC WT [12]. Here, we tested the level of RLC phosphorylation in ventricular myofibrils purified from the hearts of Tg-E22K, Tg-WT, and NTg mice (Fig. 7). Three hearts per group were used, and the phosphorylation of RLC was monitored by SDS/PAGE using ProQ/Coomassie (Fig. 7A) or by western blotting (Fig. 7B) using +P-RLC-specific and CT1-RLCtot antibodies [9,24,25]. Due to the N-terminal myc tag attached to human RLC, the migration of RLC in Tg-E22K and Tg-WT mice was slower on SDS/PAGE (Fig. 7A).
Fig. 7.
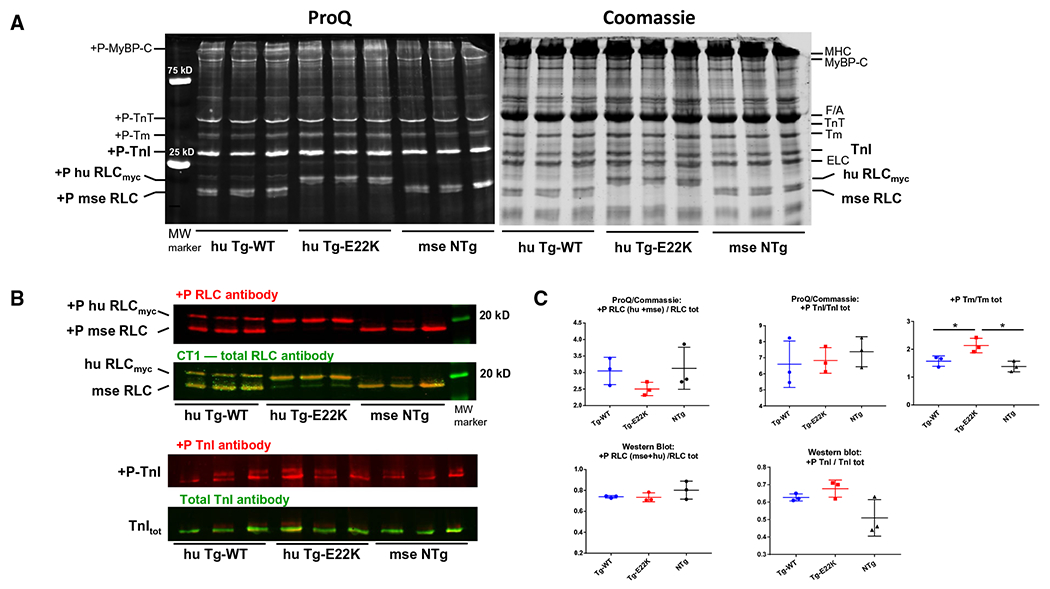
Myosin RLC, troponin-I (TnI), and tropomyosin (Tm) phosphorylation in ventricular myofibrils purified from the hearts of Tg-E22K, Tg-WT, and NTg mice. (A) SDS/PAGE and sarcomeric protein phosphorylation monitored by ProQ/Coomassie (25 μg of protein was used per well) in the hearts of n = 3 animals per group. (B) RLC and TnI phosphorylation monitored by western blotting and protein-specific antibodies (30 and 20 μg protein per well were used for detection of myosin RLC and TnI phosphorylation, respectively) in n = 3 hearts of Tg-E22K, Tg-WT, and NTg mice. (C) Quantification of protein phosphorylation in n = 3 hearts from Tg-E22K, n = 3 Tg-WT, and n = 3 NTg mice. +P-huRLCmyc, phosphorylated form of transgenic human ventricular RLC; +P-mse RLC, phosphorylated form of mouse cardiac RLC; +P-TnI, phosphorylated form of Troponin-I; MyBP-C, myosin binding protein C; F/A, F-actin; Tm, tropomyosin; TnT, troponin-T; ELC, essential myosin light chain.
The results from myofibrils stained with ProQ/Coomassie showed no effect of E22K mutation on RLC phosphorylation, and similar levels of RLC phosphorylation were noted in the hearts of the three genotypes (Fig. 7C, upper left panel). However, compared with controls, enhanced tropomyosin (Tm) phosphorylation was found in Tg-E22K myofibrils (Fig. 7C, upper right panel). Tg-E22K myofibrils blotted with +P-RLC/CT1-specific antibodies (Fig. 7C, lower left panel) confirmed the lack of effect of the E22K mutation on myosin RLC phosphorylation in the myocardium of Tg-E22K mice compared with controls. Considering the combined results from both methods, we can conclude that in contrast to its effect in isolated proteins, the E22K mutation does not obstruct RLC phosphorylation in situ. In addition to myosin RLC, cardiac MLCK was recently shown to also phosphorylate the thin filament Troponin-I (TnI), a primary target of a series of protein kinases (e.g., PKA, PKD, and PKC) [26]. Therefore, we aimed to examine the effect of E22K mutation on phosphorylation of TnI (Fig. 7). As above, the phosphorylation of TnI was monitored by SDS/PAGE using ProQ/Coomassie (Fig. 7A) or by western blotting (Fig. 7B) using +P-TnI-specific and TnItot antibodies. We found no effect of E22K mutation on TnI phosphorylation by ProQ/Coomassie (Fig. 7C, upper middle panel) and a slightly enhanced TnI phosphorylation in Tg-E22K myofibrils by western blotting (Fig. 7C, lower right panel), but the differences between the three genotypes were not statistically significant.
E22K mutation enhances actin-activated myosin ATPase activity
Considering the differences in the expression of human ventricular RLC in Tg-E22K vs Tg-WT (Fig. 1C), and functional similarities between NTg and Tg-E22K mice in calcium-activated tension (Fig. 2E) and stiffness (Figs 3B and 6A), we proceeded to test the ability of myosin to interact with actin by examining the actin-activated myosin ATPase activity of myosin purified from Tg-E22K, Tg-WT, and NTg mice (Fig. 8). Specifically for this experiment, we included an additional control, the myosin purified from Tg-WTL2 RLC mice that express near 100% of human ventricular RLC [24]. In Fig. 8, this myosin is labeled as huRLC. Despite differences in human RLC expression between both WT controls, no differences were noted in their actin-activated ATPase activity and the plots of Tg-WT (~ 26% huRLC) overlapped with those of huRLC (~ 100% huRLC) (Fig. 8A, green and blue curves). A faster turnover rate of myosin expressing the E22K mutation was observed, as manifested by the increased ATPase activity with Vmax (s−1) ~ 1.2-fold higher in Tg-E22K mice compared with controls (Fig. 8B).
Fig. 8.
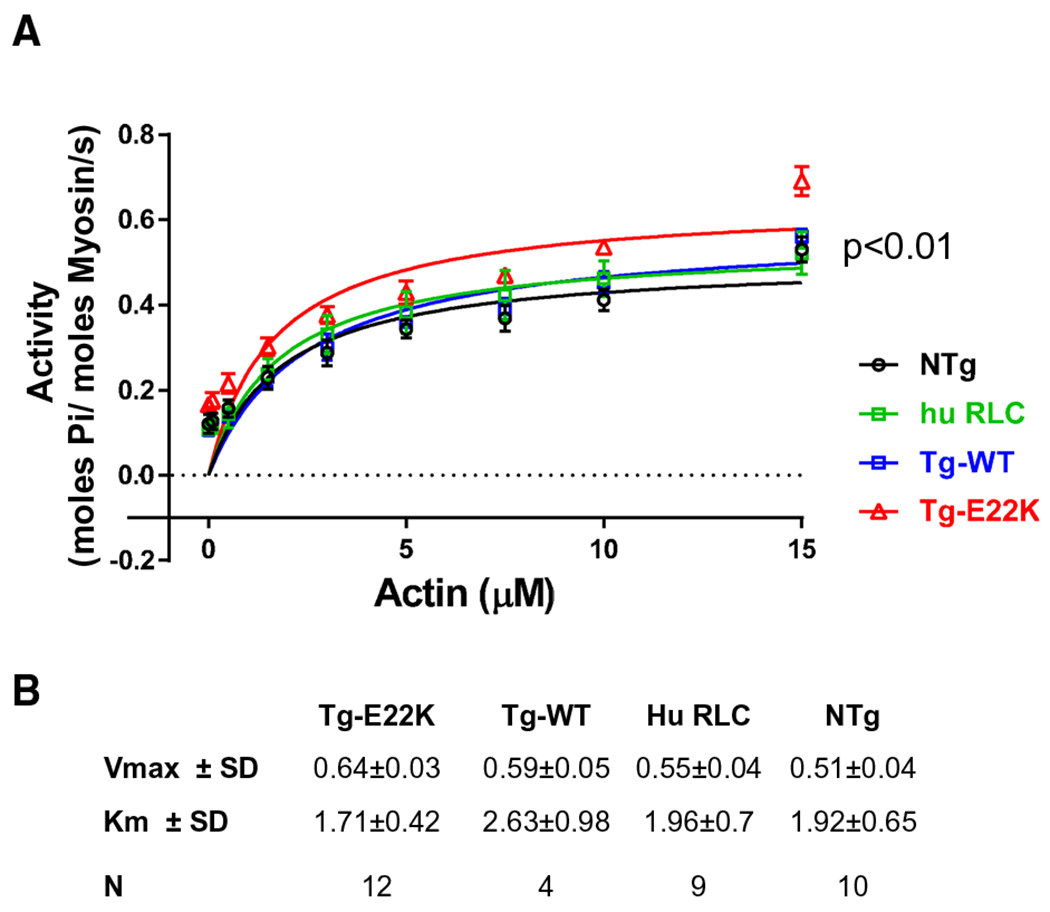
Actin-activated ATPase activity of cardiac myosin purified from the hearts of mice. (A) ATPase is plotted as a function of actin concentration. In addition to Tg-E22K, Tg-WT, and NTg hearts, myosin purified from Tg-RLCL2 mice expressing ~ 100% of human cardiac RLC (huRLC) was used. (B) Vmax and Km values obtained from the fit of experimental points to the Michaelis–Menten equation. Data are the mean ± SD, and the significance was analyzed by one-way ANOVA.
The results of Fig. 8 and the lack of differences in the actin-activated myosin ATPase activity between Tg-WT and huRLC myosins seem to justify using Tg-WT as a proper control for Tg-E22K in fiber mechanics experiments. The results suggest that the observed phenotypic differences between the genotypes originate from the disease-causing mutation instead of the higher human RLC expression measured in Tg-E22K compared with Tg-WT. However, while there are no differences between the preparations at the myosin level, the force-producing fibers that comprise regulated thin filaments may potentially be sensitive to force-generating myosin CBs containing human vs mouse RLC.
Discussion
Hypertrophic cardiomyopathy mutations in the RLC of myosin are quite rare, but they are also of great significance given the importance of RLC for muscle contraction and heart function. The RLC plays an essential structural and functional role by supporting the architecture of the myosin neck region and fine-tuning the kinetics of the actin–myosin interaction [27,28]. It wraps around the α-helical neck region of the myosin head by binding to a 35 amino acid-long IQ motif in the MHC [29]. This domain of α-MHC acts as a lever arm, amplifying small conformational changes that originate at the catalytic site into large movements, thus allowing myosin to generate force and motion [30,31]. This region of the myosin head has been hypothesized to act as the compliant element of the myosin CB with the RLC contributing to the stiffness of the lever arm [32]. The rigidity against flexion of the lever arm is important because the force is applied in an oblique angle to the lever arm rather than along the length of the lever arm.
Myofilament dysfunction due to structural and functional reasons is a hallmark of HCM caused by genetic mutations in cardiac sarcomere and has been also implicated in the pathogenesis of RLC-E22K-associated HCM [9,11]. In this study, we tested the hypothesis that E22K causes structural abnormalities in the myosin lever arm that ultimately triggers functional remodeling and heart dysfunction in Tg-E22K mice. The main findings of our study are as follows: (a) E22K-induced reduction in the active and rigor stiffness; (b) E22K-triggered decrease in active tension and the Ca2+ sensitivity of tension; (c) E22K-elicited enhancement in CB detachment rate 2πc, and Vmax of actin-activated myosin ATPase; and (d) no E22K-mediated changes in myofilament protein phosphorylation. These findings are analyzed and discussed considering potential mechanisms by which the E22K-RLC causes abnormalities in myofilament function that may ultimately lead to heart dysfunction and HCM.
E22K-mediated decrease in myofilament stiffness
Rigor is a condition in which all possible CBs are firmly attached to the thin filament; hence, it is a measure of the stiffness (σ) of the entire structure. Rigor stiffness, measured in Tg-E22K vs Tg-WT and NTg mice, originates from the serial combination of structures that include Z-line, thin filament, thick filament, and head and neck regions of myosin. One can assume that σ depicts stiffness of two serial structures, shown as the second term in Eq. 4 (Eq. 1.12 in [33]):
| (4) |
where σ1 denotes the serially combined stiffness of Z-line, thin filament, thick filament, myosin head, and the myosin essential light chain (ELC) association with the IQ1 of α-MHC (ELC-IQ1 assembly) forming structure 1. The σ2 represents the stiffness of the RLC-IQ2 assembly (structure 2). The fourth term is based on Taylor expansion for σ1 > σ2. (Eq. 5.10 in [33]). The first approximation (the fifth term after ≅) is when σ1 > σ2, and the second approximation (after ≈) is when σ1 >> σ2. Thus, Eq. 4 demonstrates that the stiffness of the weakest structure (σ2) limits the stiffness of the entire structure (σ). Because there is no change in the amino acid sequences that make up structure 1, the stiffness of this structure (σ1) is not expected to be altered by the E22K mutation. On the other hand, RLC is altered by the E22K mutation; thus, stiffness of structure 2 (σ2) is expected to change. Because we observe an E22K-related reduction of rigor stiffness (σ to half) (Fig. 6A), we can conclude that this decrease in stiffness is caused by the weakened RLC-IQ2 interaction/assembly (σ2) in the neck region of the myosin head (Eq. 4) that constitute the major part of the lever arm. The reduced stiffness during activation (Fig. 3B) is consistent with this conclusion. Similarly, according to Hooke’s Law (Eq. 5), the reduced active tension in Tg-E22K fibers (Fig. 3A) is consistent with the reduced σ2, because elementary force generation (ΔF) is the product of σ and Δx (CB step size).
| (5) |
Two variables are influencing the elementary force (ΔF) production: series stiffness (σ) and the step size (Δx), and a decrease in either one can lead to reduced force (Eq. 5). Considering a change in σ, our findings are consistent with the hypothesis that the binding of RLC to the IQ2 motif of α-MHC (Fig. 6B) strengthens the lever arm structure (increases in σ2, thereby increases in σ according to Eq. 4) [23,29,30,32]. The E22K mutation may weaken the lever arm structure (decreased σ2) by changing the interaction of E22K-RLC with the IQ2 motif of α-MHC due to the electrostatic repulsive forces between RLC’s mutated Lys22 and several Lys residues present in the vicinity of the IQ2 motif (Fig. 6B, K residues labeled in blue). Since these Lys-Lys repulsive forces are not present in Tg-WT fibers containing E22-RLC, the interaction of WT-RLC with the IQ motif of α-MHC and the integrity of the lever arm are unchanged.
The unitary myosin step size Δx depicting a linear actin displacement due to one lever arm rotation is less likely to change with the E22K mutation on myosin RLC because the mutation is not expected to change the length of the lever arm, neither the ATP hydrolysis-driven rotary movement of the lever arm. However, as elegantly shown by the Burghardt group, myosin step size measured using a novel quantum dot assay, phosphorylation of myosin RLC at Ser15 (Fig. 1A) was shown to change step size distribution by advancing the 8-nm step frequency [34]. Single-molecule experiments would have to be performed to address the question of whether the E22 to K22 mutation of myosin RLC affects unitary myosin step-size distribution.
There may be an additional possibility for the lower stiffness observed in Tg-E22K compared with Tg-WT and NTg fibers (Fig. 3B). The observation that 2πc increases in Tg-E22K vs Tg-WT/NTg (Fig. 5B) implies that more CBs are detached, thus reducing the number of strongly attached, force-generating CBs (i.e. lower duty ratio). Therefore, an E22K-mediated increase in myosin detachment rate 2πc contributes to lower force production (Fig. 3A) and reduced stiffness (Fig. 3B) in the Tg-E22K myocardium. The fact that the rigor stiffness of NTg fibers was only 61% of that measured in Tg-WT (Fig. 6A) suggests that the association of mouse RLC with IQ2 motif may not be as strong as the association of human RLC with IQ2. This weaker interaction of mouse RLC with α-MHC in mice may be the reason for the lack of HCM phenotype observed in the mouse model expressing mouse RLC-E22K [8], whereas mice expressing the human RLC-E22K demonstrated enlarged inter-ventricular septa and papillary muscles indicating hypertrophic remodeling in Tg-E22K hearts compared with Tg-WT and/or NTg littermates [9].
Force, myofilament Ca2+ sensitivity, and CB kinetics are affected by RLC-E22K
The E22K-related decrease in maximum force observed in this study (Figs 2E and 3A) is consistent with a previous observation of decreased steady-state tension monitored in skinned papillary muscle fibers from Tg-E22K vs Tg-WT mice by Szczesna-Cordary et al. [11]. As argued above, the lower level of tension observed in Tg-E22K compared with Tg-WT and NTg fibers (Fig. 3A) could be, at least in part, due to increased CB detachment rate (2πc) observed in Tg-E22K mice compared with Tg-WT/NTg controls (Fig. 5B). This would lead to a reduced number of strongly attached, force-generating CBs (lower duty ratio) resulting in decreased tension (Fig. 3A) and consequently reduced stiffness (Fig. 3B) in the Tg-E22K myocardium compared with Tg-WT and NTg mice.
While the calcium sensitivity of force (pCa50) was not significantly different between NTg and Tg-WT, it was significantly lower in Tg-E22K fibers compared with Tg-WT, with a difference of 0.107 pCa units (Fig. 2C). This result is in accord with the solution study by Szczesna et al. [12], where a mutation-induced decrease in the Ca2+ binding affinity to recombinant human cardiac E22K-RLC was monitored. However, the apparent Ca2+ dissociation constants (Kapp) of E22K-RLC-reconstituted porcine cardiac myofibrils and fibers were unchanged compared with WT-RLC-reconstituted preparations [13]. Studies with humanized transgenic Tg-E22K mice carried out by the Szczesna-Cordary laboratory [9,11] also reported different effects of E22K mutation on the Ca2+ sensitivity of force, which either increased [9] or was unchanged [11] in Tg-E22K vs Tg-WT hearts. The difference in the ΔpCa50 between the previous [9] and current study executed on the same Tg-E22K mice is puzzling, and the possibility exists that the addition of 8 mm Pi in the pCa solutions used in the current investigation that was absent in previous studies could be the reason. However, it is also possible that the myocardium of Tg-E22K underwent a cardiomyopathy remodeling over time and no longer manifests an HCM-related sensitization of myofilaments to calcium.
Considering the contribution of binding of calcium to the RLC Ca2+-Mg2+ sites to myofilament Ca2+ sensitivity, we have previously proposed that these sites, which are of equal intracellular concentration to the regulatory low-affinity Ca2+-specific sites of troponin C, are capable of buffering Ca2+ in the muscle, and can function as delayed Ca2+-binding sites along with those of TnC Ca2+-specific sites, which have a fast on-rate of Ca2+ compared with the RLC sites [24]. The effect of RLC Ca2+-binding sites would be to act as a delayed Ca2+ buffer and shorten the intracellular [Ca2+] transient by assisting the sarcoplasmic reticulum SERCA pump in the uptake of myoplasm Ca2+ during relaxation. In accord with this hypothesis, faster Ca2+ reuptake and the shorter duration of [Ca2+] and force transients were observed in electrically stimulated intact papillary muscle fibers from Tg-E22K hearts [11]. The data of these previous investigations are consistent with this report showing the E22K-mediated increase in CB detachment rate 2πc and the reduced Ca2+ sensitivity of force (Fig. 2B,C).
As presented in Fig. 5, we also assessed the kinetic constants of standard activation in Tg-E22K, Tg-WT, and NTg mice. The apparent rate constant 2πb was only slightly higher in Tg-WT compared with NTg fibers, but no changes in 2πb were observed in Tg-E22K mice compared with NTg and Tg-WT controls (Fig. 5A). However, the apparent rate constant 2πc, representing the CB detachment step, was significantly faster in Tg-E22K fibers compared with Tg-WT and NTg preparations (Fig. 5B) indicating that the E22K mutation speed up the CB dissociation kinetics contributing to decreased tension and stiffness and increased Ca2+ sensitivity of force in the Tg-E22K myocardium. In support of the E22K-mediated increase in 2πc, the actin-activated myosin ATPase activity, and hence CB turnover rate of E22K myosin, was also higher than Vmax of WT and NTg controls (Fig. 8).
Mechanisms associated with HCM-linked E22K mutation in myosin RLC: hypocontractility vs hypercontractility
Many genetic mutations in sarcomeric proteins, including myosin RLC, have been implicated in HCM disease [2,3]. Yet, the molecular mechanisms by which these mutant proteins regulate cardiac muscle mechanics in health and disease remain insufficiently understood. Evidence has been accumulating that sarcomeric protein phosphorylation has an influential role in striated muscle contraction, and in addition to the conventional modulation via Ca2+ and Tn/Tm, it can regulate cardiac muscle function and affect calcium sensitivity of contraction. For example, phosphorylation of TnI secondary to beta-adrenergic activation is known to reduce myofilament Ca2+ sensitivity. However, in muscle samples from explanted failing human hearts, TnI phosphorylation was found to be very low and Ca2+ sensitivity high, while in animal models of heart failure, the opposite results of high levels of TnI phosphorylation and low Ca2+ sensitivity were observed confounding the true effects of TnI phosphorylation in heart disease [35]. Our studies demonstrate no E22K-mediated changes in the phosphorylation level of TnI in myofibrils purified from the hearts of mice (Fig. 7).
Myosin light chain kinase-dependent phosphorylation of Ser15 in huRLC has been widely recognized to play an important role in cardiac muscle contraction under both normal and disease conditions [28,36–40]. Significantly depressed Ser15-huRLC phosphorylation was reported in heart failure patients [41] and was also observed in experimental animal models of cardiac diseases [25,40,42]. Studies in mice showed that reduced RLC phosphorylation resulted in abnormal heart performance, presumably through morphological and/or myofibrillar functional alterations (e.g., changes in force, myofilament calcium sensitivity, ATPase activity, CB kinetics) [43–46]. Likewise, attenuation of RLC phosphorylation in cardiac MLCK knockout mice was demonstrated to cause ventricular hypertrophy, fibrosis, and dilated cardiomyopathy [47]. As for TnI, no significant changes in myosin RLC phosphorylation were noted in myofibrils from Tg-E22K compared with control mice (Fig. 7).
Many HCM-associated mutations lead to the hypercontractility of cardiac myofilaments that is manifested clinically as preserved systolic performance of the heart with diminished relaxation capacity. Even though E22K is classified as an HCM-causing mutation, the phenotypes associated with this mutation that we observe in papillary muscle fibers do not fall into the typical HCM-related hypercontractile category. Similar to our data on E22K-RLC, studies on HCM-causing mutations in the MHC’s converter domain also suggested hypo- rather than hypercontractile response, shown by lowered pCa50 [48]. Our recent work on myosin ELC mutations also demonstrated that two different HCM mutations exerted opposite effects on power output, yet led to the same HCM phenotype in vivo in mice [49]. Likewise, our studies with another HCM-associated R58Q mutation in myosin RLC showed a hypo- rather than hypercontractile behavior [50]. Therefore, the effects of HCM-causing mutations on force generation, ATPase, and myofilament Ca2+ sensitivity can be quite different and thus difficult to generalize [51]. Structural work from the Houdusse laboratory reconciles these hyper- and hypocontractile models of action and explains how different HCM mutations can have disparate effects on the mechanochemical properties, yet result in a similar HCM phenotype in vivo [52]. The group has built an optimized molecular model of the sequestered state of myosin and analyzed how certain HCM mutations may destabilize the interactive head motif or affect myosin motor activity. The analysis of high-resolution cardiac myosin structures explained how HCM-associated mutations modify myosin power and conformational plasticity of the heads and how they may modify the number of heads available for force production by destabilizing the sequestered state, providing insights for the development of the HCM pathology.
Conclusions
We conclude that the integrity of the N-terminal domain of RLC is essential for its interaction with the MHC-IQ motif region located in the neck of the myosin head and any structural modifications, such as amino acid substitutions, may compromise this interaction and thus the stability of the lever arm domain. It is likely that the electrostatic interactions between RLC and MHC are weakened by the charge change from glutamic acid to lysine (E22K) in myosin RLC and that this change may activate some repulsive forces between RLC-Lys22 and the lysine residues located in the MHC-IQ domain of the myosin head. The proximity of the E22K mutation to the RLC Ca2+ binding site may also affect the Ca2+ binding properties of the RLC and weaken its interaction with MHC. In conclusion, the HCM phenotype is triggered by E22K mutation-induced decrease in the lever arm stiffness and a subsequent decline in the active force. When placed in vivo, the E22K mutation is expected to result in reduced contractility and decreased cardiac output whereby leading to HCM.
Materials and methods
Mice
All animal procedures and experiments were performed in accordance with the ‘Guide for the Care and Use of Laboratory Animals’ (NIH Publication 85–23, revised 2011). All the protocols were approved by the Institutional Animal Care and Use Committee at the University of Miami Miller School of Medicine, which has an Animal Welfare Assurance on file with the Office of Laboratory Animal Welfare (OLAW), NIH. The assurance number is #A-3224-01, approved through November 30, 2019. We are registered with USDA APHIS, registration # 58-R-007, approved through December 3, 2020. We have full accreditation with the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC International), site 001069, latest effective date, November 8, 2016. Mice were euthanized through CO2 inhalation followed by cervical dislocation. IACUC protocol # is 18-110-LF.
All animal experiments were approved by the Ethic Committee of Soochow University (reference number: BK20150353) with research governance by Hua-Gang Hu (School of Nursing, seuboyh@163.com ), and Chen Ge (Soochow University, chge@suda.edu.cn ), and experiments were conducted according to institutional animal ethics guidelines for the Care and Use of Research Animals established by Soochow University, Suzhou, China.
Nontransgenic (NTg) refers to natural B6SJL mice, which served as controls to mouse models of HCM (Tg-E22K) and Tg-WT expressing the human ventricular RLC proteins [9]. Both human Tg-E22K and Tg-WT models expressed the myc-tagged RLCs: myc-E22K and myc-WT [9]. The expression of both transgenes has been made under the control of the murine α-MHC promoter [53]. Based upon the in vitro and in vivo assays, it has been established that myc-RLCs proteins expressed in these Tg mouse models do not cause functional alterations [9]. Transgenic mice were exported from D. Szczesna-Cordary’s laboratory (Miami, FL, USA) to perform muscle contractile mechanics study in L. Wang’s laboratory (Suzhou, China). Mice were euthanized by cervical dislocation, and ventricular papillary muscles were isolated and processed in mechanical experiments as described earlier [14–16,54]. The animal protocols were approved by respective Institutional Review Committees at both universities.
For mechanical experiments, a total of 21 mice of three genotypes (eight NTg: nontransgenic, six Tg-WT: transgenic wild-type, and seven Tg-E22K: transgenic mutant) were used. All mice were 4 months old. Biochemical experiments were performed on flash-frozen hearts stored in −80 °C, and the number of mice used for biochemical analyses is listed under each specific experiment. Male and female mice were used in all experiments.
Mechanical analyses
Papillary muscles from both ventricles were chemically skinned as described previously and split into small bundles (average diameter: 134 ± 3 μm, Mean ± SEM, N = 159 preparations). Skinned fibers were mounted in the experimental apparatus by two pairs of micro-forceps: One pair was connected to the galvanometer-based length driver and the other pair to the tension transducer. The instrument used for experiments is called ’Skinned Muscle Station’ and manufactured by Myotronics (Heidelberg, Germany, https://www.myotronic.de/muscle-station-skinned.htm). Fibers were further treated in 1% Triton X-100 in the relaxing solution for 20 min to remove remaining membranes surrounding myofilaments. Then, the strips were washed again in the relaxing solution and their length was adjusted to remove the slack. This procedure resulted in sarcomere length of ~ 2.1 μm as judged by the first-order optical diffraction using He–Ne laser (wavelength 0.6328 μm) [16]. The average fiber length used for this report was 0.89 ± 0.02 mm (mean S±SEM). Studies of isometric tension during Ca2+ activation, tension-pCa, rigor, and sinusoidal analysis (range: 0.35–70 Hz in 15 frequencies at 0.25% amplitude) were carried out as described previously [14–16]. Measurements at 100 Hz were avoided due to coherent noise pickup caused by the power line, supplied at 50 Hz. While our experimental program (Dcoll.exe) and equipment are specifically designed to eliminate 50–60 Hz power line pickup, it did not eliminate 100 Hz noise caused by the second harmonic originated from the power supply. The sinusoidal analysis was performed as described in Kawai and Brandt [19], and the resulting complex modulus Y(f) data were fitted to Eq. 2.
Tension–pCa and rigor studies
The experimental protocol for tension–pCa measurements in fibers from Tg-E22K, Tg-WT, and NTg mice is schematically presented in Fig. 2A. Fibers were first relaxed (R) in the solution containing (in mm) K2H2EGTA (6), Na2H2ATP (7), H1.5K1.5Pi (8), MgAc2 (2), NaAc (41), KAc (70.5), and MOPS (10) where Ac = acetate and Pi = phosphate [16]. Then, they were immersed in the standard activating (A) solution containing 5 mm MgATP, 8 mm Pi, and 6 mm CaEGTA; pCa = 4.55 (28 μm Ca2+). Following a brief relaxation, the force–pCa dependence was measured in a solution of increasing Ca2+ concentrations (in pCa units): 7.0, 6.2, 6.0, 5.8, 5.7, 5.6, 5.5, 5.4, 5.2, 4.8, and 4.55, where pCa ≡ −log10[Ca2+]. The isometric tension was recorded, and the results were fitted to 4-parameter Hill equation (Eq. 1) using our program F_pCaTc.exe as described in previous paper [19]. Following tension–pCa and full activation study, the rigor was induced, and the stiffness of fibers was measured at 70 Hz (Fig. 2A, Rigor’). The rigor solution contained (in mm) the following: K2HPO4 (4), NaAc (55), KAc (122), and MOPS (10). The ionic strength of all solutions was 190 mm, and pH was adjusted to 7.00 by KOH. All experiments were carried out at room temperature (19 ± 1 °C).
Mouse myosin preparation
Cardiac myosin was isolated from the hearts of mice as previously reported [11,55]. One batch of myosin for NTg, Tg-WT, and Tg-E22K and two batches for HuWT were obtained. One myosin preparation was obtained from a pool of 7–10 hearts/group. Briefly, after euthanasia, whole hearts were isolated and the atria were removed. Left and right ventricles varying from 0.1 to 0.2 g were flash-frozen and stored at −80 °C until processed. The ventricular tissue was later thawed in an ice-cold Guba Straub-type buffer (pH 6.5) consisting of 300 mm NaCl, 100 mm NaH2PO4, 50 mm Na2HPO4, 1 mm MgCl2, 10 mm EDTA, 0.1% NaN3, 10 mm Na4P2O7, 1 mm DTT, and protease inhibitor cocktail in a volume of 0.75 mL buffer per 0.2 g tissue. Ventricles kept on ice were first minced by hand and then homogenized for 2 min at a frequency of 27Hz in a Mixer-Mill MM301 (Retsch USA, Newtown, PA, USA). The homogenate was then incubated on ice for 40 min before centrifugation at 200 000 g for 1 h. The supernatant was then diluted 60-fold (by volume) with 2 mm DTT and incubated on ice for 30 min with stirring and left standing without stirring for an additional 30 min. The samples were centrifuged again at 8000 g for 10 min, and resultant pellets were then resuspended in a minimal volume of buffer containing 0.4 m KCl, 10 mm MOPS (pH 7.0), 5 mm DTT, and protease inhibitor cocktail. Samples were then diluted 1 : 1 with glycerol, mixed gently, and stored at −20 °C. For ATPase assays, myosins were precipitated with 13 volumes of ice-cold 2 mm DTT and collected by centrifugation at 8000 g for 10 min. The myosin pellets were resuspended in a minimal volume of ATPase buffer consisting of 0.4 m KCl, 10 mm MOPS (pH 7.0), and 1 mm DTT and dialyzed overnight against the same buffer. The concentration of myosin was determined using a Coomassie Plus Assay (Pierce/Thermo Fisher Scientific, Waltham, MA, USA), and all myosins were diluted to a concentration of ~ 1.5 mg-mL−1 for the assay.
Actin-activated myosin ATPase activity
Rabbit skeletal F-actin, prepared according to Kazmierczak et al. [55], was used in combination with the preparations of mouse myosin in the actin-activated myosin ATPase assays [56]. Briefly, 0.6 μm myosin dissolved in 0.4 m KCl was added to a 96-well microplate containing increasing concentrations of F-actin (in μm): 0.1, 0.5, 1.5, 3, 5, 7.5, 10 and 15. The assay was performed in a final volume of 120 μL in a buffer consisting of 25 mm imidazole pH 7.0, 4 mm MgCl2, 1 mm EGTA, and 1 mm DTT. The final KCl concentration was 77 mm. Protein mixtures were first incubated on ice for 10 min and then for another 10 min at 30 °C. The reactions (run in duplicates) were initiated with the addition of 2.5 mm ATP with mixing in a Jitterbug incubator shaker (Boekel, Feasterville, PA, USA), allowed to proceed for 15 min at 30°C, and then terminated by the addition of 4% trichloroacetic acid. Precipitated proteins were cleared by centrifugation, and the inorganic phosphate was determined as described in Fiske and Subbarow. Data were analyzed using the Michaelis–Menten equation yielding Vmax and Km.
Analysis of RLC protein expression, and RLC and TnI phosphorylation
The phosphorylation level of RLC and TnI was determined in myofibrils prepared from flash-frozen hearts from 6- to 12-month-old NTg, Tg-WT, and Tg-E22K mice (three hearts per group, two female and one male) and stored in −80 °C as described in [56]. Briefly, the tissue was thawed in the CMF (cardiac myofibril) buffer consisting of 5 mm NaH2PO4, 5 mm Na2HPO4 (pH 7.0), 0.1 mm NaCl, 5 mm MgCl2, 0.5 mm EGTA, 5 mm ATP, 5 nm microcystin, 0.1% Triton X-100, 10 μL·mL−1 of phosphatase inhibitor cocktail 2 and 3 (Sigma), 5 mm DTT, and 1 μL·mL−1 protease inhibitor cocktail. The tissue was then homogenized in a Mixer-Mill MM301 until homogenous. The homogenate was then centrifuged for 4 min at 8000 g, and the supernatant was discarded. After centrifugation, the pellets were left on ice for 4 min. This step was repeated three times with CMF buffer without Triton. The pellets were then resuspended in the CMF buffer (without Triton), and the samples were subsequently mixed at 1 : 1 ratio with Laemmli buffer consisting of 62.5 mm Tris/HCl, pH 6.8, 25% glycerol, 2% SDS, 0.01% bromophenol blue, and 5% β-mercaptoethanol (β-ME); the samples were heated and loaded on 15% SDS/PAGE.
Phosphorylation of troponin-I and myosin RLC was determined using ProQ Diamond phosphoprotein gel stain reagent (Invitrogen/Thermo Fisher Scientific, Waltham, MA, USA) as described in the manufacturer’s manual. The total protein was further detected in the same gel using the Coomassie brilliant blue staining. Myofilament protein phosphorylation ratio was calculated relative to the corresponding Coomassie brilliant blue staining (ProQ/Coomassie) using the imagej software. Additionally, RLC phosphorylation was detected by western blotting using a phospho-specific RLC antibody, which recognizes the +P form of the RLC and does not react with nonphosphorylated RLC [9,24,25], followed by a secondary goat anti-rabbit antibody conjugated with IR red 800. TnI phosphorylation was detected by western blotting using a +P Ser23/24-TnI (Mab14 MMS-418R; Covance, Brekeley, CA, USA), followed by a secondary goat anti-mouse antibody conjugated with IR red 800. Total RLC protein was detected with the CT-1 antibody followed by a secondary goat anti-rabbit antibody conjugated with Cy5.5, while total TnI protein was detected with the TnI (6F9; Research Diagnostics Inc., Flanders, NJ, USA ) antibody followed by a secondary goat anti-mouse antibody conjugated with Cy5.5. Band densities were analyzed using the imagej (U. S. National Institutes of Health, Bethesda, Maryland, USA) software. RLC protein expression was calculated from western blots detecting total RLC and calculated from Tg huRLC/ (Tg huRLC + mse RLC).
Statistical analysis
Unless otherwise stated, all values are shown as means ± SD. Statistically significant differences between multiple groups were assessed using one-way ANOVA and Tukey’s multiple comparison test using the GRAPHPAD PRISM software version 7.0 (San Diego, CA, USA) for Windows. Significance was defined as P < 0.05.
Acknowledgements
We would like to thank members of the Wang laboratory (Ms. Shiqin Pan, Ms. Qianqian Zhang, Ms. Jing Xi, Ms. Biyue Hu) for maintaining mouse colonies needed for the present study. This work was supported by the Natural Science Foundation of Jiangsu Province of China BK20150353 (to LW) and the National Institutes of Health [Grant number R01-HL143830 and R56-HL146133 (to DSC)].
Abbreviations
- +P
phosphorylated
- 2πb
apparent (=measured) rate constants of exponential process B
- 2πc
apparent (=measured) rate constants of exponential process C
- b
characteristic frequency of exponential process B
- B
magnitude of exponential process B
- c
characteristic frequency of exponential process C
- C
magnitude of exponential process C
- CB
cross-bridge
- EM
elastic modulus
- f
frequency
- H
elastic modulus extrapolated to the zero frequency (f→0)
- HCM
hypertrophic cardiomyopathy
- huRLC
human isoform of RLC
- i
imaginary number, i = √−1
- IQ
light chain binding domain of MHC
- KCa
Ca2+-binding constant
- Km
actin concentration at half maximum ATPase
- LV
left ventricle
- MHC
myosin heavy chain
- MLC
myosin light chain
- MLCK
myosin light chain kinase
- N
number of experiments
- nH
cooperativity (Hill coefficient)
- NTg
non-Tg (B6SJL) mice
- P
probability of coincidence
- pCa
−log10[Ca2+]
- pCa50
concentration of Ca2+ causing half maximal activation, Ca2+ sensitivity
- PKA
Protein kinase A
- P-site
phosphorylation site
- RLC
regulatory light chain of myosin
- Tact
Ca2+ activatable tension
- Tg
transgenic
- Tg-E22K
Tg mice expressing human ventricular RLC E22K mutation
- Tg-WT
Tg mice expressing human ventricular RLC wild-type
- TLC
low [Ca2+] tension, relaxed tension level
- Tm
tropomyosin
- Tn
troponin
- VM
viscous modulus
- Vmax
maximum rate of ATP hydrolysis at large actin concentration
- Y(f)
complex modulus (frequency-dependent)
- Y∞
elastic modulus extrapolated to the infinite frequency (f→∞)
- ΔF
elementary force generated by a CB
- Δx
step size of a CB
- σ
stiffness
- σ1
serially combined stiffness of Z-line, thin filament, thick filament, myosin head, and the myosin essential light chain (ELC) associated with the IQ1 of α-MHC forming structure 1
- σ2
stiffness of the RLC-IQ2 assembly (structure 2)
Footnotes
Sub-Discipline
Bioenergetics.
Database
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
Animal protocol
BK20150353 (Soochow University).
Research governance
School of Nursing: Hua-Gang Hu: seuboyh@163.com; Soochow University: Chen Ge chge@suda.edu.cn.
Conflict of interest
The authors declare no conflicts of interest.
Data Accessibility
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
References
- 1.Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT & Bild DE (1995) Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation 92, 785–789. [DOI] [PubMed] [Google Scholar]
- 2.Semsarian C, Ingles J, Maron MS & Maron BJ (2015) New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol 65, 1249–1254. [DOI] [PubMed] [Google Scholar]
- 3.Alcalai R, Seidman JG & Seidman CE (2008) Genetic basis of hypertrophic cardiomyopathy: from bench to the clinics. J Cardiovasc Electrophysiol 19, 104–110. [DOI] [PubMed] [Google Scholar]
- 4.Yadav S, Sitbon YH, Kazmierczak K & Szczesna-Cordary D (2019) Hereditary heart disease: pathophysiology, clinical presentation, and animal models of HCM, RCM, and DCM associated with mutations in cardiac myosin light chains. Pflugers Arch 471, 683–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Poetter K, Jiang H, Hassanzadeh S, Master SR, Chang A, Dalakas MC, Rayment I, Sellers JR, Fananapazir L & Epstein ND (1996) Mutations in either the essential or regulatory light chains of myosin are associated with a rare myopathy in human heart and skeletal muscle. Nat Genet 13, 63–69. [DOI] [PubMed] [Google Scholar]
- 6.Kabaeva ZT, Perrot A, Wolter B, Dietz R, Cardim N, Correia JM, Schulte HD, Aldashev AA, Mirrakhimov MM & Osterziel KJ (2002) Systematic analysis of the regulatory and essential myosin light chain genes: genetic variants and mutations in hypertrophic cardiomyopathy. Eur J Hum Genet 10, 741–748. [DOI] [PubMed] [Google Scholar]
- 7.Claes GR, van Tienen FH, Lindsey P, Krapels IP, Helderman-van den Enden AT, Hoos MB, Barrois YE, Janssen JW, Paulussen AD, Sels JW et al. (2016) Hypertrophic remodelling in cardiac regulatory myosin light chain (MYL2) founder mutation carriers. Eur Heart J 37, 1815–1822. [DOI] [PubMed] [Google Scholar]
- 8.Sanbe A, Nelson D, Gulick J, Setser E, Osinska H, Wang X, Hewett TE, Klevitsky R, Hayes E, Warshaw DM et al. (2000) In vivo analysis of an essential myosin light chain mutation linked to familial hypertrophic cardiomyopathy. Circ Res 87, 296–302. [DOI] [PubMed] [Google Scholar]
- 9.Szczesna-Cordary D, Guzman G, Zhao J, Hernandez O, Wei J & Diaz-Perez Z (2005) The E22K mutation of myosin RLC that causes familial hypertrophic cardiomyopathy increases calcium sensitivity of force and ATPase in transgenic mice. J Cell Sci. 118, 3675–3683. [DOI] [PubMed] [Google Scholar]
- 10.Dumka D, Talent J, Akopova I, Guzman G, Szczesna-Cordary D & Borejdo J (2006) E22K mutation of RLC that causes familial hypertrophic cardiomyopathy in heterozygous mouse myocardium: effect on cross-bridge kinetics. Am J Physiol Heart Circ Physiol 291, H2098–H2106. [DOI] [PubMed] [Google Scholar]
- 11.Szczesna-Cordary D, Jones M, Moore JR, Watt J, Kerrick WGL, Xu Y, Wang Y, Wagg C & Lopaschuk GD (2007) Myosin regulatory light chain E22K mutation results in decreased cardiac intracellular calcium and force transients. FASEB J 21, 3974–3985. [DOI] [PubMed] [Google Scholar]
- 12.Szczesna D, Ghosh D, Li Q, Gomes AV, Guzman G, Arana C, Zhi G, Stull JT & Potter JD (2001) Familial hypertrophic cardiomyopathy mutations in the regulatory light chains of myosin affect their structure, Ca2+ binding, and phosphorylation. J Biol Chem 276, 7086–7092. [DOI] [PubMed] [Google Scholar]
- 13.Szczesna-Cordary D, Guzman G, Ng SS & Zhao J (2004) Familial hypertrophic cardiomyopathy-linked alterations in Ca2+ binding of human cardiac myosin regulatory light chain affect cardiac muscle contraction. J Biol Chem 279, 3535–3542. [DOI] [PubMed] [Google Scholar]
- 14.Wang L, Kazmierczak K, Yuan CC, Yadav S, Kawai M & Szczesna-Cordary D (2017) Cardiac contractility, motor function, and cross-bridge kinetics in N47K-RLC mutant mice. FEBS J 284, 1897–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang L, Muthu P, Szczesna-Cordary D & Kawai M (2013) Diversity and similarity of motor function and cross-bridge kinetics in papillary muscles of transgenic mice carrying myosin regulatory light chain mutations D166V and R58Q. J Mol Cell Cardiol 62, 153–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang L, Muthu P, Szczesna-Cordary D & Kawai M (2013) Characterizations of myosin essential light chain’s N-terminal truncation mutant Delta43 in transgenic mouse papillary muscles by using tension transients in response to sinusoidal length alterations. J Muscle Res Cell Motil 34, 93–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawai M, Kido T, Vogel M, Fink RH & Ishiwata S (2006) Temperature change does not affect force between regulated actin filaments and heavy meromyosin in single-molecule experiments. J Physiol 574, 877–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oguchi Y, Ishizuka J, Hitchcock-DeGregori SE, Ishiwata S & Kawai M (2011) The role of tropomyosin domains in cooperative activation of the actin-myosin interaction. J Mol Biol 414, 667–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kawai M & Brandt PW (1980) Sinusoidal analysis: a high resolution method for correlating biochemical reactions with physiological processes in activated skeletal muscles of rabbit, frog and crayfish. J Muscle Res Cell Motil 1, 279–303. [DOI] [PubMed] [Google Scholar]
- 20.Kawai M & Halvorson HR (1989) Role of MgATP and MgADP in the cross-bridge kinetics in chemically skinned rabbit psoas fibers. Study of a fast exponential process (C). Biophys J 55, 595–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Palmer BM, Suzuki T, Wang Y, Barnes WD, Miller MS & Maughan DW (2007) Two-state model of actomyosin attachment-detachment predicts C-process of sinusoidal analysis. Biophys J 93, 760–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Campbell KB, Chandra M, Kirkpatrick RD, Slinker BK & Hunter WC (2004) Interpreting cardiac muscle force-length dynamics using a novel functional model. Am J Physiol Heart Circ Physiol 286, H1535–H1545. [DOI] [PubMed] [Google Scholar]
- 23.Geeves MA (2002) Molecular motors: stretching the lever-arm theory. Nature 415, 129–131. [DOI] [PubMed] [Google Scholar]
- 24.Wang Y, Xu Y, Kerrick WGL, Wang Y, Guzman G, Diaz-Perez Z & Szczesna-Cordary D (2006) Prolonged Ca2+ and force transients in myosin RLC transgenic mouse fibers expressing malignant and benign FHC mutations. J Mol Biol 361, 286–299. [DOI] [PubMed] [Google Scholar]
- 25.Yuan CC, Muthu P, Kazmierczak K, Liang J, Huang W, Irving TC, Kanashiro-Takeuchi RM, Hare JM & Szczesna-Cordary D (2015) Constitutive phosphorylation of cardiac myosin regulatory light chain prevents development of hypertrophic cardiomyopathy in mice. Proc Natl Acad Sci USA 112, E4138–E4146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sevrieva IR, Brandmeier B, Ponnam S, Gautel M, Irving M, Campbell KS, Sun YB & Kampourakis T (2020) Cardiac myosin regulatory light chain kinase modulates cardiac contractility by phosphorylating both myosin regulatory light chain and troponin I. J Biol Chem 295, 4398–4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muthu P, Huang W, Kazmierczak K & Szczesna-Cordary D. (2012) Functional consequences of mutations in the myosin regulatory light chain associated with hypertrophic cardiomyopathy. In Cardiomyopathies – From Basic Research to Clinical Management (Veselka J, ed.), Ch. 17, pp. 383–408.InTech, Croatia. [Google Scholar]
- 28.Szczesna-Cordary D (2003) Regulatory light chains of striated muscle myosin. Structure, function and malfunction. Curr Drug Targets Cardiovasc Haematol Disord 3, 187–197. [DOI] [PubMed] [Google Scholar]
- 29.Rayment I, Rypniewski WR, Schmidt-Base K, Smith R, Tomchick DR, Benning MM, Winkelmann DA, Wesenberg G & Holden HM (1993) Three-dimensional structure of myosin subfragment-1: a molecular motor. Science 261, 50–58. [DOI] [PubMed] [Google Scholar]
- 30.Geeves MA & Holmes KC (1999) Structural mechanism of muscle contraction. Annu Rev Biochem 68, 687–728. [DOI] [PubMed] [Google Scholar]
- 31.Lowey S, Waller GS & Trybus KM (1993) Function of skeletal muscle myosin heavy and light chain isoforms by an in vitro motility assay. J Biol Chem 268, 20414–20418. [PubMed] [Google Scholar]
- 32.Howard J & Spudich JA (1996) Is the lever arm of myosin a molecular elastic element? Proc Natl Acad Sci USA 93, 4462–4464. [PubMed] [Google Scholar]
- 33.Kawai M (2018) Mathematics needed to solve problems of contraction. In Biomechanics, Muscle Fibers, and How to Interface Experimental Apparatus to a Computer (Kawai M, ed), pp. 65–76.Springer International Publishing, Cham. [Google Scholar]
- 34.Wang Y, Ajtai K & Burghardt TP (2014) Ventricular myosin modifies in vitro step-size when phosphorylated. J Mol Cell Cardiol 72, 231–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marston SB & de Tombe PP (2008) Troponin phosphorylation and myofilament Ca2+-sensitivity in heart failure: increased or decreased? J Mol Cell Cardiol 45, 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kamm KE & Stull JT (2011) Signaling to myosin regulatory light chain in sarcomeres. J Biol Chem 286, 9941–9947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang W & Szczesna-Cordary D (2015) Molecular mechanisms of cardiomyopathy phenotypes associated with myosin light chain mutations. J Muscle Res Cell Motil 36, 433–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tarigopula M, Davis RT 3rd, Mungai PT, Ryba DM, Wieczorek DF, Cowan CL, Violin JD, Wolska BM & Solaro RJ (2015) Cardiac myosin light chain phosphorylation and inotropic effects of a biased ligand, TRV120023, in a dilated cardiomyopathy model. Cardiovasc Res 107, 226–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dias FAL, Walker LA, Arteaga GM, Walker JS, Vijayan K, Pena JR, Ke Y, Fogaca RTH, Sanbe A, Robbins J et al. (2006) The effect of myosin regulatory light chain phosphorylation on the frequency-dependent regulation of cardiac function. J Mol Cell Cardiol 41, 330–339. [DOI] [PubMed] [Google Scholar]
- 40.Sheikh F, Ouyang K, Campbell SG, Lyon RC, Chuang J, Fitzsimons D, Tangney J, Hidalgo CG, Chung CS, Cheng H et al. (2012) Mouse and computational models link Mlc2v dephosphorylation to altered myosin kinetics in early cardiac disease. J Clin Invest 122, 1209–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van der Velden J, Papp Z, Zaremba R, Boontje NM, de Jong JW, Owen VJ, Burton PBJ, Goldmann P, Jaquet K & Stienen GJM (2003) Increased Ca2+-sensitivity of the contractile apparatus in end-stage human heart failure results from altered phosphorylation of contractile proteins. Cardiovasc Res 57, 37–47. [DOI] [PubMed] [Google Scholar]
- 42.Scruggs SB, Hinken AC, Thawornkaiwong A, Robbins J, Walker LA, de Tombe PP, Geenen DL, Buttrick PM & Solaro RJ (2009) Ablation of ventricular myosin regulatory light chain phosphorylation in mice causes cardiac dysfunction in situ and affects neighboring myofilament protein phosphorylation. J Biol Chem 284, 5097–5106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang J, Shelton JM, Richardson JA, Kamm KE & Stull JT (2008) Myosin regulatory light chain phosphorylation attenuates cardiac hypertrophy. J Biol Chem 283, 19748–19756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kerrick WGL, Kazmierczak K, Xu Y, Wang Y & Szczesna-Cordary D (2009) Malignant familial hypertrophic cardiomyopathy D166V mutation in the ventricular myosin regulatory light chain causes profound effects in skinned and intact papillary muscle fibers from transgenic mice. FASEB J 23, 855–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Warren SA, Briggs LE, Zeng H, Chuang J, Chang EI, Terada R, Li M, Swanson MS, Lecker SH, Willis MS et al. (2012) Myosin light chain phosphorylation is critical for adaptation to cardiac stress. Circulation 126, 2575–2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Toepfer CN, West TG & Ferenczi MA (2016) Revisiting Frank-Starling: regulatory light chain phosphorylation alters the rate of force redevelopment (ktr ) in a length-dependent fashion. J Physiol 594, 5237–5254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ding P, Huang J, Battiprolu PK, Hill JA, Kamm KE & Stull JT (2010) Cardiac myosin light chain kinase is necessary for myosin regulatory light chain phosphorylation and cardiac performance in vivo. J Biol Chem 285, 40819–40829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kawana M, Sarkar SS, Sutton S, Ruppel KM & Spudich JA (2017) Biophysical properties of human beta-cardiac myosin with converter mutations that cause hypertrophic cardiomyopathy. Sci Adv 3, e1601959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang Y, Yuan CC, Kazmierczak K, Szczesna-Cordary D & Burghardt TP (2018) Single cardiac ventricular myosins are autonomous motors. Open Biol 8, 170240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yadav S, Kazmierczak K, Liang J, Sitbon YH & Szczesna-Cordary D (2019) Phosphomimetic-mediated in vitro rescue of hypertrophic cardiomyopathy linked to R58Q mutation in myosin regulatory light chain. FEBS J 286, 151–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brenner B, Seebohm B, Tripathi S, Montag J & Kraft T (2014) Familial hypertrophic cardiomyopathy: functional variance among individual cardiomyocytes as a trigger of FHC-phenotype development. Front Physiol 5, 392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Robert-Paganin J, Auguin D & Houdusse A (2018) Hypertrophic cardiomyopathy disease results from disparate impairments of cardiac myosin function and auto-inhibition. Nat Commun 9, 4019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sanbe A, Gulick J, Hanks MC, Liang Q, Osinska H & Robbins J (2003) Reengineering inducible cardiac-specific transgenesis with an attenuated myosin heavy chain promoter. Circ Res 92, 609–616. [DOI] [PubMed] [Google Scholar]
- 54.Muthu P, Wang L, Yuan CC, Kazmierczak K, Huang W, Hernandez OM, Kawai M, Irving TC & Szczesna-Cordary D (2011) Structural and functional aspects of the myosin essential light chain in cardiac muscle contraction. FASEB J 25, 4394–4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kazmierczak K, Xu Y, Jones M, Guzman G, Hernandez OM, Kerrick WGL & Szczesna-Cordary D (2009) The role of the N-terminus of the myosin essential light chain in cardiac muscle contraction. J Mol Biol 387, 706–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kazmierczak K, Paulino EC, Huang W, Muthu P, Liang J, Yuan CC, Rojas AI, Hare JM & Szczesna-Cordary D (2013) Discrete effects of A57G-myosin essential light chain mutation associated with familial hypertrophic cardiomyopathy. Am J Physiol Heart Circ Physiol 305, H575–H589. [DOI] [PMC free article] [PubMed] [Google Scholar]