

Abstract
Tumor lesions comprise multiple subpopulations of cells including those endowed with “stemness” properties. The latter cells are responsible of tumor initiation, metastasis formation, resistance to conventional therapies and disease recurrence. These relatively rare cells denominated cancer stem cells (CSCs) or cancer initiating cells (CICs) are defined based on self-renewing, multipotency and tumorigenicity. These cells through their immunomodulating features can evade from immunesurveillance, persisting in the form of quiescence and dormancy. They can drive the neoplastic growth and recurrence, even after long latency. Moreover, CSCs/CICs due to their ability to modulate and shape immune responses can represent the component of a tumor causing immunotherapy resistance in cancer patients. In this review a general overview of immunological properties of CSCs/CICs is provided, with a special focus on the mechanisms of modulation of T cell mediated responses. The need to further dissect the mechanisms regulating the immunological profile of CSCs/CICs and their interactions with immune cells and tumor microenvironment is discussed. An improved characterization of the immunological properties of CSCs/CICs will contribute to the rationale design of immunotherapeutic interventions which target these cells and may lead to the eradication of malignant diseases.
Keywords: Cancer stem cells, Immunological profiling, Immune escape mechanisms, Tumor dormancy, Immune-based interventions
1. Introduction
As shown for the first time by Thomas and Burnet (Thomas L., Hoeber-Harper, 1959 New York) [1,2], the immune system can control tumor growth. The prognostic role of immune responses for cancer patients has been described [3] and, more recently this concept has been validated by the association of nature, location and density of tumor-infiltrating lymphocytes (TILs) with clinical outcome in colorectal cancer (CRC) patients [4,5]. The concept of “immunoscore’ has been created to classify CRC patients based on the extent of immune cell infiltrate; it represents a more informative prognostic biomarker than the traditional TNM staging in CRC patients [5–10]. Nevertheless, a variety of adaptive phenomena can shape the tumor-immune cell interplay enabling a dynamic equilibrium state in which cancer cells can survive in a dormant state [11]. The immune system can eliminate tumor cells while subsequently can maintain them in a state of equilibrium or, upon the induction of immune evasion mechanisms in tumor microenvironment (TME), resulting in its impairment and tumor progression. These phases of anti-tumor immune responses have been denominated the three “Es” theory of cancer [12]. Additionally, tumor cells adapt to survive in a hostile immune microenvironment by developing mechanisms which reduce their immunogenicity [13]. Anti-tumor immune responses can also select immune-resistant tumor cells, that will not be eliminated by cognate tumor antigen-specific T cells and by NK cells, resulting in tumor immunoediting [12]. This scenario can be described as immune system-mediated tumor dormancy, in which tumor cells can evade immune responsiveness through either the activation of intrinsic mechanisms or the shaping of an immunosuppressive TME.
Advances in immunotherapy have lead to the development of therapeutic strategies aimed at inducing systemic tumor antigen-specific immune responses [14–18]. Importantly, the clinical development of immune checkpoint inhibitors which unleash tumor antigen-specific T cells has changed the paradigm of cancer therapy [19–24]. However, a significant proportion of patients is unresponsive or develops resistance to these therapeutic strategies.
Tumor lesions are composed of cell subpopulations heterogeneous for genetic, phenotypic and functional properties, that can differ in their ability to generate and propagate tumors as well as in their susceptibility to therapeutic interventions. A minor subpopulation is represented by cancer stem cells or cancer initiating cells (CSCs/CICs) which are endowed with high-rate self-renewal, multi-potency and tumorigenicity [25–31]. These cells have been identified as responsible of metastatic spread, tumor recurrence, therapy resistance and disease progression [32,33]. CSCs/CICs are likely to play a crucial role in cancer patients’ immune and clinical responses to immunotherapy.
The efficient targeting of CSCs/CICs could contribute to the eradication of the component of the tumors endowed with quiescence, therapeutic resistance and dormant phenotype, all of which sustain long-term maintenance and progression of tumors. For this reason, the molecular heterogeneity, plasticity and evolution of genetic, phenotypic and immunological dynamic variations of these cells should be fully characterized. In this review we first describe the immunological properties of CSCs/CICs and the experimental evidence documenting host’s immune responses against these cells, with major emphasis on T cell mediated responses. Then we review the immune evasion mechanisms utilized by CSCs/CICs; we emphasize their role in mediating the impairment of cancer immunesurveillance, and as possible mechanisms governing tumor dormancy. Lastly, we discuss the immunotherapeutic approaches and their combinations to overcome immunomodulation mediated by CSCs/CICs.
2. Biological properties of CSCs/CICs
Cancer cells with “stemness” properties have been isolated from many hematological and solid tumors with different histological origins [34–38]. These cells can be isolated ex vivo and their CSC/CIC properties can be characterized based on their self-renewal, multipotency differentiating into cell lineages of the tissues of origin and tumorigenicity in immune deficient mice [29,39–41]. Therefore, they share some similarities with normal stem cells. The intrinsic “stemness” properties of these cells, through replication, can perpetuate CSC/CIC subpopulations and generate all the differentiated cell types that compose a tumor bulk [42]. The first report of CSC identification occurred for hematological malignancies, showing a hierarchical organization of tumor cells [35,43]. Engraftment of leukemic cells could successfully occur only upon transplantation of CD34+CD38− cell fractions leading to the identification of stem-like cancer cells in leukemia [35,43]. Interestingly, this tumor model provided the opportunity to extensively investigate the functional behavior of different leukemic cell subpopulations. These studies showed that pluripotent CSCs have a very slow cycling rate while differentiated bulk tumor cells mostly generate highly dividing cells [44].
CSC/CIC tumorigenic properties have been shown through xeno-transplantation in immune deficient mice [42]. These models have demonstrated that upon transplantation, cells with “stemness” properties can give rise to tumors resembling the original malignancies [41]. Xenograft assays have been used also to demonstrate the hierarchical organization of solid tumors, leading to the identification of tumorigenic populations, that upon serial transplantation could generate tumors comprising tumorigenic “stem-like cells” and phenotypically diverse non-tumorigenic cells [34]. These subpopulations with distinct functional properties cannot be discriminated based on morphological criteria, although upon transplantation in immunodeficient mice cells with “stemness” properties can give rise to tumors that recapitulate the heterogeneity of the original tumor lesions. Common motif of CSC studies is represented by the isolation from tumor tissues of rare cells based on the expression of single or multiple markers. Upon transplantation in immunodeficient mice these cells display tumorigenic properties, although with variable efficiency, indicating that indeed these cells are endowed with CSC/CIC properties [29,36,45,46].
The hierarchical organization of tumor cells has been clearly found in hematological malignancies where CSCs/CICs have been undoubtedly isolated based on the expression of specific markers. More controversial is the characterization of CSCs/CICs in solid tumors. Many lines of evidence have shown that differentiated tumor cells can dedifferentiate into multipotent cells with “stemness” properties [47]. Nevertheless, progress in the biological characterization of CSCs/CICs has highlighted high grade of heterogeneity, depending on their tissue derivation and on their plasticity [29,39,40,48,49]. Plasticity of CSCs/CICs has been shown in melanoma, where individual cancer cells sorted on the basis of CD133 expression can generate tumors in immunodeficient mice with similar frequency as CD133− cells [50,51]. Similarly, differential expression of demethylase has been associated with variable tumorigenicity of melanoma cells; however the expression of this marker was observed to be modulated in xenograft tumors independently on its expression by the tumor cells injected into immunodeficient mice [52]. Nevertheless, the lack of standardization of methods and reagents to isolate CSCs can also affect the high extent of variability of tumorigenic assays.
One of the variables influencing functional properties and high plasticity of CSCs/CICs is represented by their interactions and crosstalk with TME. Changes in TME can influence also the fate and biological properties of CSCs/CICs, since it comprises the “niche” needed for their maintenance and survival [37,53–59].
Multiple cell surface makers, e.g., CD24, CD44, CD133, CD166, Lgr5, etc., have been found to be expressed by cancer cells with “stemness” properties, depending on their histological origins [29,40] (Table 1). Because of their plasticity and continuous evolution in relationship with TME interplay CSCs/CICs can modulate the expression of some “bona fide” CSCs markers [60,61]. Most of the markers used to isolate and functionally characterize cells with “stemness” properties are overexpressed by these cells and shared with differentiated cellular counterparts [29,39,40,42] (Table 1). CSCs-associated markers, such as aldehyde dehydrogenase isoform 1 (ALDH1), CD44v6 and Lgr5, have been used either to isolate cells with “stemness” behavior from human tumor tissues or to localize these cells within neoplastic malignancies [62–65]. In some cases, the identification of cells expressing these markers in tumor tissues has been claimed to be correlated with disease’prognosis [62–65]. The usage of CSC/CIC-associated surface marker expression as a probe to detect these cells in human tumors and to investigate their impact on the fate of neoplastic lesions, has yielded inconclusive results. Whether this reflects the modulation of the phenotype of these cells along with changes in TME [66,67] and/or the lack of standardization of the reagents and methodology used to identify and isolate CSCs/CICs remains to be determined. In this regard, it is useful to point out that the field of CSCs/CICs would greatly benefit from an exchange of reagents used to identify these cells and by wet workshops to compare “under the same roof” methodologies used to identify and characterize CSCs/CICs. This type of approach was crucial to unravel the genetic organization of a very complex system like the HLA system, to dissect the specificity of HLA alloantisera and to define the role of the HLA system in clinical medicine.
Table 1.
Markers associated with CSCs/CICs.
| Markera | Tumor type |
|---|---|
| CD34/CD38 | AML |
| CD34/CD38/CD19; CD34/CD38−/CD19 | Leukaemia |
| ALDH1 | AML; colorectal, breast, gastric and ovarian cancer; melanoma |
| CD133 | Brain, colorectal, pancreatic, lung, ovarian, prostate, liver and gastric cancer |
| EpCAM | liver and colorectal cancer |
| CD44 | colorectal and head and neck cancer |
| CD24 | breast, pancreatic and colorectal cancer |
| CBX3-ABCA5 | osteosarcoma |
| LGR5 | colorectal cancer |
| ABCB1, ABCB5, ABCG2 | melanoma |
| CD271 | melanoma |
| SOX10 | melanoma and pancreatic cancer |
| CD166 | Colorectal cancer, NSCLC er |
| CD90 | Liver cancer |
| DNAJB8 | Renal cell and colrectal cancer |
The epithelial-to-mesenchymal transition (EMT), a developmental program which drives epithelial cells to acquire mesenchymal properties, can be influenced by inflammation and immune responses. This program that regulates embryogenesis and wound healing, is the main example of physiological transdifferentiation and becomes re-activated in cancer cells, promoting cell migration, cell dissemination and metastatic spread [68–73]. Through EMT epithelial cells acquire stem cell properties [71–75]. Interestingly, immune cells can induce EMT in both breast and melanoma cells [72,76]. Immunoediting can also either regulate EMT activation in cancer cells or select cancer cells that have already undergone EMT process [77]. Chronic inflammation in the gut can promote tissue damages and generation of intestinal fibrosis, which seems to result from epithelial-to-mesenchymal transition (EMT) [78–80]. The latter phenomenon is determined by the loss of polarization and adhesiveness by epithelia cells, and the acquisition of mesenchymal-like/myofibroblast phenotype, with increase of motility and resistance to apoptosis [78–80]. Patients with inflammatory bowel disease (IBD) have an increased risk of developing CRC depending on the duration and severity of inflammatory disease. Moreover, IBD-related CRC can display increased resistance to standard therapies and severity as compared to sporadic CRC. Therefore, this is an interesting model where inflammation, EMT and tumorigenesis are strictly related. Many other findings have linked the presence in tissues of inflammatory cells with EMT in tumors [68,77]. Consequently immune responses may play a determining role in the induction and immune selection of CSCs/CICs depending on the immune permissive or immunomodulating status of the microenvironment.
These data altogether emphasize the need to consider CSC/CIC plasticity when tracking or identifying these cells based on marker expression within tumor tissues. To this purpose xenograft models have limited usefulness since they cannot accurately predict the actual fate of cells in the tissue of origin and also their interactions with TME. No available evidence demonstrates that the behavior of tumor cells upon mouse transplantation corresponds to the hierarchical organization and actual fate of these cells in the tumor of origin. Moreover, hierarchical organization of tissues and tumorigenic properties of cellular components can vary depending on their histological source [81]. The observation by the group of Quintana that most of melanoma cells display “stemness” properties in a reversible way argues that tumor development and maintenance are mostly regulated by clonal evolution of cells rather than by a hierarchical structure [50,51]. Furthermore the extent of immunodeficiency of mice can affect the tumorigenicity and fate of cancer cells [51]. Therefore, the plasticity and de-differentiation ability of cancer cells can explain variable and conflicting observations related to the identification and role of CSCs/CICs in solid tumors.
By combining gene expression and functional analyses of tumor-derived cell subpopulation, Dick and colleagues showed that “stemness” core program associated with acute myeloid leukemia (AML)-derived CSCs represented an independent predictor marker for patients’ survival [32]. Similarly, “stemness’ gene signature could predict patients’ prognosis and contribute to classify breast cancer patients based on susceptibility to defined drug treatment [82]. Therefore, surface markers allow the selection of few cell subpopulations within tumors that can be endowed with stem cell-like properties. However, the identification of “stemness” core program could provide insights into the heterogeneity of tumor lesions and lead to a more accurate identification of CSCs that perpetuate tumors and are responsible of therapeutic resistance. Dynamic accumulation of genetic and epigenetic alterations in tumor cells leads to increased cellular heterogeneity, plasticity and prevalence of cell variants with CSC/CIC features. Moreover, the genetic pressure is a key driver of generation of distinct CSC/CIC subpopulations. Several altered signaling pathways, such as Sonic Hedgehog (SHH), Notch and Wnt/β-catenin regulate self-renewal, proliferation, and differentiation, representing therefore crucial signaling associated with “stemness” properties [29,39,40,46,83,84].
Importantly, CSCs/CICs have been identified as responsible of tumor resistance to standard therapies, such as chemotherapy and radiotherapy [85–89]. Although initially clinical responses are observed, CSCs/CICs can remain in minimal residual disease and can give rise to new tumors in the same organ site or through the metastatic colonization in other anatomic sites. Indeed, it has been reported both in pre-clinical models and through the analysis of cancer patients’ tissues, that the frequency of cells with “stemness-associated” properties is increased in post-therapy tumors as compared to pre-treatment lesions [90–93]. Some of the molecular pathways underlying therapeutic resistance have been identified, including impairment of apoptotic pathways, DNA damage repair, maintenance of quiescence, altered cell cycle regulation and upregulation of membrane-associated efflux pumps that mediate the cellular extrusion of drugs [86,94,95]. These mechanisms are orchestrated by altered activation or silencing of molecules that are members of SHH, Notch and Wnt/β-catenin pathways [96]. Resistance of CSCs to immunotherapy has been also reported in a glioma model of vaccination with dendritic cells (DCs) [97]. Thus, specific and unique functional characteristics including immunological properties, of CSCs/CICs should be identified in order to assess their role as prognostic and predictive biomarkers of responsiveness to therapies.
3. Are CSCs/CICs the Achille’s heel of T cell-based immunotherapy?
3.1. Potential mechanisms underlying the defective recognition of CSCs/CICs by cognate tumor associated antigen (TAA)-specific T cells
T lymphocytes recognize TAAs in the form of MHC/peptide complexes [16,98,99]. Most of the molecularly identified TAAs that are recognized by T cells belong to three categories: i. “self” antigens shared with lineage-related normal cells. ii. Cancer testis (CT) antigens that are over-expressed by tumor cells and are not detected in normal cells except for testis and trophoblasts. iii. Neoantigens or tumor-specific antigens that are generated by non-synonymous mutations in tumor cells.
Many TAAs have been shown to be effective targets of T-cell mediated immune responses; this information has provided the rationale to develop cancer vaccines for patients with different types of solid cancer [16]. Although TAA-specific immune responses have been detected in cancer patients vaccinated with TAAs, the common motif of these studies has been the poor clinical efficacy [16]. This limitation could be related to the inefficient targeting of CSCs/CICs. The expression of TAAs used for cancer vaccination by CSCs/CICs is the required prerogative for their eradication by cognate T lymphocytes. Early studies dedicated to the immunological profiling of CSCs/CICs showed that both “self” and CT antigens are expressed, although at low levels, by these cells [100–102]. T cell-mediated immune responses targeting ALDH1, CD133, CEP55, COA-1, DNAJB8, EpCam, HEATR1, IL-13Rα2, or SOX expressing CSCs/CICs have been documented by few studies [63] [103–112]. However, none of these TAAs is a CSC/CIC-specific marker; they represent overexpressed TAAs, shared with normal tissues. Their immunogenicity is low, due to the presence of tolerogenic T cells recognizing these antigens. The limited immunogenicity of this type of TAAs has been identified as one of the mechanisms underlying the low therapeutic efficacy of cancer vaccines [[[[16]]. In addition, effector cells could fail to react with both differentiated tumor cells and CSCs/CICs.
Defective HLA class I antigen processing machinery (APM) component expression and/or functions in CSCs/CICs have been reported by several investigators [100,101,112,113]. HLA class I APM comprises many molecules including proteasome and immunoproteasome subunits, subunits of transporters associated with antigen processing, and chaperone molecules which monitor the assembly of β2microglobulin (β2m) with HLA class I heavy chains, the loading of HLA class I molecules with TAA-derived peptides and the migration of the β2m-HLA class I heavy chain-TAA-derived peptide trimolecular complex to the cell membrane. Defective HLA class I APM component expression has been found frequently in CSCs/CICs in glioblastoma (GBM), melanoma and CRC [39,101,112–115]. Cordon-Cardo and collaborators have reported that in many types of solid tumors cells with CSCs/CICs properties failed to express HLA class I molecules. In some patients following chemotherapeutic treatments, cells with this phenotype were enriched in tumor lesions in association with reduced patients’ overall survival [113,116]. This finding may reflect the outgrowth of CSCs/CICs in tumors because of their resistance to chemotherapy. Cordon-Cardo et al., have suggested that lack of HLA class I antigen expression may represent a useful marker to identify CSCs/CICs [113,116]. They also suggested that lack of HLA class I antigen expression may be utilized to isolate CSCs/CICs from a tumor cell population [113,116]. We are skeptical about the usefulness of this strategy to isolate CSC/CICs since plenty of evidence has convincingly showed that defects in HLA class I antigen expression are present in differentiated cancer cells as well [117]. Therefore, lack of HLA class I antigen expression would not discriminate CSCs/CICs from differentiated malignant cells [118].
Limited, but convincing experimental evidence indicates that defects in HLA class I APM component expression in CSCs/CICs have functional relevance, since they provide them with an escape mechanism from recognition and destruction by cognate TAA-specific T cells. The cause-effect relationship between these two phenomena is indicated by the fact that correction of HLA class I abnormalities in CSCs/CICs restores their susceptibility to recognition and destruction by cognate TAA-specific T cells. If these in vitro findings have in vivo relevance, defects in HLA class I APM component expression and/or function in CSCs/CICs may provide them with an escape mechanism from recognition and destruction by host’s immune system. As a result they may represent a mechanism of patients’ resistance to immunotherapy.
The molecular mechanism(s) underlying defects in HLA class I APM component expression and/or function in CSCs/CICs have been characterized only to a limited extent at best. In some cases the HLA class I APM component downmodulation in CSCs/CICs can be corrected by treating these cells with IFN–α or -γ [101, 112]. This finding is compatible with the possibility that the defective HLA class I APM component expression is caused by abnormalities in the mechanism(s) which regulate their expression. Patients with this type of abnormalities are likely to benefit from therapies with epigenetic strategies. On the other hand, in other cases the HLA class I APM component expression and/or function in CSCs/CICs could not be restored by treating these cells with IFN–α or -γ [101, 112]. This finding is compatible with the possibility that the defective HLA class I APM component expression is caused by structural mutations in the gene(s) required for the expression and/or function of HLA class I APM. In these cases the expression and function of HLA class I APM can be restored only by replacing the mutated gene(s) with their wild type counterpart(s). Since gene therapy may be challenging in a clinical setting, T cell-based immunotherapy may not be the therapy of choice in patients with this type of abnormalities. These patients should be evaluated for therapies which do not utilize TAA-specific T cells as an effector mechanism.
Favorable selection of TH2 type T cells was observed following in vitro co-culture of peripheral blood lymphocytes (PBLs) with autologous CSCs/CICs [101,112]. Therefore, like normal stem cells [119], cancer cells endowed with “stemness’ features represent the immune-privileged component of a tumor allowing tumor initiation and tumor escape from immunesurveillance as well as its propagation. Nevertheless, heterogenity of CSCs/CICs is shown by the observation that detectable HLA class I expression has been reported in glioma”stemlike” cells [114,115,120,121].
Low HLA class I molecule expression levels may increase the susceptibility of CSCs/CICs to Natural Killer (NK) cells although only when ligands of NKG2D or activating NK cell receptors are efficiently expressed by these cells. In vitro recognition by NK cells of CSCs/CICs has been reported in glioma, melanoma, and CRC [114,115,120,122]. However, down-modulation of NKG2D ligands on CSCs/CICs has been documented, e.g., in GBM patients [101] suggesting that the suboptimal expression of NK cell activating ligands can impair innate immune responses against cells with “stemness” properties. Therefore, the plasticity of CSCs/CICs in conjunction with their crosstalk with TME could affect also the immunogenic profile of these cells and their susceptibility to NK cell-mediated responses.
The concept that highly immunogenic antigens, denominated neoantigens, can be generated by tumor-specific somatic mutations, that was initially proposed in animal models [14] has been recently confirmed in human studies [,123–125]. These TAAs, differently from tumor antigens shared with normal cells, are better recognized by T cells as “non-self”, driving CTL activation and expansion [126]. Indeed, neoantigens could represent novel tools for efficient targeting of CSCs/CICs. A similar frequency and type of somatic mutations in “driver” genes were detected in both differentiated cells and CSCs/CICs isolated from CRC [127]. Non-synonymous mutations in SMAD4 gene could generate neoepitopes recognized by T cells efficiently targeting CSCs/CICs [127]. Nevertheless, this study emphasized that the induction and isolation of anti-CSC T cells recognizing neoantigens required an upregulation of HLA class I APM components. In this study this was achieved by treating cells with IFN-γ [127]. These data, although need to be validated in larger cohorts of patients suggest that neoantigens could represent valuable tools to design effective immunotherapy interventions aimed at targeting CSCs/CICs.
The advent in clinical practice of immune checkpoint blockade agents registered for the first time improved overall survival of cancer patients, even for highly aggressive tumors, resistant to different therapeutic regimens [128–131]. These studies have provided the definitive demonstration that T cells unleashed by immune checkpoint-specific mAbs, which activate patients’ immune system against their own tumor can counterattack tumor growth and disease progression. As a result, dramatic regression of metastatic tumors and long-lasting clinical responses have been documented in the fraction of patients who respond to this type of therapy.
The proportion of responding patients differs markedly in various types of cancer; the reason(s) for this variability remains to be determined. Interestingly, tumor mutational burden, generation of neoantigens and/or detection of T cells reactive against neoepitopes in patients’ peripheral blood have been shown in at least some types of cancer to be correlated with patients’ clinical responses to immune checkpoint therapies [132–136]. It would be worthy in future studies to monitor whether the frequency of CSCs/CICs within tumor tissues changes along with treatments and cancer patients’ responsiveness to immune checkpoint blockade.
The characterization of CSCs/CICs immunological properties is still limited. Nevertheless, the evidence of sub-optimal expression of TAAs, that have been commonly used for vaccination of cancer patients, and of defective HLA class I APM molecules can explain the difficulties in targeting these cells with T cell-based immunotherapy. Therefore, a better understanding of the molecular profile of CSCs/CICs together with the identification of agents that can up-regulate their immunogenicity will contribute to design strategies which increase the ability of a patient’s immune system to recognize and eradicate these cells.
3.2. Mechanisms of immunomodulation associated with CSCs/CICs
3.2.1. Cytokines, growth factors, chemokines and enzymes
Analysis of the immune profiling of CSCs/CICs has highlighted their similarities with normal stem cells, including embryonic, hematopoietic and mesenchymal stem cells [29,119,137]. CSCs/CICs isolated from solid tumors with different histological origins have been shown to secrete a variety of cytokines and factors displaying immunomodulatory functions, such as Galectin-3, GDF-15, IL-10, IL-13, PGE2, TGF-β, leading to the protection of themselves and TME from immune attack [29,39,100,138,139] (Table 2). IL-10, IL-13 and TGF-β can regulate the differentiation of T cells with regulatory functions (Tregs) or myeloid derived suppressor cells (MDSCs) [29,39,102,118,139,140]. MDSCs are cells with heterogeneous granulocytic morphology deriving from myeloid progenitors that display potent immune-suppressive activity [141–144]. All the factors mentioned above play a crucial role in the accumulation in the TME of MDSCs, monocyte/macrophage and DCs with suppressive functions [145]. The production of inflammatory mediators by tumor cells drives the recruitment of immunosuppressive cells that also release cytokines therefore contributing to the maintenance of the inflammatory environment [145]. Thus, this represents one of the mechanisms for CSCs/CICs to sustain their niche. Components of innate immunity such as macrophages, and DCs depending on their morphological and phenotypic subtypes can either induce anti-tumor immune responses or promote tumor growth and progression [146]. The presence of tumor infiltrating macrophages (TAM) in tumor lesions can promote “stemness” properties of cancer cells in a variety of solid tumor models [147]. STAT3 plays a crucial role in maintenance and proliferation of CSCs/CICs [148,149]. STAT3 signaling is central in the cross-talk between TAM and CSCs/CICs; TAM can enhance activation of this molecule and induce the development of CSCs/CICs. On the other hand, these cells can up-modulate the immunosuppressive properties of TAM, leading to the impairment of T-cell mediated responses [150]. These cells have been shown to strongly modulate TAA-specific T cell responses, with increased frequency either at tumor site or in the peripheral blood of cancer patients [141]. Of note, the presence of MDSCs in TME can be associated with patients’ survival [141], and these cells have been shown also to be a predictive marker of clinical activity of immunotherapy in melanoma patients [151,152]. STAT3 activation can induce monocytes to acquire MDSC properties in pancreatic cancer tissues [153]. These cells are also critical to up-regulate CSCs/CICs and EMT in pancreatic ductal adenocarcinoma [153].
Table 2.
Expression of immunomodulatory mechanisms/molecules in CSCs/CICs vs. differentiated tumor cells.
| Molecule | CSCs/CICs vs. differentiated tumor cells | Reference(s) |
|---|---|---|
| APM | ⇓/negative | [101,102,112,118] |
| HLA molecules | ⇓/negative | [101,102,112,118] |
| IL-4 | ⇑ | [112] |
| IL-10 | ⇑ | [102,118] |
| IL-13 | ⇑ | [102] |
| TGF-β | ⇑ | [102] |
| B7-H3 | ⇑/differential localization | [101,112,159] [Maccalli et al., manuscript in preparation] |
| B7-H4 | ⇑ | [219] |
| PD-L1 | ⇑/differential localization | [101,112] [Maccalli et al., manuscript in preparation] |
| PGE2 | Detected | [219] |
| GDF-15 | ⇑ | [220] |
| STAT3 | ⇑ | [221] |
| Galectin-3 | ⇑ | [102] |
| IDO | ⇑ | [101,112] [Maccalli et al., manuscript in preparation] |
| CD200 | ⇑ | [158] |
| miRNAs | Differential expression | [165,166,167] [Maccalli et al., manuscript in preparation] |
⇑over-expressed.
⇓down-modulated.
APM: antigen processing machinery.
HLA: human leukocyte antigen.
IDO: indoleamine 2,3-dioxigenase.
IL-4: interleukin 4.
IL-10: interleukin 10.
IL-13: interleukin 13.
GDF-15: growth differentiation factor 15.
PGE2: Prostaglandin E2.
STAT3: Signal transducer and activator of transcription 3.
miRNAs: microRNAs.
Along this line T cell immunoglobulin mucin-3 (TIM-3)/Galectin 9 pathway is expressed by leukemia-derived CSCs/CICs leading to impairment of T cell responses through the differentiation and expansion of MDSCs and TAM in TME [154,155]. Additionally, these immunosuppressive cells can also promote the proliferation and survival of leukemia stem cells [154,155].
Therefore, further investigations of the cross-talk between innate and adaptive immune responses will provide insights into the immunogenic profile of CSC/CICs and its underlying mechanisms.
Interestingly, the chemokine receptor CXCL12, that is expressed by tumor and stromal cells is involved on one hand in sustaining CSC/CIC self-renewal and, on the other hand, in the recruitment in TME of MDSCs and suppressive DCs as well as in the differentiation of Tregs [156]. The inhibition of CXCL12/CXCR4 signaling has led to the decrease of tumor growth and induction of anti-tumor immune responses [156].
CD47 has been found overexpressed in CSCs/CICs from AML as compared to normal hematopoietic stem cells [157]. This molecule represents the ligand of signal regulatory protein alpha (SIRPα) that is a member of the signaling pathway of phagocytic cells such as macrophages and DCs. Through the binding of CD47 to SIRPα AML can inhibit cell-mediated phagocytosis, representing an additional mechanism to drive the impairment of innate immune responses [157].
Membrane-bound IL-4 and CD200 on CSCs/CICs isolated form epithelial tumors can also negatively modulate T cell proliferation and anti-tumor activity [112,158]. Of note, the neutralization of IL-4 expressed by CSCs/CICs can rescue the generation in vitro of CRC antigen-specific T lymphocytes [112]. One of us (Maccalli et al., manuscript in preparation) has showed that indoleamine 2,3-dioxigenase (IDO), that regulates the catabolism of tryptophan, is up-regulated, upon IFN-γ pretreatment of cells, in CSCs/CICs vs. differentiated cellular counterparts isolated from both GBM and CRC (Table 2). IDO is also used by MDSCs as an immunosuppressive mediator [145], indicating that a cross talk among CSCs/CICs and innate immunity can promote evasion from immune recognition. It was also shown that IDO represented the major player in the CSC-mediated impairment of proliferation and anti-tumor reactivity of T cells; this phenomenon could be overcome by blocking IDO activity in the presence of 1-methyl tryptophan (1-MT) (Maccalli et al., manuscript in preparation). These results suggest that the inhibition of IDO pathway could represent an effective strategy to overcome, at least in part, the immunomodulating role of CSCs/CICs. However, a phase III clinical study in melanoma patients testing the clinical efficacy of the combination of immune checkpoint blockade (anti-PD-1 mAb) with the IDO inhibitor epacadostat failed the endpoints to improve either progression-free survival or overall survival of patients. Therefore, additional investigations are required to identify the optimal clinical setting and drug administration for IDO inhibitors.
In addition, immune checkpoints molecules, such as PD-L1, B7-H3 and B7-H4 can be expressed by CSCs/CICs [101,112,159] (Table 2). The interaction of CSCs/CICs with T lymphocytes through the engagement of PD-1/PD-L1 signaling can down-modulate their differentiation and effector functions. In the last decades many advances on multiple molecular mechanisms underlying the differentiation and functional activity of immune cells have led to the clinical development of agents targeting immune checkpoints [20,160]. Some of these agents, such as antagonistic mAbs targeting CTLA-4 or PD-1/PD-L1 have shown unprecedented successful clinical results, either as monotherapy or in combination, in patients with different type of cancer refractory to standard therapies [24,161–164]. The usage of immune checkpoint blockade might represent a tool to target CSCs/CICs; nevertheless, the molecular profiling of tumor tissues including CSCs/CICs represents a requirement to predict whether immune checkpoint blockade could efficiently lead to tumor eradication.
In leukemic models it has been shown that immunosuppressive TME, with the presence of MDSCs and Tregs, activated TGF-β signaling, as well as the expression of immunoregulatory molecules, e.g. PD-L1, by CSCs/CICs protect these cells from recognition by T-cell mediated immune responses [154,155].
3.2.2. MicroRNA
MicroRNA (miRNAs) are short non-coding single-stranded mRNA that regulate through post-transcriptional gene silencing major cellular functions, such as proliferation, self-renewal, differentiation etc. [165]. miRNAs exerting gene silencing functions, bind to target mRNAs and through complementary based pairing degrade mRNA encoding target genes. The generation of miRNAs is regulated by cell-and tissue-specific transcription factors as well as proteins involved in the processing of miRNA, both of which can be influenced by chronic inflammation. They can act as suppressors of either tumor-related genes or oncogene and are commonly altered in tumors. Multiple miRNAs with altered levels have been described in cancers with different origins [165]. Some of these miRNAs have been identified as either down- or up -modulated in CSCs/CICs with relevant role in regulating self-renewal, apoptosis and differentiation [165]. For example, the expression of miRNAs MiR34a, MiR31 or MiR205, that can target either oncogenes (c-Met, Notch, CD6K), CD44 or other tumor suppressors has been found in CSCs/CICs isolated from glioma, prostate or head and neck cancer, respectively [165] (Table 2). Moreover, miRNAs 140 and 205 have been shown to confer chemotherapy resistance to CRC-derived cells with “stemness” properties [165]. Down-modulation of miRNAs 451 and 199b-5p reduced the sphere formation upon in vitro cell culture and expression of CD133+ and other stem-cell associated proteins, such as OCT4 and Nanog, in cells isolated from GBM and medulloblastoma [165,166]. High expression of MiR199b-5 was associated with patients’ survival in medulloblastoma [166].
Importantly, miRNAs can represent key regulators of immune functions and T cell differentiation [167] (Table 2). Therefore, the altered expression pattern of miRNAs in CSCs/CICs, represents one of the mechanisms regulating their immunological profile. One of us (Maccalli et al., manuscript in preparation) has found a link between differential cellular localization in CSCs/CICs vs. differentiated tumor cells of few immunomodulating agents, (e.g., PD-L1, B7-H3 and IDO; see Table 2) and the miRNAs profiling in these cells. Some of these molecules were preferentially detected in microvescicle compartment of CSCs/CICs while were mainly localized in the cytoplasms in differentiated tumor cells (Table 2).
It has been reported that miRNA-199a promotes breast cancer “stemness” properties by inhibiting the nuclear receptor corepressor LCOR, which is involved in interferon (IFN)-mediated responses. This leads to the protection of breast cancer-derived stem-like cells from differentiation and senescence that is mediated by IFNs released by immune cells [168]. MiRNA-124 that plays a regulatory role in the expression of STAT3 was not detected in glioma tissues. Its up-regulation in glioma-derived CSCs/CICs lead to inhibition of STAT3 and to the subsequent recovery of T cell-mediated immune responses [169].
3.2.3. Exosomes
Exosomes represent small vesicles (50–100 nm in diameter) that are generated from large multivesicular endosomes. The secretion of these vesicles by cells occurs upon fusion of multivesicular endosomes with the plasma membrane [172]. Exosomes have been shown to be released by a variety of immune cells, including B and T cells, DCs and macrophages and to be involved in some diseases such as infectionsus diseases, tumors and Alzheimer disease [172].
Exosomes represent cellular “messengers” and under physiological conditions mediate cell-to-cell interactions [173,174]. Although the mechanisms regulating exosomes release and their functions have not been dissected yet, it appears that their secretion can be modulated in different cell types upon environmental changes, as an adaptive cellular mechanism.
Exosomes can play a relevant role in regulating immune modulating mechanisms. Exosomes isolated from either immune cells or tumor cells can carry TAAs and MHC/peptide complexes, heat-shock proteins, which can also act as TAA carriers, adhesion molecules, membrane trafficking molecules, cytokines, chemokines, signal transduction proteins, etc. [175]. Tumor cells through the release of these microvesicles can promote invasion and migration [176,177] as well as modulate anti-tumor immune responses [172,178]. Apoptotic release of vesicle by tumor cells can also mediate cellular interplay to activate repair and regeneratione of neoplastic tissues [179]. Depending on the content of exosomes and on the type of cells involved in their up-take, these macrovesicles can either activate immune responses or act as negative immune modulators [180,181]. For example, exosomes can either redirect the differentiation of antigen processing cells, DCs and monocytes toward MDSCs or block T cell cytotoxic function or promote their apoptosis when enriched in Galectin-9, FASL and/or TRAIL [180,181].
Exosomes can contain miRNAs and can shape the TME and regulate malignant cell functions [170]. Noman et al., observed that up-regulation of miRNA-210 in hypoxic regions of lung cancer can regulate immune escape mechanisms [171]. They showed that miRNA-210 can abolish the susceptibility of tumor cells to T cell-mediated cytotoxicity, establishing an association between tumor hypoxia and immunosuppression. Notably, CSCs/CICs are generated and promoted in area of tumors and TME (tumor niche) with oxygen deprivation, indicating that indeed these rare cells could impair immune responses against tumor cells.
Through the paracrine release of exosomes normal stem cells and mesenchymal stem cells can subvert immune responses, rendering these microvesicles candidate therapeutic tools for the treatment of auto-immune and inflammatory diseases [180].
CSCs/CICs can release microvesicles promoting angiogenesis, cellular invasion and cancer progression [182,183]. These microvesicles might also exert immunosuppressive functions given their similarities with normal stem cells.
One of us has copared the cellular distribution of B7-H3, IDO and PD-L1 in CIS/CSCs vs. differentaited tumor counterpart. In the latter these molecules were preferentially localizedin the cytoplasm while in CSC/CICs they were mostly detected in microvesicles (Maccalli et al., manuscript in preparation) (Table 2). These observations suggest that cancer cells with “stemness” properties display a variety of mechanisms to achieve immune evasion from cell-mediated immune responses. Deep genomic, epigenetic and immunological investigations are required in order to achieve comprehensive profiling of immunological properties of cancer cells with “stemness” core program and, possibly, to identify interventional strategies to subvert their susceptibility to immunotherapy.
All together, these results provide evidence of immunomodulatory activity of CSCs/CICs that can impair T cell differentiation, proliferation and anti-tumor cytotoxic activity. Further insights into these immune regulatory mechanisms need to be gained to develop interventions that can overcome immunomodulating mechanisms. The resulting immunotherapy strategies are expected to display superior clinical efficacy in cancer pateints.
4. Targeting CSCs/CICs with mAb
With a few exceptions, antibody (Ab)-defined TAAs are distinct from T cell-defined TAAs. They differ from each other in terms of chemical nature, cellular localization and need of HLA class I APM for their presentation to their corresponding recognition unit. Specifically, T cell defined antigens are protein in nature. In contrast, antibody-defined TAAs can be proteins, carbohydrates and/or lipids. Furthermore, T cell-defined TAAs have prevalent intracellular expression, while antibody-defined TAAs, with few exceptions, are expressed on the membrane of cells. Lastly, T cell-defined TAAs need HLA class I APM to generate the fragment(s) recognized by cognate T cells and to transport them to cell membrane. In contrast, antibody-defined TAAs migrate to cell membrane independently of HLA class I APM.
Utilizing the conventional hybridoma technology and phage display antibody libraries a large number of TAA-specific antibodies have been developed. Many of them have been used in clinical settings and have become useful therapeutic reagents. However, information about the expression of these TAAs on CSCs/CICs is surprisingly limited in spite of the general agreement that eradication of CSCs/CICs is required to “cure” a malignant disease. We will mention our own findings in this area with special emphasis on clinically relevant TAAs.
Utilizing the hybridoma technology in the late 70′s we identified the chondroitin sulfate proteoglycan 4 (CSPG4). Like CD44, this molecule is a member of the CSPG family. CSPG members are key bioactive molecules which are directly implicated in a variety of human diseases, and especially in tumor growth and migration. CSPG4 is over-expressed by different types of cancers. They include GBM, head and neck cancer, triple negative breast cancer (TNBC), mesothelioma, chordoma, chondrosarcoma and melanoma [184,185]. This antigen has been shown to be expressed on CSCs/CICs in GBM, head and neck cancer, TNBC and melanoma.
CSPG4 is upregulated by hypoxia. This mechanism may explain its selective expression on activated pericytes in the tumor microenvironment [186], since hypoxia is a hallmark of solid tumor microenvironment. Immunotargeting of CSPG4 could selectively inhibit tumor-associated angiogenesis [187] and could contribute to the elimination of cancer cells with low or lack of expression of CSPG4. CSPG4-specific mAbs have been shown to eliminate CSCs/CICs present in TNBC tumors grafted in immunodeficient mice. In addition T cells genetically engineered with chimeric antigen receptors (CARs), that are recombinat receptors combining an extracellular antigen-recognition single chain, derived from antibody with intracellular signaling domain (T-cell activation and co-stimulation domains) [199] have been generated to target CSPG4. CSPG4-specific CARs have been shown to be very effective in eliminating neurospheres which express CSPG4 [188].
The TAAs B7-H3 and Grp94, which we are evaluating as targets of CAR T cell-based immunotherapy, have been shown to be expressed not only on differentiated cancer cells, but also on CSCs/CICs in head and neck cancer, TNBC and pancreatic ductal adenocarcinoma. More importantly, they have been shown to mediate the destruction of CSCs/CICs by CAR T cells. These results suggest that these two molecules have the potential to be useful targets to implement antibody-based immunotherapy for the treatment of various types of cancer.
5. CSCs/CICs and immunological/tumor dormancy
The majority of cancer deaths are due to metastases that can also occur years or decades after primary tumor diagnosis and treatment. In these cases, tumor cells cannot be detected before the development of recurrence or metastasis due to the phenomenon of tumor dormancy. The dormant state of tumor cells is mostly unknown. Some of the mechanisms that might account for it include cellular quiescence, altered cell signaling pathways, angiogenesis, interaction with tumor microenvironment and immune responses. Dormant cells with altered immunogenic potency and/ or with immunosuppressive functions, escaping from immune recognition may increase and cause tumor recurrence.
Clevers has recently summarized the CSC hypothesis as “the cancer patients who cannot be considered cured although initial responses to standard therapies” [42]. CSCs/CICs, representing the component of tumors resistant to standard therapy and immune recognition, can represent relevant component of tumor dormancy and responsible of the lack of complete cure of cancer patients. Moreover, stem cells properties include the ability to maintain themselves in transient or long-term quiescence [42]. Therefore, through their ability of switching from dormancy to malignancy they can give rise to new tumors in the same organ site or, through the metastatic colonization, in other anatomic sites [42]. Gene signature associated with normal quiescent mammary stem cells was identified to be shared with CSCs isolated from breast cancer, suggesting that these cells can represent the dormant component of tumors [189]. Additionally, through their low immunogenicity and heterogeneous immunomodulating properties, CSCs/CICs represent the tumor component escaping from immune recognition and from treatments with immunotherapy [29,39,40]. These cells can remain dormant and, subsequently give rise to tumor recurrence or driving the progression of the disease (Fig. 1). Leukemia stem cells have been shown to be responsible of disease relapse through their dormancy and self-renewal [190–192]. The identification of different cellular patterns of relapse endowed with “stemness” properties in AML represent useful tools to monitor the evolution of the disease [193]. Although, CSCs/CICs may acquire further genomic and epigenetic aberrations that might lead to the generation of highly immunogenic neoantigens, their low immunogenic profile including sub-optimal expression of HLA class I APM molecules and the expression of immunomodulating factors, can subvert tumor immunesurveillance and favor immune evasion [194]. Moreover, key factors for the immunological dormancy of CSCs are their up-regulation of signaling pathways regulating self-renewal, antiapoptotic mechanisms and their cross talk with inflammatory TME.
Fig. 1.
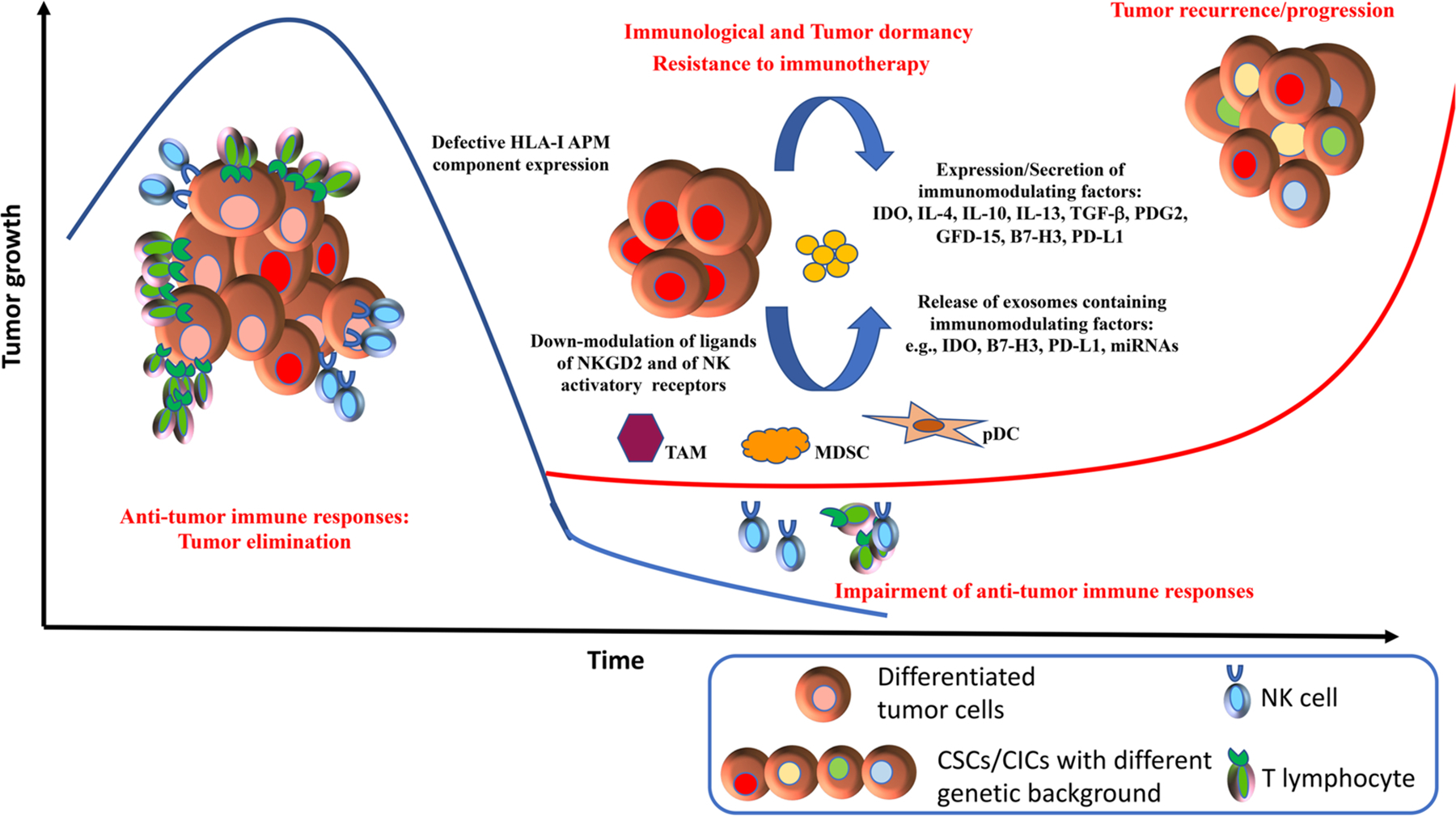
Immunological dormancy and evasion of CSCs/CICs from immune surveillance.
Tumor elimination is governed by active immune responses. Differentiated tumor cells can be recognized and killed by innate immune cells and TAA-specific T cells. The low immunogenicity of CSCs/CICs and their immunomodulating properties drive the evasion of these cells from immune recognition. Failure of immune cells to control tumor growth lead to selection of dormant cells (CSCs). The switch from quiescence to self-renewal of dormant cells, that can acquire also different genetic background and phenotype, determine tumor growth and therapy resistance, including immunotherapy, leading to tumor recurrence and progression.
A better understanding of CSCs’ contribution to clinical tumor dormancy and to evasion from immune responses will provide insights to design new and more effective therapeutic interventions for cancer patients.
6. Immunological intervention to overcome CSC-mediated immune regulation and tumor dormancy
Continuing advances in the characterization of biological and immunological properties of CSCs/CICs will allow the development of efficient strategies for the immunological targeting of these cells.
Vaccination studies in glioma patients with DCs loaded with mRNA or cell lysates from CICs showed safety and prolongation of patients’ survival [195,196]. These studies have highlighted that immunotherapy can represent a valid therapeutic option for cases in which suboptimal antigen processing and presentation by CSCs/CICs could be overcome through immunization interventions. On the other hand, the evidence that impaired APM and sub-optimal HLA expression are associated with CSCs/CICs argues against vaccination as the strategy of choice to treat cancer patients. Interventions to induce NK cell-mediated responses should be more extensively investigated [114,120]. Tumor-specific antigens generated by somatic mutations, representing strong immunogenic target molecule for immunotherapy [125,197], could be the source of TAA to generate T cells that can efficiently recognize and kill CSCs/CICs as reported by one of us [198]. These types of antigens could be exploited to either develop new cancer vaccines or to generate T cell engineered with high avidity TCR for adoptive cell therapy (ACT) [199]. However, their limitation in achieving the successful targeting of CSCs by CTL is represented by the evidence that antigen presentation can be suboptimal. Therefore, immunomodulating agents, such as IFNs or demethylating [200] agents should be combined, to up-regulate HLA class I APM components. To this aim the identification of CSC/CIC-specific signature could be exploited to identify these cells within tumor tissues and to obtain a snapshot of their immunological profile, representing prognostic/predictive tools for therapeutic decisions. On the other hand, this information will not be sufficient to monitor in vivo the interaction of CSCs/CICs with TME along with changes occurring during therapeutic treatments. Thus, further immunological investigations of cell with “stemness” properties with particular emphasis on different cellular subpopulations within tumor lesions are warranted.
The advent of immune checkpoint blockade represented for the first-time clinical success of immunotherapy, registering durable clinical responses and improved overall survival for cancer patients with different types of tumors [161,201–208]. The evidence that CSCs/CICs express immune checkpoint molecules renders this type of intervention suitable to subvert the immunomodulating profile of these cells, possibly in therapeutic regimens in combination with cancer vaccines.
Consideration should be given to combinations of immunogenic cell death-mediated chemotherapy, that could up-regulate the immunogenicity of CSCs/CICs, with immunotherapy interventions [209,210].
Other therapeutic options could be represented by agents targeting TME, hypoxia, STAT3 that can modulate CSC/CIC cross-talk with either TME or niche components that are involved in sustaining their “stemness” properties as well immunosuppressive cellular subtypes, such as MDCS and TAMs [211–213].
The clinical development of T cells engineered to express CARs represents the most recent breakthrough of immunotherapy. FDA has recently approved CAR-T cell-based drugs targeting CD19+ leukemia cells for advanced and therapy refractory pediatric and adult B cell malignancies [214].
In solid tumor, the development of CAR-T cells is still under investigations due to observations of severe toxicities when targeted TAAs are shared also by normal cells, such as the carbonic anhydrase IX (CAIX) or ERBB2 antigens [199]. Encouraging clinical results have been registered when mesothelin-specific CAR-T cells were administered to patients with malignant mesothelioma and pancreatic cancer [215]. Targeting CSCs/CICs with CAR-T cells could represent an efficient immunotherapy strategy to eradicate cells with low immunogenic profile. CAR-T cell targeting IL13Rα2, CD133 that are overexpressed by stem-like cells have been developed for clinical investigations [61,103,199,216,217].
Further investigations are warranted to pursue the biological and immunological characterization of CSCs/CICs with the aim to isolate antigens expressed by both stem-like and differentiated cancer cells but not by normal cells. These molecules will represent appropriate targets for efficient immunotherapy strategies.
Nevertheless, interventions aimed at increasing or subverting the immunogenicity and immunosuppressive profiles of CSCs/CICs are required. The combination of cancer vaccines or cell-based therapies with immunomodulating agents, e.g., epigenetic drugs or immune checkpoint blockade or agonistic agents represent promising approaches [20,200,218], although further pre-clinical and clinical investigations are needed.
7. Conclusions
The research on CSCs/CICs has contributed to their biological characterization while advances on immunological profile of cancer patients have provided an understanding of the mechanisms underlying cancer immunesurveillance and immune evasion. Combining these two fields will contribute to explain not only the mechanisms governing tumor recurrence, progression and dormancy but also the development in cancer patients of resistance to immunotherapy interventions (Fig. 1).
Cellular heterogeneity within tumor lesions and their evolution and interactions with TME may explain differences in terms of immune responsiveness and clinical outcome to immunotherapy in cancer patients. None of the currently ongoing immunotherapy approaches showed the ability to selectively target CSCs/CICs. This implies that we are still far from defeating cancer. Along this line, stable functional anti-tumor immune responses could at least maintain tumors in an “equilibrium phase”, controlling its growth and dissemination. Therefore, we need to understand the mechanisms regulating the genomics, epigenetics and immunology of stem-like cells in order to shape immune system toward effective anti-tumor immunesurveillance. This information will provide solutions to optimize or identify novel immunotherapy strategies and their combinations.
Funding
These studies have been partially funded by a grant from Qatar National Research Fund (grant no. NPRP10–0129-170277), Qatar and from grants from DOD (grant no. W81XWMH-16–1-0500) and NCI (grants no. R01DE028172, R03CA216114, RO3CA223886 and RO3CA231766).USA.
Footnotes
Conflict of interest declaration
All the authors have no conflict of interest to disclose.
References
- [1].Burnet FM, The concept of immunological surveillance, Prog. Exp. Tumor Res 13 (1970) 1–27. [DOI] [PubMed] [Google Scholar]
- [2].Burnet FM, Immunological surveillance in neoplasia, Transplant. Rev 7 (1971) 3–25. [DOI] [PubMed] [Google Scholar]
- [3].Mihm MC Jr.et al. , Tumor infiltrating lymphocytes in lymph node melanoma metastases: a histopathologic prognostic indicator and an expression of local immune response, Lab. Invest 74 (1) (1996) 43–47. [PubMed] [Google Scholar]
- [4].Pages F, et al. , Immune infiltration in human tumors: a prognostic factor that should not be ignored, Oncogene 29 (8) (2010) 1093–1102. [DOI] [PubMed] [Google Scholar]
- [5].Fridman WH, et al. , Immune infiltration in human cancer: prognostic significance and disease control, Curr. Top. Microbiol. Immunol 344 (2011) 1–24. [DOI] [PubMed] [Google Scholar]
- [6].Angell H, Galon J, From the immune contexture to the Immunoscore: the role of prognostic and predictive immune markers in cancer, Curr. Opin. Immunol 25 (2) (2013) 261–267. [DOI] [PubMed] [Google Scholar]
- [7].Bindea G, et al. , The immune landscape of human tumors: implications for cancer immunotherapy, Oncoimmunology 3 (1) (2014) e27456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bindea G, et al. , The prognostic impact of anti-cancer immune response: a novel classification of cancer patients, Semin. Immunopathol 33 (4) (2011) 335–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Mlecnik B, et al. , Integrative analyses of colorectal Cancer show immunoscore is a stronger predictor of patient survival than microsatellite instability, Immunity 44 (3) (2016) 698–711. [DOI] [PubMed] [Google Scholar]
- [10].Tosolini M, et al. , Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer, Cancer Res 71 (4) (2011) 1263–1271. [DOI] [PubMed] [Google Scholar]
- [11].Koebel CM, et al. , Adaptive immunity maintains occult cancer in an equilibrium state, Nature 450 (7171) (2007) 903–907. [DOI] [PubMed] [Google Scholar]
- [12].Dunn GP, et al. , Cancer immunoediting: from immunosurveillance to tumor escape, Nat. Immunol. 3 (11) (2002) 991–998. [DOI] [PubMed] [Google Scholar]
- [13].Kim SY, et al. , Soluble mediators from human neural stem cells play a critical role in suppression of T-cell activation and proliferation, J. Neurosci. Res 87 (10) (2009) 2264–2272. [DOI] [PubMed] [Google Scholar]
- [14].Parmiani G, et al. , Unique human tumor antigens: immunobiology and use in clinical trials, J. Immunol 178 (4) (2007) 1975–1979. [DOI] [PubMed] [Google Scholar]
- [15].Parmiani G, et al. , Autologous versus allogeneic cell-based vaccines? Cancer J 17 (5) (2011) 331–336. [DOI] [PubMed] [Google Scholar]
- [16].Parmiani G, et al. , Peptide-based vaccines for cancer therapy, Hum. Vaccin. Immunother 10 (11) (2014) 3175–3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Rosenberg SA, Progress in the development of immunotherapy for the treatment of patients with cancer, J. Intern. Med. 250 (6) (2001) 462–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Rosenberg SA, et al. , Immunologic and therapeutic evaluation of a synthetic peptide vaccine for the treatment of patients with metastatic melanoma, Nat. Med. 4 (3) (1998) 321–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Luke JJ, Ott PA, PD-1 pathway inhibitors: the next generation of immunotherapy for advanced melanoma, Oncotarget 6 (6) (2015) 3479–3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Pardoll DM, The blockade of immune checkpoints in cancer immunotherapy, Nat. Rev. Cancer 12 (4) (2012) 252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Rajan A, et al. , Nivolumab, anti-programmed death-1 (PD-1) monoclonal antibody immunotherapy: role in Advanced Cancers, Hum. Vaccin. Immunother (2016) 1–13. [DOI] [PMC free article] [PubMed]
- [22].Soares KC, et al. , PD-1/PD-L1 blockade together with vaccine therapy facilitates effector T-cell infiltration into pancreatic tumors, J Immunother 38 (1) (2015) 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tumeh PC, et al. , PD-1 blockade induces responses by inhibiting adaptive immune resistance, Nature 515 (7528) (2014) 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wilden SM, et al. , Immune checkpoint inhibitors: a milestone in the treatment of melanoma, J. Dermatol. Ges 14 (7) (2016) 685–695. [DOI] [PubMed] [Google Scholar]
- [25].Clarke MF, Becker MW, Stem cells: the real culprits in cancer? Sci. Am 295 (1) (2006) 52–59. [DOI] [PubMed] [Google Scholar]
- [26].Clarke MF, et al. , Cancer stem cells–perspectives on current status and future directions: AACR Workshop on cancer stem cells, Cancer Res 66 (19) (2006) 9339–9344. [DOI] [PubMed] [Google Scholar]
- [27].Hadjimichael C, et al. , Common stemness regulators of embryonic and cancer stem cells, World J. Stem Cells 7 (9) (2015) 1150–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Li Y, Laterra J, Cancer stem cells: distinct entities or dynamically regulated phenotypes? Cancer Res 72 (3) (2012) 576–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Maccalli C, De Maria R, Cancer stem cells: perspectives for therapeutic targeting, Cancer Immunol. Immunother 64 (1) (2015) 91–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Visvader JE, Lindeman GJ, Cancer stem cells: current status and evolving complexities, Cell Stem Cell 10 (6) (2012) 717–728. [DOI] [PubMed] [Google Scholar]
- [31].Wicha MS, et al. , Cancer stem cells: an old idea–a paradigm shift, Cancer Res 66 (4) (2006) 1883–90; discussion 1895–6. [DOI] [PubMed] [Google Scholar]
- [32].Eppert K, et al. , Stem cell gene expression programs influence clinical outcome in human leukemia, Nat. Med 17 (9) (2011) 1086–1093. [DOI] [PubMed] [Google Scholar]
- [33].Kreso A, Dick JE, Evolution of the cancer stem cell model, Cell Stem Cell 14 (3) (2014) 275–291. [DOI] [PubMed] [Google Scholar]
- [34].Al-Hajj M, et al. , Prospective identification of tumorigenic breast cancer cells, Proc. Natl. Acad. Sci. U. S. A 100 (7) (2003) 3983–3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lapidot T, et al. , A cell initiating human acute myeloid leukaemia after transplantation into SCID mice, Nature 367 (6464) (1994) 645–648. [DOI] [PubMed] [Google Scholar]
- [36].Ricci-Vitiani L, et al. , Identification and expansion of human colon-cancer-initiating cells, Nature 445 (7123) (2007) 111–115. [DOI] [PubMed] [Google Scholar]
- [37].Vermeulen L, et al. , Wnt activity defines colon cancer stem cells and is regulated by the microenvironment, Nat. Cell Biol 12 (5) (2010) 468–476. [DOI] [PubMed] [Google Scholar]
- [38].Vermeulen L, et al. , Single-cell cloning of colon cancer stem cells reveals a multi-lineage differentiation capacity, Proc. Natl. Acad. Sci. U. S. A 105 (36) (2008) 13427–13432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Maccalli C, et al. , Immunomodulating and immunoresistance properties of cancer-initiating cells: implications for the clinical success of immunotherapy, Immunol. Invest 46 (3) (2017) 221–238. [DOI] [PubMed] [Google Scholar]
- [40].Maccalli C, et al. , Immunology of cancer stem cells in solid tumours. A review, Eur. J. Cancer 50 (3) (2014) 649–655. [DOI] [PubMed] [Google Scholar]
- [41].Arienti F, et al. , Interleukin-2 gene-transduced human melanoma cells efficiently stimulate MHC-unrestricted and MHC-restricted autologous lymphocytes, Hum. Gene Ther 5 (9) (1994) 1139–1150. [DOI] [PubMed] [Google Scholar]
- [42].Clevers H, The cancer stem cell: premises, promises and challenges, Nat. Med 17 (3) (2011) 313–319. [DOI] [PubMed] [Google Scholar]
- [43].Bonnet D, Dick JE, Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell, Nat. Med. 3 (7) (1997) 730–737. [DOI] [PubMed] [Google Scholar]
- [44].Weissman IL, Stem cells: units of development, units of regeneration, and units in evolution, Cell 100 (1) (2000) 157–168. [DOI] [PubMed] [Google Scholar]
- [45].Eramo A, et al. , Identification and expansion of the tumorigenic lung cancer stem cell population, Cell Death Differ 15 (3) (2008) 504–514. [DOI] [PubMed] [Google Scholar]
- [46].Todaro M, et al. , CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis, Cell Stem Cell 14 (3) (2014) 342–356. [DOI] [PubMed] [Google Scholar]
- [47].Meacham CE, Morrison SJ, Tumour heterogeneity and cancer cell plasticity, Nature 501 (7467) (2013) 328–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Easwaran H, et al. , Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance, Mol. Cell 54 (5) (2014) 716–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Dick JE, Stem cell concepts renew cancer research, Blood 112 (13) (2008) 4793–4807. [DOI] [PubMed] [Google Scholar]
- [50].Quintana E, et al. , Efficient tumour formation by single human melanoma cells, Nature 456 (7222) (2008) 593–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Shackleton M, et al. , Heterogeneity in cancer: cancer stem cells versus clonal evolution, Cell 138 (5) (2009) 822–829. [DOI] [PubMed] [Google Scholar]
- [52].Roesch A, et al. , A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth, Cell 141 (4) (2010) 583–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Cabarcas SM, et al. , The cancer stem cell niche–there goes the neighborhood? Int. J. Cancer 129 (10) (2011) 2315–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Calabrese C, et al. , A perivascular niche for brain tumor stem cells, Cancer Cell 11 (1) (2007) 69–82. [DOI] [PubMed] [Google Scholar]
- [55].Conley SJ, et al. , Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia, Proc. Natl. Acad. Sci. U. S. A 109 (8) (2012) 2784–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Seidel S, et al. , A hypoxic niche regulates glioblastoma stem cells through hypoxia inducible factor 2 alpha, Brain 133 (Pt 4) (2010) 983–995. [DOI] [PubMed] [Google Scholar]
- [57].Cazet AS, et al. , Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer, Nat. Commun 9 (1) (2018) 2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Cully M, Tumour microenvironment: fibroblast subtype provides niche for cancer stem cells, Nat. Rev. Cancer 18 (3) (2018) 136. [DOI] [PubMed] [Google Scholar]
- [59].Cully M, Cancer: fibroblast subtype provides niche for cancer stem cells, Nat. Rev. Drug Discov 17 (3) (2018) 163. [DOI] [PubMed] [Google Scholar]
- [60].Kemper K, et al. , The AC133 epitope, but not the CD133 protein, is lost upon cancer stem cell differentiation, Cancer Res 70 (2) (2010) 719–729. [DOI] [PubMed] [Google Scholar]
- [61].Zhou F, et al. , Alpha2,3-Sialylation regulates the stability of stem cell marker CD133, J. Biochem 148 (3) (2010) 273–280. [DOI] [PubMed] [Google Scholar]
- [62].Ginestier C, et al. , ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome, Cell Stem Cell 1 (5) (2007) 555–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Visus C, et al. , Targeting ALDH(bright) human carcinoma-initiating cells with ALDH1A1-specific CD8(+) t cells, Clin. Cancer Res 17 (19) (2011) 6174–6184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Ma F, et al. , Aldehyde dehydrogenase 1 (ALDH1) expression is an independent prognostic factor in triple negative breast cancer (TNBC), Med. (Baltim.) 96 (14) (2017) e6561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Kemper K, et al. , Monoclonal antibodies against Lgr5 identify human colorectal cancer stem cells, Stem Cells 30 (11) (2012) 2378–2386. [DOI] [PubMed] [Google Scholar]
- [66].Bar EE, et al. , Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma, Stem Cells 25 (10) (2007) 2524–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Sakariassen PO, et al. , Angiogenesis-independent tumor growth mediated by stem-like cancer cells, Proc. Natl. Acad. Sci. U. S. A 103 (44) (2006) 16466–16471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Mani SA, et al. , The epithelial-mesenchymal transition generates cells with properties of stem cells, Cell 133 (4) (2008) 704–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Scheel C, Weinberg RA, Phenotypic plasticity and epithelial-mesenchymal transitions in cancer and normal stem cells? Int. J. Cancer 129 (10) (2011) 2310–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Scheel C, Weinberg RA, Cancer stem cells and epithelial-mesenchymal transition: concepts and molecular links, Semin. Cancer Biol 22 (5–6) (2012) 396–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Fantozzi A, et al. , VEGF-mediated angiogenesis links EMT-induced cancer stemness to tumor initiation, Cancer Res 74 (5) (2014) 1566–1575. [DOI] [PubMed] [Google Scholar]
- [72].Lamouille S, et al. , Molecular mechanisms of epithelial-mesenchymal transition, Nat. Rev. Mol. Cell Biol 15 (3) (2014) 178–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Nieto MA, Epithelial plasticity: a common theme in embryonic and cancer cells, Science 342 (6159) (2013) 1234850. [DOI] [PubMed] [Google Scholar]
- [74].Morel AP, et al. , Generation of breast cancer stem cells through epithelial-mesenchymal transition, PLoS One 3 (8) (2008) e2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Pradella D, et al. , EMT and stemness: flexible processes tuned by alternative splicing in development and cancer progression, Mol. Cancer 16 (1) (2017) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Gupta PB, et al. , Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells, Cell 146 (4) (2011) 633–644. [DOI] [PubMed] [Google Scholar]
- [77].Reiman JM, et al. , Immune promotion of epithelial-mesenchymal transition and generation of breast cancer stem cells, Cancer Res 70 (8) (2010) 3005–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Kalluri R, Neilson EG, Epithelial-mesenchymal transition and its implications for fibrosis, J. Clin. Invest 112 (12) (2003) 1776–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Bataille F, et al. , Evidence for a role of epithelial mesenchymal transition during pathogenesis of fistulae in Crohn’s disease, Inflamm. Bowel Dis 14 (11) (2008) 1514–1527. [DOI] [PubMed] [Google Scholar]
- [80].Flier SN, et al. , Identification of epithelial to mesenchymal transition as a novel source of fibroblasts in intestinal fibrosis, J. Biol. Chem 285 (26) (2010) 20202–20212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Quintana E, et al. , Human melanoma metastasis in NSG mice correlates with clinical outcome in patients, Sci. Transl. Med 4 (159) (2012) 159ra149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Shats I, et al. , Using a stem cell-based signature to guide therapeutic selection in cancer, Cancer Res. 71 (5) (2011) 1772–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Todaro M, et al. , Erythropoietin activates cell survival pathways in breast cancer stem-like cells to protect them from chemotherapy, Cancer Res 73 (21) (2013) 6393–6400. [DOI] [PubMed] [Google Scholar]
- [84].Catalano V, Resistance of Cancer Stem Cells to Cell- Mediated Immune Responses, Springer, 2015. [Google Scholar]
- [85].Bao S, et al. , Glioma stem cells promote radioresistance by preferential activation of the DNA damage response, Nature 444 (7120) (2006) 756–760. [DOI] [PubMed] [Google Scholar]
- [86].Dean M, et al. , Tumour stem cells and drug resistance, Nat. Rev. Cancer 5 (4) (2005) 275–284. [DOI] [PubMed] [Google Scholar]
- [87].Diehn M, Clarke MF, Cancer stem cells and radiotherapy: new insights into tumor radioresistance, J. Natl. Cancer Inst 98 (24) (2006) 1755–1757. [DOI] [PubMed] [Google Scholar]
- [88].Francipane MG, et al. , Crucial role of interleukin-4 in the survival of colon cancer stem cells, Cancer Res 68 (11) (2008) 4022–4025. [DOI] [PubMed] [Google Scholar]
- [89].Maugeri-Sacca M, et al. , Cancer stem cells and chemosensitivity, Clin. Cancer Res 17 (15) (2011) 4942–4947. [DOI] [PubMed] [Google Scholar]
- [90].Eyler CE, Rich JN, Survival of the fittest: cancer stem cells in therapeutic resistance and angiogenesis, J. Clin. Oncol 26 (17) (2008) 2839–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Frank NY, et al. , The therapeutic promise of the cancer stem cell concept, J. Clin. Invest 120 (1) (2010) 41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Li X, et al. , Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy, J. Natl. Cancer Inst 100 (9) (2008) 672–679. [DOI] [PubMed] [Google Scholar]
- [93].Wilson BJ, et al. , ABCB5 identifies a therapy-refractory tumor cell population in colorectal cancer patients, Cancer Res 71 (15) (2011) 5307–5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Wilson A, et al. , Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair, Cell 135 (6) (2008) 1118–1129. [DOI] [PubMed] [Google Scholar]
- [95].Yu F, et al. , let-7 regulates self renewal and tumorigenicity of breast cancer cells, Cell 131 (6) (2007) 1109–1123. [DOI] [PubMed] [Google Scholar]
- [96].Sotiropoulou PA, et al. , Chemical approaches to targeting drug resistance in cancer stem cells, Drug Discov. Today 19 (10) (2014) 1547–1562. [DOI] [PubMed] [Google Scholar]
- [97].Irvin DK, et al. , T cells enhance stem-like properties and conditional malignancy in gliomas, PLoS One 5 (6) (2010) e10974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Kelderman S, Kvistborg P, Tumor antigens in human cancer control, Biochim. Biophys. Acta 1865 (1) (2016) 83–89. [DOI] [PubMed] [Google Scholar]
- [99].Seremet T, et al. , Tumor-specific antigens and immunologic adjuvants in cancer immunotherapy, Cancer J 17 (5) (2011) 325–330. [DOI] [PubMed] [Google Scholar]
- [100].Codony-Servat J, Rosell R, Cancer stem cells and immunoresistance: clinical implications and solutions, Transl. Lung Cancer Res 4 (6) (2015) 689–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Di Tomaso T, et al. , Immunobiological characterization of cancer stem cells isolated from glioblastoma patients, Clin. Cancer Res 16 (3) (2010) 800–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Schatton T, et al. , Modulation of T-cell activation by malignant melanoma initiating cells, Cancer Res 70 (2) (2010) 697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Brown CE, et al. , Stem-like tumor-initiating cells isolated from IL13Ralpha2 expressing gliomas are targeted and killed by IL13-zetakine-redirected T Cells, Clin. Cancer Res 18 (8) (2012) 2199–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Engelmann K, et al. , MCF7 side population cells with characteristics of cancer stem/progenitor cells express the tumor antigen MUC1, Cancer Res 68 (7) (2008) 2419–2426. [DOI] [PubMed] [Google Scholar]
- [105].Hatiboglu MA, et al. , Immune therapeutic targeting of glioma cancer stem cells, Target. Oncol 5 (3) (2010) 217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Hua W, et al. , The CD133+ tumor stem-like cell-associated antigen may elicit highly intense immune responses against human malignant glioma, J. Neurooncol 105 (2) (2011) 149–157. [DOI] [PubMed] [Google Scholar]
- [107].Inoda S, et al. , Cytotoxic T lymphocytes efficiently recognize human colon cancer stem-like cells, Am. J. Pathol 178 (4) (2011) 1805–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Ji J, et al. , Identification of novel human leukocyte antigen-A*0201-restricted, cytotoxic T lymphocyte epitopes on CD133 for cancer stem cell immunotherapy, Stem Cells Transl. Med 3 (3) (2014) 356–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Mitra M, et al. , EpCAM is a putative stem marker in retinoblastoma and an effective target for T-cell-mediated immunotherapy, Mol. Vis 18 (2012) 290–308. [PMC free article] [PubMed] [Google Scholar]
- [110].Morita R, et al. , Heat shock protein DNAJB8 is a novel target for immunotherapy of colon cancer-initiating cells, Cancer Sci. 105 (4) (2014) 389–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Schmitz M, et al. , Identification of SOX2 as a novel glioma-associated antigen and potential target for T cell-based immunotherapy, Br. J. Cancer 96 (8) (2007) 1293–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Volonte A, et al. , Cancer-initiating cells from colorectal cancer patients escape from T cell-mediated immunosurveillance in vitro through membrane-bound IL-4, J. Immunol 192 (1) (2014) 523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Grau JJ, et al. , Enrichment of cells with Cancer stem cell-like markers in relapses of chemoresistant patients with locally advanced head and neck squamous cell carcinoma, Oncology 90 (5) (2016) 267–272. [DOI] [PubMed] [Google Scholar]
- [114].Pietra G, et al. , Natural killer cells kill human melanoma cells with characteristics of cancer stem cells, Int. Immunol 21 (7) (2009) 793–801. [DOI] [PubMed] [Google Scholar]
- [115].Castriconi R, et al. , NK cells recognize and kill human glioblastoma cells with stem cell-like properties, J. Immunol 182 (6) (2009) 3530–3539. [DOI] [PubMed] [Google Scholar]
- [116].Subramaniam PS, et al. , Targeting nonclassical oncogenes for therapy in T-ALL, Cancer Cell 21 (4) (2012) 459–472. [DOI] [PubMed] [Google Scholar]
- [117].Seliger B, Molecular mechanisms of HLA class I-mediated immune evasion of human tumors and their role in resistance to immunotherapies, HLA 88 (5) (2016) 213–220. [DOI] [PubMed] [Google Scholar]
- [118].Wei J, et al. , Glioma-associated cancer-initiating cells induce immunosuppression, Clin. Cancer Res 16 (2) (2010) 461–473. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [119].Ichiryu N, Fairchild PJ, Immune privilege of stem cells, Methods Mol. Biol 1029 (2013) 1–16. [DOI] [PubMed] [Google Scholar]
- [120].Tallerico R, et al. , Human NK cells selective targeting of colon cancer-initiating cells: a role for natural cytotoxicity receptors and MHC class I molecules, J. Immunol 190 (5) (2013) 2381–2390. [DOI] [PubMed] [Google Scholar]
- [121].Wu A, et al. , Expression of MHC I and NK ligands on human CD133+ glioma cells: possible targets of immunotherapy, J. Neurooncol 83 (2) (2007) 121–131. [DOI] [PubMed] [Google Scholar]
- [122].Tseng HC, et al. , Increased lysis of stem cells but not their differentiated cells by natural killer cells; de-differentiation or reprogramming activates NK cells, PLoS One 5 (7) (2010) e11590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Forshew T, et al. , Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA, Sci. Transl. Med 4 (136) (2012) 136ra68. [DOI] [PubMed] [Google Scholar]
- [124].Robbins PF, Tumor-infiltrating lymphocyte therapy and neoantigens, Cancer J 23 (2) (2017) 138–143. [DOI] [PubMed] [Google Scholar]
- [125].Sahin U, et al. , Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer, Nature 547 (7662) (2017) 222–226. [DOI] [PubMed] [Google Scholar]
- [126].McGranahan N, et al. , Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade, Science 351 (6280) (2016) 1463–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Mennonna D, et al. , T cell neoepitope discovery in colorectal cancer by high throughput profiling of somatic mutations in expressed genes, Gut (2015). [DOI] [PMC free article] [PubMed]
- [128].Adachi K, Tamada K, Immune checkpoint blockade opens an avenue of cancer immunotherapy with a potent clinical efficacy, Cancer Sci 106 (8) (2015) 945–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Kohrt HE, et al. , Immunodynamics: a cancer immunotherapy trials network review of immune monitoring in immuno-oncology clinical trials, J. Immunother. Cancer 4 (2016) 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Marabelle A, Gray J, Tumor-targeted and immune-targeted monoclonal antibodies: going from passive to active immunotherapy, Pediatr. Blood Cancer 62 (8) (2015) 1317–1325. [DOI] [PubMed] [Google Scholar]
- [131].Marabelle A, et al. , Intratumoral anti-CTLA-4 therapy: enhancing efficacy while avoiding toxicity, Clin. Cancer Res. 19 (19) (2013) 5261–5263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Kvistborg P, et al. , Anti-CTLA-4 therapy broadens the melanoma-reactive CD8+ T cell response, Sci. Transl. Med 6 (254) (2014) 254ra128. [DOI] [PubMed] [Google Scholar]
- [133].Rizvi NA, et al. , Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer, Science 348 (6230) (2015) 124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Schumacher TN, Schreiber RD, Neoantigens in cancer immunotherapy, Science 348 (6230) (2015) 69–74. [DOI] [PubMed] [Google Scholar]
- [135].Snyder A, et al. , Genetic basis for clinical response to CTLA-4 blockade in melanoma, N. Engl. J. Med. 371 (23) (2014) 2189–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].van Rooij N, et al. , Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma, J. Clin. Oncol 31 (32) (2013) e439–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Ankrum JA, et al. , Mesenchymal stem cells: immune evasive, not immune privileged, Nat. Biotechnol. 32 (3) (2014) 252–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Schatton T, Frank MH, Antitumor immunity and cancer stem cells, Ann. N. Y. Acad. Sci 1176 (2009) 154–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Yoshimura A, Muto G, TGF-beta function in immune suppression, Curr. Top. Microbiol. Immunol 350 (2011) 127–147. [DOI] [PubMed] [Google Scholar]
- [140].Wan YY, Flavell RA, ‘Yin-Yang’ functions of transforming growth factor-beta and T regulatory cells in immune regulation, Immunol. Rev 220 (2007) 199–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Filipazzi P, et al. , Phenotype, function and clinical implications of myeloid-derived suppressor cells in cancer patients, Cancer Immunol. Immunother 61 (2) (2012) 255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Gabrilovich DI, et al. , Coordinated regulation of myeloid cells by tumours, Nat. Rev. Immunol. 12 (4) (2012) 253–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Nagaraj S, Gabrilovich DI, Regulation of suppressive function of myeloid-derived suppressor cells by CD4+ T cells, Semin. Cancer Biol 22 (4) (2012) 282–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Youn JI, et al. , Characterization of the nature of granulocytic myeloid-derived suppressor cells in tumor-bearing mice, J. Leukoc. Biol 91 (1) (2012) 167–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Parker KH, et al. , Myeloid-derived suppressor cells: critical cells driving immune suppression in the tumor microenvironment, Adv. Cancer Res 128 (2015) 95–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Grivennikov SI, et al. , Immunity, inflammation, and cancer, Cell 140 (6) (2010) 883–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Jinushi M, et al. , Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells, Proc. Natl. Acad. Sci. U. S. A 108 (30) (2011) 12425–12430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Sherry MM, et al. , STAT3 is required for proliferation and maintenance of multipotency in glioblastoma stem cells, Stem Cells 27 (10) (2009) 2383–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Zhou J, et al. , Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance, Proc. Natl. Acad. Sci. U. S. A 104 (41) (2007) 16158–16163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Mitchem JB, et al. , Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses, Cancer Res. 73 (3) (2013) 1128–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Martens A, et al. , Baseline peripheral blood biomarkers associated with clinical outcome of advanced melanoma patients treated with ipilimumab, Clin. Cancer Res 22 (12) (2016) 2908–2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Weide B, et al. , Myeloid-derived suppressor cells predict survival of patients with advanced melanoma: comparison with regulatory T cells and NY-ESO-1- or melan-A-specific T cells, Clin. Cancer Res. 20 (6) (2014) 1601–1609. [DOI] [PubMed] [Google Scholar]
- [153].Panni RZ, et al. , Tumor-induced STAT3 activation in monocytic myeloid-derived suppressor cells enhances stemness and mesenchymal properties in human pancreatic cancer, Cancer Immunol. Immunother 63 (5) (2014) 513–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Gao L, et al. , Hypothesis: Tim-3/galectin-9, a new pathway for leukemia stem cells survival by promoting expansion of myeloid-derived suppressor cells and differentiating into tumor-associated macrophages, Cell Biochem. Biophys. 70 (1) (2014) 273–277. [DOI] [PubMed] [Google Scholar]
- [155].Riether C, et al. , Regulation of hematopoietic and leukemic stem cells by the immune system, Cell Death Differ. 22 (2) (2015) 187–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Gil M, et al. , CXCL12/CXCR4 blockade by oncolytic virotherapy inhibits ovarian cancer growth by decreasing immunosuppression and targeting cancer-initiating cells, J. Immunol 193 (10) (2014) 5327–5337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Majeti R, et al. , CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells, Cell 138 (2) (2009) 286–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Kawasaki BT, et al. , Co-expression of the toleragenic glycoprotein, CD200, with markers for cancer stem cells, Biochem. Biophys. Res. Commun 364 (4) (2007) 778–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Yao Y, et al. , B7-H4 is preferentially expressed in non-dividing brain tumor cells and in a subset of brain tumor stem-like cells, J. Neurooncol. 89 (2) (2008) 121–129. [DOI] [PubMed] [Google Scholar]
- [160].Pardoll DM, Immunology beats cancer: a blueprint for successful translation, Nat. Immunol. 13 (12) (2012) 1129–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Kyi C, Postow MA, Immune checkpoint inhibitor combinations in solid tumors: opportunities and challenges, Immunotherapy 8 (7) (2016) 821–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Naidoo J, et al. , Immune checkpoint blockade, Hematol. Oncol. Clin. North Am 28 (3) (2014) 585–600. [DOI] [PubMed] [Google Scholar]
- [163].Rotte A, et al. , Mechanistic overview of immune checkpoints to support the rational design of their combinations in cancer immunotherapy, Ann. Oncol 29 (1) (2018) 71–83. [DOI] [PubMed] [Google Scholar]
- [164].Xia Y, et al. , Immune checkpoint blockade: releasing the brake towards hematological malignancies, Blood Rev 30 (3) (2016) 189–200. [DOI] [PubMed] [Google Scholar]
- [165].Chhabra R, Saini N, MicroRNAs in cancer stem cells: current status and future directions, Tumour Biol 35 (9) (2014) 8395–8405. [DOI] [PubMed] [Google Scholar]
- [166].Garzia L, et al. , MicroRNA-199b-5p impairs cancer stem cells through negative regulation of HES1 in medulloblastoma, PLoS One 4 (3) (2009) e4998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Chen CZ, et al. , Regulation of immune responses and tolerance: the microRNA perspective, Immunol. Rev 253 (1) (2013) 112–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Celia-Terrassa T, et al. , Normal and cancerous mammary stem cells evade interferon-induced constraint through the miR-199a-LCOR axis, Nat. Cell Biol 19 (6) (2017) 711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Wei J, et al. , miR-124 inhibits STAT3 signaling to enhance T cell-mediated immune clearance of glioma, Cancer Res 73 (13) (2013) 3913–3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Sanchez CA, et al. , Exosomes from bulk and stem cells from human prostate cancer have a differential microRNA content that contributes cooperatively over local and pre-metastatic niche, Oncotarget 7 (4) (2016) 3993–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Noman MZ, et al. , Hypoxia-inducible miR-210 regulates the susceptibility of tumor cells to lysis by cytotoxic T cells, Cancer Res 72 (18) (2012) 4629–4641. [DOI] [PubMed] [Google Scholar]
- [172].Thery C, et al. , Membrane vesicles as conveyors of immune responses, Nat. Rev. Immunol. 9 (8) (2009) 581–593. [DOI] [PubMed] [Google Scholar]
- [173].de Gassart A, et al. , Exosome secretion: the art of reutilizing nonrecycled proteins? Traffic 5 (11) (2004) 896–903. [DOI] [PubMed] [Google Scholar]
- [174].Stoorvogel W, et al. , The biogenesis and functions of exosomes, Traffic 3 (5) (2002) 321–330. [DOI] [PubMed] [Google Scholar]
- [175].Greening DW, et al. , Exosomes and their roles in immune regulation and cancer, Semin. Cell Dev. Biol 40 (2015) 72–81. [DOI] [PubMed] [Google Scholar]
- [176].Dolo V, et al. , Selective localization of matrix metalloproteinase 9, beta1 integrins, and human lymphocyte antigen class I molecules on membrane vesicles shed by 8701-BC breast carcinoma cells, Cancer Res 58 (19) (1998) 4468–4474. [PubMed] [Google Scholar]
- [177].Ginestra A, et al. , The amount and proteolytic content of vesicles shed by human cancer cell lines correlates with their in vitro invasiveness, Anticancer Res 18 (5A) (1998) 3433–3437. [PubMed] [Google Scholar]
- [178].Valadi H, et al. , Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells, Nat. Cell Biol 9 (6) (2007) 654–659. [DOI] [PubMed] [Google Scholar]
- [179].Gregory CD, Dransfield I, Apoptotic tumor cell-derived extracellular vesicles as important regulators of the onco-regenerative niche, Front. Immunol 9 (2018) 1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Robbins PD, Morelli AE, Regulation of immune responses by extracellular vesicles, Nat. Rev. Immunol 14 (3) (2014) 195–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Whiteside TL, Exosomes in Cancer: another mechanism of tumor-induced immune suppression, Adv. Exp. Med. Biol 1036 (2017) 81–89. [DOI] [PubMed] [Google Scholar]
- [182].Hannafon BN, et al. , Exosome-mediated microRNA signaling from breast cancer cells is altered by the anti-angiogenesis agent docosahexaenoic acid (DHA), Mol. Cancer 14 (2015) 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Hannafon BN, Ding WQ, Cancer stem cells and exosome signaling, Stem Cell Investig 2 (2015) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Beard RE, et al. , Multiple chimeric antigen receptors successfully target chondroitin sulfate proteoglycan 4 in several different cancer histologies and cancer stem cells, J. Immunother. Cancer 2 (2014) 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Campoli M, et al. , Functional and clinical relevance of chondroitin sulfate proteoglycan 4, Adv. Cancer Res 109 (2010) 73–121. [DOI] [PubMed] [Google Scholar]
- [186].Price MA, et al. , CSPG4, a potential therapeutic target, facilitates malignant progression of melanoma, Pigment Cell Melanoma Res 24 (6) (2011) 1148–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Maciag PC, et al. , Cancer immunotherapy targeting the high molecular weight melanoma-associated antigen protein results in a broad antitumor response and reduction of pericytes in the tumor vasculature, Cancer Res 68 (19) (2008) 8066–8075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Pellegatta S, Savoldo B, Di Ianni N, Corbetta C, Chen Y, Patané M, Sun C, Pollo B, Ferrone S, DiMeco F, Finocchiaro G, Dotti G, Constitutive and TNFα-inducible expression of chondroitin sulfate proteoglycan 4 in glioblastoma and neurospheres: Implications for CAR-T cell therapy, Sci. Transl. Med 10 (430) (2018) 10.1126/scitranslmed.aao2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Pece S, et al. , Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content, Cell 140 (1) (2010) 62–73. [DOI] [PubMed] [Google Scholar]
- [190].Costello RT, et al. , Human acute myeloid leukemia CD34+/CD38- progenitor cells have decreased sensitivity to chemotherapy and Fas-induced apoptosis, reduced immunogenicity, and impaired dendritic cell transformation capacities, Cancer Res 60 (16) (2000) 4403–4411. [PubMed] [Google Scholar]
- [191].Ishikawa F, et al. , Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region, Nat. Biotechnol 25 (11) (2007) 1315–1321. [DOI] [PubMed] [Google Scholar]
- [192].Terpstra W, et al. , Long-term leukemia-initiating capacity of a CD34-subpopulation of acute myeloid leukemia, Blood 87 (6) (1996) 2187–2194. [PubMed] [Google Scholar]
- [193].Shlush LI, et al. , Tracing the origins of relapse in acute myeloid leukaemia to stem cells, Nature 547 (7661) (2017) 104–108. [DOI] [PubMed] [Google Scholar]
- [194].Bruttel VS, Wischhusen J, Cancer stem cell immunology: key to understanding tumorigenesis and tumor immune escape? Front. Immunol 5 (2014) 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Vik-Mo EO, et al. , Therapeutic vaccination against autologous cancer stem cells with mRNA-transfected dendritic cells in patients with glioblastoma, Cancer Immunol. Immunother. 62 (9) (2013) 1499–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Finocchiaro G, Pellegatta S, Immunotherapy with dendritic cells loaded with glioblastoma stem cells: from preclinical to clinical studies, Cancer Immunol. Immunother. 65 (1) (2016) 101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Trajanoski Z, et al. , Somatically mutated tumor antigens in the quest for a more efficacious patient-oriented immunotherapy of cancer, Cancer Immunol. Immunother 64 (1) (2015) 99–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Mennonna D, et al. , T cell neoepitope discovery in colorectal cancer by high throughput profiling of somatic mutations in expressed genes, Gut 66 (3) (2017) 454–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Hinrichs CS, Rosenberg SA, Exploiting the curative potential of adoptive T-cell therapy for cancer, Immunol. Rev 257 (1) (2014) 56–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Nervi C, et al. , Epigenetic treatment of solid tumours: a review of clinical trials, Clin. Epigenetics 7 (2015) 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Antonia SJ, et al. , Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): a multicentre, open-label, phase 1/2 trial, Lancet Oncol 17 (7) (2016) 883–895. [DOI] [PubMed] [Google Scholar]
- [202].Berman D, et al. , Blockade of cytotoxic T-lymphocyte antigen-4 by ipilimumab results in dysregulation of gastrointestinal immunity in patients with advanced melanoma, Cancer Immun 10 (2010) 11. [PMC free article] [PubMed] [Google Scholar]
- [203].Boutros C, et al. , Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination, Nat. Rev. Clin. Oncol (2016). [DOI] [PubMed]
- [204].Brahmer JR, PD-1-targeted immunotherapy: recent clinical findings, Clin. Adv. Hematol. Oncol 10 (10) (2012) 674–675. [PubMed] [Google Scholar]
- [205].Brahmer JR, et al. , Nivolumab: targeting PD-1 to bolster antitumor immunity, Future Oncol 11 (9) (2015) 1307–1326. [DOI] [PubMed] [Google Scholar]
- [206].Brahmer JR, et al. , Safety and activity of anti-PD-L1 antibody in patients with advanced cancer, N. Engl. J. Med 366 (26) (2012) 2455–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Haanen JB, Robert C, Immune checkpoint inhibitors, Prog. Tumor Res 42 (2015) 55–66. [DOI] [PubMed] [Google Scholar]
- [208].Larkin J, et al. , Combined nivolumab and ipilimumab or monotherapy in untreated melanoma, N. Engl. J. Med 373 (1) (2015) 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Kroemer G, et al. , Immunogenic cell death in cancer therapy, Annu. Rev. Immunol 31 (2013) 51–72. [DOI] [PubMed] [Google Scholar]
- [210].Kroemer G, et al. , Immunological effects of chemotherapy in spontaneous breast cancers, Oncoimmunology 2 (12) (2013) e27158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Bao B, et al. , Targeting CSCs in tumor microenvironment: the potential role of ROS-associated miRNAs in tumor aggressiveness, Curr. Stem Cell Res. Ther 9 (1) (2014) 22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Siveen KS, et al. , Targeting the STAT3 signaling pathway in cancer: role of synthetic and natural inhibitors, Biochim. Biophys. Acta 1845 (2) (2014) 136–154. [DOI] [PubMed] [Google Scholar]
- [213].Di Mitri D, et al. , Molecular pathways: targeting tumor-infiltrating myeloid-derived suppressor cells for Cancer therapy, Clin. Cancer Res 21 (14) (2015) 3108–3112. [DOI] [PubMed] [Google Scholar]
- [214].Ramello MC, et al. , CAR-T cells and combination therapies: What’s next in the immunotherapy revolution? Pharmacol. Res 129 (2018) 194–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Beatty GL, Gladney WL, Immune escape mechanisms as a guide for cancer immunotherapy, Clin. Cancer Res 21 (4) (2015) 687–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Brown CE, et al. , Bioactivity and safety of IL13Ralpha2-Redirected chimeric antigen receptor CD8+ t cells in patients with recurrent glioblastoma, Clin. Cancer Res 21 (18) (2015) 4062–4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Morgan RA, et al. , Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma, Hum. Gene Ther 23 (10) (2012) 1043–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Parmiani G, et al. , Integrating immune checkpoint blockade with Anti-Neo/Mutated antigens reactivity to increase the clinical outcome of immunotherapy, Vaccines (Basel) 3 (2) (2015) 420–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Harris SG, et al. , Prostaglandins as modulators of immunity, Trends Immunol 23 (3) (2002) 144–150. [DOI] [PubMed] [Google Scholar]
- [220].Roth P, et al. , GDF-15 contributes to proliferation and immune escape of malignant gliomas, Clin. Cancer Res 16 (15) (2010) 3851–3859. [DOI] [PubMed] [Google Scholar]
- [221].Wei J, et al. , Glioblastoma cancer-initiating cells inhibit T-cell proliferation and effector responses by the signal transducers and activators of transcription 3 pathway, Mol. Cancer Ther 9 (1) (2010) 67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]