

Abstract
The global gene expression program that accompanies the adaptation of Saccharomyces cerevisiae to an abrupt transfer from a fermentable to a nonfermentable carbon source was characterized by using a cDNA microarray to monitor the relative abundances and polysomal distributions of mRNAs. Features of the program included a transient reduction in global translational activity and a severe decrease in polysome size of transcripts encoding ribosomal proteins. While the overall translation initiation of newly synthesized and preexisting mRNAs was generally repressed after the carbon source shift, the mRNA encoded by YPL250C was an exception in that it selectively mobilized into polysomes, although its relative abundance remained unchanged. In addition, splicing of HAC1 transcripts, which has previously been reported to occur during accumulation of unfolded proteins in the endoplasmic reticulum, was observed after the carbon shift. This finding suggests that the nonconventional splicing complex, composed of the kinase-endonuclease Ire1p and the tRNA ligase Rlg1p, was activated. While spliced HAC1 transcripts mobilized into polysomes, the vast majority of unspliced HAC1 RNA accumulated in nonpolysomal fractions before and after the carbon source shift, indicating that translation of unspliced HAC1 RNA is blocked at the translation initiation step, in addition to the previously reported elongation step. These findings reveal that S. cerevisiae reacts to the carbon source shift with a remarkable variety of responses, including translational regulation of specific mRNAs and activation of specific enzymes involved in a nonconventional splicing mechanism.
Saccharomyces cerevisiae ferments glucose, producing ethanol and CO2, even under aerobic conditions (24, 28). The mechanisms by which this yeast senses the presence of glucose and regulates the expression of genes required for glucose uptake and metabolism and for repression of respiratory pathways are complex and are under intense scrutiny (23).
After it consumes all available glucose, S. cerevisiae uses ethanol, the product of fermentation, as a carbon source for aerobic growth. This “diauxic shift” is characterized by a transient cell cycle arrest and a metabolic adaptation to respiratory growth (40, 41). The post-diauxic-shift growth phase is characterized by one to three doublings over a period of 1 week, after which cells enter stationary phase, during which the yeast genome remains unreplicated (40, 41). While overall rates of transcription and translation are diminished in stationary-phase cells (17, 39), the abundances of transcripts of some stress-responsive genes are increased (5, 39). Interestingly, it has been reported that certain mRNAs are translated with equivalent efficiencies during exponential growth and stationary phase (14, 17), implying that translational control may also play a role in response to starvation or cell stress. More recently, cDNA microarrays were used to examine changes in gene expression that occur during the diauxic shift (13). This analysis has identified many mRNAs whose relative abundances were upregulated, such as those involved in respiratory metabolism, or downregulated, such as those involved in protein biosynthesis (13).
Although entry into stationary phase as a result of gradual glucose exhaustion has been widely investigated, much less is known about the biochemical and physiological consequences of abrupt withdrawal of S. cerevisiae from a fermentable carbon source. When faced with such a situation, the organism must react with quick adaptive responses to ensure its survival. In addition to transcriptional changes in gene expression patterns, translational regulation would allow an immediate response to sudden environmental stresses by rapidly increasing or decreasing the production of specific proteins. For example, the mRNA encoding the transcription factor Gcn4p, which upregulates the transcription of genes involved in amino acid biosynthesis, becomes selectively translated following amino acid deprivation (reviewed in reference 19).
Translational control during glucose starvation has recently received attention because the signaling pathways that lead to global reduction in protein synthesis have begun to be deciphered (1). We therefore investigated the changes in overall abundance, as well as the translational activity of individual mRNAs, as yeast cells adapt to the shift from glucose to glycerol as the sole carbon source.
MATERIALS AND METHODS
Yeast strains and media.
Yeast strains used in this study include MBS (MATα ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 can1) (21); YCS243a (ΔIRE1) (MATα ade2-1 his3-11 leu2-3,112::LEU2 UPRE-LACZ trp1-1 ura3-1 can1-100 ire1::URA3) (obtained from P. Walter, University of California, San Francisco); YCS243a (ΔIRE1) transformed with pCS110, which contains the IRE gene and a TRP marker gene (also from P. Walter); CML240 (MATa ade2-1 his3-11 leu2-3,112::LEU2 CMVp(tetR′-SSN6) trp1-Δ2 ura3-1 can1-100) (obtained from E. Herrero, Universitat de Lleida), and CML240 transformed with pCM184 (also from E. Herrero), which contains a URA3V5H6 reporter (described below) and a TRP marker. Cells were grown at 30°C in a synthetic minimal medium composed of 1.7 g of yeast nitrogen base (Difco)/liter, lacking amino acids, supplemented with 5 g of ammonium sulfate/liter. Selected components (adenine at 40 μg/ml, histidine at 20 μg/ml, leucine at 60 μg/ml, tryptophan at 40 μg/ml, uracil at 20 μg/ml [2], and 2% [wt/vol] glucose) were added when needed. Doxycycline was added to a final concentration of 2 μg/ml (3). Yeast cells for all experiments were harvested at mid-log phase (optical density at 595 nm OD595 0.5). For carbon source shifts, cells were first sedimented at 5,000 × g and 20°C for 5 min (the lower temperature prevented overheating during centrifugation). The cell pellets were immediately resuspended in the synthetic minimal medium, prewarmed to 30°C, in which glucose had been replaced with 2% (wt/vol) glycerol.
PCR and cloning.
The HAC1 intron was amplified by PCR from yeast genomic DNA, using forward (5′-CGTGATTACGATGACCAGGAAAC-3′) and reverse (5′-CAGTACAAGCCGTCCATTTC-3′) primers, under the following cycling conditions: 65°C for 1 min, 75°C for 1 min, and 95°C for 1 min for 25 cycles. These cycling parameters were used for all PCRs.
Plasmid p(CUP-URA) was constructed by first amplifying the complete URA3 open reading frame (ORF) from yeast genomic DNA with forward (5′-CTAGCTCGAGAAAATGTCGAAAGCTACATATAAGG-3′) and reverse (5′-CTAGGCATGCTTAGTTTTGCTGGCCGCATCTTC-3′) primers. The PCR product was purified, digested with XhoI and SphI, and ligated into plasmid pSAL1 (32) previously digested with XhoI and SphI (restriction sites in primers are underlined).
Plasmid p(5′cyc-ura3-cyc3′) was constructed by first amplifying the complete URA3 ORF with forward (5′-CTAGGGATCCAAAATGTCGAAAGCTACATATAAGG-3′) and reverse (5′-CTAGGGTGACCTCGAAGCTCGCCCTTGTTTTGCTGGCCGCATCTTC-3′) primers. The PCR product was purified and digested with BamHI and BstEII. The URA3 ORF was extended 99 nucleotides by an in-frame fusion with a dual V5 six-His tag, isolated by digesting pYES2/GS (GeneStorm; Invitrogen) with BstEII and PstI (the XbaI site of pYES/GS had been changed to a PstI site by site-directed mutagenesis). The above-described DNA fragments were cloned, by three-part ligation, into pCM184 (3) previously digested with BamHI and PstI.
RNA isolation and Northern analysis.
For total-RNA isolation, 10-ml cultures were pelleted at 5,000 × g and the cell pellets were frozen on dry ice. Total RNA was extracted by the hot acid phenol method (2). Eight micrograms of total RNA was fractionated on a 1.2% agarose-formaldehyde gel (2), then transferred to a Zeta-probe membrane (Bio-Rad) for 1 h by the use of a pressure blotter (Stratalinker; Stratagene). RNA was cross-linked to the membrane by UV (254 nm) irradiation at an energy setting of 120 mJ (Stratagene). The integrity of the RNA and the efficiency of transfer were evaluated by staining the membrane with 0.3% methylene blue (2).
For Northern analysis, full-length yeast ORFs (Research Genetics) or HacI intron PCR products were radiolabeled by using a RadPrime labeling kit (Gibco BRL) and purified by PCR purification column chromatography (Qiagen). Standard hybridization was performed overnight at 65°C in a solution containing 500 mM Na2HPO4 (pH 7.2), 1 mM EDTA, and 7% (wt/vol) SDS. The blots were washed twice in 40 mM Na2HPO4 (pH 7.2) containing 1 mM EDTA, and 5% (wt/vol) sodium dodecyl sulfate (SDS) (Bio-Rad) for 15 min each time and twice in 40 mM Na2HPO4 (pH 7.2) containing 1 mM EDTA and 1% (wt/vol) SDS (Bio-Rad) for 15 min each time. All washes were performed at 65°C. For oligodeoxynucleotide hybridization, 25 pmol of the HacI “splice oligo” (5′-CTGCGCTTCTGGATTACG-3′) was labeled with T4 kinase [γ-32P]ATP at 6,000 Ci/mmol and purified by using a Qiagen nucleotide removal kit. Hybridization was performed in a solution containing 5× SSC (1× SSC is 0.15 NaCl plus 0.015 M sodium citrate), 20 mM Na2HPO4 (pH 7.2), 1× Denhardt's solution, 7% (wt/vol) SDS, and 100 mg of denatured salmon sperm DNA/ml overnight at 50°C. The blots were washed twice in a solution consisting of 3× SSC, 25 mM Na2HPO4 (pH 7.2), 10× Denhardt's solution, and 5% SDS at 50°C for 30 min each time and then once in 1× SSC–1% (wt/vol) SDS at 50°C for 30 min. Quantitation was performed with a Storm PhosphorImager and ImageQuant software (Molecular Dynamics).
Polysome analysis.
Material from 75-ml cultures (OD5950.6) was used for each sucrose gradient. At the time of harvest, cycloheximide (Sigma) was added to a final concentration of 0.1 mg/ml, and the cultures were chilled on ice for 5 min. Cells were pelleted by centrifugation at 5,000 × g and 4°C for 5 min. The cell pellet was then resuspended in 3 ml of polysome extraction buffer (20 mM Tris-HCl [pH 8.0], 140 mM KCl, 5 mM MgCl2, 0.5 mM dithiothreitol [DTT], 1% [vol/vol] Triton X-100, 0.1 mg of cycloheximide/ml, and 1 mg of heparin/ml) and sedimented. This washing step was repeated, and the final cell pellet was resuspended in 750 μl of ice-cold polysome extraction buffer and transferred to a chilled 15-ml Corex glass tube containing a 500-μl volume of acid-washed 0.45- to 0.55-mm-diameter glass beads (Sigma). All subsequent steps were performed on ice with prechilled extraction buffer. The reaction mixture was vortexed at full speed for 20 and then incubated on ice for 100 s. This process was repeated three times. After cell lysis, glass beads and excess cell debris were removed by sedimentation in a microcentrifuge at 3,500 × g and 4°C for 5 min. The supernatant (approximately 0.5 ml) was transferred to a 1.7-ml microcentrifuge tube containing 0.5 ml of extraction buffer and vortexed vigorously. The sample was clarified by sedimentation in a microcentrifuge at 8,000 × g and 4°C for 5 min. Approximately 0.8 ml of sample was layered onto an 11-ml 10 to 50% (wt/vol) sucrose gradient that contained extraction buffer lacking Triton X-100. The sample was sedimented at 35,000 rpm and 4°C in an SW41 rotor for 160 min. Gradient fractions were collected as described previously (22) with minor modifications. Briefly, 14 0.9-ml fractions were collected directly into 2-ml volumes of 8 M guanidine-HCl. After the fractions were vortexed, 3 ml of 100% ethanol (EtOH) was added to each tube, and the samples were vortexed vigorously and stored overnight at −20°C to precipitate the RNA. The pellets were collected by sedimentation, washed in 75% EtOH, resuspended in 0.4 ml of Tris-EDTA (TE; pH 8.0), and transferred to 1.7-ml Eppendorf tubes. The RNA was again precipitated by addition of 50 μl of 3 M sodium acetate (pH 5.2) and 1 ml of 100% EtOH. Each RNA pellet was washed in 75% EtOH and resuspended in TE (pH 8.0) to a final volume of 25 μl. Equal volumes of all samples were analyzed by Northern analysis.
cDNA microarray analysis.
Template RNA was converted to cDNA by reverse transcriptase, and Cy3-dUTP or Cy5-dUTP was incorporated into second-strand cDNA with the Klenow fragment of DNA polymerase as described previously (15). Briefly, purified template RNA (see below) in deionized water (dH2O) was denatured at 70°C for 10 min and then incubated on ice for 1 min. First-strand cDNA was synthesized in reverse transcription buffer, containing 6 μg oligo (dT)18 (New England Biolabs), 50 mM Tris-HCl (pH 8.3), 75 mM KCl, 3 mM MgCl2, 10 mM DTT, 0.5 mM each deoxynucleoside triphosphate and 400 U of SuperScript II RNase H-free reverse transcriptase (Gibco BRL) in a final volume of 30 μl. The reaction was performed at 42°C for 2 h. Template RNA was degraded by adding 15 μl of 0.1 M NaOH and incubating at 70°C for 10 min. The mix was neutralized by adding 15 μl of 0.1 M HCl, diluted with 500 μl of TE, and concentrated to a volume of 20 to 30 μl in a Microcon 30 microconcentrator (Amicon). The dilution and concentration steps were repeated once. The sample was then denatured at 100°C for 1 min and allowed to cool at room temperature for 5 min. A Cy3- or Cy5-deoxynucleoside triphosphate fluor was incorporated in Klenow buffer (containing 12 μg of random nanomer [Research Genetics]; 10 mM Tris-HCl [pH 7.4]; 5 mM MgCl2; 7.5 mM DTT; 0.025 mM each dATP, dCTP, and dGTP; 0.009 mM dTTP; 0.03mM Cy3- or Cy5-dUTP; and 10 U of exonuclease-free Klenow enzyme [U. S. Biochemicals] [final volume, 50 μl]). The reaction was performed at 37°C for 2 h, and the samples were purified as described above. The Cy3- or Cy5-labeled cDNAs were then combined, diluted with 500 μl of TE, and concentrated to a final volume of 10 to 20 μl. Microarray hybridization was performed overnight at 65°C in a buffer containing 1.6 μg of poly(A) (Sigma)/ml 3.5× SSC, and 0.3% (wt/vol) SDS. The microarrays were washed at room temperature for 2 min each in 2× SSC, 0.1% SDS (wash 1), 1× SSC (wash 2), and 0.2× SSC (wash 3); spun dry in a centrifuge; and scanned with a GenePix 4000A microarray scanner (Axon Instruments). Cluster analysis was performed with ScanAnalyze software (M. Eisen [16]; available at http://genome-www4.stanford.edu/MicroArray/SMD/index.html).
For comparison of total RNA abundances, 20 μg of total RNA was used as a template in the first-strand cDNA reaction. For comparison of polysomal RNA abundances, additional purification steps were necessary prior to first-strand cDNA synthesis. First, to obtain equal amounts of polysomal RNA (roughly 20 μg), polysomal fractions 10 through 14 eluted from three glucose and six glycerol gradients were pooled for each time point. This design gave a slight enrichment of mRNAs isolated from glycerol polysomes. The pooled samples were then diluted to 0.65 ml with dH2O and extracted with 0.65 ml of Tris-buffered phenol-chloroform. The resultant aqueous phase (approximately 0.5 ml) was diluted to 1 ml with H2O and LiCl (added to a final concentration of 1.5 M). RNA was allowed to precipitate overnight at −20°C. The samples were thawed at 4°C and pelleted at high speed in a 4°C microcentrifuge for 15 min. Each purified RNA pellet was washed with 75% ethanol, air dried, resuspended in 0.15 ml of dH2O by repeated freezing and thawing, and then precipitated by addition of 20 μl of sodium acetate (pH 5.2) and 0.45 ml of 100% EtOH. The final RNA pellets were each resuspended in 25 μl of 1 mM Tris (pH 8.0). For comparison of polysomal and nonpolysomal RNA abundances, RNA was isolated from fractions 2 through 6 and compared directly to RNA isolated from fractions 10 through 14 of the same gradient. For each time point, material from three gradients was pooled and processed identically to minimize enrichment (6). The microarray data are available on the World Wide Web at http://genome-www4.stanford.edu/MicroArray/SMD/publications.html.
RESULTS
Withdrawal of glucose leads to a global translational repression.
To monitor the effect of the carbon source shift on protein synthesis, the overall rate of [35S]methionine incorporation was examined at various time points after the switch from glucose to glycerol medium. Protein synthesis was transiently reduced by approximately 80% at 5 min after the shift to glycerol medium and increased again by two- to threefold at 15 min after the shift, but it did not return to the level observed in exponentially growing cells (data not shown). Thus, an abrupt shift from glucose to glycerol resulted in a rapid, transient reduction of protein synthesis, in agreement with data published by Ashe et al. (1).
To test whether decreased protein synthesis was a consequence of a decreased translation rate, we examined the global distribution of ribosomes and ribosomal subunits, as well as the polysomal distribution of several individual mRNAs, such as ACT1 and ADH1 (Fig. 1). Following the carbon source shift, there was a global redistribution of ribosomes from polysomes toward 80S monosomes and dissociated 40S and 60S subunits (Fig. 1). The majority of 80S monosomes that accumulated after the shift could be dissociated in the presence of 0.8 M KCl (K. Kuhn, unpublished data), indicating that they were not engaged in the translation of mRNAs. In addition to the global loss of polysomes, the average polysome size (number of ribosomes per mRNA) was reduced as well, as exemplified by the slower sedimentation rate of ACT1 and ADH1 mRNAs. These findings indicate that the carbon source shift resulted in a decrease in the rate of translation initiation.
FIG. 1.

Polysomal association of mRNAs in yeast cells grown exclusively in glucose medium or after a 10-min shift to glycerol medium. Absorbance profiles at 254 nm of the collected sucrose gradients are shown above the panels. Fourteen equal-volume fractions were collected from each gradient (fraction 1 is the top of the gradient), and purified RNAs were analyzed by Northern blotting. The levels of ACT1, ADH1, and URA3 mRNAs were visualized by a phosphorimage of the Northern blot. The CUP1 promoter was induced by addition of 10 mM CuSO4 to the glycerol medium (but not to the glucose medium).
To test the possibilities that translation initiation was completely inhibited and that the remaining polysomal association of mRNAs was due to residual elongating ribosomes, we examined whether a newly synthesized mRNA could be translated at the time of the carbon source shift. URA3 mRNA, expressed at low levels from an uninduced CUP1 promoter, sedimented with high-molecular-weight polysomes (Fig. 1, fraction 11) prior to glucose withdrawal. New URA3 mRNA, synthesized following induction of the CUP1 promoter at the time of the carbon source shift, was predominantly polysome associated, although it was associated with fewer ribosomes (Fig. 1, fractions 9 to 11) than in the control, glucose-grown cells. The simplest interpretation of these findings is that translation initiation was markedly reduced after the shift from a fermentable to a nonfermentable carbon source but did not completely cease.
Relative abundance and polysomal association of ribosomal mRNAs are coordinately repressed after the carbon source shift.
Other reasons for the reduced overall amount of translation after the carbon source shift might include a rapid loss of certain abundant mRNA species in the cell. To examine this possibility, the relative abundance and polysomal association of mRNAs were examined after the shift from glucose to glycerol medium, using a cDNA microarray (12, 13, 15). Of the approximately 6,275 mRNAs examined, 120 mRNA species consistently showed a greater than twofold reduction in polysomal association in at least four cDNA microarray experiments (Fig. 2); 89 of those mRNAs encode ribosomal proteins.
FIG. 2.
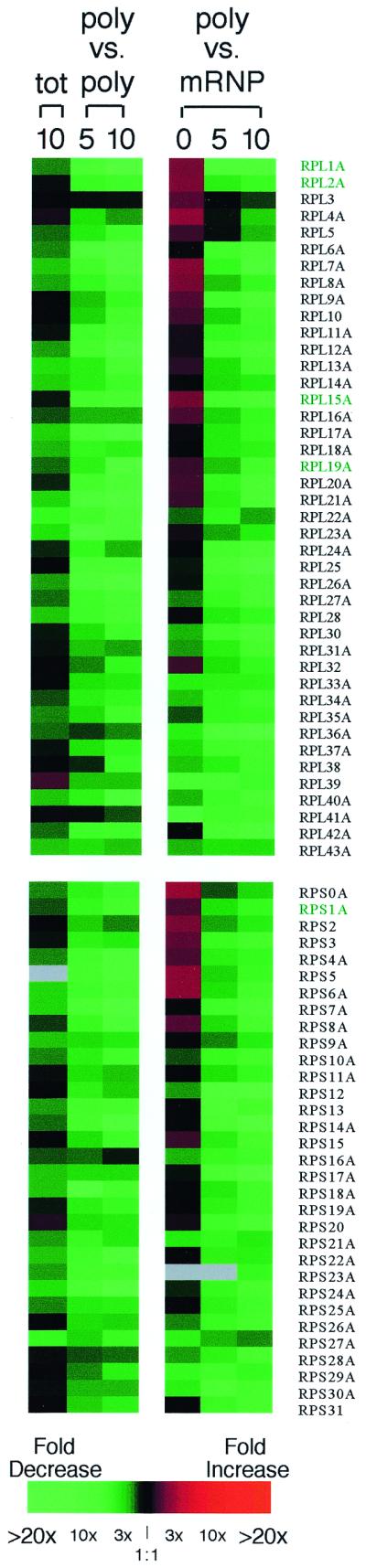
Expression of ribosomal-protein-encoding (RP) mRNAs in yeast cells after the carbon source shift. Each colored square represents the ratio of total mRNA (tot) or high-molecular-weight polysomal mRNA (poly vs. poly; fractions 10 to 14) isolated following incubation in glycerol medium for 5 or 10 min, relative to the amount of the mRNA isolated from cells grown in the presence of glucose. In addition, mRNA abundance in fractions 2 through 6, designated as mRNA protein complexes (mRNP), was compared to the abundance of mRNA present in polysomes (poly vs. mRNP). Black squares denote no significant difference in the amount of RNA isolated from glucose-grown or glycerol-shifted cells; red squares and green squares denote RNAs that are more or less abundant, respectively. The intensity of the color is proportional to the log2 of the fold increase or decrease, with maximal intensity corresponding to an eightfold increase or decrease. For duplicated RP mRNA genes (designated A and B), only data for the A gene are presented. The cDNA for RPL29 (YFR032CA) was not included in this batch of microarrays. A gray box indicates an unreadable spot on the microarray. The color intensity scale was adapted from reference 9. Selected RP mRNAs (highlighted in green) were chosen for further analysis.
It is known that the abundance of ribosomal protein (RP) mRNAs rapidly decreases when yeast cells encounter stress situations such as heat shock (18, 19, 27), diauxic shift (13, 26), or inhibition of the secretory pathway (30, 34). It has been argued that the rapid changes in intracellular concentrations of RP mRNAs are due to transcriptional repression, along with constitutively fast mRNA turnover (19, 27, 29, 35). Recently, Li et al. (29) pointed out that the 137 yeast RP genes contributed 30% of the total mRNA in the cell. Considering that the turnover rate of an average RP mRNA is 5 to 7 min (29), inhibition of transcription of RP genes might diminish the intracellular concentration of RP mRNAs within several minutes. As a result, the amount of polysomes would decrease by as much as 30% and excess 80S ribosomes and ribosomal subunits would accumulate in the cell.
The cDNA microarray analysis revealed that the total abundances of most RP mRNAs decreased rapidly, by 10 min after the carbon source shift (Fig. 2). When the partial concentrations of RP mRNAs, as a fraction of all polysomal mRNAs, of samples taken before and after the carbon source shift were directly compared, (Fig. 2), almost all RP mRNAs showed a striking decrease in their relative abundances in polysomes after the carbon source shift. To determine whether the decrease in polysome-associated RP mRNA simply reflected the decrease in total RP mRNA abundance or reflected a redistribution to nonpolysomal fractions, the relative abundance of polysomal mRNA was compared to that of mRNA isolated from the nonpolysomal fractions, designated mRNP (Fig.), following a 0-, 5-, or 10-min incubation in glycerol medium. This analysis revealed a substantial redistribution of polysomal RP mRNAs into nonpolysomal fractions after the carbon source shift.
The data obtained from the cDNA microarray analysis were corroborated by monitoring the abundance and polysomal distribution of individual RP mRNAs by Northern analysis. Many RP ORFs are rather small and are occupied by only a few translating ribosomes. For this reason, we chose to examine the polysomal association of RP mRNAs containing ORFs with more than 500 nucleotides. Such RP ORFs are occupied by four to six ribosomes when cells are grown in glucose medium (see Fig. 3B). Figure 3A shows that, in contrast to that of ADH1 mRNA, the total abundances of five individual RP mRNAs had decreased by 10 min after the carbon source shift. In addition, all five RP mRNAs relocalized from polysomal fractions 9 and 10 to monosomal fraction 6 and to nonpolysomal fraction 2 (Fig. 3B). Quantitation (Fig. 3C) showed that RP mRNAs occupied four to six ribosomes during growth in glucose medium and one ribosome after the shift to glycerol medium, suggesting a 70% reduction in the translation rate for these RP mRNAs. In contrast, ADH1 mRNA was occupied by 9 to 11 ribosomes/mRNA in glucose medium, compared to 6 to 8 ribosomes/mRNA in glycerol medium (Fig. 3B and C), indicating a reduction in translation rate of approximately 30%. Similar observations were made when the polysomal distributions of approximately 70 individual mRNA species were examined (K. Kuhn, unpublished data). ACT1 mRNA, which sediments somewhat diffusely with lower-molecular-weight polysomes after a carbon source shift (Fig. 1), contains an unusually long 5′ noncoding region which may negatively affect the translation initiation rate under global translational repression conditions. Overall, these findings indicate that the carbon source shift resulted in a rapid loss of RP mRNAs from polysome fractions. Polysomal loss of RP mRNAs was more pronounced than that of other, non-RP mRNAs.
FIG. 3.

Relative abundances and translational activities of selected RP mRNAs as a function of time following transfer to glycerol medium. (A) Total levels of RP mRNAs (RPL1, RPL2, RPL15, RPL19, and RPS1), as well as a control ADH1 mRNA, after Northern blot hybridization are displayed. (B) Polysome association of RP mRNAs. For details, see the legend to Fig. 1. The Northern blot displaying ADH1 mRNA was taken from Fig. 1. Phosphorimages of the Northern blots are shown. (C) Quantitation of polysomal distribution of averaged RP and ADH1 mRNAs after the shift to glycerol medium (panel B). The data points represent the percent intensity of each fraction relative to the total combined intensity of all fractions for each gradient. The five RP data sets from panel B were combined and averaged to give a general polysomal distribution pattern for the selected RP mRNAs.
Lowering the abundance of an mRNA after carbon shift does not decrease its translational efficiency.
It is known that transcription of RP genes is regulated under certain stress situation (34). Employing cDNA microarrays, it was found that the overall abundances of all yeast RP mRNAs diminished following a rapid shift from glucose to glycerol, suggesting that RP gene promoters were downregulated after the carbon shift. Thus, it could be argued that preexisting RP mRNAs, synthesized before the carbon source shift, were translated less efficiently after the carbon source shift. To examine this possibility, we monitored the translation of a reporter mRNA whose synthesis was controlled by a tetracycline-repressible transcription system (TetR) (3). Briefly, a URA3 ORF-containing cDNA containing the 5′ and 3′ noncoding regions of the CYC1 gene was inserted downstream of the TetR promoter. The resulting plasmid, p(5′cyc-ura3-cyc3′), was transformed into yeast strain CML240 (3). The relative abundance and polysomal association of the URA3 reporter mRNA were monitored following a shift from glucose medium to glycerol medium to which doxycycline had been added to silence the transcription of the reporter gene. Figure 4A shows that preexisting reporter mRNA decayed with a turnover rate of approximately 30 min. However, URA3 mRNA displayed similar polysomal distribution profiles in untreated cells and in doxycycline-treated cells containing smaller amounts of URA3 mRNA (Fig. 4B), indicating that the efficiency of translation of URA3 mRNA synthesized before the carbon shift was similar to that of mRNA synthesized after the carbon source switch. Thus, transcriptional inhibition after the carbon source shift does not in itself affect the efficiency with which an mRNA is translated.
FIG. 4.

Decreasing the abundance of an mRNA does not decrease its translational efficiency. (A) Total levels of URA3 and ACT1 mRNAs at various time points after addition of the tetracycline analogue doxycycline (2 μg/ml) to yeast cells grown in glycerol medium are shown. (B) Polysomal distribution of URA3 mRNA in yeast cells grown in glycerol medium in the presence (+) or absence (−) of doxycycline (doxy). The plasmid-derived URA mRNA was distinguished from the endogenous URA3 mRNA by using a probe complementary to a V5 six-His extension of the plasmid coding sequence (see Materials and Methods).
Altered abundances and polysomal distributions of individual mRNAs reveal induction of specific intracellular pathways after the carbon source shift.
The cDNA microarray analysis revealed many differences in mRNA abundance and polysomal representation in the approximately 6,275 genes examined. The relative abundances of 610 mRNAs increased by more than twofold after the shift to glycerol medium. Figure 5 lists examples of genes with known functions whose mRNA abundances had increased at least threefold by 10 min after the carbon source shift. The observed changes in gene expression were specific to the shift to glycerol medium, because expression of only 26 genes, all with unknown functions, increased more than threefold when yeast cells were shifted into freshly prepared glucose medium (data not shown).
FIG. 5.


Characterized genes whose relative mRNA abundances increase following a transfer to glycerol medium. Total and polysomal RNA levels were analyzed and displayed as described in the legend to Fig. 2. Genes selected for this figure were induced at least threefold at 10 min after a shift to glycerol medium. Exceptions were MIG1, SNF3, APE3, LAP4, PEP4, ULA1, and UBC8, all of which were induced at least 2.5-fold. The induction of YAK1 and YGP1 mRNAs was confirmed in a separate experiment. Grouping of genes was based on data available from the Yeast Protein Database (http://www.proteome.com). Gene names highlighted in red were induced specifically after an abrupt transfer from glucose to glycerol medium, but not during the diauxic shift 13. Glc7p, glycogen synthase phosphatase; TCA, tricarboxylic acid; GABA, γ-aminobutyric acid.
Although global translation was reduced, almost all of the mRNAs whose relative abundances increased were also associated with polysomes (Fig. 5). This observation was corroborated by analyzing the polysomal distributions of approximately 70 mRNA species (K. Kuhn, unpublished data). Two exceptions were the transcripts encoding flavin adenine dinucleotide-dependent glycerol-3-phosphate dehydrogenase GUT2 (36) and the Rub1p-activating protein ULA1 (31), which did not accumulate on polysomes despite exhibiting increased abundance after the carbon source shift. As a rule, virtually every mRNA species that increased in relative abundance after the shift to glycerol medium was also actively translated, although they all sedimented with fewer polysomes than did mRNAs isolated from glucose-grown cells (K. Kuhn, unpublished observation). This analysis highlights the efficiency with which S. cerevisiae responds to environmental cues. Within 5 min, new genes are expressed, processed, exported, and translated.
Interestingly, Fuge et al. (17) and Dickson and Brown (14) found that despite a global decrease in the activity of the protein synthesis machinery, the translational efficiencies of several mRNAs remained unchanged in exponentially growing and stationary-phase yeast cells. These observations prompted us to search for mRNAs whose translational efficiency either remained unchanged or actually increased following the shift to glycerol medium.
Selective mobilization of YPL250C mRNA into polysomes after the carbon source shift.
Two mRNA species showed little change in total abundance but increased abundance on polysomes after the carbon source shift (Fig. 5B). One of these mRNAs is encoded by the YPL250C gene. In cells grown continuously in glucose medium, YPL250C mRNA sedimented predominantly in fractions corresponding to mRNAs with a single bound ribosome (Fig. 6, fraction 6) and less frequently in fractions corresponding to mRNAs with two to six ribosomes (fractions 7 to 10), a biphasic distribution pattern that is indicative of a translationally repressed mRNA. After the shift to glycerol medium, however, the majority of the YPL250C mRNA sedimented in fractions corresponding to four to six ribosomes/mRNA (fractions 9 and 10). Even at 20 min after the carbon source shift, when the overall abundance of YPL250C mRNA had declined, most YPL250C mRNAs were still detected in fractions corresponding to four to six ribosomes/mRNA (Fig. 6). Addition of 10 mM EDTA to the sucrose gradient abolished cosedimentation of the YPL250C mRNA with fractions 6 to 10 (data not shown), supporting the notion that the presence of YPL250C mRNA in this region of the gradient resulted from its association with ribosomes. Furthermore, YPL250C is not translationally enhanced following resuspension in glucose medium, ruling out the possibility that activation occurs in response to sedimentation or gravity stress (K. Kuhn, unpublished data). These observations indicate that YPL250C mRNA was selectively translated at an enhanced rate after the carbon source shift, at a time when overall translation of both preexisting and newly synthesized mRNAs was reduced (Fig. 1).
FIG. 6.

YPL250C mRNA sediments with polysomes at a time when global translation rates are reduced: time course of YPL250C polysome association from cultures grown exclusively in glucose or shifted to glycerol for 20 min.
Activation of the bifunctional kinase-endonuclease Ire1p and the tRNA ligase Rlg1p after the carbon source shift.
The second example of an mRNA whose apparent polysomal association increased after a 10-min shift to glycerol, without significant changes in overall abundance, was HAC1, which encodes a transcription factor whose synthesis is stimulated as an essential part of the unfolded protein response (UPR) when unfolded proteins accumulate in the endoplasmic reticulum (ER) (7).
To verify the apparent increase in polysomal association of Hac1 mRNA following the shift to glycerol, we reexamined the polysomal distribution of Hac1 mRNA in cells that were continuously grown in glucose medium or shifted to glycerol medium. Cells grown continuously in glucose medium expressed a single 1.4-kb HAC1 mRNA species, which sedimented predominantly with nonpolysomal fraction 2 and to some extent with polysomal fractions 8 to 11 (Fig. 7). Following the carbon source shift, the polysomal distribution of the 1.4-kb HAC1 mRNA remained largely unchanged. However, a smaller, 1.2-kb species was detected predominantly in the polysomal fractions (Fig. 7). This 1.2-kb mRNA species is likely to have contributed to the increased relative abundance of polysome-associated HAC1 mRNA observed in the cDNA microarray analysis. Since polysomal fractions from three glucose gradients and six glycerol gradients were processed to obtain equal (microgram) amounts of template RNA in the cDNA microarray analysis, the sample from the glycerol gradient contained a slight enrichment of mRNA (see Materials and Methods), which explains the relatively minor HAC1 signal detected by Northern analysis (Fig. 7) compared with the more than twofold-higher signal detected by the cDNA microarray analysis (Fig. 5B).
FIG. 7.

A smaller species of HAC1 RNA associates with polysomes 10 min after transfer from glucose to glycerol medium. Northern analysis with a HAC1 ORF as a hybridization probe detected the full-length HAC1u mRNA (arrow) as well as a low-molecular-weight version of HAC1i (arrowhead) engaged with polysomes. The polysomal distribution of ADH1, taken from Fig. 1, is shown as a reference. Phosphorimages of the blots are shown below.
The appearance of the 1.2-kb HAC1 mRNA was intriguing because HAC1 expression is controlled by a regulated RNA splicing mechanism. The 1.4-kb unspliced HAC1 mRNA, known as HAC1u (uninduced), can exit the nucleus and associate with ribosomes, but its translation has been reported to be attenuated at the elongation step (8). The results in Fig. 7 indicate that HAC1u mRNA sediments predominantly in nonpolysomal fraction 2, suggesting that translation of this mRNA species may also be blocked at the translation initiation step. Upon induction of the UPR pathway, HAC1u mRNA is spliced to yield the 1.2-kb HAC1u (UPR-induced) mRNA, which is efficiently translated into Hac1pi, whose C terminus differs from that of the Hac1pu protein, encoded by HAC1u (8). Hac1pi enters the nucleus and promotes transcription of genes that contain unfolded protein response elements in their promoters, such as KAR2 and PDI1, whose products facilitate protein folding in the ER (reviewed in references 7 and 37).
To determine whether the 1.2-kb RNA species observed in the polysome profile (Fig. 7) was the spliced HAC1i mRNA, oligodeoxynucleotide primers were designed to distinguish between the unspliced and spliced versions of the HAC1 mRNA. In cells grown continuously in glucose medium, HAC1 mRNA was found to be predominantly unspliced (Fig. 8). Following transfer to glycerol medium, both unspliced and spliced (7, 37) HAC1 mRNAs could be detected (Fig. 8). The spliced HAC1 mRNA was transient, however, and it could not be detected by 45 min after the shift to glycerol medium. None of the known targets of Hac1pi (e.g., KAR2 or PDI) was detectably induced at this time after the carbon source shift (K. Kuhn, unpublished data). Thus, although the appearance of polysome-associated HAC1i mRNA suggests that some Hac1pi may have been produced, its production was not accompanied by a transcriptional induction of the UPR response. Splicing of HAC1u mRNA is tightly regulated in the cell; it is induced by the accumulation of misfolded proteins in the ER and occurs by a nonconventional splicing mechanism mediated by two proteins, a bifunctional Ire1p kinase-endonuclease encoded by IRE1 and a tRNA ligase encoded by RLG1 (10, 25, 38). To determine whether Ire1p is required for the accumulation of HAC1i following the carbon source shift, splicing of HAC1 mRNA in strains that contained or lacked IRE1 was monitored. In contrast to the Ire1p-expressing strain MBS, the IRE1 deletion strain ΔIRE1 failed to accumulate spliced HAC1i mRNA after the shift to glycerol medium (Fig. 9). Transformation of this strain with a plasmid expressing IRE1 restored the accumulation of spliced HAC1i mRNA. All strains expressed detectable levels of unspliced HAC1u mRNA, indicating that the failure of the ΔIRE1 strain to produce spliced HAC1i mRNA was not due to low levels of unspliced HAC1u precursor mRNA. Thus, splicing of HAC1u mRNA after the carbon source shift appears to be mediated by the same Ire1p-dependent pathway that is involved in the UPR.
FIG. 8.

HAC1 mRNA is transiently spliced following glucose depletion. Total RNA was isolated from glycerol-shifted cultures at intervals during a 1-h time course and hybridized either with radiolabeled Hac1 intron, with a small deoxyoligonucleotide that recognizes the Hac1 splice junction (“Splice”), or with a mixture of intron and splice probes (Intron + Splice). The “splice” deoxyoligonucleotide is 18 nucleotides in length, with 9 nucleotides of complementarity to the spliced termini of each exon. 32P-labeled probes are displayed. Phosphorimages of the blots are at top. Full-length HAC1 mRNA (arrow) and the spliced HAC1 (arrowhead) are indicated in the phosphorimage.
FIG. 9.

The presence of IRE1 is required for the transient splicing of HAC1 mRNA. Total RNAs were isolated from strain MBS, a ΔIRE1 strain (yCS243a), and the ΔIRE1/p(IRE1) strain transformed with a functional copy of IRE1 (pCS110). Northern blots were hybridized with either radiolabeled full-length IRE1 cDNA or the “splice” oligodeoxynucleotide described in the legend to Fig. 8. Phosphorimages of the blots are shown.
DISCUSSION
Following an abrupt shift from a fermentable to a nonfermentable carbon source, S. cerevisiae displays a diverse array of adaptive changes in gene expression at both the transcriptional and translational levels. The shift from a fermentable carbon source, glucose, to a nonfermentable carbon source, glycerol, resulted in a marked reduction in overall translation of mRNAs. The accumulation of 80S monosomes and ribosomal subunits after the carbon source shift indicated that the initiation phase of translation was inhibited. Two major mechanisms of downregulating global translation initiation in S. cerevisiae have been described. In the first case, amino acid starvation induces phosphorylation of initiation factor eIF2α at serine 51 by the Gcn2p kinase, which results in a reduced concentration of 40S subunits, which carry initiator tRNA-eIF2 complexes, and subsequent derepression of GCN4 mRNA translation (20). In the second case, induction of a diauxic shift by gradual glucose exhaustion leads to degradation of initiation factor eIF4G (4), resulting in limiting concentrations of the cap-binding protein complexes needed to recruit 40S subunits onto mRNAs. In the case of the rapid shift from glucose to glycerol medium described here, translational inhibition was still observed in a yeast strain that expressed a mutated eIF2α whose phosphorylation site at serine 51 had been changed to alanine. In addition, eIF4G was not degraded after shifting S. arevisiae from glucose to glycerol medium at a time when translation was markedly depressed (K. Kuhn, unpublished data). Therefore, the mechanism of global translational repression that occurs following a rapid withdrawal of glucose must differ from that described during amino acid starvation or the diauxic shift. Recently, mutants in the cyclic-AMP-dependent kinase pathway have been shown to be resistant to translational inhibition after glucose withdrawal (1), suggesting that this signaling pathway is involved in translational regulation. We noted an increased abundance of several mRNAs encoding catalytic subunits of the cyclic-AMP-dependent kinase protein kinase A (PKA) upon carbon source shift (TPK1 [4.1-fold], TPK2 [3.8-fold], and TPK3 [1.5-fold]). In addition, the relative abundance of the BCY1 mRNA, which encodes the PKA regulatory subunit, was also induced twofold. These findings support the hypothesis by Ashe et al. (1) that increased concentration and activity of PKA can play a role in the inhibition of translation following a shift from a fermentable to a nonfermentable carbon source.
Loss of polysome-associated RP mRNAs after a shift to glycerol.
Mammalian RP mRNAs contain terminal oligopyrimidine (5′ TOP) sequence elements in their 5′ noncoding regions that can negatively regulate translation initiation, particularly during serum starvation (reviewed in reference 33). In contrast, yeast RP mRNAs do not contain 5′ TOP elements, nor do they contain any obvious consensus sequences in their 5′ noncoding regions. With the exception of L30 mRNA, whose translation is negatively regulated by its encoded product Rpl30p (11), yeast RP mRNAs are generally not thought to be under translational control (33).
Curiously, all yeast RP mRNAs redistributed from polysomes to monosomes and untranslated mRNPs within 5 min after the shift to glycerol medium, suggesting that RP mRNAs are coordinately regulated at the translational level. Furthermore, we have shown that the reduced rate of ribosomal loading is not due to an inhibition of RP gene transcription. To gain information about the mechanism of the marked translational repression of RP mRNAs after a carbon shift, translation studies using a reporter mRNA containing the noncoding regions of RPL15 were initiated. Preliminary experiments showed that the 3′ noncoding region of RPL15, but not its short 5′ noncoding region, mimicked the overall 80S and polysomal distribution pattern seen with endogenous RPL15 mRNA during growth in glycerol (K. Kuhn, unpublished observation). Thus, signals for translational repression after a carbon shift may reside in the 3′ noncoding regions of RP mRNAs.
Translational regulation of YPL250C mRNA.
YPL250C mRNAs were predominantly associated with an increased number of ribosomes following the transfer from glucose to glycerol medium. The mechanism by which YPL250C mRNAs selectively recruit ribosomes when the overall activity of the translational apparatus is diminished is being investigated. Preliminary results have indicated that the 3′ noncoding region of the YPL250C mRNA is sufficient to mobilize a reporter mRNA into polysomes during glycerol-induced translational inhibition (K. Kuhn, unpublished data). Detailed characterization of the 3′ noncoding region in YPL250C mRNA will likely provide clues to the mechanism by which this mRNA confers preferential polysomal association to YPL250C mRNAs after the carbon source shift. A BY4742 strain with a YPL250C gene knockout mutation (Research Genetics) was viable and, when grown on glucose or glycerol medium, displayed a phenotype similar to that of the parent BY4742 and MBS strains used in this study. However, overexpression of YPL250C protein led to a slow-growth phenotype (K. Kuhn, unpublished data), implying that YPL250C gene expression may be tightly regulated under normal growth conditions. Nevertheless, the predicted 136 ORF-encoded YPL250C gene product is not absolutely essential for adaptation to a nonfermentable carbon source.
Splicing of HAC1u mRNA.
Following the shift from glucose to glycerol medium, HAC1u transcripts were spliced by an Ire1p-dependent mechanism. HAC1i mRNA accumulation was transient, however, and none of the known genes in the UPR pathway was detectably induced (data not shown). Alternatively, the apparent activation of Ire1p following the carbon source shift, with no obvious consequence to the UPR pathway, raises the possibility that the regulatory function of Ire1p, and perhaps Hac1p, may not be solely devoted to the UPR and may influence expression of previously unrecognized target genes. The transient nature of HAC1 mRNA splicing and translation may reflect an adaptive response to environmental stress. Transient translational events may occur in a stressed organism as an intermediate response toward adaptation to a new equilibrium state. Nevertheless, if one considers that the average cell cycle of S. cerevisiae, when grown in minimal medium, lasts approximately 120 min, then a significant amount of protein may have been synthesized within 15 min.
Translation of HAC1u has been shown to be regulated during the elongation step (reviewed in references 7 and 37). It is interesting, however, that the polysomal distribution of HAC1u mRNA in Fig. 7 differs from that detailed in previously published reports, in which the majority of HAC1u mRNA sedimented with polysomes while only a minor fraction (∼20%) sedimented with low-density mRNP fractions (8, 10). In contrast, we have routinely observed an accumulation of HAC1u mRNA in the mRNP fractions (Fig. 7, fractions 2 and 3) and very little sedimentation of this unspliced mRNA with polysome fractions. We are uncertain about the origin of this discrepancy. HAC1u mRNA is known to be distributed throughout the cytoplasm in punctate clusters (8). Although the composition of these clusters is presently unknown, the HAC1u mRNA within these clusters may be difficult to extract. Our findings argue that translation of HAC1u mRNA is blocked at the initiation step, in addition to the previously reported elongation step, and that there may be a cytoplasmic pool of HAC1u mRNA which is sequestered as a translationally inactive mRNP complex.
An interesting observation has connected the protein secretory pathway with ribosome biosynthesis. The continued functioning of the secretory pathway has been shown to be essential for ribosome biosynthesis, because inhibition of the secretory pathway reduces transcription of genes encoding RP mRNAs (34). The fact that components of the ER are affected by the carbon source shift is exemplified by the activation of the bifunctional kinase-endonuclease Ire1p and the tRNA ligase Rlg1p.
The genomic response of S. cerevisiae to nutritional change was very rapid. By combining polysomal fractionation with cDNA microarray analysis, we have primarily focused on the translational activity of thousands of individual mRNAs after a rapid depletion of glucose. Identification of individual mRNAs that are translationally controlled has historically relied on cumbersome analyses of suspected mRNA species. The cDNA microarray analysis has uncovered an mRNA species (YPL250C) that can be selectively translated during a global translational inhibition, as well as a coordinate regulation of an entire class of mRNAs (RP mRNAs). Activation of the bifunctional kinase-endonuclease Ire1p and the tRNA ligase Rlg1p after a carbon source shift was confirmed by the appearance of spliced HAC1 mRNAs in polysomal fractions. This latter finding exemplifies the effectiveness of the cDNA microarray, which can allow the detection of multiple levels of regulation that operate in the genomic response of an organism to nutritional change.
ACKNOWLEDGMENTS
We thank Karla Kirkegaard for critical reading of the manuscript. We also thank Gregg Johannes for many stimulating discussions during the course of this work. We are grateful to Peter Walter (University of California at San Francisco) for providing plasmid pIRE and the ΔIRE strain and to Enrique Herrero (Universitat de Lleida, Lleida, Spain) for providing the TetR system.
This work was supported by NIH grants RO1 GM55979 (P.S.) and T32 GM07276 (K.M.K. and J.L.D.) and by the Howard Hughes Medical Institute (J.L.D. and P.O.B.).
REFERENCES
- 1.Ashe M P, De Long S K, Sachs A B. Glucose depletion rapidly inhibits translation initiation in yeast. Mol Biol Cell. 2000;11:833–848. doi: 10.1091/mbc.11.3.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K, editors. Current protocols in molecular biology. New York, N.Y: Greene Publishing Associates and John Wiley & Sons; 1994. [Google Scholar]
- 3.Belli G, Gari E, Piedrafita L, Aldea M, Herrero E. An activator/repressor dual system allows tight tetracycline-regulated gene expression in budding yeast. Nucleic Acids Res. 1998;26:942–947. doi: 10.1093/nar/26.4.942. . (Erratum, 26:1855.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berset C, Trachsel H, Altmann M. The TOR (target of rapamycin) signal transduction pathway regulates the stability of translation initiation factor eIF4G in the yeast Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 1998;95:4264–4269. doi: 10.1073/pnas.95.8.4264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boucherie H. Protein synthesis during transition and stationary phases under glucose limitation in Saccharomyces cerevisiae. J Bacteriol. 1985;161:385–392. doi: 10.1128/jb.161.1.385-392.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carter M S, Kuhn K M, Sarnow P. Cellular internal ribosome entry site elements and the use of cDNA microarrays in their investigation. In: Sonenberg N, Hershey J W B, Mathews M B, editors. Translational control of gene expression. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 2000. pp. 615–635. [Google Scholar]
- 7.Chapman R, Sidrauski C, Walter P. Intracellular signaling from the endoplasmic reticulum to the nucleus. Annu Rev Cell Dev Biol. 1998;14:459–485. doi: 10.1146/annurev.cellbio.14.1.459. [DOI] [PubMed] [Google Scholar]
- 8.Chapman R E, Walter P. Translational attenuation mediated by an mRNA intron. Curr Biol. 1997;7:850–859. doi: 10.1016/s0960-9822(06)00373-3. [DOI] [PubMed] [Google Scholar]
- 9.Chu S, DeRisi J, Eisen M, Mulholland J, Botstein D, Brown P O, Herskowitz I. The transcriptional program of sporulation in budding yeast. Science. 1998;282:699–705. doi: 10.1126/science.282.5389.699. [DOI] [PubMed] [Google Scholar]
- 10.Cox J S, Walter P. A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell. 1996;87:391–404. doi: 10.1016/s0092-8674(00)81360-4. [DOI] [PubMed] [Google Scholar]
- 11.Dabeva M D, Warner J R. Ribosomal protein L32 of Saccharomyces cerevisiae regulates both splicing and translation of its own transcript. J Biol Chem. 1993;268:19669–19674. [PubMed] [Google Scholar]
- 12.DeRisi J L, Iyer V R. Genomics and array technology. Curr Opin Oncol. 1999;11:76–79. doi: 10.1097/00001622-199901000-00015. [DOI] [PubMed] [Google Scholar]
- 13.DeRisi J L, Iyer V R, Brown P O. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science. 1997;278:680–686. doi: 10.1126/science.278.5338.680. [DOI] [PubMed] [Google Scholar]
- 14.Dickson L M, Brown A J. mRNA translation in yeast during entry into stationary phase. Mol Gen Genet. 1998;259:282–293. doi: 10.1007/s004380050814. [DOI] [PubMed] [Google Scholar]
- 15.Eisen M B, Brown P O. DNA arrays for analysis of gene expression. Methods Enzymol. 1999;303:179–205. doi: 10.1016/s0076-6879(99)03014-1. [DOI] [PubMed] [Google Scholar]
- 16.Eisen M B, Spellman P T, Brown P O, Botstein D. Cluster analysis and display of genome-wide expression patterns Proc. Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fuge E K, Braun E L, Werner-Washburne M. Protein synthesis in long-term stationary-phase cultures of Saccharomyces cerevisiae. J Bacteriol. 1994;176:5802–5813. doi: 10.1128/jb.176.18.5802-5813.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gorenstein C, Warner J R. Coordinate regulation of the synthesis of eukaryotic ribosomal proteins. Proc Natl Acad Sci USA. 1976;73:1547–1551. doi: 10.1073/pnas.73.5.1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Herruer M H, Mager W H, Raue H A, Vreken P, Wilms E, Planta R J. Mild temperature shock affects transcription of yeast ribosomal protein genes as well as the stability of their mRNAs. Nucleic Acids Res. 1988;16:7917–7929. doi: 10.1093/nar/16.16.7917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hinnebusch A G. Translational regulation of yeast GCN4. A window on factors that control initiator-tRNA binding to the ribosome. J Biol Chem. 1997;272:21661–21664. doi: 10.1074/jbc.272.35.21661. [DOI] [PubMed] [Google Scholar]
- 21.Iizuka N, Najita L, Franzusoff A, Sarnow P. Cap-dependent and cap-independent translation by internal initiation of mRNAs in cell extracts prepared from Saccharomyces cerevisiae. Mol Cell Biol. 1994;14:7322–7330. doi: 10.1128/mcb.14.11.7322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johannes G, Sarnow P. Cap-independent polysomal association of natural mRNAs encoding c-myc, BiP, and eIF4G conferred by internal ribosome entry sites. RNA. 1998;4:1500–1513. doi: 10.1017/s1355838298981080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnston M. Feasting, fasting and fermenting. Glucose sensing in yeast and other cells. Trends Genet. 1999;15:29–33. doi: 10.1016/s0168-9525(98)01637-0. [DOI] [PubMed] [Google Scholar]
- 24.Johnston M, Carlson M. Regulation of carbon and phosphate utilization. In: Jones E W, Pringle J R, Broach J R, editors. The molecular and cellular biology of the yeast Saccharomyces. 2. Gene expression. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1992. pp. 193–281. [Google Scholar]
- 25.Kawahara T, Yanagi H, Yura T, Mori K. Endoplasmic reticulum stress-induced mRNA splicing permits synthesis of transcription factor Hac1p/Ern4p that activates the unfolded protein response. Mol Biol Cell. 1997;8:1845–1862. doi: 10.1091/mbc.8.10.1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kief D R, Warner J R. Coordinate control of syntheses of ribosomal ribonucleic acid and ribosomal proteins during nutritional shift-up in Saccharomyces cerevisiae. Mol Cell Biol. 1981;1:1007–1015. doi: 10.1128/mcb.1.11.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim C H, Warner J R. Mild temperature shock alters the transcription of a discrete class of Saccharomyces cerevisiae genes. Mol Cell Biol. 1983;3:457–465. doi: 10.1128/mcb.3.3.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lagunas R. Energetic irrelevance of aerobiosis for S. cerevisiae growing on sugars. Mol Cell Biochem. 1979;27:139–146. doi: 10.1007/BF00215362. [DOI] [PubMed] [Google Scholar]
- 29.Li B, Nierras C R, Warner J R. Transcriptional elements involved in the repression of ribosomal protein synthesis. Mol Cell Biol. 1999;19:5393–5404. doi: 10.1128/mcb.19.8.5393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li B, Warner J R. Mutation of the Rab6 homologue of Saccharomyces cerevisiae, YPT6, inhibits both early Golgi function and ribosome biosynthesis J. Biol Chem. 1996;271:16813–16819. doi: 10.1074/jbc.271.28.16813. [DOI] [PubMed] [Google Scholar]
- 31.Liakopoulos D, Doenges G, Matuschewski K, Jentsch S. A novel protein modification pathway related to the ubiquitin system. EMBO J. 1998;17:2208–2214. doi: 10.1093/emboj/17.8.2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mascorro-Gallardo J O, Covarrubias A A, Gaxiola R. Construction of a CUP1 promoter-based vector to modulate gene expression in Saccharomyces cerevisiae. Gene. 1996;172:169–170. doi: 10.1016/0378-1119(96)00059-5. [DOI] [PubMed] [Google Scholar]
- 33.Meyuhas O, Avni D, Shama S. Translational control of ribosomal protein mRNAs in eukaryotes. In: Hershey J W B, Mathews M B, Sonenberg N, editors. Translational control. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1996. pp. 363–388. [Google Scholar]
- 34.Mizuta K, Warner J R. Continued functioning of the secretory pathway is essential for ribosome synthesis. Mol Cell Biol. 1994;14:2493–2502. doi: 10.1128/mcb.14.4.2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Planta R J. Regulation of ribosome synthesis in yeast. Yeast. 1997;13:1505–1518. doi: 10.1002/(SICI)1097-0061(199712)13:16<1505::AID-YEA229>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 36.Ronnow B, Kielland-Brandt M C. GUT2, a gene for mitochondrial glycerol 3-phosphate dehydrogenase of Saccharomyces cerevisiae. Yeast. 1993;9:1121–1130. doi: 10.1002/yea.320091013. [DOI] [PubMed] [Google Scholar]
- 37.Sidrauski C, Chapman R, Walter P. The unfolded protein response: an intracellular signalling pathway with many surprising features. Trends Cell Biol. 1998;8:245–249. doi: 10.1016/s0962-8924(98)01267-7. [DOI] [PubMed] [Google Scholar]
- 38.Sidrauski C, Cox J S, Walter P. tRNA ligase is required for regulated mRNA splicing in the unfolded protein response. Cell. 1996;87:405–413. doi: 10.1016/s0092-8674(00)81361-6. [DOI] [PubMed] [Google Scholar]
- 39.Werner-Washburne M, Becker J, Kosic-Smithers J, Craig E A. Yeast Hsp70 RNA levels vary in response to the physiological status of the cell. J Bacteriol. 1989;171:2680–2688. doi: 10.1128/jb.171.5.2680-2688.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Werner-Washburne M, Braun E, Johnston G C, Singer R A. Stationary phase in the yeast Saccharomyces cerevisiae. Microbiol Rev. 1993;57:383–401. doi: 10.1128/mr.57.2.383-401.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Werner-Washburne M, Braun E L, Crawford M E, Peck V M. Stationary phase in Saccharomyces cerevisiae. Mol Microbiol. 1996;19:1159–1166. doi: 10.1111/j.1365-2958.1996.tb02461.x. [DOI] [PubMed] [Google Scholar]