

Abstract
The nitroreductase superfamily of enzymes encompasses many flavin mononucleotide (FMN)-dependent catalysts promoting a wide range of reactions. All share a common core consisting of an FMN-binding domain, and individual subgroups additionally contain one to three sequence extensions radiating from defined positions within this core to support their unique catalytic properties. To identify the minimum structure required for activity in the iodotyrosine deiodinase subgroup of this superfamily, attention was directed to a representative from the thermophilic organism Thermotoga neapolitana (TnIYD). This representative was selected based on its status as an outlier of the subgroup arising from its deficiency in certain standard motifs evident in all homologues from mesophiles. We found that TnIYD lacked a typical N-terminal sequence and one of its two characteristic sequence extensions, neither of which was found to be necessary for activity. We also show that TnIYD efficiently promotes dehalogenation of iodo-, bromo-, and chlorotyrosine, analogous to related deiodinases (IYDs) from humans and other mesophiles. In addition, 2-iodophenol is a weak substrate for TnIYD as it was for all other IYDs characterized to date. Consistent with enzymes from thermophilic organisms, we observed that TnIYD adopts a compact fold and low surface area compared with IYDs from mesophilic organisms. The insights gained from our investigations on TnIYD demonstrate the advantages of focusing on sequences that diverge from conventional standards to uncover the minimum essentials for activity. We conclude that TnIYD now represents a superior starting structure for future efforts to engineer a stable dehalogenase targeting halophenols of environmental concern.
Keywords: flavoprotein, dehalogenase, halotyrosine, reductase, superfamily, nitroreductase
Abbreviations: DmIYD, IYD from Drosophila melanogaster; FMN, flavin mononucleotide; HhIYD, IYD from Haliscomenobacter hydrossis; IYD, iodotyrosine deiodinase; NTR, nitroreductase superfamily; SSN, sequence similarity network; TCEP, tris(2-carboxyethyl)phosphine; TnIYD, IYD from T. neapolitana
The rapid accumulation of genomic information has far outstripped our ability to anticipate the function of the proteins encoded. Predicting structure is a bit less daunting if homologous sequences are available, but predicting function is typically very difficult without experimental characterization of many related sequences. The process of selecting the most appropriate representatives to examine has been significantly advanced by the introduction of sequence similarity networks (SSNs) that parse large number of sequences within a structural superfamily and identify clusters of related members (1, 2). Even entire metabolic pathways can be discovered by this approach (3). Such analysis of the nitroreductase superfamily (NTR) encompassing over 24,000 sequences successfully identified numerous clusters of uncharacterized sequences among those with previously determined activity (4). The predominant features of each subgroup may guide identification of related catalysts but do not necessarily define the essential requirements for sustaining a particular function. As described below, distinct representatives that are remote from the norm best define the minimal features necessary to support reductive dehalogenation by a subgroup within the NTR superfamily. Such information is critical for refining gene annotations and selecting the most expedient parent sequence for engineering new enzyme specificities.
The NTR superfamily extends well beyond nitroreductases and includes the dehalogenase above as well as quinone and quinoline reductases, flavin destructases, and various dehydrogenases. As described by the SSN analysis, a central hub of representatives contain only minimal sequences to form α2-homodimers that generate equivalent binding sites for two flavin mononucleotides (FMNs) within the interface of the subunits (Fig. 1) (4). Radiating away from this hub are more than 14 distinct subgroups that support various activities by the added presence of one, two, or three extensions labeled previously as E1, E2, and E3 that emanate from distinct positions within the central homodimer (Fig. 1) (4). For example, the subgroup associated with the well-characterized nitroreductase NfsB contains only an E2 extension, whereas the subgroup of flavin destructases associated with BluB contains E1, E2, and E3 extensions. The vast majority of sequences ascribed to iodotyrosine deiodinase (IYD) contain E1 and E2. These extensions have been postulated to control substrate recognition since they are typically distal to FMN (4). The central core α+β fold is then likely responsible for controlling the redox chemistry of the FMN required for the various mechanisms of substrate transformation (4). IYD provides an interesting exception since its substrate halotyrosine establishes many of the polar contacts to FMN directly (5, 6).
Figure 1.

The minimal scaffold for binding FMN in the NTR superfamily.A, a two-dimensional schematic for one of two polypeptides in the α2-homodimer indicates regions of α-helix (α), β-strand (β) and the sites for sequence extensions (E1, E2, E3). B, a three-dimensional representation of a native homodimer in which the individual polypeptides are depicted in gray and wheat. This structure is based on a protein (pdb 3E39) within the central hub as described by Akiva et al. (4). FMN is illustrated as a stick drawing with carbon atoms in orange.
Structural characterization of IYDs indicates that E1 and E2 are relatively dynamic (5, 6, 7, 8, 9). E1 appears disordered in the absence of substrate and forms a helix-linker-helix upon substrate coordination. This fold provides an active site lid that sequesters substrate from solvent and activates single electron transfer pathways of FMN as necessary for substrate dehalogenation (6, 7, 10). Active site ligands that are unable to stabilize the lid cannot induce dehalogenation and instead allow for an innate nitroreduction by a single two electron transfer (10). The role of E2 is less clear but is present in mammalian and bacterial IYDs alike (8). The structure of IYD implies a possible supporting role for E2 based on its contacts to, and presumably stabilization of, the lid structure of E1. Together, E1 and E2 were expected to control the specificity of substrates. IYD was first discovered in humans based on its ability to salvage iodide from iodotyrosine generated in the thyroid although this enzyme and its homologues also promote debromination and dechlorination (Fig. 2) (11, 12, 13). The general specificity for halotyrosines is surprisingly consistent throughout metazoa (14) and has prompted efforts to engineer IYDs for accepting halophenols as substrates for the goal of bioremediation (9). These investigations began using human IYD (HsIYD) as the parent sequence based on the availability of its crystal structure but homologues from thermophilic organisms offer an attractive alternative (15, 16). Additionally, there was hope that IYD from thermophiles might accept a broad array of substrates since the need for a halotyrosine-specific enzyme is not easily rationalized in Bacteria or Archaea. Even the ability of thermophiles to promote dehalogenation could be questioned since none contained E2 sequences in contrast to the homologues from mesophiles. However, studies presented below demonstrate that the minimal structure provided by the thermophilic organism sustains dehalogenation and should offer a robust platform for future engineering of a reductive dehalogenase active in an aerobic environment.
Figure 2.

Reductive dehalogenation promoted by iodotyrosine deiodinase (IYD).
Results
Selection of an outlier sequence within the IYD subgroup of the NTR superfamily
The IYD subgroup is evident in all three domains of life, but to date most attention has been directed toward the mammalian representatives based on their role in iodide homeostasis (17, 18, 19). The function of this enzyme in other metazoa is not yet known although a link to spermatogenesis has been identified in Drosophila (20), and a role in regulating a potassium channel in Caenorhabditis has also been suggested (21). From the SSN illustrated in Figure 3, metazoan sequences cluster together with a notable unique grouping that includes the Caenorhabditis sequence. In general, these sequences all contain significant N-terminal extensions beyond the common catalytic core and often include a single transmembrane anchor. A large number of representatives also derive from mesophilic Bacteria. These contain only short N-terminal extensions and lack an anchor for membrane association. IYDs from bacterial and archaeal thermophiles have no N-terminal sequence beyond the catalytic core and are also truncated by ten amino acids in E2 and one amino acid in E1 (Figs. 3 and S1). Examples from Pyrococcus furiosus and Thermotoga neapolitana share less than 40% identity with HsIYD and less than 58% identity between each other. We were encouraged to focus on the T. neapolitana variant, which represented one of the most distant and relatively unique outliers since preliminary data already suggested that even the P. furiosus protein maintained some activity as a deiodinase (A. Phatarphekar, N. Ingavat, and S. E. Rokita, unpublished observations) (8). These putative dehalogenases from thermophiles are far from the clusters populated by the mesophilic and psychrophilic organisms from all three domains of life. The structure and catalytic activity of the protein from T. neapolitana (TnIYD) are now reported to define the minimal structural requirements for dehalogenation.
Figure 3.

Diversity of the IYD subgroup within the NTR superfamily.A, prototypic variations in polypeptide length for a few representative homologues of Human IYD (HsIYD). Amino acid identity with respect to HsIYD is included in parenthesis. B, sequence similarity network of 625 putative IYD sequences from the NTR superfamily (e value of 5, edge alignment of 62) (4). Red diamonds, green ovals, light blue rectangles, and dark blue ovals represent sequences within archaea, prokaryotes, eukaryotes, and unknown origins, respectively.
Expression and purification of TnIYD
TnIYD was expressed in Escherichia coli with a C-terminal (His)6 tag under conditions equivalent to those used to generate IYD from Drosophila melanogaster (DmIYD) (14) and Haliscomenobacter hydrossis (HhIYD) (7). Purification included the addition of FMN to the cell lysate to boost FMN occupancy in IYD. Typically, the addition yields a bright yellow solution due to excess FMN. However, the mixture containing TnIYD and excess FMN rapidly turned dark green. After separation on Ni-NTA resin, the TnIYD fraction appeared purple and exhibited absorption bands at 350, 450, and 590 nm that are typical for a neutral flavin semiquinone (Figs. 4 and S2) (22). After the column-bound TnIYD was washed extensively with air saturated buffer, the purple color dissipated along with its absorbance at 590 nm while absorbance at 450 nm increased. This response is consistent with the expected oxidation of a flavin semiquinone under aerobic conditions. The resulting TnIYD still retained a greenish tint to the expected yellow color of an oxidized flavoprotein (Fig. S2). This tint likely results from the persistence of a weak and broad absorbance near 590 nm (Fig. 4B). No such absorbance was detected previously after preparing IYD from mesophiles, but such an observation has been noted for a few other flavoproteins after their formation of charge-transfer complexes between the flavin and various aromatic ligands (22).
Figure 4.

UV-vis spectra of TnIYD isolated after heterologous expression. A, purification of TnIYD on Ni-NTA resin produced a purple solution. B, extensive washing with air-saturated buffer prior to elution produced a yellow-green solution. The insert expands the scale to illustrate the low absorbance at long wavelengths. Enzymes were diluted in 110 mM potassium phosphate pH 7.4 with 220 mM potassium chloride under ambient conditions prior to recording the absorbance spectra (see Fig. S2 for more details).
Identification of the ligand copurifying with TnIYD and preparation of ligand-free TnIYD
The greenish-yellow preparation of TnIYD was denatured and precipitated with formic acid to release all noncovalent species. Along with FMN, a relatively hydrophilic compound was observed by reverse-phase (C18) HPLC from its absorbance at 275 nm. Subsequent examination by ultraperformance liquid chromatography–high-resolution mass spectrometry (UPLC-HRMS) identified this compound to be Tyr (Fig. S3). This discovery was quite surprising since no other IYD had previously expressed any measurable affinity for Tyr despite its obvious similarities to the substrate I-Tyr (6, 12). Aromatic ligands copurifying with other flavoproteins had been released upon FMN reduction (23), and thus TnIYD bound to Ni-NTA resin was treated with dithionite to release the bound Tyr. Re-oxidation of this protein with air-saturated buffer generated the standard yellow flavoprotein without Tyr (Figs. S2B and S3). The various states of TnIYD were also recapitulated individually. First, the purple form was generated by reduction of TnIYD in the presence of Tyr, NADH, and an E. coli flavin reductase Fre (Fig. S4). Second, the greenish-yellow form with a weak absorbance around 590 nm was formed after adding 1.7 eq. of Tyr to an otherwise Tyr-free preparation of TnIYD containing FMN in its resting, oxidized form (Fig. S5).
Binding and catalysis of TnIYD
Ligand binding to IYD is routinely monitored by the characteristic quenching of fluorescence emitted by the active site FMN in its oxidized form (6, 12). The high affinity of Tyr for TnIYD was verified by this method, and the Kd was determined to be 0.45 ± 0.07 μM at 25 °C. The Kd for I-Tyr was even smaller with a value of 0.05 ± 0.02 μM. This high affinity is similar to that of the mammalian HsIYD and more than 160-fold greater than that of HhIYD from a mesophilic bacteria. Consistent with all other IYDs characterized to date, TnIYD binds 2-iodophenol (2-IP, Kd = 31 ± 1 μM) significantly weaker than its amino acid derivative, I-Tyr. This suggests that even these thermophilic bacteria maintain a preference for halotyrosines rather than simple halophenols. Such specificity is even more apparent in the Km values for I-Tyr and 2-IP that differ by 19,000-fold (25 °C, Table 1). Surprisingly, 2-IP is dehalogenated by TnIYD with a kcat value that differs by only 2-fold relative to the kcat value of I-Tyr, but kcat/Km values still indicate a great preference for the halogenated amino acid. Overall, the kinetic constants are best used only for qualitative comparisons since the very low Km value of I-Tyr and the enzyme’s sensitivity to substrate inhibition limit the reliability of these values.
Table 1.
Steady-state properties of TnIYDa
| T °C | Substrate | kcat (min−1) | KM (μM) | kcat/KM (μM−1 min−1) |
|---|---|---|---|---|
| 25 °C | I-Tyr | 1.6 ± 0.1 | 0.22 ± 0.07 | 7.2 ± 2.4 |
| 2-IP | 0.84 ± 0.06 | (4.2 ± 0.7) × 103 | (2.0 ± 0.4) × 10−4 | |
| 60 °C | I-Tyr | 27 ± 11 | 3.5 ± 2.5 | 10 ± 6 |
| Br-Tyr | 5.1 ± 0.3 | 0.14 ± 0.12 | 36 ± 31 | |
| Cl-Tyr | 2.9 ± 0.6 | 5 ± 2 | 0.6 ± 0.3 | |
| 2-IP | 6.5 ± 0.3 | (6.6 ± 0.5) × 103 | (9.9 ± 0.9) × 10−4 |
All data and error were obtained by fitting a minimum of two independent measurements to standard Michaelis–Menten kinetics.
Enzyme turnover was also measured at an elevated temperature to approach more natural conditions for the thermophile T. neapolitana. As expected, TnIYD (Tm = 88 °C) maintained structural integrity at much higher temperatures than HsIYD (Tm = 45 °C) as observed by CD (Fig. S6). At 60 °C, the kcat and Km for I-Tyr increased by more than 20-fold relative to the values at 25 °C (Table 1). The large error associated with these values is again due to in part the limited range of substrate concentrations available before substrate inhibition dominates (Fig. S7) (17). Such inhibition was not observed during 2-IP turnover and instead high concentrations of this substrate were necessary to characterize catalysis. The kcat for 2-IP increased by less than 8-fold after the temperature was raised from 25 °C to 60 °C. The milder effect of temperature on the kcat for 2-IP versus that for I-Tyr may reflect the difficulty of 2-IP to induce a productive orientation of active site side chains, but this difference may alternatively result from only the large uncertainty of the values. The kcat/Km for 2-IP remained more than 10,000-fold smaller than that for I-Tyr at both temperatures. Similar to IYD from mesophiles (HsIYD and DmIYD), TnIYD also promotes debromination and dechlorination of the suitable halotyrosines. Again, the large uncertainty in the kinetic constants for these substrates limits possible comparisons.
Structural characterization of TnIYD
Crystals of TnIYD bound alternatively to Tyr and I-Tyr were grown by vapor diffusion and diffracted X-rays to 1.8 Å and 1.6 Å, respectively. The crystal structure of TnIYD bound to Tyr was phased by molecular replacement using a ligand-free model generated from the structure of HhIYD bound to I-Tyr (PDB 5KO8) (7). Initial electron-density maps were improved by prime-and-switch density modification (24) followed by iterative rounds of model building and refinement. Bound FMN and Tyr in each active site along with the entire TnIYD dimer except for the first two residues were visualized in these maps. The final model was refined to an Rfactor of 15.8% and Rfree of 20.1% (Table 2). The structure of the I-Tyr·TnIYD complex was determined by difference Fourier using a ligand-free model of TnIYD based on the Tyr-bound structure of TnIYD. The resulting difference maps had clear density corresponding to I-Tyr and the final occupancy of I-Tyr refined to 0.8 (Fig. S8). Overall, this structure refined to an Rfactor of 17.0% and Rfree of 20.3% (Table 2). Both crystals had the same unit cell in which the asymmetric unit contained a single copy of the native α2-homodimer. The two active sites within each dimer were essentially identical, thus only one representative of each is discussed below.
Table 2.
Data collection and refinement statistics of TnIYD structures
| Process | Tyr bound to TnIYD | I-Tyr bound to TnIYD |
|---|---|---|
| PDB ID | 6PZ0 | 6Q1L |
| Data collection | ||
| Space group | P212121 | P212121 |
| Unit Cell | ||
| a,b,c (Å) | 42.88, 81.82, 103.75 | 42.86, 81.23, 103.90 |
| α = β = γ (°) | 90 | 90 |
| Number of unique reflectionsa | 34,678 (1965) | 47,126 (2264) |
| Number of observed reflectionsa | 225,230 (11,584) | 211,852 (9484) |
| Resolution (Å)a | 35.67–1.80 (1.84–1.80) | 37.91–1.60 (1.63–1.60) |
| Rpim (%)a,b | 4.3(13.8) | 3.9 (38.0) |
| I/σa | 15.1(5.1) | 10.7 (2.1) |
| Completeness (%)a | 99.8 (97.7) | 96.9 (95.5) |
| Redundancya | 6.5 (5.9) | 4.5 (4.2) |
| Refinement | ||
| Rcryst (%)a,c | 15.83 (16.63) | 17.04 (22.607) |
| Rfree (%)a,c | 20.10 (21.59) | 20.26 (25.27) |
| Ramachandran analysis | ||
| Favored (%) | 97.85 | 98.63 |
| Allowed (%) | 2.15 | 1.37 |
| rmsd bonds | 0.007 | 0.010 |
| rmsd angles | 1.051 | 1.3 |
| Average B-factor for protein (Å2) | 23.55 | 21.81 |
| Average B-factor for water (Å2) | 31.15 | 31.30 |
| Average B-factor for ligands (Å2) | 15.21 | 14.49 |
The values in parentheses are for the highest resolution shell.
, where Ii is the intensity of an individual reflection and <I> is the mean intensity obtained from multiple observations of symmetry-related reflections.
, where Fo is an observed amplitude and Fc a calculated amplitude; Rfree is the same statistic calculated over a subset of the data that has not been used for refinement.
TnIYD has many structural features common to HhIYD and HsIYD despite sharing less than 40% sequence identity (6, 7). The backbone of TnIYD forms the same signature α+β-fold of the NTR superfamily (Fig. 5) and superposes with RMSD values of 1.5 and 1.4 Å to analogous structures of HsIYD (269 Cα atoms) and HhIYD (274 Cα atoms), respectively (6, 7). As typical for the IYD subgroup, the active site lid (E1) of TnIYD coordinates directly to I-Tyr but unlike the vast majority of representatives in this subgroup, TnIYD lacks the neighboring E2 domain that interacts with E1 (Figs. 3A, 5, and S1). No other sequence in TnIYD replaces the structural role of the E2 domains present in IYD from mesophiles. This result suggests that the IYD subgroup within the NTR superfamily only requires an E1 sequence to form an active site lid and the core FMN domain to achieve catalytic activity. In addition, the E1 domain of TnIYD is truncated by one amino acid relative to its mesophilic counterparts without a change in catalytic function or substrate specificity (Figs. 3 and S1). Similarly, the lack of an N-terminal extension in TnIYD relative to other IYDs (Fig. 3) does not adversely affect its catalytic activity nor the thermal stability of its α2-homodimer (Fig. S6). Thus, the extended N-terminal region of HsIYD that interacts extensively with its partner subunit is not required for stabilization of the native dimer (see helix of HsIYD highlighted in dark gray, Fig. 5).
Figure 5.

Comparison of TnIYD·I-Tyr (PDB 6Q1L) and HsIYD·I-Tyr (PDB 4TTC). Polypeptides of the α2-dimer are colored in white and wheat. FMN and I-Tyr are colored with orange and yellow carbons, respectively. E1 and E2 extensions are colored in blue and read. The extended N-terminus of HsIYD is colored in dark gray.
The E1-based active site lid of TnIYD shares the same motif of two α-helices and a short intervening linker that is observed in IYD from mesophiles. Nearly all interactions between the enzyme, FMN, and I-Tyr identified in prior structures are also present in the structure of TnIYD (Fig. 6A). These include polar contacts between the zwitterion of I-Tyr and both the isoalloxazine ring of FMN and the near-universal triad of Glu, Tyr, and Lys within E1 (Figs. S1 and S9). Similarly, the phenol oxygen of I-Tyr interacts with the 2′-hydroxyl of FMN and a backbone amide proton. In IYD from mesophiles, this amide proton derives from an Ala residue, and the presence of this Ala was initially thought to be sufficiently conserved to help parse sequences for the IYD subgroup (Fig. S9) (25). However, variations have since been observed particularly among IYDs from thermophiles. Only 27% of this latter group contains Ala at this position. Replacement of Ala with Met is most typical (62.5%) in IYD from bacterial and archaeal thermophiles including TnIYD (Fig. 6A) and its homologue in P. furiosus. Less frequent substitutions at this site include Ser, Lys, and Leu. In no case yet described does the side chain at this position play a significant role in substrate recognition or structural organization of the active site (see below). Accordingly, the outlier status of TnIYD now demonstrates that neither the E2 domain nor the side chain of the hydrogen-bonding donor amide is fundamental to catalysis and substrate specificity.
Figure 6.

Comparison of the active sites TnIYD bound alternatively with I-Tyr (PDB6Q1L) and Tyr (PDB6PZ0). Cartoon representations of (A) I-Tyr and (B) Tyr bound to TnIYD. Individual polypeptides of the α2-dimer are colored white and wheat. E1 is colored blue. Carbons of FMN are colored orange and carbons of I-Tyr and Tyr are colored yellow. Sulfur of the active site Met41 is also indicated in yellow and was observed in two orientations with bound Tyr. Interactions between ligands and TnIYD (≤3.5 Å) are indicated by dashed lines.
The structure of TnIYD with Tyr bound at its active site is nearly identical to that with I-Tyr bound (RMSD of 0.09 Å over 307 Cα) (Figs. 6B and S13). Remarkably, all polar contacts between Tyr and TnIYD are equivalent to those between I-Tyr and TnIYD as well as those detected in structures of HsIYD and HhIYD bound with I-Tyr (6, 7). This observation was not anticipated since the affinity of Tyr to IYD from mesophiles has been too weak to measure, whereas binding of Tyr to TnIYD is highly favorable. Still, no structural changes are evident to account for the difference in affinity as only one minor perturbation of a proximal Tyr (Y112, see below) can be detected after Tyr association with TnIYD (Fig. S10). Note too that the complex of Tyr and TnIYD containing the oxidized FMN may represent a final intermediate of a catalytic cycle. Only the halide is absence from this complex, and the order of its departure is not yet known.
Molecular basis for the unusually tight binding of Tyr to TnIYD
A small shift in Y112 from a position labeled “out” to one labeled “in” is evident when comparing the structures of TnIYD bound with I-Tyr and Tyr, respectively (Fig. S10). While this minor conformational change is unlikely to be the sole basis for the high affinity of TnIYD for Tyr, its contribution was examined with a Y112A mutation. This variant bound both I-Tyr and Tyr with nearly the same affinities as the native TnIYD, and thus the position of the aromatic side chain was not sufficient to stabilize Tyr association (Table 3).
Table 3.
Binding affinity of I-Tyr and Tyr as a function of side chain substitution (25 °C)a
| TnIYD |
Kd (μM) |
|
|---|---|---|
| I-Tyr | Tyr | |
| wt | 0.05 ± 0.02 | 0.45 ± 0.07 |
| Y112A | 0.03 ± 0.02 | 0.9 ± 0.1 |
| M41A | 0.25 ± 0.06 | 1.6 ± 0.2 |
| M41A/Y112A | 0.024 ± 0.002 | 0.95 ± 0.09 |
| M41K | 1.1 ± 0.1 | 7.4 ± 0.2 |
| M41F | 0.5 ± 0.1 | 0.09 ± 0.06 |
| Y112A/W82A/F88A | 0.9 ± 0.1 | 1200 ± 20 |
All data and error were obtained by fitting of two independent measurements.
The role of M41 was next examined by an M41A mutation. As noted above, Ala is the predominant residue at this position for IYDs from mesophiles. Additionally, the side chain of Met41 adopts multiple conformations in the crystal structure of the TnIYD⋅Tyr complex (Fig. 6B). The M41A substitution did weaken the enzyme’s affinity for Tyr by 3.6-fold, but its effect on I-Tyr was slightly greater (5-fold, Table 3). None of these individual substitutions in TnIYD could recreate the high selectivity of other IYDs for I-Tyr relative to Tyr and a variant with substitutions of M41A and Y112A in TnIYD also failed at limiting Tyr binding. The affinity for Tyr by this variant decreased by only 2-fold while the affinity for I-Tyr increased by 50%. Lys is a natural variant of M41 in IYD and offers the contrast of a charged side chain. Substitution of this residue in an M41K variant of TnIYD decreased affinity for I-Tyr and Tyr by 22-fold and 16-fold, respectively (Table 3). An alternative M41F variant was examined to test if a nonpolar aromatic side chain could selectively impede the binding of Tyr. Indeed, this variant affected ligand binding but in the opposite manner than originally anticipated. Substitution of Phe for the native Met, enhanced binding of Tyr by 5-fold and concurrently decreased binding of I-Tyr by 10-fold resulting in a protein with an inverted selectivity that now favored Tyr over I-Tyr by 5-fold. Thus, none of these individual changes could be responsible for loss of the high selectivity of I-Tyr over Tyr for IYDs from mesophilic organisms.
The ability of TnIYD to bind Tyr could instead derive from a decrease in surrounding polarity relative to HsIYD. Besides Y112, Tyr and I-Tyr are surrounded by W82 and F88 in the active site of TnIYD (Fig. S11). Y212 in HsIYD acts as an equivalent to Y112 in TnIYD, but the two remaining aromatic residues in TnIYD are replaced with D172 and T178 in HsIYD. To examine the role of these aromatic amino acids in TnIYD, a triple mutant was prepared in which all three aromatic amino acids were substituted with Ala (W82A/F88A/Y112A). This modification noticeably weakened binding of I-Tyr by 18-fold but also reestablished selectivity for this substrate by a much more substantial destabilization of Tyr binding of more than 2600-fold (Table 3). Thus, the aromatic residues in the TnIYD active site provide little specificity for I-Tyr and instead create a nonpolar environment for nonspecific association of an aromatic group.
Discussion
The NTR superfamily encompasses an FMN-binding core that has accumulated one or more extensions at common sites to create a number of subgroups promoting a range of reactions (4). The full diversity of catalysis remains to be described, but knowledge to date has already allowed application of this superfamily to numerous topics such as synthetic chemistry (26, 27), prodrug therapy (28), biomedical imaging (29), cell biology (30), and bioremediation (9, 31). Curation of this superfamily through an SSN provides a powerful guide to discover the structural and evolutionary basis for the FMN-dependent processes (4). In particular, interest in IYD derives from its unique ability to promote reductive dehalogenation under aerobic conditions in the absence of metals and chalcogens (8, 32). This dehalogenase also has the potential to serve a variety of applications, but its narrow range of substrates currently restricts its utility. To guide future manipulation of this system, we examined a representative on the periphery of the IYD subgroup to establish the minimal requirements for catalysis. A variety of predominant features of this subgroup have now been deemed nonessential by this approach.
Substrate selection and ligand affinity
How Bacteria and Archaea may be exposed to halotyrosines has not yet been studied, but the presence of halophenol derivatives in the environment is widespread (33, 34, 35). Still, TnIYD demonstrates a stringent specificity for halotyrosines that is consistent with its three conserved amino acids (E68, Y72, K92) within the E1 domain that coordinate the zwitterion of these amino acids (Fig. 6). For comparison, 2-IP is dehalogenated with an efficiency (kcat/Km) of less than 0.01% of that for iodotyrosine at 25 °C (Table 1). This difference is likely due to the necessity of the substrate’s zwitterion to organize the active site for efficient turnover as manifest in the Km value that extends over four orders of magnitude from 2-IP to I-Tyr.
TnIYD defied expectations by its ability to bind Tyr with high affinity. No other IYD has yet to demonstrate a measurable affinity for tyrosine (6, 12). Previously, high affinity of an active site ligand was predicated by substitution on the phenolic ring with a halogen or other substituent that enhanced the acidity of the phenolic proton (8). These observations were considered indicative of a preference for IYD to bind the phenolate form of its ligands. Only minute quantities of the ionized form of Tyr are present under standard conditions of pH 7.4. The affinity of TnIYD for the neutral form of Tyr is likely based on the introduction of aromatic side chains (W82, F88) that enhance the nonpolar properties of its binding site relative to that of IYDs examined from mesophiles (Fig. S11). Although a phenolic hydrogen is modeled in the crystal structure of TnIYD, the exact protonation state of bound Tyr remains to be determined. The distances between its phenolic oxygen and the hydrogen bond donor from the polypeptide backbone are all between 2.7 and 3.0 Å in the crystal structures of I-Tyr bound to either HsIYD, HhIYD, TnIYD or IYD from Mus musculus (MmIYD) (Fig. 6) (5, 6, 7). The phenolic oxygen of Tyr is also within 2.6 Å of the hydrogen bond donating 2′-hydroxy of FMN, a distance identical to that observed in all of the structures noted above for the phenolic oxygen of I-Tyr.
A corollary to the high affinity of TnIYD for Tyr is readily observed after heterologous expression in E. coli. As isolated, TnIYD appears purple rather than the usual yellow of a flavoprotein. The origins of this derive from the ability of TnIYD to scavenge endogenous Tyr from the host bacteria. Binding of Tyr stabilizes closure of the active site lid formed by the E1 sequence, and this in turn should stabilize the one electron reduced neutral semiquinone form of FMN that is apparent in the UV-vis spectrum (Fig. 4). Such stabilization was observed previously after fluorotyrosine (F-Tyr), an inert substrate analogue, bound to HsIYD (6, 10). The surprising longevity of the semiquinone form of TnIYD under aerobic conditions suggests that active site lid closure is highly efficient at protecting the cofactor from solvent and its accompanying dissolved molecular oxygen. Otherwise, reaction between the semiquinone and molecular oxygen would be extremely rapid (36).
Minimal structure required for reductive dehalogenation
E2 sequences are nearly omnipresent in the IYD subgroup of the NTR superfamily (4). Based on this observation alone, such a domain would seem essential (Fig. 3). In HsIYD, E2 forms a loop that protrudes from the otherwise rather globular profile of the homodimer (Fig. 5). A catalytic function for E2 was not expected since this loop extends away from the active site and lacks residues that are directly involved in binding substrate or FMN. A structural role is more likely due to the observed π-π stacking at a distance of 3.3 Å between a highly conserved Tyr (Y211) proximal to E2 and an equally conserved Trp (W169) within the active site lid formed by the E1 sequence (Fig. 7A). This interaction has the potential to stabilize closure of the lid and facilitate the disorder to order transition of E1 upon substrate binding. Characterization of TnIYD has now demonstrated that such an interaction is not necessary. Moreover, the entire E2 is dispensable for substrate recognition and turnover. Again, no compensatory changes in the structure of TnIYD replace the interactions lost by the absence of an E2 sequence. The closest approach between the E1 lid and remnants of E2 in TnIYD is between a Pro (P111) and Leu (L79) at a distance of 4.7 Å (Fig. 7B). Not surprisingly, neither equivalents of Y211 or W169 are conserved in TnIYD.
Figure 7.
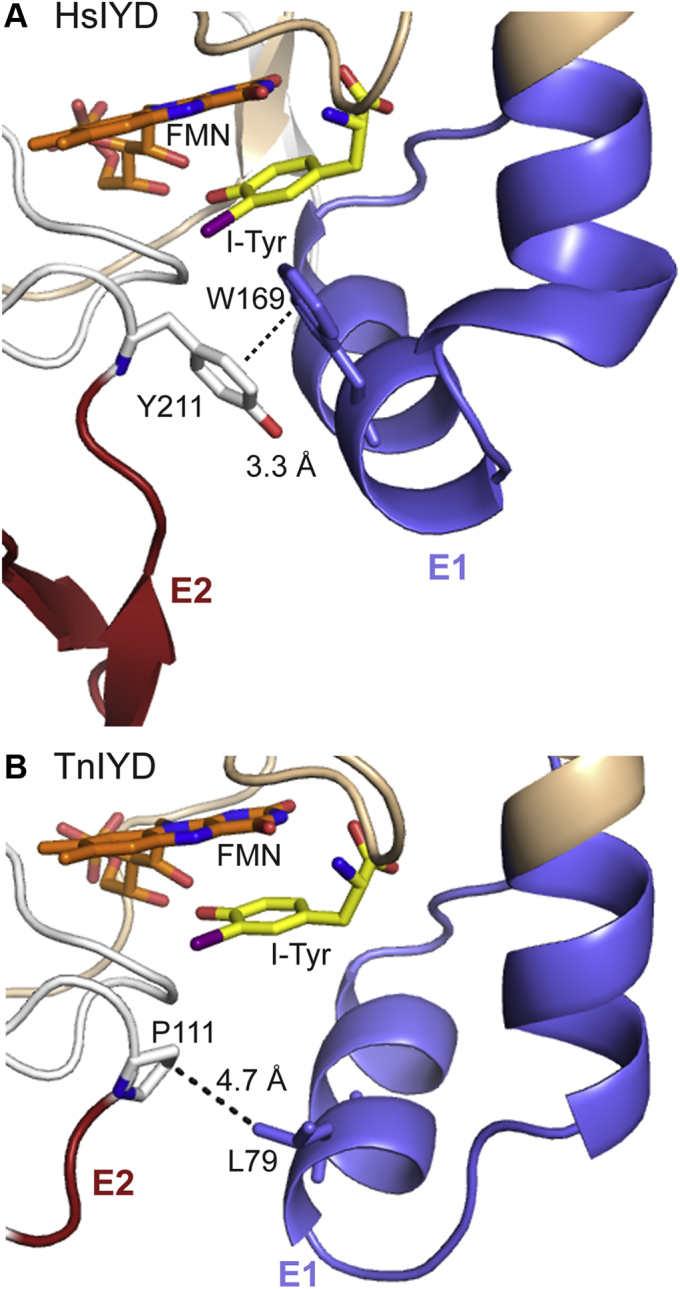
Contact between E1 and E2. Individual polypeptides of the α2-dimer in (A) HsIYD·ITyr (PDB 4TTC) and (B) TnIYD·ITyr (PDB 6Q1L) are colored gray and wheat. Carbons of FMN are colored orange and carbons of I-Tyr are colored in yellow. E1 and E2 are colored with blue and red, respectively, and the shortest distance between these two regions is indicated by a dotted line.
Unlike the domains above, E1 is essential for all eukaryotic, bacterial, and archaeal IYDs regardless of the preferred growth conditions. This region is dynamic in the absence of substrate but adopts a helix-linker-helix in the presence of substrate to form an active site lid (5, 6, 7). This organization also brings a Thr side chain proximal to the N5 position of FMN for stabilizing an intermediate FMN semiquinone and promoting a series of single-electron transfers necessary for reductive dehalogenation (6, 7, 10). Likewise, dehalogenation is very inefficient for substrates such as 2-IP that are unable to organize E1 into a lid structure (Table 1). Without the conformational changes associated with lid closure, the FMN is only capable of hydride transfer and will instead promote nitroreduction in analogy to other subgroups within the NTR family (7, 10). Most important within the E1 sequence are the universally conserved Lys and Glu (E68, K92 in TnIYD, Fig. 6) and the highly conversed Tyr (Y72) that form polar bonds to the zwitterion of halotyrosine. The significance of these interactions had previously been demonstrated by mutagenesis (14, 37). In particular, replacement of this Glu to Gln suppresses dehalogenation severely for both MmIYD and DmIYD.
Thermophilic adaptation
Many factors contribute to thermal stability of proteins. In comparison to mesophilic counterparts, proteins of thermophiles are often enriched in hydrophobic and β-branched amino acids as well as Pro and depleted in uncharged polar residues (38). Such trends are not apparent when comparing TnIYD to HsIYD and HhIYD other than the gain of hydrophobic residues (W82, F88) in the active site of TnIYD, and even these substitutions are not universal in all homologues from thermophiles. An increase in the number of salt bridges is another common strategy for achieving thermal stability (38, 39, 40), but a similar number of salt bridges is observed within HsIYD and TnIYD. Instead, the thermal stability of TnIYD likely derives from its minimal sequence that is shared by homologues from other thermophilic Bacteria as well as thermophilic Archaea and distinct from those of mesophiles from all three domains of life (4). Unique to IYD from thermophiles are (i) the lack of an N-terminal sequence extending from its catalytic core, (ii) loss of one amino acid from the E1 region, and (iii) loss of ten amino acids that form the E2 region (Fig. 3). The absence of these residues diminishes the overall surface area of the homodimer for TnIYD (16,230 Å2) relative to HsIYD (18,550 Å2) by 14% (Fig. 8 and S12). Furthermore, the absence of E2 removes a relatively dynamic region that is likely most sensitive to thermal denaturation. In the crystal structure of HsIYD, the average B-factor of E2 (64 Å2) is 40% larger than that of its core and in HhIYD the corresponding average B-factor for E2 (80 Å2) is almost 80% larger than that of its core (Fig. S13). Thermophilic adaptation within the IYD subgroup of the NTR superfamily therefore provides instruction for generating stable IYD derivatives and illustrates the minimal structure required for catalytic activity despite their status as outliers from the vast majority of IYD homologues identified to date.
Figure 8.

Increasing surface area from a protein of the NTR hub to TnIYD and HsIYD.A, the core FMN-binding domain is represented by the structure of a protein within the NTR hub (PDB 3E39) (4). B, TnIYD (PDB 6Q1L) and (C) HsIYD (PDB 4TTC) are represented by their complexes with I-Tyr. The positions of E1, E2, and E3 are noted in blue, red, and green as introduced in Figure 1. FMN is colored with orange carbon atoms. See Figure S10 for full view of the front and back of these structures.
Conclusion
Analyzing large superfamilies of proteins to create SSN is a powerful tool for discovering structurally distinct subgroups that can reveal key determinants for diagnosing function (1, 2). Recent evaluation of the NTR superfamily revealed radial development of function emanating from a central hub of proteins with a minimal FMN-binding domain (4). Each functional subgroup was dominated by a specific combination of extensions (E1, E2, and/or E3) that may help to determine catalytic specificity. At least for the IYD subgroup, however, the structural requirements for activity are not defined by the prevalence of particular features. Instead, these have been identified from the exceptional sequences of a small set of IYDs from thermophiles. Neither an extended N-terminal extension, a loop formed by E2, nor an Ala at the site of substrate–backbone hydrogen bonding is necessary for dehalogenating halotyrosines. The minimal presence of an E1 sequence forming an active site lid and the core FMN-binding domain are instead sufficient to support catalysis. Prior analysis of IYD from P. furiosus and more detailed studies on TnIYD now illustrate the conservation of catalytic function and substrate specificity for IYD ranging from mesophiles to thermophiles in all three domains of life. This far exceeded the original expectation for IYD's distribution based on its initial association with iodide homeostasis in chordata (4, 25).
Experimental procedures
Plasmids
The original plasmid (pET28) encoding TnIYD (NCBI accession: ACM22745.1) was a generous gift from Dr Janine Copp (University of British Columbia, Vancouver). The TnIYD gene was subcloned into a pET24a vector (Novagen) via restriction sites NdeI and XhoI that were introduced by PCR (see Table S1 for primers). A C-terminal (His)6 tag was then added to the TnIYD sequence by mutagenesis using standard protocols (41, 42). The resulting construct was used as the template for further mutagenesis to generate the variants listed on Table S1. The gene encoding flavin reductase (Fre) was amplified from the E. coli genome (Genehogs, Invitrogen) with a C-terminal (His)6 tag and cloned into a pET24a vector via overlap extension PCR (43). All oligonucleotides were synthesized by IDT and Sigma, and all PCR reactions utilized Phusion High-Fidelity DNA polymerase (Thermo Fisher Scientific). The desired genes were all verified by Sanger sequencing (Genewiz).
Protein expression and purification
Plasmids encoding the desired TnIYD gene and its variants were transformed into Rosetta2 chemically competent E. coli (Novagen). The culture was then grown in 500 ml LB media at 37 °C with vigorous shaking (220 rpm) before cooling to 16 °C after an OD600 of ∼1 was observed. Isopropyl thio-β-galactoside (IPTG, 25 μM final, Goldbio) was added, and the cultures were shaken (220 rpm) for ∼15 h at 16 °C. Cells were harvested by centrifugation (5000g, 10 min) at 4 °C, flash-frozen in liquid nitrogen, and stored at −80 °C. Fre was expressed using the same conditions as TnIYD. Frozen cell pellets containing TnIYD (∼4 g) were thawed at room temperature and resuspended by vortexing in ∼25 ml cell lysis buffer (50 mM sodium phosphate pH 8.0 with 500 mM sodium chloride, 10% glycerol, 25 mM imidazole, and 0.5 mM tris(2-carboxyethyl)phosphine (TCEP)). All subsequent procedures were performed at 4 °C unless otherwise noted. FMN was added to the suspension to a final concentration of ∼300 μM and subsequent cell lysis used an EmulsiFlex-C3 homogenizer (Avestin, 3 passes at ∼17,000 psi). Cell debris was removed by centrifugation at 45,000g for 30 min. The resulting supernatant was loaded onto Ni-NTA resin (5 ml) (Thermo Fisher Scientific, preequilibrated with ten column volumes of lysis buffer) by a bench top Econo pump (Bio-rad) at a flow rate of 1 ml/min. The column was then washed with 16 column volumes of air-saturated washing buffer (50 mM sodium phosphate pH 8.0 with 500 mM sodium chloride, 10% glycerol, 60 mM imidazole, and 0.5 mM TCEP) using an ÄKTA protein purification system (GE Healthcare) at 1 ml/min. Tyr-bound TnIYD was eluted with elution buffer (50 mM sodium phosphate pH 8.0 with 500 mM sodium chloride, 10% glycerol, 250 mM imidazole, and 0.5 mM TCEP). To isolate Tyr-free TnIYD, the column was alternatively washed with four column volumes of cell lysis buffer supplemented with sodium dithionite (2% final, technical grade, Sigma-Aldrich) with an Econo pump at 1 ml/min. The resulting colorless protein was then washed with air-saturated washing buffer to reoxidized TnIYD (bright yellow). This species was eluted as described above with elution buffer. Samples were then incubated with ∼300 μM FMN on ice for 1 h before an exchange to storage buffer (50 mM sodium phosphate pH 7.4 with 100 mM sodium chloride, 1 mM TCEP, and 10% glycerol) using a gravity driven PD-10 desalting column (GE healthcare) following manufacturer’s instructions. Purified protein was finally concentrated with a 30 kD molecular weight cutoff centrifugal filter unit (Amicon), flash-frozen with liquid nitrogen, and stored at -80 °C. Fre was purified by the same method and all enzymes were analyzed by 12% SDS-PAGE with Coomassie brilliant blue staining. Enzyme concentration was determined by UV-vis absorption of enzyme-bound FMN (ε450 12,500 M−1 cm−1) (44). FMN occupancy was estimated by the ratio of enzyme-bound FMN over the total protein concentration that was calculated from A280 (extinction coefficient estimated by the ExPASy ProtParam) (45) as described previously (6).
Identification of the low-molecular-weight ligand that copurifies with TnIYD expressed in E. coli
The greenish-yellow form of TnIYD (80 μM) in storage buffer was diluted with 1 equiv. of water and supplemented with 4.4% formic acid. This solution was incubated at room temperature for 5 min before centrifugation at 14,000g for 5 min. The supernatant (200 μl) was fractionated using a Jasco HPLC with two PU-980 Intelligent pumps, an MD-1510 photo-diode array detector and a solvent gradient listed in Table S2. The ligand was collected after chromatographic separation and dried under reduced pressure (Savant Speed Vac, SC100A). The resulting residue was dissolved in water (120 μl) and analyzed with a Waters Acquity/XevoG2 UHPLC-MS system using an Acquity UPLC HSS T3 column (100 Å, 1.8 μm, 2.1 mm × 100 mm) and a binary mobile phase of water (A) and acetonitrile (B) with 0.1% v/v formic acid in each at a flow rate of 0.3 μl/min. Compound separation was achieved with an initial B from 0 to 5% over 1 min and then continued to 60% B for the next 6 min with a column temperature of 35 °C. Positive electrospray ionization mode was used for detection with a capillary voltage, sampling cone, and extraction cone set to 3 kV, 40 V, and 4 V, respectively. The source temperature was 130 °C and desolvation gas temperature and flow rate were 400 °C and 400 l/h, respectively. All mass spectrometer data were analyzed by Masslynx v4.1 (Waters).
Crystallization and structure determination of TnIYD
Complexes between TnIYD and ligands were formed by incubating approximately 10 mg/ml of TnIYD with 2- and 3-fold molar excess of Tyr and I-Tyr respectively in 10 mM Tris pH 7.4 with 100 mM sodium chloride for 3 h at 4 °C. Crystals were grown by hanging-drop vapor diffusion after mixing ratios (1:1 and 2:1) of complex to well solution containing 100 mM sodium citrate pH 4.5 with 3 M NaCl. Plates were then incubated at 20 °C. Bright yellow plate-shaped crystals appeared after 1 day and grew to their maximum size in a week with approximate dimensions of 400 × 150 × 40 μm. Crystals were then transferred directly from the drop into a cryo-stabilization buffer (100 mM citric acid pH 4.5 with ∼2 mM of ligand, 3 M sodium chloride and 30% PEG400) and flash frozen in liquid nitrogen. Diffraction data were collected at beamline 12-2 at the Stanford Synchrotron Radiation Lightsource and processed with XDS/Aimless (46). For the initial structure, phases were determined by iterative rounds of molecular replacement with a search model corresponding to a ligand-free, trimmed, polyalanine monomeric model of HhIYD (PDB 5KO8) (7) in Phaser (47). Initial electron-density maps were improved by prime-and-switch density modification (24). Subsequent structures were determined by difference Fourier. Models were built by iterative rounds of manual-building in Coot (48) and refinement in Phenix (49). The occupancy of the ligands was refined manually by setting the occupancy and evaluating the difference Fourier maps. Polder omit maps were calculated in Phenix.
Structural analysis
Distance measurements were calculated and structure figures were rendered in Pymol (50). Buried surface area calculations were performed in PDBePISA (51, 52).
Binding affinity of active site ligands
Active site binding is reported as dissociation constants (Kd) with TnIYD and its variants and determined by ligand-dependent quenching of the fluorescence of FMNox as described previously (12, 53). Briefly, a solution of ligand was titrated with mild stirring into 2–4 μM enzyme in 100 mM potassium phosphate pH 7.4 and 200 mM potassium chloride at 25 ± 1 °C. Fluorescence emission was measured at 516–526 nm when excited at 450 nm. The ratio of observed to initial fluorescence (after correcting for dilution) versus ligand concentration was fit to a two-state binding isothermal.
Catalytic dehalogenation
Rates of catalysis were measured by formation of the dehalogenated products Tyr or phenol using reverse-phase HPLC as previously described (7). Briefly, 50 nM–2.5 μM enzyme was incubated with various concentrations of halotyrosine or 2-IP in 220 mM potassium chloride and 110 mM potassium phosphate pH 7.4 (900 μl) at the assay temperature for 5 min. Sodium dithionite (5%) in 5% sodium bicarbonate (100 μl) was then added to initiate catalysis. Reaction was quenched by adding a 1:1 mixture (100 μl) of aq. formic acid (88%) and Cl-Tyr (0.55 μM final in the assay buffer as an internal standard). The resulting mixture was separated and quantified (A271) by reverse-phase C18 (Agilent, Microsorb-MV 300 C18, 250 × 4.6 mm) HPLC using solvent gradients (1 ml/min) summarized in Table S2. Rate versus substrate concentration was fit to the Michaelis–Menten equation (Origin 9.0) (Fig. S7).
Data availability
The structures presented in this paper have been deposited in the Protein Data Bank (PDB) with the codes 6PZ0 and 6Q1L.
Supporting information
This article contains supporting information (54).
Conflict of interest
The authors declare that they have no conflicts of interest with the contents of this article.
Acknowledgments
We are grateful to Janine Copp (Department of Biochemistry & Molecular Biology, University of British Columbia) for helpful discussions and the original plasmid encoding TnIYD. This work was supported in part by National Science Foundation (SusChEM 90079121) and National Institutes of Health (GM130937 and T32GM080189). Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393).
Author contributions
Z. S. and B. X. conceptualization; Z. S. and J. M. K. investigation; S. S. methodology; S. E. R. supervision.
Funding and additional information
The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH.
Edited by F. Peter Guengerich
Supporting information
References
- 1.Gerlt J.A. Genomic enzymology: Web tools for leveraging protein family sequence-function space and genome context to discover novel functions. Biochemistry. 2017;56:4293–4308. doi: 10.1021/acs.biochem.7b00614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Copp J.N., Anderson D.W., Akiva E., Babbit P.C., Tokuriki N. Exploring the sequence, function, and evolutionary space of protein superfamilies using sequence similarity networks and phylogenetic reconstructions. Methods Enzymol. 2018;620:315–347. doi: 10.1016/bs.mie.2019.03.015. [DOI] [PubMed] [Google Scholar]
- 3.Zhao S., Kumar R., Sakai A., Vetting M.W., Wood B.M., Brown S., Bonanno J.B., Hillerich B.S., Seidel R.D., Babbitt P.C., Almo S.C., Sweedler J.V., Gerlt J.A., Cronan J.E., Jacobson M.P. Discovery of new enzymes and metabolic pathways by using structure and genome context. Nature. 2013;502:698–702. doi: 10.1038/nature12576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Akiva E., Copp J.N., Tokuriki N., Babbit P.C. Evolutionary and molecular foundations of multiple contemporary functions of the nitroreductase superfamily. Proc. Nat. Acad. Sci. U. S. A. 2017;114:E9549–E9558. doi: 10.1073/pnas.1706849114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thomas S.R., McTamney P.M., Adler J.M., LaRonde-LeBlanc N., Rokita S.E. Crystal structure of iodotyrosine deiodinase, a novel flavoprotein responsible for iodide salvage in thyroid glands. J. Biol. Chem. 2009;284:19659–19667. doi: 10.1074/jbc.M109.013458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hu J., Chuenchor W., Rokita S.E. A switch between one- and two-electron chemistry of the human flavoprotein iodotyrosine deiodinase is controlled by substrate. J. Biol. Chem. 2015;290:590–600. doi: 10.1074/jbc.M114.605964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ingavat N., Kavran J.M., Sun Z., Rokita S.E. Active site binding is not sufficient for reductive deiodination by iodotyrosine deiodinase. Biochemistry. 2017;56:1130–1139. doi: 10.1021/acs.biochem.6b01308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun Z., Su Q., Rokita S.E. The distribution and mechanism of iodotyrosine deiodinase defied expectations. Arch. Biochem. Biophys. 2017;632:77–87. doi: 10.1016/j.abb.2017.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun Z., Rokita S.E. Towards a halophenol dehalogenase from iodotyrosine deiodinase via computational design. ACS Catal. 2018;8:11783–11793. [Google Scholar]
- 10.Hu J., Su Q., Schlessman J.L., Rokita S.E. Redox control of iodotyrosine deiodinase. Protein Sci. 2019;28:68–78. doi: 10.1002/pro.3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rokita S.E., Adler J.M., McTamney P.M., Watson J.A., Jr. Efficient use and recycling of the micronutrient iodide in mammals. Biochimie. 2010;92:1227–1235. doi: 10.1016/j.biochi.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McTamney P.M., Rokita S.E. A mammalian reductive deiodinase has broad power to dehalogenate chlorinated and brominated substrates. J. Am. Chem. Soc. 2009;131:14212–14213. doi: 10.1021/ja906642n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bobyk K.D., Ballou D.P., Rokita S.E. Rapid kinetics of dehalogenation promoted by iodotyrosine deiodinase from human thyroid. Biochemistry. 2015;54:4487–4494. doi: 10.1021/acs.biochem.5b00410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Phatarphekar A., Rokita S.E. Functional analysis of iodotyrosine deiodinase from Drosophila melanogaster. Protein Sci. 2016;25:2187–2195. doi: 10.1002/pro.3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Atomi H., Sato T., Kanai T. Application of hyperthermophiles and their enzymes. Curr. Opin. Biotechnol. 2011;22:618–626. doi: 10.1016/j.copbio.2011.06.010. [DOI] [PubMed] [Google Scholar]
- 16.Bommarius A.S., Blum J.K., Abrahamson M.J. Status of protein engineering for biocatalysts: How to design an industrially useful biocatalyst. Curr. Opin. Chem. Biol. 2011;15:194–200. doi: 10.1016/j.cbpa.2010.11.011. [DOI] [PubMed] [Google Scholar]
- 17.Rosenberg I.N., Goswami A. Purification and characterization of a flavoprotein from bovine thyroid with iodotyrosine deiodinase activity. J. Biol. Chem. 1979;254:12318–12325. [PubMed] [Google Scholar]
- 18.Moreno J.C., Klootwijk W., van Toor H., Pinto G., D'Alessandro M., Lèger A., Goudie D., Polak M., Grüters A., Visser T.J. Mutations in the iodotryosine deiodinase gene and hypothyroidism. N. Engl. J. Med. 2008;358:1811–1818. doi: 10.1056/NEJMoa0706819. [DOI] [PubMed] [Google Scholar]
- 19.Afink G., Kulik W., Overmars H., de Randamie J., Veenboer T., van Cruchten A., Craen M., Ris-Stalpers C. Molecular characterization of iodotyrosine dehalogenase deficiency in patients with hypothyroidism. J. Clin. Endocinol. Metab. 2008;93:4894–4901. doi: 10.1210/jc.2008-0865. [DOI] [PubMed] [Google Scholar]
- 20.Phatarphekar A., Su Q., Eun S.H., Chen X., Rokita S.E. The importance of a halotyrosine dehalogenase for Drosophila fertility. J. Biol. Chem. 2018;293:10314–10321. doi: 10.1074/jbc.RA118.003364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de la Cruz I.P., Ma L., Horvitz H.R. The Caenorhabditis elegans iodotyrosine deiodinase ortholog SUP-18 functions through a conserved channel SC-Box to regulate the muscle two-pore domain potassium channel SUP-9. PLoS Genet. 2014;10:1004175. doi: 10.1371/journal.pgen.1004175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Massey V., Ghisla S. Role of charge-transfer interactions in flavoprotein catalysis. Ann. N.Y. Acad. Sci. 1974;227:446–465. [Google Scholar]
- 23.Matthews R.G., Massey V., Sweeley C.C. Identification of p-hydroxybenzaldehyde as the ligand in the green form of Old Yellow Enzyme. J. Biol. Chem. 1975;250:9294–9298. [PubMed] [Google Scholar]
- 24.Terwilliger T.C. Using prime-and-switch phasing to reduce model bias in molecular replacement. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2144–2149. doi: 10.1107/S0907444904019535. [DOI] [PubMed] [Google Scholar]
- 25.Phatarphekar A., Buss J.M., Rokita S.E. Iodotyrosine deiodinase: A unique flavoprotein present in organisms of diverse phyla. Mol. Biosyst. 2014;10:86–92. doi: 10.1039/c3mb70398c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Durchschein K., Hall M., Faber K. Unusual reactions mediated by FMN-dependent ene- and nitro-reductases. Green. Chem. 2013;15:1764–1772. [Google Scholar]
- 27.Miller A.-F., Park J.T., Ferguson K.L., Pitsawong W., Bommarius A.S. Informing efforts to develop nitroreductases for amine production. Molecules. 2018;23:211. doi: 10.3390/molecules23020211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Copp J.N., Williams E.M., Rich M.H., Patterson A.V., Smaill J.B., Ackerley D.F. Toward a high-throughput screening platform for directed evolution of enzymes that activate genotoxic prodrugs. Protein Eng. Des. Sel. 2014;27:399–403. doi: 10.1093/protein/gzu025. [DOI] [PubMed] [Google Scholar]
- 29.Williams E.M., Rich M.H., Mowday A.M., Ashoorzadeh A., Copp J.N., Guise C.P., Anderson R.F., Flanaga J.U., Smaill J.B., Patterson A.V., Ackerley D.F. Engineering Escherichia coli NfsB to activate a hypoxia-resistant analogue of the PET probe EF5 to enable non-Invasive imaging during enzyme prodrug therapy. Biochemistry. 2019;58:3700–3710. doi: 10.1021/acs.biochem.9b00376. [DOI] [PubMed] [Google Scholar]
- 30.Mathias J.R., Zhang Z., Saxena M.T., Mumm J.S. Enhanced cell-specific ablation in Zebrafish using a triple mutant of Escherichia coli Nitroreductase. Zebrafish. 2014;11:85–97. doi: 10.1089/zeb.2013.0937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ackerley D.F., Gonzalez C.F., Keyhan M., Blake R., II, Matin A. Mechanism of chromate reduction by the Escherichia coli protein, NfsA, and the role of different chromate reductases in minimizing oxidative stress during chromate reduction. Environ. Microbiol. 2004;6:851–860. doi: 10.1111/j.1462-2920.2004.00639.x. [DOI] [PubMed] [Google Scholar]
- 32.Rokita S.E. In: Comprehensive Natural Products III: Chemistry and Biology. Liu H.-w., Begley T.P., editors. Wiley; Hoboken: 2020. Reductive dehalogenases; pp. 157–186. [Google Scholar]
- 33.Vetter W. Marine halogenated natural products of environmental relevance. Rev. Environ. Contam. Toxicol. 2006;188:1–57. doi: 10.1007/978-0-387-32964-2_1. [DOI] [PubMed] [Google Scholar]
- 34.Zhai H., Zhang X., Zhu X., Liu J., Ji M. Formation of brominated disinfection byproducts during chloramination of drinking water: New polar species and overall kinetics. Environ. Sci. Technol. 2014;48:2579–2588. doi: 10.1021/es4034765. [DOI] [PubMed] [Google Scholar]
- 35.Agarwal V., Moore B.S. Enzymatic synthesis of polybrominated dioxins from the marine environment. ACS Chem. Biol. 2014;9:1980–1984. doi: 10.1021/cb5004338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Romero E., Castellanos J.R.G., Gadda G., Fraaije M.W., Mattevi A. Same substrate, many reactions: Oxygen activation in flavoenzymes. Chem. Rev. 2018;114:1742–1769. doi: 10.1021/acs.chemrev.7b00650. [DOI] [PubMed] [Google Scholar]
- 37.Buss J.M., McTamney P.M., Rokita S.E. Expression of a soluble form of iodotyrosine deiodinase for active site characterization by engineering the native membrane protein from Mus musculus. Protein Sci. 2012;21:351–361. doi: 10.1002/pro.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sterner R., Liebl W. Thermophilic adaptation of proteins. Crit. Rev. Biochem. Mol. Biol. 2001;36:39–106. doi: 10.1080/20014091074174. [DOI] [PubMed] [Google Scholar]
- 39.Baker P.J. From hyperthermophile to psychrophile: The structural basis of temperature stability of the amino acids dehydrogenases. Biochem. Soc. Trans. 2004;32:264–268. doi: 10.1042/bst0320264. [DOI] [PubMed] [Google Scholar]
- 40.Petsko G.A. Structural basis of thermostability in hyperthermophilic proteins, or “there's more than one way to skin a cat”. Methods Enzymol. 2001;334:469–478. doi: 10.1016/s0076-6879(01)34486-5. [DOI] [PubMed] [Google Scholar]
- 41.Zheng L., Baumann U., Reymond J.-L. An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 2004;32:e115. doi: 10.1093/nar/gnh110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu H.-w., Naismith J.H. An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 2008;8:91. doi: 10.1186/1472-6750-8-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bryksin A.V., Matsumura I. Overlap extension PCR cloning: A simple and reliable way to create recombinant plasmids. Biotechniques. 2010;48:463–465. doi: 10.2144/000113418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koziol J. Fluorometric analyses of riboflavin and its coenzymes. Methods Enzymol. 1971;18:235–285. [Google Scholar]
- 45.Gasteiger E., Gattiker A., Hoogland C., Ivanyi I., Appel R.D., Bairoch A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003;31:3784–3788. doi: 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry land cell constants. J. Appl. Crystallogr. 1993;26:795–800. [Google Scholar]
- 47.McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Emsley P., Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 49.Adams P.D., Afonine P.V., Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.-W., Kapral G.J., Grosse-Kunstleve R.W., Mc- Coy A.J., Moriarty N.W., Oeffner R., Read R.J., Richardson D.C., et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.The PyMOL Molecular Graphics System, Version 2.0. Schrödinger, LLC; New York City, NY: 2017. [Google Scholar]
- 51.'Protein interfaces, surfaces and assemblies' service PISA at the European Bioinformatics Institute. European Bioinformatics Institute; Hinxton, UK: 2021. [Google Scholar]
- 52.Krissinel E., Henrick K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 53.Warner J.R., Copley S.D. Pre-steady-state kinetic studies of the reductive dehalogenation catalyzed by tetrachlorohydroquinone dehalogenase. Biochemistry. 2007;46:13211–13222. doi: 10.1021/bi701069n. [DOI] [PubMed] [Google Scholar]
- 54.Sievers F., Wilm A., Dineen D.G., Gibson T.J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J.D., Higgins D.G. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The structures presented in this paper have been deposited in the Protein Data Bank (PDB) with the codes 6PZ0 and 6Q1L.