

Abstract
Advancing reaction rates for electrochemical CO2 reduction in membrane electrode assemblies (MEAs) have boosted the promise of the technology while exposing new shortcomings. Among these is the maximum utilization of CO2, which is capped at 50% (CO as targeted product) due to unwanted homogeneous reactions. Using bipolar membranes in an MEA (BPMEA) has the capability of preventing parasitic CO2 losses, but their promise is dampened by poor CO2 activity and selectivity. In this work, we enable a 3-fold increase in the CO2 reduction selectivity of a BPMEA system by promoting alkali cation (K+) concentrations on the catalyst’s surface, achieving a CO Faradaic efficiency of 68%. When compared to an anion exchange membrane, the cation-infused bipolar membrane (BPM) system shows a 5-fold reduction in CO2 loss at similar current densities, while breaking the 50% CO2 utilization mark. The work provides a combined cation and BPM strategy for overcoming CO2 utilization issues in CO2 electrolyzers.
The field of electrochemical CO2 reduction (ECO2R) has advanced substantially in the past decade. Activity, selectivity, and stability have been improved due to the deployment of gas diffusion electrodes as a catalytic support in flowing catholyte cells and membrane electrode assemblies (MEAs).1−4 Despite improvements, the intrinsic homogeneous reactions that occur alongside the desired ECO2R make the process less favorable with over half of all reacted CO2 lost to carbonate instead of value-added products.5−8
The loss of CO2 occurs when the required protons for ECO2R are provided by water-splitting, which results in OH– being produced in equal proportion to the electrons transferred. In the presence of OH–, CO2 reacts chemically to form HCO3– and CO32– ions (eqs S1–S5). These reactions not only decrease utilization of the inputted CO2 but also lower system conductivity and result in salt precipitation in the CO2 gas channel in the presence of alkali cations.9,10 Unless this issue can be resolved, CO2 utilization efficiency (eq 1) will inevitably plateau at a maximum of ∼50% for CO production for neutral and alkaline media.11−13 Here, CO2 utilization efficiency is defined as the ratio of the reacted CO2 that is converted to the target product carbon monoxide to that of the total CO2 reacted in the system (CO2 → {CO, HCOO–, HCO3–, CO32–}). The CO2 utilization efficiency is considered independent of flow rate and is not to be confused with the total single-pass conversion of CO2 within the system for which the reader is referred elsewhere.8,12
| 1 |
In order to reduce CO2 consumption by OH–, a promising approach is to introduce excess H+ near the cathode’s surface. Protons can be provided either directly from an acidic catholyte or via the membrane. Both approaches allow for the neutralization of OH– and regeneration of CO2, which has already been converted to (bi)carbonates. For example, recently, Huang et al. reported ECO2R on Cu in an acid environment, which increased single-pass CO2 utilization to 77% in a GDE flow cell.14 Here, the protons required for CO2 electrolysis are still envisioned to come from water-splitting, resulting in OH– formation similar to neutral and alkaline electrolytes. However, the excess protons in the surrounding electrolyte both neutralize excess OH– and reclaim CO2 that was lost to (bi)carbonate. While high CO2 utilizations are reached in this case, the dominant reaction remains to be H2 at ∼40% Faradaic efficiency (FE) because of the excess number of protons. Importantly, the excess protons provided in this system are not linked to the current density applied, implying that different optimal input acidities and flow rates are required for different current densities. A more recent work was able to reach higher FEs of ∼90% for CO2 to CO on Au in acidic media,15 but the maximum utilizations achievable and homogeneous reactions were not discussed. It is then unclear if these demonstrated high FEs can be simultaneously achieved with high utilizations.
Alternatively to using acidic catholytes, protons can be internally generated proportionally to the applied current density through ion exchange membranes. Using a cation exchange membrane (CEM) coupled with an acidic anolyte or pure water16−18 would permit protons to be efficiently transferred to the cathode only in the amount required to offset the formed OH–. With proper interface engineering of the catalyst/electrolyte/membrane, these protons could be used to regenerate CO2 rather than undergoing direct proton reduction to H2. For instance, recent work from O’Brien et al. demonstrated a CO2 single pass conversion of 85% using pure water and an IrO2 catalyst on the anode side with a CEM for proton shuttling.18
A final approach to provide protons to the cathode is to use a bipolar membrane (BPM) operating in reversed bias, which results in water dissociation at the sandwiched cation and anion membrane interfaces.19−21 Under such operation, a proton is sent to the cathode and hydroxide, to the anode. In addition to providing a proton source to the cathode, a BPM further allows for the use of an alkaline anolyte and Ni anode at the penalty of higher membrane voltages. Previous efforts to employ BPMs in an MEA (BPMEA) configuration without a liquid catholyte, however, have been unable to limit excess H2 production, giving poor CO2 reduction selectivities and subsequently low CO2 utilizations. Researchers have attributed excess H2 to both low hydration of the membrane22 and too high concentrations of H+ at the cathode/membrane interface.23
In all of the above scenarios, however, researchers have separately determined the importance of having alkali cations present at the electrode–electrolyte interface when performing ECO2R. Unlike alkaline conditions where high ECO2R Faradaic efficiencies can be achieved over a range of cation concentrations, recent work in acidic or neutral pH cathode conditions highlights that a special consideration of cation concentrations is required to achieve high CO2 reduction selectivities.24−28 Combining these observations with previous BPMEA demonstrations that have traditionally suffered from poor CO2 reduction selectivities, we hypothesized that the low selectivity in a BPMEA system could be overcome by increasing cation concentrations at the cathode.17,18,27 Thus, if the low parasitic CO2 loss of BPM’s can be achieved simultaneously with improved CO2 reduction performance, high CO2 utilization efficiencies would be possible as a result.
In this work, we first took advantage of the traditionally undesired ion crossover in BPMs to increase the concentrations of cations at the cathode in a BPMEA configuration. The large concentration gradient of cations from the anolyte to the cathode provided a diffusion of K+ ions to the cathode’s surface, resulting in an ECO2R selectivity improvement of 3-fold as a result of increased anolyte concentrations. Then, we compared the CO2 converted to CO and CO2 lost to electrolyte in both BPMEA and an anion exchange membrane (AEM) employed MEA system (AEMEA). Results show that the CO2 lost in a BPMEA cell is around 5 times lower than that in an AEMEA cell in a high alkaline environment. As a consequence, with increased Faradaic efficiencies, the resulting CO2 utilization efficiency is 2 times higher in a BPMEA system.
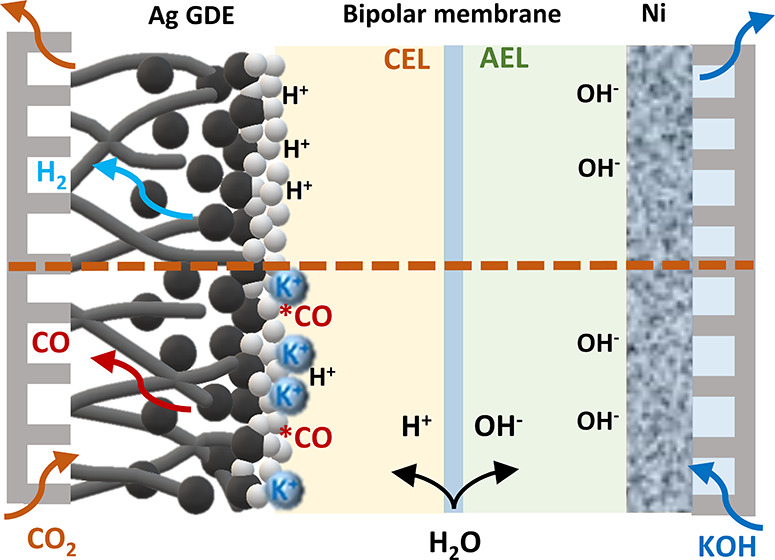
Within a BPM operating under reversed bias (Figures 1 and 2a), the current transported across the membrane is not unidirectional as is the case for a CEM or AEM but is rather bidirectional due to the production of both H+ and OH– to transport charge equivalent to the system current density. While H+/OH– transport is the desired operational effect, researchers have noted ion crossover as an important property of BPMs, and generally described, this as an unwanted effect especially at low current densities.19,20,29 Here, we sought to use concentration-dependent ion crossover as a beneficial effect to provide varying concentrations of K+ to the cathode/membrane interface of a BPMEA.
Figure 1.
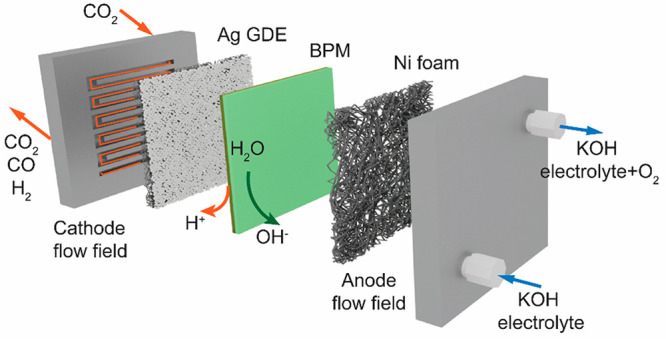
Illustration of BPM under reversed bias in a MEA cell.
Figure 2.

Illustration of the BPMEA system (a) and Faradaic efficiency and cell voltage as a function of current density in different concentrations of KOH solution in a BPMEA (b–d).
In the BPMEA configuration shown in Figure 2a, ion crossover of K+ from the anode to the cathode will occur as a result diffusion and migration, both of which are concentration dependent.30,31 In order to promote further cation flux to the cathode in a BPMEA cell for ECO2R, we varied the concentration of the KOH anolyte from 0.2 to 3 M and subsequently performed electrochemical CO2 reduction at various current densities. In the low cation concentration case (0.2 M KOH) shown in Figure 2b, CO Faradaic efficiencies remain low and decrease from 23% at 50 mA/cm2 to 16% at 200 mA/cm2. As ample CO2 is available because of the gaseous CO2 phase in close proximity to the catalyst layer, the decreasing trend in CO is due to favorable hydrogen evolution kinetics rather than limited CO2. Without the presence of a catholyte buffer, this is likely due to excess H+ flux providing a high proton concentration at elevated current densities. Upon increasing the anolyte concentration, the CO selectivity steadily rises, however, becoming on par with H2 at 200 mA/cm2 for the 3 M KOH case (Figure 2d). At even lower current densities of 50 mA/cm2, a CO selectivity of 68% is reached. The upward trend in ECO2R selectivity then tracks that of increased K+ concentrations (see Figure S1). The activity obtained in 3 M KOH here is very similar to what was reported by Lees et al. for direct 3 M KHCO3 reduction in a MEA cell equipped with a BPM.32
To investigate whether the observed selectivity changes are instead a function of increased OH– concentration or system conductivity, we utilized a 0.2 M KOH + 0.4 M K2CO3 anolyte mixture to decouple K+ and OH– effects (chronopotentiometry in Figure S2). The anolyte solution of 0.2 M KOH + 0.4 M K2CO3 then has the same pH of 13.5 as 0.2 M KOH (Table S1) but a K+ concentration of 1 M. Figure S3 shows that the CO Faradaic efficiency in this mixture solution (53%) is similar to the 1 M KOH case at 50 mA/cm2 and much higher than the 0.2 M KOH case (24%). As current densities are increased further to 200 mA/cm2, the 0.2 M KOH + 0.4 M K2CO3 case actually achieves the highest performance at a CO FE of 42%. The hypothesis for this increase of performance is that the crossover of K+ is probably higher in the mixture than in 1 M KOH. Although the K+ concentration is the same in both solutions, measurements investigating the crossover in BPMs have shown dependence on the property of ions (cations and anions) existing in the solutions, which leads to different K+ crossover rate as a result.29 The combined results in Figures 2 and S3 then show that ECO2R can be improved in a BPMEA system via increased K+ flux to the cathode instead of the higher local pH provided by higher concentrations of OH–. In all cases, the flux of potassium in the system is expected to reach a steady state between the anode and cathode compartments, which equilibrates within the first few minutes of an experiment as indicated by the stable CO selectivity after the first GC injection (Figure S4).
As shown in Figures 2 and S5, the cell voltage is also increasing as the anolyte concentration increases. To account for the large ohmic resistance of the FumaTech BPM used in these experiments (130–160 μm), we subtracted the voltage, which is induced by the ohmic resistance (Table S2). After correction, the cell voltages show similar values at the same current densities in all electrolytes (Figure S6). However, the high voltage is not the cause for a higher ECO2R performance. At the same cell voltage of around 4 V, CO partial current density is still the highest in 3 M KOH solution (Figure S7). Furthermore, ECO2R was also conducted using pure water as an anolyte in the BPMEA system. All CO Faradaic efficiencies show less than 10% (Figure S8), which is significantly lower than the performance in 0.2 M KOH solution. During the whole course of the experiment, the pH of the anolyte remained the same in the BPMEA system (Table S1). Such high stability by maintaining the pH of the electrolyte is another advantage of a BPM and indicates that CO2 crossover is relatively low compared to that of an AEM.33 The stable pH also suggests that water dissociation occurred during ECO2R, and OH– produced by BPM could supplement OH– lost during the oxygen evolution reaction (OER).
In order to compare the CO2 utilization efficiency of the BPMEA case, we also need a point of comparison for a high CO selectivity configuration. For this, we reproduced experiments for a Ag-sputtered catalyst in an MEA configuration with an AEM (Figure 3a). We performed AEM experiments over a similar range of anolyte concentrations for comparison purposes and to observe any effects from increased cation concentrations. As shown in Figure 3c,d, ECO2R activity is higher overall when using an AEM than using a BPM, which is consistent with what is reported in the literature.34 Over the range of tested current densities, CO always remains the dominant product, ranging from 70% to 90% in selectivity. No strong dependence on the anolyte concentration is observed over the 0.2 to 3 M range tested. The results show that, in neutral or alkaline media such as in an AEMEA system, high ECO2R activity still can be achieved even when cation concentrations are low. Dioxide Materials even reported high CO Faradaic efficiency using 10 mM KHCO3 as anolyte in the same AEMEA cell.34 The improved selectivity has been explained by the better kinetics of ECO2R than HER in such environment, where the proton donor in both cases is from water and cations play less of a role in selectivity as compared to acidic media.14,35
Figure 3.

Illustration of an AEMEA system (a) and Faradaic efficiency and cell voltage as a function of current density in different concentrations of KOH solution in an AEMEA (b–d).
CO Faradaic efficiency decreases as observed in the AEM case as a function of current density and KOH concentration. Upon an increase in both, the drop in CO selectivity is replaced by a higher Faradaic efficiency of HCOO–, which we observed in the anolyte stream through high-pressure liquid chromatograph (HPLC) tests.6 The higher HCOO– FE at these conditions has been observed elsewhere and linked to increases in local reaction pH.36
With both the BPMEA and AEM selectivity results acquired, we compared the overall CO2 utilization efficiency (eq 1) toward CO of the two systems, now taking into account the amount of CO2 that is lost to an unwanted (bi)carbonate reaction. Here, Figure 4a–c shows the conversion of CO2 to CO as well as the overall CO2 loss in absolute terms of flow rate, while the CO2 utilization efficiency is presented in Figure 4d–f. The results in Figure 4a–c are useful to highlight the differences between the BPMEA and AEM systems. In particular by observing the gray areas of these images, we can see that in an AEM configuration the amount of CO2 consumed by OH– and formate production increases with current density and KOH concentration. Meanwhile, the green areas for the AEM remain similar to anion concentration, collectively leading to CO2 utilization efficiencies below 40% in all cases (Figure 4d–f). Conversely, when one observes the behavior of the BPMEA system, CO2 loss does not vary significantly with varying anolyte concentrations. For all concentrations studied, the unwanted loss of CO2 is roughly 1 mL/min at 50 mA/cm2 and 3 mL/min at 200 mA/cm2. When paired with the improved Faradaic efficiencies with increasing K+ concentrations, the overall CO2 utilization efficiency increases for the BPMEA system, leading to a high of 60% in 3 M KOH (Figure 4f) with unwanted CO2 loss 4–5 times lower than for the AEM case. We note that the experimental results show that CO2 is still consumed in a BPMEA cell, albeit to a much lesser extent. This indicates that the amount of H+ flux from BPM is not large enough to neutralize all the OH– produced or regenerate all CO2 converted to (bi)carbonate during electrolysis. As for conversion of CO2 to CO (green area in Figure 4a–c), AEMEA outperforms BPMEA in all KOH concentrations due to its higher FE for ECO2R. The CO2 single pass conversion efficiency in the BPMEA cell is also calculated accordingly (Figure S9).
Figure 4.

CO2 converted to CO and lost CO2 in flow rate (a–c) and CO2 utilization efficiency (d–f) as a function of current density in both BPMEA and AEMEA systems. CO2 inflow is 50 mL/min.
Combined, the results show the simultaneous benefit of using a BPMEA with increased cation flux to the cathode. We maintain low parasitic reactions by providing a proton flux from the BPM to the cathode, while increasing Faradaic efficiency by also providing higher K+ concentrations to the cathode. The dependency can be viewed clearly in Figure 5a where the lost CO2 remains flat due to the BPM, while increased cations improve CO2 selected toward CO, even in a likely acidic reaction environment.
Figure 5.

(a) CO2 converted to CO and CO2 lost in electrolyte in all concentrations at 200 mA/cm2 in a BPMEA system. CO2 inflow is 50 mL/min. (b) Carbon balance in a BPMEA cell with lower CO2 utilization and (c) increased CO2 utilization by H+ neutralizing OH– (1) or H+ regenerating CO2 through the reaction with (bi)carbonates (2).
The utilization efficiency is the lowest in 0.2 M KOH in the BPMEA system, which can be explained by the low performance of ECO2R when cations are less available on the catalyst’s surface (CO FE < 20%). This is explained by the hydrogen evolution reaction (HER) maintaining favorable kinetics over ECO2R under cation-limited acidic conditions.15 In the absence of cations and ECO2R, the H+ from the BPM is assumed to be directly reduced to H2 on the catalyst’s surface instead of neutralizing all OH– generated from water and CO2 reduction.27,35 Thus, CO2 utilization efficiencies have been shown to be even lower than the theoretical amount of 50% as shown in Figure 5b. With an increased availability of cations in the 1 and 3 M KOH cases, ECO2R kinetics, which requires protons to come from water-splitting, are however more favored than HER. The H+ from the BPM is then hypothesized to partially neutralize the OH– and regenerate CO2 from (bi)carbonates (Figure 5c) instead of directly being reduced to H2. Thus, CO2 utilization efficiencies are improved. For a deeper understanding of the mechanisms, localized concentrations, and pathways of H+ in such a system, detailed modeling is required in the future, which accounts for both the acid and base versions of homogeneous and heterogeneous reactions.
From the presented results, a number of operational comparisons can also be made about the BPMEA and AEMEA cases independent of CO2 utilization. We would like to point out that the cell voltages needed are smaller in an AEMEA than a BPMEA system. After correcting the voltages for cell resistance (Figures S6 and S10), the voltages in the BPMEA are around 0.9–1 V higher than in the AEMEA cell. This extra voltage for the BPMEA is explained by the minimum voltage needed for the water dissociation inside a BPM, which is around 0.83 V, as well as an additional driving force at the given current density.19 Nevertheless, BPMs have the potential of reaching higher CO2 utilization as demonstrated in this work, which under a proper technoeconomic analysis could be evaluated to determine if it offsets the added required voltage. In addition, BPMs can maintain stable anolyte pH values without electrolyte replenishment. The usage of non-noble catalysts as anodes is then a possible option, which can add extra merit to a BPMEA system.
In a further assessment, there is a substantial reduction in salt precipitation for the BPMEA case, which supports the reduced consumption of CO2 by the electrolyte. A major cause for the failure of CO2 electrolyzer when using an AEM is salt precipitation and blockage of the gas flow field. That happens typically in around 1 h (Figure S11). In an AEMEA cell, salt accumulation partially blocked the gas flow field in 1 M KOH and fully blocked the gas channel in the 3 M KOH case after 80 min of operational time. However, in a BPMEA system, no salt was observed after the same duration in 1 M KOH, and there was little salt formed in 3 M KOH. A further run of the BPMEA cell with 1 M KOH anolyte at 100 mA/cm2 showed stable cell voltage, CO Faradaic efficiency, and anolyte pH for 5.5 h without any salt formation on the gas channel as shown in Figure S12. These results imply that the potassium and carbonate concentrations reach a steady-state value below the salt precipitation limit at this current density and anolyte concentration. Thus, once a certain potassium concentration is reached on the cathode side, potassium is not expected to accumulate indefinitely and will instead form a balance. Similarly, the formed carbonate in the BPMEA case will also not continuously build up and is expected to physically crossover the CEM of the BPMEA. Less salt formation also indicates that less CO2 is converted to (bi)carbonate, which is consistent with results shown in Figure 4. The morphology of the Ag catalyst after the ECO2R test in both the BPMEA and AEMEA cells did not change (Figure S13), suggesting a good stability of Ag in both cells during the CO2 reduction process.
We hypothesize that with a future modification of the BPMEA system the catalyst could further favor CO2 reduction over HER with reduced cell voltages. Under such circumstances, H+ created from the BPM could neutralize OH– (Figure 5b) or quickly react with (bi)carbonate ions (Figure 5c), instead of resulting in HER. One way of doing so is by further promoting CO2 reduction selectivity. In a recent example, Endrődi et al. mixed the inputted CO2 gas feed with alkali-cation containing solutions as a way to improve the cation concentration at the catalyst surface in a MEA cell with a pure water anolyte.17 With this treatment, ECO2R to CO reached several times higher activity than without any treatment, although it required repeat implementation. Other solutions could be coating the catalyst layer with an ionomer that has suitable cation groups in favor of ECO2R.18 Using catalysts that have better kinetics for ECO2R than Ag could also favor ECO2R over HER.37 Further developments may also allow for a reduction in cell voltages. As demonstrated by Oener et al.,38 the incorporation of catalysts within the BPM architecture can decrease the overpotential needed for water dissociation and thus minimize the overall BPMEA cell voltages for high-rate CO2 electrolysis. Further reductions are also foreseen by optimizing the full contact between the cathode, membrane, and anode to ensure that all electrochemical surfaces are fully functional. A challenge in such a system is to ensure that the proton flux from the BPM can function optimally with a potentially thicker cathode layer. With a beneficial local environment and better catalysts in a BPMEA cell, however, there is upward potential for CO2 utilization efficiency and energy efficiency with the given approach.
In this work, we reported an increased ECO2R performance in a MEA system coupled with a BPM under reversed bias. This was achieved by allowing a higher cation concentration to be transported to the catalyst surface. Our results showed that CO Faradaic efficiency improved from less than 20%, as reported in the literature, by 3-fold to 68%. With the current-dependent H+ produced from the BPM, lost CO2 was also reduced by 5-fold in BPMEA cell. Thus, a CO2 utilization efficiency was achieved, which was 2 times higher than in an AEMEA cell. Furthermore, the BPMEA cell also showed better stability than AEMEA by maintaining a stable pH of the anolyte and preventing rapid salt precipitation at the cathode. With further advancement in the commercial BPM, we anticipate the BPMEA could be a promising option for higher CO2 utilization efficiencies. In addition, this work addresses the importance of cation embedment while using a MEA configuration, which leads to an advanced design for next generation CO2 electrolyzers.
Acknowledgments
K.Y. acknowledges funding from China Scholarship Council (CSC) for this work. T.B. would like to acknowledge the European Union’s Horizon 2020 research and innovation program under grant agreement No. 85144 (SELECT-CO2) and the NWO for an individual Veni grant.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsenergylett.1c02058.
Description of the experimental details regarding the setup and parameters, calculation of CO2 conversion and consumption during electrochemical CO2 reduction, further detailed measurements of the performance in the used electrolyzers, image of CO2 gas channel after the reaction, and SEM image of Ag/GDE before and after the reaction (PDF)
Author Contributions
K.Y. conceived the project and completed all of the electrochemical experiments. M.L. and S.S. helped with the experimental setup and scientific discussion. M.A.B and W.A.S. helped with the scientific discussion. K.Y. and T.B wrote the manuscript with editing contributions from all coauthors.
The authors declare no competing financial interest.
Supplementary Material
References
- Dinh C.-T.; Burdyny T.; Kibria M. G.; Seifitokaldani A.; Gabardo C. M.; De Arquer F. P. G.; Kiani A.; Edwards J. P.; De Luna P.; Bushuyev O. S. CO2 electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interface. Science 2018, 360 (6390), 783–787. 10.1126/science.aas9100. [DOI] [PubMed] [Google Scholar]
- Burdyny T.; Smith W. A. CO 2 reduction on gas-diffusion electrodes and why catalytic performance must be assessed at commercially-relevant conditions. Energy Environ. Sci. 2019, 12 (5), 1442–1453. 10.1039/C8EE03134G. [DOI] [Google Scholar]
- Higgins D.; Hahn C.; Xiang C.; Jaramillo T. F.; Weber A. Z. Gas-diffusion electrodes for carbon dioxide reduction: A new paradigm. ACS Energy Letters 2019, 4 (1), 317–324. 10.1021/acsenergylett.8b02035. [DOI] [Google Scholar]
- Endrődi B.; Kecsenovity E.; Samu A.; Halmágyi T.; Rojas-Carbonell S.; Wang L.; Yan Y.; Janáky C. High carbonate ion conductance of a robust PiperION membrane allows industrial current density and conversion in a zero-gap carbon dioxide electrolyzer cell. Energy Environ. Sci. 2020, 13 (11), 4098–4105. 10.1039/D0EE02589E. [DOI] [Google Scholar]
- Rabinowitz J. A.; Kanan M. W. The future of low-temperature carbon dioxide electrolysis depends on solving one basic problem. Nat. Commun. 2020, 11 (1), 5231. 10.1038/s41467-020-19135-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrazábal G. O.; Strøm-Hansen P.; Heli J. P.; Zeiter K.; Therkildsen K. T.; Chorkendorff I.; Seger B. Analysis of mass flows and membrane cross-over in CO2 reduction at high current densities in an MEA-type electrolyzer. ACS Appl. Mater. Interfaces 2019, 11 (44), 41281–41288. 10.1021/acsami.9b13081. [DOI] [PubMed] [Google Scholar]
- Ma M.; Clark E. L.; Therkildsen K. T.; Dalsgaard S.; Chorkendorff I.; Seger B. Insights into the carbon balance for CO 2 electroreduction on Cu using gas diffusion electrode reactor designs. Energy Environ. Sci. 2020, 13 (3), 977–985. 10.1039/D0EE00047G. [DOI] [Google Scholar]
- Dinh C.-T.; Li Y. C.; Sargent E. H. Boosting the single-pass conversion for renewable chemical electrosynthesis. Joule 2019, 3 (1), 13–15. 10.1016/j.joule.2018.10.021. [DOI] [Google Scholar]
- Endrodi B.; Kecsenovity E.; Samu A.; Darvas F.; Jones R.; Török V.; Danyi A.; Janáky C. Multilayer electrolyzer stack converts carbon dioxide to gas products at high pressure with high efficiency. ACS energy letters 2019, 4 (7), 1770–1777. 10.1021/acsenergylett.9b01142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripatti D. S.; Veltman T. R.; Kanan M. W. Carbon monoxide gas diffusion electrolysis that produces concentrated C2 products with high single-pass conversion. Joule 2019, 3 (1), 240–256. 10.1016/j.joule.2018.10.007. [DOI] [Google Scholar]
- Weng L.-C.; Bell A. T.; Weber A. Z. Towards membrane-electrode assembly systems for CO 2 reduction: a modeling study. Energy Environ. Sci. 2019, 12 (6), 1950–1968. 10.1039/C9EE00909D. [DOI] [Google Scholar]
- Jeng E.; Jiao F. Investigation of CO 2 single-pass conversion in a flow electrolyzer. Reaction Chemistry Engineering 2020, 5 (9), 1768–1775. 10.1039/D0RE00261E. [DOI] [Google Scholar]
- Kas R.; Star A. G.; Yang K.; Van Cleve T.; Neyerlin K. C.; Smith W. A. Along the Channel Gradients Impact on the Spatioactivity of Gas Diffusion Electrodes at High Conversions during CO2 Electroreduction. ACS Sustainable Chem. Eng. 2021, 9 (3), 1286–1296. 10.1021/acssuschemeng.0c07694. [DOI] [Google Scholar]
- Huang J. E.; Li F.; Ozden A.; Rasouli A. S.; de Arquer F. P. G.; Liu S.; Zhang S.; Luo M.; Wang X.; Lum Y. CO2 electrolysis to multicarbon products in strong acid. Science 2021, 372 (6546), 1074–1078. 10.1126/science.abg6582. [DOI] [PubMed] [Google Scholar]
- Monteiro M. C. O.; Philips M. F.; Schouten K. J. P.; Koper M. T. M. Efficiency and selectivity of CO2 reduction to CO on gold gas diffusion electrodes in acidic media. Nat. Commun. 2021, 12, 4943. 10.1038/s41467-021-24936-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Z.; Peng H.; Wei X.; Zhou H.; Gong J.; Huai M.; Xiao L.; Wang G.; Lu J.; Zhuang L. An alkaline polymer electrolyte CO 2 electrolyzer operated with pure water. Energy Environ. Sci. 2019, 12 (8), 2455–2462. 10.1039/C9EE01204D. [DOI] [Google Scholar]
- Endrődi B.; Samu A.; Kecsenovity E.; Halmágyi T.; Sebők D.; Janáky C. Operando cathode activation with alkali metal cations for high current density operation of water-fed zero-gap carbon dioxide electrolysers. Nature Energy 2021, 6 (4), 439–448. 10.1038/s41560-021-00813-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien C. P.; Miao R. K.; Liu S.; Xu Y.; Lee G.; Robb A.; Huang J. E.; Xie K.; Bertens K.; Gabardo C. M. Single Pass CO2 Conversion Exceeding 85% in the Electrosynthesis of Multicarbon Products via Local CO2 Regeneration. ACS Energy Letters 2021, 6, 2952–2959. 10.1021/acsenergylett.1c01122. [DOI] [Google Scholar]
- Vermaas D. A.; Wiegman S.; Nagaki T.; Smith W. A. Ion transport mechanisms in bipolar membranes for (photo) electrochemical water splitting. Sustainable Energy Fuels 2018, 2 (9), 2006–2015. 10.1039/C8SE00118A. [DOI] [Google Scholar]
- Bui J. C.; Digdaya I.; Xiang C.; Bell A. T.; Weber A. Z. Understanding Multi-Ion Transport Mechanisms in Bipolar Membranes. ACS Appl. Mater. Interfaces 2020, 12 (47), 52509–52526. 10.1021/acsami.0c12686. [DOI] [PubMed] [Google Scholar]
- Blommaert M. A.; Aili D.; Tufa R. A.; Li Q.; Smith W. A.; Vermaas D. A. Insights and Challenges for Applying Bipolar Membranes in Advanced Electrochemical Energy Systems. ACS Energy Letters 2021, 6, 2539–2548. 10.1021/acsenergylett.1c00618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvatore D. A.; Weekes D. M.; He J.; Dettelbach K. E.; Li Y. C.; Mallouk T. E.; Berlinguette C. P. Electrolysis of Gaseous CO2 to CO in a Flow Cell with a Bipolar Membrane. ACS Energy Letters 2018, 3 (1), 149–154. 10.1021/acsenergylett.7b01017. [DOI] [Google Scholar]
- Chen Y.; Vise A.; Klein W. E.; Cetinbas F. C.; Myers D. J.; Smith W. A.; Deutsch T. G.; Neyerlin K. C. A robust, scalable platform for the electrochemical conversion of CO2 to formate: identifying pathways to higher energy efficiencies. ACS Energy Letters 2020, 5 (6), 1825–1833. 10.1021/acsenergylett.0c00860. [DOI] [Google Scholar]
- Resasco J.; Chen L. D.; Clark E.; Tsai C.; Hahn C.; Jaramillo T. F.; Chan K.; Bell A. T. Promoter effects of alkali metal cations on the electrochemical reduction of carbon dioxide. J. Am. Chem. Soc. 2017, 139 (32), 11277–11287. 10.1021/jacs.7b06765. [DOI] [PubMed] [Google Scholar]
- Ringe S.; Clark E. L.; Resasco J.; Walton A.; Seger B.; Bell A. T.; Chan K. Understanding cation effects in electrochemical CO2 reduction. Energy Environ. Sci. 2019, 12 (10), 3001–3014. 10.1039/C9EE01341E. [DOI] [Google Scholar]
- Fink A. G.; Lees E. W.; Zhang Z.; Ren S.; Delima R. S.; Berlinguette C. Impact of alkali cation identity on the conversion of HCO3–to CO in bicarbonate electrolyzers. ChemElectroChem 2021, 8, 2094. 10.1002/celc.202100408. [DOI] [Google Scholar]
- Monteiro M. C.; Dattila F.; Hagedoorn B.; García-Muelas R.; López N.; Koper M. T. Absence of CO2 electroreduction on copper, gold and silver electrodes without metal cations in solution. Nature Catalysis 2021, 4, 654–662. 10.1038/s41929-021-00655-5. [DOI] [Google Scholar]
- Chen L. D.; Urushihara M.; Chan K.; Nørskov J. K. Electric field effects in electrochemical CO2 reduction. ACS Catal. 2016, 6 (10), 7133–7139. 10.1021/acscatal.6b02299. [DOI] [Google Scholar]
- Blommaert M. A.; Verdonk J. A.; Blommaert H. C.; Smith W. A.; Vermaas D. A. Reduced ion crossover in bipolar membrane electrolysis via increased current density, molecular size, and valence. ACS Applied Energy Materials 2020, 3 (6), 5804–5812. 10.1021/acsaem.0c00687. [DOI] [Google Scholar]
- Tang L. Concentration dependence of diffusion and migration of chloride ions: Part 1. Theoretical considerations. Cem. Concr. Res. 1999, 29 (9), 1463–1468. 10.1016/S0008-8846(99)00121-0. [DOI] [Google Scholar]
- Tang L. Concentration dependence of diffusion and migration of chloride ions: Part 2. Experimental evaluations. Cem. Concr. Res. 1999, 29 (9), 1469–1474. 10.1016/S0008-8846(99)00120-9. [DOI] [Google Scholar]
- Lees E. W.; Goldman M.; Fink A. G.; Dvorak D. J.; Salvatore D. A.; Zhang Z.; Loo N. W.; Berlinguette C. P. Electrodes designed for converting bicarbonate into CO. ACS Energy Letters 2020, 5 (7), 2165–2173. 10.1021/acsenergylett.0c00898. [DOI] [Google Scholar]
- Ma M.; Kim S.; Chorkendorff I.; Seger B. Role of ion-selective membranes in the carbon balance for CO 2 electroreduction via gas diffusion electrode reactor designs. Chemical science 2020, 11 (33), 8854–8861. 10.1039/D0SC03047C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z.; Yang H.; Kutz R.; Masel R. I. CO2 electrolysis to CO and O2 at high selectivity, stability and efficiency using sustainion membranes. J. Electrochem. Soc. 2018, 165 (15), J3371. 10.1149/2.0501815jes. [DOI] [Google Scholar]
- Bondue C. J.; Graf M.; Goyal A.; Koper M. T. Suppression of hydrogen evolution in acidic electrolytes by electrochemical CO2 reduction. J. Am. Chem. Soc. 2021, 143 (1), 279–285. 10.1021/jacs.0c10397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifitokaldani A.; Gabardo C. M.; Burdyny T.; Dinh C.-T.; Edwards J. P.; Kibria M. G.; Bushuyev O. S.; Kelley S. O.; Sinton D.; Sargent E. H. Hydronium-induced switching between CO2 electroreduction pathways. J. Am. Chem. Soc. 2018, 140 (11), 3833–3837. 10.1021/jacs.7b13542. [DOI] [PubMed] [Google Scholar]
- Yang K.; Kas R.; Smith W. A.; Burdyny T. Role of the carbon-based gas diffusion layer on flooding in a gas diffusion electrode cell for electrochemical CO2 reduction. ACS Energy Letters 2021, 6 (1), 33–40. 10.1021/acsenergylett.0c02184. [DOI] [Google Scholar]
- Oener S. Z.; Foster M. J.; Boettcher S. W. Accelerating water dissociation in bipolar membranes and for electrocatalysis. Science 2020, 369 (6507), 1099–1103. 10.1126/science.aaz1487. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.