

Abstract
Chimeric antigen receptor (CAR) T cell therapy is a relatively new form of immunotherapy that has had success in treating patients with hematologic malignancies, leading to three recent United States Food and Drug Administration approvals. However, several challenges hinder the widespread use of CAR-T therapy. Here, we review the application of functional nucleic acids such as aptamers and ribozymes as novel tools to improve a variety of steps in CAR-T cell therapy development. We critically examine key studies that highlight the benefits of functional nucleic acids at different stages of cell-based therapy and discuss the feasibility of their practical clinical application. Finally, we offer insights into potential opportunities where chemists can significantly contribute to the innovative incorporation of functional nucleic acids to overcome challenges associated with this cutting-edge immunotherapy.
Keywords: functional nucleic acids, CAR-T, aptamer, ribozyme, T cells, biosensing
1. Successes and Challenges of CAR-T Cell Therapy
Chimeric antigen receptor (CAR)-T cell therapy has emerged as a powerful clinical strategy in the field of cancer immunotherapy over the past decade. It is a form of adoptive cell therapy which involves reprogramming of patients’ immune cells ex vivo to specifically target and kill cancer cells in vivo. CAR-T cells first reported in 1992, now referred to as first generation CAR-Ts,1 were composed of four domains (Figure 1A).2 The antigen recognition domain is a single chain variable fragment (scFv) derived from the variable region of monoclonal antibodies. Different scFvs can be selected for their binding ability to target specific antigens. The hinge domain is commonly an IgG-based spacer that links the scFv with the rest of CAR construct, thus improving antigen binding by reducing spatial constraints between the CAR and target antigen. Consequently, the transmembrane domain, usually obtained from glycoproteins CD4, CD8α, or CD28, bridges and transduces ligand recognition signal from the ectodomain to the endodomain. Lastly, the intracellular domain initiates a signaling cascade in the T cell via a CD3ζ chain containing immuno-tyrosine activation motifs (ITAMs). An antigen-binding signal initiates the intracellular cascade of phosphorylation reactions by lymphocyte-specific protein tyrosine kinases, leading to various intracellular effector functions, such as the release of cytokines and destruction of tumor cells.3
Figure 1.

CAR-T generations and their modifications. (A) First-generation CARs consisted of the scFv antigen recognition domain and the CD3ζ intracellular signaling domain. (B) Second generation CARs added intracellular costimulatory domains CD28 or 4-1BB to the already present scFv and CD3 ζ chain. (C) Third generation CARs combined two costimulatory domains such as 4-1BB and CD28 with the CD3ζ domain. (D) Fourth generation CARs added costimulatory transgenes or cytokine release to second generation CARs. Created with Biorender.com.
First-generation CAR-T therapy failed to show clinical success due to poor activation and persistence in patients.4 The original CAR design used only one CD3ζ signaling motif to provide the activation signal and lacked costimulatory signals to sustain the signaling cascade, prevent anergy and promote proliferation.5 Therefore, a second-generation CAR-T with one additional intracellular costimulatory domain (i.e., CD28, 4-1BB) was developed (Figure 1B). The second-generation CAR-T showed greater activation and cancer clearance in clinical trials for hematological cancers, leading to the first breakthrough FDA approvals of Kymriah (tisagenlecleucel) and Yescarta (axicabtagene ciloleucel).6 Although addition of a costimulatory domain in the second-generation CAR-T greatly improved antitumor efficacy, it was unable to sufficiently improve persistence of the CAR-T cells. In fact, the commonly used costimulatory endodomains CD28 and 4-1BB cause T cell exhaustion at different rates by decreasing inner tonic signaling.7 Interestingly, CD28 caused faster expansion of CAR-T cells, while 4-1BB showed relatively slow but higher expansion and longer persistence.8 Therefore, third generation CAR-T cells were developed by combining these complementary costimulatory domains, resulting in varying success in clinical trials.9,10 A fourth generation, focusing on targeting solid tumors, is also being developed by including inducible transgenic immune modifiers and cytokines.11 These modifications are expected to better shape the tumor microenvironment and thus impact antitumor effects of CAR-T cells in solid tumors. Development of future generation CAR-Ts, particularly by incorporating new technology such as functional nucleic acids, may further improve efficacy of CAR-Ts.
CAR-T cells are “living” drugs which theoretically could provide long-term surveillance and protection against target cancer cells as well as combat relapse due to residual cancer cells or metastasis. Interestingly, early phase clinical trials indicate that peripherally infused CAR-T cells are also able to cross the blood–brain barrier and target brain cancers and metastases that are usually difficult to target noninvasively.12,13 Despite these breakthroughs, many factors hamper the widespread use of CAR-T therapy. A major challenge results from the severe toxicities, including cytokine release syndrome (CRS), neurotoxicity, and on-target off-tumor toxicity that is commonly caused by CAR-T cell therapy.5 Notably, CRS, which is caused by rapid and high-volume release of cytokines, including tumor necrotic factors (TNF), interferons, and interleukin (IL)-2, IL-6, IL-8, and IL-10,14 can lead to detrimental organ dysfunction and neurotoxicity.15 Additionally, CAR-T therapy requires suitable surface level targets that are universally expressed in the tumor cells, but are not expressed on healthy cells. Identifying target antigens that meet both criteria is challenging and often leads to development of CARs that have potential to damage healthy cells through on-target off-tumor effects.16 CAR-T cell therapy has also performed poorly in treating solid tumors due to the complex immunosuppressive tumor microenvironment and immense heterogeneity of the tumor cells.17 Together, these limitations highlight the need for new innovative strategies to tackle the challenges and wield the full benefits of CAR-T cell therapy.
2. Application of Functional Nucleic Acids in CAR-T Cell Production and Therapy
Nucleic acid technology is currently experiencing an explosion of successes for many therapies18,19 and may offer improvements for immunotherapeutic strategies including CAR-T cell therapy. Indeed, nucleic acids can function beyond the conversion and storage of genetic material. Importantly, both DNA and RNA can be employed as “enzymes” and for ligand binding through the development of DNAzymes/ribozymes and aptamers, respectively.20 Since the first discovery of catalytic RNA in nature,21,22 many ribozymes have been discovered, engineered, studied, and applied to a multitude of applications ranging from biosensing to gene editing and control.23,24
In contrast to ribozymes, aptamers were first developed synthetically before their discovery in nature.25 Like antibodies, which have been the industry standard for decades, aptamers bind to targets ranging from simple inorganic ions to cells with high selectivity and nanomolar affinities (Table 1).26 However, aptamers offer several advantages over antibodies. In particular, aptamers are produced in vitro through a controlled process called Systematic Evolution of Ligands by Exponential Enrichment (SELEX),27,28 which enables their selection against a wide range of targets under nonphysiological conditions.29 Furthermore, their small size results in improved tissue distributation compared to antibodies.30 Moreover, antibodies often trigger an immune response, leading to deleterious side effects, while aptamers are inherently nonimmunogenic. Finally, the solid-phase synthesis of aptamers allows for easy and direct modification with fluorescent tags.29 As such, aptamers are emerging as cutting-edge tools and therapies.31 Several aptamers that bind to immunomodulatory-relevant targets (Table 1) have been described.
Table 1. Examples of Aptamers That Bind to Immunomodulatory-Relevant Targets, Nucleic Acid Type, Binding Affinity (Kd), and References.
| Target | Aptamer Type | Kd (nM)a | Reference |
|---|---|---|---|
| 4-1BB costimulatory receptor | RNA | 40 | (40) |
| OX40 cell surface receptor | RNA | 8 | (41) |
| cytotoxic T cell antigen-4 | RNA | 10 | (42) |
| programmed cell death-1 | DNA | 112 | (43) |
| tumor necrosis factor-α | DNA | 7 | (44) |
| interleukin-2 receptor-α | DNA | 13.4 | (45) |
| interleukin-6 receptor | DNA | 0.2 | (46) |
| interleukin-8 | RNA | 0.00172 | (47) |
| interleukin-10 receptor | RNA | 12 | (48) |
| CD8+ T cell | DNA | 18.3 | (32) |
| CD8+ T cell | DNA | 1.9 | (32) |
| CD8+ T cell | DNA | 2.4 | (32) |
Only targets with reported Kd values are indicated
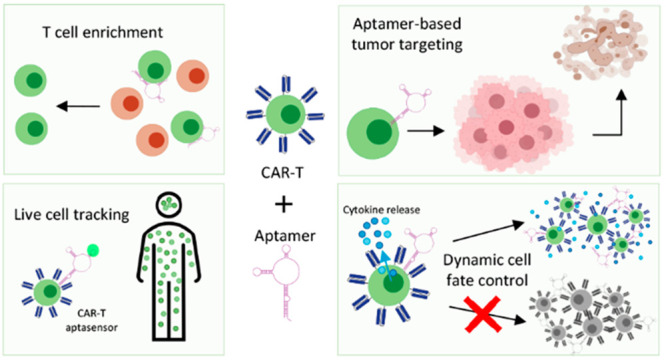
Here, we discuss how functional nucleic acids can be used in CAR-T therapy to improve T cell enrichment in CAR-T production,32,33 reduce and manage the possible toxicities associated with CAR construct,34 improve cell proliferation capacity,35,36 and enable live in vivo cell tracking.37−39
2.1. Aptamers for Cell Isolation
The first step in CAR-T cell production is collecting cytotoxic T cells from the patient. Peripheral blood mononuclear cells (PBMCs) are harvested by leukapheresis, a centrifugation-based method for separating the cellular components from the soluble components of cell suspensions.49,50 Subsequently, red blood cells and platelet contaminants are removed from the cellular component, and lymphocytes are isolated using size and density-based cell fractionation.51 The purity and yield of the starting T cell isolate is crucial for the quality of the final CAR-T product. Defined composition of CD4+:CD8+ CAR-T cells confer more potent and superior antitumor reactivity in vivo.52 Therefore, there is a need for highly specific cell isolation methods. However, pure T cell yields vary significantly based on the patient and the type of disease;49 thus, collection of sufficient T cell yields for CAR-T cell manufacturing is often difficult for patients with advanced malignancies and an extensive treatment history. Furthermore, it can be challenging to ensure the quality and specificity of the final T cell isolate with the commonly used cell separation methods.53
Immunomagnetic cell separation, combining positive and negative selection, is one of the best methods to achieve specific T cell isolation. In the positive selection, cells are labeled with antibodies that target specific cytotoxic T cell surface proteins (e.g., CD4+, CD8+, or CD62L).54 The antibodies are linked to magnetic particles, allowing the labeled cells to be retained in the final isolation fraction after incubation of the sample in a magnetic field.55 In contrast, the negative selection involves labeling unwanted cell types with immunomagnetic particles and depleting them by applying a magnetic field. While combining positive and negative selection improves specific isolation of T cells, the nearly irreversible antibody labeling compromises the function of the T cells and thus reduces CAR-T cell therapy efficacy.33 Therefore, a highly promising opportunity is to replace antibodies with aptamers. These nucleic acid alternatives can be used as selective cell labels since they share the highly selective nature of antibodies and yet are functionally reversible.
Several aptamers have been selected that reversibly bind to target cells, allowing for removal following the aptamer-based cell isolation. One report used RNA aptamers targeting the epidermal growth factor receptor (EGFR) to purify EGFR+ cells.33 As a proof-of-concept, EGFR+ cells were incubated either with fluorescently tagged aptamers and subjected to fluorescence-activated cell sorting (FACS) or with aptamer-conjugated magnetic beads and subjected to magnetic-activated cell sorting (MACS). Both strategies enabled successful isolation of EGFR+ cells from a cell mixture consisting of EGFR+ and EGFR- cells. To reversibly remove the aptamer, a 15-nucleotide DNA sequence complementary to the RNA aptamer, called an “antidote”, was used to disrupt aptamer binding. This strategy resulted in the aptamer-free capture of 15% of EGFR+ cells out of the mixture. Moreover, the aptamer–antidote system was able to specifically isolate and release 33% of the EGFR+ cells from human blood spiked with EGFR+ cells. Interestingly, the recovery rate was dependent upon the ratio of EGFR+ cells and leukocytes, suggesting that tuning the aptamer:antidote ratio could be potentially optimized to increase both specificity and yield. In all cases, addition of antidote to cell–aptamer–magnetic bead complexes generated 95–99% pure population of aptamer-free EGFR+ cells. Critically, the EGFR signaling was restored to a native state after RNA aptamer removal, confirming the utility of aptamers for cell enrichment, while ensuring the targets remain functional.
Despite many successes developing cell-binding-aptamers capable of isolating various cell types,56 little research has been done to specifically isolate T cells for immunotherapy. One exception is the report by Kacherovsky et al.32 describing the exploration of aptamer-based cell sorting in T cell isolation and CAR-T cell therapy by using a similar approach as Gray et al.33 Specifically, magnetic bead conjugated aptamers that bind to CD8 on T cells were developed. These DNA aptamers were used to positively purify CD8+ T cells out of a complex cell mixture of PBMCs (Figure 2). Subsequent incubation with the complementary DNA “antidote” permitted label-free elution. Remarkably, greater than 70% CD8+ T cell was released from the aptamers with approximately 95% purity of CD8+ T cells, confirming the effectiveness of aptamer-based isolation. One drawback was that the function of the CD8 receptor after aptamer removal was not assessed to verify the restoration of receptor signaling to its native state. However, CAR-T cells were generated with the isolated T cells and compared head-to-head with commonly used antibody-isolated T cells. Fortunately, aptamer-enriched CAR-T cells displayed similar outgrowth, phenotype, gene expression, and effector function as the antibody-enriched CAR-T cells.
Figure 2.

Specific isolation of CD8+ T cells from a heterogeneous mixture using aptamers bound to magnetic beads. CD8+ T cell-specific aptamers are shown being bound to magnetic beads. The aptamers bind specifically to CD8+ T cells in a mixture containing both CD4+ and CD8+ T cells. The bound cells are then magnetically isolated from the unbound cells and released, allowing for specific isolation of CD8+ T cells. Created with Biorender.com.
To further refine isolation of specific T cells, additional aptamers could be selected and employed. For example, more rigorous rounds of positive and negative selection of cells, similar to the immunomagnetic cell separation protocol described previously, could result in improved T cell samples for therapy. In this case, aptamers for the targets of interest such as CD4+ and CD3+ T cells must be employed (see, for example, refs (57 and 58)), as well as for targets on cells that need to be removed from the apheresis cell suspension, including B lymphocytes and natural killer cells. Many aptamers have been described for these targets and additional aptamers can be selected using protein-based SELEX, cell-SELEX,30 and perhaps with in vivo SELEX.59 In one early example, Mayer et al.60 used cell-SELEX to identify aptamers that bind to CD19+ Burkitt’s lymphoma cells. These isolated aptamers distinguished between Burkitt’s lymphoma B-cells and primary B-cells, highlighting the selectivity of cell-binding aptamers. There are also dozens of cell-SELEX reports describing the selection of new aptamers to specific cell types, often displaying binding affinity as low as the picomolar range,61 highlighting an important opportunity to select novel aptamers of relevance for CAR-T therapy.
2.2. Tumor Targeting with Aptamers as an Alternative Therapy
The second stage in CAR-T cell production is the genetic modification of isolated T cells to express CARs against antigens present on target cancer cells. Currently, CAR gene delivery is mainly achieved using viral transduction with retro- or lentiviruses.62 Viral-mediated gene insertion into the host cell genome permits stable expression of CAR gene, but has high potential for carcinogenicity and is more likely to induce an immune response.63 Comparatively, the alternative CRISPR-Cas 9 system may damage certain genes, such as the tumor suppressor gene p53.64 Therefore, there is a need for tools that can reprogram T cells to augment their antitumor efficacy, while minimizing adverse side effects.
Rather than generating virally transduced CAR-T cells, Liu et al.34 demonstrated that aptamers can be used for T cell targeting of tumor cells and thus eliminate the threat of carcinogenicity associated with virally inserted CARs (Figure 3). As proof-of-principle, DNA aptamers specific to gastric cancer cells SGC-7901 and colon cancer cells CT26,65−67 were conjugated to CD3+ T cells (aptamer-CD3+ T cells) using metabolic cell labeling and click chemistry. The resulting aptamer conjugated cells bound to SGC-7901 gastric cancer cells, as well as CT26 colon carcinoma cells with high specificity in vitro and in vivo. Importantly, adoptive transfer of the aptamer-CD3+ T cells into mouse tumor models led to significant regression in tumor volume. The aptamer-CD3+ T cells were enriched at the tumor microenvironment and produced strong cytotoxicity with enhanced biomarkers of cell death. Interestingly, the aptamer-CD3+ T cells displayed antitumor effects, which were stronger than an anti-PD1 immune-checkpoint monoclonal antibody (mAb) treatment in mice. Furthermore, when the aptamer-CD3+T cells were combined with an anti-PD1 mAb, synergistic antitumor effects were observed, highlighting the usefulness of a combination therapy. Therefore, there is a strong potential for adoptive aptamer-based T cell strategy as a feasible and efficacious approach for tumor-targeted immunotherapy.
Figure 3.

Carcinogenicity risk with regular CAR-T cell manufacturing using lentiviral vectors compared to aptamers. The CAR viral vector inserts itself in the genome of the T cell. This results in a possible proto-oncogene insertion, causing uncontrollable cell division. The aptamer is inserted into the membrane of the T cell and therefore does not impact the cell genome. Created with Biorender.com.
In addition to eliminating carcinogenicity potential upon transfection,68 aptamer-T cell immunotherapy has several advantages over virally transfected CAR-T cells. There is a reduced potential for a cytokine storm response since aptamers are less immunogenic than the antibodies employed in the antigen binding region.30,34,69 Moreover, if CRS does occur, aptamers can be rapidly inactivated with complementary aptamer antidotes.70 However, it is important to note that CAR-T cells also have several advantages that would make replacing CARs with aptamer-T cells difficult. For example, CAR-Ts have long half-lives, allowing for less frequent treatments71,72 whereas DNA and RNA aptamers are easily degraded by nucleases and are rapidly eliminated in vivo.73 To address this challenge, the use of alternate backbone chemistries74 and aptamer cyclization are showing improved stabilities in vivo.75 Notably, a recent study made use of stable cyclized aptamers in vivo to perform a “recognition-then-activation” aptamer system for T cell immunotherapy, to then treat cancer cells more specifically.76 Nonetheless, aptamers inserted on the surface of T cells, rather than engineered to express on the surface, are not replicated with each cell division and would be diluted over time, reducing treatment efficacy. Therefore, future prospects of aptamer-based T cell therapy requires further investigation.
2.3. Live CAR-T Cell Biosensing
To date, there are no standardized methods to monitor in vivo behaviors and targeting efficacy of injected CAR-T cells. There is a need for new tools that can interrogate the pharmacokinetic and pharmacodynamic properties of CAR-T cells in patients. This enabling technology would help address several ongoing issues including the poor proliferation and persistence that leads to relapse in 40–60% of patients77 and the challenges in activation/targeting efficiency.15 One approach is to use radiolabeling or protein-based reporter genes coupled with imaging technologies, such as Positron Emission Tomography (PET), to obtain real-time distribution and in vivo persistence information of CAR-T cells.78 Although these protein or radiolabeling-based biosensors have greatly assisted researchers to visualize the biodistribution of cells, few can reveal current interactions, bound states, and function of cells with high spatiotemporal resolution.
Cell tracking strategies should efficiently label cells of interest, persist within the cells with minimal transfer to bystanders, and provide a detectable change in signal to reflect changes in cell status and function. Given that CAR-T cells are present in the patient’s blood, a complex heterogeneous environment, the highly specific binding enabled by the development of chemically modified aptamers and rigorous selection methods79 is expected to be beneficial for CAR-T monitoring in vivo. Furthermore, aptamers can easily be released from their target using a competitive antidote ligand if the tracking interferes with treatment. Finally, aptamers can easily be conjugated with fluorophores or other conjugates to produce readable signals via imaging methods including fluorescence optical imaging, magnetic resonance imaging and PET. Below, we compare three approaches in which aptamers could be applied for live-cell tracking of CAR-T cells. Aptamers developed to date (Table 1) as well as novel selection of aptamers could be applied for these purposes.
2.3.1. Biosensing CAR-T Cell Surface Interactions within the Cellular Niche Environment
The CAR construct needs to interact with many different cells before it finds its cell surface target to achieve activation, persistence, and long-term memory. Aptamers can be used to both sense and modulate cell surface interactions80,81 and thus provide a unique opportunity to study and probe CAR-T interactions. For example, You et al.37 developed DNA aptamer-based biosensors for four unknown proteins on the surface of Ramos cells. These aptamer biosensors were able to transduce transient membrane encounters into readable fluorescence signals, enabling the measurement of rate and interaction preference of targets to receptors during live cell membrane signaling events. While this particular example was applied to interrogate proteins coexpressed on a Ramos cell surface, a cell model for Burkitt’s lymphoma, applying this same tracking system to CAR-T cell surface domains may help uncover the underlying structure of CAR-T cell signaling networks from the point of administration and throughout the duration of therapy, including long-term memory development. Indeed, methods such as Ligand Guided Selection82 have been described that will enable new aptamer selection to relevant cell surface domains.
2.3.2. Biosensing CAR-T Activation and Cytokine Release Profiles
Live-cell aptamer-based biosensors may also be valuable to measure the T cell signaling profile. Specifically, insight into immune cell behavior, such as cytokine secretion, would help define cell-to-cell variation and provide a better understanding of how CAR-T uniquely induces CRS within the bulk population of highly heterogeneous native immune cells.83 Although antibody-based flow-cytometry assays have been applied to obtain single-cell cytokine measurements,38 these are end-point assays and cannot monitor cytokine production with high spatiotemporal resolution. As such, there is a need for real-time tracking tools to elucidate the complex signaling by CAR-T cells. One recent approach described by Qiu et al.38 uses a membrane-anchored DNA aptamer sensor to probe the single cell secretion of type II interferon (IFN-gamma), a cytokine that is crucial for both innate and adaptive immunity. In this study, an IFN-gamma specific aptamer was labeled with a fluorophore and a quencher and anchored to the T cell phospholipid layer using a cholesterol tail. Upon IFN-gamma binding, the aptamer structure was altered, spatially separating the fluorophore from the quencher, resulting in a strong, concentration-dependent fluorescent signal. The binding of the aptamer to interferon was then assessed by adding IFN-gamma and inactivated T cells to a buffer and observing fluorescence. Finally, single aptamer-decorated T cells were encapsulated in microfluidic droplets to allow direct measurement of IFN-gamma concentrations in the extracellular microenvironment at the single cell level. The effectiveness in a more complex environment such as the case with CAR-T cells circulating the human cardiovascular system must be explored. Nonetheless, future application of this aptamer biosensor strategy to detect signaling molecules from CAR-T cells is exciting, potentially incorporating aptamers that bind to IL-2 and IL-6 in addition to IFN to observe CRS toxicity in CAR-T patients.
2.3.3. Quantitative Real Time Biosensing of CAR-T Cell Expansion, Biodistribution, and Persistence
Real time CAR-T cell tracking would provide information on the distribution of CAR-T cells within the body, as well as their overall survival, thus providing useful information on the current state of the treatment. Indeed, aptamers have been applied for live cell tracking demonstrating promise for their application to CAR-T cells.84 In one of the first examples, a DNA aptamer was able to specifically track its target, Ramos cells, directly in a mouse model.39 Specifically, the fluorescently labeled aptamers were injected into the tail vein of Ramos tumor-bearing mice, as well as mice with acute lymphocytic leukemia cell line (CCRF-CEM) to serve as a tumor xenograft mouse control. Four hours post injection, red fluorescence was distinctly visible only at the Ramos tumor site and no fluorescence was seen in the mice bearing the CCRF-CEM tumors. Over the past few years, several more examples have been described, making use of aptamers as imaging probes for a variety of imaging techniques reviewed extensively by others.85,86
Despite the potential benefits of applying aptamers to live in vivo CAR-T cell tracking, there are currently no studies that have developed aptamer biosensors to CAR-T cells. One challenge may be the susceptibility of aptamers to plasma exonucleases.75 However, several strategies exist to improve their stability such as including modifications to the sugar–phosphate backbone post-SELEX.87 Notably, there are several feasible targets on CAR-T cells that can used to generate aptamers. Indeed, most of the known receptors on activated T cells including the 4-1BB receptor, OX40 receptor, and CTLA-4 receptors have reported aptamers (Table 1). Furthermore, it is plausible to imagine that during CAR-T development, the same aptamers used to isolate the T cells by targeting any of the above receptors can be further modified to include live cell tracking features for a multiplexed use. As such, we expect that the future use of aptamers for personalized noninvasive monitoring of CAR-T cell surface interactions, signaling profile, and expansion and persistence rates could have an impact on increasing the efficacy and reducing toxicity of the therapy.
2.4. Controlling CAR-T Cell Therapy with Functional Nucleic Acids
Once CAR-Ts are produced and injected into the patient, treatment can still fail due to an inability to survive and persist in the host as a result of insufficient homeostatic cytokines and stimulatory antigen presenting cells.35 Current strategies that improve CAR-T cell persistence include myeloablative total body irradiation or chemotherapy combined with high levels of added proliferative cytokine (IL-2)88 which can all cause toxic side-effects. Alternative strategies that upregulate expression of growth-related genes suffer from lack of tight regulation and risk uncontrolled lymphoproliferation and leukemic transformation.89 As such, there is an unmet need in which functional nucleic acids may offer tightly controlled regulatory systems for growth-stimulatory gene-expression.
To improve T cell proliferation and persistence, Chen et al.35 applied a functional nucleic acid switch as a cell-intrinsic control system for growth-stimulating cytokine production. The switch was designed so that aptamer-target binding activity was directly coupled to another functional nucleic acid, called a ribozyme that can self-cleave.90 When the ribozyme is integrated into the 3′ UTR of the target transgene encoding proliferative cytokines, its self-cleavage leaves the mRNA transcript susceptible to nucleases. As a result, the mRNA is degraded, resulting in no proliferative cytokine production.91 In this work, an aptamer that binds to the small molecule theophylline was coupled to the hammerhead ribozyme. In this way, binding of the target, theophylline, to the aptamer inhibits the catalytic activity of the ribozyme therefore stabilizing the mRNA transcript, allowing for translation (Figure 4). In the absence of theophylline, the ribozyme is active and can self-cleave, resulting in rapid degradation of the cytokine mRNA transcript. These aptamer-based switches were therefore used to alter expression levels of cytokines in direct response to exogenously delivered theophylline, thereby achieving dynamic control of T cell proliferation.
Figure 4.

Small molecule-responsive ribozyme switches dynamically regulate T cell proliferation. When there is no theophylline drug binding to the aptamer, the ribozyme self-cleaves, leading to IL-2 mRNA degradation, cytokine production block, and eventually cell death. When theophylline binds to the aptamer, the ribozyme is inactivated, and the IL-2 mRNA transcript is stabilized, thus leading to cytokine production and cell proliferation. Created with Biorender.com.
The efficacy of the theophylline-responsive aptamer-based switch system for tightly regulating cytokines IL-2 or IL-15 was tested in the mouse T cell line, CTLL-2, as well as primary human T cells.35 In the absence of theophylline, the T cells exhibited proliferation levels similar to those of cells growing in the absence of cytokines. However, an increase in cytokine production was measured via fluorescence every time the cytokine producing gene was switched on. A titratable increase in cytokine production occurred over a range of theophylline concentrations, demonstrating the utility of dynamic control by functional nucleic acids. Impressively, these switches also enabled long-term, dynamic control over IL-15 gene expression and therefore in vivo T cell expansion in a mouse model. One drawback is that the clinical translation of theophylline-responsive switches for CAR-T cell therapy in humans is limited by the narrow therapeutic index of theophylline.
Building on this work, Wong et al.36 developed a drug-responsive, microRNA (miRNA)-based gene regulatory system that alters endogenous cytokine receptor subunits, rather than requiring the use of trans-genes. Specifically, an aptamer-controlled microRNA switch was developed to IL-2Rβ, a cytokine receptor that is displayed on the surface of T cells and is necessary for T cell activation and proliferation. In this design, binding of the aptamer target prevents proper maturation of the miRNA, thereby leading to a reduction in RNAi-mediated gene silencing and a subsequent increase in target gene expression. To additionally address toxicity issues associated with the previous theophylline switches, these microRNA switches were controlled using (6R)-folinic acid ((6R)-FA), the biologically inactive and nontoxic enantiomer of the FDA-approved drug leucovorin. Several (6R)-FA aptamers with nanomolar binding affinities that were identified and characterized were integrated into this new switch platform92 at the basal segment domain of an IL-2 receptor-targeting miRNA hairpin36 (Figure 5). The resulting switches were highly specific and could be activated using drug levels with no measurable toxicity. Impressively, the miRNA switch system enabled dynamic regulation of autonomous mouse T cell growth following the addition and withdrawal of (6R)-FA.
Figure 5.

CAR-T cell control using a ribozyme switch responsive to (6R)-FA). Lack of ligand addition causes the formation of miRNA duplex from the pre miRNA. The miRNA prevents the ligand from binding to the receptor, leading to cell death. When (6R)-FA is added, it binds to the aptamer and prevents the formation of the miRNA duplex. This allows for cytokines to bind the receptor and form a dimer, which leads to cell proliferation. Created with Biorender.com.
These nucleic acid-based proliferation control studies demonstrate that the limited off-target effects of the functional nucleic acid switches and their stringent control over gene expression help address critical limitations in T cell proliferation. Importantly, these functional nucleic acid switches may provide an important strategy for modulating CAR-T persistence and proliferation, providing the ability to shut off CAR-T activity if toxicities become severe. Compared to suicide switches that exhibit an all or nothing response, nucleic acid-based switches can dynamically regulate and modulate T cell growth and production to tune for appropriate therapeutic activity while reducing toxicity. To date, the functional nucleic acid-based switches have been tested in T cells; therefore, further studies are required to assess the applicability to CAR-T cells.
3. Future Opportunities
Combination Immunotherapy
The small size of aptamers provides a beneficial advantage in accessing difficult to reach tumor sites and can be useful as a cotreatment with CAR-T. As one example, researchers recently demonstrated the use of aptamer-drug conjugate nanomicelles that bind to nucleolin overexpressed on many cancer cell membranes to boost antitumor responses of checkpoint blockade immunotherapy. This promising approach, while not directly related to CAR-T therapy, could be a useful combination approach to help treat difficult cancers or hard to reach solid tumors.93 Another example where aptamers synergistically operate with CAR-T cell therapy involves the use of anti-CTLA-4 and PD-L1 aptamers to block signaling pathways in solid tumors.94 Excitingly, the aptamers bound to the receptors specifically, recruited T cells to the tumor site, and increased T cell proliferation. Furthermore, antitumor activity and slowed tumor progression was shown in mice. Therefore, applying similar combination therapy of CAR-T cells with relevant functional nucleic acids can help increase recruitment of immune cells to the tumor site and help overcome limitations of targeting solid tumors.
Reducing Treatment Loss
Several issues also limit the CAR-T cell targeting. For example, tumor cells typically downregulate their antigens. In particular, patients with B-cell acute lymphoblastic leukemias suffer from CD19 antigen loss after relapse from CAR-T cell treatment.95 As a result, targeting these cells with cytotoxic T cells and T cell therapies is less effective and reduces CAR-T treatment efficacy.96 One-way nucleic acids could help solve this issue would be by replacing the scFv region of the CAR with aptamers that could be generated in vitro to bind to other targets on cancer cells and prevent treatment efficacy loss.
Long-Term Biosensing of CAR-T Therapy
Many of the functional nucleic acid studies reviewed here show promise but have only been tested in T cells or Ramos cells. As such, it will be interesting to explore the use of aptamers for isolating T cells with high affinity in clinical settings and specifically on CAR-T cells. Given that CAR-T therapy is still relatively new, there are many opportunities to explore functional nucleic acids as biosensing tools to study CAR-T over time in patients.
New Functional Nucleic Acids
There is always a need for additional functional nucleic acid tools that are specifically optimized for the application of interest. An important untapped opportunity is the use of DNAzymes in CAR-T therapy. Similar to the ribozyme and microRNA strategy used to control cytokine production,35 DNAzymes have recently been developed to alter endogenous gene expression directly in cells97 and could be useful to potentiate or control CAR-T therapy. Furthermore, DNAzymes have been successfully applied for a wide variety of biosensing applications98 and therefore may be useful to monitor CAR-T therapy. Finally, we emphasized several examples throughout the review where the selection of new aptamer candidates to CAR-T cells and their components or cancer surface markers will further enable their use in CAR-T therapy.
4. Conclusion
We described several applications in which functional nucleic acids could be beneficial for improving CAR-T therapy. The applications span from using high affinity aptamers for effective isolation of T cells, for biosensing, and for tuning ongoing CAR-T therapy. For example, functional nucleic acids that enable tunable and dynamic control of genes relevant in CAR-T growth and persistence are a promising strategy to reduce toxic effects and help boost proliferation. While no studies have directly tracked CAR-Ts with functional nucleic acids, proof-of-concept studies using other systems have demonstrated the potential for aptamers to measure cell-surface interactions, to track important proliferation markers, and to track the CAR-T cells themselves. While much work is needed to expand the proof-of-concept studies to demonstrate their feasibility in animal models and the clinic, these promising diverse efforts indicate a bright future for functional nucleic acids in CAR-T cell research and other ongoing immunotherapy strategies.
Acknowledgments
We thank Professor Jason Tanny for proofreading the paper and members of the McKeague lab for useful discussions on this work. All graphics were created using Biorender.com.
Glossary
Abbreviations
- (6R)-FA
(6R)-folinic acid
- ALL
acute lymphoblastic leukemia
- CAR
chimeric antigen receptor
- CD
cluster of differentiation
- CRS
cytokine release syndrome
- EGFR
epidermal growth factor receptor
- FasL
Fas ligand
- IFN
type II interferon
- IgG
immunoglobulin G
- IL
interleukin
- Kd
dissociation constant
- mAb
monoclonal antibody
- MHC
major histocompatibility complex
- miRNA
microRNA
- nM
nanomolar
- PD-L1
programmed death-ligand 1
- PET
positron emission tomography
- scFv
single chain variable fragment
- SELEX
Systematic Evolution of Ligands by Exponential Enrichment
- TCR
T cell receptor
- TIL
tumor infiltrating lymphocytes
- TNF
tumor necrosis factor
This work is supported by the Natural Science and Engineering Research Council of Canada (NSERC) Grant to M.M. and Undergraduate Student Research Award to L.N., the Canada Institutes of Health Research (CIHR) Graduate Scholarship Program to B.M., and the Canada Research Chair Program to M.M.
The authors declare no competing financial interest.
References
- Eshhar Z.; Waks T.; Gross G.; Schindler D. G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T cell receptors. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 720–724. 10.1073/pnas.90.2.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guedan S.; Calderon H.; Posey A. D. Jr.; Maus M. V. Engineering and Design of Chimeric Antigen Receptors. Mol. Ther.--Methods Clin. Dev. 2019, 12, 145–156. 10.1016/j.omtm.2018.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech S. M.; Wherry E. J.; Ahmed R. Effector and memory T cell differentiation: implications for vaccine development. Nat. Rev. Immunol. 2002, 2, 251–262. 10.1038/nri778. [DOI] [PubMed] [Google Scholar]
- Firor A. E.; Jares A.; Ma Y. From humble beginnings to success in the clinic: Chimeric antigen receptor-modified T cells and implications for immunotherapy. Exp. Biol. Med. 2015, 240, 1087–1098. 10.1177/1535370215584936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majzner R. G.; Mackall C. L. Clinical lessons learned from the first leg of the CAR T cell journey. Nat. Med. 2019, 25, 1341–1355. 10.1038/s41591-019-0564-6. [DOI] [PubMed] [Google Scholar]
- Ma S.; Li X.; Wang X.; Cheng L.; Li Z.; Zhang C.; Ye Z.; Qian Q. Current Progress in CAR-T Cell Therapy for Solid Tumors. Int. J. Biol. Sci. 2019, 15, 2548–2560. 10.7150/ijbs.34213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long A. H.; Haso W. M.; Shern J. F.; Wanhainen K. M.; Murgai M.; Ingaramo M.; Smith J. P.; Walker A. J.; Kohler M. E.; Venkateshwara V. R.; Kaplan R. N.; Patterson G. H.; Fry T. J.; Orentas R. J.; Mackall C. L. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015, 21, 581–590. 10.1038/nm.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P.; Lu X.-a.; Zhang X.; Xiong M.; Zhang J.; Zhou X.; Qi F.; Yang J.; He T. Which is better in CD19 CAR-T treatment of r/r B-ALL, CD28 or 4-1BB? A parallel trial under the same manufacturing process. J. Clin. Oncol. 2018, 36, 3041–3041. 10.1200/JCO.2018.36.15_suppl.3041. [DOI] [Google Scholar]
- Tang X. Y.; Sun Y.; Zhang A.; Hu G. L.; Cao W.; Wang D. H.; Zhang B.; Chen H. Third-generation CD28/4-1BB chimeric antigen receptor T cells for chemotherapy relapsed or refractory acute lymphoblastic leukaemia: a non-randomised, open-label phase I trial protocol. BMJ. Open 2016, 6, e013904 10.1136/bmjopen-2016-013904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enblad G.; Karlsson H.; Gammelgård G.; Wenthe J.; Lövgren T.; Amini R. M.; Wikstrom K. I.; Essand M.; Savoldo B.; Hallböök H.; Höglund M.; Dotti G.; Brenner M. K.; Hagberg H.; Loskog A. A Phase I/IIa Trial Using CD19-Targeted Third-Generation CAR T Cells for Lymphoma and Leukemia. Clin. Cancer Res. 2018, 24, 6185–6194. 10.1158/1078-0432.CCR-18-0426. [DOI] [PubMed] [Google Scholar]
- Chmielewski M.; Abken H. TRUCKs: the fourth generation of CARs. Expert Opin. Biol. Ther. 2015, 15, 1145–1154. 10.1517/14712598.2015.1046430. [DOI] [PubMed] [Google Scholar]
- O’Rourke D. M., Nasrallah M. P., Desai A., Melenhorst J. J., Mansfield K., Morrissette J. J. D., Martinez-Lage M., Brem S., Maloney E., Shen A., Isaacs R., Mohan S., Plesa G., Lacey S. F., Navenot J. M., Zheng Z., Levine B. L., Okada H., June C. H., Brogdon J. L., Maus M. V.. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma, Sci. Transl. Med.(2017) 9, 10.1126/scitranslmed.aaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukari B.; Samarasinghe R. M.; Noibanchong J.; Shigdar S. L. Non-Invasive Delivery of Therapeutics into the Brain: The Potential of Aptamers for Targeted Delivery. Biomedicines 2020, 8, 120. 10.3390/biomedicines8050120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brudno J. N.; Kochenderfer J. N. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood 2016, 127, 3321–3330. 10.1182/blood-2016-04-703751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santomasso B.; Bachier C.; Westin J.; Rezvani K.; Shpall E. J. The Other Side of CAR T-Cell Therapy: Cytokine Release Syndrome, Neurologic Toxicity, and Financial Burden. American Society of Clinical Oncology Educational Book 2019, 433–444. 10.1200/EDBK_238691. [DOI] [PubMed] [Google Scholar]
- Sun S.; Hao H.; Yang G.; Zhang Y.; Fu Y. Immunotherapy with CAR-Modified T Cells: Toxicities and Overcoming Strategies. J. Immunol. Res. 2018, 2018, 2386187. 10.1155/2018/2386187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R.; Li X.; He Y.; Zhu W.; Gao L.; Liu Y.; Gao L.; Wen Q.; Zhong J. F.; Zhang C.; Zhang X. Recent advances in CAR-T cell engineering. J. Hematol. Oncol. 2020, 13, 86. 10.1186/s13045-020-00910-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni J. A.; Witzigmann D.; Thomson S. B.; Chen S.; Leavitt B. R.; Cullis P. R.; van der Meel R. The current landscape of nucleic acid therapeutics. Nat. Nanotechnol. 2021, 16, 630–643. 10.1038/s41565-021-00898-0. [DOI] [PubMed] [Google Scholar]
- Dolgin E. How COVID unlocked the power of RNA vaccines. Nature 2021, 589, 189–191. 10.1038/d41586-021-00019-w. [DOI] [PubMed] [Google Scholar]
- Alsaafin A.; McKeague M. Functional nucleic acids as in vivo metabolite and ion biosensors. Biosens. Bioelectron. 2017, 94, 94–106. 10.1016/j.bios.2017.02.030. [DOI] [PubMed] [Google Scholar]
- Zaug A. J.; Grabowski P. J.; Cech T. R. Autocatalytic cyclization of an excised intervening sequence RNA is a cleavage-ligation reaction. Nature 1983, 301, 578–583. 10.1038/301578a0. [DOI] [PubMed] [Google Scholar]
- Guerrier-Takada C.; Gardiner K.; Marsh T.; Pace N.; Altman S. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell 1983, 35, 849–857. 10.1016/0092-8674(83)90117-4. [DOI] [PubMed] [Google Scholar]
- Maurel M. C.; Leclerc F.; Herve G. Ribozyme Chemistry: To Be or Not To Be under High Pressure. Chem. Rev. 2020, 120, 4898–4918. 10.1021/acs.chemrev.9b00457. [DOI] [PubMed] [Google Scholar]
- Park S. V.; Yang J. S.; Jo H.; Kang B.; Oh S. S.; Jung G. Y. Catalytic RNA, ribozyme, and its applications in synthetic biology. Biotechnol. Adv. 2019, 37, 107452. 10.1016/j.biotechadv.2019.107452. [DOI] [PubMed] [Google Scholar]
- McKeague M.; DeRosa M. C. Challenges and Opportunities for Small Molecule Aptamer Development. J. Nucleic Acids 2012, 2012, 748913. 10.1155/2012/748913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeague M.; McConnell E. M.; Cruz-Toledo J.; Bernard E. D.; Pach A.; Mastronardi E.; Zhang X.; Beking M.; Francis T.; Giamberardino A.; Cabecinha A.; Ruscito A.; Aranda-Rodriguez R.; Dumontier M.; DeRosa M. C. Analysis of In Vitro Aptamer Selection Parameters. J. Mol. Evol. 2015, 81, 150–161. 10.1007/s00239-015-9708-6. [DOI] [PubMed] [Google Scholar]
- Tuerk C.; Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- Ellington A. D.; Szostak J. W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- Wu Y. X.; Kwon Y. J. Aptamers: The ″evolution″ of SELEX. Methods 2016, 106, 21–28. 10.1016/j.ymeth.2016.04.020. [DOI] [PubMed] [Google Scholar]
- Zhou J.; Rossi J. Aptamers as targeted therapeutics: current potential and challenges. Nat. Rev. Drug Discovery 2017, 16, 181–202. 10.1038/nrd.2016.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigdar S.; Schrand B.; Giangrande P. H.; de Franciscis V. Aptamers: Cutting edge of cancer therapies. Mol. Ther. 2021, 29, 2396. 10.1016/j.ymthe.2021.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kacherovsky N.; Cardle I. I.; Cheng E. L.; Yu J. L.; Baldwin M. L.; Salipante S. J.; Jensen M. C.; Pun S. H. Traceless aptamer-mediated isolation of CD8(+) T cells for chimeric antigen receptor T cell therapy. Nature biomedical engineering 2019, 3, 783–795. 10.1038/s41551-019-0411-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray B. P.; Requena M. D.; Nichols M. D.; Sullenger B. A. Aptamers as Reversible Sorting Ligands for Preparation of Cells in Their Native State. Cell Chemical Biology 2020, 27, 232–244. 10.1016/j.chembiol.2019.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C.-G.; Wang Y.; Liu P.; Yao Q.-L.; Zhou Y.-Y.; Li C.-F.; Zhao Q.; Liu G.-H.; Zhang X.-L. Aptamer-T Cell Targeted Therapy for Tumor Treatment Using Sugar Metabolism and Click Chemistry. ACS Chem. Biol. 2020, 15, 1554–1565. 10.1021/acschembio.0c00164. [DOI] [PubMed] [Google Scholar]
- Chen Y. Y.; Jensen M. C.; Smolke C. D. Genetic control of mammalian T cell proliferation with synthetic RNA regulatory systems 2010, 107, 8531. 10.1073/pnas.1001721107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong R. S.; Chen Y. Y.; Smolke C. D. Regulation of T cell proliferation with drug-responsive microRNA switches. Nucleic Acids Res. 2018, 46, 1541–1552. 10.1093/nar/gkx1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You M.; Lyu Y.; Han D.; Qiu L.; Liu Q.; Chen T.; Sam Wu C.; Peng L.; Zhang L.; Bao G.; Tan W. DNA probes for monitoring dynamic and transient molecular encounters on live cell membranes. Nat. Nanotechnol. 2017, 12, 453–459. 10.1038/nnano.2017.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu L.; Wimmers F.; Weiden J.; Heus H. A.; Tel J.; Figdor C. G. A membrane-anchored aptamer sensor for probing IFNgamma secretion by single cells. Chem. Commun. (Cambridge, U. K.) 2017, 53, 8066–8069. 10.1039/C7CC03576D. [DOI] [PubMed] [Google Scholar]
- Shi H.; Tang Z.; Kim Y.; Nie H.; Huang Y. F.; He X.; Deng K.; Wang K.; Tan W. In vivo fluorescence imaging of tumors using molecular aptamers generated by cell-SELEX. Chem. - Asian J. 2010, 5, 2209–2213. 10.1002/asia.201000242. [DOI] [PubMed] [Google Scholar]
- McNamara J. O.; Kolonias D.; Pastor F.; Mittler R. S.; Chen L.; Giangrande P. H.; Sullenger B.; Gilboa E. Multivalent 4-1BB binding aptamers costimulate CD8+ T cells and inhibit tumor growth in mice. J. Clin. Invest. 2008, 118, 376–386. 10.1172/JCI33365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dollins C. M.; Nair S.; Boczkowski D.; Lee J.; Layzer J. M.; Gilboa E.; Sullenger B. A. Assembling OX40 aptamers on a molecular scaffold to create a receptor-activating aptamer. Chem. Biol. 2008, 15, 675–682. 10.1016/j.chembiol.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santulli-Marotto S.; Nair S. K.; Rusconi C.; Sullenger B.; Gilboa E. Multivalent RNA Aptamers That Inhibit CTLA-4 and Enhance Tumor Immunity. Cancer Res. 2003, 63, 7483. [PubMed] [Google Scholar]
- Prodeus A.; Abdul-Wahid A.; Fischer N. W.; Huang E. H. B.; Cydzik M.; Gariépy J. Targeting the PD-1/PD-L1 Immune Evasion Axis With DNA Aptamers as a Novel Therapeutic Strategy for the Treatment of Disseminated Cancers. Mol. Ther.--Nucleic Acids 2015, 4, e237–e237. 10.1038/mtna.2015.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orava E. W.; Jarvik N.; Shek Y. L.; Sidhu S. S.; Gariépy J. A Short DNA Aptamer That Recognizes TNFα and Blocks Its Activity in Vitro. ACS Chem. Biol. 2013, 8, 170–178. 10.1021/cb3003557. [DOI] [PubMed] [Google Scholar]
- Shahdordizadeh M.; Taghdisi S. M.; Sankian M.; Ramezani M.; Abnous K. Design, isolation and evaluation of the binding efficiency of a DNA aptamer against interleukin 2 receptor alpha, in vitro. Int. Immunopharmacol. 2017, 53, 96–104. 10.1016/j.intimp.2017.10.011. [DOI] [PubMed] [Google Scholar]
- Gupta S.; Hirota M.; Waugh S. M.; Murakami I.; Suzuki T.; Muraguchi M.; Shibamori M.; Ishikawa Y.; Jarvis T. C.; Carter J. D.; Zhang C.; Gawande B.; Vrkljan M.; Janjic N.; Schneider D. J. Chemically modified DNA aptamers bind interleukin-6 with high affinity and inhibit signaling by blocking its interaction with interleukin-6 receptor. J. Biol. Chem. 2014, 289, 8706–8719. 10.1074/jbc.M113.532580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung H. J.; Choi S.; Lee J. W.; Ok C. Y.; Bae Y.-S.; Kim Y.-H.; Lee W.; Heo K.; Kim I.-H. Inhibition of human neutrophil activity by an RNA aptamer bound to interleukin-8. Biomaterials 2014, 35, 578–589. 10.1016/j.biomaterials.2013.09.107. [DOI] [PubMed] [Google Scholar]
- Berezhnoy A.; Stewart C. A.; McNamara J. O. 2nd; Thiel W.; Giangrande P.; Trinchieri G.; Gilboa E. Isolation and optimization of murine IL-10 receptor blocking oligonucleotide aptamers using high-throughput sequencing. Mol. Ther. 2012, 20, 1242–1250. 10.1038/mt.2012.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalabi H.; Khuu H.; Fry T. J.; Shah N. N.. Chapter 12 - Cell-Based Therapies: A New Frontier of Personalized Medicine. In Novel Designs of Early Phase Trials for Cancer Therapeutics, Kummar S., Takimoto C., Eds.; Academic Press, 2018, pp 175–191. [Google Scholar]
- Padmanabhan A. Cellular collection by apheresis. Transfusion 2018, 58, 598–604. 10.1111/trf.14502. [DOI] [PubMed] [Google Scholar]
- Wang X.; Rivière I. Clinical manufacturing of CAR T cells: foundation of a promising therapy. Mol. Ther Oncolytics 2016, 3, 16015. 10.1038/mto.2016.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommermeyer D.; Hudecek M.; Kosasih P. L.; Gogishvili T.; Maloney D. G.; Turtle C. J.; Riddell S. R. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 2016, 30, 492–500. 10.1038/leu.2015.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BØyum A.; Brincker Fjerdingstad H.; Martinsen I.; Lea T.; LØvhaug D. Separation of Human Lymphocytes from Citrated Blood by Density Gradient (NycoPrep) Centrifugation: Monocyte Depletion Depending upon Activation of Membrane Potassium Channels. Scandinavian Journal of Immunology 2002, 56, 76–84. 10.1046/j.1365-3083.2002.01102.x. [DOI] [PubMed] [Google Scholar]
- Vormittag P.; Gunn R.; Ghorashian S.; Veraitch F. S. A guide to manufacturing CAR T cell therapies. Curr. Opin. Biotechnol. 2018, 53, 164–181. 10.1016/j.copbio.2018.01.025. [DOI] [PubMed] [Google Scholar]
- Bhattacharjee J.; Das B.; Mishra A.; Sahay P.; Upadhyay P. Monocytes isolated by positive and negative magnetic sorting techniques show different molecular characteristics and immunophenotypic behaviour. F1000Research 2017, 6, 2045. 10.12688/f1000research.12802.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn M. R.; Jimenez R. M.; Chaput J. C. Analysis of aptamer discovery and technology. Nature Reviews Chemistry 2017, 1, 0076. 10.1038/s41570-017-0076. [DOI] [Google Scholar]
- Zumrut H. E.; Ara M. N.; Maio G. E.; Van N. A.; Batool S.; Mallikaratchy P. R. Ligand-guided selection of aptamers against T cell Receptor-cluster of differentiation 3 (TCR-CD3) expressed on Jurkat.E6 cells. Anal. Biochem. 2016, 512, 1–7. 10.1016/j.ab.2016.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zumrut H.; Yang Z.; Williams N.; Arizala J.; Batool S.; Benner S. A.; Mallikaratchy P. Ligand-Guided Selection with Artificially Expanded Genetic Information Systems against TCR-CD3epsilon. Biochemistry 2020, 59, 552–562. 10.1021/acs.biochem.9b00919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sola M.; Menon A. P.; Moreno B.; Meraviglia-Crivelli D.; Soldevilla M. M.; Carton-Garcia F.; Pastor F. Aptamers Against Live Targets: Is In Vivo SELEX Finally Coming to the Edge?. Mol. Ther.--Nucleic Acids 2020, 21, 192–204. 10.1016/j.omtn.2020.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer G.; Ahmed M. S.; Dolf A.; Endl E.; Knolle P. A.; Famulok M. Fluorescence-activated cell sorting for aptamer SELEX with cell mixtures. Nat. Protoc. 2010, 5, 1993–2004. 10.1038/nprot.2010.163. [DOI] [PubMed] [Google Scholar]
- Kumar Kulabhusan P.; Hussain B.; Yüce M. Current Perspectives on Aptamers as Diagnostic Tools and Therapeutic Agents. Pharmaceutics 2020, 12, 646. 10.3390/pharmaceutics12070646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan R. A.; Boyerinas B. Genetic Modification of T Cells. Biomedicines 2016, 4, 9. 10.3390/biomedicines4020009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X.; Cai H.; Zhao L.; Ning L.; Lang J. CAR-T cell therapy in ovarian cancer: from the bench to the bedside. Oncotarget 2017, 8, 64607–64621. 10.18632/oncotarget.19929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haapaniemi E.; Botla S.; Persson J.; Schmierer B.; Taipale J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018, 24, 927–930. 10.1038/s41591-018-0049-z. [DOI] [PubMed] [Google Scholar]
- Ding F.; Guo S.; Xie M.; Luo W.; Yuan C.; Huang W.; Zhou Y.; Zhang X. L.; Zhou X. Diagnostic applications of gastric carcinoma cell aptamers in vitro and in vivo. Talanta 2015, 134, 30–36. 10.1016/j.talanta.2014.09.036. [DOI] [PubMed] [Google Scholar]
- Lai W. Y.; Huang B. T.; Wang J. W.; Lin P. Y.; Yang P. C. A Novel PD-L1-targeting Antagonistic DNA Aptamer With Antitumor Effects. Mol. Ther.--Nucleic Acids 2016, 5, e397 10.1038/mtna.2016.102. [DOI] [PubMed] [Google Scholar]
- Sun X.; Pan Q.; Yuan C.; Wang Q.; Tang X. L.; Ding K.; Zhou X.; Zhang X. L. A Single ssDNA Aptamer Binding to Mannose-Capped Lipoarabinomannan of Bacillus Calmette-Guérin Enhances Immunoprotective Effect against Tuberculosis. J. Am. Chem. Soc. 2016, 138, 11680–11689. 10.1021/jacs.6b05357. [DOI] [PubMed] [Google Scholar]
- Milone M. C.; O’Doherty U. Clinical use of lentiviral vectors. Leukemia 2018, 32, 1529–1541. 10.1038/s41375-018-0106-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldevilla M. M.; Villanueva H.; Pastor F. Aptamers: A Feasible Technology in Cancer Immunotherapy. J. Immunol. Res. 2016, 2016, 1083738–1083738. 10.1155/2016/1083738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai C.; Gao S.; Hu S.; Liu X.; Li H.; Dong J.; Huang A.; Zhu L.; Zhou P.; Li S.; Shao N. Self-Assembled Multivalent Aptamer Nanoparticles with Potential CAR-like Characteristics Could Activate T Cells and Inhibit Melanoma Growth. Mol. Ther Oncolytics 2020, 17, 9–20. 10.1016/j.omto.2020.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter D. L.; Levine B. L.; Kalos M.; Bagg A.; June C. H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011, 365, 725–733. 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awasthi R.; Pacaud L.; Waldron E.; Tam C. S.; Jäger U.; Borchmann P.; Jaglowski S.; Foley S. R.; van Besien K.; Wagner-Johnston N. D.; Kersten M. J.; Schuster S. J.; Salles G.; Maziarz R. T.; Anak Ö.; Del Corral C.; Chu J.; Gershgorin I.; Pruteanu-Malinici I.; Chakraborty A.; Mueller K. T.; Waller E. K. Tisagenlecleucel cellular kinetics, dose, and immunogenicity in relation to clinical factors in relapsed/refractory DLBCL. Blood Adv. 2020, 4, 560–572. 10.1182/bloodadvances.2019000525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita Y.; Leslie M.; Kameyama H.; Volk D. E.; Tanaka T. Aptamer Therapeutics in Cancer: Current and Future. Cancers 2018, 10, 80. 10.3390/cancers10030080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratschmer C.; Levy M. Effect of Chemical Modifications on Aptamer Stability in Serum. Nucleic Acid Therapeutics 2017, 27, 335–344. 10.1089/nat.2017.0680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuai H.; Zhao Z.; Mo L.; Liu H.; Hu X.; Fu T.; Zhang X.; Tan W. Circular Bivalent Aptamers Enable in Vivo Stability and Recognition. J. Am. Chem. Soc. 2017, 139, 9128–9131. 10.1021/jacs.7b04547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.; Sun X.; Xu J.; Cui C.; Safari Yazd H.; Pan X.; Zhu Y.; Chen X.; Li X.; Li J.; Tan W. Circular Bispecific Aptamer-Mediated Artificial Intercellular Recognition for Targeted T Cell Immunotherapy. ACS Nano 2020, 14, 9562–9571. 10.1021/acsnano.9b09884. [DOI] [PubMed] [Google Scholar]
- Ghorashian S.; Kramer A. M.; Onuoha S.; Wright G.; Bartram J.; Richardson R.; Albon S. J.; Casanovas-Company J.; Castro F.; Popova B.; Villanueva K.; Yeung J.; Vetharoy W.; Guvenel A.; Wawrzyniecka P. A.; Mekkaoui L.; Cheung G. W.-K.; Pinner D.; Chu J.; Lucchini G.; Silva J.; Ciocarlie O.; Lazareva A.; Inglott S.; Gilmour K. C.; Ahsan G.; Ferrari M.; Manzoor S.; Champion K.; Brooks T.; Lopes A.; Hackshaw A.; Farzaneh F.; Chiesa R.; Rao K.; Bonney D.; Samarasinghe S.; Goulden N.; Vora A.; Veys P.; Hough R.; Wynn R.; Pule M. A.; Amrolia P. J. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat. Med. 2019, 25, 1408–1414. 10.1038/s41591-019-0549-5. [DOI] [PubMed] [Google Scholar]
- Lee S. H.; Soh H.; Chung J. H.; Cho E. H.; Lee S. J.; Ju J.-M.; Sheen J. H.; Kim H.; Oh S. J.; Lee S.-J.; Chung J.; Choi K.; Kim S.-Y.; Ryu J.-S. Feasibility of real-time in vivo 89Zr-DFO-labeled CAR T cell trafficking using PET imaging. PLoS One 2020, 15, e0223814–e0223814. 10.1371/journal.pone.0223814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold L.; Ayers D.; Bertino J.; Bock C.; Bock A.; Brody E. N.; Carter J.; Dalby A. B.; Eaton B. E.; Fitzwater T.; Flather D.; Forbes A.; Foreman T.; Fowler C.; Gawande B.; Goss M.; Gunn M.; Gupta S.; Halladay D.; Heil J.; Heilig J.; Hicke B.; Husar G.; Janjic N.; Jarvis T.; Jennings S.; Katilius E.; Keeney T. R.; Kim N.; Koch T. H.; Kraemer S.; Kroiss L.; Le N.; Levine D.; Lindsey W.; Lollo B.; Mayfield W.; Mehan M.; Mehler R.; Nelson S. K.; Nelson M.; Nieuwlandt D.; Nikrad M.; Ochsner U.; Ostroff R. M.; Otis M.; Parker T.; Pietrasiewicz S.; Resnicow D. I.; Rohloff J.; Sanders G.; Sattin S.; Schneider D.; Singer B.; Stanton M.; Sterkel A.; Stewart A.; Stratford S.; Vaught J. D.; Vrkljan M.; Walker J. J.; Watrobka M.; Waugh S.; Weiss A.; Wilcox S. K.; Wolfson A.; Wolk S. K.; Zhang C.; Zichi D. Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS One 2010, 5, e15004 10.1371/journal.pone.0015004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batool S.; Bhandari S.; George S.; Okeoma P.; Van N.; Zumrut H. E.; Mallikaratchy P. Engineered Aptamers to Probe Molecular Interactions on the Cell Surface. Biomedicines 2017, 5, 54. 10.3390/biomedicines5030054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallikaratchy P. Evolution of Complex Target SELEX to Identify Aptamers against Mammalian Cell-Surface Antigens. Molecules 2017, 22, 215. 10.3390/molecules22020215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zumrut H. E.; Mallikaratchy P. R. Ligand Guided Selection (LIGS) of Artificial Nucleic Acid Ligands against Cell Surface Targets. ACS Appl. Bio Mater. 2020, 3, 2545–2552. 10.1021/acsabm.9b00938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy H.; Iqbal M.; Chavez J. C.; Kharfan-Dabaja M. A. Cytokine Release Syndrome: Current Perspectives. ImmunoTargets Ther. 2019, 8, 43–52. 10.2147/ITT.S202015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L.; Wang Y.; Xu X.; Liu Y.; Lin B.; Zhang M.; Zhang J.; Wan S.; Yang C.; Tan W. Aptamer-Based Detection of Circulating Targets for Precision Medicine. Chem. Rev. 2021, 121, 12035. 10.1021/acs.chemrev.0c01140. [DOI] [PubMed] [Google Scholar]
- Bouvier-Muller A.; Duconge F. Application of aptamers for in vivo molecular imaging and theranostics. Adv. Drug Delivery Rev. 2018, 134, 94–106. 10.1016/j.addr.2018.08.004. [DOI] [PubMed] [Google Scholar]
- Yoon S.; Rossi J. J. Targeted Molecular Imaging Using Aptamers in Cancer. Pharmaceuticals 2018, 11, 71. 10.3390/ph11030071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freage L.; Jamal D.; Williams N. B.; Mallikaratchy P. R. A Homodimeric Aptamer Variant Generated from Ligand-Guided Selection Activates the T Cell Receptor Cluster of Differentiation 3 Complex. Mol. Ther.--Nucleic Acids 2020, 22, 167–178. 10.1016/j.omtn.2020.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gattinoni L.; Powell D. J. Jr.; Rosenberg S. A.; Restifo N. P. Adoptive immunotherapy for cancer: building on success. Nat. Rev. Immunol. 2006, 6, 383–393. 10.1038/nri1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leen A. M.; Rooney C. M.; Foster A. E. Improving T cell therapy for cancer. Annu. Rev. Immunol. 2007, 25, 243–265. 10.1146/annurev.immunol.25.022106.141527. [DOI] [PubMed] [Google Scholar]
- Xiang J. S.; Kaplan M.; Dykstra P.; Hinks M.; McKeague M.; Smolke C. D. Massively parallel RNA device engineering in mammalian cells with RNA-Seq. Nat. Commun. 2019, 10, 4327. 10.1038/s41467-019-12334-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeague M.; Wong R. S.; Smolke C. D. Opportunities in the design and application of RNA for gene expression control. Nucleic Acids Res. 2016, 44, 2987–2999. 10.1093/nar/gkw151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeague M.; Wang Y.-H.; Smolke C. D. In Vitro Screening and in Silico Modeling of RNA-Based Gene Expression Control. ACS Chem. Biol. 2015, 10, 2463–2467. 10.1021/acschembio.5b00518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng Z.; Wang L.; Liu K.; Liu J.; Tan W. Enhancing anti-PD-1 immunotherapy by nanomicelles self-assembled from aptamer-multivalent-drug conjugates. Angew. Chem., Int. Ed. 2021, 60, 15459. 10.1002/anie.202102631. [DOI] [PubMed] [Google Scholar]
- Du Y.; Zhang D.; Wang Y.; Wu M.; Zhang C.; Zheng Y.; Zheng A.; Liu X. A highly stable multifunctional aptamer for enhancing antitumor immunity against hepatocellular carcinoma by blocking dual immune checkpoints. Biomater. Sci. 2021, 9, 4159–4168. 10.1039/D0BM02210A. [DOI] [PubMed] [Google Scholar]
- Majzner R. G.; Mackall C. L. Tumor Antigen Escape from CAR T cell Therapy. Cancer Discovery 2018, 8, 1219–1226. 10.1158/2159-8290.CD-18-0442. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Wu Z.; Liu Y.; Han W. New development in CAR-T cell therapy. J. Hematol. Oncol. 2017, 10, 53. 10.1186/s13045-017-0423-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Nguyen K.; Spitale R. C.; Chaput J. C. A biologically stable DNAzyme that efficiently silences gene expression in cells. Nat. Chem. 2021, 13, 319–326. 10.1038/s41557-021-00645-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell E. M.; Cozma I.; Mou Q.; Brennan J. D.; Lu Y.; Li Y. Biosensing with DNAzymes. Chem. Soc. Rev. 2021, 50, 8954. 10.1039/D1CS00240F. [DOI] [PMC free article] [PubMed] [Google Scholar]