

Abstract
Lysine-specific demethylase 1 (LSD1 or KDM1A) is a chromatin modifying enzyme playing a key role in the cell cycle and cell differentiation and proliferation through the demethylation of histones and nonhistone substrates. In addition to its enzymatic activity, LSD1 plays a fundamental scaffolding role as part of transcription silencing complexes such as rest co-repressor (CoREST) and nucleosome remodeling and deacetylase (NuRD). A host of classical amine oxidase inhibitors such as tranylcypromine, pargyline, and phenelzine together with LSD1 tool compounds such as SP-2509 and GSK-LSD1 have been extensively utilized in LSD1 mechanistic cancer studies. Additionally, several optimized new chemical entities have reached clinical trials in oncology such as ORY-1001 (iadademstat), GSK2879552, SP-2577 (seclidemstat), IMG-7289 (bomedemstat), INCB059872, and CC-90011 (pulrodemstat). Despite this, no single study exists that characterizes them all under the same experimental conditions, preventing a clear interpretation of published results. Herein, we characterize the whole LSD1 small molecule compound class as inhibitors of LSD1 catalytic activity, disruptors of SNAIL/GFI1 (SNAG)-scaffolding protein–protein interactions, inducers of cell differentiation, and potential anticancer treatments for hematological and solid tumors to yield an updated, unified perspective of this field. Our results highlight significant differences in potency and selectivity among the clinical compounds with iadademstat being the most potent and reveal that most of the tool compounds have very low activity and selectivity, suggesting some conclusions derived from their use should be taken with caution.
Keywords: LSD1 inhibitors, cancer, acute myeloid leukemia, small cell lung cancer
Epigenetics is reaching maturity in the oncology field as a first wave of drugs have already been approved,1,2 and several novel drug classes are progressing in clinical trials.3,4 One of the most prominent and studied epigenetics targets in the cancer field is lysine-specific demethylase I (LSD1 or KDM1A). LSD1 is a member of the flavine adenine dinucleotide-dependent (FAD-dependent) amine oxidase (AO) family of demethylases integrated in several chromatin modifying multiprotein complexes such as rest co-repressor (CoREST) and nucleosome remodeling and deacetylase (NuRD).5−7 It is known to demethylate lysines 4 and 9 on the tail of histone 38,9 as well as other nonhistone substrates such as p53.10 When methyl groups are removed from mono- and dimethylated H3K4, LSD1 is known to repress gene expression.8 Conversely, it has been reported to activate gene transcription by demethylating H3K9 in complex with nuclear hormone receptors.9LSD1 is an essential gene in mammalian biology, and many different specific roles have been reported.11 LSD1 is also involved in the generation of hematopoietic stem cells12 and its knockout results in the reduction of granulopoiesis, erythropoiesis, and platelet production.13 These effects must be factored in when designing pharmacological applications for LSD1 inhibitors.
LSD1 is overexpressed in many proliferative diseases including hematological, lung, breast, and prostate cancers,14−16 highlighting it as an attractive target in oncology. In addition, since LSD1 is recruited to target genes via interaction with transcription factors, a catalysis-independent protein–protein interaction (PPI) with SNAIL/growth factor independent 1 (SNAG)-domain transcription factors (TFs) has been found to be essential for LSD1 as a cancer driver,17,18 silencing gene expression, blocking cellular differentiation, and promoting cancer stemness. SNAG-domain TFs feature an N-terminal domain that mimics the histone H3 tail, interacting with the active site of LSD1 in a similar fashion to its substrates.19,20 This SNAG-domain TF/LSD1 PPI is key for the structural integrity of many transcription silencing complexes and is, in fact, the driver promoting differentiation block in acute myeloid leukemia (AML) and promoting the neuroendocrine phenotype in small cell lung cancer (SCLC), where the TFs involved are growth factor independent 1 (GFI1)21−23 and insulinoma-associated protein 1 (INSM1),23,24 respectively. Other neuroendocrine tumor types where GFI1 and INSM1 play pivotal roles are medulloblastoma25 and Merkel cell carcinoma.26−28
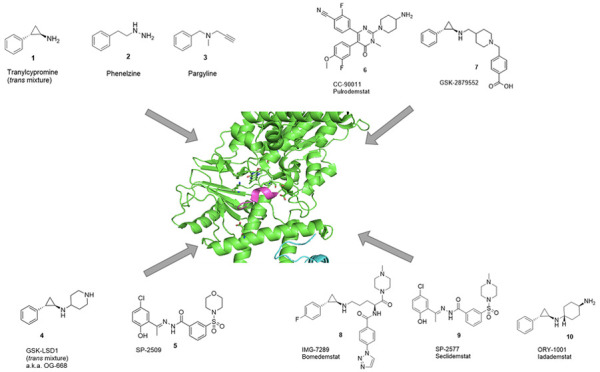
A growing body of knowledge has been built over the last 15 years on LSD1 inhibitors. The literature describes the development of active-site interacting linear peptide inhibitors,29 cyclic peptide inhibitors,30,31 and a variety of reversible and irreversible low molecular weight ligands,32−34 many of which have been supported by structure-based approaches.35,36 In the irreversible class, several AO inhibitors have been described to covalently bind to the FAD cofactor of LSD1; tranylcypromine (TCP or Parnate, 1), phenelzine (PLZ, 2), and pargyline (PRG, 3) are among them. TCP, PLZ, and PRG together with GSK-LSD1 (4) (a.k.a. OG-668, as it is a compound first described in Oryzon’s patent application WO2013/057320 (example 1)) and SP-2509 (5) (Figure 1) have been extensively used as tool compounds in the literature to explore the biological roles of LSD1 and the potential of LSD1 inhibition as a therapeutic approach in oncology. Among other roles, TCP has been described to induce differentiation and impair the survival of myeloid blasts in AML in combination with all-trans retinoic acid (ATRA).37 PLZ has been described to target FAD and to additionally interfere with the assembly of the CoREST multiprotein complex in triple-negative breast cancer (TNBC).38 By the inhibition of LSD1, PRG has been hypothesized to block epithelial-to-mesenchymal transition (EMT) in kidney tumors39 and to slow the growth of xenografts in oral and prostate cancer.40,41 OG-668 has been found to inhibit differentiation block in AML42 and to have therapeutic synergy in combination with ATRA.43 SP-2509 has been described as a reversible LSD1 tool compound, which is reported to have anticancer properties in prostate tumors44 and in Ewing’s sarcoma.45 Some of these chemical probes have also been used as starting points for the design of selective, next-generation LSD1 inhibitors.
Figure 1.

2D structures of representative LSD1 ligands with tool compounds 1–5 and clinical stage inhibitors 6–10.
Several reversible and covalent LSD1 inhibitors have entered clinical trials in oncology, namely: CC-90011 (pulrodemstat) (6),46 GSK-2879552 (7),47,48 IMG-7289 (bomedemstat) (8),49 SP-2577 (seclidemstat) (9),50 ORY-1001 (iadademstat) (10)51,52 (Figure 1), and INCB05987253 (structure undisclosed, therefore not included in the current analysis). CC-90011 is currently in clinical trials for advanced SCLC and AML. GSK-2879552, discontinued, was tested in clinical trials for AML, SCLC, and myelodysplastic syndrome (MDS). Bomedemstat is currently in clinical trials for myeloid-related malignancies. Seclidemstat is also in clinical trials for MDS and Ewing’s sarcoma. Iadademstat is currently in clinical trials for AML and SCLC. Thus, the efficacy of LSD1 inhibitors is currently being validated in the clinic for both hematological and solid tumors.
Despite the wealth of information published, a common analysis of how this compound class modulates LSD1’s dual enzymatic/scaffolding role is missing: in particular, the relationship between catalytic inhibitory potency and disruption of PPI with TFs remains to be explored. The differences in relative potency and selectivity between the tool compounds and the clinical trial candidates in biochemical and cell-based assays should also be clarified, especially in view of the multiple mechanistic cancer studies where these tool compounds have been used. Thus, this paper represents an attempt to characterize in vitro, under the same experimental conditions, the whole of the small molecule LSD1 inhibitor class in oncology. For this purpose, 1–10 were first tested for biochemical activity and selectivity, followed by efficacy in cell-based assays for hematological (AML) and solid (SCLC) tumors and in the associated mechanistic analyses of LSD1 target engagement, biomarker expression, and the disruption of SNAG-TF/LSD1 interactions.
Results
Solubility and Stability of LSD1 Inhibitors
Before biological testing, all the compounds under study were characterized in terms of physicochemical properties. To this end, the stability and solubility in aqueous buffer of 1–10 were determined experimentally, as fast degradation and poor solubility could lead to potential artifacts in all subsequent assays.54
Regarding stability, all compounds were incubated for 6 days in aqueous buffer at 37 °C (temperature used in cell-based assays) and their integrity was checked at days 0, 4, and 6. As shown in Table S1, some of the tool compounds show remarkable stabilities with >90% of parent compound remaining up to day 6. PLZ and SP-2509 were exceptions, as only 4% and 68%, respectively, remained at day 6, pointing at a low stability under the conditions tested. Similarly, all the clinical candidates exhibited excellent stabilities (>90% up to day 6), seclidemstat being the only exception with 72% remaining at day 6. Inspection of the structures reveals that all the unstable compounds (PLZ, SP-2509, and seclidemstat) feature either a hydrazine or an acylhydrazone chemical function. We hypothesize that the latter might be the cause of chemical instability in aqueous buffer for these compounds.55
Compounds were also tested for their turbidimetric solubility to establish the maximum concentrations at which each compound could be evaluated in vitro. As shown in Table S2, the solubility of the whole class is good overall; compounds 1–4, 6–8, and 10 did not precipitate at the maximal concentration tested. The only two compounds displaying high absorbance at 100 μM were SP-2509 and seclidemstat, pointing at solubility issues under the conditions tested. For this reason, the latter compounds were tested at a maximum concentration of 30 μM in the in vitro assays.
Catalytic Inhibitory Potency and Selectivity
As previously stated, the main goal of the current study was to place compounds 1–10 in the same reference frame. For this purpose, they were tested for biochemical activity, first on their primary target (LSD1) and then on structurally related amine oxidases, to determine their selectivity.
LSD1 IC50 was determined with two different methodologies: horseradish peroxidase (HRP) coupled assay56 and homogeneous time-resolved fluorescence assay (HTRF).57 Results of the HTRF assay are reported in Figure 2, while IC50’s obtained with both methodologies are reported in Table S2.
Figure 2.

Biochemical characterization of LSD1 inhibitors. Dose–response curves of LSD1 inhibition for tool compounds (A) and clinical stage inhibitors (B) determined with the HTRF assay. Selectivity of compounds vs other FAD-dependent enzymes (C). aValues obtained with the HTRF assay. bCompound not soluble at 100 μM. Data are represented as mean ± SD of N = 2, n = 2 or 3.
Among the tool compounds, the IC50’s of TCP and SP-2509 are in the micromolar range (5.6 and 2.5 μM, respectively), whereas the IC50’s of PLZ and PRG are in the millimolar range. Only OG-668 shows an IC50 in the low nanomolar range (7.6 nM). Regarding the clinical candidates, their IC50’s are significantly lower than those of the tool compounds (except for OG-668). Seclidemstat displays the highest IC50 (1.3 μM), followed by GSK-28799552 and bomedemstat, which feature an IC50 in the double-digit nanomolar range (56.8 nM), and only iadademstat and pulrodemstat have subnanomolar potencies of 0.33 and 0.66 nM, respectively, very close to the detection limit of the assay.
The data obtained with the HTRF assay were corroborated by the HRP coupled assay (see Table S2), giving the same ranking of potency among the compounds, although all values are generally higher because of the lower sensitivity of the latter assay.
The best measure of inhibitory potency for an irreversible inhibitor is the second-order rate constant obtained from the ratio kinact/KI, since it considers both the affinity of initial noncovalent binding (KI) and the rate of the subsequent covalent bond formation (kinact). TCP has been described to inhibit LSD1 through the formation of a covalent adduct with FAD with a kinact/KI of 22 M–1 s–1.58 PLZ has been reported to be 35-fold more efficient as an LSD1 irreversible inactivator than TCP,56 while irreversible inhibition of LSD1 by PRG has been detected only at millimolar concentrations, demonstrating that this compound is 120-fold less potent than TCP.59 We determined the kinetic parameters of LSD1 irreversible inhibitors using the well-established horseradish peroxidase coupled assay (Table S3). In our hands, TCP displayed a kinact/KI of 34 M–1 s–1, while we could not observe a time-dependent inhibition of LSD1 by PRG during the time course of the assay, even using millimolar concentrations of inhibitor. PLZ could not be tested due to compound fluorescence at the wavelength used for the HRP coupled assay. Among the irreversible tool compounds, OG-668 displayed the highest inactivation efficiency (kinact/KI = 2.57 × 104 M–1 s–1), mostly due to a high noncovalent affinity toward LSD1 (KI = 626 nM).
Among the clinical stage compounds that display an irreversible mechanism of inhibition, iadademstat was by far the most active compound (kinact/KI = 1.19 × 106 M–1 s–1): it is 177-fold more potent than GSK-2879552 and 73-fold more potent than bomedemstat. Iadademstat features not only the highest rate of inactivation among the irreversible LSD1 inhibitors tested but also the highest noncovalent affinity, being the only inhibitor displaying a low nanomolar KI.
SP-250960 and the clinical stage compounds seclidemstat61 and pulrodemstat46 have been described as reversible LSD1 inhibitors. Jump dilution analysis62 applied to the HRP coupled assay confirmed the reversible nature of inhibition. As shown in Figure S1, after incubation with pulrodemstat, LSD1 recovered 82% of activity upon jump dilution, while with SP-2509 and seclidemstat the recovery was 40% and 62%, respectively. As a control, we used TCP for which there was no recovery of activity, as expected with an irreversible inhibitor.
The selectivity of 1–10 in the amine oxidase class was determined by checking the inhibition of three other FAD-dependent enzymes, namely, the closely related LSD2 (KDM1B) and monoamine oxidases A and B (MAO-A and MAO-B). LSD2 inhibition was evaluated with both the HRP coupled assay and the HTRF assay, as for LSD1, while MAO-A and MAO-B inhibition was determined with the kynuramine assay.63 As shown in Table S2, neither the tool compounds nor the clinical candidates inhibit LSD2 with IC50 values around or above 100 μM. Due to solubility issues, SP-2509 and seclidemstat were tested at 10 μM: at this concentration, both compounds inhibit LSD2 less than 50%. As shown in Figure S2 and Table S2, several tool compounds inhibit MAO-A and MAO-B with IC50’s that go as low as submicromolar. TCP inhibits MAO-A and MAO-B with IC50’s of 2.84 and 0.73 μM, respectively; PLZ inhibits both with IC50’s of 0.42 and 0.83 μM and PRG, with IC50’s of 3.84 and 0.24 μM, respectively. Only OG-668 and SP-2509 do not inhibit MAOs with IC50’s over 100 μM. Thus, as previously reported, TCP, PLZ, and PRG were confirmed to be more active on MAO-A/B than on LSD1.
Taken together, our biochemical data confirm that most of the tool compounds, except for OG-668, are weak and nonselective LSD1 inhibitors as they inhibit other AOs. This calls for caution as TPC, PLZ, PRG, and SP-2509 have been used in multiple studies to decode the involvement of LSD1 in several physiological and pathological processes, especially cancer. Our analysis also shows that, although the clinical stage compounds are on average more potent, they vary widely as LSD1 inhibitors. Seclidemstat is the least potent LSD1 inhibitor among the clinical candidates in our hands (1.3 μM), followed by GSK-2879552 and bomedemstat, which are 160 and 57 nM in the HTRF assay, respectively. Clearly, the two most potent LSD1 clinical candidates are Oryzon’s iadademstat (0.33 nM) and Bristol Myers Squibb’s pulrodemstat (0.66 nM). Collectively, our data strongly recommend using the tool compound OG-668 and the clinical candidates pulrodemstat or iadademstat as specific LSD1 inhibitors in pharmacological studies involving LSD1 inhibition.
In Vitro Efficacy in Hematological Tumors (AML)
AML has been extensively established as one of the tumors driven by the interaction between LSD1 and the transcription factor GFI1.21−23 The direct binding of the two proteins is known to cause differentiation block and promote cancer stemness via repression of a series of target genes including cluster of differentiation molecule 11b/integrin alpha-M (CD11b/ITGAM).64 Since numerous studies suggest high sensitivity of AML to LSD1 inhibition,65 we decided to test 1–10 for impact on AML viability, LSD1 TE, and expression of cell differentiation markers in a panel of AML cell lines. Figure 3 shows the effect of the tool and clinical compound sets on a representative AML line (additional cell lines in Figure S3).
Figure 3.

Efficacy in AML. (A) Effect of tool (left) and clinical stage (right) LSD1 inhibitors on cell viability in the AML cell line TF-1a following 96 h of incubation at concentrations ranging from 0.002 to 2000 nM (N = 2, n = 3). (B) Assessment of LSD1 target engagement in the AML cell line MV(4;11) treated with tool (left) and clinical stage (right) LSD1 inhibitors following 24 h of incubation at 2000, 200, 20, and 2 nM (N = 1, n = 3). (C) CD11b protein levels were assessed by flow cytometry in THP-1 cells following 96 h of incubation with tool (left) and clinical stage (right) LSD1 inhibitors at concentrations ranging from 0.02 to 2000 nM (N = 2 for iadademstat, N = 1 for the rest). (D) The effect of tool (left) and clinical stage (right) compounds on SNAG-domain/LSD1 interaction was assessed using a modified ELISA assay testing the binding of recombinant LSD1 previously incubated with LSD1 inhibitors to a peptide containing the GFI1B SNAG domain (N = 1, n = 3). All data are expressed as relative to the vehicle condition and represented as mean ± SD.
Among the tool compounds, OG-668 has the strongest antileukemic activity (subnanomolar EC50 for 2 out of 3 cell lines tested), followed by SP-2509, which displays an EC50 of 1624.50 nM for TF-1a and 203.35 and 422.00 nM for MOLM-13 and MV(4;11), respectively (Figures 3A and S3 and Table S4). The EC50 of TCP, PLZ, and PRG could not be determined, as they are above 2 μM, the highest concentration tested in the assay. Among the clinical candidates, seclidemstat behaves similarly to its parent tool compound SP-2509, displaying low activity across the panel of cell lines (Figures 3A and S3 and Table S4). The rest of the clinical candidates show nanomolar EC50 for the three AML lines with the following ranking from more to less potent: iadademstat > pulrodemstat > bomedemstat > GSK-2879552. The inspection of EC50’s clearly points at iadademstat as the most potent in antileukemic activity, as it is the only LSD1 inhibitor tested with subnanomolar activity in the whole panel (Figures 3A and S3 and Table S4). As noted above, the mechanism of action of the LSD1 inhibitor class involves inhibition of differentiation block rather than displaying a clear cytotoxic effect; this explains why, in cell-based assays, even the most potent inhibitors show a viability reduction of ca. 60% instead of an absolute 100% inhibition.
To assess whether the impact on AML cell viability is dependent on the efficiency of cellular LSD1 inhibition, we used a proprietary LSD1 target engagement (TE) assay based on the use of a biotinylated chemoprobe, which covalently inhibits the LSD1 enzyme.66 Among the tool compounds, OG-668 is the only compound achieving high LSD1 TE at all concentrations tested (Figure 3B). TCP achieves acceptable (>70%) LSD1 TE only at the highest concentration (2 μM), whereas we observe no LSD1 TE for PLZ, PRG, and SP-2509.
The clinical candidates reveal higher LSD1 TE values than the tool compounds (other than OG-668) with a ranking that is perfectly in line with the effects on cell viability. In particular, the most active compound is iadademstat, followed by pulrodemstat, bomedemstat, and GSK-2879552 (Figure 3B). Surprisingly, seclidemstat displays no LSD1 TE, in line with the limited effect on AML cell viability. These results point at LSD1 TE as the key factor determining the efficacy observed in the AML viability assay.
To assess whether the efficacy observed in the viability assays is correlated with overcoming the differentiation block in AML induced by LSD1 inhibition, we used a flow cytometry assay measuring the induction of the CD11b/ITGAM marker in THP-1 cells. As shown in Figure 3C and Table S5, TCP shows a modest induction with an EC50 of ∼1.4 μM, again with OG-668 being the only tool compound with potent activity (0.62 nM). PLZ, PRG, and SP-2509 show no induction of this marker at the concentrations tested; thus, their EC50 could not be determined. The clinical candidates show higher potency with a ranking that is in line with the viability and the LSD1 TE data. Thus, CD11b induction ranks candidates, from more to less potent, in the following order: iadademstat > pulrodemstat > bomedemstat > GSK-2879552 (Figure 3C and Table S5). In agreement with the above data (Figure 3A,B), seclidemstat does not induce the CD11b marker, adding further evidence that this compound has limited effects on AML cell lines.
Finally, we assessed the effect of the compounds on the disruption of the LSD1/GFI1 PPI by using an in vitro assay measuring the interaction between recombinant LSD1 and a peptide containing the GFI1B SNAG domain sequence. Among the tool compounds, again only OG-668 shows reduction of the LSD1/GFI1B interaction in the 2 μM to 2 nM range, whereas TCP, PLZ, PRG, and SP-2509 are essentially inactive at the concentrations tested (only TCP shows some activity at 2 μM) (Figure 3D). Among the clinical candidates, iadademstat and pulrodemstat are the most potent in disrupting the LSD1/GFI1B PPI, followed by bomedemstat and by GSK-2879552, in line with their respective percentages of LSD1 TE (compare Figure 3B,D). On the other hand, seclidemstat does not disrupt the LSD1/GFI1B interaction (Figure 3D), in agreement with the lack of LSD1 target engagement.
Taken together, our data reveal that the tool compounds have very low efficacy on AML cell lines with the exception of OG-668. Among the clinical candidates, iadademstat and pulrodemstat are the most potent in reducing AML viability, inducing differentiation and disrupting the LSD1/GFI1 PPI. Bomedemstat shows intermediate potency in all tests, while seclidemstat exhibits a limited impact on viability, which seems to be through mechanisms that are not linked to the inhibition of LSD1 catalytic activity nor to the disruption of the LSD1/GFI1 complex.
In Vitro Efficacy in Solid Tumors (SCLC)
SCLC is also a well-established tumor type driven by the interaction of LSD1 with a SNAG-domain transcription factor, in this case INSM1.23 The disruption of this complex results in the modulation of genes leading to NOTCH signaling activation and consequent downregulation of neuroendocrine factors such as ASCL1, a main driver of the neuroendocrine transcriptional program, ultimately inhibiting SCLC progression.67 In a homologous fashion to AML, 1–10 were tested for their impact on cell viability, LSD1 TE, expression of the neuroendocrine marker gastrin-releasing peptide (GRP), and the disruption of the LSD1/INSM1 interaction in the SCLC cell line NCI-H510A.
As seen for AML, the impact on SCLC cell line viability also shows great variation. Among the tool compounds, only OG-668 displays strong anticancer activity (EC50 of 1.17 nM). EC50’s for TCP, PLZ, and PRG could not be determined (above 2 μM), and SP-2509 displays an EC50 of 56.24 nM (Figure 4A and Table S4). Among the clinical candidates, seclidemstat behaves similarly to its parent SP-2509, displaying moderate activity (EC50 of 184.35 nM). The rest of the clinical candidates display low nanomolar EC50 activities with a ranking that perfectly matches that obtained for AML: iadademstat > pulrodemstat> bomedemstat > GSK-2879552. Iadademstat is again the most potent clinical candidate with subnanomolar (EC50 = 0.16 nM) antiproliferative activity, being around 10-fold more potent than the second best, pulrodemstat (1.77 nM) (Figure 4A and Table S4).
Figure 4.

Efficacy in SCLC. (A) Effect of tool (left) and clinical stage (right) LSD1 inhibitors on cell viability in the SCLC cell line NCI-H510A following 10 days of incubation at concentrations ranging from 0.002 to 2000 nM (N = 2, n = 3). (B) Assessment of LSD1 target engagement in the NCI-H510A cells treated with tool (left) and clinical stage (right) LSD1 inhibitors following 24 h of incubation at 2000, 200, 20, and 2 nM (N = 1, n = 3). (C) GRP gene expression levels were assessed by qRT-PCR in NCI-H510A cells following 6 days of incubation with tool (left) and clinical stage (right) LSD1 inhibitors at 2000, 200, 20, and 2 nM (N = 1, n = 3). Data were calculated using the 2–ΔΔCt method, normalized by GUSB. (D) The effect of tool (left) and clinical stage (right) compounds on the SNAG-domain/LSD1 interaction was assessed using an INSM1/LSD1 interaction ELISA assay performed on nondenaturing cell lysates of NCI-H510A cells following 24 h of incubation with compounds at 2000, 200, 20, and 2 nM (N = 1, n = 3). All data are expressed as relative to the vehicle condition and represented as mean ± SD.
The analysis of LSD1 TE confirms the viability results. Among the tool compounds, OG-668 shows high LSD1 TE (>80%) at 20 nM, followed by TCP, which displays LSD1 TE only at the highest concentration of 2 μM (Figure 4B). None of the other tool compounds show any LSD1 TE at the concentrations used. Among the clinical stage compounds, the irreversible inhibitors show strong target engagement with a potency that correlates perfectly with the viability reduction, as does the reversible pulrodemstat. Seclidemstat, however, did not show any significant LSD1 TE at the concentrations tested.
Subsequently, we assessed the effect of the compounds on the expression of the neuroendocrine gene GRP, an established SCLC marker.68 OG-668 is the only tool compound able to reduce GRP expression at all concentrations (Figure 4C). PLZ, PRG, and SP-2509 show no impact on the GRP expression at the concentrations tested, whereas TCP shows activity only at the highest concentration (2 μM). Among the clinical candidates, iadademstat is the compound that most potently inhibits the expression of the neuroendocrine biomarker (including at 2 nM), followed by pulrodemstat, bomedemstat, and finally GSK-2879552 (Figure 4C). In line with all the above, seclidemstat does not impact GRP expression as LSD1 target engagement was negligible.
Finally, we assessed the impact of the compounds on the disruption of the LSD1/INSM1 PPI, a well-established driver of SCLC.24 As with the AML panel, PLZ, PRG, and SP-2509 are essentially inactive at the concentrations tested. TCP shows moderate activity only at the highest concentration (2 μM) in line with the moderate 60% LSD1 TE value. Among the tool compounds, only OG-668 shows a reduction of the LSD1/INSM1 interaction at all four concentrations (Figure 4D). The clinical candidates display much higher activities with iadademstat and pulrodemstat again being more potent than bomedemstat and GSK-2879552 due to their higher LSD1 target engagement. Seclidemstat is totally inactive at disrupting LSD1/INSM1 PPI in line with the lack of significant LSD1 target engagement (Figure 4D).
Taken together, the cell-based results for SCLC follow closely those obtained for AML cell lines. Most tool compounds tested have very low activities as inhibitors of SCLC viability. OG-668 was an exception and ranked second only to iadademstat in most cellular assays. Among the clinical candidates, iadademstat ranks first as the most potent in reducing viability, downregulating the expression of the neuroendocrine marker GRP, and reducing the LSD1/INSM1 PPI interaction. Seclidemstat is devoid of LSD1 TE and of any activity as LSD1/INSM1 disruptor and does not impact gene expression of the neuroendocrine marker GRP.
Discussion
With the goal of characterizing the whole LSD1 inhibitor compound class (Figure 1), we provide data comparing potency, selectivity, and cellular activity of a set of LSD1 inhibitor tool compounds and clinical stage candidates in oncology. Our global analysis allowed us to run a series of highly interesting pairwise comparisons that so far could be neither found nor derived from the literature, such as (i) activity as inhibitors of catalysis vs activity as PPI disruptors; (ii) activities of tool compounds vs clinical candidates; (iii) activities of irreversible vs reversible compounds; (iv) activity as PPI disruptors vs expression of cellular differentiation markers; (v) activity as PPI disruptors vs antiproliferative activity in cell viability assays. Additionally, the in vitro data obtained here could be used to partly rationalize clinical developments of this compound class.
Overall, the potency ranking of catalytic inhibition of the tested inhibitors paralleled their LSD1 TE and consequently their capacity to disrupt the SNAG-domain/LSD1 PPI. The compounds with the highest SNAG-domain/LSD1 PPI-disrupting activities exhibited the highest antiproliferative properties, LSD1 TE, and regulation of cell differentiation markers (induction of CD11b in AML and repression of GRP in SCLC) in the selected cell lines, thus confirming the established hypothesis.21−23
Iadademstat and OG-668 were the most potent irreversible inhibitors in all in vitro assays performed. OG-668 was confirmed to be a robust tool compound for in vitro cellular studies. It is less potent than iadademstat in vitro (and also in in vivo models of SCLC, data not shown), but it is selective for LSD1 over other AOs, and has low nanomolar activity as anti-proliferative in AML and SCLC, in line with a high activity in LSD1/GFI1 and LSDI/INSM1 PPI assays and impact on both differentiation markers. The quality of chemical probes in cancer biology has been recently discussed in Blagg and Workman,69 where OG-668 (GSK-LSD1) is described as a reliable tool compound.
GSK-2879552 and bomedemstat are notably less potent, displaying medium activities in all LSD1 related assays. For example, GSK-2879552 and bomedemstat are 491-fold and 174-fold less active than iadademstat in the LSD1 HTRF assay, respectively, and 849-fold and 345-fold less active in the (TF-1a) AML viability assays, respectively, in line with their lower LSD1 TE values.
Although they have been extensively used in LSD1 mechanistic studies, TCP, PLZ, and PRG are poor to extremely poor and nonselective LSD1 inhibitors with low PPI-disrupting activities, low impact on cellular differentiation markers, and low activity as antiproliferative treatments for AML and SCLC. TCP has been described to impact AML viability in combination with ATRA,37 in accordance with previous data; in our hands, it showed micromolar activity in LSD1 biochemical assays, activity in the LSD1/GFI1 binding assay only at the highest concentration tested (2 μM), a modest induction of the differentiation marker CD11b, and no activity as an AML antiproliferative agent at concentrations up to 2 μM. PLZ has also been described to be an LSD1 inhibitor and to irreversibly inactivate FAD, altering LSD1 catalytic activity and its interaction with RCOR proteins.38 PRG has been reported to impact viability by inhibiting LSD1 in nonsmall cell lung cancer (NSCLC)70 to inhibit growth in an LSD1-dependent manner in breast cancer,71 to inhibit EMT in prostate cancer,41 and to reduce kidney cancer progression via LSD1 inhibition.72 However, these compounds inhibited LSD1 only at millimolar concentrations in our HTRF assay and were inactive in the cell-based assays at 2 μM. Most data in the literature attributing biological effects of TCP, PLZ, and PRG to LSD1 inhibition were generated at concentrations at least 100-fold beyond the doses required to fully inhibit the targets for which they were originally developed. Off-target effects alternative to LSD1 inhibition may thus significantly contribute to the reported biological activities, as it was recently shown for PLZ in enzalutamide-resistant prostate cancer,73 and therefore, these compounds are far from ideal to assess LSD1 function and should thus be avoided.
Regarding reversible inhibitors, tool compound SP-2509 and related clinical candidate seclidemstat behaved as clear outliers. Unlike what was found for the other tested compounds, which yield ≈5- to 30-fold lower IC50 values in the HTRF compared to the HRP assay, SP-2509 (2.5 and 2.6 μM) and seclidemstat (1.3 and 2.4 μM) had similar potencies in both assays. The moderate antiproliferative activity of SP-2509 and seclidemstat in AML and SCLC cells was not accompanied by significant LSD1 TE. While it could be argued that the LSD1 TE assay used in this study was optimized primarily for irreversible inhibitors,66 target engagement of (reversible) pulrodemstat was readily detected by our method. We did not detect the disruption of LSD1/GFI1 or LSD1/INSM1 PPI interactions for SP-2509 or seclidemstat nor the induction of differentiation markers in AML or the reduction of neuroendocrine markers in SCLC at the concentrations tested. Therefore, our results do not support the hypothesis that these compounds bind LSD1 in any productive manner to impact catalytic activity or SNAG-domain interactions. SP-2509 has been hypothesized to allosterically disrupt the interaction of LSD1 with a series of protein partners (i.e., Zinc Finger protein 217, ZNF217) by binding at the enzyme’s active site,44,74 which is at odds with our results given its nil target engagement. Of note, in silico efforts to generate productive binding modes of this compound in the LSD1 active site failed (both rigid docking approaches such as Schrodinger’s Glide75 and flexible Monte Carlo simulations with the Pele platform76 were used), whereas productive poses could be generated for pulrodemstat (data not shown). SP-2509 has been suggested to be a pan-assay interference inhibitor77 (PAINs78,79) and its antiproliferative activity, a result of unidentified off-target effects,80 which could be consistent with the results presented herein. Unlike seclidemstat, we confirmed pulrodemstat to be a bona fide, active site targeted reversible LSD1 inhibitor. Pulrodemstat is almost as potent as iadademstat in the HRP and HRTF assays, although less potent in cellular assays.
Up to date, 7 different inhibitors have been tested in the clinic in oncology, most of which are included in the present study. It is difficult to anticipate to what extent the preclinical features of these drugs may predict their clinical success. Iadademstat was the first inhibitor to enter clinical trials. It was found to be safe and well-tolerated and produced clear differentiating events and some antileukemic activity in the first in human trial in RR-AML.52 In an ongoing Phase II in combination with the hypomethylating agent azacytidine, the 30 month data of the study showed high rates of antileukemic activity, fast time to responses, and long-lasting remissions.81 The drug is also being explored in SCLC. More than 100 patients have been treated so far with iadademstat, and due to its high activity, doses in human patients range from 60 to 90 μg/m2/day (100 to 150 μg/day).
GSK-2879552, a compound with intermediate biochemical and cell potency, was the second selective LSD1 inhibitor to enter the clinic and to be explored in AML, MDS, and SCLC. Unexpectedly, in the NCT02034123 SCLC trial, the drug produced 10% G3/4 encephalopathy with also one G5 encephalopathy death.82 To the best of our knowledge, encephalopathies have not been reported with any other LSD1i and may therefore represent a compound-specific effect rather than on-target toxicity. GSK-2879552 was ultimately discontinued in the three indications as its risk-benefit profile did not favor further development.
Pulrodemstat is currently being tested in five trials in AML, Non-Hodgkin’s lymphomas, and prostate and small cell lung cancers, alone or in different combinations and regimens. Preliminary data showed encouraging antitumor activity and favorable tolerability in patients with unresectable solid tumors and relapsed and/or refractory Non-Hodgkin’s lymphoma.83 Cyclopropylamine compounds like iadademstat irreversibly inactivate LSD1 by generating an adduct with the FAD cofactor,32,33,51 whereas pulrodemstat resides reversibly in the active site cavity.46 The theoretical advantage of the reversible nature of the interaction would be an increased control of excessive on-target activity. However, due to a long half-life (≈70 h) reported in humans, the target occupancy of pulrodemstat is expected to be similar to that of covalent molecules,83 and the reported dosing scheme in the trials (1QW) would seem to confirm this. Thrombocytopenia and neutropenia events are consistent with the target biology and were indeed reported for this compound in monotherapy.83,84
Bomedemstat, a notably less potent irreversible LSD1 inhibitor, is being explored in essential thrombocythemia and myelofibrosis (MF). The Phase II doses (40–60 mg/day) are about 500-fold higher than iadademstat, exceeding the 10 mg/day threshold managed commonly in the risk assessment for idiosyncratic toxicity of covalent binders and increasing the risk for off-target activities.85,86 Preliminary results in MF are encouraging although less promising than those obtained using other epigenetic compounds, like the BET-1 inhibitor CPI-0610, in terms of Total Symptom Score 50 (TSS50) and Spleen Volume Reduction (SVR35) readouts.87 This might indicate a lower actionability in MF by targeting LSD1 instead of BET-1 or be a consequence of its more modest LSD1 inhibitory activity.
In summary, this extensive chemical, biochemical, and cell-based characterization of the LSD1 inhibitor class for the first time placed most of the tool compounds and clinical candidates under the same reference frame for biochemical inhibition, PPI, and antiproliferative activities. The data at multiple levels are consistent and robust. A visual summary is provided in Figure 5. Globally, our profiling highlights OG-668 as the preferred in vitro tool compound and iadademstat as the most potent clinical stage candidate. The work presented here is expected to be of use to guide the selection of inhibitors for use in mechanistic cancer studies and to better understand the incoming clinical data from the clinical stage LSD1 inhibitors.
Figure 5.

Summary of compound performance. (A) LSD1 IC50 values obtained with the HTRF assay. (B) Efficacy in AML cell lines. (C) Efficacy in the SCLC cell line. The graphs were generated with GraphPad Prism 9 using data discussed previously. Clinical stage compounds are represented as circles; tool compounds are represented as triangles.
Methods
Commercially available compounds were purchased directly from vendors: compounds 2, 5, 6, and 9 (MedChemExpress, refs. HY-B1018A (2), HY-12635 (5), HY-129388B (6), and HY-103713 (9)), 1 (Selleckchem, ref. S4246), and 3 (Sigma-Aldrich, ref. P8013). Compounds 4 (OG-668), 7 (GSK-2879552), 8 (bomedemstat), and 10 (iadademstat) were obtained by custom synthesis at Oryzon following the procedures described in the corresponding patents: 4 (WO2013/057320 A1), 7 (WO2012/135113 A2), 8 (WO2018/035259), and 10 (WO2013/057322 A1).
Turbidimetric Solubility Assay
The kinetic solubility of the compounds was determined by the turbidimetric solubility assay. The DMSO stock solution of the compounds was serially diluted 1:3 in DMSO in duplicate and then transferred to phosphate buffered saline, pH 7.4. The tested compound concentrations were the same as those used in the biochemical assays. The final DMSO concentration was 0.5%. Absorbance at 620 nm was measured on a Tecan Infinite F200 microplate reader and plotted against compound concentration. A precipitation was indicated by a slope change of the segmental linear regression.
Stability in Aqueous Buffer
A 20 mM DMSO solution of each compound was prepared. 50 μL of this 20 mM DMSO solution was diluted with 150 μL of PBS buffer, and the solution was stored at 37 °C. Samples were analyzed at T = 0, 4, and 6 days by LCMS. Compounds 2, 4, 5, and 9 were analyzed by HPLC method conditions A. Compound 8 was analyzed by HPLC method conditions B, and compounds 1, 3, 6, 7, and 10 were analyzed by HPLC method conditions C.
HPLC Method Conditions A
Column: AQUITY UPLC BEH C18 1.7 μm, 2.1 × 50 m. Mobile phase A: 10 mM ammonium bicarbonate in water. Mobile phase B: 100% ACN. Time (min) and % B: 0−5; 0.3−5; 7.0−90; 8.6−90; 9.2−5; 10.0−5. Flow rate: 0.5 mL/min.
HPLC Method Conditions B
Column: XBridge C18 4.6 × 150 mm, 3.5 μm. Mobile phase A: 10 mM ammonium bicarbonate in water. Mobile phase B: acetonitrile. Gradient (T/% B): 0/10, 07/90, 15/90, 15.01/10. Flow rate: 1.0 mL/min.
HPLC Method Conditions C
Column: Kinetex EVO C18 50 × 4.6 mm, 2.6 μm. Mobile phase: A: 10 mM NH4HCO3, pH 8; B: ACN (95:5). Gradient (A/B): (95:5), 0.5 min; (95:5), 6.5 min; (0:100), 2 min; (0:100), post run 1.5 min. Flow rate: 1.5 mL/min.
Biochemical Assays
LSD1 Biochemical Assays
LSD1 inhibition was measured by the peroxidase coupled assay and the HTRF assay.
LSD1 Peroxidase Coupled Assay
The peroxidase coupled assay was performed as previously described.51 Briefly, 3-fold serial dilutions of compounds at different concentrations in duplicate were preincubated on ice with 38.5 nM human recombinant LSD1 enzyme (BPS Bioscience, ref. 50100) in 50 mM sodium phosphate, pH 7.4, for 15 min in black 96-well plates. An enzymatic reaction was initiated by the addition of dimethyl H3(1-21)K4 peptide substrate (Anaspec, ref. 63677) at a concentration corresponding to the Km. The final concentration of DMSO in the assay was 0.5%. After 30 min of incubation at 37 °C, the detection mix of Amplex Red and horseradish peroxidase (Invitrogen) was added according to the manufacturer’s recommendations, and the sample was incubated for 5 min at room temperature in the dark. The conversion of Amplex Red to resorufin was monitored by fluorescence (excitation: 540 nm; emission: 590 nm) using a Tecan Infinite F200 microplate reader. The control of LSD1 activity (100% activity) was obtained in the absence of inhibitor and corrected for background fluorescence in the absence of both LSD1 and substrate. Enzyme inhibition at each compound concentration was calculated as a percentage of inhibition of control of LSD1 enzyme activity. IC50 was determined by fitting the percentage of inhibition at each log compound concentration with a nonlinear 4-parameter equation using GraphPad PRISM version 9.0.1 (GraphPad Software, Inc., La Jolla, CA/USA).
LSD1 HTRF Assay
Serial dilutions of compounds at different concentrations in triplicate were preincubated for 15 min on ice with 0.45 nM human recombinant LSD1 enzyme (Active Motif, ref. 31426) in 50 mM Tris-HCl, pH 8.5, 50 mM NaCl, 0.01% Tween-20, and 1 mM DTT in white 384-well plates. The enzymatic reaction was initiated by the addition of a mix of 10 μM flavin adenine dinucleotide and biotinylated monomethyl H3(1-21)K4 peptide substrate (Anaspec, ref. 64355) at a concentration corresponding to the Km. The final concentration of DMSO in the assay was 0.5%. After 1 h of incubation at 25 °C, a detection mix containing 0.75 nM Eu3+-cryptate-labeled anti-H3K4me0 detection antibody (Cisbio, ref. 61KA0KAE), 20.8 nM XL665-conjugated streptavidin (Cisbio, ref. 610SAXLF), and 500 μM TCP (Selleckchem, ref. S4246) was added, and after 1 h of incubation at 25 °C, the TR-FRET signal was measured with a Tecan Infinite FPlex microplate reader (excitation: 320 nm; emission: 620 nm/665 nm). The HTRF ratio for each individual well was calculated as 104 × (emission at 665 nm/emission at 620 nm). The control of the LSD1 activity (100% activity) was obtained in the absence of inhibitor and corrected for background in the absence of LSD1. Enzyme inhibition at each compound concentration was calculated as a percentage of inhibition of control of LSD1 enzyme activity. IC50 was determined by fitting the percentage inhibition at each log compound concentration with a nonlinear 4-parameter equation using GraphPad PRISM version 9.0.1 (GraphPad Software, Inc., La Jolla, CA/USA). Potential compound interference with TR-FRET signal was inspected by incubating each compound at the highest concentration tested in the inhibition assay in the presence of 3 nM biotinylated H3(1-21) peptide product (Anaspec, ref. 61702) with all the components of the assay except for substrate and enzyme.
LSD2 Biochemical Assays
LSD2 inhibition was measured by the peroxidase coupled assay and HTRF assay.
LSD2 Peroxidase Coupled Assay
The peroxidase coupled assay was performed as previously described51 with minor modifications. 10-fold serial dilutions of compounds were preincubated on ice with 86.2 nM human recombinant LSD2 enzyme (Active Motif, ref. 31879) in 40 mM Hepes–NaOH, pH 8.5, for 15 min in duplicate in black 96-well plates. Dimethyl H3(1-21)K4 peptide substrate (Anaspec, ref. 63677) was then added at a concentration corresponding to the Km. The final concentration of DMSO in the assay was 0.5%. After 20 min of incubation at 25 °C, Amplex Red and horseradish peroxidase (Invitrogen) were added according to manufacturer’s recommendations and incubated for 5 min at room temperature in the dark. The conversion of Amplex Red to resorufin was monitored by fluorescence (excitation: 540 nm; emission: 590 nm) using a Tecan Infinite F200 microplate reader. The maximum of the LSD2 activity was obtained in the absence of inhibitor and corrected for background fluorescence in the absence of both LSD2 and substrate. Enzyme inhibition at each compound concentration was calculated as a percent of inhibition of LSD2 enzyme activity.
LSD2 HTRF Assay
10-fold serial dilutions of compounds were preincubated for 15 min on ice with 150 nM human recombinant LSD2 enzyme (Active Motif, ref. 31879) in 50 mM Tris-HCl, pH 8.0, and 0.02% Triton X-100 in white 384-well plates. The enzymatic reaction was initiated by the addition of a mix of 1 μM flavin adenine dinucleotide and biotinylated monomethyl H3(1-21)K4 peptide substrate (Anaspec, ref. 64355) at a concentration corresponding to the Km. The final concentration of DMSO in the assay was 0.5%. After 2 h of incubation at 25 °C, a detection mix containing 0.75 nM Eu3+-cryptate-labeled anti-H3K4me0 detection antibody (Cisbio, ref. 61KA0KAE) and 20.8 nM XL665-conjugated streptavidin (Cisbio, ref. 610SAXLF) was added, and after 1 h of incubation at 25 °C, the TR-FRET signal was measured with a Tecan Infinite FPlex microplate reader (excitation: 320 nm; emission: 620 nm/665 nm). The HTRF ratio for each individual well was calculated as 104 × (emission at 665 nm/emission at 620 nm). The control of LSD2 activity (100% activity) was obtained in the absence of inhibitor and corrected for background in the absence of LSD2. Enzyme inhibition at each compound concentration was calculated as a percentage of inhibition of control of LSD2 enzyme activity. IC50 was determined by fitting the percentage of inhibition at each log compound concentration with a nonlinear 4-parameter equation using GraphPad PRISM version 9.0.1 (GraphPad Software, Inc., La Jolla, CA/USA).
MAO-A and MAO-B Kynuramine Assay
The inhibition of human MAO-A and MAO-B enzymes was determined with the kynuramine assay as described by Maes et al.51 with minor modifications. Briefly, serial dilutions of compounds were preincubated in duplicate with recombinant MAO protein (Sigma-Aldrich, refs. M7316 and M7441) for 15 min on ice in 100 mM Hepes–NaOH, pH 7.5, in black 96-well plates. The final concentration of DMSO in the assay was 0.5%. Kynuramine was added at the corresponding Km, and the reaction was incubated for 1 h at 37 °C. The oxidative deamination of kynuramine was stopped with 2 N NaOH, and the conversion of kynuramine to 4-hydroxyquinoline was monitored at an excitation of 320 nm and emission of 380 nm with a Tecan Infinite F200 microplate reader. The maximum of MAO activity was obtained in the absence of inhibitor and corrected for background fluorescence in the absence of both enzyme and substrate. Enzyme inhibition at each compound concentration was calculated as a percent of inhibition of enzyme activity. IC50 was determined by fitting the percentage of inhibition at each log compound concentration with a nonlinear 4-parameter equation with GraphPad PRISM version 9.0.1 (GraphPad Software, Inc., La Jolla, CA/USA).
Irreversible Inactivation of LSD1
The kinetic parameters of irreversible inhibition of LSD1 were determined using the peroxidase coupled assay as previously described.51 Briefly, 1.5-fold serial dilutions of the compounds were mixed with dimethyl H3(1-21)K4 peptide substrate (Anaspec, ref. 63677) at a concentration corresponding to 1.3× Km, 50 μM Amplex Red, and 0.1 U/mL horseradish peroxidase (Invitrogen, ref. A22188) in 50 mM sodium phosphate, pH 7.4, in black 96-well plates. The final concentration of DMSO in the assay was 0.7%. After the addition of 10 nM LSD1 (BPS Bioscience, ref. 50100), the conversion of Amplex Red reagent to resorufin was continuously monitored by fluorescence (excitation: 540 nm; emission: 590 nm) with a Tecan Infinite F200 microplate reader at 25 °C. Background corrected progress curves obtained for each compound concentration were fit to the following equation for slow-binding inhibitors88
with vs = 0.
kobs values obtained at each compound concentration were fit to the following equation
where Kiapp = Ki (1 + [S]/Km).58
Determination of LSD1 Reversible Inhibition
The reversibility of inhibition was assessed by applying the jump dilution analysis62 to the peroxidase coupled assay. Human recombinant LSD1 enzyme (BPS Bioscience, ref. 50100) was incubated for 15 min at 25 °C with a concentration of compound equivalent to 10-fold the IC50 and then rapidly diluted 100-fold with a mixture containing the peroxidase coupled assay reagents and dimethyl H3(1-21)K4 peptide substrate (Anaspec, ref. 63677) at 1.3× Km. The progress curve was then monitored for 15 min by fluorescence (excitation: 540 nm; emission: 590 nm) with a Tecan Infinite F200 microplate reader at 25 °C.
After background subtraction, the recovery of enzyme activity upon jump dilution was expressed as a percentage of activity of LSD1 incubated and diluted in the absence of inhibitor and compared with the activity of LSD1 incubated and diluted with the compound at 10-fold the IC50 (10× control).
Cell Viability Assays
AML
Mycoplasma-free AML cells were seeded in 96-well plates at the optimal density to guarantee linear growth throughout the assay (8000 cells/well for MV(4;11), 4000 cells/well for MOLM-13, and 2000 cells/well for TF-1a) in 50 μL of complete RPMI-1640 medium (Sigma) supplemented with 10% FBS and 2 mM l-glutamine. Each experimental condition was tested in technical triplicates, including medium-only and vehicle-treated controls for background correction and normalization, respectively. After seeding, 50 μL of medium containing 7 serial dilutions (1:10) of 2×-concentrated compounds were added to the cells to obtain 100 μL with 1×-concentrated compound at each dilution (concentration range: 2000–0.002 nM). Cells were then incubated for 96 h at 37 °C in a controlled 5% CO2 atmosphere prior to evaluation of cell viability using alamarBlue cell viability reagent (ThermoFisher Scientific, Waltham, MA/USA). alamarBlue is a cell viability indicator that uses the natural reducing power of living cells to convert resazurin to the fluorescent molecule resorufin. Briefly, alamarBlue stock solution was diluted 1:20 in the culture medium, and after 3 h of incubation, fluorescence was measured using a TECAN Infinity 2000 plate reader (Tecan Group Ltd., Männedorf, CH; 540–570 nm excitation wavelength; 580–610 nm emission wavelength). For each condition, the average fluorescence was calculated from the 3 technical replicates of 2 independent biological replicates; background was calculated from the fluorescence of medium-only controls and subtracted from each data point. Data were analyzed using the GraphPad PRISM version 9.0.1 (GraphPad Software, Inc., La Jolla, CA/USA) to calculate the best-fitting curves and the EC50 values.
SCLC
Mycoplasma-free NCI-H510A SCLC cells were seeded in 96-well plates at the optimal density to guarantee linear growth throughout the assay (3000 cells/well) in 50 μL of complete RPMI-1640 medium (Sigma) supplemented with 10% FBS and 2 mM l-glutamine. Each experimental condition was tested in technical triplicates, including medium-only and vehicle-treated controls for background correction and normalization, respectively. After seeding, 50 μL of medium containing 7 serial dilutions (1:10) of 2×-concentrated compounds were added to the cells to obtain 100 μL of 1×-concentrated compound at each dilution (concentration range: 2000–0.002 nM). Cells were then incubated at 37 °C in a controlled 5% CO2 atmosphere for a total of 10 days with medium replenishment supplemented with 1×-concentrated compounds at day 5, and cell viability was evaluated with the alamarBlue cell viability reagent (ThermoFisher Scientific, Waltham, MA/USA). Briefly, alamarBlue stock solution was diluted 1:20 in the culture medium, and after 3 h of incubation, fluorescence was measured using a TECAN Infinity 2000 plate reader (Tecan Group Ltd., Männedorf, CH; 540–570 nm excitation wavelength; 580–610 nm emission wavelength). For each condition, the average fluorescence was calculated from the 3 technical replicates of 2 independent biological replicates; background was calculated from the fluorescence of medium-only controls and subtracted from each data point. Data were analyzed using the GraphPad PRISM version 9.0.1 (GraphPad Software, Inc., La Jolla, CA/USA) to calculate the best-fitting curves and the EC50 values.
Analysis of AML Differentiation
Mycoplasma-free THP1 cells were seeded in 6-well plates at the optimal cell density to guarantee linear growth throughout the assay (150 000 cells/well) in 1 mL of complete RPMI-1640 medium (Sigma) supplemented with 10% FBS and 2 mM l-glutamine. After seeding, cells were treated with 1 mL of 2×-concentrated compounds at 1:10 dilution steps (concentration range: 2000–0.02 nM; final DMSO concentration: 0.1%) and incubated at 37 °C in a controlled 5% CO2 atmosphere for a total of 96 h. After incubation, cells from each well were washed twice in cold 1× PBS and resuspended in 200 μL of 1× PBS + 1% FBS, and 100 μL was transferred in duplicate to a V-bottomed 96-well plate. The plate was centrifuged at 400g for 5 min; the supernatants were removed and cell pellets, resuspended in 100 μL of either CD-11b/Mac-1-PE (BD Biosciences, ref. 555388) or mouse IgG1-PE (BD Biosciences, ref. 555749) at 1/5 dilution in 1× PBS + 1% FBS and incubated at 4 °C for 45 min (protected from light). After 3× washes with cold 1× PBS + 1% FBS, stained cells were resuspended in 600 μL of cold 1× PBS + 1% FBS and analyzed by flow cytometry (Gallios, Beckman Coulter). Data were analyzed with FlowJo software and represented as the % of CD11b positive cells (differentiated) in the total analyzed live cell population.
Analysis of LSD1 Target Engagement
The assessment of LSD1 target engagement in native protein extracts of MV(4;11) and NCI-H510A cells previously treated with vehicle or compounds was performed according to the protocol described by Mascaró et al.66 Briefly, mycoplasma-free MV(4;11) and NCI-H510A cells were seeded in T75 and T25 flasks, respectively (5 million cells in 15 mL and 5 million cells in 5 mL of complete RPMI-1640 medium supplemented with 10% FBS and 2 mM l-glutamine, respectively), and treated with 2000, 200, 20, and 2 nM of the selected compounds for 24 h at 37 °C in a controlled 5% CO2 atmosphere. After incubation, pellets of 1.5 million cells were generated by centrifugation at 200g for 5 min. After supernatant aspiration, pellets were washed with 1 mL of 1× PBS and centrifuged again at 200g for 5 min, and after supernatant removal, the pellets were stored at −80 °C until use. Cell pellets were lysed in the presence of cell lysis buffer (Cell Signaling, ref. 9803) supplemented with cOmplete Mini-Protease Inhibitor Cocktail (Sigma-Aldrich, ref. 11836153001) and 25 nM biotinylated LSD1 chemoprobe (OG-881), and total and free LSD1 were measured by luminescent ELISAs. For the quantification of total LSD1 protein (TOTAL LSD1), LumiNunc Plates MaxiSorp (Nunc, ref. 436110) were coated with a monoclonal anti-LSD1 antibody (Abcam, ref. ab53269) at 2 μg/mL in PBS, whereas for the quantification of the fraction not bound to inhibitor (FREE LSD1), the plates were coated with streptavidin (Promega Biotech Ibérica, ref. Z7041) at 10 μg/mL in PBS. All coating was performed at 4 °C overnight. The following day, wells were washed 3 times in PBS and 0.1% Tween-20 and blocked with 1% PBS–BSA (Sigma, ref. A3059) for 1 h and 30 min at RT. An LSD1 calibration curve using a full-length recombinant LSD1 (Active Motif, ref. 31334) diluted in PBS was included in each plate. Plates were then incubated for 1 h at RT and washed five times, and afterward, a monoclonal anti-LSD1 antibody (Cell Signaling, ref. 2184) diluted at 0.125 μg/mL in PBS was added to the plate and incubated for 1 h at RT. After six washes, a peroxidase-conjugated secondary antibody (Jackson Inmunoresearch, ref. 711-035-152) diluted 1:5000 was added to the plates, and they were incubated for 1 h at RT and washed six times again. Finally, 100 μL/well of chemiluminescent substrate (Invitrogen, ref. 37074) was added; the plates were centrifuged for 30 s at 1500 rpm, shaken for 1 min at 100 rpm, and incubated inside the microplate reader (Infinite 200, Tecan) for 3 min at 25 °C. The relative luminescence unit (RLU) readouts were acquired using a 1000 ms integration time and 150 ms settle time. All assays were run in technical triplicates, and the percentage of target engagement to LSD1 was calculated relative to the vehicle treatment.
GFI1B-Peptide/LSD1 Interaction Assay
The day before the assay, LumiNunc Plates MaxiSorp (Nunc, ref. 436110) were coated with 100 μL/well of either 10 μg/mL streptavidin (Promega Biotech Ibérica, ref. Z7041) in PBS or 2 μg/mL of a monoclonal anti-LSD1 antibody (Abcam, ref. ab53269) in PBS for the quantification of the GFI1B-peptide/LSD1 interaction and total LSD1 levels, respectively. The plates were incubated at 4 °C overnight. On the day of the assay, the plates were washed three times with PBS supplemented with 0.1% Tween-20 (PBS-T) and blocked with 1% BSA in PBS for 1 h and 30 min. After the blocking, streptavidin-coated plates were washed with PBS-T and incubated for 1 h at RT with 1.786 ng/mL of a biotinylated peptide containing the SNAG domain sequence from the human GFI1B protein (sequence: PRSFLVKSKKAHTYHQPRVQ-Ttds-Lys(biotin); custom peptide synthesis, JPT peptide technologies); plates incubated with the anti-LSD1 antibody were instead washed and incubated with PBS for 1 h at RT. During this incubation, a solution containing recombinant LSD1 (Active Motif, ref. 31334), RCOR1 (BPS Bioscience, ref. 50274), and each compound diluted at one of four dilutions (2 μM, 200 nM, 20 nM, and 2 nM) was prepared in PBS supplemented with 2% BSA and incubated for 1 h at 23 °C. After this incubation, the LSD1/RCOR1/compound incubations were diluted 1:3.8 with 1% BSA in PBS to a final LSD1 concentration of 6.6 ng/mL, added to the plates (previously washed three times with PBS-T) at a volume of 100 μL/well, and incubated for 1 h at RT. After washing five times with PBS-T, 0.125 μg/mL of a rabbit anti-LSD1 antibody (Cell Signaling, ref. 2184) was added to all plates, which were incubated for 1 h at RT. The plates were then washed six times and incubated with a secondary peroxidase-conjugated donkey antirabbit IgG antibody (Jackson Immunoresearch, ref. 711-035-152) for 1 h at RT and washed 6 times again. Finally, 100 μL/well of chemiluminescent substrate (Thermo Fisher Scientific, ref. 37074) was added to the plates. These were centrifuged for 30 s at 1500 rpm in order to eliminate bubbles and shaken for 1 min at 100 rpm. Finally, plates were incubated inside an Infinite 200-Tecan Microplate reader at 25 °C for 3 min, and the luminescence was read at 1000 ms of integration time and 150 ms of settle time.
INSM1/LSD1 Interaction Assay
Mycoplasma-free NCI-H510A cells were seeded in T25 flasks (5 million cells in 5 mL of complete RPMI-1640 medium supplemented with 10% FBS and 2 mM l-glutamine) and treated with 2000, 200, 20, and 2 nM of the selected compounds for 24 h at 37 °C in a controlled 5% CO2 atmosphere. After incubation, pellets of 2.5 million cells were generated by centrifugation at 200g for 5 min. After supernatant aspiration, pellets were washed with 1 mL of 1× PBS and centrifuged again at 200g for 5 min, and after supernatant removal, the pellets were stored at −80 °C until use for protein extraction. The day before the INSM1/LSD1 interaction assay, LumiNunc Plates MaxiSorp (Nunc, ref. 436110) were coated with 100 μL/well of either 1 μg/mL of an anti-INSM1 antibody (Santa Cruz Biotechnology, ref. sc-271408) in PBS or 2 μg/mL of a monoclonal anti-LSD1 antibody (Abcam, ref. ab53269) in PBS for the quantification of the INSM1/LSD1 interaction and total LSD1 levels, respectively. Plates were incubated at 4 °C overnight . On the day of the assay, NCI-H510A frozen cell pellets were thawed and resuspended in 200 μL of cell lysis buffer (Cell Signaling, ref. 9803) supplemented with cOmplete Mini-Protease Inhibitor Cocktail (Sigma-Aldrich, ref. 11836153001). Samples were kept on ice for 10 min prior to high-speed centrifugation to remove cellular debris. Cell lysates were kept on ice. The plates coated overnight were then washed 3 times with PBS supplemented with 0.1% Tween-20 (PBS-T) and blocked in PBS with 1% BSA for 1 h and 30 min at RT. Total protein concentration was determined by the Bradford assay, and either 100 μg/well (INSM1/LSD1) or 2 μg/well (total LSD1) was added to the plates (previously washed once with PBS-T) and incubated for 1 h at RT. After washing five times with PBS-T, 0.125 μg/mL of a rabbit anti-LSD1 antibody (Cell Signaling, ref. 2184) was added to all plates and incubated for 1 h at RT. The plates were then washed six times and incubated with a secondary peroxidase-conjugated donkey antirabbit IgG antibody (Jackson Immunoresearch, ref. 711-035-152) for 1 h at RT and washed six times again. Finally, 100 μL/well of chemiluminescent substrate (Thermo Fisher Scientific, ref. 37074) was added to the plates. These were centrifuged for 30 s at 1500 rpm in order to eliminate bubbles and shaken for 1 min at 100 rpm. Finally, the plates were incubated inside an Infinite 200-Tecan Microplate reader at 25 °C for 3 min, and the luminescence was read at 1000 ms of integration time and 150 ms of settle time.
RNA Extraction and qRT-PCR
Mycoplasma-free NCI-H510A cells were seeded in T25 flasks (0.5 million cells in 5 mL of complete RPMI-1640 medium supplemented with 10% FBS and 2 mM l-glutamine) and treated with 2000, 200, 20, and 2 nM of the selected compounds for 6 days at 37 °C in a controlled 5% CO2 atmosphere. After incubation, pellets of 0.5 million cells were generated by centrifugation at 200g for 5 min. After supernatant aspiration, pellets were washed with 1 mL of 1× PBS and centrifuged again at 200g for 5 min, and after supernatant removal, pellets were stored at −80 °C until use for total RNA extraction. Total RNA was extracted using the RNeasy Mini kit (Qiagen, ref. 74106) according to the manufacturer’s instructions, and the evaluation of RNA quality was performed with the Nanodrop ONE (Thermo Scientific) and the Agilent 2100 bioanalyzer. First strand synthesis was performed with the iScript Reverse Transcription Supermix (Bio-Rad, ref. 1708841) using 1 μg of total RNA. qRT-PCR reactions were performed in technical triplicates (n = 3) in a Roche Lightcycler II 480 using 10 ng of first strand DNA and off-the-shelf Taqman assays (GRP, ThermoFisher, ref. Hs01107047_m1; GUSB, ThermoFisher, ref. Hs00939627_m1). Mean Cp values for each data point were calculated after outlier elimination using the Grubbs test (applied if the standard deviation of three technical PCR replicates was higher than 0.25 Cp). GRP gene expression values were calculated using the 2–ΔΔCt method, normalized by GUSB expression and using the vehicle condition as the calibrator.
Acknowledgments
The authors would like to thank colleagues at Oryzon for fruitful discussions and feedback on the manuscript. They would also like to thank all biology lab technicians for their excellent work on the experimental assays: kinetic solubility and biochemical assays (Rosa Adrados and Helena Sarret), cell-based assays (Ewelina Maria Strzemieczna, Sergi Purcet, and Mireia Verdú), and protein and gene expression assays (Susana Coca, Laia Hernández, and Joaquim Vila).
Glossary
Abbreviations
- LSD1/KDM1A
lysine-specific demethylase 1
- CoREST
rest co-repressor
- NuRD
nucleosome remodeling and deacetylase
- SNAG
SNAIL/GFI1
- GFI1
growth factor independent 1
- INSM1
insulinoma associated protein 1
- LSD2
lysine-specific demethylase 2
- H3K4
histone 3 lysine 4
- H3K9
histone 3 lysine 9
- AO
amine oxidase
- MAO-A
monoamine oxidase A
- MAO-B
monoamine oxidase B
- AML
acute myeloid leukemia
- SCLC
small cell lung cancer
- NSCLC
nonsmall cell lung cancer
- TF
transcription factor
- PPI
protein–protein interaction
- NCE
new chemical entity
- HTRF
homogeneous time resolved fluorescence assay
- HRP
horseradish peroxidase
- EMT
epidermal mesenchymal transition
- FAD
flavin adenine dinucleotide
- TNBC
triple negative breast cancer
- CD11B/ITGAM
cluster of differentiation molecule 11b/Integrin alpha-M
- GRP
gastrin-releasing peptide
- ATRA
all-trans retinoic acid
- MDS
myelodysplastic syndrome
- MAO
monoamine oxidase
- TCP
tranylcypromine
- PLZ
phenelzine
- PRG
pargyline
- TE
target engagement
- ZNF217
zinc Finger protein 217
- PAIN
pan assay interference inhibitor
- TSS50
total symptom score 50
- SVR35
spleen volume reduction 35
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.1c00223.
Figure S1, jump dilution analysis of reversible LSD1 inhibitors; Figure S2, dose–response curves of MAO-A and MAO-B inhibition for selected tool compounds; Figure S3, efficacy in AML; Table S1, stability of compounds in aqueous buffer at 37 °C; Table S2, kinetic solubility and inhibition of LSD1 and other FAD-dependent enzymes; Table S3, kinetic parameters of LSD1 irreversible inhibition; Table S4, EC50 viability values for AML and SCLC cell lines; Table S5, EC50 differentiation values for the AML cell line THP1 (PDF)
Author Contributions
⊥ N.S. and P.D. contributed equally.
The authors declare no competing financial interest.
Special Issue
Published as part of the ACS Pharmacology & Translational Science special issue “Epigenetics 2022”.
Supplementary Material
References
- Ganesan A.; Arimondo P. B.; Rots M. G.; Jeronimo C.; Berdasco M. The Timeline of Epigenetic Drug Discovery: From Reality to Dreams. Clin. Epigenet. 2019, 11 (1), 174. 10.1186/s13148-019-0776-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates S. E. Epigenetic Therapies for Cancer. N. Engl. J. Med. 2020, 383 (7), 650–663. 10.1056/NEJMra1805035. [DOI] [PubMed] [Google Scholar]
- Verma M.; Kumar V.. Epigenetic Drugs for Cancer and Precision Medicine. In Epigenetics of Aging and Longevity; Elsevier, 2018; pp 439–451; 10.1016/B978-0-12-811060-7.00021-8. [DOI] [Google Scholar]
- Lundstrom K. Epigenetics: Rethinking of Drug Research and Development. Future Med. Chem. 2019, 10.4155/fmc-2019-0174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y.-J.; Matson C.; Lan F.; Iwase S.; Baba T.; Shi Y. Regulation of LSD1 Histone Demethylase Activity by Its Associated Factors. Mol. Cell 2005, 19 (6), 857–864. 10.1016/j.molcel.2005.08.027. [DOI] [PubMed] [Google Scholar]
- Lee M. G.; Wynder C.; Cooch N.; Shiekhattar R. An Essential Role for CoREST in Nucleosomal Histone 3 Lysine 4 Demethylation. Nature 2005, 437 (7057), 432–435. 10.1038/nature04021. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Zhang H.; Chen Y.; Sun Y.; Yang F.; Yu W.; Liang J.; Sun L.; Yang X.; Shi L.; Li R.; Li Y.; Zhang Y.; Li Q.; Yi X.; Shang Y. LSD1 Is a Subunit of the NuRD Complex and Targets the Metastasis Programs in Breast Cancer. Cell 2009, 138 (4), 660–672. 10.1016/j.cell.2009.05.050. [DOI] [PubMed] [Google Scholar]
- Shi Y.; Lan F.; Matson C.; Mulligan P.; Whetstine J. R.; Cole P. A.; Casero R. A.; Shi Y. Histone Demethylation Mediated by the Nuclear Amine Oxidase Homolog LSD1. Cell 2004, 119 (7), 941–953. 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Metzger E.; Wissmann M.; Yin N.; Müller J. M.; Schneider R.; Peters A. H. F. M.; Günther T.; Buettner R.; Schüle R. LSD1 Demethylates Repressive Histone Marks to Promote Androgen-Receptor-Dependent Transcription. Nature 2005, 437 (7057), 436–439. 10.1038/nature04020. [DOI] [PubMed] [Google Scholar]
- Huang J.; Sengupta R.; Espejo A. B.; Lee M. G.; Dorsey J. A.; Richter M.; Opravil S.; Shiekhattar R.; Bedford M. T.; Jenuwein T.; Berger S. L. P53 Is Regulated by the Lysine Demethylase LSD1. Nature 2007, 449 (7158), 105–108. 10.1038/nature06092. [DOI] [PubMed] [Google Scholar]
- Maiques-Diaz A.; Somervaille T. C. LSD1: Biologic Roles and Therapeutic Targeting. Epigenomics 2016, 8 (8), 1103–1116. 10.2217/epi-2016-0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thambyrajah R.; Mazan M.; Patel R.; Moignard V.; Stefanska M.; Marinopoulou E.; Li Y.; Lancrin C.; Clapes T.; Möröy T.; Robin C.; Miller C.; Cowley S.; Göttgens B.; Kouskoff V.; Lacaud G. GFI1 Proteins Orchestrate the Emergence of Haematopoietic Stem Cells through Recruitment of LSD1. Nat. Cell Biol. 2016, 18 (1), 21–32. 10.1038/ncb3276. [DOI] [PubMed] [Google Scholar]
- Sprüssel A.; Schulte J. H.; Weber S.; Necke M.; Händschke K.; Thor T.; Pajtler K. W.; Schramm A.; König K.; Diehl L.; Mestdagh P.; Vandesompele J.; Speleman F.; Jastrow H.; Heukamp L. C.; Schüle R.; Dührsen U.; Buettner R.; Eggert A.; Göthert J. R. Lysine-Specific Demethylase 1 Restricts Hematopoietic Progenitor Proliferation and Is Essential for Terminal Differentiation. Leukemia 2012, 26 (9), 2039–2051. 10.1038/leu.2012.157. [DOI] [PubMed] [Google Scholar]
- Karakaidos P.; Verigos J.; Magklara A. LSD1/KDM1A, a Gate-Keeper of Cancer Stemness and a Promising Therapeutic Target. Cancers 2019, 11 (12), 1821. 10.3390/cancers11121821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch J. T.; Harris W. J.; Somervaille T. C. P. LSD1 Inhibition: A Therapeutic Strategy in Cancer?. Expert Opin. Ther. Targets 2012, 16 (12), 1239–1249. 10.1517/14728222.2012.722206. [DOI] [PubMed] [Google Scholar]
- Kahl P.; Gullotti L.; Heukamp L. C.; Wolf S.; Friedrichs N.; Vorreuther R.; Solleder G.; Bastian P. J.; Ellinger J.; Metzger E.; Schüle R.; Buettner R. Androgen Receptor Coactivators Lysine-Specific Histone Demethylase 1 and Four and a Half LIM Domain Protein 2 Predict Risk of Prostate Cancer Recurrence. Cancer Res. 2006, 66 (23), 11341–11347. 10.1158/0008-5472.CAN-06-1570. [DOI] [PubMed] [Google Scholar]
- Vinyard M. E.; Su C.; Siegenfeld A. P.; Waterbury A. L.; Freedy A. M.; Gosavi P. M.; Park Y.; Kwan E. E.; Senzer B. D.; Doench J. G.; Bauer D. E.; Pinello L.; Liau B. B. CRISPR-Suppressor Scanning Reveals a Nonenzymatic Role of LSD1 in AML. Nat. Chem. Biol. 2019, 15 (5), 529–539. 10.1038/s41589-019-0263-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari-Amorotti G.; Fragliasso V.; Esteki R.; Prudente Z.; Soliera A. R.; Cattelani S.; Manzotti G.; Grisendi G.; Dominici M.; Pieraccioli M.; Raschellà G.; Chiodoni C.; Colombo M. P.; Calabretta B. Inhibiting Interactions of Lysine Demethylase LSD1 with Snail/Slug Blocks Cancer Cell Invasion. Cancer Res. 2013, 73 (1), 235–245. 10.1158/0008-5472.CAN-12-1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C.; Ayyanathan K. Snail/Gfi-1 (SNAG) Family Zinc Finger Proteins in Transcription Regulation, Chromatin Dynamics, Cell Signaling, Development, and Disease. Cytokine Growth Factor Rev. 2013, 24 (2), 123–131. 10.1016/j.cytogfr.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron R.; Binda C.; Tortorici M.; McCammon J. A.; Mattevi A. Molecular Mimicry and Ligand Recognition in Binding and Catalysis by the Histone Demethylase LSD1-CoREST Complex. Structure 2011, 19 (2), 212–220. 10.1016/j.str.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleque S.; Kim J.; Rooke H. M.; Orkin S. H. Epigenetic Regulation of Hematopoietic Differentiation by Gfi-1 and Gfi-1b Is Mediated by the Cofactors CoREST and LSD1. Mol. Cell 2007, 27 (4), 562–572. 10.1016/j.molcel.2007.06.039. [DOI] [PubMed] [Google Scholar]
- van Bergen M. G. J. M.; van der Reijden B. A. Targeting the GFI1/1B—CoREST Complex in Acute Myeloid Leukemia. Front. Oncol. 2019, 9, 1027. 10.3389/fonc.2019.01027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi S.; Ishikawa Y.; Mizutani A.; Iwasaki S.; Matsumoto S.; Kamada Y.; Nomura T.; Nakamura K. LSD1 Inhibitor T-3775440 Inhibits SCLC Cell Proliferation by Disrupting LSD1 Interactions with SNAG Domain Proteins INSM1 and GFI1B. Cancer Res. 2017, 77 (17), 4652–4662. 10.1158/0008-5472.CAN-16-3502. [DOI] [PubMed] [Google Scholar]
- Mahalakshmi B.; Baskaran R.; Shanmugavadivu M.; Nguyen N. T.; Velmurugan B. K. Insulinoma-Associated Protein 1 (INSM1): A Potential Biomarker and Therapeutic Target for Neuroendocrine Tumors. Cell. Oncol. 2020, 43 (3), 367–376. 10.1007/s13402-020-00505-9. [DOI] [PubMed] [Google Scholar]
- Lee C.; Rudneva V. A.; Erkek S.; Zapatka M.; Chau L. Q.; Tacheva-Grigorova S. K.; Garancher A.; Rusert J. M.; Aksoy O.; Lea R.; Mohammad H. P.; Wang J.; Weiss W. A.; Grimes H. L.; Pfister S. M.; Northcott P. A.; Wechsler-Reya R. J. Lsd1 as a Therapeutic Target in Gfi1-Activated Medulloblastoma. Nat. Commun. 2019, 10 (1), 332. 10.1038/s41467-018-08269-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park D. E.; Cheng J.; McGrath J. P.; Lim M. Y.; Cushman C.; Swanson S. K.; Tillgren M. L.; Paulo J. A.; Gokhale P. C.; Florens L.; Washburn M. P.; Trojer P.; DeCaprio J. A. Merkel Cell Polyomavirus Activates LSD1-Mediated Blockade of Non-Canonical BAF to Regulate Transformation and Tumorigenesis. Nat. Cell Biol. 2020, 22 (5), 603–615. 10.1038/s41556-020-0503-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCaprio J. A. Molecular Pathogenesis of Merkel Cell Carcinoma. Annu. Rev. Pathol.: Mech. Dis. 2021, 16 (1), 69–91. 10.1146/annurev-pathmechdis-012419-032817. [DOI] [PubMed] [Google Scholar]
- Leiendecker L.; Jung P. S.; Krecioch I.; Neumann T.; Schleiffer A.; Mechtler K.; Wiesner T.; Obenauf A. C. LSD 1 Inhibition Induces Differentiation and Cell Death in Merkel Cell Carcinoma. EMBO Mol. Med. 2020, 12 (11), e12525. 10.15252/emmm.202012525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tortorici M.; Borrello M. T.; Tardugno M.; Chiarelli L. R.; Pilotto S.; Ciossani G.; Vellore N. A.; Bailey S. G.; Cowan J.; O’Connell M.; Crabb S. J.; Packham G.; Mai A.; Baron R.; Ganesan A.; Mattevi A. Protein Recognition by Short Peptide Reversible Inhibitors of the Chromatin-Modifying LSD1/CoREST Lysine Demethylase. ACS Chem. Biol. 2013, 8 (8), 1677–1682. 10.1021/cb4001926. [DOI] [PubMed] [Google Scholar]
- Kumarasinghe I. R.; Woster P. M. Synthesis and Evaluation of Novel Cyclic Peptide Inhibitors of Lysine-Specific Demethylase 1. ACS Med. Chem. Lett. 2014, 5 (1), 29–33. 10.1021/ml4002997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumarasinghe I. R.; Woster P. M. Cyclic Peptide Inhibitors of Lysine-Specific Demethylase 1 with Improved Potency Identified by Alanine Scanning Mutagenesis. Eur. J. Med. Chem. 2018, 148, 210–220. 10.1016/j.ejmech.2018.01.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y.; Liao G.; Yu B. LSD1/KDM1A Inhibitors in Clinical Trials: Advances and Prospects. J. Hematol. Oncol. 2019, 12 (1), 129. 10.1186/s13045-019-0811-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes T.; Carceller E.; Salas J.; Ortega A.; Buesa C. Advances in the Development of Histone Lysine Demethylase Inhibitors. Curr. Opin. Pharmacol. 2015, 23, 52–60. 10.1016/j.coph.2015.05.009. [DOI] [PubMed] [Google Scholar]
- Fu X.; Zhang P.; Yu B. Advances toward LSD1 Inhibitors for Cancer Therapy. Future Med. Chem. 2017, 9 (11), 1227–1242. 10.4155/fmc-2017-0068. [DOI] [PubMed] [Google Scholar]
- Niwa H.; Umehara T. Structural Insight into Inhibitors of Flavin Adenine Dinucleotide-Dependent Lysine Demethylases. Epigenetics 2017, 12 (5), 340–352. 10.1080/15592294.2017.1290032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marabelli C.; Marrocco B.; Mattevi A. The Growing Structural and Functional Complexity of the LSD1/KDM1A Histone Demethylase. Curr. Opin. Struct. Biol. 2016, 41, 135–144. 10.1016/j.sbi.2016.07.011. [DOI] [PubMed] [Google Scholar]
- Schenk T.; Chen W. C.; Göllner S.; Howell L.; Jin L.; Hebestreit K.; Klein H.-U.; Popescu A. C.; Burnett A.; Mills K.; Casero R. A. Jr; Marton L.; Woster P.; Minden M. D.; Dugas M.; Wang J. C. Y.; Dick J. E.; Müller-Tidow C.; Petrie K.; Zelent A. Inhibition of the LSD1 (KDM1A) Demethylase Reactivates the All-Trans-Retinoic Acid Differentiation Pathway in Acute Myeloid Leukemia. Nat. Med. 2012, 18 (4), 605–611. 10.1038/nm.2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan A. H. Y.; Tu W.; McCuaig R.; Hardy K.; Donovan T.; Tsimbalyuk S.; Forwood J. K.; Rao S. Lysine-Specific Histone Demethylase 1A Regulates Macrophage Polarization and Checkpoint Molecules in the Tumor Microenvironment of Triple-Negative Breast Cancer. Front. Immunol. 2019, 10, 1351. 10.3389/fimmu.2019.01351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K.-H.; Kim B.-C.; Jeong S.-H.; Jeong C. W.; Ku J. H.; Kwak C.; Kim H. H. Histone Demethylase LSD1 Regulates Kidney Cancer Progression by Modulating Androgen Receptor Activity. Int. J. Mol. Sci. 2020, 21 (17), 6089. 10.3390/ijms21176089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Zhu Y.; Wang Q.; Hu H.; Li Z.; Wang D.; Zhang W.; Qi B.; Ye J.; Wu H.; Jiang H.; Liu L.; Yang J.; Cheng J. The Histone Demethylase LSD1 Is a Novel Oncogene and Therapeutic Target in Oral Cancer. Cancer Lett. 2016, 374 (1), 12–21. 10.1016/j.canlet.2016.02.004. [DOI] [PubMed] [Google Scholar]
- Wang M.; Liu X.; Guo J.; Weng X.; Jiang G.; Wang Z.; He L. Inhibition of LSD1 by Pargyline Inhibited Process of EMT and Delayed Progression of Prostate Cancer in Vivo. Biochem. Biophys. Res. Commun. 2015, 467 (2), 310–315. 10.1016/j.bbrc.2015.09.164. [DOI] [PubMed] [Google Scholar]
- Cusan M.; Cai S. F.; Mohammad H. P.; Krivtsov A.; Chramiec A.; Loizou E.; Witkin M. D.; Smitheman K. N.; Tenen D. G.; Ye M.; Will B.; Steidl U.; Kruger R. G.; Levine R. L.; Rienhoff H. Y. Jr; Koche R. P.; Armstrong S. A. LSD1 inhibition exerts its antileukemic effect by recommissioning PU.1- and C/EBPα-dependent enhancers in AML. Blood 2018, 131 (15), 1730–1742. 10.1182/blood-2017-09-807024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smitheman K. N.; Severson T. M.; Rajapurkar S. R.; McCabe M. T.; Karpinich N.; Foley J.; Pappalardi M. B.; Hughes A.; Halsey W.; Thomas E.; Traini C.; Federowicz K. E.; Laraio J.; Mobegi F.; Ferron-Brady G.; Prinjha R. K.; Carpenter C. L.; Kruger R. G.; Wessels L.; Mohammad H. P. Lysine specific demethylase 1 inactivation enhances differentiation and promotes cytotoxic response when combined with all-trans retinoic acid in acute myeloid leukemia across subtypes. Haematologica 2019, 104 (6), 1156–1167. 10.3324/haematol.2018.199190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehrawat A.; Gao L.; Wang Y.; Bankhead A. III; McWeeney S. K.; King C. J.; Schwartzman J.; Urrutia J.; Bisson W. H.; Coleman D. J.; Joshi S. K.; Kim D.-H.; Sampson D. A.; Weinmann S.; Kallakury B. V. S.; Berry D. L.; Haque R.; Van Den Eeden S. K.; Sharma S.; Bearss J.; Beer T. M.; Thomas G. V.; Heiser L. M.; Alumkal J. J. LSD1 Activates a Lethal Prostate Cancer Gene Network Independently of Its Demethylase Function. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (18), E4179–E4188. 10.1073/pnas.1719168115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pishas K. I.; Drenberg C. D.; Taslim C.; Theisen E. R.; Johnson K. M.; Saund R. S.; Pop I. L.; Crompton B. D.; Lawlor E. R.; Tirode F.; Mora J.; Delattre O.; Beckerle M. C.; Callen D. F.; Sharma S.; Lessnick S. L. Therapeutic Targeting of KDM1A/LSD1 in Ewing Sarcoma with SP-2509 Engages the Endoplasmic Reticulum Stress Response. Mol. Cancer Ther. 2018, 17 (9), 1902–1916. 10.1158/1535-7163.MCT-18-0373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanouni T.; Severin C.; Cho R. W.; Yuen N. Y.-Y.; Xu J.; Shi L.; Lai C.; Del Rosario J. R.; Stansfield R. K.; Lawton L. N.; Hosfield D.; O’Connell S.; Kreilein M. M.; Tavares-Greco P.; Nie Z.; Kaldor S. W.; Veal J. M.; Stafford J. A.; Chen Y. K. Discovery of CC-90011: A Potent and Selective Reversible Inhibitor of Lysine Specific Demethylase 1 (LSD1). J. Med. Chem. 2020, 63 (23), 14522–14529. 10.1021/acs.jmedchem.0c00978. [DOI] [PubMed] [Google Scholar]
- Mohammad H. P.; Smitheman K. N.; Kamat C. D.; Soong D.; Federowicz K. E.; Van Aller G. S.; Schneck J. L.; Carson J. D.; Liu Y.; Butticello M.; Bonnette W. G.; Gorman S. A.; Degenhardt Y.; Bai Y.; McCabe M. T.; Pappalardi M. B.; Kasparec J.; Tian X.; McNulty K. C.; Kruger R. G. A DNA Hypomethylation Signature Predicts Antitumor Activity of LSD1 Inhibitors in SCLC. Cancer Cell 2015, 28 (1), 57–69. 10.1016/j.ccell.2015.06.002. [DOI] [PubMed] [Google Scholar]
- Mohammad H. P.; Kruger R. G. Antitumor activity of LSD1 inhibitors in lung cancer. Molecular & Cellular Oncology 2016, 3 (2), e1117700. 10.1080/23723556.2015.1117700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jutzi J. S.; Kleppe M.; Dias J.; Staehle H. F.; Shank K.; Teruya-Feldstein J.; Gambheer S. M. M.; Dierks C.; Rienhoff H. Y.; Levine R. L.; Pahl H. L. LSD1 Inhibition Prolongs Survival in Mouse Models of MPN by Selectively Targeting the Disease Clone. HemaSphere 2018, 2 (3), e54. 10.1097/HS9.0000000000000054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey C. P.; Figueroa M.; Gangadharan A.; Lee D. A.; Chandra J. Scaffolding LSD1 Inhibitors Impair NK Cell Metabolism and Cytotoxic Function Through Depletion of Glutathione. Front. Immunol. 2020, 11, 2196. 10.3389/fimmu.2020.02196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes T.; Mascaró C.; Tirapu I.; Estiarte A.; Ciceri F.; Lunardi S.; Guibourt N.; Perdones A.; Lufino M. M. P.; Somervaille T. C. P.; Wiseman D. H.; Duy C.; Melnick A.; Willekens C.; Ortega A.; Martinell M.; Valls N.; Kurz G.; Fyfe M.; Buesa C. ORY-1001, a Potent and Selective Covalent KDM1A Inhibitor, for the Treatment of Acute Leukemia. Cancer Cell 2018, 33 (3), 495–511.E12. 10.1016/j.ccell.2018.02.002. [DOI] [PubMed] [Google Scholar]
- Salamero O.; Montesinos P.; Willekens C.; Pérez-Simón J. A.; Pigneux A.; Récher C.; Popat R.; Carpio C.; Molinero C.; Mascaró C.; Vila J.; Arévalo M. I.; Maes T.; Buesa C.; Bosch F.; Somervaille T. C. P. First-in-Human Phase I Study of Iadademstat (ORY-1001): A First-in-Class Lysine-Specific Histone Demethylase 1A Inhibitor, in Relapsed or Refractory Acute Myeloid Leukemia. J. Clin. Oncol. 2020, 38 (36), 4260–4273. 10.1200/JCO.19.03250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston G.; Ramsey H. E.; Liu Q.; Wang J.; Stengel K. R.; Sampathi S.; Acharya P.; Arrate M.; Stubbs M. C.; Burn T.; Savona M. R.; Hiebert S. W. Nascent Transcript and Single-Cell RNA-Seq Analysis Defines the Mechanism of Action of the LSD1 Inhibitor INCB059872 in Myeloid Leukemia. Gene 2020, 752, 144758. 10.1016/j.gene.2020.144758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di L.; Kerns E. H.. Solubility Issues in Early Discovery and HTS. In Solvent Systems and Their Selection in Pharmaceutics and Biopharmaceutics; Springer: New York, 2007; pp 111–136; 10.1007/978-0-387-69154-1_4. [DOI] [Google Scholar]
- Jumde V. R.; Mondal M.; Gierse R. M.; Unver M. Y.; Magari F.; van Lier R. C. W.; Heine A.; Klebe G.; Hirsch A. K. H. Design and Synthesis of Bioisosteres of Acylhydrazones as Stable Inhibitors of the Aspartic Protease Endothiapepsin. ChemMedChem 2018, 13 (21), 2266–2270. 10.1002/cmdc.201800446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culhane J. C.; Wang D.; Yen P. M.; Cole P. A. Comparative Analysis of Small Molecules and Histone Substrate Analogues as LSD1 Lysine Demethylase Inhibitors. J. Am. Chem. Soc. 2010, 132 (9), 3164–3176. 10.1021/ja909996p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu V.; Fisch T.; Long A. M.; Tang J.; Lee J. H.; Hierl M.; Chen H.; Yakowec P.; Schwandner R.; Emkey R. High-Throughput TR-FRET Assays for Identifying Inhibitors of LSD1 and JMJD2C Histone Lysine Demethylases. J. Biomol. Screening 2012, 17 (1), 27–38. 10.1177/1087057111418228. [DOI] [PubMed] [Google Scholar]
- Yang M.; Culhane J. C.; Szewczuk L. M.; Jalili P.; Ball H. L.; Machius M.; Cole P. A.; Yu H. Structural Basis for the Inhibition of the LSD1 Histone Demethylase by the Antidepressant Trans-2-Phenylcyclopropylamine. Biochemistry 2007, 46 (27), 8058–8065. 10.1021/bi700664y. [DOI] [PubMed] [Google Scholar]
- Schmidt D. M. Z.; McCafferty D. G. Trans-2-Phenylcyclopropylamine Is a Mechanism-Based Inactivator of the Histone Demethylase LSD1. Biochemistry 2007, 46 (14), 4408–4416. 10.1021/bi0618621. [DOI] [PubMed] [Google Scholar]
- Sorna V.; Theisen E. R.; Stephens B.; Warner S. L.; Bearss D. J.; Vankayalapati H.; Sharma S. High-Throughput Virtual Screening Identifies Novel N′-(1-Phenylethylidene)-Benzohydrazides as Potent, Specific, and Reversible LSD1 Inhibitors. J. Med. Chem. 2013, 56 (23), 9496–9508. 10.1021/jm400870h. [DOI] [PubMed] [Google Scholar]
- Soldi R.; Ghosh Halder T.; Weston A.; Thode T.; Drenner K.; Lewis R.; Kaadige M. R.; Srivastava S.; Daniel Ampanattu S.; Rodriguez del Villar R.; Lang J.; Vankayalapati H.; Weissman B.; Trent J. M.; Hendricks W. P. D.; Sharma S. The Novel Reversible LSD1 Inhibitor SP-2577 Promotes Anti-Tumor Immunity in SWItch/Sucrose-NonFermentable (SWI/SNF) Complex Mutated Ovarian Cancer. PLoS One 2020, 15 (7), e0235705. 10.1371/journal.pone.0235705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland R. A.Evaluation of Enzyme Inhibitors in Drug Discovery; John Wiley & Sons, Inc., 2013; 10.1002/9781118540398. [DOI] [Google Scholar]
- Morinan A.; Garratt H. M. An Improved Fluorimetric Assay for Brain Monoamine Oxidase. J. Pharmacol. Methods 1985, 13 (3), 213–223. 10.1016/0160-5402(85)90021-X. [DOI] [PubMed] [Google Scholar]
- Fang J.; Ying H.; Mao T.; Fang Y.; Lu Y.; Wang H.; Zang I.; Wang Z.; Lin Y.; Zhao M.; Luo X.; Wang Z.; Zhang Y.; Zhang C.; Xiao W.; Wang Y.; Tan W.; Chen Z.; Lu C.; Atadja P.; Li E.; Zhao K.; Liu J.; Gu J. Upregulation of CD11b and CD86 through LSD1 Inhibition Promotes Myeloid Differentiation and Suppresses Cell Proliferation in Human Monocytic Leukemia Cells. Oncotarget 2017, 8 (49), 85085–85101. 10.18632/oncotarget.18564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S.; Liu M.; Yao Y.; Yu B.; Liu H. Targeting LSD1 for Acute Myeloid Leukemia (AML) Treatment. Pharmacol. Res. 2021, 164, 105335. 10.1016/j.phrs.2020.105335. [DOI] [PubMed] [Google Scholar]
- Mascaró C.; Ortega A.; Carceller E.; Ruiz Rodriguez R.; Ciceri F.; Lunardi S.; Yu L.; Hilbert M.; Maes T. Chemoprobe-Based Assays of Histone Lysine Demethylase 1A Target Occupation Enable in Vivo Pharmacokinetics and Pharmacodynamics Studies of KDM1A Inhibitors. J. Biol. Chem. 2019, 294 (20), 8311–8322. 10.1074/jbc.RA118.006980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augert A.; Eastwood E.; Ibrahim A. H.; Wu N.; Grunblatt E.; Basom R.; Liggitt D.; Eaton K. D.; Martins R.; Poirier J. T.; Rudin C. M.; Milletti F.; Cheng W.-Y.; Mack F.; MacPherson D. Targeting NOTCH Activation in Small Cell Lung Cancer through LSD1 Inhibition. Sci. Signaling 2019, 12 (567), eaau2922. 10.1126/scisignal.aau2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Wang D.; Zheng G.; Yang Y.; Du L.; Dong Z.; Zhang X.; Wang C. Clinical Evaluation and Therapeutic Monitoring Value of Serum Tumor Markers in Lung Cancer. Int. J. Biol. Markers 2016, 31 (1), 80–87. 10.5301/jbm.5000177. [DOI] [PubMed] [Google Scholar]
- Blagg J.; Workman P. Choose and Use Your Chemical Probe Wisely to Explore Cancer Biology. Cancer Cell 2017, 32 (1), 9–25. 10.1016/j.ccell.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv T.; Yuan D.; Miao X.; Lv Y.; Zhan P.; Shen X.; Song Y. Over-Expression of LSD1 Promotes Proliferation, Migration and Invasion in Non-Small Cell Lung Cancer. PLoS One 2012, 7 (4), e35065. 10.1371/journal.pone.0035065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.; Vasilatos S. N.; Boric L.; Shaw P. G.; Davidson N. E. Inhibitors of Histone Demethylation and Histone Deacetylation Cooperate in Regulating Gene Expression and Inhibiting Growth in Human Breast Cancer Cells. Breast Cancer Res. Treat. 2012, 131 (3), 777–789. 10.1007/s10549-011-1480-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K.-H.; Kim B.-C.; Jeong S.-H.; Jeong C. W.; Ku J. H.; Kwak C.; Kim H. H. Histone Demethylase LSD1 Regulates Kidney Cancer Progression by Modulating Androgen Receptor Activity. Int. J. Mol. Sci. 2020, 21 (17), 6089. 10.3390/ijms21176089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K.; Luo J.; Yeh S.; You B.; Meng J.; Chang P.; Niu Y.; Li G.; Lu C.; Zhu Y.; Antonarakis E. S.; Luo J.; Huang C.-P.; Xu W.; Chang C. The MAO Inhibitors Phenelzine and Clorgyline Revert Enzalutamide Resistance in Castration Resistant Prostate Cancer. Nat. Commun. 2020, 11 (1), 2689. 10.1038/s41467-020-15396-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin X.; Du Y.; Liu X.; Wang L. LSD1 Promotes Prostate Cancer Cell Survival by Destabilizing FBXW7 at Post-Translational Level. Front. Oncol. 2021, 10, 3441. 10.3389/fonc.2020.616185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shelley M.; Perry J. K.; Shaw D. E.; Francis P.; Shenkin P. S. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47 (7), 1739–1749. 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- Iglesias J.; Saen-oon S.; Soliva R.; Guallar V. Computational Structure-based Drug Design: Predicting Target Flexibility. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2018, 8 (5), e1367. 10.1002/wcms.1367. [DOI] [Google Scholar]
- Mould D. P.; McGonagle A. E.; Wiseman D. H.; Williams E. L.; Jordan A. M. Reversible Inhibitors of LSD1 as Therapeutic Agents in Acute Myeloid Leukemia: Clinical Significance and Progress to Date. Med. Res. Rev. 2015, 35 (3), 586–618. 10.1002/med.21334. [DOI] [PubMed] [Google Scholar]
- Baell J. B.; Nissink J. W. M. Seven Year Itch: Pan-Assay Interference Compounds (PAINS) in 2017—Utility and Limitations. ACS Chem. Biol. 2018, 13 (1), 36–44. 10.1021/acschembio.7b00903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z.-Y.; Yang Z.-J.; He J.-H.; Lu A.-P.; Liu S.; Hou T.-J.; Cao D.-S. Benchmarking the Mechanisms of Frequent Hitters: Limitation of PAINS Alerts. Drug Discovery Today 2021, 26, 1353. 10.1016/j.drudis.2021.02.003. [DOI] [PubMed] [Google Scholar]
- Sonnemann J.; Zimmermann M.; Marx C.; Ebert F.; Becker S.; Lauterjung M.-L.; Beck J. F. LSD1 (KDM1A)-Independent Effects of the LSD1 Inhibitor SP2509 in Cancer Cells. Br. J. Haematol. 2018, 183 (3), 494–497. 10.1111/bjh.14983. [DOI] [PubMed] [Google Scholar]
- Salamero O.; Somervaille T.; Molero A.; Acuña E.; Perez-Simon J. A.; Rodriguez-Arbolí E.; Marchan B.; Garcia-Avila S.; Coll-Jorda R.; Quiñones-Roces T.; Pérez A.; Cano I.; Rodriguez-Veira R.; Gutierrez S.; Buesa C.; Bosch F.; Montesinos P.. ALICE maintains high clinical response rates supporting the efficacy of iadademstat combination with azacitidine in AML management. European Hematology Association conference 2021 (EHA-2021); https://www.oryzon.com/sites/default/files/events/20210611_EHA2021_poster_1.pdf.
- Bauer T. M.; Besse B.; Martinez-Marti A.; Trigo J. M.; Moreno V.; Garrido P.; Ferron-Brady G.; Wu Y.; Park J.; Collingwood T.; Kruger R. G.; Mohammad H. P.; Ballas M. S.; Dhar A.; Govindan R. Phase I, Open-Label, Dose-Escalation Study of the Safety, Pharmacokinetics, Pharmacodynamics, and Efficacy of GSK2879552 in Relapsed/Refractory SCLC. J. Thorac. Oncol. 2019, 14 (10), 1828–1838. 10.1016/j.jtho.2019.06.021. [DOI] [PubMed] [Google Scholar]
- Hollebecque A.; Salvagni S.; Plummer R.; Isambert N.; Niccoli P.; Capdevila J.; Curigliano G.; Moreno V.; Martin-Romano P.; Baudin E.; Arias M.; Mora S.; de Alvaro J.; Di Martino J.; Parra-Palau J. L.; Sánchez-Pérez T.; Aronchik I.; Filvaroff E. H.; Lamba M.; Nikolova Z.; de Bono J. S. Phase I Study of Lysine-Specific Demethylase 1 Inhibitor, CC-90011, in Patients with Advanced Solid Tumors and Relapsed/Refractory Non-Hodgkin Lymphoma. Clin. Cancer Res. 2021, 27 (2), 438–446. 10.1158/1078-0432.CCR-20-2380. [DOI] [PubMed] [Google Scholar]
- Paz-Ares L.; Juan-Vidal O.; Carcereny E.; Greillier L.; Navarro A.; Bennouna J.; Santoro A.; Berardi R.; Besse B.; Salvagni S.; Gonzalez H.; de Alvaro J.; Parra-Palau J.; Sánchez-Pérez T.; Aronchik I.; Filvaroff E.; Lamba M.; Nikolova Z.; Ponce S. 559P Phase Ib Study of CC-90011 plus Etoposide and Cisplatin (EP) in Patients with First-Line Extensive-Stage (ES) Small Cell Lung Cancer (SCLC). Annals of Oncology 2020, 31, S482–S483. 10.1016/j.annonc.2020.08.673. [DOI] [Google Scholar]
- Baillie T. A. Approaches to Mitigate the Risk of Serious Adverse Reactions in Covalent Drug Design. Expert Opin. Drug Discovery 2021, 16 (3), 275–287. 10.1080/17460441.2021.1832079. [DOI] [PubMed] [Google Scholar]
- Lammert C.; Einarsson S.; Saha C.; Niklasson A.; Bjornsson E.; Chalasani N. Relationship between Daily Dose of Oral Medications and Idiosyncratic Drug-Induced Liver Injury: Search for Signals. Hepatology 2008, 47 (6), 2003–2009. 10.1002/hep.22272. [DOI] [PubMed] [Google Scholar]
- Pettit K.; Gerds A. T.; Yacoub A.; Watts J. M.; Tartaczuch M.; Bradley T. J.; Shortt J.; Stevenson W. S.; Curtin N. J.; Rossetti J. M.; Burbury K.; Natsoulis G.; Jones A.; Talpaz M.; Peppe J.; Ross D. M.; Rienhoff H. Y. Jr. A Phase 2a Study of the LSD1 Inhibitor Img-7289 (Bomedemstat) for the Treatment of Myelofibrosis. Blood 2019, 134 (Supplement_1), 556. 10.1182/blood-2019-123244. [DOI] [Google Scholar]
- Morrison J. F. The Slow-Binding and Slow, Tight-Binding Inhibition of Enzyme-Catalysed Reactions. Trends Biochem. Sci. 1982, 7 (3), 102–105. 10.1016/0968-0004(82)90157-8. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.