

Abstract
The macro- and microvascular complications of type 1 and 2 diabetes lead to increased disease severity and mortality. The receptor for advanced glycation end products (RAGE) can bind AGEs and multiple proinflammatory ligands that accumulate in diabetic tissues. Preclinical studies indicate that RAGE antagonists have beneficial effects on numerous complications of diabetes. However, these antagonists target the extracellular domains of RAGE, which bind distinct RAGE ligands at diverse sites in the immunoglobulin-like variable domain and two constant domains. The cytoplasmic tail of RAGE (ctRAGE) binds to the formin, Diaphanous-1 (DIAPH1), and this interaction is important for RAGE signaling. To comprehensively capture the breadth of RAGE signaling, we developed small-molecule antagonists of ctRAGE-DIAPH1 interaction, termed RAGE229. We demonstrated that RAGE229 is effective in suppressing RAGE-DIAPH1 binding, Förster resonance energy transfer, and biological activities in cellular assays. Using solution nuclear magnetic resonance spectroscopy, we defined the molecular underpinnings of the interaction of RAGE229 with RAGE. Through in vivo experimentation, we showed that RAGE229 assuaged short- and long-term complications of diabetes in both male and female mice, without lowering blood glucose concentrations. Last, the treatment with RAGE229 reduced plasma concentrations of TNF-α, IL-6, and CCL2/JE-MCP1 in diabetic mice, in parallel with reduced pathological and functional indices of diabetes-like kidney disease. Targeting ctRAGE-DIAPH1 interaction with RAGE229 mitigated diabetic complications in rodents by attenuating inflammatory signaling.
INTRODUCTION
The receptor for advanced glycation end products (RAGE), a member of the immunoglobulin (Ig) superfamily, binds to and transduces the signals triggered by a diverse group of ligands. The protein encoded by AGER, RAGE, is composed of three extracellular domains: one Ig-like variable (V)–type domain followed by two Ig-like constant (C)–type domains, named C1 and C2; a single transmembrane–spanning domain; and a short, highly charged cytoplasmic tail (ct) domain or ctRAGE. The receptor is named for its ligand families, the advanced glycation end products (AGEs), which accumulate in obesity, diabetes, aging, and oxidative and inflammatory stresses (1–3). RAGE is also a receptor for the S100/calgranulins (such as S100A12 and S100B); amphoterin, also known as high mobility group box 1 (HMGB1); amyloid-beta peptide; phosphatidylserine (PS); and lysophosphatidic acid (LPA), as examples (4–9). Although it was earlier shown that RAGE ligands bound at the V-type domain (and/or to the V-C1 domain), numerous studies have demonstrated that certain RAGE ligands may bind on the C1- and C2-type extracellular domains that do not involve ligation of the Ig-like V-type domain (10, 11).
Given the evidence that the ctRAGE is required for RAGE ligand–stimulated signaling (12, 13), it was logical to probe the mechanisms by which this domain recruited signal transduction cascades to modulate gene expression and cellular properties. Toward this end, a yeast two-hybrid assay, used to discover ctRAGE binding molecules, identified Diaphanous-1 (DIAPH1) as a putative interacting partner (14). Immunoprecipitation experiments confirmed this interaction and identified that ctRAGE bound to the formin homology 1 (FH1) domain of DIAPH1; reduction of Diaph1 expression in cultured cells through silencing RNA approaches blocked RAGE ligand–mediated activation of multiple signaling pathways (14). RAGE and DIAPH1 are highly expressed in the human and murine diabetic kidney, and recently, it was shown that deletion of Diaph1 in vivo imparted similar benefits to that of Ager deletion in murine models of vascular injury, cardiac ischemia, and diabetes-associated kidney disease (15–17).
Nuclear magnetic resonance (NMR) experiments revealed that the N terminus of ctRAGE domain (residues 2 to 15) is ordered, whereas residues 16 to 43 of the C terminus are disordered and without discrete structure (18). The structure of ctRAGE is dynamic and likely composed of multiple conformers. Accordingly, mutation of amino acid residues Arg5 and Gln6 to alanine (Ala) residues of the N terminus of ctRAGE eliminated critical electrostatic interactions between Arg5 and Glu10 and disrupted the tertiary structure. In distinct work, super-resolution stochastic optical reconstruction microscopy (STORM) and single-particle tracking (SPT) revealed that in the presence of mutation of these same amino acids in ctRAGE or reduction in DIAPH1 expression, cell membrane RAGE clusters and diffusion are modulated, thereby providing additional support for the ctRAGE-DIAPH1 interactions (19).
The mutation of Arg5/Gln6 to Ala residues in ctRAGE limited the functional consequences of its interaction with the FH1 domain of DIAPH1 (18). When this mutation was introduced into primary murine aortic smooth muscle cells (SMCs), RAGE ligand S100B-stimulated phosphorylation of AKT (protein kinase B), migration, and proliferation were suppressed compared with cells transfected with control vector. However, these functional responses to non–RAGE ligand platelet-derived growth factor (PDGF) were not affected (18).
We previously screened a library of >58,000 known small molecules and identified 13 small-molecule competitive inhibitors of ctRAGE interaction with DIAPH1; these molecules reduced RAGE ligand–mediated signaling and suppressed the acute inflammatory effects of RAGE ligands in vivo (20). In the current study, upon critical review of the properties of these 13 identified antagonists, we aimed to optimize structure-activity relationships (SARs) of one of the classes emerging from that screen, the 2-phenylquinoline scaffold, for testing in murine models of complications of diabetes. We report the discovery of RAGE229, N-(4-(7-cyano-4-(morpholin-4ylmethyl)quinolin-2-yl)phenyl)acet-amide, as a ctRAGE-DIAPH1 inhibitor. Compared to vehicle controls, RAGE229 blocked ctRAGE-DIAPH1 interaction and reduced the consequences of in flammation, ischemia, and long-term diabetes in mice with type 1– and/or type 2-like diabetes, as tested in in vivo models of cardiac ischemia, wound healing, and diabetes-associated kidney disease. Collectively, this work identifies a potent antagonist of ctRAGE-DIAPH1 interaction and identifies a potential approach for targeting the consequences of RAGE pathological activation in chronic diseases such as diabetic complications.
RESULTS
Identification of small molecules that bind to ctRAGE derived from the phenylquinoline scaffold: RAGE203, RAGE208, and RAGE229
To identify highly potent antagonists of the ctRAGE-DIAPH1 interaction, the phenylquinoline scaffold (based on compound 11 in the original report) (20) was selected for further development on account of the multiple opportunities for molecule optimization from the 13 small-molecule antagonists of ctRAGE-DIAPH1 interaction. The medicinal chemistry strategy led to the identification of two early-stage molecules, RAGE203 and RAGE208, in which tryptophan fluorescence quenching assays illustrated the following affinity constants of the small molecules to the ctRAGE, KD of 30 ± 10 nM and 24 ± 6 nM, respectively (Fig. 1, A and B). Subsequent SAR studies revealed that improvements were achievable by adding substituents at the quinoline 7 positions, the acetamide and quinoline 4-methylene amino groups, which led to the discovery of RAGE229, KD of 2 ± 1 nM (Fig. 1C).
Fig. 1. RAGE203, RAGE208, and RAGE229 bind to ctRAGE with nanomolar affinities.
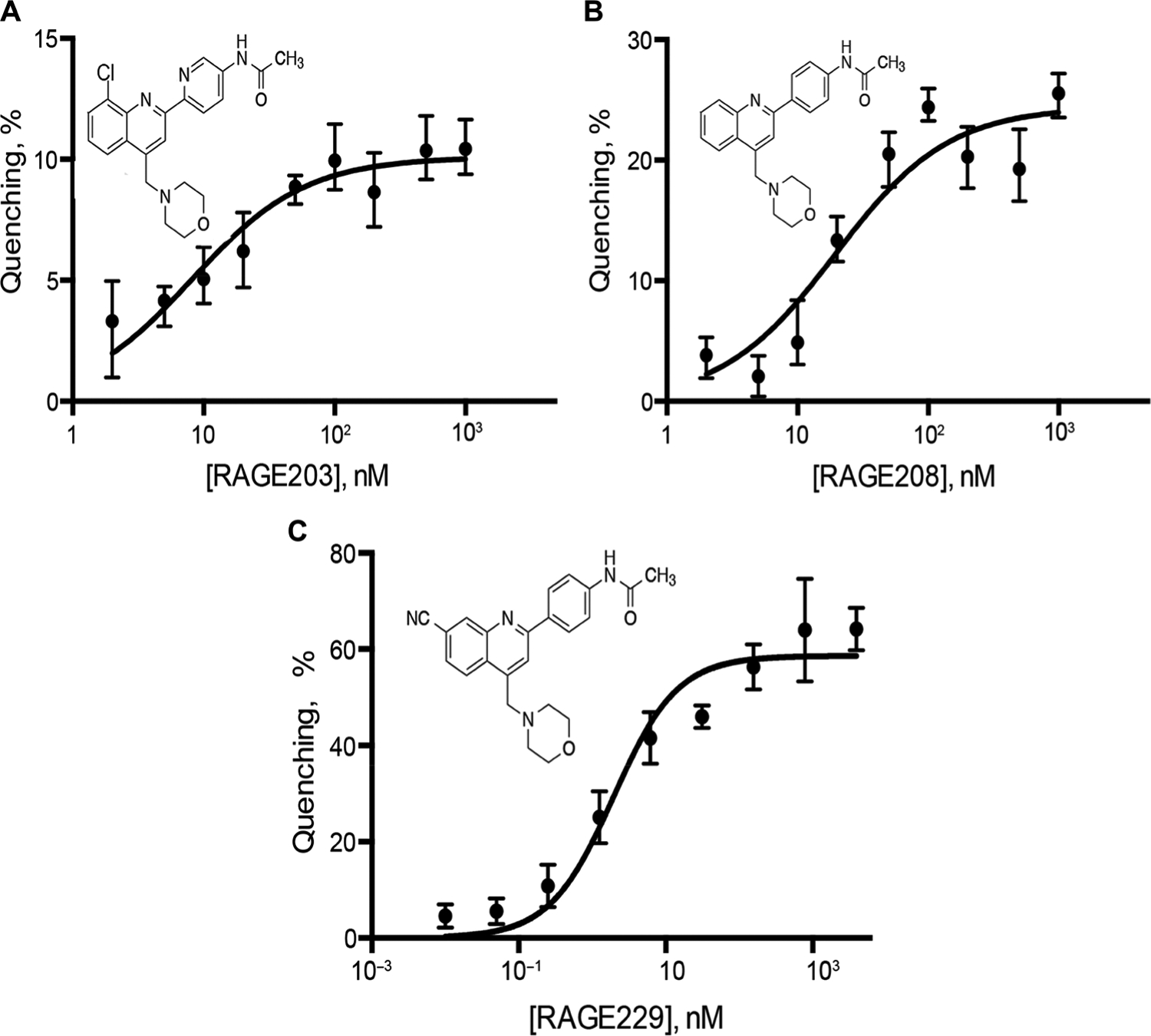
Fluorescence titration of the compounds into the solution of ctRAGE resulted in the fluorescence quenching. The affinities of RAGE203 (A), RAGE208 (B), and RAGE229 (C) for the ctRAGE were estimated by fitting the data with the single-site binding isotherm and determined to be 30 ± 10, 24 ± 6, and 2 ± 1 nM, respectively. All experiments were performed in triplicate, and the results are expressed as the means ± SD.
RAGE229: NMR spectroscopy and Förster resonance energy transfer
Solution NMR spectroscopy was used to gain structural understanding of the RAGE229-ctRAGE complex. Adding 50 μM RAGE229 into the 50 μM solution of [U-15N] ctRAGE resulted in small but reproducible changes in the ctRAGE chemical shifts (Fig. 2A). The most affected residues were Gln3, Arg7, Arg8, and Gly9. These residues lie in the partially structured region of ctRAGE, amino acids 2 to 15, and overlap with the region that was shown to bind to DIAPH1 (18, 20). This result suggests that RAGE229 can directly compete with DIAPH1 for binding. The solution structure of the RAGE229-ctRAGE complex was determined with the precision of 0.5 Å based on the 384 distance constraints including 13 intermolecular constraints and 268 dihedral constraints (Fig. 2B, fig. S1, and table S1). Major contacts in the complex are between the acetamide moiety of RAGE229 and Arg8 and Glu16 and between the cyano group of RAGE229 and the guanadino group of Arg12 (Fig. 2C). The quinoline ring of RAGE229 is positioned against the ctRAGE surface formed by the methylene group of Arg4 and Arg5 (Fig. 2C). The morpholino group is facing the solvent and is likely to be important for RAGE229 solubility. The lack of the cyano group at the 7 position on the quinolone ring is the likely reason for the lower affinities of RAGE203 and RAGE208 for the ctRAGE (Fig. 1).
Fig. 2. Binding of RAGE229 to the ctRAGE is mostly due to polar interactions and antagonizes RAGE-DIAPH1 interaction in human embryonic kidney 293T.
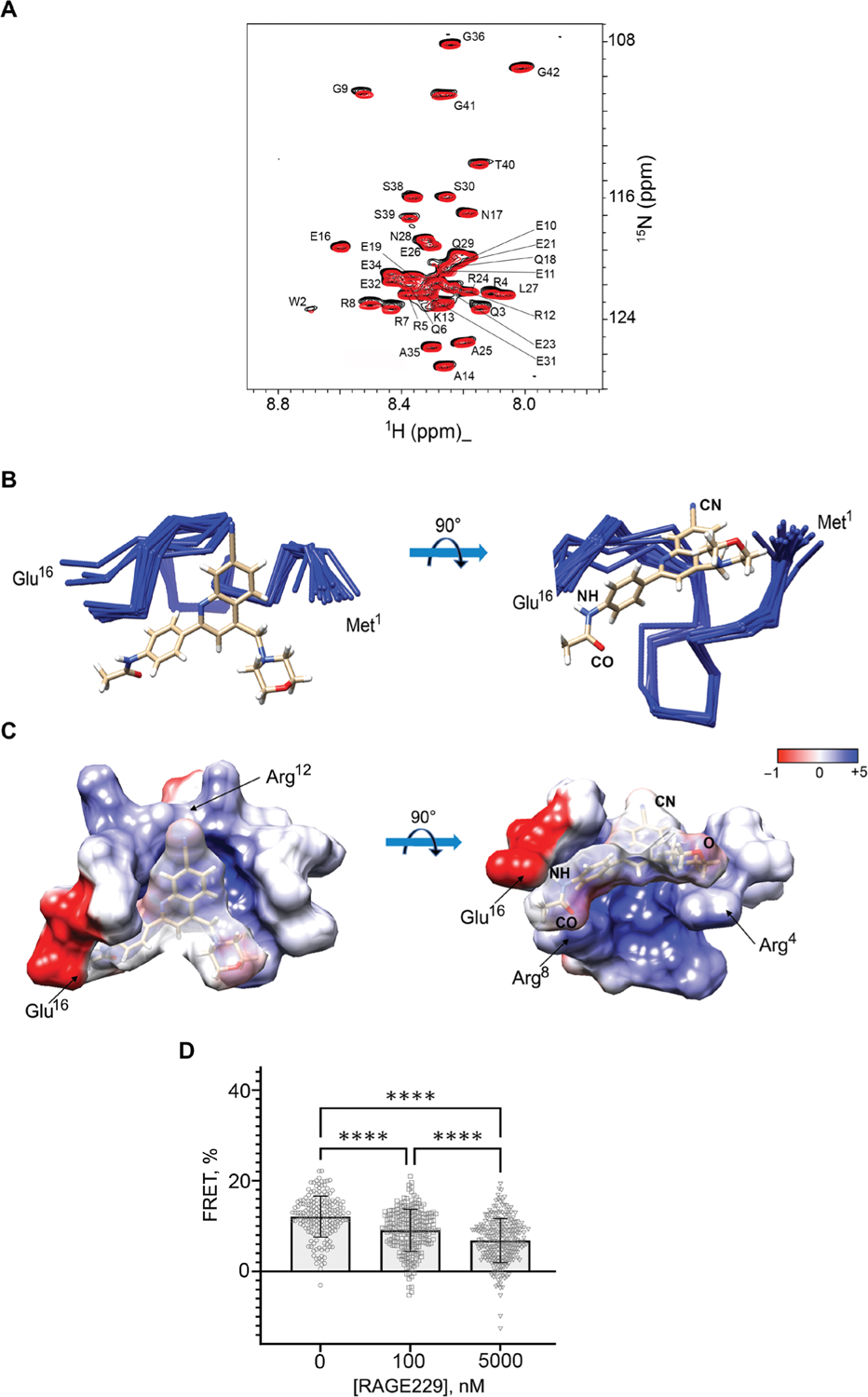
(A) Overlay of the two-dimensional 1H-15N heteronu-clear single-quantum coherence (HSQC) NMR spectra of 50 μM [U-15N] ctRAGE and the complex of 50 μM [U-15N] ctRAGE and RAGE229 with molar ratio of 1:1:2. The assignments of ctRAGE amide protons and nitrogens are indicated. (B) Overlay of the 10 best solution structures of the ctRAGE-RAGE229 complex. Cyano (CN) and acetamide (CO and NH) groups of RAGE229 are indicated. (C) Electrostatic potential was mapped on the ctRAGE surface of the ctRAGE-RAGE229 complex. Amino acids responsible for the interactions with cyano and acetamide moieties of RAGE229 are indicated. The coordinates and chemical shift assignments of the ctRAGE-RAGE229 complex have been deposited in the Protein Data Bank with accession number 6VXG. (D) Bar graph shows the decrease in the average FRET values between RAGE-eCFP and constitutively active DIAPH1-eYFP after cells were incubated with 100 nM and 5 μM RAGE229. FRET is indicative of RAGE-DIAPH1 interaction and is determined as the increase in donor emission. Data are presented as means ± SD. Statistical analysis was performed by unpaired t test. ****P < 0.0001.
Förster resonance energy transfer (FRET) between RAGE-eCFP and constitutively active DIAPH1 (DIAPH1-eYFP) (fig. S2) was used to demonstrate that RAGE229 directly competes with DIAPH1 for RAGE binding. Both RAGE-eCFP and DIAPH1-eYFP were transfected into human embryonic kidney 293T cells. RAGE-DIAPH1 interaction results in the increase in the RAGE-eCFP fluorescence, the FRET value of 12.1 ± 4.5%, after photobleaching DIAPH1-eYFP (21). Adding 100 nM or 5 μM RAGE229 decreased the FRET value to 9.1 ± 4.5% and 6.8 ± 4.8%, respectively (P < 0.0001) (Fig. 2D). The minimal value of FRET between a particular pair of interactors depends on a concentration of fluorescent proteins in the immediate proximity of a plasma membrane (10). Thus, RAGE229 antagonizes the RAGE-DIAPH1 complex in a dose-dependent manner. On the basis of these findings, we sought to determine whether the small molecules described above exerted effects on functional consequences of RAGE ligands.
Effects of RAGE203, RAGE208, and RAGE229 on SMC migration
We previously developed an in vitro cell–based assay to test the effects of small-molecule antagonists on ctRAGE-DIAPH1 interaction. Murine aortic SMCs endogenously express RAGE and DIAPH1, and previous work has shown that the expression of RAGE and DIAPH1 in these cells affects RAGE ligand–stimulated migration in vitro (20) and affects the in vivo response to denudation of the femoral artery in mice, which is a potent stimulus for SMC migration (15, 22). In this assay, RAGE ligand carboxymethyllysine (CML)–AGE is used to stimulate cultured SMCs in the presence of a scratch wound. Escalating doses of small-molecule antagonist are incubated with the SMCs for 1.5 hours (and then removed), followed by addition of CML-AGE and the scratch wounding for 4 hours, after which time the percent inhibition is measured at each dose. As illustrated in Fig. 3 (A to C) and fig. S3, the IC50 for inhibition of SMC migration was 950 ± 80 nM for RAGE203, 70 ± 20 nM for RAGE208, and 26 ± 9 nM for RAGE229. To determine the specificity of the small molecules to the ctRAGE-DIAPH1 interaction, SMCs were treated with PDGF-BB, which is not a RAGE ligand. As illustrated in Fig. 3D and fig. S3, RAGE203, RAGE208, and RAGE229 exerted no effect on PDGFBB–stimulated SMC migration compared with vehicle treatment.
Fig. 3. Small-molecule antagonists of RAGE affect scratch wounding in murine and human SMCs.
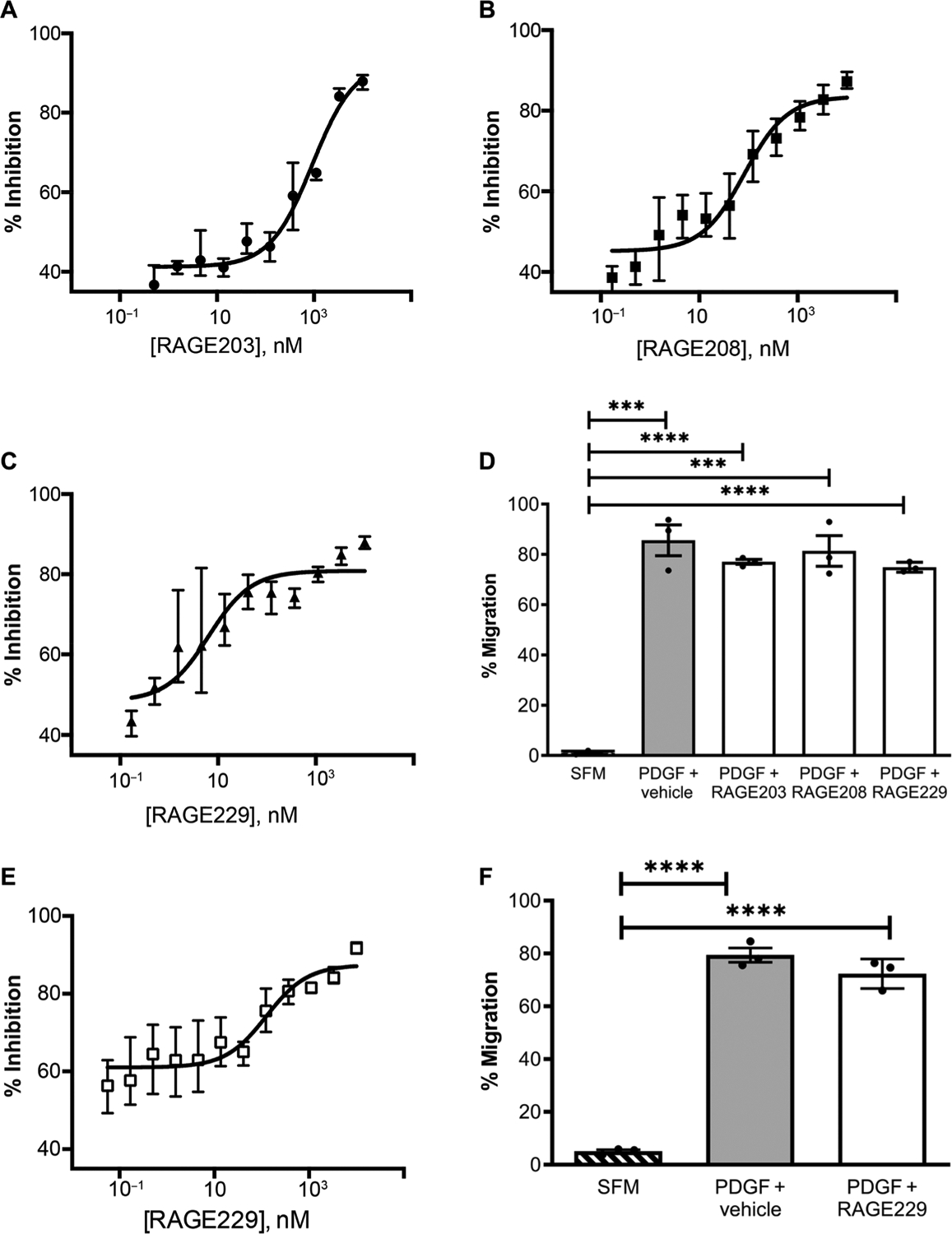
Small-molecule antagonists (12-point dose response; 1, 10 μM; 2, 3.333 μM; 3, 1.111 μM; 4, 0.370 μM; 5, 0.123 μM; 6, 0.041 μM; 7, 0.014 μM; 8, 0.005 μM; 9, 0.002 μM; 10, 0.001 μM; 11, 0.0002 μM; and 12, 0.00006 μM) tested in an SMC scratch wounding experiment. The IC50 values for inhibition for the small-molecule antagonists in murine SMCs are 950 ± 80 nM for RAGE203 (A), 70 ± 20 nM for RAGE208 (B), and 26 ± 9 nM for RAGE229 (C). (D) To verify the specificity of the small molecules tested, the percent migration in response to a non–RAGE ligand, PDGF-BB, was used as a positive control. SFM (serum-free medium) is the negative control. (E and F) In human SMCs, the IC50 value for inhibition of the small-molecule antagonist RAGE229 was 120 ± 60 nM (E). The percent migration induced by the non–RAGE ligand PDGF-BB and the effects of RAGE229 were tested (F). Compound incubation (concentration, 1 μM) and study design are as described for (A) to (F). Assays were performed in triplicate, and results are representative of three independent experiments. Data are presented as means ± SEM. After confirmation that the data passed the Shapiro-Wilk test for normality, statistical analysis was performed by unpaired t test. ***P < 0.001 and ****P < 0.0001.
We probed the relevance of our findings to human RAGE and DIAPH1 by testing RAGE229 in primary human aortic SMCs. The IC50 for inhibition of human SMC migration was 120 ± 60 nM (Fig. 3E). In human SMCs treated with PDGF-BB, RAGE229 had no effect on migration (Fig. 3F and fig. S4).
Effects of RAGE203, RAGE208, and RAGE229 on short-term models of inflammation, cardiac ischemia, and diabetes in mice
Previous work illustrated that RAGE is a receptor for damage-associated molecular pattern (DAMP) molecules; among them are the S100/calgranulins and amphoterin (HMGB1) (5, 23). In our precedent studies, we showed that a soluble receptor decoy of RAGE (soluble or sRAGE), or blocking antibodies to RAGE, suppressed inflammation in nondiabetic mice subjected to a delayed-type hypersensitivity (DTH) test using immunization followed by challenge (foot pad) with methylated bovine serum albumin (mBSA) 3 weeks later (5). Here, we tested whether treatment with the small molecules affected inflammation responses in mice undergoing the DTH study. The small-molecule antagonists and equal volumes of vehicle, dimethyl sulfoxide (DMSO), were administered twice daily by oral gavage, 5 mg/kg per dose for a total of four doses. The last dose was administered 12 hours before the foot pad scoring. As shown in Fig. 4A, treatment with the small molecules significantly reduced inflammation score for RAGE203, RAGE208, and RAGE229 compared with vehicle (DMSO) administration (P < 0.01, P < 0.05, and P < 0.01, respectively).
Fig. 4. Effects of RAGE203, RAGE208, and RAGE229 in DTH and cardiac ischemia-reperfusion injury.
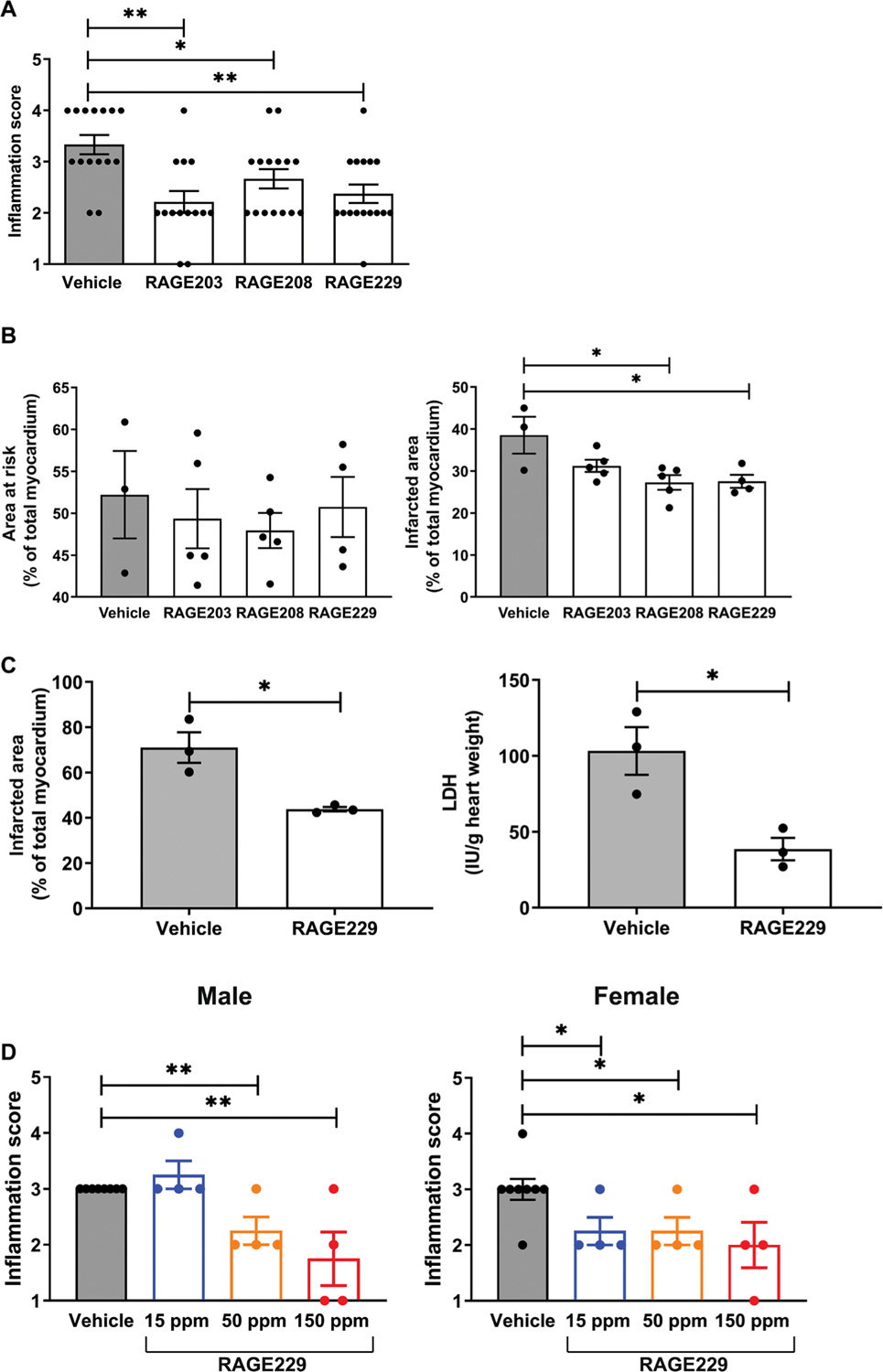
(A) Effect of ctRAGE-DIAPH1 compounds on DTH scores in female CF-1 mice. Footpad inflammation scores are shown in mice treated with compounds compared to vehicle. Statistical analysis was performed by Cochran-Armitage test. (B) Effect of RAGE203, RAGE208, and RAGE229 on myocardial infarct area in C57BL/6J mice. Area at risk (left) and infarct area (right) are shown. After confirmation that data passed the Shapiro-Wilk test for normality, statistical analysis was performed by unpaired t test. (C) Effect of RAGE229 (1 μM) in the isolated perfused diabetic heart. Infarct area (left) and lactate dehydrogenase (LDH) measurements (right) were performed. After confirmation of normality, data were analyzed by unpaired t test. (D) Effect of RAGE229 medicated on DTH scores in male and female CD-1 mice. Inflammation score is shown for male and female mice fed one of three RAGE229-specially modified chows (15, 50, or 150 ppm) or vehicle chow for 7 days before being challenged in the hind paw with mBSA. The number of mice per group is indicated in each figure. Statistical analysis was performed by Cochran-Armitage test. *P < 0.05 and **P < 0.01.
In separate short-term experiments, we administered RAGE203, RAGE208, or RAGE229 to male mice with diabetes undergoing 30 minutes of transient ligation and 48 hours of reperfusion of the left anterior descending coronary artery. Mice were rendered type 1–like diabetic with streptozotocin (STZ) for 2 months and pretreated with RAGE203, RAGE208, or RAGE229 with twice daily oral gavage treatment at 5 mg/kg per dose for 4 days before the induction of infarction. Treatment was continued twice daily until euthanasia, 48 hours after the infarction. Mice were euthanized, and the hearts were retrieved for staining with triphenyl tetrachloride (TTC). As shown in Fig. 4B, although there were no differences in the area at risk (left) among the groups, when compared with vehicle treatment, the infarct area was significantly lower in mice treated with either RAGE208 or RAGE229 (P < 0.05). RAGE203 had no effect. There were no differences in serum glucose concentrations among the four groups of diabetic mice (table S2).
In addition, we probed the effects of RAGE229 in the ex vivo isolated perfused hearts of male type 1–like diabetic Akita mice (Ins2Akita/+) (24) subjected to 30 min of normoxia, followed by zero-flow ischemia (30 min) and then by 60 min of reoxygenation. RAGE229 (1 μM) or vehicle incubation of the isolated heart was begun 10 min before zero low-flow ischemia and continued through the end of the experiment. Perfusion of the isolated heart with RAGE229 resulted in a significant attenuation of the infarct area (P < 0.05) (Fig. 4C, left). The release of lactate dehydrogenase (LDH), a marker of tissue injury, was significantly lower in the RAGE229-treated compared with vehicle-treated diabetic hearts (P < 0.05) (Fig. 4C, right). There were no differences in serum glucose or body weight (table S3). Thus, small-molecule antagonism of ctRAGE-DIAPH1 interaction attenuates injury in ex vivo and in vivo models of cardiac ischemia-reperfusion injury in diabetic mice.
On the basis of the evidence that RAGE229 demonstrated superior properties when compared with RAGE203 or RAGE208, and in preparation for eventual long-term administration of RAGE229 in murine models of diabetic complications, we prepared RAGE229-containing chow. Pharmacokinetic studies verified the bioavailability of RAGE229 after either oral (gavage) or intravenous administration (table S4 and fig. S5). On the basis of these data, RAGE229 (>95% purity) was prepared in standard rodent chow. Three distinct concentrations of RAGE229-containing chow were prepared, 150, 50, and 15 parts per million (ppm), to deliver about 30, 10, and 3 mg/kg of RAGE229-containing chow, respectively, per day per mouse. Male and female CF-1 mice were subjected to DTH; administration of RAGE229-containing chow was begun 7 days before the foot pad challenge and continued through euthanasia. In male mice, compared with vehicle, administration of RAGE229-containing chow at 150 or 50 ppm resulted in significant reduction in inflammation score (P < 0.01) (Fig. 4D, left). In contrast, administration of RAGE229-containing chow at 15 ppm had no effect on inflammation score (Fig. 4D, left). In female mice, compared with vehicle treatment, administration of RAGE229-containing chow at 150, 50, or 15 ppm significantly reduced foot pad score (P < 0.05) (Fig. 4D, right). There were no differences in body weight in male or female mice when comparing RAGE229-treated to vehicle-treated mice (table S5). The plasma concentrations of RAGE229 at euthanasia suggested increasing plasma concentrations of RAGE229 as the dose was escalated from 15 to 50 to 150 ppm in the RAGE229-containing chow in both male and female mice (table S5). Last, we tested RAGE229, administered by intraperitoneal injection, to female mice undergoing DTH. As shown in fig. S6, compared with vehicle, female mice receiving RAGE229 [5 mg/kg, intraperitoneally (ip)] every 12 hours for four total doses displayed significantly lower inflammation score (P < 0.05). Thus, RAGE229 was able to attenuate inflammation in mice.
Diabetic wound healing: Effects of topical administration of RAGE229
A major challenge was to test the effects of RAGE229 in long-term diabetes complications. First, we tested whether these small molecules accelerated wound closure in diabetes. In human subjects, diabetic foot ulcers are a major complication of type 1 and 2 diabetes, which may lead to limb/digit amputation (25). Food and Drug Administration–approved devices remain limited and include agents such as local cell-based matrices to stimulate wound healing (26) and ointments (27, 28). We tested these concepts in the BTBR ob/ob mice, a model of type 2–like diabetes, in which multiple previous studies illustrated impaired wound healing when compared with nondiabetic control mice (29–34). Full-thickness excisional wounds (1.5 cm by 1.5 cm) were created on the backs of 8-week-old male and female BTBR ob/ob mice, and the wounds were covered with sterile plastic dressing (35). On days 3 to 10 after wounding, RAGE229 (5 mg/kg per mouse) or equal volumes of DMSO were injected topically underneath the dressing two times daily and directly onto the surface of the wound. Wounds were photographed on days 0, 7, 14, and 21, and wound closure was determined. As shown in Fig. 5 (A and B), on day 21, the percent wound closure was significantly higher in male BTBR ob/ob mice treated with RAGE229 versus vehicle (P < 0.01); in female mice, no improvements in wound closure on day 21 with RAGE229 treatment were shown. We examined the rates of change of wound closure and the effects of RAGE229. In male mice, although there were no differences in percent wound closure between days 0 and 7, days 7 and 14, and days 14 and 21, significant improvements in percent wound closure were noted in RAGE229 versus vehicle treatment (P = 0.008 and P = 0.007, respectively) (Fig. 5C). In female BTBR ob/ob mice, there were no differences observed at any time point considered (P values are indicated in the figure) (Fig. 5D).
Fig. 5. Administration of topical RAGE229 accelerates wound healing in diabetic mice.
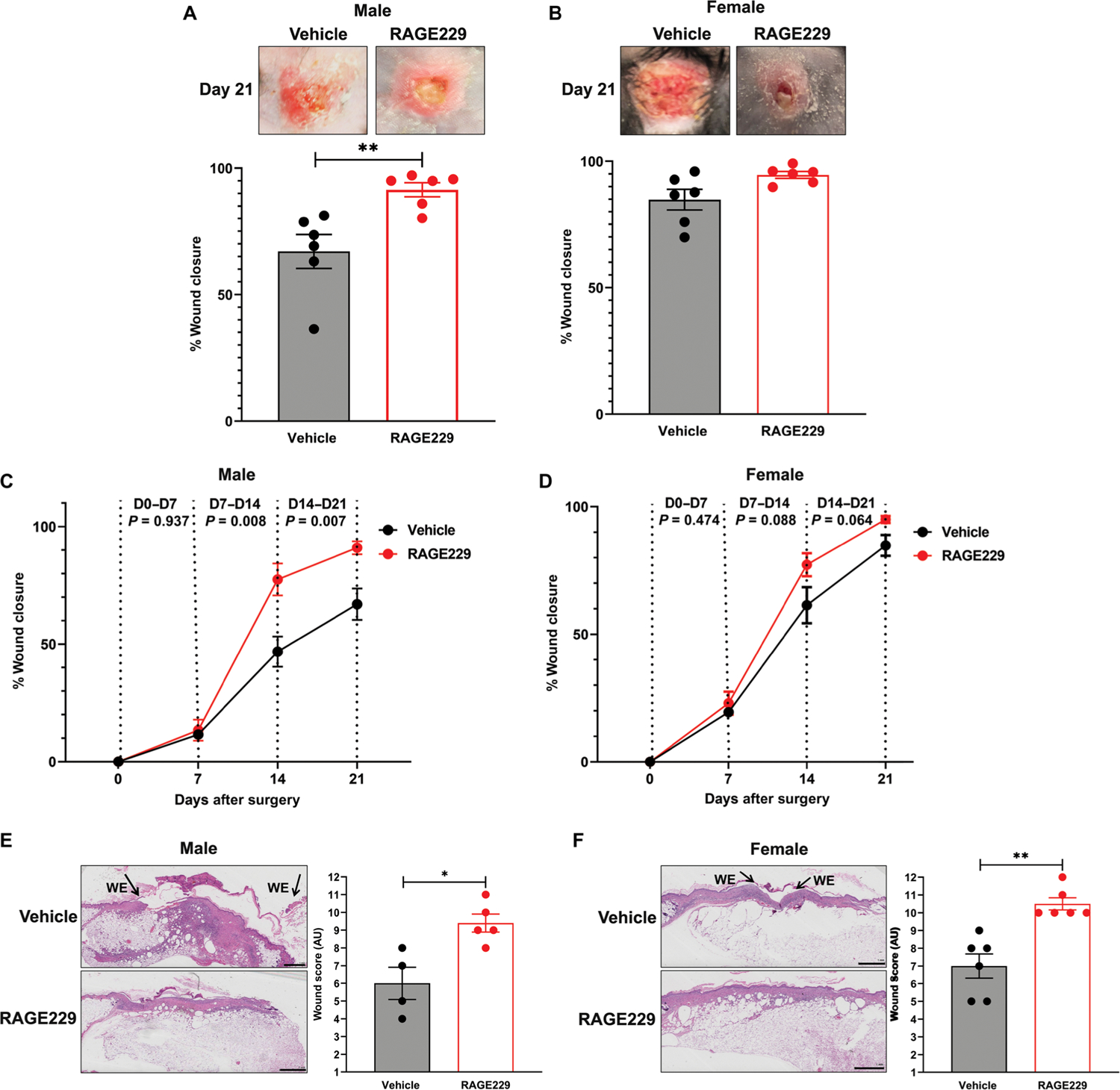
At age 8 weeks, male and female BTBR ob/ob mice were subjected to full-thickness excisional wounding. % Wound closure from baseline is shown for males (A, C, and E) and females (B, D, and F) (N = 6). In (E) and (F), histological score was obtained from day 21 wounds. WE, wound edge. Original magnification, ×20; image magnification, ×1.25. Scale bars, 1 mm. The number of mice per group is indicated in each figure. Data are presented as means ± SEM, and statistical analysis was performed by unpaired t test after the Shapiro-Wilk test for normality. *P < 0.05 and **P < 0.01. AU, arbitrary units.
In addition, we used a scoring system to evaluate wound healing based on a maximum of 12 points upon evaluation of hematoxylin and eosin–stained slides with analysis performed by investigators unaware of the experimental treatment (36). As shown in Fig. 5 (E and F), on day 21, in male and female BTBR ob/ob mice, topical treatment with RAGE229 resulted in significantly higher wound score versus vehicle-treated mice (P < 0.05 and P < 0.01, respectively) (Fig. 5F). In the figure, “WE” indicates wound edge. In male and female mice, there were no treatment-related differences in body weight or in the concentrations of blood glucose (table S6). Thus, topical administration of RAGE229 in type 2–like diabetic mice accelerated wound healing responses and set the stage for testing in long-term models of diabetic kidney complications.
RAGE229 and long-term type 1–like diabetes kidney complications
We tested the effects of RAGE229 in murine models of long-term diabetes–associated kidney disease. The leading cause of chronic kidney disease (CKD) is diabetes (37–40). CKD increases risk for all-cause mortality and cardiovascular disease in diabetic subjects (40). Even with optimal diabetes care, treatment with angiotensin-converting enzyme inhibitors or angiotensin receptor blockers does not eliminate diabetes-associated kidney disease (41). Hence, antagonism of ctRAGE-DIAPH1 may address an important gap for the management of diabetic complications.
We began by probing the effects of RAGE229 in a type 1–like diabetes model, in which hyperglycemia was induced via STZ at age 8 weeks in C57BL/6J mice. After documentation of diabetes or the control state, diabetic male and female mice were treated for an additional 6 months with RAGE229-containing chow at 150, 50, or 15 ppm/day or control chow. Only mice that demonstrated serum glucose ≥250 mg/dl on at least two separate occasions before random allocation to treatment or vehicle groups were eligible for study participation. Multiple endpoints were studied. First, we examined mesangial sclerosis and tubular atrophy and interstitial fibrosis, which were determined by a renal pathologist naïve to the experimental conditions. In male and female mice, compared with the nondiabetic state, diabetes resulted in a significant increase in mesangial sclerosis score (P < 0.0001 and P < 0.01, respectively) (Fig. 6, A and E). In male diabetic mice, treatment with 150 or 50 ppm resulted in significant reduction in mesangial sclerosis score (P < 0.05); treatment with 15 ppm had no effect (Fig. 6, A and E). In female diabetic mice, treatment with 150, 50, or 15 ppm resulted in a significant reduction in mesangial sclerosis score (P < 0.05) (Fig. 6, A and E). Tubular atrophy and interstitial fibrosis were examined in male and female mice. In both sexes, vehicle-treated diabetic mice displayed higher tubular atrophy and interstitial fibrosis (P < 0.01) (fig. S7). In male and female mice, compared with vehicle or RAGE229, 15 ppm, the administration of 150 or 50 ppm resulted in significantly lower tubular atrophy and interstitial fibrosis (P < 0.05) (fig. S7).
Fig. 6. Treatment with RAGE229 protects from mesangial sclerosis, podocyte effacement, GBM thickening, and UAE in male and female type 1–like diabetic C57BL/6J mice.
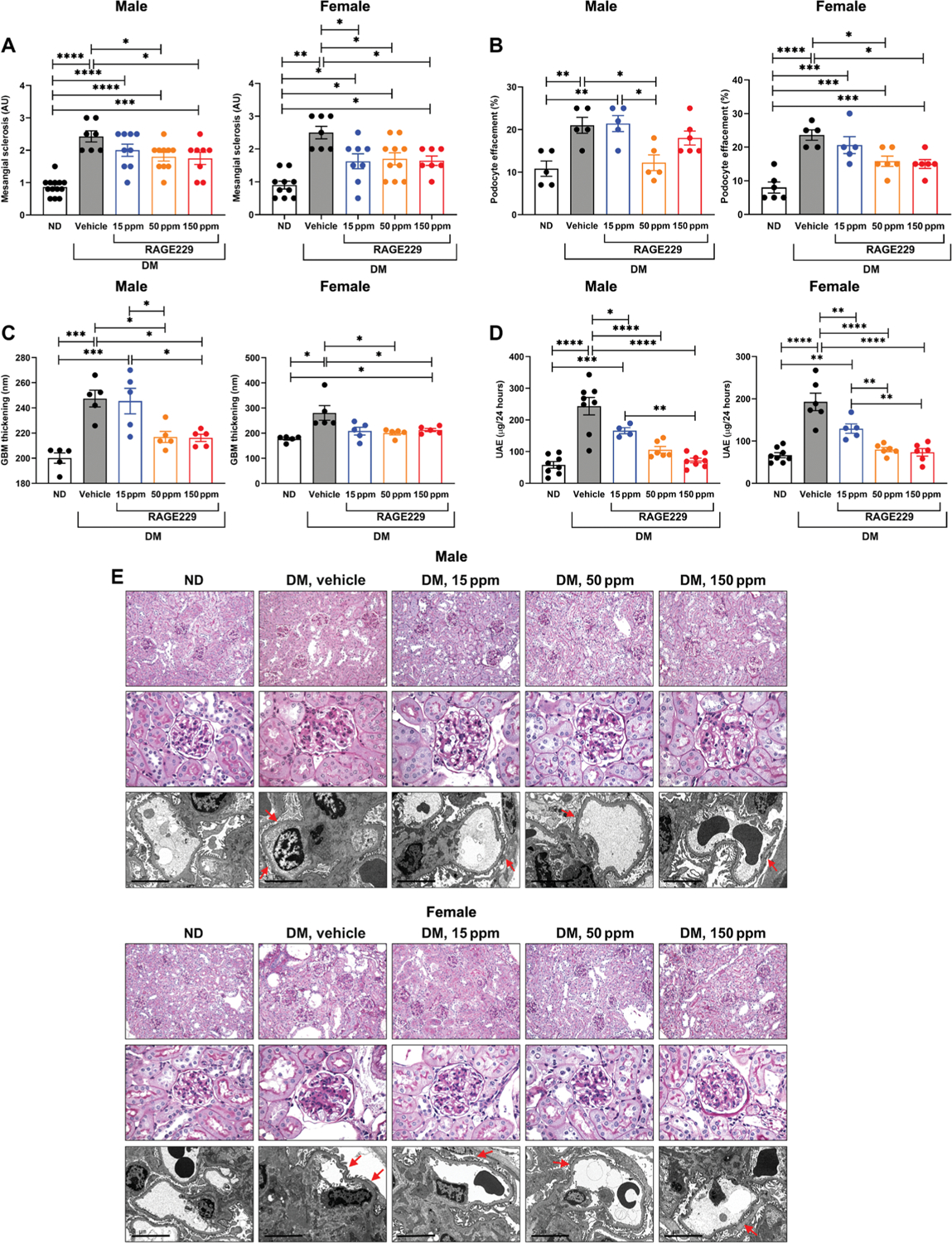
Diabetic animals (DM) were further randomized to either receive one of three concentrations (15, 50, and 150 ppm) of RAGE229-containing chow or vehicle chow (vehicle) and euthanized after 6 months of STZ-induced hyperglycemia. Non-diabetic (ND) control animals only received vehicle chow. (A) Mesangial sclerosis score. For male mice, statistical significance was determined by ANOVA with post hoc Tukey’s test after passing the Shapiro-Wilk test for normality. For female mice, the data did not pass the Shapiro-Wilk test for normality, and the Wilcoxon rank sum test was used. (B) Podocyte effacement. For both male and female mice, statistical significance was determined by ANOVA with post hoc Tukey’s test after passing the Shapiro-Wilk test for normality. (C) Thickness of the GBM. For male mice, statistical significance was determined by ANOVA with post hoc Tukey’s test after passing the Shapiro-Wilk test for normality. For female mice, the data did not pass the Shapiro-Wilk test for normality, and the Wilcoxon rank sum test was used. (D) Measurement of UAE after 6 months of diabetes. For both male and female mice, statistical significance was determined by ANOVA with post hoc Tukey’s test after passing the Shapiro-Wilk test for normality. (E) Representative images of periodic acid–Schiff–stained sections (top row, ×200 magnification; middle row, ×600 magnification) and electron microscopy images (bottom row, ×10,000 magnification; scale bars, 5 μm) of kidney cortex tissue in male and female mice. Arrows indicate examples of podocyte effacement. The number of mice per group is indicated in each figure. Data are presented as means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Second, we examined podocyte effacement by electron microscopy. In both male and female mice, when comparing vehicle chow–treated diabetic mice with nondiabetic mice, a significant increase in podocyte effacement was observed (P < 0.01 and P < 0.0001, respectively) (Fig. 6, B and E). In diabetic male mice, treatment with RAGE229-containing chow at 50 ppm, but not 150 or 15 ppm, resulted in reduction in podocyte effacement (P < 0.05) (Fig. 6, B and E). In female diabetic mice, treatment with RAGE229-containing chow at 150 and 50 ppm resulted in a significant attenuation of podocyte effacement (P < 0.05), whereas RAGE229 at 15 ppm had no effect (Fig. 6, B and E).
Third, by electron microscopy, we measured the thickness of the glomerular basement membrane (GBM). In both male and female mice, an increase in GBM thickness was observed in diabetic versus nondiabetic mice (P < 0.001 and P < 0.05, respectively) (Fig. 6, C and E). In male diabetic mice, treatment with RAGE229-containing chow at 150 or 50 ppm resulted in a significantly lower GBM thickness (P < 0.05) (Fig. 6, C and E). Treatment with RAGE229 at 15 ppm had no effect (Fig. 6, C and E). In female diabetic mice, treatment with RAGE229 at 150 and 50 ppm resulted in lower GBM thickness (P < 0.05) (Fig. 6, C and E). Treatment with RAGE229 at 15 ppm had no effect (Fig. 6, C and E).
Fourth, we examined urinary albumin excretion (UAE) over 24 hours. In both male and female mice, diabetes resulted in increased UAE compared with nondiabetic mice (P < 0.0001) (Fig. 6D). In male diabetic mice, treatment with RAGE229 at 150, 50, and 15 ppm resulted in significantly lower UAE (P < 0.0001, P < 0.0001, and P < 0.05, respectively) (Fig. 6D). In female diabetic mice, treatment with RAGE229 at 150, 50, and 15 ppm resulted in significantly lower UAE (P < 0.0001, P < 0.0001, and P < 0.01, respectively) (Fig. 6D).
Table S7 illustrates that there were no RAGE229 treatment effects on body weight or on blood glucose concentrations in male and female mice. Table S7 also displays the mean RAGE229 concentrations for each treatment group of male and female mice.
Collectively, these data indicate that in both male and female type 1–like diabetic mice, treatment with RAGE229 reduced pathological and functional (UAE) measures of diabetic kidney disease. Next, we evaluated the effects of RAGE229 on kidney complications in a murine model of type 2 diabetes.
RAGE229 and long-term type 2–like diabetes kidney complications
Previous studies have shown that BTBR ob/ob mice develop pathological and functional indices of diabetic kidney disease (42). The wild-type offspring were enrolled as the nondiabetic control mice, and the homozygous BTBR ob/ob obese mice were used as the diabetic model. We treated the diabetic mice with 150 ppm RAGE229-containing chow or vehicle chow from 8 weeks of age for 4 months. Table S8 illustrates the body weights and serum glucose concentrations. Statistical analyses revealed increased concentrations of blood glucose in male ob/ob mice treated with RAGE229 compared with controls. In female ob/ob mice, although there were no differences in blood glucose concentrations, the RAGE229-treated mice displayed significantly lower body weight compared with control animals.
In both male and female mice, diabetes was associated with a significant increase in mesangial score, podocyte effacement, GBM thickness, and UAE (P < 0.01) (Fig. 7, A to E). Mesangial sclerosis was reduced in male and female diabetic mice treated with RAGE229-containing chow at 150 ppm (P < 0.01 and P < 0.001, respectively) (Fig. 7, A and E). Male diabetic mice showed significantly greater tubular atrophy and interstitial fibrosis compared with nondiabetic control mice (P < 0.001); this increase was significantly attenuated by RAGE229 at 150 ppm (P < 0.05) (fig. S8). In female mice, there were no diabetes-related changes in these endpoints and no difference in RAGE229 150 ppm–treated mice versus vehicle-treated mice (fig. S8). Treatment with RAGE229-containing chow significantly reduced podocyte effacement compared with vehicle-treated diabetic male and female mice (P < 0.01 and P < 0.05, respectively) (Fig. 7, B and E). The treatment also reduced GBM thickness in both male and female diabetic mice compared with vehicle-treated controls (P < 0.01 and P < 0.05, respectively) (Fig. 7, C and E). Moreover, UAE was significantly reduced in female animals treated with RAGE229-containing chow (P < 0.01) (Fig. 7D). No effect of the treatment on UAE was observed in male diabetic mice (Fig. 7D). Overall, RAGE229 treatment in type 2–like diabetic mice with long-term kidney complications reduced multiple pathological and functional parameters relevant to diabetic kidney disease, although with some sex-dependent differences.
Fig. 7. Treatment with RAGE229 protects from mesangial sclerosis, podocyte effacement, GBM thickening, and UAE in male and female BTBR ob/ob mice.
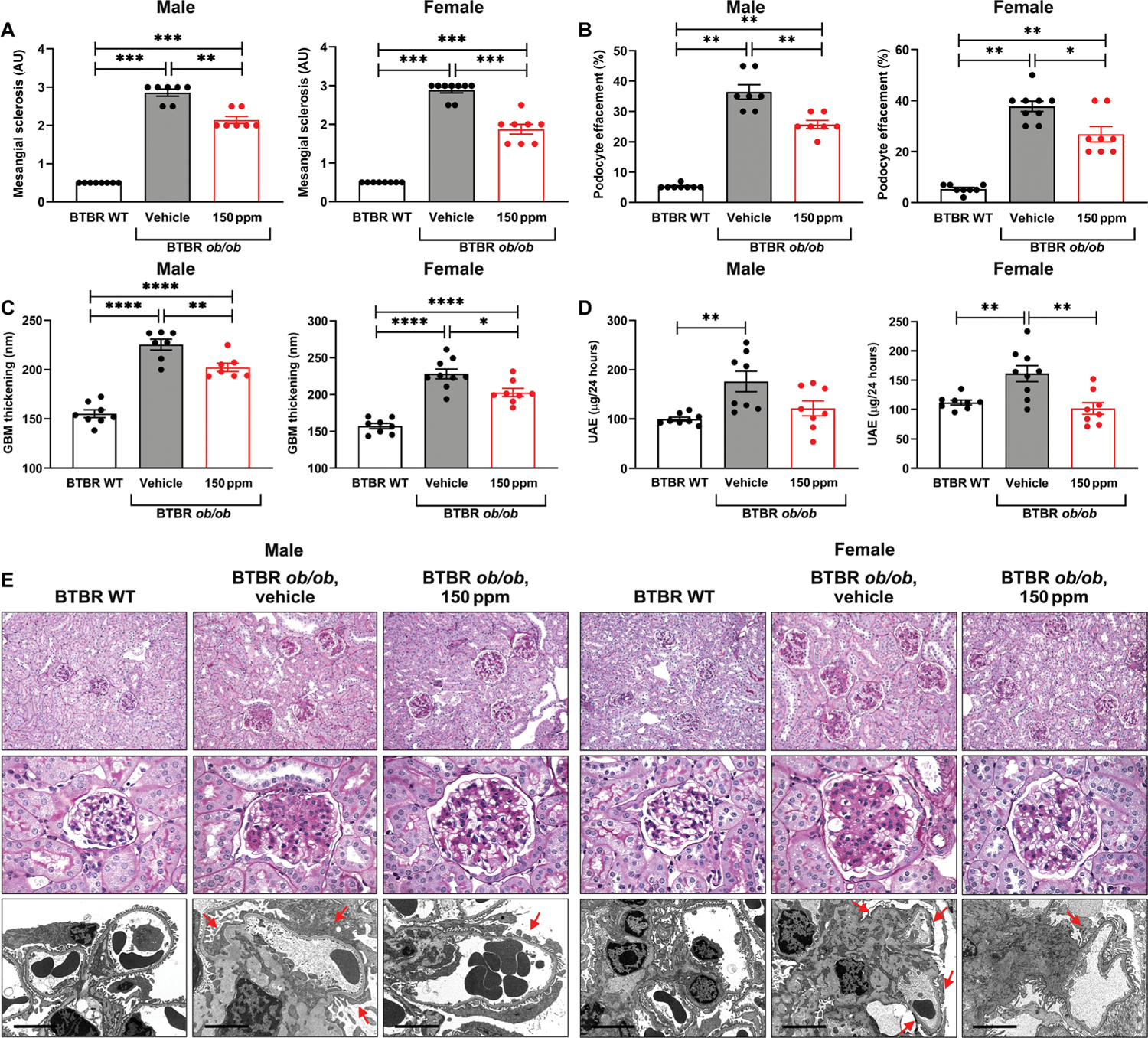
(A) Mesangial sclerosis score. For male and female mice, the data did not pass the Shapiro-Wilk test for normality, and the Wilcoxon rank sum test was used. (B) Podocyte effacement. For male and female mice, the data did not pass the Shapiro-Wilk test for normality, and the Wilcoxon rank sum test was used. (C) Thickness of the GBM. For male and female mice, statistical significance was determined by ANOVA with post hoc Tukey’s test after passing the Shapiro-Wilk test for normality. (D) Measurement of UAE after 4 months of treatment. For both male and female mice, statistical significance was determined by Welch’s ANOVA and t test with pooled SD after passing the Shapiro-Wilk test for normality. (E) Representative images of periodic acid–Schiff–stained sections (top row, ×200 magnification; middle row, ×600 magnification) and electron microscopy images (bottom row, ×8000 magnification; scale bars, 5 μm) of kidney cortex tissue in male and female mice. Arrows indicate examples of podocyte effacement in the representative images. The number of mice per group is indicated in each figure. Data are presented as means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. WT, wild type.
Signaling pathways such as activation of AKT and ERK1/2 MAPK (extracellular-regulated mitogen-activated protein kinase) are important in processes linked to diabetic kidney disease (43). Human aortic SMCs were treated with the RAGE ligand CML-AGE. Compared to treatment with serum-free medium (SFM), CML-AGE resulted in significant increases in phospho-Ser473 AKT/total AKT and phospho-Thr202/Tyr204 ERK1/2 MAPK (P < 0.01 and P < 0.0001, respectively) (fig. S9). Pretreatment with RAGE229 before CML-AGE prevented the CML-AGE–mediated increase in phosphorylation of these signaling molecules (P < 0.01 and P < 0.0001, respectively) (fig. S9). These studies suggest that the findings in preclinical models might also apply to humans and highlight the ability of RAGE229 to suppress pathological signaling mediated by CML-AGE, which is relevant to diabetic kidney disease.
Biomarkers of treatment outcome with RAGE229-containing chow in diabetic mice
Last, a critical element of long-term studies in diabetic mice and, ultimately, in human subjects is the evidence of successful modulation of the disease process after pharmacological intervention. Recent studies in human subjects with type 1 and type 2 diabetes probed for circulating inflammatory proteins that were associated with a 10-year risk of end-stage renal disease (44). It was reported that a highly robust “kidney risk inflammatory signature” consisted of 17 proteins highly enriched in tumor necrosis factor (TNF) receptor superfamily members that met these prognostic criteria. In other studies, elevated concentrations of TNF-α were observed in diabetic patients with evidence of kidney disease (45, 46). Accordingly, we tested the hypothesis that TNF-α concentrations were elevated in diabetic mice compared with nondiabetic mice and that treatment with RAGE229-containing chow might attenuate inflammation.
First, we used a cell-based model of human macrophage-like cells in which treatment with RAGE ligand CML-AGE was probed, along with the potential effects of the RAGE small-molecule antagonists of ctRAGE-DIAPH1 interaction. As shown in Fig. 8A, treatment of human THP1 macrophage-like cells with RAGE ligand CML-AGE increased TNF mRNA compared with cells incubated with SFM (P < 0.0001). Preincubation with RAGE203, RAGE208, or RAGE229 at 1 μM resulted in a significant attenuation of TNF mRNA transcripts (P < 0.0001) (Fig. 8A). Having established that RAGE ligands up-regulated TNF mRNA in THP1 cells, we probed whether treatment of diabetic mice with RAGE229-containing chow would affect plasma TNF-α concentrations.
Fig. 8. Effect of RAGE229 on TNF gene expression in THP1 cells and plasma TNF, CCL2, and IL-6 concentrations in mice.
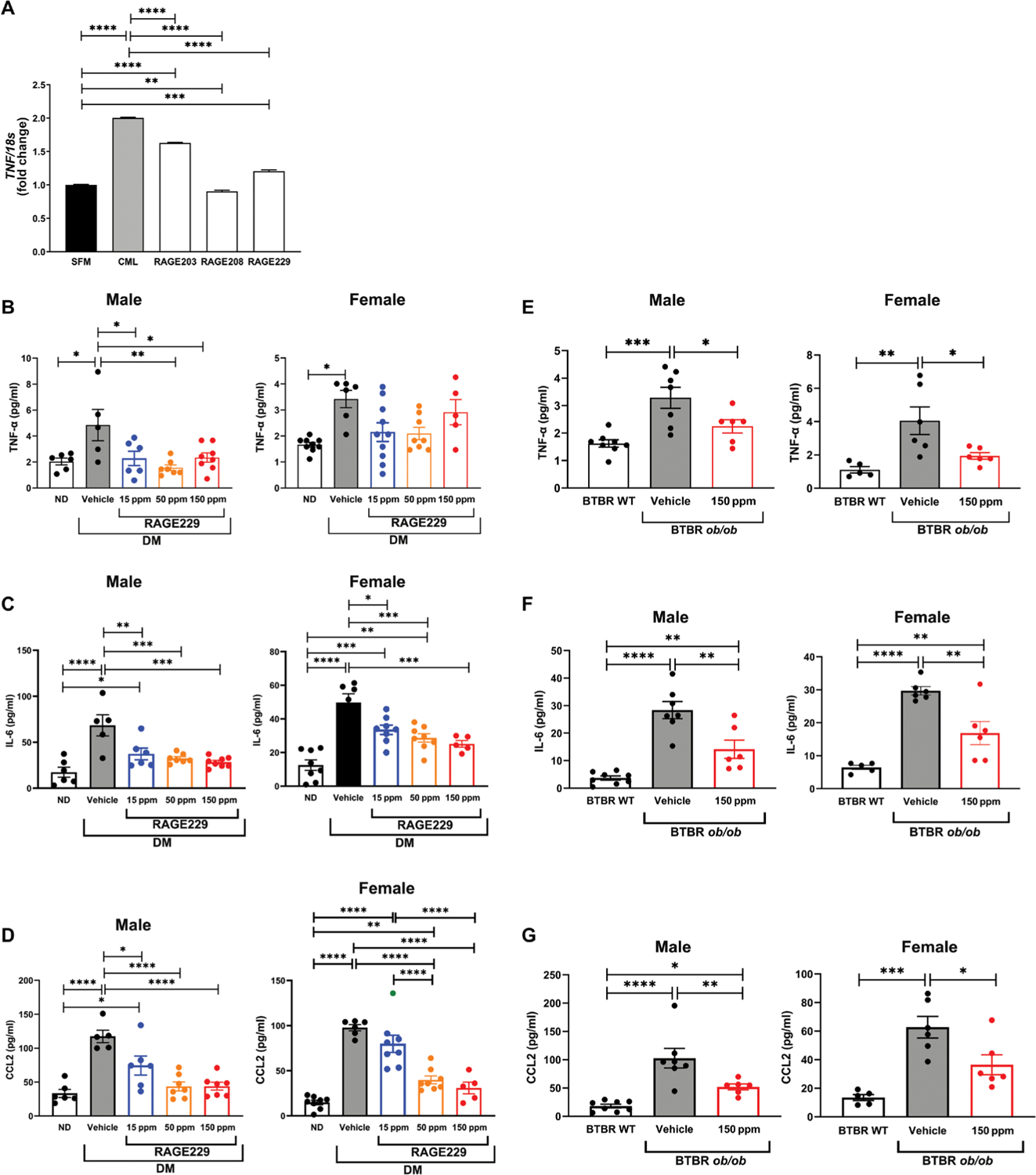
(A) Gene expression of TNF was determined in human THP1 cells treated with RAGE203, RAGE208, or RAGE229 (1 μM) and CML-AGE. Data are presented as means ± SEM. Data from three independent replicate experiments are shown. Statistical analysis was performed by unpaired t test. (B) Plasma concentrations of TNF-α were measured in male and female C57BL/6J mice at euthanasia. For male mice, statistical significance was determined by ANOVA with post hoc Tukey’s test after passing the Shapiro-Wilk test for normality. For female mice, the data did not pass the Shapiro-Wilk test for normality, and the Wilcoxon rank sum test was used. (C) Plasma concentrations of IL-6 were measured in male and female C57BL/6J mice at euthanasia. For male mice, statistical significance was determined by Welch’s ANOVA, followed by a t test with pooled SD after passing the Shapiro-Wilk test for normality. For female mice, statistical significance was determined by ANOVA with post hoc Tukey’s test after passing the Shapiro-Wilk test for normality. (D) Plasma concentrations of CCL2 were measured in male and female C57BL/6J mice at euthanasia. For male mice, statistical significance was determined by ANOVA with post hoc Tukey’s test after passing the Shapiro-Wilk test for normality. For female mice, Welch’s ANOVA was used, followed by a t test with pooled SD, after passing the Shapiro-Wilk test for normality. (E) Plasma concentrations of TNF-α were measured in male and female BTBR ob/ob mice at euthanasia. For male and female mice, statistical significance was determined by ANOVA with post hoc Tukey’s test after passing the Shapiro-Wilk test for normality. (F) Plasma concentrations of IL-6 were measured in male and female BTBR ob/ob mice at euthanasia. For male and female mice, statistical significance was determined by Welch’s ANOVA with post hoc t test with pooled SD test after passing the Shapiro-Wilk test for normality. (G) Plasma concentrations of CCL2 were measured in male and female BTBR ob/ob mice at euthanasia. For male mice, statistical significance was determined by Welch’s ANOVA with post hoc t test with pooled SD test after passing the Shapiro-Wilk test for normality. For female mice, statistical significance was determined by ANOVA with post hoc Tukey’s test after passing the Shapiro-Wilk test for normality. The number of mice per group is indicated in each figure. Data are presented as means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
In the type 1–like diabetic mice (STZ model), diabetes resulted in a significant increase in plasma TNF-α concentrations in both male and female mice (P < 0.05) (Fig. 8B). Treatment of the male diabetic mice with RAGE229-containing chow at 150, 50, or 15 ppm reduced plasma TNF-α versus vehicle treatment (P < 0.05, P < 0.01, and P < 0.05, respectively) (Fig. 8B). No effects on TNF-α concentrations were found in the female diabetic mice treated with RAGE229-containing chow compared with vehicle-treated animals (Fig. 8B).
We also tested plasma concentrations of distinct inflammatory markers. In both male and female mice, plasma interleukin-6 (IL-6) was increased in vehicle-treated diabetic mice compared with non-diabetic mice (P < 0.0001) (Fig. 8C). In both male and female diabetic mice, treatment with RAGE229 at 150, 50, or 15 ppm resulted in significantly lower IL-6 concentrations compared with vehicle-treated animals (Fig. 8C).
In the male and female type 1–like diabetic mice, plasma C-C motif chemokine ligand 2 (CCL2)/JE monocyte chemoattractant peptide-1 (JE/MCP1) (noted as “CCL2” in the figure) was significantly higher when compared with nondiabetic controls (P < 0.0001) (Fig. 8D). In male mice, treatment with RAGE229 at 150, 50, or 15 ppm resulted in significantly lower concentrations of plasma CCL2 compared with controls (P < 0.0001, P < 0.0001, and P < 0.05, respectively) (Fig. 8D). In female mice, treatment with 150 or 50 ppm, but not 15 ppm, resulted in significant attenuation of CCL2 compared with control vehicle (P < 0.0001) (Fig. 8D).
We tested the effects of diabetes and RAGE229-containing chow in the type 2–like diabetic BTBR ob/ob mice. In both the male and female mice, diabetic BTBR ob/ob mice displayed significantly higher plasma TNF-α concentrations compared with nondiabetic control mice (P < 0.001 and P < 0.01, respectively) (Fig. 8E). In both male and female BTBR ob/ob diabetic mice, treatment with RAGE229-containing chow (150 ppm) reduced plasma TNF-α concentrations compared with vehicle treatment (P < 0.05) (Fig. 8E). In the case of IL-6, male and female BTBR ob/ob mice demonstrated significantly higher IL-6 compared with nondiabetic control mice (P < 0.0001) (Fig. 8F). RAGE229 at 150 ppm reduced plasma IL-6 in both male and female BTBR ob/ob mice (P < 0.01) (Fig. 8F).
Last, both diabetic male and female BTBR ob/ob mice showed higher plasma CCL2 concentrations compared with nondiabetic control mice (P < 0.0001 and P < 0.001) (Fig. 8G), and RAGE229 at 150 ppm reduced this increase in male and female animals (P < 0.01 and P < 0.05, respectively) (Fig. 8G). Together, these data indicate that diabetes increases plasma concentrations of TNF-α and other inflammatory molecules in both type 1–like and type 2 murine diabetes models and that such elevations are reduced by RAGE229 in both models.
DISCUSSION
Elevated concentrations of blood glucose directly contribute to the complications of diabetes in human subjects, as deduced from landmark studies in which rigorous management of hyperglycemia with intensive glucose-lowering strategies reduced long-term complications in type 1 and 2 diabetes (47, 48). Recent studies have revealed that treatment with antidiabetes agents, such as the glucagon-like peptide 1 (GLP-1) agonists, dipeptidyl peptidase 4 (DPP4) inhibitors, and sodium glucose cotransporter 2 (SGLT2) inhibitors (49, 50), variably exerted effects on hyperglycemia, as well as on cardiovascular and renal sequelae (51–58). Some of these agents have caused complications, such as ketoacidosis, gastrointestinal upset, worsening of diabetic retinopathy, and urinary and other infections (59). Because these agents are largely recommended for patients with type 2 and not type 1 diabetes, the lack of their generalizability for targeting diabetic complications may limit any potential benefits for patients with type 1 diabetes. Hence, there remain important gaps in the therapeutic armamentarium for diabetic complications.
The present work reveals that the benefits of small-molecule antagonism of ctRAGE-DIAPH1 interaction with RAGE229 are not dependent on blood glucose–lowering effects. The only differences noted in these studies were observed in the male BTBR ob/ob mice used in the RAGE-containing chow kidney disease studies. In that experimental model, the concentrations of blood glucose were higher in the RAGE229-treated compared with vehicle-treated male but not female mice. We speculate that the higher concentrations of blood glucose at age 6 months in mice treated with RAGE229 compared with vehicle reflected a beneficial effect on the attenuation of aging-associated general decline in blood glucose concentrations that is reported in these mice, and that parallels an overall reduced health and reduced survival; these aging effects were assuaged by RAGE229. Previous data reported that ob/ob mice are indistinguishable from their lean littermates at birth, but within 2 weeks, they become heavier and develop hyperinsulinemia, with overt hyperglycemia becoming manifest by the fourth week of life (60); concentrations of blood glucose have been shown to rise and reach a peak after 3 to 5 months of age; after that, these concentrations decrease and eventually become nearly normal in old animals. Thus, we speculate that RAGE229 improved the general health of the mice and prevented the aging-associated decline in blood glucose concentrations. It is essential to note that in the C57BL/6 type 1–like diabetes model, RAGE229 exerted no effect on blood glucose concentrations, and in the BTBR ob/ob mice subjected to wound healing and RAGE229, no differences in blood glucose were observed, likely because these mice were younger than 6 months at the time of euthanasia, and hence, the vehicle-treated BTBR ob/ob mice had not yet begun the age-associated decline.
Beyond the direct effects of high concentrations of blood glucose, one of the downstream sequelae of hyperglycemia is the production of AGEs. AGEs, the first defined ligands of RAGE, exert maladaptive effects in the vasculature and end organs in diabetes (4, 61, 62). Type 1 and 2 diabetes cause stimulation of inflammatory mechanisms in sites such as the pancreatic islets and the adipose tissue, respectively, and set the stage for systemic inflammatory stress linked to the numerous macro- and microvascular complications that target nearly every organ in the body. Specifically, beyond advanced glycation, multiple distinct pathways have been implicated in these inflammatory phenomena, including autoimmunity (type 1 diabetes), modulation of the epigenome, activation of the inflammasome, and the generation of pro-oxidative and tissue-damaging stress responses (63–66). In type 2 diabetes, the underlying inflammatory response in adipose and other metabolic tissues unleashes mediators such as TNF-α, IL-1, IL-6, IL-10, and MCP1 (CCL2), all of which may contribute to inflammatory stress, potentially linked to the pathogenesis of complications (67, 68). Collectively, these considerations support the premise that anti-inflammation–directed treatments might beneficially modulate the course of diabetic complications (69).
Multiple studies have linked RAGE to inflammation and, in particular, to expression of TNF-α, at least in part through nuclear factor κB (NF-κB) (70–74). In the specific case of diabetes-associated kidney disease, a role for members of the TNF family as biomarkers of kidney disease has recently been suggested (44–46). Preclinical data have shown that treatment of STZ-induced diabetic mice with an anti-murine TNF-α antibody reduced macrophage recruitment and damage to the kidney (75). To specifically link TNF-α to macrophage biology, TNF-α was selectively ablated in macrophages using macrophage-specific TNF-α–deleted mice and showed similar degrees of protection as noted with the pan-pharmacological inhibition approach (75).
The study suggests that immune cells are a likely source of proinflammatory cytokines, such as TNF-α. Consistent with this premise, the present work revealed that RAGE ligands up-regulated TNF mRNA in human macrophage-like THP1 cells and that this up-regulation was suppressed by treatment with RAGE229. In vivo, in models of type 1–like and type 2–like diabetes, in parallel with reduction in histopathological, ultrastructural, and functional indices of reduced diabetic kidney disease, long-term treatment of the mice with RAGE229-containing chow resulted in decreased plasma concentrations of TNF-α. Although the experiments in diabetic male C57BL/6J mice revealed attenuation of TNF-α concentrations by RAGE229, data in the female mice displayed greater variability and overall no effect in lowering TNF-α concentrations. Of note, however, TNF-α concentrations were lower in the plasma of both male and female BTBR ob/ob mice treated with RAGE229 compared with vehicle-treated animals, in parallel with improvement in multiple diabetic kidney disease–related parameters. In human patients with diabetes, both plasma and circulating white blood cell concentrations of TNF-α were often beneficially affected by a variety of pharmacological or lifestyle interventions (76–80). We tested additional inflammatory target molecules in our preclinical models and found that plasma concentrations of CCL2/JE-MCP1 and IL-6 were also reduced in the long-term kidney studies in type 1–like and type 2–like diabetes.
It is important to consider that the main goal of the present study was to test whether small-molecule antagonists, such as RAGE229, that block the interaction of ctRAGE with DIAPH1 would be therapeutic in murine models of long-term chronic complications. Our data using NMR spectroscopy and FRET indicated that RAGE229 directly antagonized the interaction of ctRAGE with DIAPH1, thereby supporting the mechanism of action. Hence, because RAGE229 is an in vivo chemical probe (81), not a clinically enabled or readied agent, its efficacy in murine models of type 1–like and type 2–like diabetes provides support for the ongoing refinement of the molecule through subsequent SAR studies.
There are limitations to this study. First, the dose-dependent effects of RAGE229 in some of the endpoints varied. It is possible that among the endpoints tested, individual mice food intake may have varied over time. It is not possible to single house these mice to measure precisely how much food each individual mouse consumed. Single housing would add stress to the animals that might have resulted in altered food intake and general health decline. Although the animals were obligatorily co-housed, they were well cared for with respect to frequent cage changes and care to keep their bodies as dry and clean as possible. Despite these efforts, certain mice may become dominant, leading to varied food availability and intake.
Second, it is possible that sex differences relevant to RAGE229 exist. Although some differences were identified in the short-term feeding study (7-day treatment), in DTH, wound healing in type 2–like diabetic mice (wound healing closure), and in multiple endpoints in the type 1–like and type 2–like diabetic kidney, we showed that both male and female mice experienced benefits. We note that in the BTBR ob/ob studies, measurements of tubular atrophy and interstitial fibrosis showed sex differences when comparing diabetic to nondiabetic mice; hence, such underlying sex differences might have affected the response to RAGE229.
These present considerations notwithstanding, the plasma detectability, efficacy, and ability to affect multiple disease-relevant and inflammatory biomarkers of the diabetic state establish through RAGE229 strong pharmacological proof of concept that small-molecule antagonism of ctRAGE-DIAPH1 interaction, known as inhibitors of the protein-protein interaction, may present a viable opportunity for the treatment of diabetic complications. Future studies using more refined and clinically ready molecules will need to test the cotreatment of ctRAGE-DIAPH1 small-molecule antagonists with inhibitors of the angiotensin-converting enzyme or the inhibitors of SGLT2 to establish clinically relevant doses, particularly after determining that these latter agents do not interfere with the metabolism and/or activity of small-molecule ctRAGE-DIAPH1 antagonists.
In summary, these results provide proof of concept and highlight the efficacy of pharmacological antagonism of ctRAGE-DIAPH1 for the treatment of the short-term and long-term complications of type 1–like and type 2–like diabetes in mouse models. The present findings indicate that the benefits of these small-molecule antagonists were independent of lowering the concentrations of blood glucose, thereby suggesting that the effects are due to modulation of disease-specific activities in the target organs, be it the heart, the dermis, or the kidney. Furthermore, these data illustrate that small-molecule antagonism of the ctRAGE/DIAPH1 interaction reduced the concentrations of circulating inflammation-related mediators, thereby identifying trackable markers of their activity in long-term models of diabetes-associated complications.
MATERIALS AND METHODS
Study design
Predefined study endpoints
Endpoint selection criteria were based on the published literature and our experience in using the indicated techniques and mouse models. Sample sizes were selected on the basis of our previous studies in which similar experimental endpoints were tested (5, 17, 20, 21, 35, 82–85). All available data were included in the statistical analyses, and no data points were considered outliers.
Rationale and study design
The overall objective of the study was to test the hypothesis that small-molecule antagonism of the binding of the ctRAGE with the intracellular formin molecule DIAPH1 would block the effects of RAGE ligand–stimulated signaling. We designed multiple studies, including in vitro, cell culture based, ex vivo, and in vivo endpoints, to test these small-molecule antagonists. In in vivo studies, we designed short-term and long-term experiments; where possible, we tested mice with type 1–like and type 2–like diabetes. Multiple measurement techniques were used, including binding assays, cell culture–based signal transduction, and molecular and activity endpoints, and in vivo, multiple endpoints were tested, including pathology, mRNA or protein, and function and plasma biomarker analyses.
Randomization
In the case of in vitro work, treatment groups for sets of test tubes or cell culture wells were randomly assigned. In the case of murine studies, both those purchased and born in house or cages were randomly assigned to treatment groups.
Blinding
In the case of small-molecule versus vehicle treatments, the conduct and administration of the agents were performed in a blinded manner in nearly all cases. At the conclusion of the experiments, all samples in all experiments were analyzed by experimenters who were unaware of the experimental groups or treatments.
Replication
For in vitro studies, the number of biological and technical replicates is clearly indicated in the legends. For the in vivo studies, the number of biological replicates (i.e., the number of mice) is clearly indicated as the data are presented in the bar graph representative of individual mice. Hence, the sum of the data units is the number of biological replicate (mice) in the in vivo studies.
Statistical analyses
Data are shown as means ± SEM or the means ± SD, which is indicated in each figure legend. Normality of the data was assessed using the Shapiro-Wilk normality test. If normality assumption was met, data were subsequently evaluated by independent two-sample t tests or analysis of variance (ANOVA) with post hoc Tukey’s test. Welch’s ANOVA was applied when homogeneity of variance was violated to control for multiple comparisons, as indicated. Nonparametric Wilcoxon rank sum tests were implemented instead to assess differences (Kruskal-Wallis tests were applied for group tests) if normality assumption was violated. Cochran-Armitage tests were used to assess the association between a variable with two categories and an ordinal variable. All analyses were performed with GraphPad Prism 8 and R 3.6.3. P values <0.05 were used to denote statistical significance.
Supplementary Material
Acknowledgments:
We acknowledge the expert assistance of L. Woods of the Diabetes Research Program, Department of Medicine, NYU Grossman School of Medicine, in the preparation of this manuscript.
Funding:
This work was supported by the U.S. Public Health Service (1R24DK103032, 1R01DK122456-01A1, and P01HL146367) to A.M.S., A.S., and R.R., and 1P01HL131481 to A.M.S. and R.R.; the Department of Defense award (W81XWH-17-1-0201 and W81XWH-17-1-0202) to A.M.S. and R.R.; and the NYU Histology Core, which is partially supported by NYU Cancer Institute Cancer Center Support Grant 5P30CA016087-31. Support was also provided by the Diabetes Research Program, NYU Grossman School of Medicine.
Footnotes
SUPPLEMENTARY MATERIALS
Competing interests: M.B.M., R.R., A.S., and A.M.S. have patents and patent applications through NYU Grossman School of Medicine that have been submitted/published and that are directly related to the work detailed in this manuscript. These include the following: (i) Patent Number 10,729,695 (United States), Amino, amido, and heterocyclic compounds as modulators of RAGE activity and uses thereof; (ii) patent number 10,265,320 (United States), Amino, amido, and heterocyclic compounds as modulators of RAGE activity and uses thereof; (iii) application number 14851038.1 (pending, Europe), Amino, amido, and heterocyclic compounds as modulators of RAGE activity and uses thereof; (iv) application number 16/094,720 (pending but the application has been allowed, United States), Quinoline compounds as modulators or RAGE activity and uses thereof; (v) application number 17786436.0 (pending, Europe), Quinoline compounds as modulators or RAGE activity and uses thereof; and (vi) application number PCT/US2020/044926 (pending, United States), Indole compounds as modulators of RAGE activity and uses thereof. R.J.D. is a principal and consultant of RJD Medicinal Chemistry and Drug Discovery Consulting LLC. R.J.D. is a consultant and an advisor for NYU Office of Therapeutic Alliances and was a compensated consultant and advisor on this research project. R.R. is a consultant for Applied Therapeutics. A.M.S. is a consultant for Peles Therapeutics (unpaid).
Data and materials availability:
All data associated with this study are present in the paper or the Supplementary Materials. Any materials reported in this research are available through a material transfer agreement (MTA) with New York University Grossman School of Medicine. The coordinates and chemical shift assignments of the ctRAGE-RAGE229 complex have been deposited in the Protein Data Bank with accession number 6VXG.
REFERENCES AND NOTES
- 1.Duncan BB, Heiss G, Nonenzymatic glycosylation of proteins—A new tool for assessment of cumulative hyperglycemia in epidemiologic studies, past and future. Am. J. Epidemiol 120, 169–189 (1984). [DOI] [PubMed] [Google Scholar]
- 2.Gaens KH, Goossens GH, Niessen PM, van Greevenbroek MM, van der Kallen CJ, Niessen HW, Rensen SS, Buurman WA, Greve JW, Blaak EE, van Zandvoort MA, Bierhaus A, Stehouwer CD, Schalkwijk CG, Nε-(carboxymethyl)lysine-receptor for advanced glycation end product axis is a key modulator of obesity-induced dysregulation of adipokine expression and insulin resistance. Arterioscler. Thromb. Vasc. Biol 34, 1199–1208 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Anderson MM, Requena JR, Crowley JR, Thorpe SR, Heinecke JW, The myeloperoxidase system of human phagocytes generates Nepsilon-(carboxymethyl) lysine on proteins: A mechanism for producing advanced glycation end products at sites of inflammation. J. Clin. Invest 104, 103–113 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Egaña-Gorroño L, López-Díez R, Yepuri G, Ramirez LS, Reverdatto S, Gugger PF, Shekhtman A, Ramasamy R, Schmidt AM, Receptor for advanced glycation end products (RAGE) and mechanisms and therapeutic opportunities in diabetes and cardiovascular disease: Insights from human subjects and animal models. Front. Cardiovasc. Med 7, 37 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, Avila C, Kambham N, Bierhaus A, Nawroth P, Neurath MF, Slattery T, Beach D, McClary J, Nagashima M, Morser J, Stern D, Schmidt AM, RAGE mediates a novel proinflammatory axis: A central cell surface receptor for S100/calgranulin polypeptides. Cell 97, 889–901 (1999). [DOI] [PubMed] [Google Scholar]
- 6.Hori O, Brett J, Slattery T, Cao R, Zhang J, Chen JX, Nagashima M, Lundh ER, Vijay S, Nitecki D, Morser J, Stern D, Schmidt AM, The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J. Biol. Chem 270, 25752–25761 (1995). [DOI] [PubMed] [Google Scholar]
- 7.Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A, Nawroth P, Stern D, Schmidt AM, RAGE and amyloid-β peptide neurotoxicity in Alzheimer’s disease. Nature 382, 685–691 (1996). [DOI] [PubMed] [Google Scholar]
- 8.He M, Kubo H, Morimoto K, Fujino N, Suzuki T, Takahasi T, Yamada M, Yamaya M, Maekawa T, Yamamoto Y, Yamamoto H, Receptor for advanced glycation end products binds to phosphatidylserine and assists in the clearance of apoptotic cells. EMBO Rep. 12, 358–364 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rai V, Touré F, Chitayat S, Pei R, Song F, Li Q, Zhang J, Rosario R, Ramasamy R, Chazin WJ, Schmidt AM, Lysophosphatidic acid targets vascular and oncogenic pathways via RAGE signaling. J. Exp. Med 209, 2339–2350 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xie J, Reverdatto S, Frolov A, Hoffmann R, Burz DS, Shekhtman A, Structural basis for pattern recognition by the receptor for advanced glycation end products (RAGE). J. Biol. Chem 283, 27255–27269 (2008). [DOI] [PubMed] [Google Scholar]
- 11.Leclerc E, Fritz G, Vetter SW, Heizmann CW, Binding of S100 proteins to RAGE: An update. Biochim. Biophys. Acta 1793, 993–1007 (2009). [DOI] [PubMed] [Google Scholar]
- 12.Harja E, Bu DX, Hudson BI, Chang JS, Shen X, Hallam K, Kalea AZ, Lu Y, Rosario RH, Oruganti S, Nikolla Z, Belov D, Lalla E, Ramasamy R, Yan SF, Schmidt AM, Vascular and inflammatory stresses mediate atherosclerosis via RAGE and its ligands in apoE−/− mice. J. Clin. Invest 118, 183–194 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bucciarelli LG, Ananthakrishnan R, Hwang YC, Kaneko M, Song F, Sell DR, Strauch C, Monnier VM, Yan SF, Schmidt AM, Ramasamy R, RAGE and modulation of ischemic injury in the diabetic myocardium. Diabetes 57, 1941–1951 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hudson BI, Kalea AZ, Del Mar Arriero M, Harja E, Boulanger E, D’Agati V, Schmidt AM, Interaction of the RAGE cytoplasmic domain with diaphanous-1 is required for ligand-stimulated cellular migration through activation of Rac1 and Cdc42. J. Biol. Chem 283, 34457–34468 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Touré F, Fritz G, Li Q, Rai V, Daffu G, Zou YS, Rosario R, Ramasamy R, Alberts AS, Yan SF, Schmidt AM, Formin mDia1 mediates vascular remodeling via integration of oxidative and signal transduction pathways. Circ. Res 110, 1279–1293 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O’Shea KM, Ananthakrishnan R, Li Q, Quadri N, Thiagarajan D, Sreejit G, Wang L, Zirpoli H, Aranda JF, Alberts AS, Schmidt AM, Ramasamy R, The formin, DIAPH1, is a key modulator of myocardial ischemia/reperfusion injury. EBioMedicine 26, 165–174 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Manigrasso MB, Friedman RA, Ramasamy R, D’Agati V, Schmidt AM, Deletion of the formin Diaph1 protects from structural and functional abnormalities in the murine diabetic kidney. Am. J. Physiol. Renal Physiol 315, F1601–F1612 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rai V, Maldonado AY, Burz DS, Reverdatto S, Yan SF, Schmidt AM, Shekhtman A, Signal transduction in receptor for advanced glycation end products (RAGE): Solution structure of C-terminal rage (ctRAGE) and its binding to mDia1. J. Biol. Chem 287, 5133–5144 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu Q, Smith EA, Diaphanous-1 affects the nanoscale clustering and lateral diffusion of receptor for advanced glycation endproducts (RAGE). Biochim. Biophys. Acta Biomembr 1861, 43–49 (2019). [DOI] [PubMed] [Google Scholar]
- 20.Manigrasso MB, Pan J, Rai V, Zhang J, Reverdatto S, Quadri N, DeVita RJ, Ramasamy R, Shekhtman A, Schmidt AM, Small molecule inhibition of ligand-stimulated RAGE-DIAPH1 signal transduction. Sci. Rep 6, 22450 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xue J, Manigrasso M, Scalabrin M, Rai V, Reverdatto S, Burz DS, Fabris D, Schmidt AM, Shekhtman A, Change in the molecular dimension of a RAGE-ligand complex triggers RAGE signaling. Structure 24, 1509–1522 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sakaguchi T, Yan SF, Yan SD, Belov D, Rong LL, Sousa M, Andrassy M, Marso SP, Duda S, Arnold B, Liliensiek B, Nawroth PP, Stern DM, Schmidt AM, Naka Y, Central role of RAGE-dependent neointimal expansion in arterial restenosis. J. Clin. Invest 111, 959–972 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taguchi A, Blood DC, del Toro G, Canet A, Lee DC, Qu W, Tanji N, Lu Y, Lalla E, Fu C, Hofmann MA, Kislinger T, Ingram M, Lu A, Tanaka H, Hori O, Ogawa S, Stern DM, Schmidt AM, Blockade of RAGE-amphoterin signalling suppresses tumour growth and metastases. Nature 405, 354–360 (2000). [DOI] [PubMed] [Google Scholar]
- 24.Wang J, Takeuchi T, Tanaka S, Kubo SK, Kayo T, Lu D, Takata K, Koizumi A, Izumi T, A mutation in the insulin 2 gene induces diabetes with severe pancreatic beta-cell dysfunction in the Mody mouse. J. Clin. Invest 103, 27–37 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. https://www.medicalnewstoday.com/articles/324875.php#how-common-is-it.
- 26.Domaszewska-Szostek A, Krzyzanowska M, Siemionow M, Cell-based therapies for chronic wounds tested in clinical studies: Review. Ann. Plast. Surg 83, e96–e109 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Wieman TJ, Smiell JM, Su Y, Efficacy and safety of a topical gel formulation of recombinant human platelet-derived growth factor-BB (becaplermin) in patients with chronic neuropathic diabetic ulcers. A phase III randomized placebo-controlled double-blind study. Diabetes Care 21, 822–827 (1998). [DOI] [PubMed] [Google Scholar]
- 28.Raghav A, Khan ZA, Labala RK, Ahmad J, Noor S, Mishra BK, Financial burden of diabetic foot ulcers to world: A progressive topic to discuss always. Ther. Adv. Endocrinol. Metab 9, 29–31 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao H, Lu S, Chai J, Zhang Y, Ma X, Chen J, Guan Q, Wan M, Liu Y, Hydrogen sulfide improves diabetic wound healing in ob/ob mice via attenuating inflammation. J. Diabetes Complications 31, 1363–1369 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Azar YM, Green R, Niesler CU, van de Vyver M, Antioxidant preconditioning improves the paracrine responsiveness of bone marrow mesenchymal stem cells to diabetic wound fluid. Stem Cells Dev. 27, 1646–1657 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Zhao H, Chen J, Chai J, Zhang Y, Yu C, Pan Z, Gao P, Zong C, Guan Q, Fu Y, Liu Y, Cytochrome P450 (CYP) epoxygenases as potential targets in the management of impaired diabetic wound healing. Lab. Invest 97, 782–791 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Zhang J, Li L, Li J, Liu Y, Zhang CY, Zhang Y, Zen K, Protein tyrosine phosphatase 1B impairs diabetic wound healing through vascular endothelial growth factor receptor 2 dephosphorylation. Arterioscler. Thromb. Vasc. Biol 35, 163–174 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schurmann C, Goren I, Linke A, Pfeilschifter J, Frank S, Deregulated unfolded protein response in chronic wounds of diabetic ob/ob mice: A potential connection to inflammatory and angiogenic disorders in diabetes-impaired wound healing. Biochem. Biophys. Res. Commun 446, 195–200 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Schurmann C, Linke A, Engelmann-Pilger K, Steinmetz C, Mark M, Pfeilschifter J, Klein T, Frank S, The dipeptidyl peptidase-4 inhibitor linagliptin attenuates inflammation and accelerates epithelialization in wounds of diabetic ob/ob mice. J. Pharmacol. Exp. Ther 342, 71–80 (2012). [DOI] [PubMed] [Google Scholar]
- 35.Goova MT, Li J, Kislinger T, Qu W, Lu Y, Bucciarelli LG, Nowygrod S, Wolf BM, Caliste X, Yan SF, Stern DM, Schmidt AM, Blockade of receptor for advanced glycation end-products restores effective wound healing in diabetic mice. Am. J. Pathol 159, 513–525 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Greenhalgh DG, Sprugel KH, Murray MJ, Ross R, PDGF and FGF stimulate wound healing in the genetically diabetic mouse. Am. J. Pathol 136, 1235–1246 (1990). [PMC free article] [PubMed] [Google Scholar]
- 37.Levey AS, Coresh J, Chronic kidney disease. Lancet 379, 165–180 (2012). [DOI] [PubMed] [Google Scholar]
- 38. https://www.niddk.nih.gov/health-information/diabetes/overview/preventing-problems.
- 39. https://www.cdc.gov/diabetes/pdfs/data/statistics/national-diabetes-statistics-report.pdf.
- 40.Coresh J, Astor BC, Greene T, Eknoyan G, Levey AS, Prevalence of chronic kidney disease and decreased kidney function in the adult US population: Third National Health and Nutrition Examination Survey. Am. J. Kidney Dis 41, 1–12 (2003). [DOI] [PubMed] [Google Scholar]
- 41.Levin A, Tonelli M, Bonventre J, Coresh J, Donner JA, Fogo AB, Fox CS, Gansevoort RT, Heerspink HJL, Jardine M, Kasiske B, Kottgen A, Kretzler M, Levey AS, Luyckx VA, Mehta R, Moe O, Obrador G, Pannu N, Parikh CR, Perkovic V, Pollock C, Stenvinkel P, Tuttle KR, Wheeler DC, Eckardt K-U; ISN Global Kidney Health Summit participants, Global kidney health 2017 and beyond: A roadmap for closing gaps in care, research, and policy. Lancet 390, 1888–1917 (2017). [DOI] [PubMed] [Google Scholar]
- 42.Hudkins KL, Pichaiwong W, Wietecha T, Kowalewska J, Banas MC, Spencer MW, Mühlfeld A, Koelling M, Pippin JW, Shankland SJ, Askari B, Rabaglia ME, Keller MP, Attie AD, Alpers CE, BTBR Ob/Ob mutant mice model progressive diabetic nephropathy. J. Am. Soc. Nephrol 21, 1533–1542 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Whiteside CI, Dlugosz JA, Mesangial cell protein kinase C isozyme activation in the diabetic milieu. Am. J. Physiol. Renal Physiol 282, F975–F980 (2002). [DOI] [PubMed] [Google Scholar]
- 44.Niewczas MA, Pavkov ME, Skupien J, Smiles A, Md Dom ZI, Wilson JM, Park J, Nair V, Schlafly A, Saulnier PJ, Satake E, Simeone CA, Shah H, Qiu C, Looker HC, Fiorina P, Ware CF, Sun JK, Doria A, Kretzler M, Susztak K, Duffin KL, Nelson RG, Krolewski AS, A signature of circulating inflammatory proteins and development of end-stage renal disease in diabetes. Nat. Med 25, 805–813 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Macisaac RJ, Ekinci EI, Jerums G, Markers of and risk factors for the development and progression of diabetic kidney disease. Am. J. Kidney Dis 63, S39–S62 (2014). [DOI] [PubMed] [Google Scholar]
- 46.Pichler R, Afkarian M, Dieter BP, Tuttle KR, Immunity and inflammation in diabetic kidney disease: Translating mechanisms to biomarkers and treatment targets. Am. J. Physiol. Renal Physiol 312, F716–F731 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nathan DM; DCCT/EDIC Research Group, The diabetes control and complications trial/epidemiology of diabetes interventions and complications study at 30 years: Overview. Diabetes Care 37, 9–16 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Implications of the United Kingdom prospective diabetes study. American diabetes association. Diabetes Care 21, 2180–2184 (1998). [DOI] [PubMed] [Google Scholar]
- 49.Yaribeygi H, Sathyapalan T, Sahebkar A, Molecular mechanisms by which GLP-1 RA and DPP-4i induce insulin sensitivity. Life Sci. 234, 116776 (2019). [DOI] [PubMed] [Google Scholar]
- 50.Yaribeygi H, Sathyapalan T, Maleki M, Jamialahmadi T, Sahebkar A, Molecular mechanisms by which SGLT2 inhibitors can induce insulin sensitivity in diabetic milieu: A mechanistic review. Life Sci. 240, 117090 (2020). [DOI] [PubMed] [Google Scholar]
- 51.Filion KB, Azoulay L, Platt RW, Dahl M, Dormuth CR, Clemens KK, Hu N, Paterson JM, Targownik L, Turin TC, Udell JA, Ernst P, A multicenter observational study of incretin-based drugs and heart failure. N. Engl. J. Med 374, 1145–1154 (2016). [DOI] [PubMed] [Google Scholar]
- 52.Guthrie R, Practice pearl: Liraglutide and cardiovascular and renal events in type 2 diabetes. Postgrad. Med 130, 154–158 (2018). [DOI] [PubMed] [Google Scholar]
- 53.Scirica BM, Bhatt DL, Braunwald E, Steg PG, Davidson J, Hirshberg B, Ohman P, Frederich R, Wiviott SD, Hoffman EB, Cavender MA, Udell JA, Desai NR, Mosenzon O, McGuire DK, Ray KK, Leiter LA, Raz I, Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N. Engl. J. Med 369, 1317–1326 (2013). [DOI] [PubMed] [Google Scholar]
- 54.Guthrie R, Canagliflozin and cardiovascular and renal events in type 2 diabetes. Postgrad. Med 130, 149–153 (2018). [DOI] [PubMed] [Google Scholar]
- 55.Marso SP, Daniels GH, Brown-Frandsen K, Kristensen P, Mann JF, Nauck MA, Nissen SE, Pocock S, Poulter NR, Ravn LS, Steinberg WM, Stockner M, Zinman B, Bergenstal RM, Buse JB; LEADER Steering Committee; LEADER Trial Investigators, Liraglutide and cardiovascular outcomes in type 2 diabetes. N. Engl. J. Med 375, 311–322 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mann JFE, Ørsted DD, Brown-Frandsen K, Marso SP, Poulter NR, Rasmussen S, Tornøe K, Zinman B, Buse JB, Liraglutide and renal outcomes in type 2 diabetes. N. Engl. J. Med 377, 839–848 (2017). [DOI] [PubMed] [Google Scholar]
- 57.Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, Mattheus M, Devins T, Johansen OE, Woerle HJ, Broedl UC, Inzucchi SE, Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N. Engl. J. Med 373, 2117–2128 (2015). [DOI] [PubMed] [Google Scholar]
- 58.Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, Shaw W, Law G, Desai M, Matthews DR, Canagliflozin and cardiovascular and renal events in type 2 diabetes. N. Engl. J. Med 377, 644–657 (2017). [DOI] [PubMed] [Google Scholar]
- 59.Secrest MH, Udell JA, Filion KB, The cardiovascular safety trials of DPP-4 inhibitors, GLP-1 agonists, and SGLT2 inhibitors. Trends Cardiovasc. Med 27, 194–202 (2017). [DOI] [PubMed] [Google Scholar]
- 60.Lindström P, The physiology of obese-hyperglycemic mice [ob/ob mice]. ScientificWorldJournal 7, 666–685 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Paul S, Ali A, Katare R, Molecular complexities underlying the vascular complications of diabetes mellitus—A comprehensive review. J. Diabetes Complications 34, 107613 (2020). [DOI] [PubMed] [Google Scholar]
- 62.Yamagishi SI, Matsui T, Role of hyperglycemia-induced advanced glycation end product (AGE) accumulation in atherosclerosis. Ann. Vasc. Dis 11, 253–258 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tsalamandris S, Antonopoulos AS, Oikonomou E, Papamikroulis GA, Vogiatzi G, Papaioannou S, Deftereos S, Tousoulis D, The role of inflammation in diabetes: Current concepts and future perspectives. Eur. Cardiol 14, 50–59 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pang M, Li Y, Gu W, Sun Z, Wang Z, Li L, Recent advances in epigenetics of macrovascular complications in diabetes mellitus. Heart Lung Circ. 30, 186–196 (2021). [DOI] [PubMed] [Google Scholar]
- 65.Curley S, Gall J, Byrne R, Yvan-Charvet L, McGillicuddy FC, Metabolic inflammation in obesity—At the crossroads between fatty acid and cholesterol metabolism. Mol. Nutr. Food Res e1900482 (2020). [DOI] [PubMed] [Google Scholar]
- 66.Menini S, Iacobini C, Vitale M, Pugliese G, The inflammasome in chronic complications of diabetes and related metabolic disorders. Cell 9, 1812 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hotamisligil GS, Mechanisms of TNF-α-induced insulin resistance. Exp. Clin. Endocrinol. Diabetes 107, 119–125 (1999). [DOI] [PubMed] [Google Scholar]
- 68.Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, Charo I, Leibel RL, Ferrante AW Jr., CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J. Clin. Invest 116, 115–124 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yaribeygi H, Atkin SL, Pirro M, Sahebkar A, A review of the anti-inflammatory properties of antidiabetic agents providing protective effects against vascular complications in diabetes. J. Cell. Physiol 234, 8286–8294 (2019). [DOI] [PubMed] [Google Scholar]
- 70.Miyata T, Hori O, Zhang J, Yan SD, Ferran L, Iida Y, Schmidt AM, The receptor for advanced glycation end products (RAGE) is a central mediator of the interaction of AGE-beta2microglobulin with human mononuclear phagocytes via an oxidant-sensitive pathway. Implications for the pathogenesis of dialysis-related amyloidosis. J. Clin. Invest 98, 1088–1094 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lalla E, Lamster IB, Feit M, Huang L, Spessot A, Qu W, Kislinger T, Lu Y, Stern DM, Schmidt AM, Blockade of RAGE suppresses periodontitis-associated bone loss in diabetic mice. J. Clin. Invest 105, 1117–1124 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang Z, Li DD, Liang YY, Wang DS, Cai NS, Activation of astrocytes by advanced glycation end products: Cytokines induction and nitric oxide release. Acta Pharmacol. Sin 23, 974–980 (2002). [PubMed] [Google Scholar]
- 73.Hofmann MA, Drury S, Hudson BI, Gleason MR, Qu W, Lu Y, Lalla E, Chitnis S, Monteiro J, Stickland MH, Bucciarelli LG, Moser B, Moxley G, Itescu S, Grant PJ, Gregersen PK, Stern DM, Schmidt AM, RAGE and arthritis: The G82S polymorphism amplifies the inflammatory response. Genes Immun. 3, 123–135 (2002). [DOI] [PubMed] [Google Scholar]
- 74.Cataldegirmen G, Zeng S, Feirt N, Ippagunta N, Dun H, Qu W, Lu Y, Rong LL, Hofmann MA, Kislinger T, Pachydaki SI, Jenkins DG, Weinberg A, Lefkowitch J, Rogiers X, Yan SF, Schmidt AM, Emond JC, RAGE limits regeneration after massive liver injury by coordinated suppression of TNF-α and NF-κB. J. Exp. Med 201, 473–484 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Awad AS, You H, Gao T, Cooper TK, Nedospasov SA, Vacher J, Wilkinson PF, Farrell FX, Brian Reeves W, Macrophage-derived tumor necrosis factor-α mediates diabetic renal injury. Kidney Int. 88, 722–733 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Adeshara KA, Bangar NS, Doshi PR, Diwan A, Tupe RS, Action of metformin therapy against advanced glycation, oxidative stress and inflammation in type 2 diabetes patients: 3 months follow-up study. Diabetes Metab. Syndr 14, 1449–1458 (2020). [DOI] [PubMed] [Google Scholar]
- 77.Mo D, Liu S, Ma H, Tian H, Yu H, Zhang X, Tong N, Liao J, Ren Y, Effects of acarbose and metformin on the inflammatory state in newly diagnosed type 2 diabetes patients: A one-year randomized clinical study. Drug Des. Devel. Ther 13, 2769–2776 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Anholm C, Kumarathurai P, Pedersen LR, Samkani A, Walzem RL, Nielsen OW, Kristiansen OP, Fenger M, Madsbad S, Sajadieh A, Haugaard SB, Liraglutide in combination with metformin may improve the atherogenic lipid profile and decrease C-reactive protein level in statin treated obese patients with coronary artery disease and newly diagnosed type 2 diabetes: A randomized trial. Atherosclerosis 288, 60–66 (2019). [DOI] [PubMed] [Google Scholar]
- 79.Dong L, Li J, Lian Y, Tang ZX, Zen Z, Yu P, Li Y, Long-term intensive lifestyle intervention promotes improvement of stage III diabetic nephropathy. Med. Sci. Monit 25, 3061–3068 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Annibalini G, Lucertini F, Agostini D, Vallorani L, Gioacchini A, Barbieri E, Guescini M, Casadei L, Passalia A, Del Sal M, Piccoli G, Andreani M, Federici A, Stocchi V, Concurrent aerobic and resistance training has anti-inflammatory effects and increases both plasma and leukocyte levels of IGF-1 in late middle-aged type 2 diabetic patients. Oxid. Med. Cell. Longev 2017, 3937842 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Arrowsmith CH, Audia JE, Austin C, Baell J, Bennett J, Blagg J, Bountra C, Brennan PE, Brown PJ, Bunnage ME, Buser-Doepner C, Campbell RM, Carter AJ, Cohen P, Copeland RA, Cravatt B, Dahlin JL, Dhanak D, Edwards AM, Frederiksen M, Frye SV, Gray N, Grimshaw CE, Hepworth D, Howe T, Huber KV, Jin J, Knapp S, Kotz JD, Kruger RG, Lowe D, Mader MM, Marsden B, Mueller-Fahrnow A, Müller S, O’Hagan RC, Overington JP, Owen DR, Rosenberg SH, Roth B, Ross R, Schapira M, Schreiber SL, Shoichet B, Sundström M, Superti-Furga G, Taunton J, Toledo-Sherman L, Walpole C, Walters MA, Willson TM, Workman P, Young RN, Zuercher WJ, The promise and peril of chemical probes. Nat. Chem. Biol 11, 536–541 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Aleshin A, Ananthakrishnan R, Li Q, Rosario R, Lu Y, Qu W, Song F, Bakr S, Szabolcs M, D’Agati V, Liu R, Homma S, Schmidt AM, Yan SF, Ramasamy R, RAGE modulates myocardial injury consequent to LAD infarction via impact on JNK and STAT signaling in a murine model. Am. J. Physiol. Heart Circ. Physiol 294, H1823–H1832 (2008). [DOI] [PubMed] [Google Scholar]
- 83.Bucciarelli LG, Kaneko M, Ananthakrishnan R, Harja E, Lee LK, Hwang YC, Lerner S, Bakr S, Li Q, Lu Y, Song F, Qu W, Gomez T, Zou YS, Yan SF, Schmidt AM, Ramasamy R, Receptor for advanced-glycation end products: Key modulator of myocardial ischemic injury. Circulation 113, 1226–1234 (2006). [DOI] [PubMed] [Google Scholar]
- 84.Reiniger N, Lau K, McCalla D, Eby B, Cheng B, Lu Y, Qu W, Quadri N, Ananthakrishnan R, Furmansky M, Rosario R, Song F, Rai V, Weinberg A, Friedman R, Ramasamy R, D’Agati V, Schmidt AM, Deletion of the receptor for advanced glycation end products reduces glomerulosclerosis and preserves renal function in the diabetic OVE26 mouse. Diabetes 59, 2043–2054 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wendt TM, Tanji N, Guo J, Kislinger TR, Qu W, Lu Y, Bucciarelli LG, Rong LL, Moser B, Markowitz GS, Stein G, Bierhaus A, Liliensiek B, Arnold B, Nawroth PP, Stern DM, D’Agati VD, Schmidt AM, RAGE drives the development of glomerulosclerosis and implicates podocyte activation in the pathogenesis of diabetic nephropathy. Am. J. Pathol 162, 1123–1137 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Staley JP, Kim PS, Formation of a native-like subdomain in a partially folded intermediate of bovine pancreatic trypsin inhibitor. Protein Sci. 3, 1822–1832 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cavanagh J, Fairbrother WJ, Palmer AG, Rance M, Skelton NJ. Cavanagh J, Fairbrother WJ, Palmer AG, Rance M, Skelton NJ, Eds., Protein NMR Spectroscopy (Academic Press, ed. 2, 2007), p. 912. [Google Scholar]
- 88.Iwahara J, Wojciak JM, Clubb RT, An efficient NMR experiment for analyzing sugar-puckering in unlabeled DNA: Application to the 26-kDa dead ringer-DNA complex. J. Magn. Reson 153, 262–266 (2001). [DOI] [PubMed] [Google Scholar]
- 89.Zwahlen C, Legault P, Vincent SJF, Greenblatt J, Konrat R, Kay LE, Methods for measurement of intermolecular NOEs by multinuclear NMR spectroscopy: Application to a bacteriophage λ N-peptide/boxB RNA complex. J. Am. Chem. Soc 119, 6711–6721 (1997). [Google Scholar]
- 90.Masse JE, Keller R, AutoLink: Automated sequential resonance assignment of biopolymers from NMR data by relative-hypothesis-prioritization-based simulated logic. J. Magn. Reson 174, 133–151 (2005). [DOI] [PubMed] [Google Scholar]
- 91.Güntert P, Automated NMR structure calculation with CYANA. Methods Mol. Biol 278, 353–378 (2004). [DOI] [PubMed] [Google Scholar]
- 92.Cornilescu G, Delaglio F, Bax A, Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR 13, 289–302 (1999). [DOI] [PubMed] [Google Scholar]
- 93.Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas M, Ulrich EL, Markley JL, Ionides J, Laue ED, The CCPN data model for NMR spectroscopy: Development of a software pipeline. Proteins 59, 687–696 (2005). [DOI] [PubMed] [Google Scholar]
- 94.Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM, AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8, 477–486 (1996). [DOI] [PubMed] [Google Scholar]
- 95.Ai HW, Hazelwood KL, Davidson MW, Campbell RE, Fluorescent protein FRET pairs for ratiometric imaging of dual biosensors. Nat. Methods 5, 401–403 (2008). [DOI] [PubMed] [Google Scholar]
- 96.van Rheenen J, Langeslag M, Jalink K, Correcting confocal acquisition to optimize imaging of fluorescence resonance energy transfer by sensitized emission. Biophys. J 86, 2517–2529 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Xia Z, Liu Y, Reliable and global measurement of fluorescence resonance energy transfer using fluorescence microscopes. Biophys. J 81, 2395–2402 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Youvan DC, Silva CM, Bylina EJ, Coleman WJ, Dilworth MR, Yang MM, Calibration of fluorescence resonance energy transfer in microscopy using genetically engineered GFP derivates on nickel chelating beads. Biotechnol. Alia 3, 1–18 (1997). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or the Supplementary Materials. Any materials reported in this research are available through a material transfer agreement (MTA) with New York University Grossman School of Medicine. The coordinates and chemical shift assignments of the ctRAGE-RAGE229 complex have been deposited in the Protein Data Bank with accession number 6VXG.