

Abstract
Epidemiologic studies in diabetic patients as well as research in model organisms have indicated the potential of metformin as a drug candidate for the treatment of various types of cancer, including breast cancer. To date most of the anti-cancer properties of metformin have, in large part, been attributed either to the inhibition of mitochondrial NADH oxidase complex (Complex I in the electron transport chain) or the activation of AMP-activated kinase (AMPK). However, it is becoming increasingly clear that AMPK activation may be critical to alleviate metabolic and energetic stresses associated with tumor progression suggesting that it may, in fact, attenuate the toxicity of metformin instead of promoting it. Here, we demonstrate that AMPK opposes the detrimental effects of mitochondrial complex I inhibition by enhancing glycolysis at the expense of, and in a manner dependent on, pyruvate availability. We also found that metformin forces cells to rewire their metabolic grid in a manner that depends on AMPK, with AMPK-competent cells upregulating glycolysis and AMPK-deficient cell resorting to ketogenesis. In fact while the killing effects of metformin were largely rescued by pyruvate in AMPK competent cells, AMPK-deficient cells required instead acetoacetate, a product of fatty acid catabolism indicating a switch from sugar to fatty acid as a central source of ATP for these cells. In summary, our results indicate that AMPK activation is not responsible for metformin anticancer activity and may instead alleviate energetic stress by activating glycolysis.
INTRODUCTION
Metformin has emerged as a pharmacologic tool to manage insulin resistance and type II diabetes due to its effects in stimulating glucose uptake by insulin resistant cells[1]. Mechanistic studies in animals and model systems have established that, indirectly, metformin promotes the activation of AMP-activated kinase (AMPK)[2, 3], which in turn, stimulates glucose uptake[4, 5]. Metformin, via AMPK, compensates for deficiencies in Akt/PkB-dependent signaling that normally drives Glut4 translocation to the plasma membrane in response to insulin[6].
Interest in metformin as an anticancer drug was sparked by epidemiologic observations of lower incidence of cancer in metformin-treated diabetic patients compared to those not taking the drug[5, 7, 8]. In these patients, the anticancer effects of metformin were hypothesized to be a secondary effect of reducing insulin secretion and lowering blood glucose. Hence, metformin, either via acting as an inhibitor of mitochondrial respiration or through its effects on insulin-dependent glucose uptake, has emerged as an attractive drug candidate to generate cellular energetic deficits in cancer cells resulting in their selective targeting, apoptosis and death.
Physiologically, deficits in mitochondrial-dependent ATP production are alleviated by the rapid activation of AMPK both in healthy cells[9, 10] and cancer[11–13] suggesting that under conditions of deficient ATP synthesis AMPK-dependent signaling contributes to avert energetic crises[14–16]. However, the simultaneous observations that metformin activates AMPK and promotes cancer cell death led to the proposition that the anti-cancer effects of metformin might result, at least in some part, from AMPK activation[17]. The contrast of this idea with the aforementioned role of AMPK in alleviating metabolic and energetic stress is noteworthy, particularly in light of previous studies from our group that indicated AMPK may be important to support breast cancer progression via stimulating glycolysis in the face of progressive mitochondria deterioration[13]. This apparent discrepancy motivated the present study comparing the susceptibility of AMPK-competent and AMPK-deficient breast cancer cells to metformin as well as their mechanisms of resistance to it. In brief, our results indicate that AMPK activation is a mechanism of metformin resistance in MCF7 breast cancer cells. Also, that AMPK activation is important to stimulate compensatory glycolysis under conditions of mitochondrial complex I inhibition by metformin. Interestingly, we also found that AMPK seems to be required for NAD+ recycling via pyruvate reduction to lactate. In its absence, cancer cell metabolic activity, in the presence of metformin, was better supported by carbonyls, like acetoacetate, derived from acetyl-CoA, a breakdown product of fatty acid catabolism. We also found divergence in the way AMPK-deficient and AMPK-competent cells reprogram amino acid metabolism pathways under metformin challenge. We contend, based on these results, that AMPK is not directly responsible for the anticancer effects of metformin but alleviates the energetic crisis caused by it via activating glycolysis, enabling pyruvate usage for NAD+ recycling and promoting metabolic flexibility under mitochondrial electron transport chain deficiency.
RESULTS
Metformin treatment causes cytotoxicity to breast cancer cell lines independently of estrogen receptor expression status.
An initial evaluation of the sensitivity of different breast cancer cell lines to killing by metformin showed indiscriminate susceptibility independent of estrogen receptor (ER) or progesterone receptor (PR) status (Figure 1a–d). The analysis of AMPKα1 expression in these cells indicated only slight variation across cell lines (Figure 1e). In addition, cell lines showed various degrees of AMPK activation (as indicated by either Thr172 and AMPK-target acetylCoA carboxylase phosphorylation) in response to metformin (Figure 1f). MCF7 and MDA-MB231 were the cell lines showing the largest differential in AMPK activation, as judged by both parameters, among those tested and were chosen as prototype ER+/PR+ luminal and ER−/PR− basal breast cancer cells for further studies.
Figure 1.
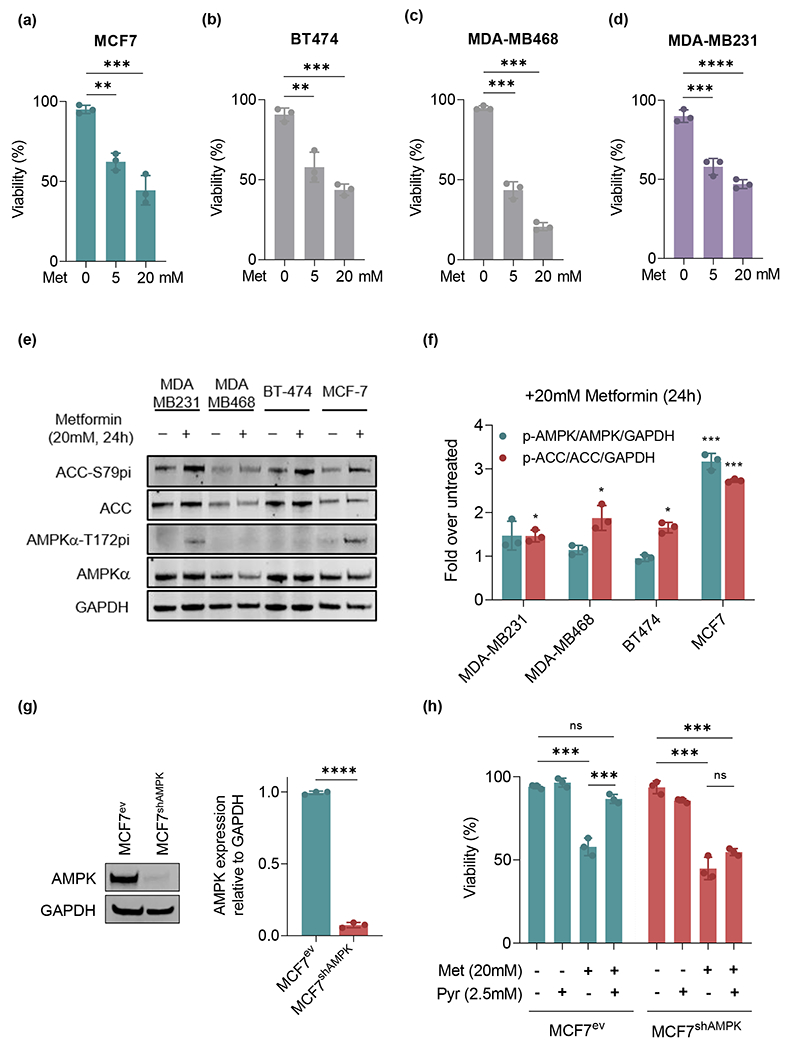
Breast cancer cell cytotoxicity and AMPK activation by metformin are largely independent of ER or PR status. (a-d): MCF7, BT-474, MDA-MB468 and MDA-MB-231 were tested for their sensitivity to 5mM and 20mM metformin. (e-f): AMPKα activation was assessed using the phosphorylation of active site Thr172 (AMPKα-T172pi / AMPKα) and downstream target acetylCoA-carboxylase (ACC-S79pi / ACC) as proxy assayed by Western blot. (g): MCF7 cells were stably transfected with a shRNA targeted to AMPKα1 catalytic subunit (MCF7shAMPK) or empty plasmid carrying a geneticin resistance gene (MCF7ev). Western blots were performed and confirmed the target silencing. (h): Pyruvate fails to rescue AMPKα1-deficient from metformin killing. MCF7ev and MCF7shAMPK were treated with 20mM metformin and/or 2.5mM pyruvate for 24 h followed by staining with 1μM calcein acetoxymethyl (alive) and 2.5μM ethidium homodimer 1 (dead) to test their viability. All experiments are representative of 3 independent biological replicates. Data are presented as mean ± SEM. Statistical analysis was performed using One-way ANOVA. ns not significant; * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001.
Silencing of AMPKα1 had only minimal effects on MCF7 cell killing by metformin
To examine the effect of AMPK activation on the response of MCF7 cells to metformin additional studies were performed using cells knocked down for the catalytic α1 subunit of AMPK. AMPK depletion was achieved via the stable expression of shRNA targeting AMPKα1 transfected into MCF7 cells followed by selection with geneticin to recover resistant clones silenced for the expression of AMPK (Figure 1g). Cells expressing shRNA targeting AMPK (heretofore referred as MCF7shAMPK) or counterparts transfected with an empty vector expressing a geneticin resistance gene (referred to as MCF7ev) were compared for their capacity to survive a metformin challenge. Figure 1h indicates that, under basal conditions, silencing AMPKα1 affected MCF7shAMPK and MCF7ev similarly with roughly 40% cell death after 24h exposure to 20 mM metformin. However, the effect of 2.5 mM pyruvate in rescuing survival in the presence of metformin was AMPK-dependent with MCF7shAMPK remaining susceptible. This result suggested that the inhibition of complex I by metformin eliminates the mitochondrial recycling of NAD+ that is simultaneously required for glycolysis and could be rescued by pyruvate in AMPK-competent cells but not in AMPK-deficient MCF7shAMPK. To gather further support for this idea, we measured ratios of NAD+/NADH and ATP in AMPK-competent (MCF7ev), AMPK-deficient (MCF7shAMPK) and MDA-MB231 cells. Results shown in Figure 2a show that the ratio NAD+/NADH was significantly decreased in all three cell lines treated with metformin. They also indicated that while pyruvate restores NAD+ levels in AMPK-competent cells (MCF7ev, MDA-MB231), MCF7shAMPK remained with a lower NAD+/NADH indicating that AMPK activity is required for the regeneration of NAD+ from pyruvate in MCF7 cells. Metformin-induced reduction in the NAD+/NADH ratio resulted, as expected, in lower steady state ATP levels (Figure 2B). In all cell lines metformin reduced steady state levels of ATP by ca. 50 – 80%, indicating a potential ATP shortage as a contributor to the killing effects of metformin. To verify this possibility, we also examined glutathione levels (GSH), glutathione peroxidase (GPx) activity and the NADP+/NADPH levels that regulate antioxidant capacity in these cells (Figure S1). AMPK knockdown had little effect on MCF7 NAD+/NADPH ratios but significantly affected MDA-MB231 that is notoriously among the most glycolytic breast cancer cell lines. Metformin slightly affected NADP+/NADPH across cell lines and conditions with more robust effects observed with MDA-MB231shAMPK cells (Figure S1a). The reduction in NADP+/NADPH ration had, in all cases, minimal effect on GPx activity (Figure S1b) and GSH levels (Figure S1c) that remained relatively unchanged suggesting only minor roles for AMPK or metformin in suppressing the ability of cells to detoxify ROS under the tested conditions.
Figure 2.

AMPK is required for pyruvate-dependent NAD+ regeneration. (a): NAD+/ NADH ratio was measured in three different cell lines (MCF7ev, MCF7shAMPK and MDA-MB231). NAD+ levels in AMPK-competent cells (MCF7ev and MDA-MB231) were rescued by pyruvate after challenge with 20mM metformin. In contrast, pyruvate failed to rescue NAD+ in MCF7shAMPK. (b): ATP levels decrease with metformin challenge in all cell lines and is partially rescued by pyruvate only in AMPK-competent cells. All experiments are representative of 3 independent biological replicates. Data are presented as mean ± SEM. Statistical analysis was performed using One-way ANOVA. ns not significant; * p < 0.05; ** p < 0.01; **** p < 0.0001.
Similar to the results obtained with NAD+/NADH measurements, pyruvate rescued ATP steady state levels in AMPK-competent cells but not in MCF7shAMPK (Figure 2b). Hence, these results confirmed previous findings[18] that pyruvate can be an alternate electron acceptor (in lieu of oxygen) maintaining ATP production from glycolysis when mitochondrial respiration is impaired (i.e. by metformin) and extend them by showing pyruvate utilization to regenerate NAD+ requires AMPK.
AMPK activity is required to activate compensatory glycolysis.
Based on previous studies that showed AMPK activates glycolysis via phosphorylating phosphofructokinase II (PFKII)[19], we tested the idea that cells lacking AMPK are restricted in their ability to upregulate glycolysis even if NAD+ recycling is enabled by pyruvate supplementation. Figure 3a shows extracellular flow analysis (Seahorse) of MCF7shAMPK vs. MCF7ev performed using 2x104 cells per well. Results indicated that MCF7shAMPK have lower rates of oxygen consumption (Figure 3a, upper panel), indicating reduced mitochondrial respiration as well as a significant lower extracellular acidification rates (ECAR) (Figure 3a, lower panel) reflecting lower glycolytic activity both under basal conditions as well as under conditions of disabled mitochondrial oxidative phosphorylation (i.e. after oligomycin as well as FCCP treatment). In addition, the measurement of glycolytic rates both under basal conditions and after oligomycin treatment showed that MCF7shAMPK cells have lower glycolytic activity at baseline as well as are less capable of activating glycolysis once the ETC is inhibited (i.e. with oligomycin), Figure 3b. Experiments with MDA-MB231 and MDA-MB231shAMPK led to similar observations (Figure S2a–b). The defect in sustaining or activating glycolysis in MCF7shAMPK cells seemed not to be related to the capacity of these cells to uptake glucose. Experiments using fluorophore-labeled glucose or (palmitate) analogs indicated that the uptake of these nutrients by MCF7shAMPK is either increased (glucose) or the similar (palmitate) compared to MCF7ev (Figure 3c) indicating that it is the capacity to catabolize glucose and not the ability to internalize it that accounts for the reduced glycolytic activity in MCF7shAMPK.
Figure 3.
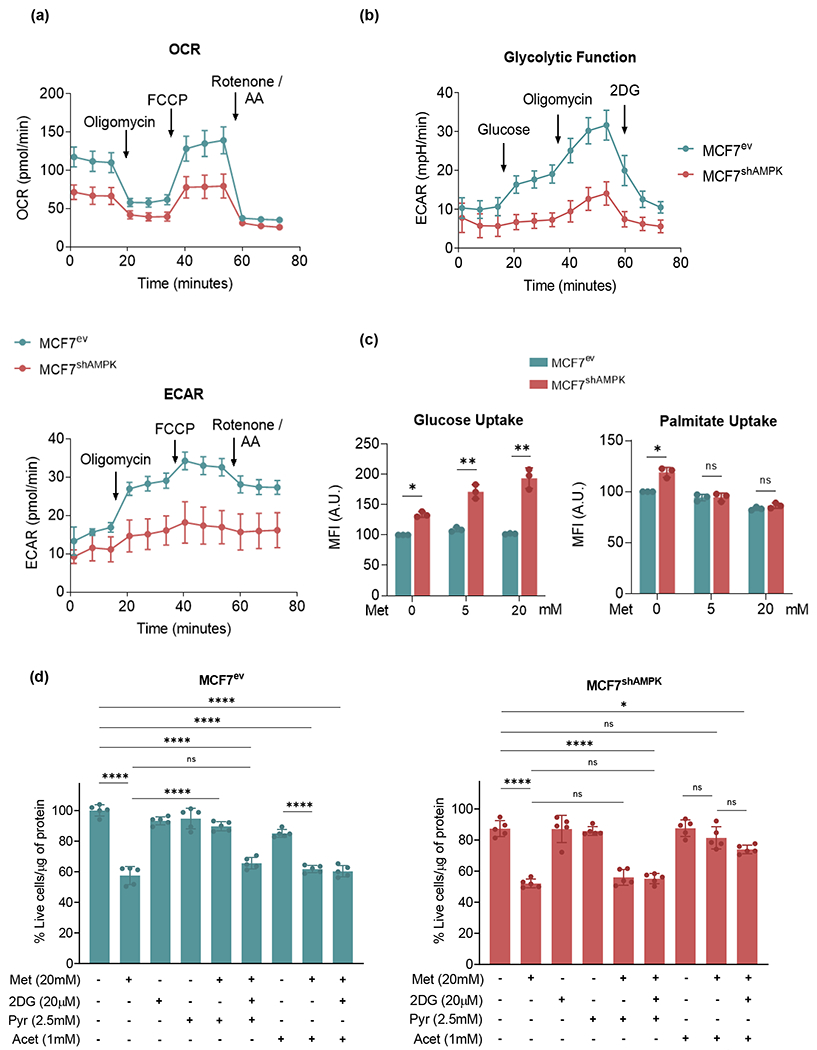
AMPK is critical to upregulate glycolysis in response to metformin challenge. (a): Oxygen consumption rate (OCR) (upper panel) and extracellular acidification rate (ECAR) (lower panel) of MCF7ev and MCF7shAMPK cells were measured using a Seahorse XF96 analyzer. MCF7shAMPK cells show lower OCR and ECAR rates under basal conditions as well as after oligomycin and FCCP treatments. (b): MCF7shAMPK cells show lower glycolytic activity at baseline and are less capable of activating glycolysis after ETC inhibition with oligomycin. (c): Cells were incubated with fluorophore-labeled glucose (2-NBDG) and bodipy-palmitate and intracellular fluorescence was measured. Glucose uptake was increased in MCF7shAMPK cells while palmitate uptake was similar when compared to MCF7ev cells both under basal and metformin-challenge conditions. (d): MCF7ev and MCF7shAMPK cells viability was tested with a 20mM metformin (Met) challenge with or without glycolysis inhibitor 2-DG (20 mM), 2.5mM pyruvate, 1mM acetoacetate. Pyruvate was capable of rescuing cell survival in an AMPK-dependent way in MCF7ev cells. MCF7shAMPK, in turn, were responsive to being rescued by acetoacetate but not pyruvate. All experiments are representative of at least 3 independent biological replicates. Data are presented as mean ± SEM. Statistical analysis was performed using One-way ANOVA. ns not significant; * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.001.
Based on the findings that the killing effects of metformin are only alleviated by pyruvate in AMPK-competent cells, experiments were performed to test the idea that AMPK-deficiency imposes the need for an alternate electron acceptor that can be used to oxidize NADH regenerating NAD+. Acetoacetate, a dicarbonyl compound produced by the ketogenic pathway was examined as a possibility. Results shown in Figure 3d indicated that acetoacetate was capable of rescuing survival of MCF7shAMPK cells but could not be used by AMPK-competent MCF7 even when glycolysis was inhibited with 2-DG. These results indicate that AMPK serves as a switch determining what alternate electron acceptors are of utility for cells that cannot use oxygen as a primary oxidant.
AMPK activity is a determinant of electron acceptor preference for NAD+ recycling
Results shown above as well as previous findings by others support the idea that the ETC, and in particular complex I, is the major cellular system recycling NAD+ from NADH[20, 21] and that in its absence, pyruvate may be used as an alternate electron acceptor[18]. Our results indicated however that the ability to use pyruvate as a replacement for the ETC in NAD+ generation requires AMPK activity. To further test this idea, we performed experiments blocking the ETC at the level of complex III using antimycin (a complex III inhibitor). Both MCF7ev as well as MCF7shAMPK were sensitive to antimycin as indicated by a massive reduction in viability (>50%) in both cell lines (Figure 4a). As expected, treatment with antimycin A, decreased the NAD+/NADH ratio in both cell lines reflecting the successful inhibition of the ETC whose activity is required for NADH oxidation (Figure 4b). It was noted, however, that while pyruvate was capable of restoring NAD+ levels in MCF7ev cells, levels of NAD+ remained nearly unchanged in antimycin A-treated MCF7shAMPK whether in the presence or in the absence of pyruvate (Figure 4b). Interestingly, acetoacetate, restored NAD+/NADH ratios in MCF7shAMPK but not in MCF7ev indicating that even when AMPK and the ETC are impaired cells can adapt to use electron acceptors derived from alternate sources (such as fatty acid-derived ketones) to regenerate NAD+. These results indicate that although the ETC in both cell lines is a major mechanism of NADH oxidation, in its absence, the capacity of cells to use specific β-oxo-carboxylic metabolites (as alternative oxidants) strongly depends on AMPK activity. As shown in Figure 4c, ATP levels closely followed the availability of NAD+, indicating that in both cell lines the inhibition of ETC with antimycin restricts ATP production from glycolysis that requires NAD+. Figure 4d confirms this idea by showing that in the presence of metformin, ATP production becomes largely dependent on glycolysis with an overall reduction in the total ATP levels in MCF7shAMPK cell line likely reflecting the reduced capacity of MCF7shAMPK cells to use glucose to support their energetic metabolism. In this regard, metabolic flexibility analysis shown in Figure 4e, indicates that MCF7ev cells are largely dependent on glucose and fatty acid oxidation either in the absence or in the presence of metformin suggesting these cells maintain their capacity to use glucose (and glycolysis) even when complex I is inhibited. Differently, MCF7shAMPK cells switch to glutamine and fatty acid oxidation with cells becoming nearly absolutely dependent on amino acids and fatty acids in the presence of 20 mM metformin. Together, these results indicate that the capacity to use glucose to support energy production is maintained by and requires AMPK activity. Similar results were obtained with MDA-MB231 cells with some notable differences. First, metformin reduced ATP production rates both in MDA-MB231 and MDA-MB231shAMPK indicating that in comparison with MCF7 derived cell lines, MDA-MB231 are metabolically maxed out in their ability to produce ATP and unable to compensate, as efficiently as MCF7 cells, for the inhibition of complex I by metformin (Figure S3a). In addition, MCF7 and MDA-MB231 seemed to use different compensatory strategies to cope with metformin, which became more evident under conditions of AMPK deficiency. While MCF7 seemed to rely more heavily on fatty acid-derived metabolites for metformin resistance, MDA-MB231 cells showed stronger dependence on glutamine catabolism to support the energetic metabolism (Figure S3b).
Figure 4.
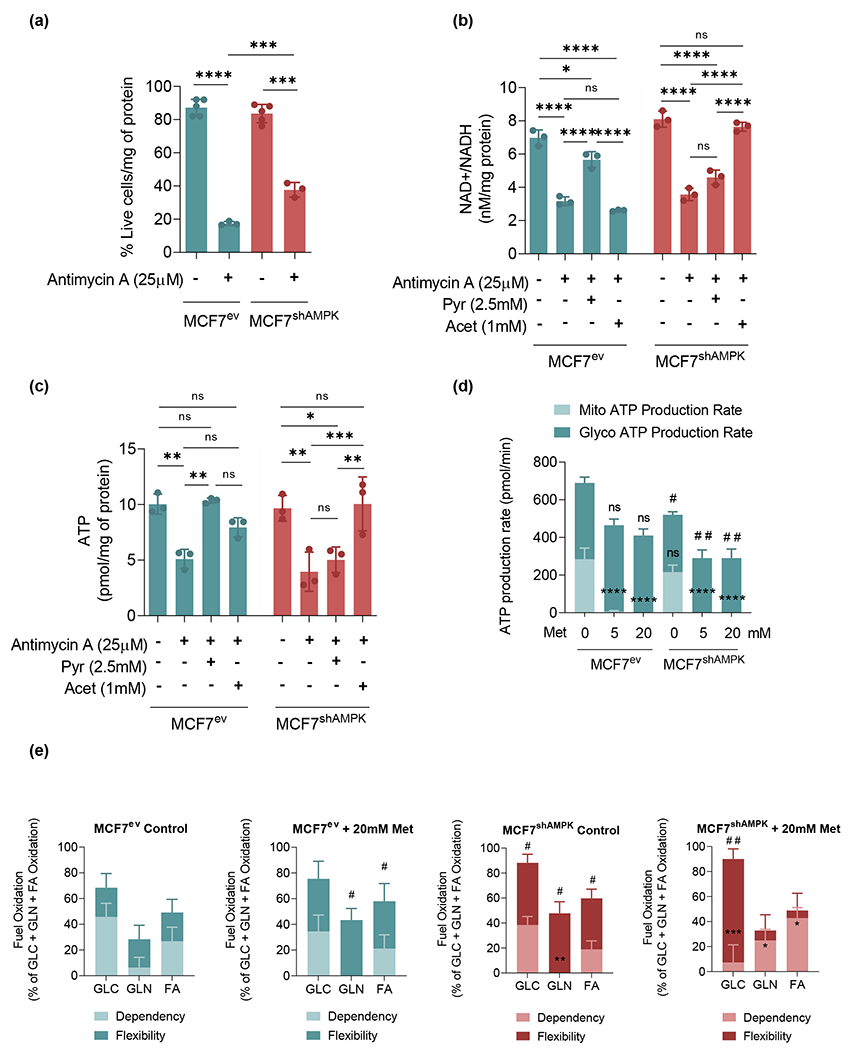
Metformin differentially affects metabolic flexibilities and dependencies of cancer cells depending on their AMPK status. (a): Antimycin A (complex III inhibitor) decreases the viability of both MCF7ev and MCF7shAMPK cells. (b): NAD+ / NADH ratio was reduced by Antimycin A and restored by pyruvate in MCF7ev cells and acetoacetate in MCF7shAMPK cells. (c): ATP levels were decreased by Antimycin A and rescued by pyruvate in MCF7ev cells or acetoacetate in the case of MCF7shAMPK cells. (d): ATP production rate was measured using a Seahorse XF96 analyzer. Metformin challenge largely increased glycolysis-dependent ATP production in both MCF7ev and MCF7shAMPK cells. (e): Percentage of glucose, glutamine and fatty acids oxidation was measured using a Seahorse XF96 analyzer. MCF7ev cells depend on glucose and fatty acid oxidation either in the absence or in the presence of metformin. MCF7shAMPK instead tends to switch to glutamine and fatty acid oxidation when in the presence of metformin. All experiments are representative of at least 3 independent biological replicates. Data are presented as mean ± SEM. Statistical analysis was performed using One-way or Two-way ANOVA. For panels A, B and C: ns not significant; * p < 0.05; ** p < 0.01; *** p < 0.001. For panel D: ns not significant; **** p < 0.0001 for comparison with Mito ATP production rate in MCF7ev untreated cells. ns not significant; # p < 0.05; ## p < 0.01 for comparison with Glyco ATP production rate in MCF7ev untreated cells. For panel E: * p < 0.05; ** p < 0.01, ***p < 0.001 for comparison with the relative fuel Dependency in MCF7ev Control. # p < 0.05; ## p < 0.01 for comparison with the relative fuel Flexibility in MCF7ev Control.
Metabolome studies indicate metformin affects different pathways depending on AMPK activity
Based on evidence obtained from extracellular analysis studies, we surmised that different metabolic strategies are used by AMPK-competent and AMPK-deficient cells to survive metformin. To test this idea, we performed a metabolite profiling analysis focusing on a hydrophilic metabolite panel comparing MCF7ev and MCF7shAMPK cells challenged with metformin (20 mM) for 24h (Figure 5). PCA analysis results indicated that cells belonging to same group clustered well together with amino acid metabolism, glycolysis/TCA metabolites as well as products of the ketogenic pathway being the largest contributors to the separation of the groups (principal component 1, PC1). (Figure 5a). Also, heat map plots shown in Figure 5b indicated distinct clusters of metabolites affected differently by metformin, the silencing of AMPK or both. Most compounds in cluster 1 represented metabolites that were in lower abundance in cells treated with metformin or AMPK-deficient cells compared to controls. These included amino acids (i.e. Gly, Asp, Leu, Val, Met, Ser/Tyr, Ala and Gln) as well as the glycolysis/TCA cycle metabolites glucose-6-phosphate, citrate, succinate. Cluster 2 showed metabolites most heavily influenced by AMPK with lower abundance in AMPK-deficient cells and included purines and pyrimidines like AMP, GMP, cytosine, cytidine, thymidine, uridine and the TCA cycle metabolites fumarate and α-ketoglutarate that notably are downstream of glutamine entry point into the cycle. Cluster 3 featured metabolites most heavily impacted by metformin. Redox active metabolites such as GSSG and cysteine were found in this group and showed reduced abundances in cells treated with metformin. Another notable metabolite in this group was NAD+ that was reduced in both MCF7 and MCF7shAMPK treated with metformin compared to untreated controls. Cluster 4 grouped metabolites generally increased in shAMPK cells compared to AMPK-competent MCF7 and included several metabolites associated with lipid metabolism including methyl acetoacetate, butyric acid, 2-hydroxy-butyrate as well as amino-group metabolism including citrulline, spermine and spermidine. Collectively, the comparison of pathways most affected by metformin in AMPK-deficient cells vs. untreated AMPK-competent MCF7 indicated that amino acid and nitrogen group metabolism as well as fatty acid catabolism were the most represented (Figure 5c) in agreement with results shown in Figure 4e that indicated a switch from glucose to glutamine and fatty acid metabolism induced by the silencing of AMPK and enhanced by metformin. The plot of selected metabolites shown in Figure 5d reinforces the idea that metformin suppresses the ability of MCF7 cells to compensate for OXPHOS inhibition via activating glycolysis with pyruvate and lactate (glycolysis end products) and α-KG, succinate and citrate (TCA cycle metabolites) being reduced in metformin treated-AMPK deficient cells compared with metformin-treated controls. They also indicate that AMPK-deficient rely more heavily on fatty acid dependent metabolism with methyl acetoacetate and butyric acid being elevated in MCF7shAMPK compared to MCF7 cells. Taken together with results shown in Figure 4e, data in Figure 5 indicate that AMPK-deficient cells are less metabolically flexible and have a heavier dependence on amino acid and lipid catabolism compared to AMPK-cells both normally and under conditions of metformin challenge.
Figure 5.
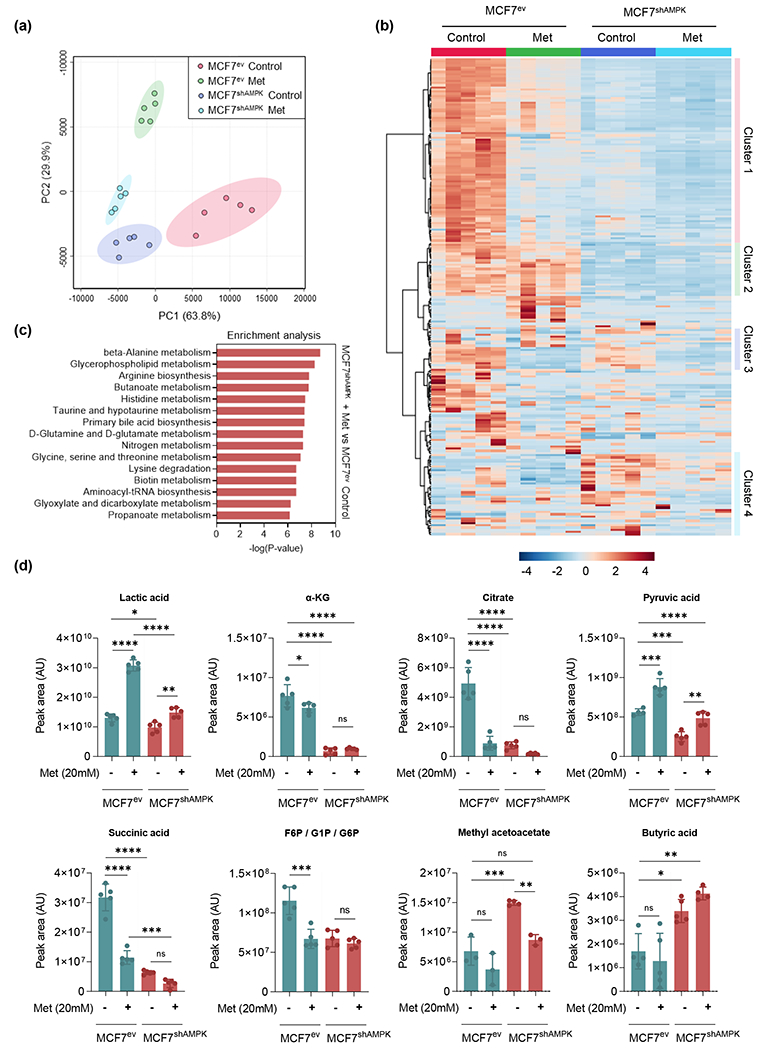
Targeted metabolome analysis indicate clusters of metabolites differentially impacted by metformin, AMPK-silencing and both combined. AMPK competent and deficient cells challenged with metformin were submitted to metabolome-wide association study (MWAS). Analysis was performed by using MetaboAnalyst ver 5.0. (a): PCA plot of metabolomics datasets of MCF7ev control, MCF7ev + 20mM metformin, MCF7shAMPK control and MCF7shAMPK + 20mM metformin. (b): Heat map of differentially abundant metabolites among the four groups. (c): Differentially enriched pathways in MCF7shAMPK + 20mM metformin vs MCF7ev. (d): Relative peak areas of representative metabolites involved in glucose, aminoacids and fatty acids metabolism. All experiments are representative of 5 independent biological replicates. Data are presented as mean ± SEM. Statistical analysis was performed using One-way ANOVA. ns not significant; * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001.
AMPK knockdown differentially affected the expression of genes affecting amino acid handling in MCF7 and MDA-MB231
Based on results obtained from metabolome studies, further analysis of pathways involved in aminoacid metabolism were examined in MCF7 and MDA-MB231 cells under basal and metformin-treated conditions. We focused on genes regulated by mTOR (a direct AMPK target)[22] involved in the metabolism of glutamine, glycine, serine and asparagine which were among differentially affected metabolites on the comparative comprehensive studies of hydrophilic metabolites quantified in MCF7 and MCF7shAMPK cells under basal and metformin-treated conditions. These genes included phosphoglycerate dehydrogenase (PHGDH) and phosphoserine aminotransferase-1 (PSAT1) that connect glycolysis to the biosynthesis of serine, glycine, cysteine as well as the generation of αKG from glutamine/glutamate, pyruvate carboxylase (PC) that carboxylates pyruvate to produce the TCA cycle metabolite oxaloacetate and asparagine synthetase (ASNS) involved in asparagine synthesis from aspartate and oxaloacetate. Importantly, asparagine has been shown to function as an aminoacid exchange factor regulating the ability of cancer cells to absorb exogenous aminoacids such as histidine, serine as well as export amino groups removed from other aminoacids [23] to generate carbonyls needed to support energetic metabolism. Results shown in Figure 6A indicate that metformin caused more significant changes to genes regulating aminoacid utilization than AMPK knockdown. PHGDH, PSAT1 and ASNS were significantly upregulated by metformin in MCF7 cells indicating a switch towards the utilization of glycolysis intermediates for the synthesis of serine (and derived aminoacids) as well as an increase in the rate of asparagine for exchange for exogenous aminoacids while relying on fatty acids for energy production (Figure 4e). In these cells, only PC seemed to respond more robustly to the AMPK status. Because PC catalyzes the conversion of pyruvate into oxaloacetate this result suggests that AMPK-deficient MCF7 may utilize the oxaloacetate/malate arm of the TCA to generate some NAD+. While metformin slightly increased the expression of genes regulating aminoacid metabolism in MDA-MB231, AMPK knockdown had a significantly more robust effect in driving the expression of PHGDH, PSAT1 and PC down (Figure 6b) indicating a major role for AMPK in regulating enzymes catalyzing aminoacid synthesis and catabolism in these cells.
Figure 6.
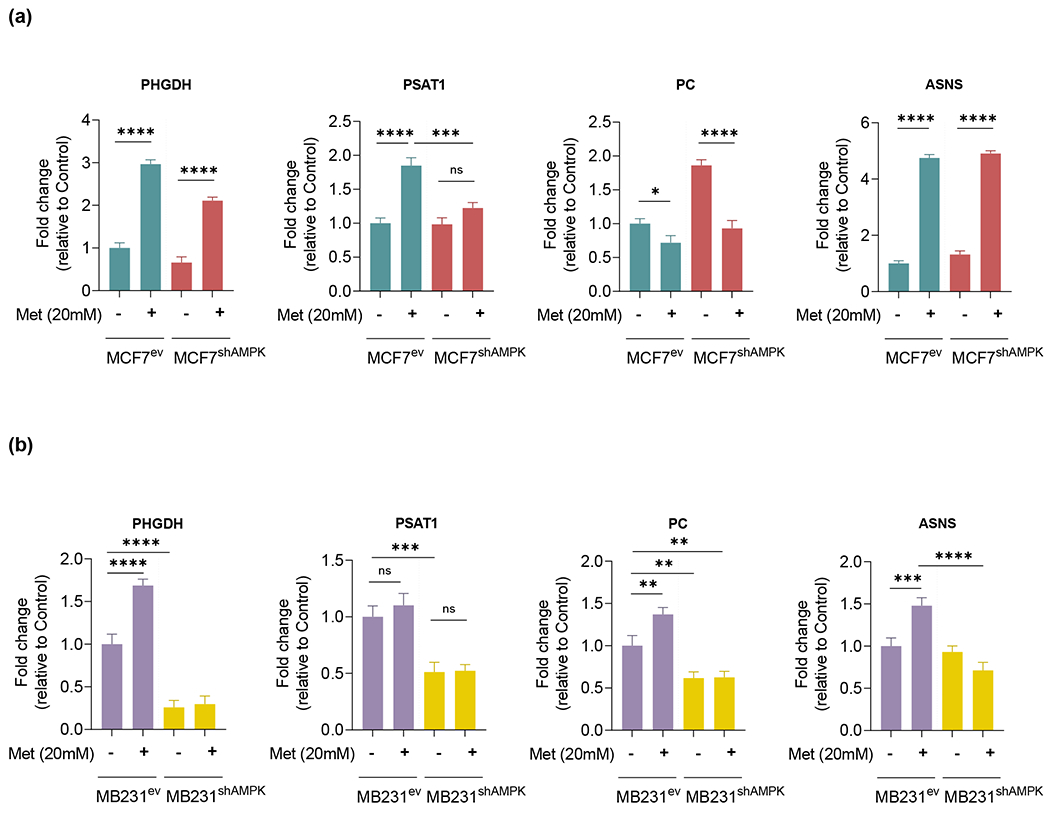
AMPK knockdown and metformin challenge affect the expression of genes encoding enzymes involved in the regulation of aminoacid utilization. PHGDH, PSAT1, PC and ASNS relative gene expression levels were measured by qRT-PCR. (a): MCF7ev and MCF7shAMPK cells challenged with 20mM Metformin. Metformin challenge had a significant effect on the expression of aminoacid utilization genes, while a more discrete effect of AMPK knockdown was observed. (b): MDA-MB231ev and MDA-MB231shAMPK cells challenged with 20mM Metformin. For MDA-MB231 cells silencing of AMPK had a more robust effect on PHGDH, PSAT1 and PC expression. All experiments are representative of 3 independent biological replicates. Data are presented as mean ± SEM. Statistical analysis was performed using One-way ANOVA. ns not significant; * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001.
Proliferation and clonogenicity are regulated by AMPK and differentially rescued by carbonylic metabolites
The effect of AMPK knockdown as well as pyruvate and acetoacetate on the proliferation and clonogenic growth of breast cancer cells was examined. Results in Figure 7a and 7b show that proliferation in MCF7 and MDA-MB231, as anticipated, was significantly reduced across cell lines by metformin. While pyruvate rescued proliferation rates of AMPK-competent metformin-treated cells to near baseline levels, AMPK-deficient cells treated with metformin were rescued to a significant lesser degree. Reflecting results presented above, acetoacetate was effective in rescuing AMPK-deficient cells but not AMPK-competent ones. Likewise, when cells were grown under anchorage independent conditions, metformin significantly reduced the number of colonies formed from both AMPK-competent and AMPK-deficient cells. Similar to the results obtained in the proliferation studies, pyruvate partially rescued colony formation from AMPK-competent MCF7 cells while acetoacetate but not pyruvate partially rescued colony formation in the case of AMPK-deficient ones (Figure 7b). Taken together these results indicate that AMPK directs the capacity of cells to use different carbonyls as alternate electron acceptors to support growth and proliferation.
Figure 7:
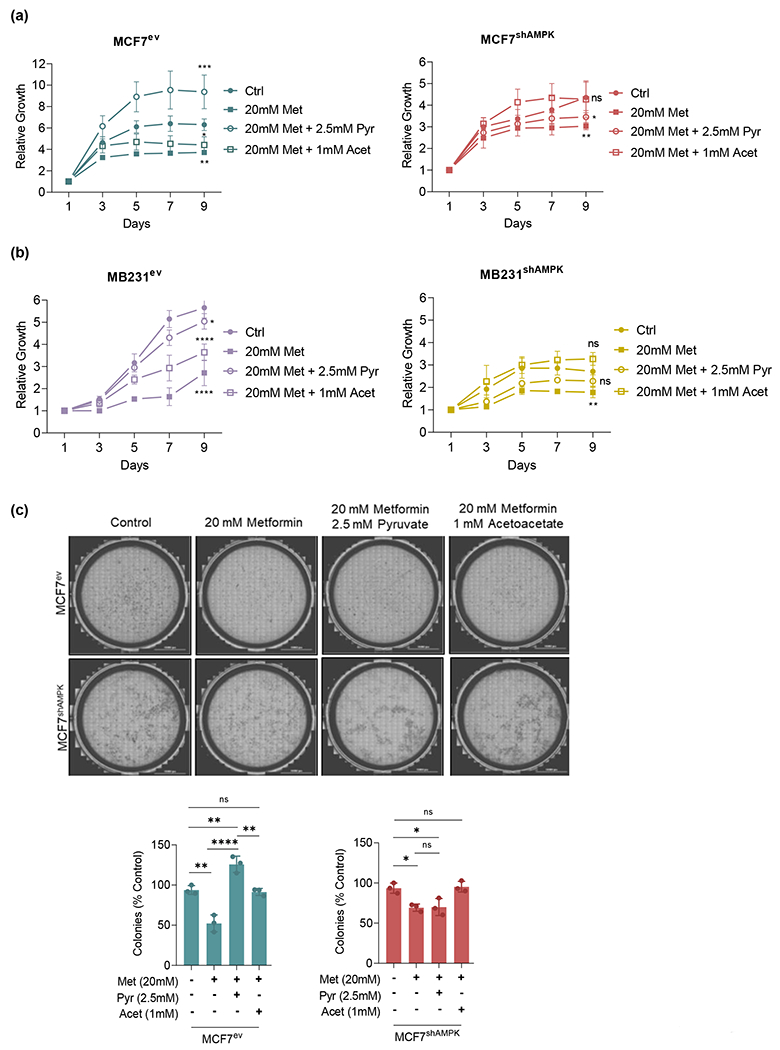
AMPK and metformin play an important role in cell proliferation and clonogenicity. Proliferation of AMPK-deficient MCF7 and MDA-MB231 cells challenged with 20mM metformin with or without 2.5mM pyruvate assessed for 9 days. (a): MCF7 proliferation was reduced by metformin and rescued by pyruvate in AMPK-competent or acetoacetate in AMPK-deficient cells. (b): MDA-MB231 proliferation was similarly affected by metformin and rescued by pyruvate or acetoacetate in a manner dependent on AMPK. (c): Colony formation using MCF7 cells. Metformin reduced the ability of MCF7 to form colonies under anchorage independent conditions. Pyruvate rescued the clonogenicity of AMPK-competent cells only, while acetoacetate rescued this ability in both cell lines. All experiments are representative of 3 independent biological replicates. Data are presented as mean ± SEM. Statistical analysis was performed using One-way ANOVA. ns not significant; * p < 0.05; ** p < 0.01; **** p < 0.0001.
DISCUSSION
Studies by various groups contributed to the current view that AMPK is a context-dependent tumor suppressor, at times inhibiting tumor evolution to more lethal phenotypes while relieving energetic and metabolic barriers associated with tumor growth and metastasis[11–13, 16, 24, 25]. Metformin is a metabolic activator of AMPK and a compound proven to be effective towards killing cancer cells[17]. The current study using cells devoid of AMPK1 activity shows that the anti-cancer effects of metformin are not dependent on AMPK either in the case of prototype luminal (MCF7) or basal (MDA-MB231) breast cancer cell lines. The results show, however, that AMPK functions as a determinant of what compensatory metabolic strategies cancer cells can activate to maintain energetic and biosynthetic metabolism in the face of complex I inhibition by metformin.
In this regard, we found that in the case of MCF7, AMPK is required for the utilization of pyruvate, an alternate β-oxocarboxylic acid electron acceptor used by AMPK-competent cells to recycle NAD+. Results obtained in this study indicated that although AMPK-deficient cells are unable to use pyruvate, they can use acetoacetate, a β-ketoacid derived primarily from acetylCoA, as an oxidant to replenish NAD+ under conditions of ETC inhibition by metformin. Because a major source of acetylCoA is fatty acid oxidation (particularly when glycolysis is compromised), we propose that AMPK-deficient cells switch to a primarily fatty acid-catabolic metabolism to maintain NAD+ levels and glycolytic ATP production. This idea is supported by data shown in Figure 4e indicating that the metabolic dependencies of MCF7ev on glucose and fatty acids do not change qualitatively in the presence of metformin except that glycolysis becomes the major source of ATP. In the case of MCF7shAMPK metabolic dependency switches to glutamine and fatty acids in a manner that depends on metformin concentration. AMPK-competent MDA-MB231 cells seem to display a similar switch to fatty acid oxidation when challenged with metformin, but differently than MCF7, AMPK knockdown leads to a stronger dependency on glutaminolysis suggesting major differences in how basal and luminal cancer cells deal with ETC inhibition furthered by AMPK knockdown. Regardless, these results indicate that AMPK is a determinant of the metabolic preferences as well as dependencies of cancer cells and suggest AMPK-deficient cells may be particularly sensitive to fatty acid and/or glutamine withdrawal compared to AMPK competent ones.
Metabolome studies of AMPK-competent compared to AMPK-deficient cells further support a central role for AMPK in serving as a determinant of metabolic preferences. Pathways most differentially affected by metformin relate to amino group metabolism, which enables glutamine utilization to fuel energetic metabolism. These studies also showed that amino acid pathways supporting energy metabolism and the carnitine shuttle functioning in the import of fatty acids to mitochondria are critical to support the energetic metabolism of MCF7shAMPK cells particularly in the presence of metformin. In mitochondria, fatty acids such as palmitate (the substrate used in extracellular flow analysis assays) are broken down by β-oxidation to acetyl-CoA. Our results are consistent with the idea that acetoacetate derived from acetyl-CoA accumulation, via the ketogenic pathway, supports the energetic viability of MCF7shAMPK cells by providing an electron acceptor (i.e. acetoacetate) that regenerates NAD+ from NADH.
In summary, the studies presented here indicate that AMPK affords considerable metabolic flexibility to breast cancer cells enabling them to maintain glucose utilization as major source of carbon to support their energetic metabolism even when the ETC is functioning precariously or not operational at all. In the absence of AMPK, cancer cells become dependent on glutamine and fatty acids for ATP production. This inability to use glucose imposes a significant limitation for cancer cells to survive and proliferate. In this context, AMPK is an important agent supporting cancer progression via the relief of significant metabolic limitations that arise when AMPK activity is absent.
METHODS
Reagents.
Metformin hydrochloride (Sigma Aldrich, #PHR1084), sodium pyruvate (Thermo Fisher Scientific, #11360070), 2-deoxy-D-glucose (Sigma Aldrich, #D8375), methyl acetoacetate (Sigma Aldrich, #537365), Antimycin A (Sigma Aldrich, A8674), RIPA buffer (Thermo Fosher Scientific, #89900), 2-NBDG (Thermo Fisher Scientific, #N13195), bodipy-palmitate (Thermo Fisher Scientific, #D3821).
Cell culture.
MCF7 and MDA-MB231 cells expressing an empty vector or constitutively expressing shAMPKα1 were a kind gift from Dr. Kevin Claffey (University of Connecticut) and Dr. Costas Koumenis (University of Pennsylvania) respectively. BT-474 and MDA-MB468 cells were obtained from ATCC. All cell lines were grown in Dulbecco’s modified Eagle’s medium (DMEM) (Thermo Fisher Scientific, #11965118) supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% antibiotic. Cells were regularly teste for mycoplasma and maintained at 37°C with 5% CO2 and plated for experiments in pyruvate-free and glutamine-free media before treatments.
Viability assay.
2 x 104 cells were treated with 20mM metformin, supplemented or not with 2.5mM pyruvate, 50mM 2-deoxy-D-glucose (2DG) and 1mM methyl acetoacetate, a cell permeable precursor of acetoacetate, for 72 h. After treatments, cells were washed with PBS and then stained with 1μM calcein acetoxymethyl and 2.5μM ethidium homodimer 1 (LIVE/DEAD® Viability/Cytotoxicity kit, Life Technologies Inc, #L3224) according to the manufacturer’s instructions. Viability/Cytotoxicity readings were obtained using a Celigo cytometer (Celigo). N = 3 independent experiments with 3 technical replicates for each cell line.
Protein extraction and immunoblot.
1 x 106 cells were treated with 20mM metformin for 24h and lysed with RIPA buffer, separated in SDS polyacrylamide gels (NuPAGE™ 4-12% Bis-Tris Protein Gels) and transferred to 0.22μm nitrocellulose membranes (BIO-RAD). Membranes were incubated with the following primary antibodies: acetyl CoA carboxylase (Cell Signaling, #3662), phospho-acetyl-CoA carboxylase (Ser79) (Cell Signaling, #11818), AMPKα (Cell Signaling, #2793), phospho-AMPKα (Thr172) (Cell Signaling, #2535), GAPDH (Cell Signaling, #97166). Membranes were then incubated with IRdye secondary antibodies (LI-COR) and imaged on an Odyssey FC (LI-COR) imaging station. N = 3 independent experiments for each cell line.
ATP production assay.
2 x 104 cells were treated with 20mM metformin for 24h, supplemented or not with 2.5mM pyruvate, 1mM methyl acetoacetate or 25μM antimycin A and then analyzed for ATP production using the ENLITEN ATP assay system kit (Promega, #FF2000) according to the manufacturer’s instructions. N = 3 independent experiments with 3 technical replicates for each cell line.
NAD+/NADH assay.
2 x 104 cells were treated with 20mM metformin for 24h, supplemented or not with 2.5mM pyruvate, 1mM methyl acetoacetate or 25μM antimycin A and then analyzed for NAD+/NADH using cell-Based Assay Kit (Cayman Chemical, #600480) according to the manufacturer’s instructions. N = 3 independent experiments with 3 technical replicates for each cell line.
NADP+/NADPH assay.
2 x 106 cells were treated with 20mM metformin for 24h and then analyzed for NADP+/NADPH using the NADP/NADPH Assay Kit (Abcam, #ab65349) according to the manufacturer’s instructions. N = 3 independent experiments with 3 technical replicates for each cell line.
GPx activity.
2 x 106 cells were treated with 20mM metformin for 24h and the activity of GPx was measured by using the Glutathione Peroxidase Assay Kit (Abcam, #ab102530) according to the manufacturer’s instructions. N = 3 independent experiments with 3 technical replicates for each cell line.
GSH levels.
2 x 104 cells were treated with 20mM metformin for 24h and the GSH levels were measured by using the GSH-Glo™ Glutathione Assay (Promega, #V6911) according to the manufacturer’s instructions. N = 3 independent experiments with 3 technical replicates for each cell line.
Measurement of cellular respiration and extracellular acidification.
2 x 104 cells were grown as previously described in a XF96 culture plate (Agilent, #101085-004). Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured using a Seahorse XF96 analyzer (Agilent) under basal conditions in the presence of 2μM oligomycin, 1.5μM FCCP and 0.5μM rotenone/antimycin A according to manufacturer’s instructions (Seahorse XF Cell Mito Stress Test Kit, Agilent, #103010-100). N = 3 independent experiments with 12 technical replicates for each cell line.
Glycolysis stress assay.
2 x 104 cells were grown as previously described. ECAR was measured using a Seahorse XF96 analyzer (Agilent) under basal conditions in the presence of 10mM glucose, 1μM oligomycin, 50mM 2-Deoxy-D-Glucose according to the manufacturer’s instructions (Seahorse XF Glycolysis Stress Test Kit, Agilent, #103020-100). N = 3 independent experiments with 12 technical replicates for each cell line.
ATP rate assay.
2 x 104 cells were grown as previously described. ATP production rate was measured using a Seahorse XF96 analyzer (Agilent) under basal conditions and after 24h treatment with 20mM metformin in the presence of 150μM oligomycin and 50μM rotenone/antimycin A according to the manufacturer’s instructions (Seahorse XF Real-Time ATP Rate Assay Kit, Agilent, #103592-100). N = 3 independent experiments with 12 technical replicates for each cell line.
Mitochondrial fuel oxidation assay.
2 x 104 cells were grown as previously described. Glucose, Glutamine and Fatty Acid oxidation was measured using a Seahorse XF96 analyzer (Agilent) under basal conditions and after treatment with 20mM metformin in the presence of 3μM BPTES, 4μM Etomoxir and 2μM UK5099 according to the manufacturer’s instructions (Seahorse XF Mito Fuel Flex Test Kit, Agilent, #103260-100). N = 3 independent experiments with 3 technical replicates for each cell line.
Glucose Uptake Assays.
1 x 106 cells were incubated with 10μM 2-NBDG at 37°C for 1h, 3h and 5h. Cells were then lifted by scraping and suspended in 200μL buffer (PBS with 5% FBS and 0.1% sodium azide) for flow cytometry. Intracellular fluorescence was quantified using the FITC channel in a BD LSR II instrument using FACSDiva software. Live cells were gated on the basis of cell side and forward scatter. Doublets were excluded based on SSC-H vs. SSC-W plots. FlowJo software 9.3.2 (Tree Star, OR) was used to analyze the data. N = 3 independent experiments with 3 technical replicates for each cell line.
Fatty Acid Uptake Assays.
1 x 106 cells were incubated with 1μg/mL bodipy-palmitate at 37°C for 5 min, 15 min and 30 min. Cells were then lifted by scraping and suspended in 200μL buffer (PBS with 5% FBS and 0.1% sodium azide) for flow cytometry. Intracellular fluorescence was quantified using the FITC channel in a BD LSR II instrument using FACSDiva software. FlowJo software 9.3.2 (Tree Star, OR) was used to analyze the data. N = 3 independent experiments with 3 technical replicates for each cell line.
RNA extraction and qRT-PCR.
1 x 106 cells were treated with 20mM metformin for 24h. Total RNA was extracted using RNeasy Kit (Qiagen, #74104) according to the manufacturer’s instructions. cDNA was synthesized using High Capacity cDNA Reverse Transcription kit (Applied Biosystem, #4368814). Quantitative PCR was performed on an Applied Biosystems Quantstudio 6 Flex Real-Time PCR System using Fast SYBR Green Master Mix (Applied Biosystems, #4385612). The relative gene expression was calculated using the 2−ΔΔCt method with GAPDH as endogenous control for normalization. The primer sequences are: PHGDH forward 5’-TGCGGAAAGTGCTCATCAGT-3’, PHGDH reverse 5’-GGTTCTGCTTTTCCACCACC-3’, PSAT1 forward 5’-TGCGGCCAGTTCAGTGCT-3’, PSAT1 reverse 5’-GCTGACCAAGCTCCTGTCAC-3’, PC forward 5’-GGCTACACCTACCCAGAC-3”, PC reverse 5’-GGAGTCAAACACACGGAAGAC-3’, ASNS forward 5’-CCAGACCAAAGTGGATGGTG-3’, ASNS reverse 5’-GCAAACACACCATCCAACATAC-3’. N = 3 independent experiments with 6 technical replicates for each cell line.
Proliferation assay.
2 x 103 cells were seeded in 96-well plates and treated with 20mM metformin, 2.5mM pyruvate or 1mM methyl acetoacetate and the proliferation/viability was measured for 9 days using a Celigo cytometer (Celigo). Medium (with or without treatment) was changed every three days. The results are shown as growth relative to day 1. N = 3 independent experiments with 6 technical replicates for each cell line.
Soft agar colony assay.
1 x 104 cells were treated with 20mM metformin, 2.5mM pyruvate or 1mM methyl acetoacetate during the experiment. Cells were mixed in 0.3% noble agar and laid on top of 0.5% noble agar in a 6-well plate. The cells were incubated for 21 days with addition of 200μL medium (with or without treatment) on top of the agar every three days. Colonies were stained with 0.1% crystal violet in 10% ethanol for 30 min and counted under a microscope. N = 3 independent experiments for each cell line.
Metabolomics.
1 x 106 cells were treated with 20mM metformin for 24h. Following the treatment, cells were washed with ice-cold 0.9% NaCl, overlaid with ultra-cold HPLC grade-methanol/water (80/20, v/v) and incubated at −80°C for 20 min. Cells were scraped and centrifuged at 16,000 x g for 15 min at 4 °C. The supernatant was collected and dried using SpeedVac. Samples were reconstituted in 50% acetonitrile and centrifuged at 20,000 x g for 30 min at 4°C. Samples were analyzed by High-Performance Liquid Chromatography and High-Resolution Mass Spectrometry and Tandem Mass Spectrometry (HPLC-MS/MS). Specifically, system consisted of a Thermo Q-Exactive in line with an electrospray source and an Ultimate3000 (Thermo) series HPLC consisting of a binary pump, degasser, and auto-sampler outfitted with a Xbridge Amide column (Waters; dimensions of 2.3mm × 100mm and a 3.5μm particle size). The mobile phase A contained 95% (vol/vol) water, 5% (vol/vol) acetonitrile, 10mM ammonium hydroxide, 10mM ammonium acetate, pH = 9.0; B was 100% Acetonitrile. The gradient was as following: 0 min, 15% A; 2.5 min, 30% A; 7 min, 43% A; 16 min, 62% A; 16.1-18 min, 75% A; 18-25 min, 15% A with a flow rate of 150μL/min. The capillary of the ESI source was set to 275°C, with sheath gas at 35 arbitrary units, auxiliary gas at 5 arbitrary units and the spray voltage at 4.0 kV. In positive/negative polarity switching mode, an m/z scan range from 60 to 900 was chosen and MS1 data was collected at a resolution of 70,000. The automatic gain control (AGC) target was set at 1 × 106 and the maximum injection time was 200 ms. The top 5 precursor ions were subsequently fragmented, in a data-dependent manner, using the higher energy collisional dissociation (HCD) cell set to 30% normalized collision energy in MS2 at a resolution power of 17,500. Besides matching m/z, metabolites are identified by matching either retention time with analytical standards and/or MS2 fragmentation pattern. Data acquisition and analysis were carried out by Xcalibur 4.1 software and Tracefinder 4.1 software, respectively (both from Thermo Fisher Scientific). N = 5 biological replicates for each cell line.
Metabolomics analysis.
The analysis of metabolites was performed in MetaboAnalyst 5.0. For the PCA and heatmap analysis peak of each metabolite was normalized by the total ion current (TIC). Ward clustering algorithm, Euclidian distance measures were used to construct the heatmap of significantly changed metabolites (One-way ANOVA). For the enrichment analysis, the peaks of two groups were normalized by the total ion current (TIC) and submitted to Enrichment Analysis tool in MetabolAnalyst. The enrichment was plotted using −log10(p-value). For the relative quantification of peak areas of representative metabolites involved in glucose, aminoacids and fatty acids metabolism each metabolite was normalized by the dilution factor and the final peak area obtained was plotted using GraphPad Prism v8 (GraphPad Software Inc.).
Statistical analysis.
Statistical analysis was performed with GraphPad Prism v8 (GraphPad Software Inc.) by using One-way or Two-way analysis of variance (ANOVA) with Tukey post hoc test. Data with a normal distribution are expressed as the mean ± SEM. A value of p < 0.05 was considered significant.
Supplementary Material
ACKNOWELEDGMENTS
The authors acknowledge the technical assistance of Mr. Matheus Pedrosa for his assistance with Figure designs, Dr. Juliana C. P. Calado and Metabolomics core from Robert Lurie Comprehensive Cancer Center for metabolomics analysis. The authors are grateful for funding from the U.S. National Institutes of Health, NIAID R01AI131267 (to M.G.B.); NIEHS R01028149 (to M.G.B.); NCI R01CA216882 (to M.G.B.) and DOD/ARO grant number 72983 (to M.G.B.); and American Heart Association Scientist Development Grant #17SDG33661117 (to Y.C.).
Footnotes
COMPETING INTERESTS
The authors have no conflict of interest to declare.
REFERENCES
- 1.Flory J, Lipska K. Metformin in 2019. JAMA 2019; 321: 1926–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fryer LG, Parbu-Patel A, Carling D. The Anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J Biol Chem 2002; 277: 25226–25232. [DOI] [PubMed] [Google Scholar]
- 3.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 2001; 108: 1167–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kurth-Kraczek EJ, Hirshman MF, Goodyear LJ, Winder WW. 5’ AMP-activated protein kinase activation causes GLUT4 translocation in skeletal muscle. Diabetes 1999; 48: 1667–1671. [DOI] [PubMed] [Google Scholar]
- 5.Cazzaniga M, Bonanni B, Guerrieri-Gonzaga A, Decensi A. Is it time to test metformin in breast cancer clinical trials? Cancer Epidemiol Biomarkers Prev 2009; 18: 701–705. [DOI] [PubMed] [Google Scholar]
- 6.Ferreira GD, Germeyer A, de Barros Machado A, do Nascimento TL, Strowitzki T, Brum IS et al. Metformin modulates PI3K and GLUT4 expression and Akt/PKB phosphorylation in human endometrial stromal cells after stimulation with androgen and insulin. Eur J Obstet Gynecol Reprod Biol 2014; 175: 157–162. [DOI] [PubMed] [Google Scholar]
- 7.Libby G, Donnelly LA, Donnan PT, Alessi DR, Morris AD, Evans JM. New users of metformin are at low risk of incident cancer: a cohort study among people with type 2 diabetes. Diabetes Care 2009; 32: 1620–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bo S, Benso A, Durazzo M, Ghigo E. Does use of metformin protect against cancer in Type 2 diabetes mellitus? J Endocrinol Invest 2012; 35: 231–235. [DOI] [PubMed] [Google Scholar]
- 9.Zhao H, Li T, Wang K, Zhao F, Chen J, Xu G et al. AMPK-mediated activation of MCU stimulates mitochondrial Ca(2+) entry to promote mitotic progression. Nat Cell Biol 2019; 21: 476–486. [DOI] [PubMed] [Google Scholar]
- 10.Hardie DG, Schaffer BE, Brunet A. AMPK: An Energy-Sensing Pathway with Multiple Inputs and Outputs. Trends Cell Biol 2016; 26: 190–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tennakoon JB, Shi Y, Han JJ, Tsouko E, White MA, Burns AR et al. Androgens regulate prostate cancer cell growth via an AMPK-PGC-1alpha-mediated metabolic switch. Oncogene 2014; 33: 5251–5261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eichner LJ, Brun SN, Herzig S, Young NP, Curtis SD, Shackelford DB et al. Genetic Analysis Reveals AMPK Is Required to Support Tumor Growth in Murine Kras-Dependent Lung Cancer Models. Cell Metab 2019; 29: 285–302 e287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hart PC, Mao M, de Abreu AL, Ansenberger-Fricano K, Ekoue DN, Ganini D et al. MnSOD upregulation sustains the Warburg effect via mitochondrial ROS and AMPK-dependent signalling in cancer. Nat Commun 2015; 6: 6053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feng Y, Ke C, Tang Q, Dong H, Zheng X, Lin W et al. Metformin promotes autophagy and apoptosis in esophageal squamous cell carcinoma by downregulating Stat3 signaling. Cell Death Dis 2014; 5: e1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Menendez JA, Oliveras-Ferraros C, Cufi S, Corominas-Faja B, Joven J, Martin-Castillo B et al. Metformin is synthetically lethal with glucose withdrawal in cancer cells. Cell Cycle 2012; 11: 2782–2792. [DOI] [PubMed] [Google Scholar]
- 16.Cheong JH, Park ES, Liang J, Dennison JB, Tsavachidou D, Nguyen-Charles C et al. Dual inhibition of tumor energy pathway by 2-deoxyglucose and metformin is effective against a broad spectrum of preclinical cancer models. Mol Cancer Ther 2011; 10: 2350–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ferretti AC, Hidalgo F, Tonucci FM, Almada E, Pariani A, Larocca MC et al. Metformin and glucose starvation decrease the migratory ability of hepatocellular carcinoma cells: targeting AMPK activation to control migration. Sci Rep 2019; 9: 2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.King MP, Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science 1989; 246: 500–503. [DOI] [PubMed] [Google Scholar]
- 19.Almeida A, Moncada S, Bolanos JP. Nitric oxide switches on glycolysis through the AMP protein kinase and 6-phosphofructo-2-kinase pathway. Nat Cell Biol 2004; 6: 45–51. [DOI] [PubMed] [Google Scholar]
- 20.Gutman M, Singer TP, Beinert H, Casida JE. Reaction sites of rotenone, piericidin A, and amytal in relation to the nonheme iron components of NADH dehydrogenase. Proc Natl Acad Sci U S A 1970; 65: 763–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Djavadi-Ohaniance L, Hatefi H. Oxidation of NADPH by submitochondrial particles from beef heart in complete absence of transhydrogenase activity from NADPH to NAD. J Biol Chem 1975; 250: 9397–9403. [PubMed] [Google Scholar]
- 22.Xu J, Ji J, Yan XH. Cross-talk between AMPK and mTOR in regulating energy balance. Crit Rev Food Sci Nutr 2012; 52: 373–381. [DOI] [PubMed] [Google Scholar]
- 23.Spinelli JB, Yoon H, Ringel AE, Jeanfavre S, Clish CB, Haigis MC. Metabolic recycling of ammonia via glutamate dehydrogenase supports breast cancer biomass. Science 2017; 358: 941–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A 2004; 101: 3329–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell 2005; 18: 283–293. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.