

Abstract
Here we report the first small-molecule inhibitors of human sulfide:quinone oxidoreductase (SQOR) that decrease the rate of breakdown of hydrogen sulfide (H2S), a potent cardioprotective signaling molecule. SQOR is a mitochondrial membrane-bound protein that catalyzes a two-electron oxidation of H2S to sulfane sulfur (S0), using glutathione (or sulfite) and coenzyme Q (CoQ) as S0 and electron acceptor, respectively. Inhibition of SQOR may constitute a new approach for the treatment of heart failure with reduced ejection fraction. Starting from top hits identified in a high-throughput screen, we conducted SAR development guided by docking of lead candidates into our crystal structure of SQOR. We identified potent SQOR inhibitors such as 19 which has an IC50 of 29 nM for SQOR inhibition and favorable pharmacokinetic and ADME properties required for in vivo efficacy testing.
Keywords: Sulfide:quinone oxidoreductase (SQOR), small-molecule inhibitors, heart failure, SAR development
Graphical Abstract
Hydrogen sulfide (H2S) is a gasotransmitter displaying pleiotropic functions in numerous and diverse physiological processes1–4. H2S is a universally accepted cardioprotective signaling molecule that protects against the progression of hypertension and atherosclerosis and impedes cardiac remodeling brought about by myocardial injury that results in heart failure5–7. H2S is the only gasotransmitter that is enzymatically metabolized8. The steady-state tissue concentration of H2S reflects the difference between the high rates of biosynthesis and mitochondrial metabolism of the gasotransmitter9. Human sulfide:quinone oxidoreductase (SQOR) is positioned at an optimal site of therapeutic intervention because it catalyzes the first irreversible step in H2S metabolism.
Small-molecule inhibitors of SQOR constitute a potentially new class of drugs for the treatment of pathological conditions associated with impaired H2S homeostasis. Most notably, approximately 50% of hospitalized heart failure patients are characterized by reduced ejection fraction and exhibit low levels of hydrogen sulfide (H2S)10–12. Furthermore, reduced H2S levels are also found in patients with renal dysfunction13, a frequently observed comorbidity in heart failure patients14.
SQOR is a mitochondrial membrane-bound protein that catalyzes a two-electron oxidation of H2S to sulfane sulfur (S0), using glutathione (or sulfite) and coenzyme Q (CoQ) as S0 and electron acceptor, respectively15,16. Although selective inhibitors of human SQOR were unknown at the onset of our study, the CoQ-binding site was deemed a likely druggable target, as judged by the proven efficacy of antimalarial, antifungal, antitumor, and immunosuppressive drugs that target CoQ-binding sites in mitochondrial complex III and dihydroorotate dehydrogenase17–20.
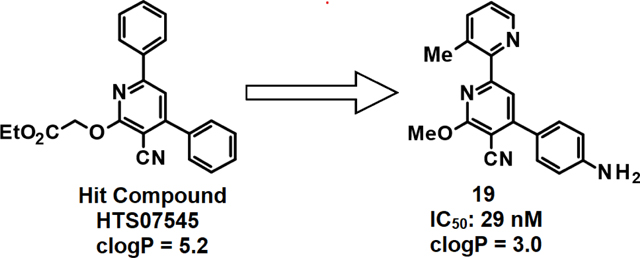
We recently conducted a high-throughput screen (HTS) of a small-molecule library and identified several classes of compounds as favorable candidates for optimization, with maximal priority given to 2,4-diphenylpyridines (class A/A’)21,22. As shown in Fig. 1, the top 3 class A/A’ HTS hits are potent SQOR inhibitors (IC50 ≤ 30 nM) but are poorly soluble in water and exhibit cLogP values considerably above the desired range (1 to 3). In this study we describe the use of medicinal chemistry and structure-based drug design in studies that produced a candidate suitable for in vivo efficacy studies.
Figure 1.
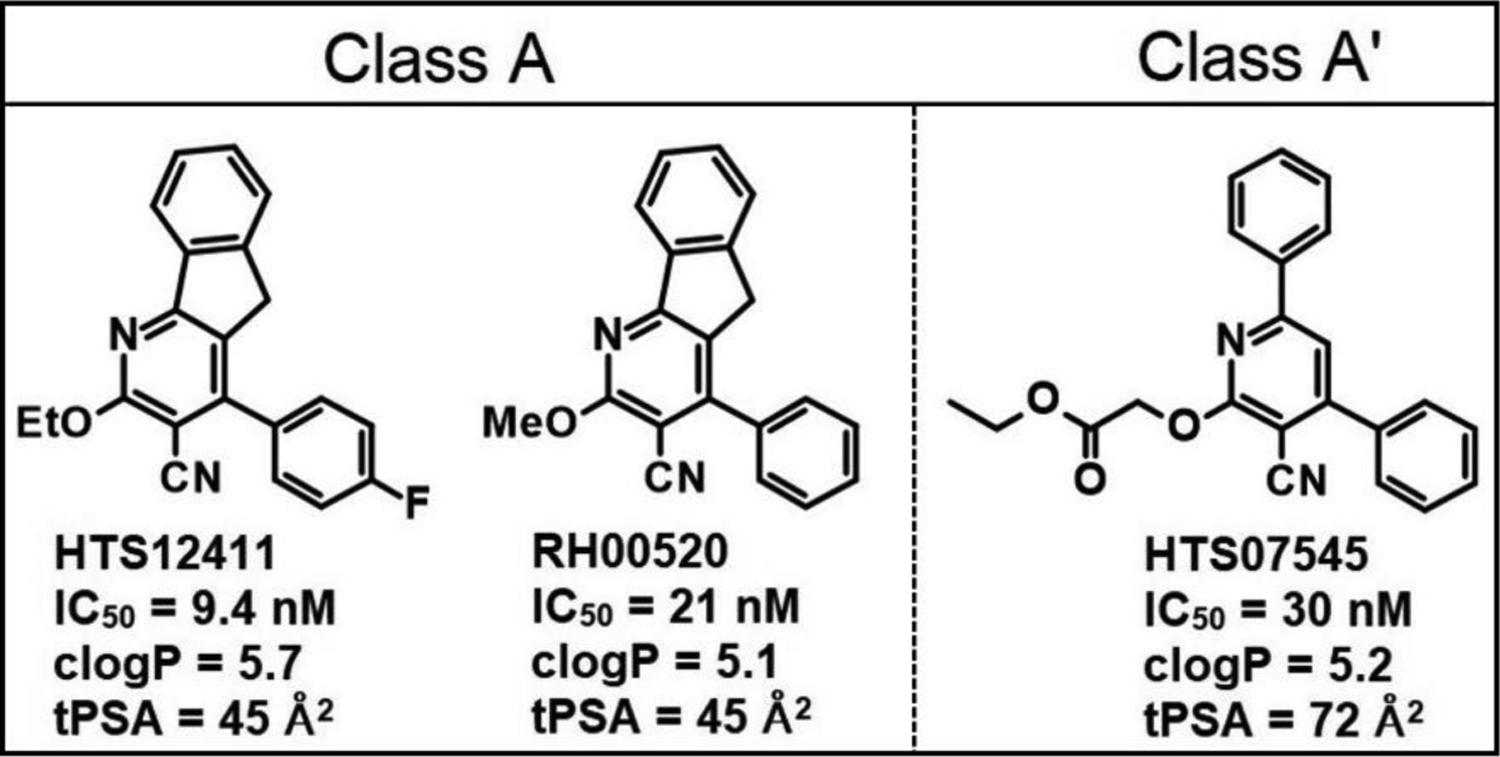
Properties of top class A/A’ HTS hits. Data from ref 21.
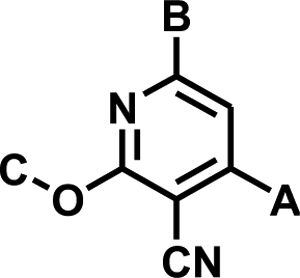
Structural data for SQOR were not available at the initiation of our hit-to-lead optimization studies, which were carried out primarily on the unconstrained and chemically more tractable diarylpyridine class A’, as exemplified by 1 (IC50 = 25 nM) (Table 1). Structure-activity relationships (SAR) among representative analogs (1 – 19), out of a total of 120 compounds that were synthesized, are shown in Table 1 and provide an accurate overview of the observed SAR.
Table 1.
SAR among representative class A’ analogs (1 – 19)
|
||||
|---|---|---|---|---|
| Compound | A | B | C | IC50 ± SE (nM) |
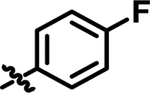
| 1 | 4-FPh | Ph | Me | 25 ± 2 |
| 2 | 4-FPh | 4-(MeO)Ph | Me | 1,360 ± 150 |
| 3 | 4-FPh | 3-(MeO)Ph | Me | 108 ± 6 |
| 4 | 4-FPh | 2-(MeO)Ph | Me | 75 ± 7.4 |
| 5 | 4-FPh | t-Bu | Me | 153 ± 14 |
| 6 | 4-ClPh | Ph | Me | 252 ± 21 |
| 7 | 2,4-Cl2Ph | Ph | Me | >100,000 |
| 8 | t-Bu | 2-Pyr | Me | 850 ± 140 |
| 9 | 4-FPh | Ph | MeO(CH2)2 | 123 ± 8.7 |
| 10 | Ph | Ph | PhCH2 | 100 ± 14 |
| 11 | 4-Pyr | Ph | Me | 541 ± 21 |
| 12 | 4-FPh | 3-Pyr | Me | 213 ± 24 |
| 13 | 4-FPh | 4-Pyr | Me | 17,400 ± 1300 |
| 14 | 4-FPh | 2-Pyr | Me | 14 ± 1.1 |
| 15 | 4-FPh | 3-Me-2-Pyr | Me | 8.7 ± 0.74 |
| 16 | 4-FPh | 2-Pyr | Allyl | 5.5 ± 0.39 |
| 17 | 4-FPh | 3-Me-2-Pyr |
|
43 ± 2.4 |
| 18 | 4-FPh | 3-Me-2-Pyr | 3-PyrCH2 | 36 ± 3.1 |
| 19 | 4-NH2Ph | 3-Me-2-Pyr | Me | 29 ± 2.7 |
For area B, our studies indicate that substitution on the para-position is detrimental to potency, and that substitutions on the meta- and ortho-positions are tolerated, as shown by 2, 3, and 4. Seeking to reduce the degree of aromaticity of the molecules, a non-aryl derivative 5 was synthesized and showed reasonably good activity, indicating tolerance in this area for non-planarity.
Systematic movement of substituents around ring A demonstrated that substitution was optimal at the para-position, as demonstrated in 1. As the size of the halogen substituent was increased, such as by replacing the 4-fluoro in 1 with a 4-chloro in 6, the potency of the resulting compounds decreased. A 2-chloro substituent was not tolerated in ring A, as shown by the inactivity of 7. A non-aryl derivative 8 was also synthesized in area A, however this variation was less well tolerated, as compared with the same non-aryl substituent in area B (5). It was also demonstrated that this ring was essential for activity, as replacement of the ring with a hydrogen resulted in an inactive compound (data not shown).
We found that a broad variety of groups are tolerated in area C. Two examples of the different area C substitutions are shown in 9 and 10. We also demonstrated that the alkoxy group can be replaced with a thioether, with only a small reduction in inhibitor potency (Fig. 2, 20 versus 21).
Figure 2.
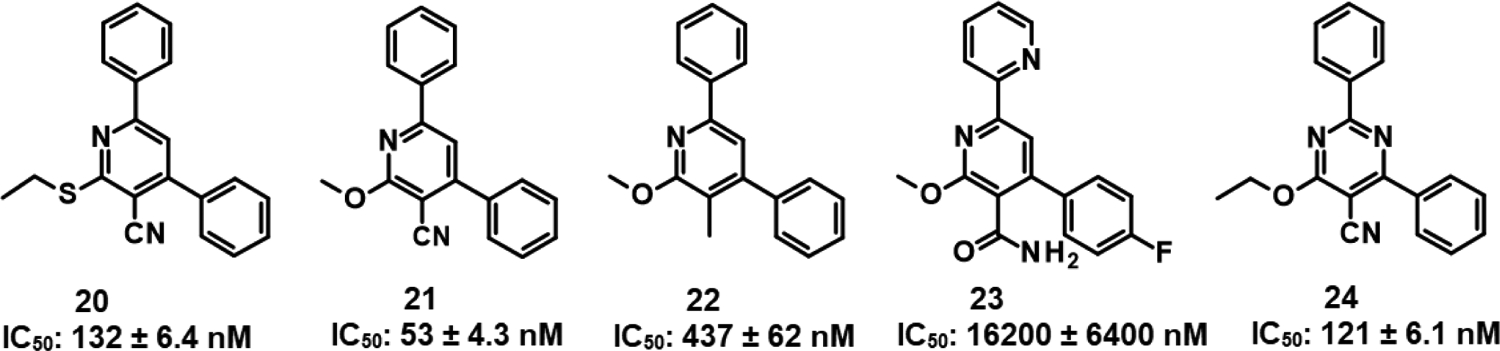
SAR for additional class A’ analogs (20 – 24).
Replacement of the cyano group with a methyl (22) or primary amide (23) (Fig. 2) results in a substantial decrease in potency as compared with e.g., 1 (IC50: 25 nM), indicating the importance of the cyano moiety. Replacement of the central pyridine ring with a pyrimidine gave a derivative (24) that was only 2-fold less active than the equivalent pyridine 21 (Fig. 2), showing that this change was well tolerated.
Heteroaromatic derivatives have also been investigated in areas A, B, and C. For area A, the 2-, 3-, and 4–pyridyl derivatives, as exemplified by 11, were much less active than the equivalent phenyl analog, 21 (IC50: 53 nM). However, for area B, although 3-pyridyl 12 and, in particular, 4-pyridyl 13 were less active than the phenyl, the 2-pyridyl-substituted 14 was well tolerated and allows for the possibility of making salts of the compounds to improve the aqueous solubility of the molecules. Combination of the 2-pyridyl substitution in area B with the previously demonstrated ortho-substitution preference for area B, e.g., 4, produced highly active compounds, such as 15. Moreover, the ortho-substitution in area B in 15 should cause the 2-pyridyl ring and central pyridyl ring to position themselves orthogonal to one another, decreasing the planarity of the system, and thus improving compound aqueous solubility23. Combining the beneficial area B 2-pyridyl substitution with variation in area C resulted in highly potent derivatives, as exemplified in 16. Seeking to improve the solubility of 16, solubilizing groups have been successfully incorporated into area C, as in 17 and 18.
The synthesis of 2-alkoxy-3-cyanopyridine class A’ derivatives was carried out by following one of three synthetic pathways. The route shown in Scheme 1 was one of two routes used for the synthesis of derivatives containing small groups in area C. Thus, for the synthesis of 6, which contains a methoxy group in area C, condensation of the requisite benzaldehyde (25) with malononitrile readily gave the versatile late-stage intermediate 26, which could then be reacted with the acetophenone of choice, using methanol as both solvent and reagent, to give the final product 6 (Scheme 1). (See Scheme 3 for an example of an alternate route used for the synthesis of compounds with a small group in area C.)
Scheme 1.
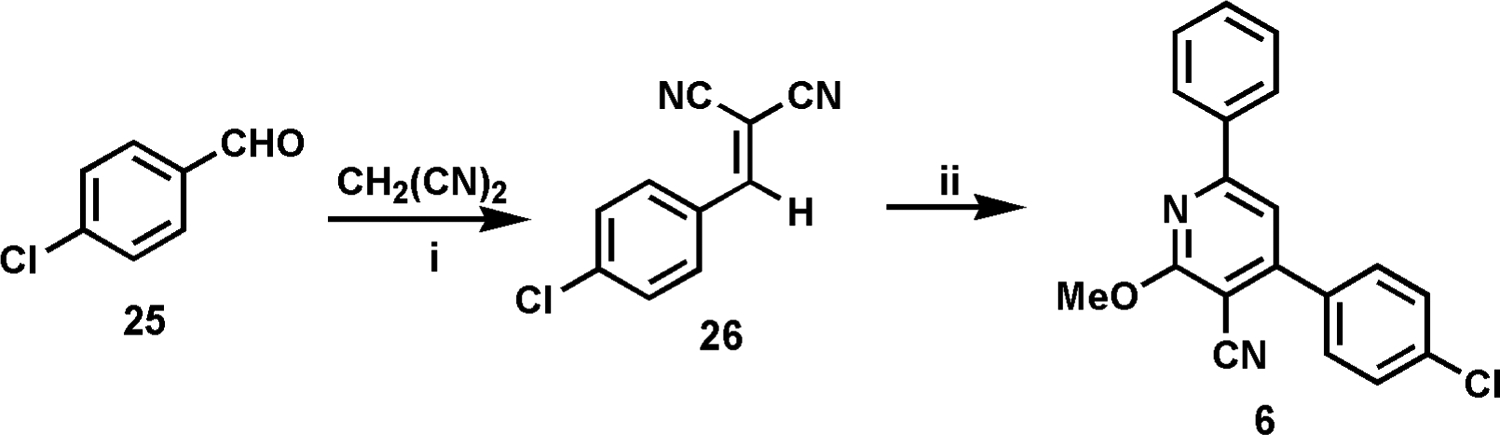
i, KOH, EtOH, H2O, 20 °C, 45 min, 89%; ii, acetophenone, NaOH, MeOH, 20 °C, 3 days, 16%.
The route shown in Scheme 2 was utilized for the incorporation of larger groups in area C. Thus, for the synthesis of 18, which contains a (pyridin-3-yl)methoxy group in area C, an initial three component condensation of 27, 28, and 29 provided the intermediate pyridone 30 (which was inactive in our IC50 assay), which could then be alkylated with the alkylating agent of choice in the presence of silver carbonate, to give the desired final product 18.
Scheme 2.
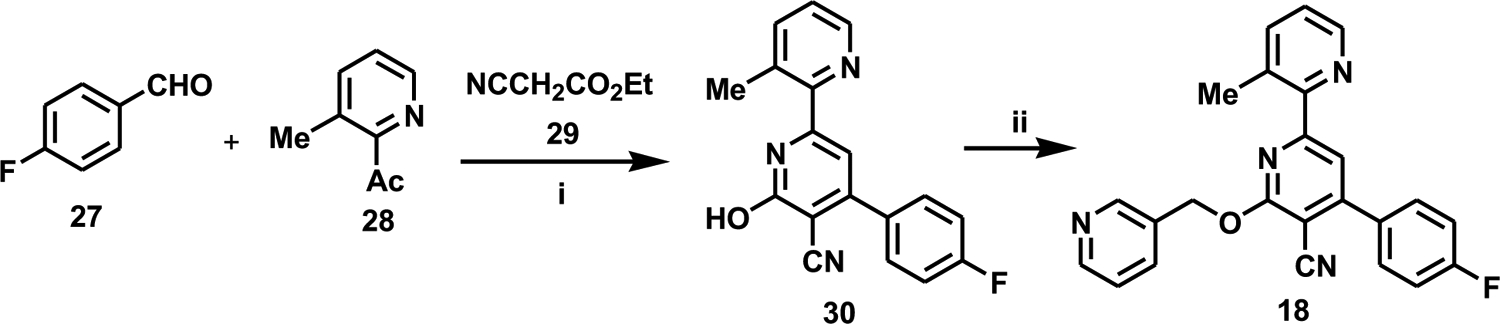
i, NH4Cl, 1,4-dioxane, reflux, 16 h, 34%; ii, 3-(BrCH2)pyridine, Ag2CO3, DMF, 140 °C, 20 h, 47%.
The most promising class A’ analogs produced in the SAR campaign described above were found to (i) contain a 2-pyridyl substituent in area B, (ii) exhibit cLogP values (cLogP ~ 4) closer to the desired range (1 to 3), and (iii) retain the high potency observed for top class A/A’ HTS hits (Table 2). At this stage of the project, we had succeeded in solving the X-ray structure of human SQOR at 2.59 Å resolution24 in studies that opened the door to structure-based drug design. The X-ray structure showed that SQOR contained two active sites, located on opposite faces of the planar flavin ring of FAD. The H2S-oxidizing active site comprises a small, hydrophilic cavity above the re-face of the flavin ring. The CoQ-binding pocket is located within a large, mainly hydrophobic, cavity above the si-face of the flavin ring. Importantly, we recently demonstrated that class A/A’ HTS hits act as competitive inhibitors with respect to CoQ21.
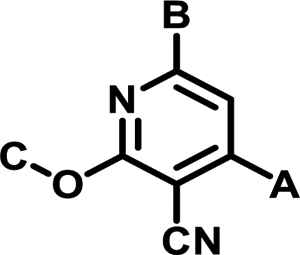
Table 2.
Calculated properties for the most potent SAR class A’ analogs
|
||||||
|---|---|---|---|---|---|---|
| Compound | A | B | C | IC50 (nM) | tPSA | clogP |
| 1 |
|
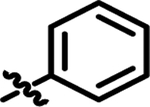
|
Me | 25 | 45.38 | 5.17 |
| 14 |
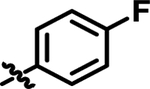
|
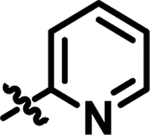
|
Me | 14 | 57.74 | 3.954 |
| 15 |
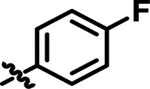
|
|
Me | 8.7 | 57.74 | 4.153 |
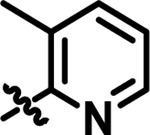
| 16 |
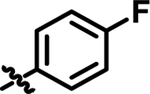
|
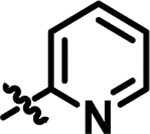
|
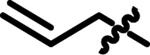
|
5.5 | 57.74 | 4.728 |
| 18 |
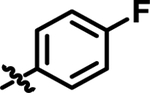
|
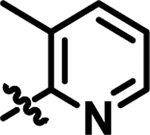
|
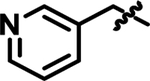
|
36 | 70.1 | 4.024 |
| 19 |
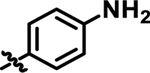
|
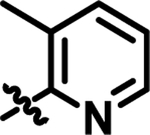
|
Me | 29 | 83.76 | 2.966 |
We conducted docking studies to obtain further insight into the observed SAR and to facilitate additional optimization of the physicochemical and ADME/PK properties of class A’ inhibitors. The observed mechanism of action suggested that class A/A’ inhibitors bind to the CoQ-binding pocket in SQOR. The CoQ-binding site is located within an internal tunnel that connects the enzyme’s membrane-binding surface to its hydrophilic H2S-oxidizing active site24. The entry to the pocket that binds CoQ is located on the surface of the enzyme, within a region that is likely to be inserted (monotopically) into the inner mitochondrial membrane. The polar quinone ring of the substrate, decylubiquinone (DCQ), is bound near the bottom of the CoQ-binding cavity (Fig. 3A). The molecule of DCQ is surrounded by mostly hydrophobic residues. A hydrogen bond is formed between Trp435:NE1 and the O2 carbonyl oxygen in the quinone ring of DCQ. The hydrophobic decyl tail of DCQ extends from the quinone ring through to the opening of the pocket on the membrane-facing surface of the protein.
Figure 3.
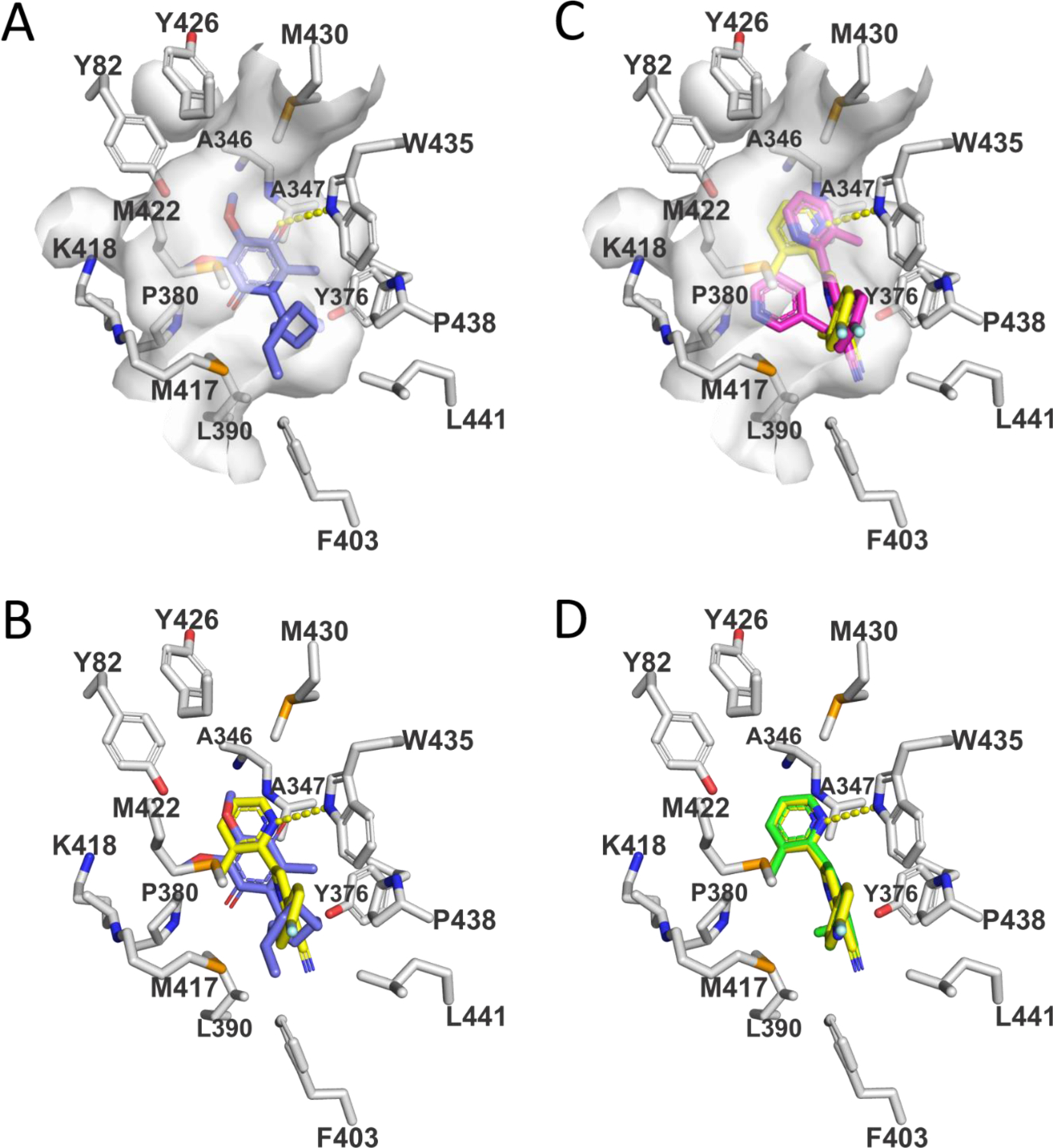
Views of the CoQ-binding pocket in SQOR complexes with substrate (DCQ) or enzyme inhibitors. Carbons in amino acid side chains are colored white. Hydrogen bonds are indicated by dashed yellow lines. (A) View of the SQOR complex with DCQ (blue carbons). The surface of the CoQ-binding cavity is shown in white. The O2 carbonyl oxygen in the quinone ring of DCQ forms a hydrogen bond with W435:NE1. (B) The model of the SQOR complex with 15 (yellow carbons) was produced using GLIDE (Schrödinger) and is compared to that observed with DCQ (blue carbons). A hydrogen bond between the 2-pyridyl ring in 15 and W435:NE1 is indicated. (C) The model of the SQOR complex with 18 (magenta carbons) was produced using GOLD (Cambridge Crystallographic Data Centre) and is compared with the model of the enzyme complex with 15 (yellow carbons). The surface of the CoQ-binding cavity is shown in white. Unlike with 18, 15 forms a strong hydrogen bond to W435:NE1, as indicated by the dashed yellow line. (D) The recently reported model of the SQOR complex with 1921,22 (green carbons) was produced using GLIDE (Schrödinger) and superimposes with the preferred docking pose obtained for 15 (yellow carbons).
Models of SQOR complexes with class A’ SAR compounds were constructed using GLIDE (Schrödinger, LLC, New York, NY) or the GOLD Suite of Programs (CCDC Software Ltd., Cambridge, UK). Docking was performing using flexible ligands and a rigid, ligand-free protein (PDB:ID 6M06). Similar models for the preferred docking pose were produced with either program. In the highest-scoring docking mode, 15 binds within the CoQ-binding pocket, occupying a binding site very similar to that observed for DCQ (Fig. 3B). Thus, the ring nitrogen of the 2-pyridyl substituent in area B in 15 superimposes with the O2 carbonyl in DCQ and both atoms are hydrogen-bonded to Trp435:NE1. The para-fluoro substituent in area A in 15 points toward entrance of the pocket and is exposed on the protein surface, as observed for the terminal carbon of the decyl tail in DCQ.
As noted above, a broad variety of groups are tolerated in area C, as illustrated by 18, in which the 2-((pyridin-3-yl)methoxy) substituent replaces the 2-methoxy group in 15. Comparison of the preferred docking poses obtained for 18 and 15 shows that a large group in area C can be readily accommodated within the CoQ-binding pocket without affecting the binding site occupied by the para-fluorophenyl substituent in area A (Fig. 3C). A potential steric clash between the substituents in areas B and C in 18 is avoided by a 180-degree rotation of the 2-pyridyl substituent in area B. The altered orientation of the 2-pyridyl substituent is stabilized by pi-pi stacking with Trp435 but precludes formation of a hydrogen bond with Trp435:NE1, as is observed with 15.
The models of SQOR complexes with 15 and 18 identified the para-position in area A as exposed on the protein surface and therefore likely to tolerate more polar substitution, such as the aniline moiety that we subsequently introduced to produce 19 (Table 1). The preferred docking pose obtained for 1921,22 superimposes with that obtained for 15 (Fig. 3D). Compound 19 retains excellent SQOR inhibition (IC50 = 29 nM) (Table 1) with improved polarity (cLogP = 3.0) and topological polar surface area (tPSA = 84), as compared with 15 (cLogP = 4.2, tPSA = 58) (Table 2).
We found that 19 exhibits 250-fold higher aqueous solubility, greatly improved cell permeability (Caco-2), and reduced plasma protein binding as compared with 15 (Table 3). Moreover, PK studies in mice indicate that 19 exhibits considerably increased exposure, as judged by a 25-fold higher AUC value, and a somewhat lower but acceptable t1/2 value (Table 3).
Table 3.
Observed properties of SAR class A’ lead compounds
|
|||||||
|---|---|---|---|---|---|---|---|
| Compound | X | Mouse Plasma Protein Binding (% bound) | Aqueous Solubility pH 7 (μM) | Caco-2 permeability (1 × 10−6 cm/s) | Mouse PK(IP)a | ||
| A→B | B→A | t1/2 (h) | AUClast (ng·h/mL) | ||||
| 15 | F | 99.64 | 0.013 | 0.455 | 0.769 | 5.96 | 42 |
| 19 | NH2 | 97.8 | 3.18 | 9.4 | 4.15 | 1.13b | 1082b |
IP dose (10 mg/kg) was formulated in N-methylpyrrolidinone (NMP): propylene glycol: 19% (w/v) 2-hydroxypropyl - beta-cyclodextran (kleptose) in water (10:40:50). Data are reported as the mean (n=3).
Data from reference 21.
We identified 19 as a potential candidate for proof-of-concept in vivo efficacy testing, based on its greatly improved physicochemical properties and cell permeability and the favorable results obtained in mouse PK studies. We designed an alternate, improved synthetic route suitable for the gram-scale synthesis of 19 in good overall yield (24%) and in the high purity (>97%) required in-vivo efficacy studies (Scheme 3). Starting from commercially available substituted benzaldehyde 31, condensation with ketone 28 gives chalcone 32 in good yield. Reaction of 32 with malononitrile in the presence of sodium methoxide gave 33, and Boc-deprotection using HCl in 1,4-dioxane allowed for the filtration of the pure desired product 19 as the bis-HCl salt in high yield, as shown by the analytic data given in Supplementary material.
Scheme 3.
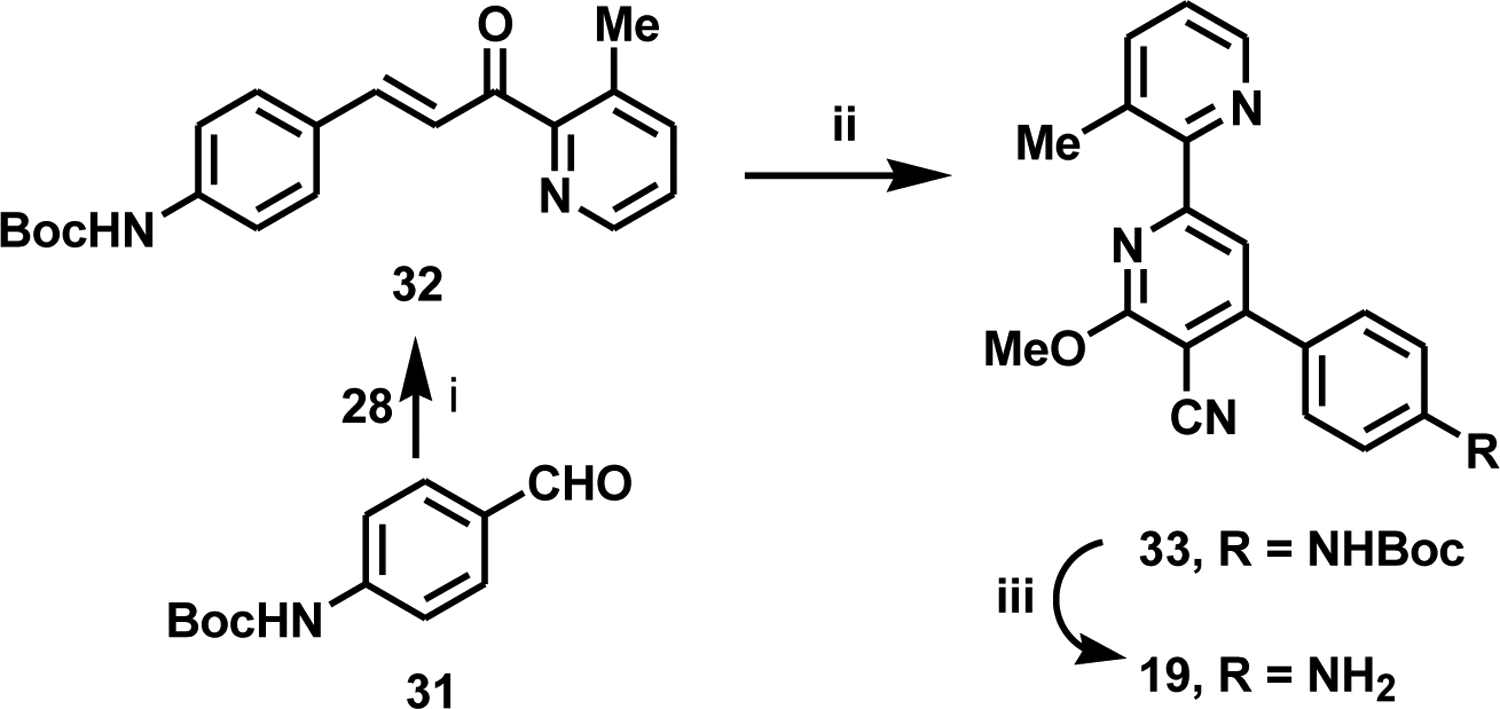
i, 1-(3-methylpyridin-2-yl)ethan-1-one, KOH, MeOH, 20 °C, 3 days, 63%; ii, malononitrile, NaOMe, MeOH, 20 °C, 16 h, 38%; iii, HCl, dioxane/CH2Cl2, MeOH, 20 °C, 16 h, 99%.
In summary, our hit-to-lead optimization SAR and structure-based design studies led to the identification of 19, a cell-permeable class A’ derivative that retains the high SQOR inhibitory potency observed for top class A/A’ HTS hits with physicochemical properties, metabolic stability, and PK properties suitable for in vivo efficacy testing. As we recently reported21,22, additional in vitro tests showed that 19 exhibits little or no toxicity in mammalian cells and a high degree of specificity for the CoQ-binding site in SQOR. Moreover, in vivo studies demonstrated that administration of 19 preserves cardiac function and prevents the progression to heart failure in a murine model21,22.
Supplementary Material
Acknowledgments
This work was supported funded by NIH National Heart, Lung, and Blood Institute grant R41 HL134435 (Jorns, PI).
Abbreviations.
- ADME
absorption, distribution, metabolism, and excretion
- CoQ
coenzyme Q
- DCIP
2,6-dichlorophenolindophenol
- DCQ
decylubiquinone
- HTS
high-throughput screen
- IC50
half-maximal inhibitory concentration
- PK
pharmacokinetics
- SAR
structure-activity relationships
- SQOR
sulfide:quinone oxidoreductase
- tPSA
topological polar surface area
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References and notes
- 1.Blackstone E, Morrison M, Roth MB, H2S induces a suspended animation–like state in mice. Science 308, 518 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Kimura H, Hydrogen sulfide as a neuromodulator. Mol. Neurobiol 26, 13–19 (2002). [DOI] [PubMed] [Google Scholar]
- 3.Li L, Rose P, Moore PK, Hydrogen sulfide and cell signaling. Annu. Rev. Pharmacol. Toxicol 51, 169–187 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Peng Y-J, Nanduri J, Raghuraman G, Souvannakitti D, Gadallab MM, Kumar GK, Snyder SH, Prabhakar NR, H2S mediates O2 sensing in the carotid body. Proc. Natl. Acad. Sci. USA 107, 10719–10724 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hackfort BT, Mishra PK, Emerging role of hydrogen sulfide-microRNA crosstalk in cardiovascular diseases. Am. J. Physiol. Heart Circ. Physiol 310, H802–H812 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mani S, Li HZ, Untereiner A, Wu LY, Yang GD, Austin RC, Dickhout JG, Lhotak S, Meng QH, Wang R, Decreased endogenous production of hydrogen sulfide accelerates atherosclerosis. Circulation 127, 2523–2534 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, Meng Q, Mustafa AK, Mu W, Zhang S, Snyder SH, Wang R, H2S as a physiologic vasorelaxant: Hypertension in mice with deletion of cystathionine γ-lyase. Science 322, 587–590 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yong R, Searcy DG, Sulfide oxidation coupled to ATP synthesis in chicken liver mitochondria. Comp. Biochem. Physiol. B, Biochem. Mol. Biol 129, 129–137 (2001). [DOI] [PubMed] [Google Scholar]
- 9.Vitvitsky V, Kabil O, Banerjee R, High turnover rates for hydrogen sulfide allow for rapid regulation of its tissue concentrations. Antioxid. Redox Signal 17, 22–31 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang HL, Wu HC, Li ZL, Geng B, Tang CS, Changes of the new gaseous transmitter H2S in patients with coronary heart disease. J. First Mil. Med. Univ 25, 951–954 (2005). [PubMed] [Google Scholar]
- 11.Kovacic D, Glavnik N, Marinsek M, Zagozen P, Rovan K, Goslar T, Mars T, Podbregar M, Total plasma sulfide in congestive heart failure. J. Card. Fail 18, 541–548 (2012). [DOI] [PubMed] [Google Scholar]
- 12.Polhemus DJ, Calvert JW, Butler J, Lefer DJ, The cardioprotective actions of hydrogen sulfide in acute myocardial infarction and heart failure. Scientifica 2014, 768607 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feliers D, Lee HJ, Kasinath BS, Hydrogen sulfide in renal physiology and disease. Antioxid. Redox Signal 25, 720–731 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schefold JC, Filippatos G, Hasenfuss G, Anker SD, Von Haehling S, Heart failure and kidney dysfunction: epidemiology, mechanisms and management. Nat. Rev. Nephrol 12, 610–623 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Augustyn KDC, Jackson MR, Jorns MS, Use of tissue metabolite analysis and enzyme kinetics to discriminate between alternate pathways for hydrogen sulfide metabolism. Biochemistry 56, 986–996 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jackson MR, Melideo SL, Jorns MS, Human sulfide:quinone oxidoreductase catalyzes the first step in hydrogen sulfide metabolism and produces a sulfane sulfur metabolite. Biochemistry 51, 6804–6815 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Capper MJ, O’Neill PM, Fisher N, Strange RW, Moss D, Ward SA, Berry NG, Lawrenson AS, Hasnain SS, Biagini GA, Antonyuk SV, Antimalarial 4(1H)-pyridones bind to the Qi site of cytochrome bc1. Proc. Natl. Acad. Sci. U. S. A 112, 755–760 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nilsen A, LaCrue AN, White KL, Forquer IP, Cross RM, Marfurt J, Mather MW, Delves MJ, Shackleford DM, Saenz FE, Morrisey JM, Steuten J, Mutka T, Li Y, Wirjanata G, Ryan E, Duffy S, Kelly JX, Sebayang BF, Noviyanti R, Sinden RE, Kocken CHM, Price RN, Avery VM, Angulo-Barturen I, Jiménez-Díaz MB, Jiménez-Díaz MB, Ferrer S, Herreros E, Sanz LM, Gamo F, Bathurst I, Burrows JN, Siegl P, Guy RK, Winter RW, Vaidya AB, Charman SA, Manetsch R, Riscoe Mi. K., Quinolone-3-diarylethers: A new class of antimalarial drug. Sci. Transl. Med 5, 177ra37 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oliver JD, Sibley GEM, Beckmann N, Dobb KS, Slater MJ, McEntee L, du Pre S, Livermore J, Bromley MJ, Wiederhold NP, Hope WW, Kennedy AJ, Law D, Birch M, F901318 represents a novel class of antifungal drug that inhibits dihydroorotate dehydrogenase. Proc. Natl. Acad. Sci. U. S. A 113, 12809–12814 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vyas VK, Ghate M, Recent developments in the medicinal chemistry and therapeutic potential of dihydroorotate dehydrogenase (DHODH) inhibitors. Mini Rev. Med. Chem 11, 1039–1055 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Jackson MR, Cox KD, Baugh SDP, Wakeen L, Rashad AA, Lam PYS, Polyak B, Jorns MS, Discovery of a first-in-class inhibitor of sulfide:quinone oxidoreductase that protects against adverse cardiac remodeling and heart failure. Cardiovasc. Res, Advance online publication. 10.1093/cvr/cvab206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jorns MS, Jackson MR, Cox KD, Lam PYS, and Baugh SDP Compounds and methods for treating or preventing heart failure. Drexel University. PCT/US2020/012938[WO 2020/146636 A1], 1–228. 7-16-2020. Pennsylvania/USA
- 23.Ishikawa M, Hashimoto Y, Improvement in aqueous solubility in small molecule drug discovery programs by disruption of molecular planarity and symmetry. J. Med. Chem 54, 1539–1554 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Jackson MR, Loll PJ, Jorns MS, X-ray structure of human sulfide:quinone oxidoreductase: Insights into the mechanism of mitochondrial hydrogen sulfide oxidation. Structure 27, 794–805 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baugh SDP, Chaly A, Weaver DG, Pelletier JC, Thanna S, Freeman KB, Reitz AB, Scott RW, Highly potent, broadly active antifungal agents for the treatment of invasive fungal infections. Bioorg. Med. Chem. Lett 33, 127727 (2021). [DOI] [PubMed] [Google Scholar]
- 26.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT, Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem 49, 6177–6196 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Jacobson MP, Pincus DL, Rapp CS, Day TJF, Honig B, Shaw DE, Friesner RA, A hierarchical approach to all-atom protein loop prediction. Proteins 55, 351–367 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Jacobson MP, Friesner RA, Xiang Z, Honig B, On the role of the crystal environment in determining protein side-chain conformations. J. Mol. Biol 320, 597–608 (2002). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.