

Abstract
Acute COVID-19, caused by SARS-CoV-2, is characterized by diverse clinical presentations, ranging from asymptomatic infection to fatal respiratory failure, and often associated with varied longer-term sequelae. Over the past 18 months, it has become apparent that inappropriate immune responses contribute to the pathogenesis of severe COVID-19. Researchers working at the intersection of COVID-19 and autoimmunity recently gathered at an American Autoimmune Related Diseases Association Noel R. Rose Colloquium to address the current state of knowledge regarding two important questions: Does established autoimmunity predispose to severe COVID-19? And, at the same time, can SARS-CoV-2 infection trigger de novo autoimmunity? Indeed, work to date has demonstrated that 10% to 15% of patients with critical COVID-19 pneumonia exhibit autoantibodies against type I interferons, suggesting that preexisting autoimmunity underlies severe disease in some patients. Other studies have identified functional autoantibodies following infection with SARS-CoV-2, such as those that promote thrombosis or antagonize cytokine signaling. These autoantibodies may arise from a predominantly extrafollicular B cell response that is more prone to generating autoantibody-secreting B cells. This Review highlights the current understanding, evolving concepts, and unanswered questions provided by this unique opportunity to determine mechanisms by which a viral infection can be exacerbated by, and even trigger, autoimmunity. The potential role of autoimmunity in post-acute sequelae of COVID-19 is also discussed.
Introduction
In December 2019, a novel coronavirus, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), emerged in Wuhan, Hubei Province, China, and caused the coronavirus disease 2019 (COVID-19) pandemic. COVID-19 presentations range from asymptomatic infection to mild flu-like symptoms to fatal respiratory failure. In addition, many patients experience long-term symptoms of COVID-19 persisting weeks to months after the initial onset of symptoms and extending beyond the original organ involvement, known as post-acute sequelae of COVID-19 (PASC) and more commonly called “long COVID.”
Over the past 18 months, researchers have sought to determine mechanisms by which an individual’s immune system may be helpful or harmful in COVID-19. In the context of vaccination, it is apparent that adaptive immunity can quite effectively negate severe COVID-19. At the same time, it appears that preexisting autoimmunity may influence, often deleteriously, the course of COVID-19 in certain individuals. Of particular note, work to date has demonstrated that 10% to 15% of patients with critical COVID-19 pneumonia exhibit autoantibodies against type I interferons (IFNs). Meanwhile, in other patients, the virus may contribute to a de novo breakdown in immune tolerance, triggering pathogenic autoantibodies in susceptible individuals. In some reports, more than 50% of patients hospitalized with moderate to severe COVID-19 have circulating autoantibodies; the extent to which these autoantibodies persist after hospital discharge is a question that has for the most part not been addressed.
In the summer of 2021, the Noel R. Rose COVID-19 and Autoimmunity Colloquium, organized by the American Autoimmune Related Diseases Association (AARDA), brought together researchers working at the intersection of COVID-19 and autoimmunity to address the current state of knowledge regarding two important questions: Does established autoimmunity predispose to severe COVID-19? And, at the same time, can SARS-CoV-2 infection trigger de novo autoimmunity? The breadth of expertise reflected the desire to create a colloquium that spanned multiple medical specialties and scientific disciplines. Participants represented diverse fields, including biobanking, cardiovascular medicine, clinical informatics, immunology, pathology, and rheumatology, among others. This Review highlights the current state of knowledge regarding the intersection of COVID-19 and autoimmunity, including work and ideas discussed during the COVID-19 and Autoimmunity Colloquium.
Immunopathology of severe COVID-19
A cardinal histopathological feature of severe COVID-19 is pulmonary microangiopathy with evidence of fibrin thrombi, activated platelets, and neutrophil extracellular traps within vessels (1, 2). Furthermore, infiltrating neutrophils, monocytes, and macrophages are observed in additional organs beyond the lungs, including the heart, central nervous system, and liver (2–4). In addition to cell activation and infiltration, local and systemic complement activation likely contributes to the microangiopathy. In patients with severe COVID-19, exaggerated complement deposition has been detected in various tissues, including the lungs (5). Meanwhile, systemic detection of alternative complement pathway activation has also been appreciated in severe disease (6).
A subset of patients with COVID-19 develop hyperinflammation with high cytokine and chemokine levels in a pattern that is similar to, but still distinct from, the autoinflammatory macrophage activation syndrome that complicates various autoimmune diseases, such as systemic juvenile idiopathic arthritis and systemic lupus erythematosus (SLE) (7–9). It is notable that immunomodulatory medications, especially dexamethasone (10), appear to improve survival in severe COVID-19. Trials of more targeted therapies have also been conducted in COVID-19. These include inhibitors of cytokines (or their receptors), such as IL-6 (11), IL-1 (12), and granulocyte-macrophage colony-stimulating factor (13). Small molecules that block cytokine-mediated signaling, for instance, Janus kinase (JAK) inhibitors (14, 15), have also been studied. The efficacy of these targeted therapies is less proven than that of dexamethasone and is likely highly dependent on the timing of administration and patient selection (16).
Role of preexisting autoimmunity
The clinical manifestations of COVID-19 are variable, ranging from asymptomatic infection in many individuals to critical pneumonia in about 2% to 3% of patients (17). The dominant epidemiological risk factor for life-threatening COVID-19 is age, with the risk of death doubling every 5 years from childhood onward. Male sex and preexisting comorbidities contribute to a lesser extent (odds ratios typically <1.5) (18). We now also know that genetic variants (such as monogenic inborn errors) and — with high relevance to this Review — preexisting immunological abnormalities may underlie the severity of disease in some individuals (19).
The role of type I IFNs in COVID-19.
Type I IFNs induce the expression of many IFN-stimulated genes (ISGs) that are essential for antiviral immunity (Figure 1A). For at least a subset of patients, SARS-CoV-2 induces only a limited type I IFN response, leading to poor control of SARS-CoV-2 replication and the transition to severe COVID-19 (20–22). At the same time, other studies have delivered a contrasting message that some patients with severe COVID-19 exhibit robust and sustained type I IFN responses that ultimately contribute to organ damage (23–26). While type I IFNs and ISGs are certainly helpful and protective in early stages of infection, additional work is needed to understand which specific subgroups of patients with severe disease are more likely to be helped or harmed by type I IFNs.
Figure 1. Some patients with severe COVID-19 exhibit autoantibodies antagonizing type I IFN immunity.

(A) Type I IFNs bind to the IFN-α/β receptor (IFNAR) to induce the expression of IFN-stimulated genes (ISGs) that are essential for antiviral immunity. (B) Anti-IFN autoantibodies block IFN binding to its receptor, preventing the upregulation of ISG expression and impairing antiviral immunity. Uncontrolled replication of SARS-CoV-2 may then result in hyperinflammation and tissue damage.
Genetics and autoantibodies that disrupt type I IFN immunity.
In an international cohort, about 3.5% of patients with severe COVID-19 carried rare loss-of-function inborn errors of TLR3- and IRF7-dependent type I IFN immunity that have previously been shown to underlie critical influenza pneumonia (27, 28). For example, four previously healthy, unrelated adults between 25 and 50 years of age had autosomal recessive, complete deficiency of the IFNAR1 chain of the type I IFN receptor (n = 2) or IFN regulatory factor 7 (IRF7) (n = 2; refs. 27, 28).
In the same cohort (but in other patients without these inborn errors), neutralizing autoantibodies against type I IFNs were detected in at least 10% of patients with critical COVID-19 pneumonia, but not in patients with asymptomatic infection (Figure 1B and ref. 29). Importantly, these autoantibodies may be causal and not a consequence of critical COVID-19, as they can be found in at least some patients before infection (29). The autoantibodies of patients with critical COVID-19 primarily targeted IFN-ω and IFN-α, but not IFN-β, IFN-κ, or IFN-ε (29); therefore, patients with these antibodies could potentially still benefit from early administration of IFN-β (30).
Neutralizing autoantibodies against type I IFNs have been identified since the 1980s in patients treated with IFN-α2 and IFN-β (31), patients with SLE (32), patients with thymoma and/or myasthenia gravis (33), and nearly all patients with autoimmune polyendocrinopathy syndrome type 1 (APS-1; refs. 34–36). Although these autoantibodies are typically clinically silent for much of life, most patients with APS-1 who were infected with SARS-CoV-2 developed severe COVID-19, further suggesting that preexisting autoantibodies against type I IFNs predispose to severe manifestations of COVID-19 (37). Moreover, the same autoantibodies against type I IFNs underlie adverse reactions to yellow fever live attenuated viral vaccine in about a third of cases (38). Notably, multiple centers have confirmed that autoantibodies against type I IFNs underlie at least 10% of cases of life-threatening COVID-19 pneumonia in the general population (29, 39–48).
More recently, autoantibodies neutralizing lower, more physiological concentrations of type I IFNs were found in at least 15% of patients with critical COVID-19 pneumonia, including 20% of patients older than 80 years (49). Furthermore, these autoantibodies were found in about 20% of COVID-19 deaths across all ages (49, 50). Analysis of more than 34,000 uninfected individuals demonstrated that these autoantibodies were present in 0.18% of individuals between 18 and 69 years of age, rising to 4% in individuals older than 70 years (49), a pattern that likely contributes to the age-associated risk of life-threatening COVID-19.
In parallel to this recent work, a genome-wide, unbiased approach found that about 1% of male patients younger than 60 years of age with critical COVID-19 pneumonia had X-linked recessive TLR7 deficiency (51). Plasmacytoid dendritic cells isolated from these patients produced negligible amounts of type I IFNs in response to SARS-CoV-2 (51). When combined with the autosomal inborn errors of TLR3-dependent type I IFN immunity that likely impact pulmonary epithelial cells (28), inborn errors may underlie critical COVID-19 in 3% to 4% of patients, especially in those younger than 60 years (whereas autoantibodies are more commonly involved in patients older than 60 years). Collectively, inborn errors (5%) and autoantibodies associated with type I IFN signaling (15%) could account for about 20% of cases of critical COVID-19 pneumonia.
A two-step model seems likely whereby some patients demonstrate inadequate type I IFN immunity during early infection (whether mediated by inborn errors, autoantibodies, or other unknown factors). This contributes to unrestrained viral replication and spread, resulting in pulmonary and systemic hyperinflammation (Figure 1B and ref. 18). Therefore, the timing of therapies enhancing type I IFN signaling is likely to be crucial, and they should be administered in the first few days of SARS-CoV-2 infection.
Role of de novo autoimmunity
Some clinical features of moderate to severe COVID-19 are reminiscent of those seen in autoimmune diseases such as antiphospholipid syndrome, inflammatory arthritis, SLE, and anti-MDA5 syndrome (52–55). In addition, there are numerous case reports of patients developing classifiable autoimmune diseases, such as rheumatoid arthritis, psoriatic arthritis, and type 1 diabetes, concomitantly with or immediately following SARS-CoV-2 infection (56–64). These various observations have led investigators to question whether de novo autoimmunity may contribute to at least a subset of patients who experience a more severe course with COVID-19.
Other examples of virus-associated autoimmunity.
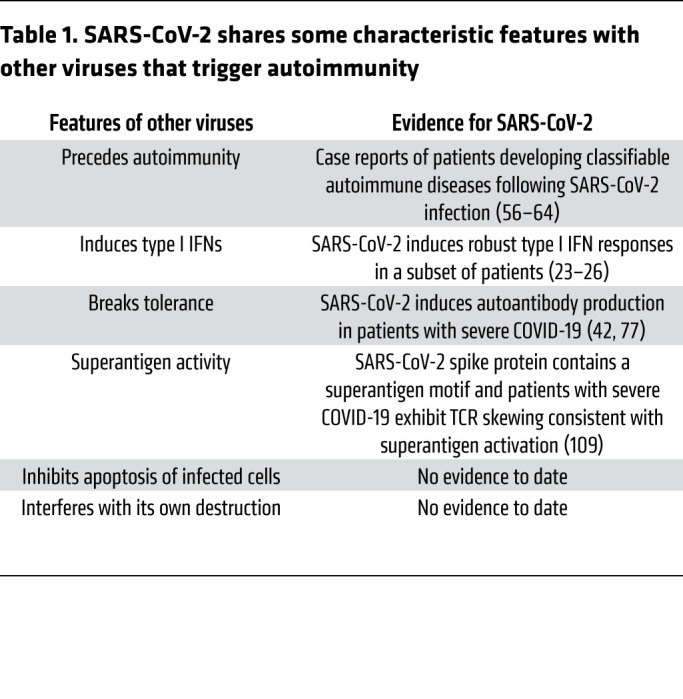
Viruses such as cytomegalovirus, parvovirus B19, and Epstein-Barr virus (EBV) have been postulated to be environmental triggers of autoimmunity in genetically predisposed individuals (65). As one example, serological evidence of EBV reactivation tracks not only with the transition to SLE, but also with increased disease activity in individuals with established SLE (66, 67); indeed, antibodies against EBV nuclear antigen-1 cross-react with the SLE-associated antigens Sm and Ro (68–70), and levels of anti-EBV antibodies correlate with SLE-associated autoantibodies (66, 71–73). Viruses that trigger autoimmunity exhibit several characteristic features (Table 1), including a tendency to cause ubiquitous and/or persistent infection, as well as an ability to tip the host immune response toward loss of tolerance via production of autoreactive lymphocytes. Mechanistically, viruses may contribute to autoimmunity-prone immune responses in various ways. Examples include molecular and functional mimicry, superantigen activity, and stimulation of inflammatory signaling, including production of type I IFNs (74–76).
Table 1. SARS-CoV-2 shares some characteristic features with other viruses that trigger autoimmunity.
Profiling the autoantigenome of COVID-19.
To determine whether COVID-19 promotes autoantibody production, several groups have endeavored to comprehensively profile the autoantigenome of COVID-19. Using established antigen arrays, Chang et al. identified autoantibodies associated with rheumatological diseases in 49% of patients hospitalized with COVID-19, compared with less than 15% of healthy controls (77). Many of these autoantibodies are traditionally associated with rare autoimmune diseases, such as autoimmune myositis (77). In addition, 60% to 80% of patients hospitalized with COVID-19 had at least one anti-cytokine autoantibody with the potential to modulate immune responses (77).
Wang et al. used a more unbiased approach and screened for autoantibodies against extracellular and secreted proteins, which were theorized to be the main targets for functional autoantibodies. Using rapid extracellular antigen profiling (REAP), in which barcoded human extracellular and secreted proteins are displayed on the surface of yeast (78), they identified a wide range of antibodies targeting immune-related antigens, such as cytokines and chemokines, in the plasma of patients with COVID-19 (42). Mouse surrogates of these autoantibodies increased disease severity in a mouse model of SARS-CoV-2 infection (42). Furthermore, patients with COVID-19 exhibited autoantibodies against tissue-associated antigens that correlated positively with disease severity (42). Importantly, some autoantibodies were clearly induced following SARS-CoV-2 infection, suggesting that COVID-19 contributes to loss of tolerance (42, 77).
Autoantigens can form affinity complexes with the glycosaminoglycan dermatan sulfate (DS). These complexes may then engage B cell receptor signaling in autoreactive B1 cells and thereby induce autoantibody production (79–81). Self-proteins with affinity to DS are therefore more likely to be autoantigens. Wang et al. identified autoantigens with DS affinity from different cell lines and compared them with proteins altered at the protein or transcript level in SARS-CoV-2 infection (82–84). Notably, many of the SARS-CoV-2–altered proteins with DS affinity were associated with COVID-19 disease manifestations, such as neurological symptoms, thrombosis, and possibly PASC (82–84).
Other functional autoantibodies.
Abnormal coagulation, along with microvascular and macrovascular thrombosis, is associated with not only severe COVID-19 (85, 86), but also the autoimmune thromboinflammatory disease antiphospholipid syndrome. Antiphospholipid syndrome is characterized by the presence of antiphospholipid autoantibodies (aPLs), which promote thrombosis by activating endothelial cells and platelets while also stimulating neutrophils to release neutrophil extracellular traps (NETs) (Figure 2A and refs. 87–90). Patients hospitalized with COVID-19 exhibit elevated levels of NETs (91), which correlate with disease severity and thrombosis (92). In one study, approximately half of patients hospitalized with COVID-19 had at least one type of aPL, while positive aPL testing was associated with neutrophil activation, more NET release, reduced oxygenation efficiency, and more severe disease (93). Importantly, total IgG fractions from patients with COVID-19 who were positive for aPLs triggered NET release from healthy neutrophils (93), activated endothelial cells to upregulate cell adhesion molecules (94), and accelerated thrombosis when transferred into mice (93). All these phenotypes are similar to those associated with IgG fractions from individuals with established antiphospholipid syndrome.
Figure 2. Potential downstream mechanisms of autoantibodies identified in patients with severe COVID-19.

(A) A subset of patients with severe COVID-19 have anti-phospholipid antibodies (aPLs) and/or anti–neutrophil extracellular trap (anti-NET) autoantibodies. aPLs may activate endothelial cells and platelets and stimulate neutrophils to release NETs. Anti-NET antibodies bind to NETs, impairing NET degradation by DNase. Together, these autoantibodies may activate complement and promote thrombosis. (B) In some patients with severe COVID-19, antibodies can prevent the expression of ISGs by antagonizing signaling through the type I IFN receptor in an FcγRIIb-dependent fashion, impairing antiviral immunity.
Patients with antiphospholipid syndrome and other rheumatological diseases have elevated levels of antibodies that bind to NETs, impairing NET degradation and likely activating complement (95, 96). Levels of anti-NET antibodies are also increased in patients hospitalized with COVID-19, with the highest levels in patients requiring mechanical ventilation (97). Anti-NET antibodies correlate with NET remnants in blood, COVID-19 severity, and platelet count and inversely correlate with oxygenation efficiency and NET clearance (97). Taken together, these findings suggest a potential role in COVID-19–associated thrombosis (Figure 2A).
Another study by Combes et al. found that immune cells from patients with mild COVID-19, including neutrophils and monocytes, expressed a strong ISG signature (98). In contrast, and in line with some of the studies mentioned above (20, 21), ISG-expressing cells were less likely to be found in patients with severe COVID-19 requiring intubation and intensive care (98). In the study, one of seven patients with severe COVID-19 exhibited autoantibodies against IFN-α (98). In the remaining six patients, total IgG fractions antagonized signaling through the monocyte type I IFN receptor in FcγRIIb-dependent fashion (Figure 2B and refs. 98, 99). Although the antigen specificity of these antibodies remains to be determined (which would allow this concept to be tested more broadly in additional cohorts), these data suggest that therapies inhibiting FcγRIIb may have the potential to restore type I IFN responses in some patients with severe COVID-19.
Potential mechanisms of de novo autoimmunity in COVID-19.
Effector B cell responses can be activated through the germinal center or extrafollicular pathways. Unlike germinal center reactions, extrafollicular maturation lacks certain checkpoints to prevent autoreactivity and, as such, is more prone to generating autoantibodies (100). In SLE, a large proportion of antibody-secreting cells originate from naive B cells (as opposed to memory B cells), which are activated via the extrafollicular pathway in a TLR7-dependent manner (101, 102). These extrafollicular B cells, known as double-negative (DN2) B cells, lack IgD, CD27, CXCR5, and CD21. They are poised to become antibody-secreting cells, tend to produce pathogenic autoantibodies, and are enriched in patients with active SLE, including patients with lupus nephritis (102).
Reminiscent of SLE, higher levels of both circulating DN2 B cells and circulating plasma cells associate with greater disease severity and poor outcomes in COVID-19 (Figure 3 and ref. 100). In addition, patients who succumb to COVID-19 lack Bcl6+ germinal centers (103), consistent with a predominantly extrafollicular response. Patients with severe COVID-19 also exhibited higher numbers of unmutated SLE-associated, autoimmune-prone IGHV4-34 antibody–secreting cells in circulation (100, 104). The mechanisms contributing to extrafollicular pathway activation in severe COVID-19 are unknown; however, TLR7 drives DN2 cell differentiation (102), and TLR7 recognizes viral single-stranded RNA genomes, such as that of SARS-CoV-2 (105). Furthermore, patients with severe COVID-19 exhibit elevated plasma IL-6 levels (106), which correlate with DN2 cell expansion (107). Notably, patients with high extrafollicular responses exhibit high titers of neutralizing antibodies against the receptor-binding domain of the SARS-CoV-2 spike protein (100), suggesting that a lack of protective antibodies is not the main driver of disease severity in these patients. Together, these studies indicate that autoimmune-prone extrafollicular B cells dominate the B cell response in many patients with severe COVID-19, likely contributing to the loss of tolerance and dysregulated humoral immunity in patients with severe disease.
Figure 3. Potential mechanisms of de novo autoimmunity in COVID-19.
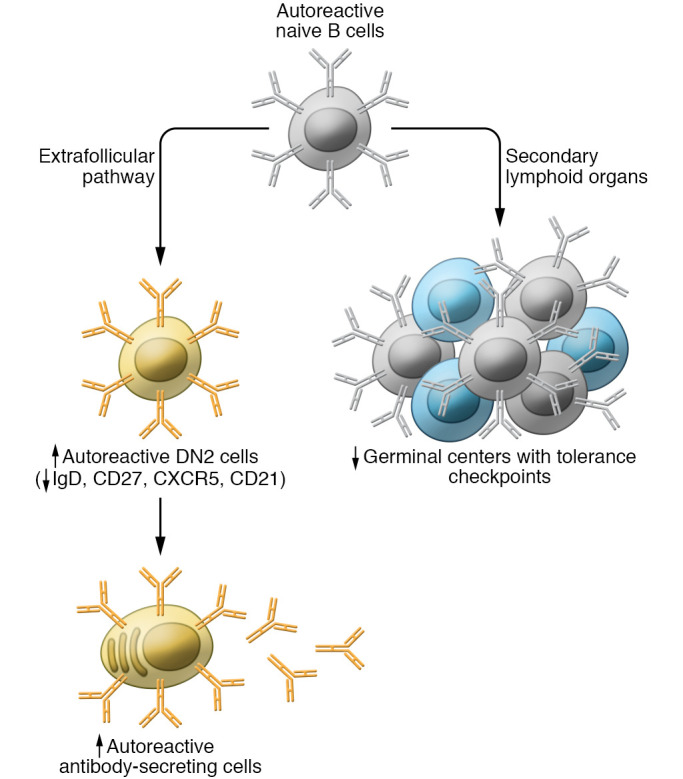
Naive B cells can be activated via both the germinal center and the extrafollicular pathway. The extrafollicular pathway lacks some tolerance checkpoints that prevent the activation and maturation of autoreactive B cells and is, therefore, more prone to generating autoantibodies. Patients with severe COVID-19 exhibit higher levels of extrafollicular B cells lacking IgD, CD27, CXCR5, and CD21 (known as double-negative [DN2] cells) and plasma cells. They may also lack germinal centers. Red arrows indicate increased or reduced levels in patients with severe COVID-19 compared with patients with mild COVID-19.
Some viruses possess superantigen activity, enabling broad nonspecific T cell activation via MHC class II or the T cell receptor (TCR) and contributing to hyperinflammation and autoimmunity (108). Using computational modeling, Cheng et al. demonstrated that the SARS-CoV-2 spike protein contains a high-affinity motif similar to bacterial superantigens that directly interacts with the TCR and may form a ternary complex with MHC class II (109). Interestingly, the authors found that some patients with severe COVID-19 have a skewed TCR repertoire consistent with superantigen activity (109). Multisystem inflammatory syndrome in children (MIS-C) is a severe inflammatory syndrome with multiorgan involvement that occurs in a small percentage of children following SARS-CoV-2 infection (110). One study of 16 children with severe MIS-C found significant expansion of TCR β chain variable gene 11-2 (TRBV11-2), TRBV24-1, and TRBV11-3 in MIS-C patients relative to febrile control patients, such that up to 24% of the clonal T cell space was taken up by clones using TRBV11-2 (111). In silico modeling indicated that polyacidic residues in the Vβ chain encoded by TRBV11-2 strongly interact with the superantigen-like motif of SARS-CoV-2 spike glycoprotein, suggesting that unprocessed SARS-CoV-2 spike may directly mediate TRBV11-2 expansion. Another study found that 24 of 32 patients (75%) with MIS-C (and none in other clinical groups) displayed TRBV11-2 (also known as Vβ21.3+) expansions (112). Notably, TRBV11-2 T cells correlate with MIS-C cytokine storm and were enriched in a cluster of patients with autoimmunity-associated immunoglobulin heavy chain variable region genes and increased autoantibodies targeting tissue-specific autoantigens (113). Therefore, superantigen-like T cells may trigger hyperinflammation and the production of autoantibodies following SARS-CoV-2 infection, contributing to de novo autoimmunity. The extent to which SARS-CoV-2 spike–mediated superantigen activity contributes to autoimmunity in adults with severe COVID-19 is a topic worthy of further research.
Does autoimmunity play a role in PASC?
Whether autoimmunity contributes to PASC is only beginning to be addressed. Furthermore, as definitions of PASC are still being established, any available data will need to be interpreted in relation to the definition used by the authors. One recent study of 96 patients detected antinuclear antibody (ANA) titers ≥1:160 in 43.6% of patients at 12 months after COVID-19 symptom onset (114). In the cohort, the frequency of neurocognitive symptoms (such as concentration problems) was significantly higher in the group with ANA titers ≥1:160 compared with the group with titers less than 1:160 (107). Outstanding questions that should be systematically considered in the coming months and years include the following: Do preexisting autoantibodies predispose someone with COVID-19 to develop PASC? How commonly do de novo autoantibodies persist beyond the acute phase of SARS-CoV-2 infection, and will these patients transition to a classifiable autoimmune disease? Meanwhile, does virus-induced autoreactivity underlie at least some of the wide spectrum of clinical phenotypes associated with PASC? If so, can patients with acute disease or PASC be immunologically profiled to identify those who might benefit from immune-modulating therapies? Answering these questions will require the generation of multiethnic biospecimen repositories from COVID-19 patients, such as the Collaborative Cohort of Cohorts for COVID-19 Research (C4R), which includes information from before, during, and after SARS-CoV-2 infection that can potentially enable the necessary longitudinal investigations (115). Analysis of large-scale electronic health record (EHR) data (116–118) will likely also be needed to determine clinical associations with COVID-19, such as autoimmune manifestations. This approach has already been used to identify hospitalization trends and clinical and laboratory features and to predict severity in patients with COVID-19 (119–121) through initiatives such as the international Consortium for Clinical Characterization of COVID-19 by EHR (4CE; https://covidclinical.net) and the National COVID Cohort Collaborative (N3C; https://ncats.nih.gov/n3c).
Conclusions
Many of the studies discussed above leveraged patient samples obtained in the early months of the COVID-19 pandemic, before the regular use of dexamethasone, and certainly before the advent of vaccination. Understanding the extent to which these interventions (and hopefully additional interventions to come) change how SARS-CoV-2 interacts with the immune system is one important future direction. It will also be valuable to see additional studies that use affinity purification to characterize downstream mechanisms of specific autoantibody species. Furthermore, longitudinal cohorts that capture patient samples at the time of acute illness and then in follow-up will be important; establishing cohorts that also include pre-COVID samples will be even more valuable.
In conclusion, data to date strongly suggest that some severe COVID-19 cases can be explained by preexisting autoantibodies (which, interestingly, confer a risk similar to that conferred by rare inborn genetic errors in the same pathways). With regard to de novo autoantibody formation, a variety of such antibodies are detected when patients are hospitalized with severe COVID-19; however, there is still work to be done to determine whether these antibodies are important contributors to severe disease or an epiphenomenon of the marked inflammation. Going forward, the COVID-19 pandemic would seem to provide a once-in-a-lifetime opportunity to more precisely determine how a viral infection can be exacerbated by, and even trigger, autoimmunity.
Acknowledgments
This Review was inspired by a Colloquium sponsored by the American Autoimmune Related Diseases Association and Macon Callicott. JSK was supported by grants from the NIH (R01HL115138), Burroughs Wellcome Fund, Lupus Research Alliance, and Rheumatology Research Foundation. RC was supported by the Lupus Research Alliance. JAJ was supported by grants from the NIH (U19AI062629, P30AR073750, U54GM104938) and the Lou Kerr Chair in Biomedical Research. YK was supported by the Intramural Research Program of the National Heart, Lung, and Blood Institute and National Institute of Allergy and Infectious Diseases, the Lasker Foundation, and the Falk Medical Research Trust Catalyst Award. AMR was supported by grants from the Ludwig and Mathers Family Foundations and the Pew Charitable Trusts.
Version 1. 10/28/2021
In-Press Preview
Version 2. 12/15/2021
Electronic publication
Footnotes
Conflict of interest: JSK received support from Jazz Pharmaceuticals for preclinical research. RC provided consulting services to GlaxoSmithKline and received support for preclinical research from Janssen Pharmaceuticals. SEF provided consulting services to Boehringer Ingelheim in 2020. JAJ received research support from Progentec Biosciences LLC. YK is an inventor on a pending patent (US20180369278A1) to the University of Michigan on the use of biogases in vascular disease. PM is a clinical training fellow within the Experimental Medicine Initiative to Explore New Therapies network (EMINENT) from the Medical Research Council and GlaxoSmithKline and receives project funding unrelated to this report. PM also receives cofunding from the National Institute for Health Research (NIHR) University College London Hospitals Biomedical Research Centre. AMR is an inventor on a pending US patent application (PCT/US21/23521) filed by Yale University describing the rapid extracellular antigen profiling (REAP) technology and is the founder of Seranova Bio, the commercial licensee of this patent. CS provided consulting services for and received speaker fees from Eli Lilly and Janssen Pharmaceuticals. PJU received research support from EMD Serono and Synairgen. PJU received income from 4DMT, Abbvie, Amarin, Gilead, Immunic, Intracellular Therapeutics, Seranova Bio, and Vir. JYW is the founder and chief scientific officer of Curandis.
Copyright: © 2021, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2021;131(24):e154886. https://doi.org/10.1172/JCI154886.
Contributor Information
Jason S. Knight, Email: jsknight@umich.edu.
Roberto Caricchio, Email: roc@temple.edu.
Alexis J. Combes, Email: alexis.combes@ucsf.edu.
Betty Diamond, Email: bdiamond@nshs.edu.
Sharon E. Fox, Email: sfox3@lsuhsc.edu.
David A. Hanauer, Email: hanauer@med.umich.edu.
Judith A. James, Email: Judith-James@omrf.org.
Yogendra Kanthi, Email: yogen.kanthi@nih.gov.
Virginia Ladd, Email: vladd@aarda.org.
Puja Mehta, Email: puja.mehta@ucl.ac.uk.
Aaron M. Ring, Email: aaron.ring@yale.edu.
Ignacio Sanz, Email: ignacio.sanz@emory.edu.
Carlo Selmi, Email: carlo.selmi@humanitas.it.
Russell P. Tracy, Email: russell.tracy@uvm.edu.
Paul J. Utz, Email: pjutz@stanford.edu.
Catriona A. Wagner, Email: catriona@wagnerscimed.com.
Julia Y. Wang, Email: juliawang13@gmail.com.
References
- 1.Fox SE, et al. Pulmonary and cardiac pathology in African American patients with COVID-19: an autopsy series from New Orleans. Lancet Respir Med. 2020;8(7):681–686. doi: 10.1016/S2213-2600(20)30243-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fox SE, et al. Unexpected features of cardiac pathology in COVID-19 infection. Circulation. 2020;142(11):1123–1125. doi: 10.1161/CIRCULATIONAHA.120.049465. [DOI] [PubMed] [Google Scholar]
- 3.Fox SE, et al. Cardiac endotheliitis and multisystem inflammatory syndrome after COVID-19. Ann Intern Med. 2020;173(12):1025–1027. doi: 10.7326/L20-0882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fox SE, et al. COVID-19 myocarditis: quantitative analysis of the inflammatory infiltrate and a proposed mechanism. Cardiovasc Pathol. 2021;54:107361. doi: 10.1016/j.carpath.2021.107361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Magro C, et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl Res. 2020;220:1–13. doi: 10.1016/j.trsl.2020.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ma L, et al. Increased complement activation is a distinctive feature of severe SARS-CoV-2 infection. Sci Immunol. 2021;6(59):eabh2259. doi: 10.1126/sciimmunol.abh2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang C, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ruan Q, et al. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. 2020;46(5):846–848. doi: 10.1007/s00134-020-05991-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Henderson LA, et al. On the alert for cytokine storm: immunopathology in COVID-19. Arthritis Rheumatol. 2020;72(7):1059–1063. doi: 10.1002/art.41285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.RECOVERY Collaborative Group. et al. Dexamethasone in hospitalized patients with Covid-19. N Engl J Med. 2020;384(8):693–704. doi: 10.1056/NEJMoa2021436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abani O, et al. Tocilizumab in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial. Lancet. 2021;397(10285):1637–1645. doi: 10.1016/S0140-6736(21)00676-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cavalli G, et al. Interleukin-1 blockade with high-dose anakinra in patients with COVID-19, acute respiratory distress syndrome, and hyperinflammation: a retrospective cohort study. Lancet Rheumatol. 2020;2(6):e325–e331. doi: 10.1016/S2665-9913(20)30127-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mehta P, et al. Granulocyte-macrophage colony stimulating factor in COVID-19: friend or foe? Lancet Rheumatol. 2021;3(6):e394–e395. doi: 10.1016/S2665-9913(21)00078-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mehta P, et al. JAK inhibitors in COVID-19: the need for vigilance regarding increased inherent thrombotic risk. Eur Respir J. 2020;56(3):2001919. doi: 10.1183/13993003.01919-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu C, et al. A decade of jakinibs: what have we learned and what may be the future. Arthritis Rheumatol. doi: 10.1002/art.41906. [published online June 27, 2021]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mehta P, et al. Therapeutic blockade of granulocyte macrophage colony-stimulating factor in COVID-19-associated hyperinflammation: challenges and opportunities. Lancet Respir Med. 2020;8(8):822–830. doi: 10.1016/S2213-2600(20)30267-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morens DM, Fauci AS. Emerging pandemic diseases: how we got to COVID-19. Cell. 2020;182(5):1077–1092. doi: 10.1016/j.cell.2020.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Q, et al. Life-threatening COVID-19: defective interferons unleash excessive inflammation. Med (N Y) 2020;1(1):14–20. doi: 10.1016/j.medj.2020.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Casanova JL, et al. A global effort to define the human genetics of protective immunity to SARS-CoV-2 infection. Cell. 2020;181(6):1194–1199. doi: 10.1016/j.cell.2020.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blanco-Melo D, et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. 2020;181(5):1036–1045. doi: 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hadjadj J, et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science. 2020;369(6504):718–724. doi: 10.1126/science.abc6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trouillet-Assant S, et al. Type I IFN immunoprofiling in COVID-19 patients. J Allergy Clin Immunol. 2020;146(1):206–208. doi: 10.1016/j.jaci.2020.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou Z, et al. Heightened innate immune responses in the respiratory tract of COVID-19 patients. Cell Host Microbe. 2020;27(6):883–890. doi: 10.1016/j.chom.2020.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilk AJ, et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat Med. 2020;26(7):1070–1076. doi: 10.1038/s41591-020-0944-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee JS, et al. Immunophenotyping of COVID-19 and influenza highlights the role of type I interferons in development of severe COVID-19. Sci Immunol. 2020;5(49):eabd1554. doi: 10.1126/sciimmunol.abd1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lucas C, et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature. 2020;584(7821):463–469. doi: 10.1038/s41586-020-2588-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ciancanelli MJ, et al. Infectious disease. Life-threatening influenza and impaired interferon amplification in human IRF7 deficiency. Science. 2015;348(6233):448–453. doi: 10.1126/science.aaa1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Q, et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science. 2020;370(6515):eabd4570. doi: 10.1126/science.abd4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bastard P, et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science. 2020;370(6515):eabd4585. doi: 10.1126/science.abd4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bastard P, et al. Interferon-β therapy in a patient with incontinentia pigmenti and autoantibodies against type I IFNs infected with SARS-CoV-2. J Clin Immunol. 2021;41(5):931–933. doi: 10.1007/s10875-021-01023-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vallbracht A, et al. Interferon-neutralizing antibodies in a patient treated with human fibroblast interferon. Nature. 1981;289(5797):496–497. doi: 10.1038/289496a0. [DOI] [PubMed] [Google Scholar]
- 32.Panem S, et al. Antibodies to alpha-interferon in a patient with systemic lupus erythematosus. J Immunol. 1982;129(1):1–3. [PubMed] [Google Scholar]
- 33.Meager A, et al. Anti-cytokine autoantibodies in autoimmunity: preponderance of neutralizing autoantibodies against interferon-alpha, interferon-omega and interleukin-12 in patients with thymoma and/or myasthenia gravis. Clin Exp Immunol. 2003;132(1):128–136. doi: 10.1046/j.1365-2249.2003.02113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meager A, et al. Anti-interferon autoantibodies in autoimmune polyendocrinopathy syndrome type 1. PLoS Med. 2006;3(7):e289. doi: 10.1371/journal.pmed.0030289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meyer S, et al. AIRE-deficient patients harbor unique high-affinity disease-ameliorating autoantibodies. Cell. 2016;166(3):582–595. doi: 10.1016/j.cell.2016.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bruserud Ø, et al. A longitudinal follow-up of autoimmune polyendocrine syndrome type 1. J Clin Endocrinol Metab. 2016;101(8):2975–2983. doi: 10.1210/jc.2016-1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bastard P, et al. Preexisting autoantibodies to type I IFNs underlie critical COVID-19 pneumonia in patients with APS-1. J Exp Med. 2021;218(7):e20210554. doi: 10.1084/jem.20210554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bastard P, et al. Auto-antibodies to type I IFNs can underlie adverse reactions to yellow fever live attenuated vaccine. J Exp Med. 2021;218(4):e20202486. doi: 10.1084/jem.20202486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.de Prost N, et al. Plasma exchange to rescue patients with autoantibodies against type I interferons and life-threatening COVID-19 pneumonia. J Clin Immunol. 2021;41(3):536–544. doi: 10.1007/s10875-021-00994-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koning R, et al. Autoantibodies against type I interferons are associated with multi-organ failure in COVID-19 patients. Intensive Care Med. 2021;47(6):704–706. doi: 10.1007/s00134-021-06392-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Troya J, et al. Neutralizing autoantibodies to type I IFNs in >10% of patients with severe COVID-19 pneumonia hospitalized in Madrid, Spain. J Clin Immunol. 2021;41(5):914–922. doi: 10.1007/s10875-021-01036-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang EY, et al. Diverse functional autoantibodies in patients with COVID-19. Nature. 2021;595(7866):283–288. doi: 10.1038/s41586-021-03631-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abers MS, et al. Neutralizing type-I interferon autoantibodies are associated with delayed viral clearance and intensive care unit admission in patients with COVID-19. Immunol Cell Biol. 2021;99(9):917–921. doi: 10.1111/imcb.12495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goncalves D, et al. Antibodies against type I interferon: detection and association with severe clinical outcome in COVID-19 patients. Clin Transl Immunology. 2021;10(8):e1327. doi: 10.1002/cti2.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Solanich X, et al. Pre-existing autoantibodies neutralizing high concentrations of type I interferons in almost 10% of COVID-19 patients admitted to intensive care in barcelona. J Clin Immunol. doi: 10.1007/s10875-021-01136-x. [published online September 27, 2021]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lopez J, et al. Early nasal type I IFN immunity against SARS-CoV-2 is compromised in patients with autoantibodies against type I IFNs. J Exp Med. 2021;218(10):e20211211. doi: 10.1084/jem.20211211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van der Wijst MGP, et al. Type I interferon autoantibodies are associated with systemic immune alterations in patients with COVID-19. Sci Transl Med. 2021;13(612):eabh2624. doi: 10.1126/scitranslmed.abh2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ziegler CGK, et al. Impaired local intrinsic immunity to SARS-CoV-2 infection in severe COVID-19. Cell. 2021;184(18):4713–4733. doi: 10.1016/j.cell.2021.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bastard P, et al. Autoantibodies neutralizing type I IFNs are present in ~4% of uninfected individuals over 70 years old and account for ~20% of COVID-19 deaths. Sci Immunol. 2021;6(62):eabl4340. doi: 10.1126/sciimmunol.abl4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chauvineau-Grenier A, et al. Autoantibodies neutralizing type I interferons in 20% of COVID-19 deaths in a French hospital [preprint]. Posted on Research Square October 1, 2021. [DOI]
- 51.Asano T, et al. X-linked recessive TLR7 deficiency in ~1% of men under 60 years old with life-threatening COVID-19. Sci Immunol. 2021;6(62):eabl4348. doi: 10.1126/sciimmunol.abl4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Novelli L, et al. The JANUS of chronic inflammatory and autoimmune diseases onset during COVID-19 — a systematic review of the literature. J Autoimmun. 2020;117:102592. doi: 10.1016/j.jaut.2020.102592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rodríguez Y, et al. Autoinflammatory and autoimmune conditions at the crossroad of COVID-19. J Autoimmun. 2020;114:102506. doi: 10.1016/j.jaut.2020.102506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Giannini M, et al. Similarities between COVID-19 and anti-MDA5 syndrome: what can we learn for better care? Eur Respir J. 2020;56(3):2001618. doi: 10.1183/13993003.01618-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang Y, et al. Similarities and differences between severe COVID-19 pneumonia and anti-MDA-5-positive dermatomyositis-associated rapidly progressive interstitial lung diseases: a challenge for the future. Ann Rheum Dis. doi: 10.1136/annrheumdis-2020-218594. [published online August 5, 2020]. [DOI] [PubMed] [Google Scholar]
- 56.Stefano LD, et al. Transient monoarthritis and psoriatic skin lesions following COVID-19. Ann Rheum Dis. doi: 10.1136/annrheumdis-2020-218520. [published online August 4, 2020]. doi:10.1136/annrheumdis-2020-218520 . [DOI] [PubMed] [Google Scholar]
- 57.Liew IY, et al. A case of reactive arthritis secondary to Coronavirus Disease 2019 infection. J Clin Rheumatol. 2020;26(6):233–233. doi: 10.1097/RHU.0000000000001560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ono K, et al. Reactive arthritis after COVID-19 infection. RMD Open. 2020;6(2):e001350. doi: 10.1136/rmdopen-2020-001350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Novelli L, et al. A case of psoriatic arthritis triggered by SARS-CoV-2 infection. Rheumatology (Oxford) 2020;60(1):e21–e23. doi: 10.1093/rheumatology/keaa691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chee YJ, et al. Diabetic ketoacidosis precipitated by Covid-19 in a patient with newly diagnosed diabetes mellitus. Diabetes Res Clin Pract. 2020;164:108166. doi: 10.1016/j.diabres.2020.108166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Talarico R, et al. Symmetric peripheral polyarthritis developed during SARS-CoV-2 infection. Lancet Rheumatol. 2020;2(9):e518–e519. doi: 10.1016/S2665-9913(20)30216-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Saricaoglu EM, et al. The first reactive arthritis case associated with COVID-19. J Med Virol. 2021;93(1):192–193. doi: 10.1002/jmv.26296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yokogawa N, et al. Case of acute arthritis following SARS-CoV-2 infection. Ann Rheum Dis. 2021;80(6):e101. doi: 10.1136/annrheumdis-2020-218281. [DOI] [PubMed] [Google Scholar]
- 64.Alivernini S, et al. Comparative analysis of synovial inflammation after SARS-CoV-2 infection. Ann Rheum Dis. 2021;80(6):e91. doi: 10.1136/annrheumdis-2020-218315. [DOI] [PubMed] [Google Scholar]
- 65.Smatti MK, et al. Viruses and autoimmunity: a review on the potential interaction and molecular mechanisms. Viruses. 2019;11(8):762. doi: 10.3390/v11080762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jog NR, et al. Association of Epstein-Barr virus serological reactivation with transitioning to systemic lupus erythematosus in at-risk individuals. Ann Rheum Dis. 2019;78(9):1235–1241. doi: 10.1136/annrheumdis-2019-215361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen CJ, et al. High prevalence of immunoglobulin A antibody against Epstein-Barr virus capsid antigen in adult patients with lupus with disease flare: case control studies. J Rheumatol. 2005;32(1):44–47. [PubMed] [Google Scholar]
- 68.Kaufman KM, et al. Peptide mimics of a major lupus epitope of SmB/B′. Ann N Y Acad Sci. 2003;987(1):215–229. doi: 10.1111/j.1749-6632.2003.tb06051.x. [DOI] [PubMed] [Google Scholar]
- 69.McClain MT, et al. An altered immune response to Epstein-Barr nuclear antigen 1 in pediatric systemic lupus erythematosus. Arthritis Rheum. 2006;54(1):360–368. doi: 10.1002/art.21682. [DOI] [PubMed] [Google Scholar]
- 70.Poole BD, et al. Lupus-like autoantibody development in rabbits and mice after immunization with EBNA-1 fragments. J Autoimmun. 2008;31(4):362–371. doi: 10.1016/j.jaut.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zandman-Goddard G, et al. Exposure to Epstein-Barr virus infection is associated with mild systemic lupus erythematosus disease. Ann N Y Acad Sci. 2009;1173(1):658–663. doi: 10.1111/j.1749-6632.2009.04754.x. [DOI] [PubMed] [Google Scholar]
- 72.Esen BA, et al. Serologic response to Epstein-Barr virus antigens in patients with systemic lupus erythematosus: a controlled study. Rheumatol Int. 2012;32(1):79–83. doi: 10.1007/s00296-010-1573-4. [DOI] [PubMed] [Google Scholar]
- 73.Vista ES, et al. Strong viral associations with SLE among Filipinos. Lupus Sci Med. 2017;4(1):e000214. doi: 10.1136/lupus-2017-000214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Quan TE, et al. Epstein-Barr virus promotes interferon-alpha production by plasmacytoid dendritic cells. Arthritis Rheum. 2010;62(6):1693–1701. doi: 10.1002/art.27408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Severa M, et al. EBV stimulates TLR- and autophagy-dependent pathways and impairs maturation in plasmacytoid dendritic cells: implications for viral immune escape. Eur J Immunol. 2013;43(1):147–158. doi: 10.1002/eji.201242552. [DOI] [PubMed] [Google Scholar]
- 76.Jog NR, James JA. Epstein Barr virus and autoimmune responses in systemic lupus erythematosus. Front Immunol. 2021;11:623944. doi: 10.3389/fimmu.2020.623944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chang SE, et al. New-onset IgG autoantibodies in hospitalized patients with COVID-19. Nat Commun. 2021;12(1):5417. doi: 10.1038/s41467-021-25509-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wang EY, et al. REAP: a platform to identify autoantibodies that target the human exoproteome [preprint]. Posted on bioRxiv February 12, 2021. [DOI]
- 79.Wang JY, et al. Dermatan sulfate interacts with dead cells and regulates CD5(+) B-cell fate: implications for a key role in autoimmunity. Am J Pathol. 2011;178(5):2168–2176. doi: 10.1016/j.ajpath.2011.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rho J, et al. Human proteins with affinity for dermatan sulfate have the propensity to become autoantigens. Am J Pathol. 2011;178(5):2177–2190. doi: 10.1016/j.ajpath.2011.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lee J, et al. Dermatan sulfate is a potential regulator of IgH via interactions with pre-BCR, GTF2I, and BiP ER complex in pre-B lymphoblasts. Front Immunol. 2021;12:680212. doi: 10.3389/fimmu.2021.680212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Wang JY, et al. An autoantigen atlas from human lung HFL1 cells offers clues to neurological and diverse autoimmune manifestations of COVID-19 [preprint]. Posted on bioRxiv January 24, 2021. [DOI] [PMC free article] [PubMed]
- 83.Wang JY, et al. An autoantigen profile of human A549 lung cells reveals viral and host etiologic molecular attributes of autoimmunity in COVID-19. J Autoimmun. 2021;120:102644. doi: 10.1016/j.jaut.2021.102644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Wang JY, et al. An autoantigen-ome from HS-Sultan B-lymphoblasts offers a molecular map for investigating autoimmune sequelae of COVID-19 [preprint]. https://dx.doi.org/10.1101%2F2021.04.05.438500 Posted on bioRxiv April 6, 2021.
- 85.Colling ME, Kanthi Y. COVID-19-associated coagulopathy: an exploration of mechanisms. Vasc Med. 2020;25(5):471–478. doi: 10.1177/1358863X20932640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wu C, et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern Med. 2020;180(7):934–943. doi: 10.1001/jamainternmed.2020.0994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yalavarthi S, et al. Release of neutrophil extracellular traps by neutrophils stimulated with antiphospholipid antibodies: a newly identified mechanism of thrombosis in the antiphospholipid syndrome. Arthritis Rheumatol. 2015;67(11):2990–3003. doi: 10.1002/art.39247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Meng H, et al. In vivo role of neutrophil extracellular traps in antiphospholipid antibody-mediated venous thrombosis. Arthritis Rheumatol. 2017;69(3):655–667. doi: 10.1002/art.39938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Thålin C, et al. Neutrophil extracellular traps. Arterioscler Thromb Vasc Biol. 2019;39(9):1724–1738. doi: 10.1161/ATVBAHA.119.312463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zuo Y, et al. Antiphospholipid syndrome: a clinical perspective. Chin Med J (Engl) 2020;133(8):929–940. doi: 10.1097/CM9.0000000000000705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zuo Y, et al. Neutrophil extracellular traps in COVID-19. JCI Insight. 2020;5(11):e138999. doi: 10.1172/jci.insight.138999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zuo Y, et al. Neutrophil extracellular traps and thrombosis in COVID-19. J Thromb Thrombolysis. 2021;51(2):446–453. doi: 10.1007/s11239-020-02324-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zuo Y, et al. Prothrombotic autoantibodies in serum from patients hospitalized with COVID-19. Sci Transl Med. 2020;12(570):eabd3876. doi: 10.1126/scitranslmed.abd3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Shi H, et al. Endothelial cell-activating antibodies in COVID-19 [preprint]. Posted on medRxiv July 9, 2021. [DOI]
- 95.Leffler J, et al. Degradation of neutrophil extracellular traps is decreased in patients with antiphospholipid syndrome. Clin Exp Rheumatol. 2013;32(1):66–70. [PubMed] [Google Scholar]
- 96.Zuo Y, et al. Anti-neutrophil extracellular trap antibodies and impaired neutrophil extracellular trap degradation in antiphospholipid syndrome. Arthritis Rheumatol. 2020;72(12):2130–2135. doi: 10.1002/art.41460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zuo Y, et al. Autoantibodies stabilize neutrophil extracellular traps in COVID-19. JCI Insight. 2021;6(15):e150111. doi: 10.1172/jci.insight.150111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Combes AJ, et al. Global absence and targeting of protective immune states in severe COVID-19. Nature. 2021;591(7848):124–130. doi: 10.1038/s41586-021-03234-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dhodapkar KM, et al. Selective blockade of the inhibitory Fcgamma receptor (FcgammaRIIB) in human dendritic cells and monocytes induces a type I interferon response program. J Exp Med. 2007;204(6):1359–1369. doi: 10.1084/jem.20062545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Woodruff MC, et al. Extrafollicular B cell responses correlate with neutralizing antibodies and morbidity in COVID-19. Nat Immunol. 2020;21(12):1506–1516. doi: 10.1038/s41590-020-00814-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tipton CM, et al. Diversity, cellular origin and autoreactivity of antibody-secreting cell population expansions in acute systemic lupus erythematosus. Nat Immunol. 2015;16(7):755–765. doi: 10.1038/ni.3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jenks SA, et al. Distinct effector B cells induced by unregulated toll-like receptor 7 contribute to pathogenic responses in systemic lupus erythematosus. Immunity. 2018;49(4):725–739. doi: 10.1016/j.immuni.2018.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kaneko N, et al. Loss of Bcl-6-expressing T follicular helper cells and germinal centers in COVID-19. Cell. 2020;183(1):143–157. doi: 10.1016/j.cell.2020.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tipton CM, et al. Understanding B-cell activation and autoantibody repertoire selection in systemic lupus erythematosus: a B-cell immunomics approach. Immunol Rev. 2018;284(1):120–131. doi: 10.1111/imr.12660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.van der Made CI, et al. Presence of genetic variants among young men with severe COVID-19. JAMA. 2020;324(7):663–673. doi: 10.1001/jama.2020.13719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jones SA, Hunter CA. Is IL-6 a key cytokine target for therapy in COVID-19? Nat Rev Immunol. 2021;21(6):337–339. doi: 10.1038/s41577-021-00553-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zumaquero E, et al. IFNγ induces epigenetic programming of human T-bethi B cells and promotes TLR7/8 and IL-21 induced differentiation. Elife. 2019;8:e41641. doi: 10.7554/eLife.41641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wucherpfennig KW. Mechanisms for the induction of autoimmunity by infectious agents. J Clin Invest. 2001;108(8):1097–1104. doi: 10.1172/JCI200114235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cheng MH, et al. Superantigenic character of an insert unique to SARS-CoV-2 spike supported by skewed TCR repertoire in patients with hyperinflammation. Proc Natl Acad Sci U S A. 2020;117(41):25254–25262. doi: 10.1073/pnas.2010722117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.McMurray JC, et al. Multisystem inflammatory syndrome in children (MIS-C), a post-viral myocarditis and systemic vasculitis—a critical review of its pathogenesis and treatment. Front Pediatr. 2020;8:626182. doi: 10.3389/fped.2020.626182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Porritt RA, et al. HLA class I-associated expansion of TRBV11-2 T cells in multisystem inflammatory syndrome in children. J Clin Invest. 2021;131(10):e146614. doi: 10.1172/JCI146614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Moreews M, et al. Polyclonal expansion of TCR Vb 21.3+ CD4+ and CD8+ T cells is a hallmark of multisystem inflammatory syndrome in children. Sci Immunol. 2021;6(59):eabh1516. doi: 10.1126/sciimmunol.abh1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Porritt RA, et al. The autoimmune signature of hyperinflammatory multisystem inflammatory syndrome in children. J Clin Invest. 2021;131(20):e151520. doi: 10.1172/JCI151520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Seeßle J, et al. Persistent symptoms in adult patients one year after COVID-19: a prospective cohort study. Clin Infect Dis. doi: 10.1093/cid/ciab611. [published online July 5, 2021]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Oelsner EC, et al. Collaborative Cohort of Cohorts for COVID-19 Research (C4R) study: study design [preprint]. https://www.medrxiv.org/content/10.1101/2021.03.19.21253986v1 Posted on medRxiv March 20, 2021. [DOI] [PMC free article] [PubMed]
- 116.Hanauer DA, et al. Exploring clinical associations using ‘-omics’ based enrichment analyses. PLoS One. 2009;4(4):e5203. doi: 10.1371/journal.pone.0005203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hanauer DA, Ramakrishnan N. Modeling temporal relationships in large scale clinical associations. J Am Med Inform Assoc. 2012;20(2):332–341. doi: 10.1136/amiajnl-2012-001117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hanauer DA, et al. Applying MetaMap to Medline for identifying novel associations in a large clinical dataset: a feasibility analysis. J Am Med Inform Assoc. 2014;21(5):925–937. doi: 10.1136/amiajnl-2014-002767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bourgeois FT, et al. International analysis of electronic health records of children and youth hospitalized with COVID-19 infection in 6 countries. JAMA Netw Open. 2021;4(6):e2112596. doi: 10.1001/jamanetworkopen.2021.12596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Weber GM, et al. International comparisons of harmonized laboratory value trajectories to predict severe COVID-19: leveraging the 4CE collaborative across 342 hospitals and 6 countries: a retrospective cohort study [preprint]. Posted on medRxiv February 5, 2021. [DOI]
- 121.Bennett TD, et al. Clinical characterization and prediction of clinical severity of SARS-CoV-2 infection among US adults using data from the US National COVID Cohort Collaborative. JAMA Netw Open. 2021;4(7):e2116901. doi: 10.1001/jamanetworkopen.2021.16901. [DOI] [PMC free article] [PubMed] [Google Scholar]