

Abstract
Drug repurposing aims to find new uses for already existing and approved drugs. Normally this has occurred through serendipity while high throughput screening of FDA approved drug libraries has been responsible for increasing its popularity. Computational repurposing is a newer credible approach to drug discovery that has been steadily building a foundation with applications and examples of experimental validation. One particular area of computational repurposing is using machine learning. We now provide a brief overview of recent developments in drug repurposing using machine learning alongside other computational approaches for comparison. In addition, we will highlight several applications of computational drug repurposing using machine learning for cancer indications targeting kinase inhibitors, Alzheimer’s disease as well as COVID-19 from many different groups as well as our own. This overview should serve to rebalance the ‘repurposing hype’ of the past year, set expectations of what is possible as well as suggest areas for future development.
Graphical Abstract
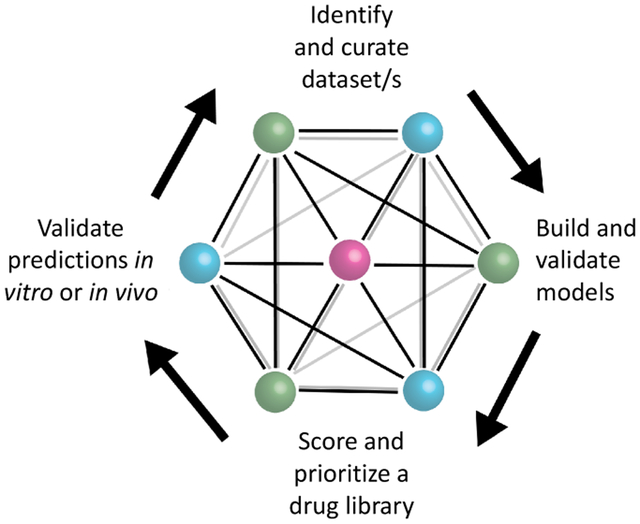
Introduction
Bringing a new small molecule drug to the clinic is a lengthy and costly process that has been widely described [1] taking anywhere from over a decade and may range from over $700M to $2.7Bn by recent estimates [2]. Naturally, any advancements in technology which could accelerate the process or decrease the cost significantly would be a true paradigm shift. Drug repurposing is a simple concept with a long history which promises a great deal in this respect [3]. Drug repurposing aims to develop a new use for a drug beyond its original purpose and has been applied more widely as FDA approved drug libraries became more commercially accessible, allowing academics and small companies to run medium to high throughput screens on thousands of molecules [4]. In several cases, new uses for existing drugs were found and some candidates have progressed to the clinic [5–7]. Drug companies also started to pay more attention to repositioning compounds that perhaps were either on the shelf or failed for another indication. Interestingly, this view of repurposing molecules is not new, a bibliometric analysis of PubMed showed that, since at least the 1940s, many molecules have been tested against a large number of diseases. For example 189 drugs were linked with over 300 diseases [3]. More recently one approach that has been swiftly gaining traction over the past 5–10 years is computational drug repurposing. This method became especially relevant in 2020 as scientists attempted to find a quick solution for treating COVID-19, and drug repurposing offered a quick path to treatment [8]. Several computational tools, such as similarity searching, docking, pharmacophores, and machine learning can be used for small molecule drug discovery. We will focus primarily on the machine learning in this review which can encompass a wide array of data resources and computational approaches (Figure 1). In our view this requires several steps such as identification and curation of dataset/s, building and validation (internal and preferably external) of the machine learning models, scoring and prioritization of a drug library (or clinical assets), before final validation of these predictions in vitro and / or in vivo (see graphical abstract). Previous reviews by us or others have described some of the earlier computational repurposing successes, data sources available to enable this [9,10] as well as some of the opportunities and limitations of repurposing industry provided clinical stage molecules [11]. As will be seen, machine learning applications can still be quite broad in how they are implemented whether with small molecule, genomics, image assay data or combinations of these or other data types (Figure 1). We will also describe several areas where there have been significant repurposing efforts, beginning with COVID-19, kinases for cancer and Alzheimer’s disease (AD) as well as for other diseases.
Figure 1.
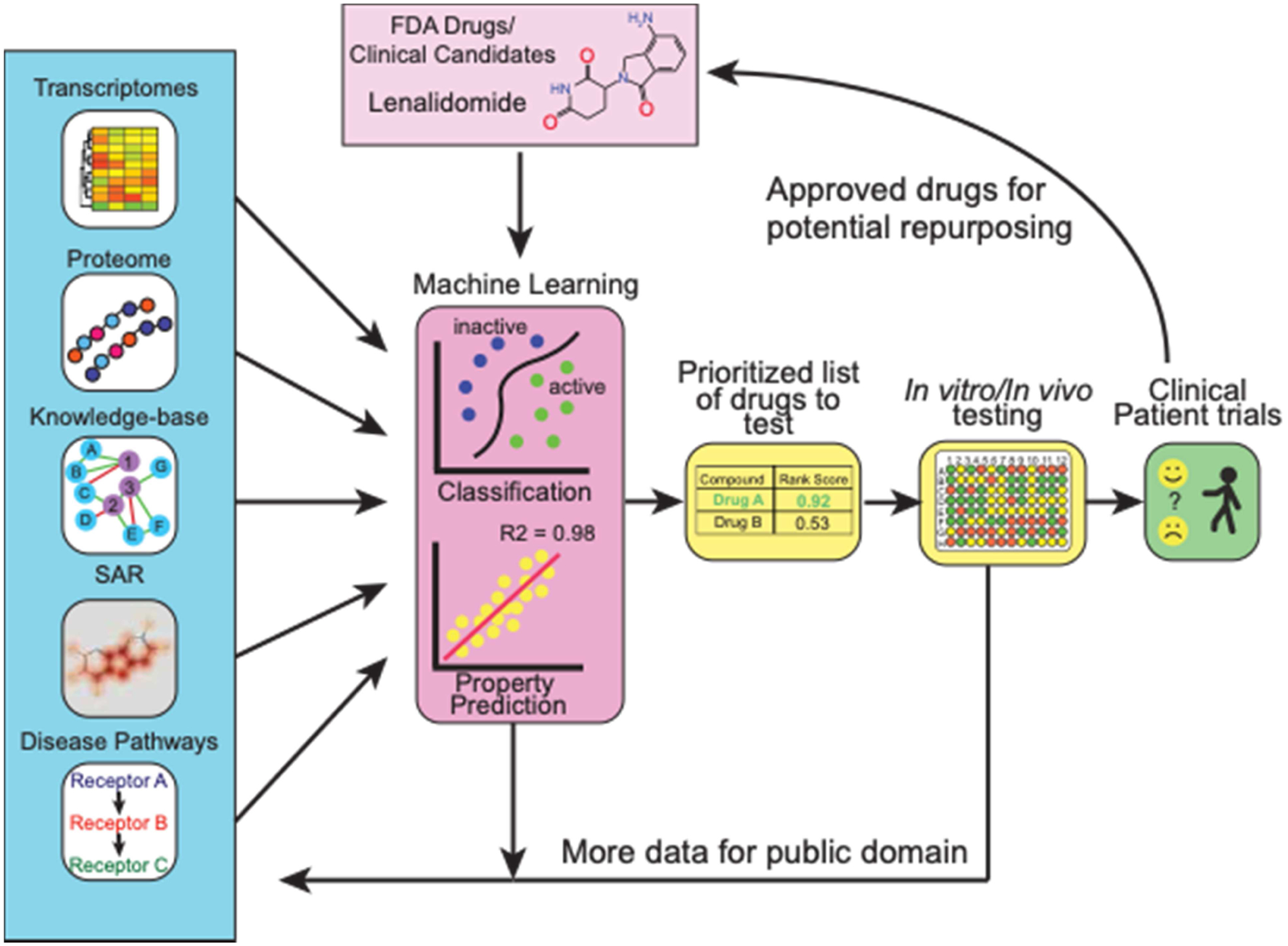
Overview of computational machine learning approaches for drug repurposing.
1. COVID-19 repurposing
Since the outbreak of SARS-CoV-2 in late 2019 there have been hundreds of papers and likely many more preprints describing computational approaches or short communications proposing compounds with likely activity against SARS-CoV-2 [8]. 2020 brought drug repurposing a level of recognition that was unimaginable before the COVID-19 epidemic. As researchers were generally isolated from their labs, this led to many using computational tools to propose compounds for possible testing against the virus. To see thousands of researchers turning their attention to the COVID-19 problem and the flood of papers that followed, the expectation is that a drug will surely follow. In the case of treatments for COVID-19, however, while there have been a few small successes, and many clinical trials ongoing, a drug candidate has yet to emerge from all this computational repurposing. A number of causes likely contributed to the lack of success stories. While researchers used computational tools to propose compounds for testing against the virus, the lack of BSL3 lab availability (during 2020) often meant little to no experimental data existed to back up their proposals other than some computational score or a docked molecule in a protein (and sometimes both). This has perhaps also sown more confusion as the general public is unable to discern the difference between a computational prediction and one that is experimentally verified. In very few of these computational derived examples, compounds were tested in vitro against a target protein or in virus infected whole cells. There were also many examples of studies in which a small number of drugs or other molecules were selected for various reasons (not computationally) and then tested [12], and these have tended to outnumber the computational repurposing papers. While not all of the following approaches use machine learning, several other computational approaches will also be described.
One of the earliest molecules to be computationally repurposed using a knowledge graph approach was the AAK1 and JAK1/2 kinase inhibitor baricitinib [13] (Table 1). The mechanism was eventually validated in vitro and in human patients [14,15] as well as in combination with hydroxychloroquine [16]. As the outbreak progressed there were also examples of high throughput screens against SARS-CoV-2 targets or in infected cells as well [17–22]. A cheminformatics and protein interaction map identified FDA and clinical stage compounds binding to sigma-1 and 2 receptors which act as host factors for COVID-19. The most potent compound identified in Vero cells was PB28 (IC90 280nM), which is not an approved drug, while several approved drugs such as clemastine and cloperastine were also identified (Table 1), albeit with much weaker μM activity [23]. To some extent the use and efficacy of atypical antipsychotics (likely targeting sigma receptors) was also shown using real world data analysis of hospitalized SARS-CoV-2 patients [24]. For all the clinical failures associated with hydroxychloroquine [25,26] it has still served as a starting point for computational repurposing. In one example it was used as a template for ligand based virtual screening of 3981 molecules that had obtained regulatory approval worldwide. Amodiaquine (antimalarial) was the most active in vitro in Vero EG cells along with zuclopenthixol (typical antipsychotic) and nebivolol (beta blocker) [27]. Docking FDA drugs in the Mpro structure has identified dipyridamole (IC50 0.53μM) and this showed activity in Vero cells (EC50~0.1 μM), subsequently it was taken to a small clinical trial with 8 patients and demonstrated positive responses [28]. There have been many other examples of computational docking in a target followed by assessment of binding to the target or assay of activity, but in most cases the activity in infected cells is not described [29,30]. Another approach has been to repurpose molecules that were previously computationally identified using machine learning for another virus then applying them to SARS-CoV-2. This is an approach we have taken and in due course demonstrated that the Ebola inhibitors tilorone, quinacrine and pyronaridine may also be sub μM inhibitors of SARS-CoV-2 infected A549-ACE2 cells [12]. As yet there has not been a “home-run” that has derived from the repurposing approach for SARS-CoV-2 although it has intensified the interest in kinase inhibitors as potential antivirals.
Table 1.
Examples of computationally derived molecules from drug repurposing for SARS-CoV-2.
| Structure | Drug | Method | In vitro/ in vivo activity | Reference |
|---|---|---|---|---|
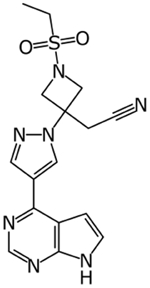
|
Baricitinib | knowledge graph | Significantly reduced viral load in human liver spheroids at 400 and 800 nM | [13–16] |
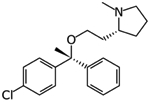
|
Clemastine | Protein interaction map | pIC50 5.67 | [23] |
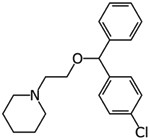
|
Cloperastine | Protein interaction map | pIC50 5.27 | [23] |
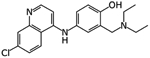
|
Amodiaquine | Ligand-based virtual screen | EC50 0.13 μM and 5.4 μM | [27] |
|
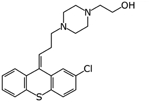
Zuclopenthixol | Ligand-based virtual screen | 1.35 μM and 15 μM | [27] |
|
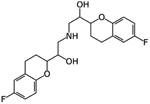
Nebivolol | Ligand-based virtual screen | 2.72 μM and 2.8 μM | [27] |
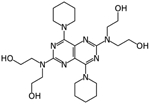
|
Dipyridamole | Docking in Mpro | IC50 0.53 μM, EC50~0.1 μM | [28] |
2. Kinase repurposing and cancer applications
The human genome encodes about 634 kinases (pseudokinases included), which are enzymes that transfer a polar phosphate group from ATP to regulatory serine, threonine, or tyrosine residues on proteins and have a role in hundreds of diseases [31,32]. To date the FDA has approved 62 small molecule protein kinase inhibitors which have a market in the tens of billions of dollars per year [31,32]. Most of these are delivered orally apart from temsirolimus (IV) and netarsudil (eye drops). As of 2019, eleven of them inhibited protein-serine/threonine protein kinases, two are dual specificity protein kinases, eleven target non-receptor protein-tyrosine kinases, and 28 block receptor protein-tyrosine kinases. 46 are used in the treatment cancers (e.g leukemias, breast and lung cancers) whereas 8 are for non-malignancies such as myelofibrosis, polycythemia vera, chronic immune thrombocytopenia, rheumatoid arthritis, renal graft vs. host disease, idiopathic pulmonary fibrosis, glaucoma, Crohn disease, and ulcerative colitis [31,32]. There are many hundreds of kinases, so this creates challenges to ensure selectivity depending on the target or the degree of polypharmacology that is permissible. The wealth of publicly available data on this target class also represents an opportunity to leverage to design and develop new classes of compounds as well as repurposing existing kinase inhibitors for other kinases not tested. The market for anticancer drugs is considerable [33]. Commercial interest in kinase inhibitors is at an all-time high in recent years with Merck acquiring Arqule for $2.7Bn for a single clinical stage candidate (ARQ531 a BTK inhibitor) and acquiring Peloton Therapeutics for $2.2Bn. Pfizer bought Array Biopharma for $10.6Bn for their pipeline which included two marketed kinase inhibitors, and in early 2019 Eli Lilly bought Loxo Oncology for $8Bn which has several kinase inhibitors in their pipeline. There still appears a significant commercial opportunity in this target area.
Kinases are therefore a very heavily studied class of proteins in cancer and yet a large part of the kinome is “dark” with no drugs or probes [34]. There is hence considerable interest in accelerating the identification of molecules that might be probes or potential drugs for proteins belonging to the protein kinase families that are currently unannotated. There have been notable efforts to create public structure activity (SAR) datasets such as the Published Kinase Inhibitor Set (PKIS) as a resource for developing new probes for kinases [35]. The illuminating the druggable genome (IDG) program began in 2014 with the goal of developing techniques and information about proteins targets for this protein class as well as G-protein coupled receptor, ion channels and nuclear receptor classes of proteins which had not been characterized or for which molecules or biologics had not been identified [36–38]. The number of proteins that are listed on the IDG website as of February 2021 is 328 (162 are kinases [39,40]). Currently there are only 18 previously dark kinases for which chemical probes have been identified. In parallel with this effort, another NIH common fund project called the library of integrated network -based cellular signatures (LINCS) consortium which has generated public data which relates to how cells respond to various genetic or environmental stressors with results obtained in cultured and primary cells whose state has been perturbed by a “perturbagen” [41]. This data collected by 15 institutions has been made available to the community through many different tools and workflows [41] which enables various different types of evaluations [42].
For example, one of the datasets and tools in the LINCS is the connectivity map (CMap) which brings together data on genes, thousands of drugs and disease states. The original basis for the CMap was 164 drugs and tool compounds tested in 3 cell lines with microarray data that was subsequently used in several repurposing projects to identify new uses for old drugs [43–45]. The approach was recently expanded with a reduced expression profile limited to 1000 landmark genes and tested with ~19,000 small molecules (as well as thousands of shRNAs, cDNAs, and biologics) in 6 cancer cell lines. A connectivity score was developed with three confidence measures [46]. Such a tool provides the capability to predict the mechanism of action of molecules based on similarity profiles of compounds of known function. 1902 of these compounds were mapped to known protein targets. When using an external set of compounds, 76% could be connected in the correct perturbagen class. Some drugs were also found to associate to multiple targets for which they were similarly active. CMap was used to discover the mechanism of action for new small molecules and this was demonstrated for kinase inhibition with several drugs. Working in the opposite direction, to discover a molecule specific for another protein kinase was also demonstrated by querying the database for signatures of what knockdown of the target would look like and then find molecules sharing a similar signature [46]. Such studies have inspired others to use compound-induced transcriptomic data generated from cell lines in order to predict compound activity to molecular targets. Random forest models generated with this data for a small subset of 69 genes had similar results to or outperformed Morgan fingerprints [47]. The same group has also demonstrated how the transcriptomic data can also be used with a generative adversarial network to design molecules that address desired targets (although it should be mentioned that these two studies were theoretical and lacked experimental verification using molecule synthesis and testing) [48].
It can be summarized that de-orphaning kinases is a very slow process involving biochemical, enzymatic and cellular profiling often followed by assessment of phosphorylation, cellular target engagement and structural analyses which may occasionally result in a single molecule illuminating more than one of the dark kinases [40]. There are now many data sources relevant to kinases that could be integrated to develop computational approaches for assisting in de-orphaning dark kinases that have not been attempted or validated. In most cases the computational tools leverage molecule similarity analysis. For example, Virtual Kinome profiler was used to profile ~37 million compound-kinase pairs and predict 151,708 compounds against 248 kinases simultaneously [49]. This approach identified 19 small-molecule inhibitors (including already approved drugs) of EGFR, HCK, FLT1, and MSK1 protein kinases [49]. Earlier work by the same group identified 4 new off targets (FRK, FYN A, ABL1, SLK) for tivozanib, an approved VEGF tyrosine kinase inhibitor approved to treat renal cell carcinoma [50]. Others have focused on developing the optimal kinase library for screening which made use of molecule similarity to existing small molecule kinase inhibitors [34]. Some researchers have used machine learning to map the activity of compounds across the kinome using the t-distributed stochastic neighbor embedding (t-SNE) algorithm. Validation of this model focused on the FLT3 kinase [51]. Some of the largest kinome-wide screening efforts have combined public and proprietary data and modeled 280 kinases, with most random forest models having AUCs > 0.7. The models were evaluated with leave out tests but not prospective testing [52]. Other groups have narrowed their efforts to modeling a single kinase, such as a model for JAK2 inhibitors which was used to select 13 compounds, out of which 6 had IC50 values less than 100nM [53]. Different algorithms have been used including deep neural networks (DNN) [54], Bayesian and Recursive partitioning models [55], other quantitative structure activity relationship (QSAR) methods [56] or deep generative models [57]. All of these computational tools could be potentially of use for drug repurposing. For example, a multitask deep learning model built with data for 391 kinases was used to find targets for clinically approved and investigational kinase drugs for which the researchers validated their predictions [58].
Most groups working on computational predictions for kinases have focused on identifying novel molecules. Some have also taken different approaches combining different data types or using whole cell or genomic data. An early study demonstrated how machine learning could be used to predict cancer cell sensitivity using genomic and chemical properties as well as proposed the potential for repurposing but without providing any examples of this [59]. A more recent version of this (although not focused on kinases) used deep learning with transcriptomic and molecular data in order to identify pimozide for repurposing for non-small cell cancer as well as tested in vitro (IC50 19.54 μM in A549 cells) which while weak, was at a comparable activity level to gemcitabine [60]. One group developed a platform called 3D-REMAP to use ligand binding site information and protein-ligand docking to propose a cardiovascular drug levosimendan as a kinase inhibitor (RIOK1, Kd 0.82 μM as well as inhibited FLT3, MAP2K5, PIP5K1A, GAK and KIT) and potential repurposing candidate [61]. We have recently generated Bayesian machine learning models for a rare cancer called chordoma using datasets from two independent cell-based screens. These were then used to prioritize a small number of clinical stage kinase inhibitors and demonstrated that one of these AZD2014 (mTOR1 and mTOR2) possessed comparable sub μM in vitro activity as the approved drug afatinib (EGFR, currently in a clinical trial for this disease) in two chordoma cell lines [62].
Machine learning approaches can also be used with multiple omics datasets to predict a therapeutics response signature which can be used to optimize patient treatment and this application was then validated with a wide array of kinase inhibitors, both approved and clinical candidates [63]. There is therefore considerable scope for using these types of computational machine learning approaches for repurposing of pre-existing kinase inhibitors or clinical stage compounds for cancer and potentially other disease indications.
3. Alzheimer’s disease repurposing
AD is widely known as the most common type of dementia, it is an irreversible neurodegenerative disease that is also the 6th leading cause of death in USA, affects over 5.1 million adults over age 65, and costs over $150 billion per year [64]. AD is a typical age-dependent neurodegenerative disease that affects 5% of individuals >65 years, 20% of those >85 years, and more than one- third of those >90 years [65]. However, approximately 200,000 individuals under age 65 have younger-onset AD, though there is greater uncertainty about the younger-onset estimate. Therefore, in the absence of proper preventative and therapeutic efforts, its prevalence will continue to increase as life expectancy increases. The mechanisms underlying the AD pathophysiology are still unclear. Aggregation of tau and amyloid beta (Aβ) proteins as well as decreased acetylcholine are hallmarks of the disease and the focus of many studies [66]. The only therapies currently on the market for treatment of AD, namely four cholinesterase inhibitors (tacrine, rivastigmine, donepezil and galantamine) and one N-methyl-D-aspartate (NMDA) receptor inhibitor (memantine), are only symptomatic and do not affect the underlying disease mechanisms or alter the disease course [64]. Therefore, drug repurposing may offer a potential avenue to identify additional clinical stage molecules.
A recent study described an approach using machine learning to quantify the pathology and AD severity as well as using neuronal cell lines treated with 80 kinase inhibitors to access the gene expression profile [67]. Some of the best scoring compounds included ruxolitinib (JAK1/2) and regorafenib (RET, VEGFRs) which were assessed for mechanism, however experimental validation against AD was not tested in vitro or in vivo [67]. Post-mortem examination of brains with Alzheimer’s disease shows increased levels of activated Glycogen Synthase Kinase 3β (GSK3β) [68,69]. GSK3β is a proline-directed serine/threonine kinase that is involved in several intracellular signaling cascades, and is involved in a host of cellular functions, including gene transcription, glucose regulation and apoptosis [70–72]. GSK3β figures prominently in Alzheimer’s disease as the main kinase responsible for the phosphorylation of the microtubule-stabilizing protein tau [73–75]. Extracellular Aβ deposits, are also connected to the activity of GSK3β because this activity is linked to the improper processing of the amyloid-precursor protein APP [76]. The β-site APP-cleaving enzyme 1 (BACE1) is responsible for the pathogenic β-cleavage of APP that results in aβ peptides, and GSK3β inhibition downregulates expression of the BACE1 gene reducing aβ production [77]. GSK3 inhibitors are of interest to the pharmaceutical industry for their potential use in various diseases, including diabetes, cancer, AD, Parkinson’s Disease, psychiatric diseases, and stem cell proliferation [78]. There are dozens of commercially available GSK3 inhibitors, of varying potency and selectivity, however most of these inhibitors are not suitable as therapeutics due to their toxicity [79]. Only a few GSK3β inhibitors have made it to clinical trials for AD and only two of these have been tested for efficacy in treating AD, and neither one has shown great effect in treating the disease [80,81]. We have curated and validated a Bayesian machine learning model (using extended connectivity fingerprints) for inhibition of GSK3β which has over 2300 molecules (from ChEMBL) and a 5-fold cross validation ROC > 0.90 [82]. This computational model was used to prioritize commercial compounds for testing. The clinical candidate ruboxistaurin was identified as an inhibitor of GSK3β and was validated using the ADP-Glo™ Kinase Assay with an IC50 of 39 nM. A secondary in vitro assay using the Z’LYTE™ Kinase Assay, demonstrated an IC50 of 155 nM [82]. The IC50 for Ruboxistaurin against GSK3β was higher than for the initial target PKCβ (6 nM) but lower than the closely related kinase PKCα (360 nM). Ruboxistaurin has been independently patented as an inhibitor of GSK3β for the treatment of neurological or psychiatric disorders with (US 9,265,764 B2). To date we have curated and validated Bayesian machine learning models for 10 AD targets (Figure 2). In general, the 5-fold cross validation statistics are ROC > 0.8 and MCC > 0.5. There are definitely limitations as some datasets are small, but those with thousands of molecules look ideal. These computational models could then be used to find novel modulators of these proteins which could be used alone or in combination to treat AD. This type of approach could also be taken for additional AD targets to develop a suite of machine learning models that will represent structure-activity data from public sources.
Figure 2.
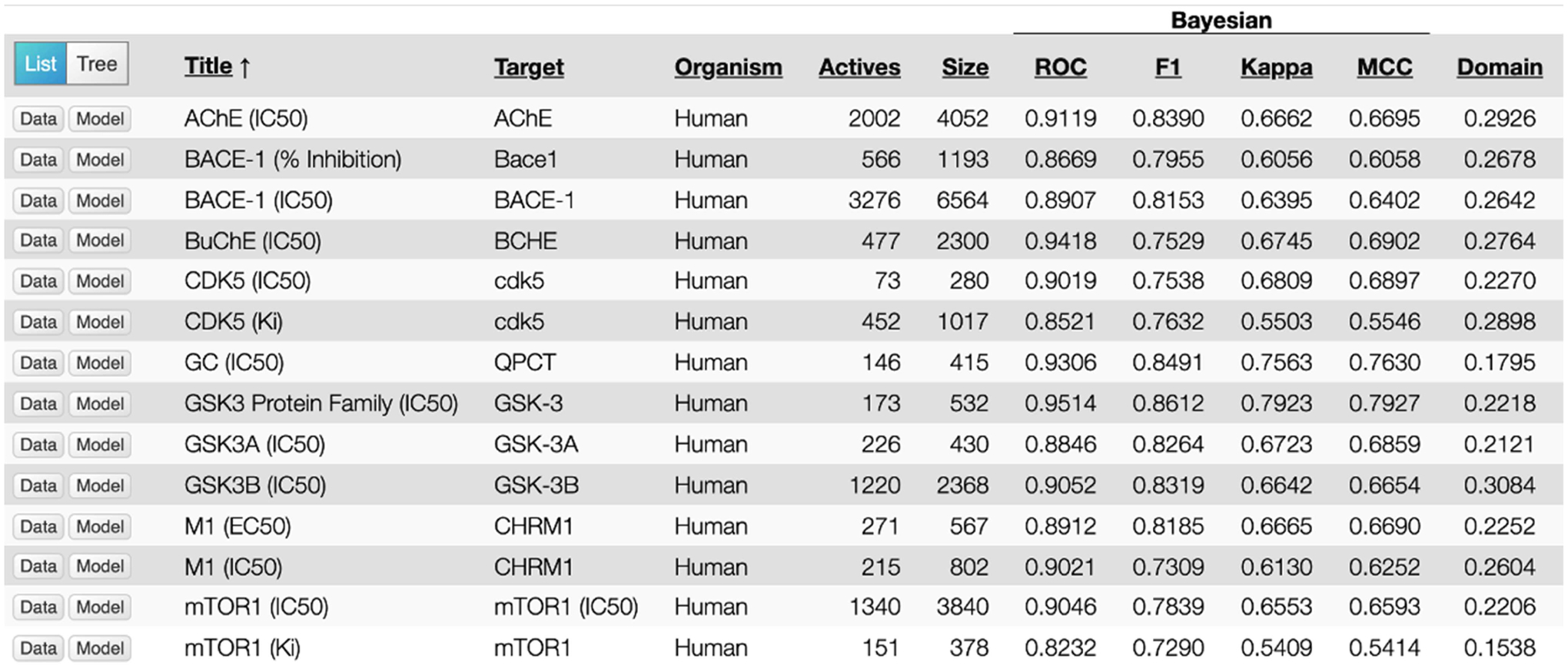
Alzheimer’s disease Bayesian machine learning models generated with ECFP6 descriptors and 5-fold cross validation statistics [82].
4. Antibacterial machine learning
As has been documented many times, antibiotic drug discovery has been profoundly difficult with very low hit rates from high throughput screening. There is also an urgent need for new drugs due to the occurrence of drug resistance. The lack of market support has also made it commercially treacherous for any companies working in this area. Computational machine learning approaches have been widely applied for antibacterial drug discovery for over a decade. We and others [83] have published extensively using the naïve Bayesian approach (with functional connectivity fingerprints descriptors) for tuberculosis drug discovery [84,85]. These models based on whole cell screening data from Mycobacterium tuberculosis were used to score (repurpose) molecules from the GSK antimalarial dataset [86] and from the seven compounds that were selected and tested, five had MIC ≤ 2 μg/mL, the most active being 0.0625 μg/mL [87]. Further examples include using two different whole cell models to score three vendor libraries and then 124 actives were found from testing 550 compounds [88], while after filtering a library of >150,000 molecules, 11 out of 48 compounds were active [89]. Overall hit rates were 15–71% for suggested compounds, much higher than the 0.6 – 1.5% from random library HTS [87–89]. We have also developed machine learning models with 18,886 molecules (with activity cut offs of 10 μM, 1 μM and 100 nM)[90] and used them to evaluate multiple machine learning methods (including deep learning and support vector machines). Bayesian machine learning models were on a par with Deep Neural Networks with external test sets [90]. These models could be used to virtually screen FDA approved drugs, although actual high throughput screens were carried out over a decade ago, since then hundreds of new non-antibacterial drugs have been approved, but it is likely many of them have never been tested against Mycobacterium tuberculosis, and this represents a future repurposing opportunity.
A recent deep neural network trained on E. coli data for 2335 molecules was used to score > 107 million molecules and one of these, halicin (a repurposed c-Jun kinase inhibitor SU3327) had an MIC of 2 μg/ml [91]. This molecule also showed broad spectrum bactericidal activity as well as activity in a mouse model of Acinetobacter baumannii infection. After screening other large compound collections, 2 further molecules with broad spectrum antibacterial activity were identified [91]. An example of a much larger library of compounds used for machine learning was 74,567 molecules tested against E. coli in order to generate models with gradient boosting, random forests, feed forward neural networks, support vector machines and Sammon mapping (average classification accuracy for internal testing 77.5–83.2%) [92]. These computational models were used to score a library of 5000 compounds that was also tested in vitro. It was not described how each method performed with this external test set, although actives that were correctly predicted were shown, of which several had MIC < 1 μg/ml [92]. While this approach was applied to identify novel antibacterials it could also be applied for repurposing, although this was not apparently attempted. These efforts further suggested how ligand-based machine learning approaches can search through the vast chemical space of molecules to prioritize small numbers of compounds to test and provide a higher hit rate than random screening.
5. Applying machine learning for drug repurposing to other diseases and applications
High throughput image-based assays used to generate ‘fingerprints’ have been used with deep neural networks on a glucocorticoid receptor translocation dataset of over half a million compounds which was then used to predict compounds against a kinase target (50-fold enrichment) and a CNS target (289-fold enrichment) [93]. Rather than a drug repurposing application per se, this is more of a data repurposing demonstration. However, this approach could clearly be used for drug repurposing projects. The therapeutic performance mapping system was used with rat proteomic data to identify repurposed molecule combinations for nerve root avulsion [94]. This systems proteomics approach combines network maps, and mathematical models and was used to select compounds for in vitro testing and the combination of acamprosate and ribavirin was found to be neuroprotective when tested in vivo as it accelerated nerve regeneration and recovery [94]. A Bayesian machine learning approach used transcriptional data and in vitro screening data to identify synergistic drug combinations for use in malaria by developing compound pathway signatures [95]. 35 compound pairs were selected and tested (precision 83.5% and recall 65.1%) demonstrating hydroxyzine and tacrolimus as well as raloxifene and thioridazine had high levels of synergy [95]. As a final example, computational target prediction approach (cheminformatic similarity ensemble approach) was used to predict the activity of pharmaceutical excipients against 3117 medically relevant proteins. Those target excipient pairs with high probability were prioritized and 69 were tested in vitro. 19 were active against one of 12 targets (36% success rate) [96]. Thus, many of these molecules had relevant activities and in some cases the concentration of excipient would be at a level to have a physiological effect (e.g. cetylpyridinium chloride and the dopamine receptor 3) [96]. This could be considered an excipient repurposing application. These repurposing applications are literally scratching the surface of what may be possible with different datasets that are used to train machine learning models for various applications relevant to drug repurposing.
5. Discussion
Drug repurposing using machine learning has grown steadily over the past decade as we have started to see the application of deep neural networks and the revolution in computer power provided by graphics processing units (GPUs). This has in turn led to the growth in venture capital backed artificial intelligence drug discovery companies using purely computational approaches to discover drugs or repurpose existing drugs. As we have described herein there has been considerable use of computational approaches (including machine learning) to COVID-19, cancer, Alzheimer’s disease and antibacterial drug discovery amongst other areas. While it could still be considered as nascent in terms of the development and applications, we are already starting to see some of the benefits, namely leveraging the large amounts of public data that is available in the public domain to identify new uses for these drugs that are already approved. This may ultimately offer faster identification of drugs for new disease applications in a manner that may also be more cost effective. It is likely that the use of drug repurposing using machine learning will become even more mainstream after episodes like COVID-19. What is of course key to continued viability of this approach is that the computational predictions are not used in isolation but are always backed up by convincing in vitro and in vivo verification of the predictions. Ultimately, repurposed drugs reaching patients will be the ultimate mark of success of this approach.
Highlights.
Machine learning for drug repurposing was increasing before 2020.
It has been applied to model small molecule screening data, transcriptomics and proteomics.
Computationally repurposed approved kinase inhibitors have been identified by several groups.
Opportunities exist for computational machine learning in areas of difficult drug discovery e.g. antibiotics and Alzheimer’s disease.
Acknowledgments
We kindly acknowledge Dr. Patricia Vignaux, Dr. Thomas Lane, Ms. Kimberley Zorn, Ms. Eni Minerali, Mr. Daniel Foil and Ms. Jennifer Klein as well as other colleagues for their assistance with some of the research on machine learning repurposing studies described by our group.
Funding
We kindly acknowledge NIH funding: R44GM122196-02A1 from NIGMS, 3R43AT010585-01S1 from NCCAM, R41GM131433-01A1 from NIGMS and 1R43ES031038-01 from NIEHS. “Research reported in this publication was supported by the National Institute of Environmental Health Sciences of the National Institutes of Health under Award Number R43ES031038. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.”
Abbreviations
- Aβ
amyloid beta
- AD
Alzheimer’s Disease
- BACE1
β-site APP-cleaving enzyme 1
- CMap
connectivity map
- DNN
deep neural networks
- FDA
food and drug administration
- GSK3β
Glycogen Synthase Kinase 3β
- IDG
illuminating the druggable genome
- LINCS
library of integrated network-based cellular signatures
- NMDA
N-methyl-D-aspartate
- PKIS
Published Kinase Inhibitor Set
- QSAR
quantitative structure activity relationship
- SAR
structure activity relationship
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest
SE is owner and FU and ACP are employees of Collaborations Pharmaceuticals, Inc.
References
- 1.Paul SM, Mytelka DS, Dunwiddie CT, Persinger CC, Munos BH, Lindborg SR, Schacht AL: How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nat Rev Drug Discov 2010, 9:203–214. [DOI] [PubMed] [Google Scholar]
- 2.Wouters OJ, McKee M, Luyten J: Estimated Research and Development Investment Needed to Bring a New Medicine to Market, 2009–2018. JAMA 2020, 323:844–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baker NC, Ekins S, Williams AJ, Tropsha A: A bibliometric review of drug repurposing. Drug Discov Today 2018, 23:661–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ekins S, Gerlach J, Zorn KM, Antonio BM, Lin Z, Gerlach A: Repurposing Approved Drugs as Inhibitors of Kv7.1 and Nav1.8 to Treat Pitt Hopkins Syndrome. Pharm Res 2019, 36:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boucherie DM, Duarte GS, Machado T, Faustino PR, Sampaio C, Rascol O, Ferreira JJ: Parkinson’s Disease Drug Development Since 1999: A Story of Repurposing and Relative Success. J Parkinsons Dis 2021, 11:421–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Toshner M, Spiekerkoetter E, Bogaard H, Hansmann G, Nikkho S, Prins KW: Repurposing of medications for pulmonary arterial hypertension. Pulm Circ 2020, 10:2045894020941494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shameer K, Readhead B, Dudley JT: Computational and experimental advances in drug repositioning for accelerated therapeutic stratification. Curr Top Med Chem 2015, 15:5–20. [DOI] [PubMed] [Google Scholar]
- 8.Ekins S, Mottin M, Ramos P, Sousa BKP, Neves BJ, Foil DH, Zorn KM, Braga RC, Coffee M, Southan C, et al. : Deja vu: Stimulating open drug discovery for SARS-CoV-2. Drug Discov Today 2020, 25:928–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ekins S, Williams AJ, Krasowski MD, Freundlich JS: In silico repositioning of approved drugs for rare and neglected diseases. Drug Discov Today 2011, 16:298–310. [DOI] [PubMed] [Google Scholar]
- 10.Li J, Zheng S, Chen B, Butte AJ, Swamidass SJ, Lu Z: A survey of current trends in computational drug repositioning. Brief Bioinform 2016, 17:2–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Southan C, Williams AJ, Ekins S: Challenges and recommendations for obtaining chemical structures of industry-provided repurposing candidates. Drug Discov Today 2013, 18:58–70. [DOI] [PubMed] [Google Scholar]
- 12.Puhl AC, Fritch EJ, Lane TR, Tse LV, Yount BL, Sacramento CQ, Fintelman-Rodrigues N, Tavella TA, Maranhão Costa FT, Weston S, et al. : Repurposing the Ebola and Marburg Virus Inhibitors Tilorone, Quinacrine, and Pyronaridine: In Vitro Activity against SARS-CoV-2 and Potential Mechanisms. ACS Omega 2021, 6:7454–7468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Richardson P, Griffin I, Tucker C, Smith D, Oechsle O, Phelan A, Rawling M, Savory E, Stebbing J: Baricitinib as potential treatment for 2019-nCoV acute respiratory disease. Lancet 2020, 395:e30–e31. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This short article highlighted the repurposing of baricitinib against SARS-CoV-2 using a knowledge graph approach.
- 14.Stebbing J, Krishnan V, de Bono S, Ottaviani S, Casalini G, Richardson PJ, Monteil V, Lauschke VM, Mirazimi A, Youhanna S, et al. : Mechanism of baricitinib supports artificial intelligence-predicted testing in COVID-19 patients. EMBO Mol Med 2020, 12:e12697. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This short article highlighted the in vitro activity of baricitinib against SARS-CoV-2.
- 15.Lenz HJ, Richardson P, Stebbing J: The Emergence of Baricitinib: A Story of Tortoises Versus Hares. Clin Infect Dis 2021, 72:1251–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Titanji BK, Farley MM, Mehta A, Connor-Schuler R, Moanna A, Cribbs SK, O’Shea J, DeSilva K, Chan B, Edwards A, et al. : Use of Baricitinib in Patients With Moderate to Severe Coronavirus Disease 2019. Clin Infect Dis 2021, 72:1247–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coelho C, Gallo G, Campos CB, Hardy L, Wurtele M: Biochemical screening for SARS-CoV-2 main protease inhibitors. PLoS One 2020, 15:e0240079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith E, Davis-Gardner ME, Garcia-Ordonez RD, Nguyen TT, Hull M, Chen E, Baillargeon P, Scampavia L, Strutzenberg T, Griffin PR, et al. : High-Throughput Screening for Drugs That Inhibit Papain-Like Protease in SARS-CoV-2. SLAS Discov 2020, 25:1152–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rothan HA, Teoh TC: Cell-Based High-Throughput Screening Protocol for Discovering Antiviral Inhibitors Against SARS-COV-2 Main Protease (3CLpro). Mol Biotechnol 2021, 63:240–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gorshkov K, Chen CZ, Xu M, Carlos de la Torre J, Martinez-Sobrido L, Moran T, Zheng W: Development of a High-Throughput Homogeneous AlphaLISA Drug Screening Assay for the Detection of SARS-CoV-2 Nucleocapsid. ACS Pharmacol Transl Sci 2020, 3:1233–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen CZ, Shinn P, Itkin Z, Eastman RT, Bostwick R, Rasmussen L, Huang R, Shen M, Hu X, Wilson KM, et al. : Drug Repurposing Screen for Compounds Inhibiting the Cytopathic Effect of SARS-CoV-2. Front Pharmacol 2020, 11:592737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dittmar M, Lee JS, Whig K, Segrist E, Li M, Kamalia B, Castellana L, Ayyanathan K, Cardenas-Diaz FL, Morrisey EE, et al. : Drug repurposing screens reveal cell-type-specific entry pathways and FDA-approved drugs active against SARS-Cov-2. Cell Rep 2021, 35:108959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, O’Meara MJ, Rezelj VV, Guo JZ, Swaney DL, et al. : A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583:459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** An early publication describing the repurposing of molecules for SARS-CoV-2 with experimental validation.
- 24.Gordon DE, Hiatt J, Bouhaddou M, Rezelj VV, Ulferts S, Braberg H, Jureka AS, Obernier K, Guo JZ, Batra J, et al. : Comparative host-coronavirus protein interaction networks reveal pan-viral disease mechanisms. Science 2020, 370. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** An early publication describing the repurposing of molecules for SARS-CoV-2 with clinical record validation.
- 25.Boulware DR, Pullen MF, Bangdiwala AS, Pastick KA, Lofgren SM, Okafor EC, Skipper CP, Nascene AA, Nicol MR, Abassi M, et al. : A Randomized Trial of Hydroxychloroquine as Postexposure Prophylaxis for Covid-19. N Engl J Med 2020, 383:517–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roomi S, Ullah W, Ahmed F, Farooq S, Sadiq U, Chohan A, Jafar M, Saddique M, Khanal S, Watson R, et al. : Efficacy of Hydroxychloroquine and Tocilizumab in Patients With COVID-19: Single-Center Retrospective Chart Review. J Med Internet Res 2020, 22:e21758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bocci G, Bradfute SB, Ye C, Garcia MJ, Parvathareddy J, Reichard W, Surendranathan S, Bansal S, Bologa CG, Perkins DJ, et al. : Virtual and In Vitro Antiviral Screening Revive Therapeutic Drugs for COVID-19. ACS Pharmacol Transl Sci 2020, 3:1278–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** An example of a study that used computational predictions for drugs with SARS-CoV-2 activity which was then validated in vitro.
- 28.Liu X, Li Z, Liu S, Sun J, Chen Z, Jiang M, Zhang Q, Wei Y, Wang X, Huang YY, et al. : Potential therapeutic effects of dipyridamole in the severely ill patients with COVID-19. Acta Pharm Sin B 2020, 10:1205–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feng S, Luan X, Wang Y, Wang H, Zhang Z, Wang Y, Tian Z, Liu M, Xiao Y, Zhao Y, et al. : Eltrombopag is a potential target for drug intervention in SARS-CoV-2 spike protein. Infect Genet Evol 2020, 85:104419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Virdi RS, Bavisotto RV, Hopper NC, Frick DN: Discovery of Drug-like Ligands for the Mac1 Domain of SARS-CoV-2 Nsp3. bioRxiv 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roskoski R: Protein Kinase Inhibitors. Edited by; 2020. vol 2020.] [Google Scholar]
- 32.Roskoski R Jr.: Properties of FDA-approved small molecule protein kinase inhibitors: A 2020 update. Pharmacol Res 2020, 152:104609. [DOI] [PubMed] [Google Scholar]
- 33.Shibata S, Matsushita M, Saito Y, Suzuki T: Anticancer Drug Prescription Patterns in Japan: Future Directions in Cancer Therapy. Ther Innov Regul Sci 2018, 52:718–723. [DOI] [PubMed] [Google Scholar]
- 34.Moret N, Clark NA, Hafner M, Wang Y, Lounkine E, Medvedovic M, Wang J, Gray N, Jenkins J, Sorger PK: Cheminformatics Tools for Analyzing and Designing Optimized Small-Molecule Collections and Libraries. Cell Chem Biol 2019, 26:765–777 e763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Elkins JM, Fedele V, Szklarz M, Abdul Azeez KR, Salah E, Mikolajczyk J, Romanov S, Sepetov N, Huang XP, Roth BL, et al. : Comprehensive characterization of the Published Kinase Inhibitor Set. Nat Biotechnol 2016, 34:95–103. [DOI] [PubMed] [Google Scholar]
- 36.Nguyen DT, Mathias S, Bologa C, Brunak S, Fernandez N, Gaulton A, Hersey A, Holmes J, Jensen LJ, Karlsson A, et al. : Pharos: Collating protein information to shed light on the druggable genome. Nucleic Acids Res 2017, 45:D995–D1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oprea TI, Bologa CG, Brunak S, Campbell A, Gan GN, Gaulton A, Gomez SM, Guha R, Hersey A, Holmes J, et al. : Unexplored therapeutic opportunities in the human genome. Nat Rev Drug Discov 2018, 17:317–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rodgers G, Austin C, Anderson J, Pawlyk A, Colvis C, Margolis R, Baker J: Glimmers in illuminating the druggable genome. Nat Rev Drug Discov 2018, 17:301–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Berginski ME, Moret N, Liu C, Goldfarb D, Sorger PK, Gomez SM: The Dark Kinase Knowledgebase: an online compendium of knowledge and experimental results of understudied kinases. Nucleic Acids Res 2021, 49:D529–D535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tamir TY, Drewry DH, Wells C, Major MB, Axtman AD: PKIS deep dive yields a chemical starting point for dark kinases and a cell active BRSK2 inhibitor. Sci Rep 2020, 10:15826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Keenan AB, Jenkins SL, Jagodnik KM, Koplev S, He E, Torre D, Wang Z, Dohlman AB, Silverstein MC, Lachmann A, et al. : The Library of Integrated Network-Based Cellular Signatures NIH Program: System-Level Cataloging of Human Cells Response to Perturbations. Cell Syst 2018, 6:13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Niepel M, Hafner M, Mills CE, Subramanian K, Williams EH, Chung M, Gaudio B, Barrette AM, Stern AD, Hu B, et al. : A Multi-center Study on the Reproducibility of Drug-Response Assays in Mammalian Cell Lines. Cell Syst 2019, 9:35–48 e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zimmer M, Lamb J, Ebert BL, Lynch M, Neil C, Schmidt E, Golub TR, Iliopoulos O: The connectivity map links iron regulatory protein-1-mediated inhibition of hypoxia-inducible factor-2a translation to the anti-inflammatory 15-deoxy-delta12,14-prostaglandin J2. Cancer Res 2010, 70:3071–3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lamb J: The Connectivity Map: a new tool for biomedical research. Nat Rev Cancer 2007, 7:54–60. [DOI] [PubMed] [Google Scholar]
- 45.Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, et al. : The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313:1929–1935. [DOI] [PubMed] [Google Scholar]
- 46.Subramanian A, Narayan R, Corsello SM, Peck DD, Natoli TE, Lu X, Gould J, Davis JF, Tubelli AA, Asiedu JK, et al. : A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171:1437–1452 e1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baillif B, Wichard J, Mendez-Lucio O, Rouquie D: Exploring the Use of Compound-Induced Transcriptomic Data Generated From Cell Lines to Predict Compound Activity Toward Molecular Targets. Front Chem 2020, 8:296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mendez-Lucio O, Baillif B, Clevert DA, Rouquie D, Wichard J: De novo generation of hit-like molecules from gene expression signatures using artificial intelligence. Nat Commun 2020, 11:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ravikumar B, Timonen S, Alam Z, Parri E, Wennerberg K, Aittokallio T: Chemogenomic Analysis of the Druggable Kinome and Its Application to Repositioning and Lead Identification Studies. Cell Chem Biol 2019, 26:1608–1622 e1606. [DOI] [PubMed] [Google Scholar]; * An example of using computational approaches to repurpose kinase inhibitors.
- 50.Cichonska A, Ravikumar B, Parri E, Timonen S, Pahikkala T, Airola A, Wennerberg K, Rousu J, Aittokallio T: Computational-experimental approach to drug-target interaction mapping: A case study on kinase inhibitors. PLoS Comput Biol 2017, 13:e1005678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Janssen APA, Grimm SH, Wijdeven RHM, Lenselink EB, Neefjes J, van Boeckel CAA, van Westen GJP, van der Stelt M: Drug Discovery Maps, a Machine Learning Model That Visualizes and Predicts Kinome-Inhibitor Interaction Landscapes. J Chem Inf Model 2019, 59:1221–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Merget B, Turk S, Eid S, Rippmann F, Fulle S: Profiling Prediction of Kinase Inhibitors: Toward the Virtual Assay. J Med Chem 2017, 60:474–485. [DOI] [PubMed] [Google Scholar]
- 53.Yang M, Tao B, Chen C, Jia W, Sun S, Zhang T, Wang X: Machine Learning Models Based on Molecular Fingerprints and an Extreme Gradient Boosting Method Lead to the Discovery of JAK2 Inhibitors. J Chem Inf Model 2019, 59:5002–5012. [DOI] [PubMed] [Google Scholar]
- 54.Xiao T, Qi X, Chen Y, Jiang Y: Development of Ligand-based Big Data Deep Neural Network Models for Virtual Screening of Large Compound Libraries. Mol Inform 2018, 37:e1800031. [DOI] [PubMed] [Google Scholar]
- 55.Fang J, Yang R, Gao L, Yang S, Pang X, Li C, He Y, Liu AL, Du GH: Consensus models for CDK5 inhibitors in silico and their application to inhibitor discovery. Mol Divers 2015, 19:149–162. [DOI] [PubMed] [Google Scholar]
- 56.Kong Y, Bender A, Yan A: Identification of Novel Aurora Kinase A (AURKA) Inhibitors via Hierarchical Ligand-Based Virtual Screening. J Chem Inf Model 2018, 58:36–47. [DOI] [PubMed] [Google Scholar]
- 57.Zhavoronkov A, Ivanenkov YA, Aliper A, Veselov MS, Aladinskiy VA, Aladinskaya AV, Terentiev VA, Polykovskiy DA, Kuznetsov MD, Asadulaev A, et al. : Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat Biotechnol 2019, 37:1038–1040. [DOI] [PubMed] [Google Scholar]
- 58.Li X, Li Z, Wu X, Xiong Z, Yang T, Fu Z, Liu X, Tan X, Zhong F, Wan X, et al. : Deep Learning Enhancing Kinome-Wide Polypharmacology Profiling: Model Construction and Experiment Validation. J Med Chem 2020, 63:8723–8737. [DOI] [PubMed] [Google Scholar]
- 59.Menden MP, Iorio F, Garnett M, McDermott U, Benes CH, Ballester PJ, Saez-Rodriguez J: Machine learning prediction of cancer cell sensitivity to drugs based on genomic and chemical properties. PLoS One 2013, 8:e61318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li B, Dai C, Wang L, Deng H, Li Y, Guan Z, Ni H: A novel drug repurposing approach for non-small cell lung cancer using deep learning. PLoS One 2020, 15:e0233112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lim H, He D, Qiu Y, Krawczuk P, Sun X, Xie L: Rational discovery of dual-indication multi-target PDE/Kinase inhibitor for precision anti-cancer therapy using structural systems pharmacology. PLoS Comput Biol 2019, 15:e1006619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Anderson E, Havener TM, Zorn KM, Foil DH, Lane TR, Capuzzi SJ, Morris D, Hickey AJ, Drewry DH, Ekins S: Synergistic drug combinations and machine learning for drug repurposing in chordoma. Sci Rep 2020, 10:12982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stetson LC, Pearl T, Chen Y, Barnholtz-Sloan JS: Computational identification of multi-omic correlates of anticancer therapeutic response. BMC Genomics 2014, 15 Suppl 7:S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mielke MM, Vemuri P, Rocca WA: Clinical epidemiology of Alzheimer’s disease: assessing sex and gender differences. Clin Epidemiol 2014, 6:37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee JE, Han PL: An update of animal models of Alzheimer disease with a reevaluation of plaque depositions. Exp Neurobiol 2013, 22:84–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.West S, Bhugra P: Emerging drug targets for Abeta and tau in Alzheimer’s disease: a systematic review. Br J Clin Pharmacol 2015, 80:221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rodriguez S, Hug C, Todorov P, Moret N, Boswell SA, Evans K, Zhou G, Johnson NT, Hyman BT, Sorger PK, et al. : Machine learning identifies candidates for drug repurposing in Alzheimer’s disease. Nat Commun 2021, 12:1033. [DOI] [PMC free article] [PubMed] [Google Scholar]; * An early application of machine learning for Alzheimer’s disease.
- 68.Leroy K, Yilmaz Z, Brion JP: Increased level of active GSK-3beta in Alzheimer’s disease and accumulation in argyrophilic grains and in neurones at different stages of neurofibrillary degeneration. Neuropathol Appl Neurobiol 2007, 33:43–55. [DOI] [PubMed] [Google Scholar]
- 69.Pei JJ, Tanaka T, Tung YC, Braak E, Iqbal K, Grundke-Iqbal I: Distribution, levels, and activity of glycogen synthase kinase-3 in the Alzheimer disease brain. J Neuropathol Exp Neurol 1997, 56:70–78. [DOI] [PubMed] [Google Scholar]
- 70.Zhang Y, Huang NQ, Yan F, Jin H, Zhou SY, Shi JS, Jin F: Diabetes mellitus and Alzheimer’s disease: GSK-3beta as a potential link. Behav Brain Res 2018, 339:57–65. [DOI] [PubMed] [Google Scholar]
- 71.Beurel E, Jope RS: The paradoxical pro- and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic apoptosis signaling pathways. Prog Neurobiol 2006, 79:173–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Turenne GA, Price BD: Glycogen synthase kinase3 beta phosphorylates serine 33 of p53 and activates p53’s transcriptional activity. BMC Cell Biol 2001, 2:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ishiguro K, Omori A, Takamatsu M, Sato K, Arioka M, Uchida T, Imahori K: Phosphorylation sites on tau by tau protein kinase I, a bovine derived kinase generating an epitope of paired helical filaments. Neurosci Lett 1992, 148:202–206. [DOI] [PubMed] [Google Scholar]
- 74.Hanger DP, Hughes K, Woodgett JR, Brion JP, Anderton BH: Glycogen synthase kinase-3 induces Alzheimer’s disease-like phosphorylation of tau: generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci Lett 1992, 147:58–62. [DOI] [PubMed] [Google Scholar]
- 75.Lovestone S, Reynolds CH, Latimer D, Davis DR, Anderton BH, Gallo JM, Hanger D, Mulot S, Marquardt B, Stabel S, et al. : Alzheimer’s disease-like phosphorylation of the microtubule-associated protein tau by glycogen synthase kinase-3 in transfected mammalian cells. Curr Biol 1994, 4:1077–1086. [DOI] [PubMed] [Google Scholar]
- 76.Uemura K, Kuzuya A, Shimozono Y, Aoyagi N, Ando K, Shimohama S, Kinoshita A: GSK3beta activity modifies the localization and function of presenilin 1. J Biol Chem 2007, 282:15823–15832. [DOI] [PubMed] [Google Scholar]
- 77.Ly PT, Wu Y, Zou H, Wang R, Zhou W, Kinoshita A, Zhang M, Yang Y, Cai F, Woodgett J, et al. : Inhibition of GSK3beta-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J Clin Invest 2013, 123:224–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Maqbool M, Hoda N: GSK3 Inhibitors in the Therapeutic Development of Diabetes, Cancer and Neurodegeneration: Past, Present and Future. Curr Pharm Des 2017, 23:4332–4350. [DOI] [PubMed] [Google Scholar]
- 79.Walz A, Ugolkov A, Chandra S, Kozikowski A, Carneiro BA, O’Halloran TV, Giles FJ, Billadeau DD, Mazar AP: Molecular Pathways: Revisiting Glycogen Synthase Kinase-3beta as a Target for the Treatment of Cancer. Clin Cancer Res 2017, 23:1891–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Matsunaga S, Fujishiro H, Takechi H: Efficacy and Safety of Glycogen Synthase Kinase 3 Inhibitors for Alzheimer’s Disease: A Systematic Review and Meta-Analysis. J Alzheimers Dis 2019, 69:1031–1039. [DOI] [PubMed] [Google Scholar]
- 81.Kuroki H, Anraku T, Kazama A, Bilim V, Tasaki M, Schmitt D, Mazar AP, Giles FJ, Ugolkov A, Tomita Y: 9-ING-41, a small molecule inhibitor of GSK-3beta, potentiates the effects of anticancer therapeutics in bladder cancer. Sci Rep 2019, 9:19977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vignaux P, Minerali E, Foil DH, Puhl AC, Ekins S: Machine Learning for Discovery of GSK3β Inhibitors. ACS Omega 2020, 5:26551–26561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Prathipati P, Ma NL, Keller TH: Global Bayesian models for the prioritization of antitubercular agents. J Chem Inf Model 2008, 48:2362–2370. [DOI] [PubMed] [Google Scholar]
- 84.Ekins S, Bradford J, Dole K, Spektor A, Gregory K, Blondeau D, Hohman M, Bunin B: A Collaborative Database And Computational Models For Tuberculosis Drug Discovery. Mol BioSystems 2010, 6:840–851. [DOI] [PubMed] [Google Scholar]
- 85.Ekins S, Kaneko T, Lipinksi CA, Bradford J, Dole K, Spektor A, Gregory K, Blondeau D, Ernst S, Yang J, et al. : Analysis and hit filtering of a very large library of compounds screened against Mycobacterium tuberculosis Mol BioSyst 2010, 6:2316–2324. [DOI] [PubMed] [Google Scholar]
- 86.Gamo F-J, Sanz LM, Vidal J, de Cozar C, Alvarez E, Lavandera J-L, Vanderwall DE, Green DVS, Kumar V, Hasan S, et al. : Thousands of chemical starting points for antimalarial lead identification. Nature 2010, 465:305–310. [DOI] [PubMed] [Google Scholar]
- 87.Ekins S, Reynolds R, Kim H, Koo M-S, Ekonomidis M, Talaue M, Paget SD, Woolhiser LK, Lenaerts AJ, Bunin BA, et al. : Bayesian Models Leveraging Bioactivity and Cytotoxicity Information for Drug Discovery. Chem Biol 2013, 20:370–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ekins S, Reynolds RC, Franzblau SG, Wan B, Freundlich JS, Bunin BA: Enhancing Hit Identification in Mycobacterium tuberculosis Drug Discovery Using Validated Dual-Event Bayesian Models PLOSONE 2013, 8:e63240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ekins S, Casey AC, Roberts D, Parish T, Bunin BA: Bayesian models for screening and TB Mobile for target inference with Mycobacterium tuberculosis. Tuberculosis (Edinb) 2014, 94:162–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lane T, Russo DP, Zorn KM, Clark AM, Korotcov A, Tkachenko V, Reynolds RC, Perryman AL, Freundlich JS, Ekins S: Comparing and Validating Machine Learning Models for Mycobacterium tuberculosis Drug Discovery. Mol Pharm 2018, 15:4346–4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Stokes JM, Yang K, Swanson K, Jin W, Cubillos-Ruiz A, Donghia NM, MacNair CR, French S, Carfrae LA, Bloom-Ackermann Z, et al. : A Deep Learning Approach to Antibiotic Discovery. Cell 2020, 180:688–702 e613. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** An important article describing how a deep learning ligand-based model was able to identify a new use for an existing molecule.
- 92.Ivanenkov YA, Zhavoronkov A, Yamidanov RS, Osterman IA, Sergiev PV, Aladinskiy VA, Aladinskaya AV, Terentiev VA, Veselov MS, Ayginin AA, et al. : Identification of Novel Antibacterials Using Machine Learning Techniques. Front Pharmacol 2019, 10:913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Simm J, Klambauer G, Arany A, Steijaert M, Wegner JK, Gustin E, Chupakhin V, Chong YT, Vialard J, Buijnsters P, et al. : Repurposing High-Throughput Image Assays Enables Biological Activity Prediction for Drug Discovery. Cell Chem Biol 2018, 25:611–618 e613. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** This article describes how image analysis data can be used to preedict activity of compounds
- 94.Romeo-Guitart D, Fores J, Herrando-Grabulosa M, Valls R, Leiva-Rodriguez T, Galea E, Gonzalez-Perez F, Navarro X, Petegnief V, Bosch A, et al. : Neuroprotective Drug for Nerve Trauma Revealed Using Artificial Intelligence. Sci Rep 2018, 8:1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.KalantarMotamedi Y, Eastman RT, Guha R, Bender A: A systematic and prospectively validated approach for identifying synergistic drug combinations against malaria. Malar J 2018, 17:160. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This article describes how a machine learning approach can be used to predict synergistic activity.
- 96.Pottel J, Armstrong D, Zou L, Fekete A, Huang XP, Torosyan H, Bednarczyk D, Whitebread S, Bhhatarai B, Liang G, et al. : The activities of drug inactive ingredients on biological targets. Science 2020, 369:403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This article described the use of similarity ensemble approach to predict the activities of inactive ingredients in drugs which were then validated in vitro.