

Abstract
Heritable thoracic aortic disease (HTAD) and familial thoracic aortic aneurysm/dissection (FTAAD) are important causes of human morbidity/mortality, most without identifiable genetic cause. In a family with FTAAD, we identified a missense p. (Ser178Arg) variant in PLOD1 segregating with disease, and evaluated PLOD1 enzymatic activity, collagen characteristics and in human aortic vascular smooth muscle cells (VSMCs), studied the effect on function. Comparison with homologous PLOD3 enzyme indicated that the pathogenic variant may affect the N-terminal glycosyltransferase (GT) domain, suggesting unprecedented PLOD1 activity. In vitro assays demonstrated that wild-type (WT) PLOD1 is capable of processing UDP-glycan donor substrates, and that the variant affects the folding stability of the GT domain and associated enzymatic functions. The PLOD1 substrate lysine was elevated in the proband, however the enzymatic product hydroxylysine and total collagen content was not different, albeit despite collagen fibril narrowing and preservation of collagen turnover. In VSMCs overexpressing WT PLOD1, there was upregulation in procollagen gene expression (secretory function) which was attenuated in the variant, consistent with loss-of-function. In comparison, si-PLOD1 cells demonstrated hypercontractility and upregulation of contractile markers, providing evidence for phenotypic switching. Together, the findings suggest that the PLOD1 product is preserved, however newly identified glucosyltransferase activity of PLOD1 appears to be affected by folding stability of the variant, and is associated with compensatory VSMC phenotypic switching to support collagen production, albeit with less robust fibril girth. Future studies should focus on the impact of PLOD1 folding/variant stability on the tertiary structure of collagen and ECM interactions.
Keywords: Aortic Dissection, Procollagen-Lysine, 2-Oxoglutarate 5-Dioxygenase 1 (PLOD1), Glycosyltransferase, Genetics, Aortopathy, Lysyl Hydroxylase-1
Introduction
Heritable thoracic aortic disease (HTAD) and FTAAD (familial thoracic aortic aneurysm and dissection) are aortopathic conditions characterized by diverse genetic heterogeneity. HTAD and FTAAD can lead to aortic aneurysm and dissection, resulting in sudden cardiac death, that conservatively affects at least 10,000 adults in the United States each year.1 Up to 20% of patients with a dilated or aneurysmal aorta have a first-degree relative similarly affected, reflecting the most common, autosomal dominant heritable pattern of disease.2, 3 The most widely used comprehensive clinical genetic panels for aortopathy test >20 genes, but are only successful at identifying a causal variant in 20% of patients.4 Thus, the majority of patients and families with a predisposition for vascular involvement, do not have a readily identifiable genetic cause. Equally concerning, is that even when the genetic basis of disease is known, the molecular mechanisms remain poorly understood.
Aortic wall integrity relies on interactions between vascular smooth muscle cells (VSMCs), elastin, collagen and specialized proteins in the extracellular matrix (ECM) which interface at the elastin-contractile unit (ECU).5, 6 Procollagen-lysine, 2-oxoglutarate 5-dioxygenase 1 (PLOD1) is a gene that encodes for the enzyme lysyl hydroxylase-1 (PLOD1, also known as LH1), which functions in procollagen formation through post-translational modification of several lysine residues into hydroxylysine. It has been implicated in kyphoscoliotic Ehlers-Danlos Syndrome (kEDS), a skeletal connective tissue disease that carries minimal to no risk for aortopathy.7 We identified a family (spanning 3 generations) with an autosomal dominant novel pathogenic variant in PLOD1 (c.534C>A (p.Ser178Arg), that resulted in the presence of highly penetrant aortic aneurysm and/or aortic dissection. Given that PLOD1 has largely been remitted as a primary source of HTAD/FTAAD, we aimed to investigate the impact of this gene on vascular integrity.
Here, we report a novel pathogenic PLOD1 missense variant in a family with autosomal dominant FTAAD. In pathologic specimens, we identified relatively preserved aortic medial structure with mild focal separation and loss of elastic fibers. Therefore, we evaluated the impact of the variant upon collagen production, enzymatic function (PLOD1) and VSMC function. We found that the amount of total collagen was preserved with no difference in turnover surrogates, however with mild narrowing in the fibrillar product and presumed compensatory upregulation of the PLOD1 substrate lysine. To study enzymatic function, we assessed molecular analyses in silico and identified the mutation site matching a critical residue for the glycosyl transferase activity of the homologous multifunctional PLOD3 (also known as LH3).8, 9 In vitro biochemical studies revealed the ability of wild-type PLOD1 to process glycosyl transferase donor substrates, supporting a possible impact of the mutation on enzyme function. Whilst the PLOD1 p. (Ser178Arg) variant could not be obtained recombinantly in sufficient amounts for biochemical characterization, homologous PLOD3 p. (Asp190Arg) showed a strong reduction in glycosyl transferase donor substrate processing and folding stability compared to the wild-type enzyme. Finally, in VSMCs we found evidence of phenotypic switching whereby silencing of PLOD1 resulted in hypercontractility and conversely, in VSMCs that overexpressed PLOD1 we demonstrated increases in procollagen genes COL1A1 and COL3A1, indicative of a secretory phenotype. Overexpression of the PLOD1 p. (Ser178Arg) variant resulted in attenuation of the secretory phenotype, indicating that the mutation leads to a loss-of-function. Together, our data identify an important genetic contribution for FTAAD, and elucidate that PLOD1 dysfunction likely contributes to aortopathy at least in part, via post-translational modification of collagen, PLOD1 enzymatic activity and VSMC function.
Methods
Family Recruitment and Clinical Evaluation
The proband in the family studied was referred for the evaluation of an unrelated cardiovascular condition (patent foramen ovale). However, upon review of the family history it was evident that aortic aneurysm and dissection were concerning across at least 3 generations. In total, five kindred were recruited to participate in the study. The institutional review board approved the human studies research, and informed consent was obtained from all members enrolled, conforming to the Declaration of Helsinki.10 We gathered a complete 3-generation pedigree evaluating for a history of connective tissue disease, aneurysms, dissection and/or sudden cardiac death. Hereto forward PLOD1 refers to Ensembl: ENST00000196061.4, NCBI: NM_000302.4 and PLOD3 refers to Ensembl: ENST000002223127.3, NCBI: NM_001084.5.
Genetic testing
Specimen Collection:
Blood from the proband and saliva from relatives was collected (Oragene Discover OGR-500 saliva collection kit) and DNA was isolated via the QIAGEN Gentra® Puregene® kit according to the manufacturer instructions. The proband submitted resected aortic tissue at the time of elective surgery, and histologic review was performed (see below).
Library Preparation and Sequencing:
DNA sample analysis contributed to the final data, and each step including sample test, library preparation, and sequencing, is known to influence the quality of the data, and in turn, data quality directly impacts the analysis of results. To guarantee the reliability of the data, quality control (QC) was performed at each step of the procedure. There are two main methods of QC for DNA samples: (1) Agarose Gel Electrophoresis: tests DNA degradation and potential contamination, (2) Qubit 2.0: quantifies the DNA concentration precisely. (From Novogene Co., Ltd)
Library Construction and Quality Control:
A total amount of 1.0μg genomic DNA per sample was used as input material for the DNA sample preparation. Sequencing libraries were generated using Agilent SureSelect Human All ExonV6 kit (Agilent Technologies, CA, USA) following manufacturer’s recommendations and x index codes were added to attribute sequences to each sample. Briefly, fragmentation was carried out by hydrodynamic shearing system (Covaris, Massachusetts, USA) to generate 180–280bp fragments. Remaining overhangs were converted into blunt ends via exonuclease/polymerase activities and enzymes were removed. After adenylation of 3’ ends of DNA fragments, adapter oligonucleotides were ligated. DNA fragments with ligated adapter molecules on both ends were selectively enriched in a PCR reaction. Captured libraries were enriched in a PCR reaction to add index tags to prepare for hybridization. Products were purified using AMPure XP system (Beckman Coulter, Beverly, USA) and quantified using the Agilent high sensitivity DNA assay on the Agilent Bioanalyzer 2100 system. (From Novogene Co., Ltd)
Sequencing:
The qualified libraries are fed into Illumina sequencers after pooling according to its effective concentration and expected data volume. (From Novogene Co., Ltd)
Analysis:
Raw whole exome sequencing data was processed via GenomeNext, LLC (Helios GenomeNext Annotation and Reporting) according to updated standards for the American College of Medical Genetics.11 Further variant prioritization based upon biologic function,12 and phenotype,13 was completed. There were 3 variants which occurred in relevant aortopathic genes, however none resulted in a mutation with functional consequence (synonymous mutation and/or splice regional variant): TGFBR1 p. (Ser69Ser), MYH11 p. (Ala1283Ala) and LOX splice region variant (c.740+6G>A). In total, 6 final potential candidate genes resulted from the search: CLTCL1, KALRN, MFN2, PDHA, PLOD1 and TTN and are outlined in Supplemental Table 1.
Cloning
Wild-type PLOD1 cDNA was made using ProtoScript® II First Strand cDNA Synthesis Kit (New England BioLabs). PCR was performed to amplify wild-type PLOD1 using the following primers: F-CACCGTCGCGAAGTTTCCAG, R-TGGTTTAAATGACCTTTAATAGAAACTCTCCAGTTTCT. The PCR product was cleaned and ligated with a pENTR™ TOPO® vector (ThermoFisher Scientific). The Q5 Site-Directed Mutagenesis Kit (NEB) was used to create the WT (primers: F-ATGCGGCCCCTGCTGCTA, R-GGTGAAGGAGCCCTGAAAATACAGG and c.534C>A variant (primers: F-ACAGCGACAGAGATCAGCTGTTTTACACC, R-CCTGGCCCTCCCACTCGG).
Human tissue specimens, Immunohistochemistry
Formalin fixed, paraffin-embedded sections of the ascending aorta were examined after the proband underwent elective surgical resection (aneurysm). Pathologic specimens were compared to control human aortic tissue obtained through Lifeline of Ohio Organ Procurement, as previously described.14 Specimens were limited to adults < 50 years of age (n=2 male and n=2 female). Specimens were embedded in optimal cutting temperature (OCT) media and slides were prepared (3 mm cuts). Slides were treated with hematoxylin and eosin (Sigma) to evaluate cellular and extracellular structure, and Russel-Movatt pentachrome stain (StatLab) to more closely evaluate connective tissue including the elastic lamellae, collagen and cellular elements. Immunohistochemistry was performed with anti-SMA (Sigma Aldrich A2547; 1:500, overnight at 4°C), anti-COL3A1 (Abcam ab7778; 1:200, 30 minutes at room temperature), and anti-PLOD1 (Novus Biologicals NBP2-38770; 1:100, overnight at 4°C) using VECTASTAIN® ABC Kit, Rabbit IgG (Vector Labs PK-6101; 1:400) and ImmPACT® DAB Substrate (Vector Labs SK-4105). Picrosirius red staining was performed by the core lab facility at Nationwide Children’s Hospital. For protein (total) quantification of PLOD1 (Ponceau), anti-PLOD1 (Novus Biologicals NBP2-38770; 1:700, overnight at 4°C) was used with a GAPDH (Fitzgerald 10R-G109a; 1:5000 overnight at 4°C) control.
Collagen Studies
Collagen preparation:
The samples have been lyophilized for 10 hours, crushed after lyophilization into powder, and weighed out 2–5 mg after crushing. 6 M HCl was then added at a ratio of 1 mL/10 mg of tissue. Samples included all four tissue samples (1 proband and 3 control) and a blank as control (clean vial with no tissue). Heated tissues in sealed glass vials at 100 °C for 24 hours and then hydrolyzed samples were centrifuged at 20,000 g for 20 minutes. Solutions are placed in 0.2 μm spin filter collected after ultracentrifugation at 20,000 g for 5 minutes. A 200 uL volume of filtered solution are then placed in a clean glass vials and dried in SpeedVac at 25 °C until the solution is dry (2 hours). Dried samples are reconstituted in 600 uL 5% MeOH with 0.1 % Formic acid and 10 μg/mL Pyridoxine as internal standard (Total mass in each vial = 2 mg (1 mL/10 mg) reconstituted in 600 uL = 2000 μg/0.6 mL = 3333.33 μg tissue/mL final solution). Calibration solutions made by diluting blank 5% MeOH with 0.1 % FA + pyridoxine (Calibration solutions: 0.0 μg/mL, 0.01. 0.1, 1.0, 10.0, and 100.0 ug/mL Hyp and Pro). Samples were lyophilized as ‘tissue sections’ and all targets analyzed from the acid hydrolyzed solutions. Lysine and hydroxylysine were quantified with the same calibration solutions as hydroxyproline and proline (calibration solutions made by diluting blank 5% MeOH with 0.1% formic acid + pyroxidine (0.0 g/mL, 0.005, 0.05, 0.5, 5.0 and 50.0 g/mL of all targets).
Liquid chromatography-Mass spectrophotometry quantification (LC-MS):
Thermo Scientific UltiMate 3000 HPLC used for LC. 5 μL of sample was injected for every run. Solvent A: 100% H2O 0.1% Formic acid. Solvent B: 100% MeOH 0.1% Formic acid. Poroshell 120 SB-C18 columns used (2 × 100 mm, 2.7 μm particle size). Flow rate of 200 μL/min with a column temperature of 40 °C with an initial gradient of 5% B, 10% B at min 2, 50% B at min 7, 100% B at minute 8.5, holding 100% B until min 10, back to 5% B at min 12, and holding 5% B until the end of run at min 15.
The samples were quantified using a Heated Electrospray ionization (HESI) source on a Thermo Scientific Q-Exactive Plus Orbitrap mass spectrometer. All transitions were performed in parallel reaction monitoring mode at a collision energy of 20 V. For proline, the transition monitored was precursor 116.07 m/z and product ion 70.06 m/z, hydroxyproline monitored at a transition of 132.07 m/z to product ion 86.05 m/z, the internal standard pyridoxine at a transition of 170.08 m/z to a product ion of 152.20, pentosidine monitored at precursor mass of 379.21 m/z, lysine at a transition of 147.11 m/z to a product ion of 84.08 m/z, and hydroxylysine at a transition of 163.107 m/z to a product ion of 128.07 m/z. For all experiments, the capillary voltage was set to 4.5 kV with a capillary temperature of 320 °C, an auxiliary temperature of 100 °C, a sheath gas of 15, auxiliary gas of 5, and 0.5 sweep gas.
Cell culture, treatment, transfection
Vascular smooth muscle cells (VSMCs) derived from human aorta (Lifeline® Cell Technology, CA, USA) were used for all VSMC studies. Vasculife ® SMC Smooth Muscle Cell Medium (Lifeline Cell Technology, MD, USA) was used for cell culture. Intentional silencing of the PLOD1 gene was performed in the human VSMCs via siRNA. Cells were transfected (Lipofectamine ® RNAiMAX Transfection Reagent, Thermo Fisher Scientific, MA, USA) into 24-well plates with 0.5 μL of 10 μM of si-PLOD1 (Qiagen SI00006636) or Silencer Select negative control (ThermoFisher Scientific 4390843, MA, USA). Four siRNAs were tested from the Qiagen FlexiTube GeneSolution GS5351 (Qiagen 1027416) for maximum PLOD1 knock-down efficiency at 10 μm siRNA without detrimental effects. Western blot was performed to confirm silencing of PLOD1. Briefly, 30 μg protein was run on a 4–15% Mini-PROTEAN TGX Stain-Free Protein Gel (BioRad 4568081) and stained at 1:1000 with anti-PLOD1 antibody (Novus Biologicals, NBP2-38770). For the wildtype and variant, transfection was accomplished with 100 ng of Effectine Qiagen Cloning methods (see above).
Contraction studies
Collagen gel-based assays were used to perform VSMC contraction studies according to previously reported methods.15 Collagen lattice at a final concentration of 1 mg/mL was prepared by mixing type-I collagen (ThermoFisher, A1048301), Vasculife ® SMC Smooth Muscle Cell Medium, Dulbecco’s Phosphate Buffered Saline (Invitrogen). 450 μL of collagen solution was added to 50 μL of VSMC suspension (4×104 cells per well). Each 500 μL collagen/cell mixture was added to one well in a 96 well plate and allowed to polymerize for 30 minutes at 37 °C in a CO2 incubator. After 30 minutes at 37 °C, 140 μL of VSMC media (2% FBS) was added to each well. Cells were incubated for 48 hours before imaging. Thirty minutes before imaging, collagen gels were released from the sides of the well with a 10 μL pipet tip and the media was replaced with VSMC media (10% FBS). The gel were imaged with a ThermoFisher EVOS FL Auto. Gel area was determined using ImageJ.
Quantitative PCR
Total RNA (500 ng) was used for first-strand complementary DNA synthesis with the SuperScript III Reverse Transcriptase VILO cDNA Synthesis Kit (Invitrogen, ThermoFisher Scientific). qPCR reactions were performed in triplicate on cDNA samples with TaqMan Gene Expression Assays (ACTA2, Hs00426835_g1; MYH11, Hs00975796_m1; CCND1, Hs00765553_m1; CCND2, Hs00153380_m1; COL1A1, Hs00164004_m1; COL3A1, Hs00943809_m1; GAPDH, Hs99999905_m1; Life Technologies, ThermoFisher Scientific) and TaqMan Universal PCR Master Mix (ThermoFisher Scientific). PCR was performed on an Applied Biosystems QuantStudio™ 3 Real-Time PCR System. Relative gene expression was calculated using the 2−ΔΔCt method. The glyceraldehydes-3-phosphate dehydrogenase (GAPDH) gene was used as the reference gene. The target genes included six genetic markers of the contractile and proliferative phenotypes of smooth muscle cells. All real-time PCR experiments were carried out with three biological replicates, and three technical replicates were performed for every sample.
In silico evaluation of the PLOD1 p. (Ser178Arg) variant
The PLOD1 p. (Ser178Arg) mutation was initially identified in the SiMPLOD database,8 as matching the residue Asp190 in the glycosyltransferase catalytic site of the homologous PLOD3 enzyme. Comparative evaluation of folding stability was carried out using the experimental crystal structure of human PLOD3 (PDB 6FXR),16 and the homology model of human PLOD1 available through the SiMPLOD website (http://fornerislab.unipv.it/SiMPLOD/). Both PDB files were used to further forecast the impact of the p. (Ser178Arg) mutation by evaluating the impact on the three-dimensional protein organization using Missense3D,17 through normal mode perturbation analysis in Dynamut,18 and to assess folding free energy changes in MaestroWeb,19 PoPMuSiC,20 mCSM,21 MuPRO,22 DUET23 and iMutant24. A summary analysis of the in silico prediction results is shown in Supplementary Table 2.
Recombinant production of wild-type PLOD1 and PLOD3 for biochemical studies
The coding sequences for human PLOD1 or PLOD3 were cloned into a pUPE.106.08 expression vector (U-protein Express BV, The Netherlands) in frame with a 6xHis-tag followed by a Tobacco Etch Virus (TEV) protease cleavage site. PLOD1 p. (Ser178Arg) and PLOD3 p. (Asp190Arg) mutants were generated using site-directed mutagenesis using oligonucleotides pairs PLOD1-S178R-Fw: CGCGATCAGCTGTTTTACACCAAGATC/PLOD1-S178R-Rv: GTCGCTGTCCTGGCCCTCCCACTCGG and PLOD3-D190R-Fw: CGCGACCAGCTGTTCTACACACGGCT/PLOD3-D190R-Rv: GTCATCATCCTTGTACTTCCACTGGCG, respectively. Suspension growing HEK293F cells (Life Technologies, UK) were transfected at a confluence of 106 cells ml−1, using 1 μg of plasmid DNA and 3 μg of linear polyethyleneimine (PEI; Polysciences, Germany).25 Cells were harvested 6 days after transfection by centrifuging the medium for 15 minutes at 1000 g. The clarified media were filtered using a 0.2 mm syringe filter and the pH was adjusted to 8.0 prior to affinity purification as previously described.16 All proteins were isolated from the medium exploiting the affinity of the 6xHis tag for the HisTrap Excel (GE Healthcare, USA) affinity column. The N-terminal 6xHis-tag was removed by incubating the purified samples for 2 hours at RT with TEV protease. The purified proteins, as assessed from SDS-PAGE analysis under reducing and non-reducing conditions, were further polished using a Superdex 200 10/300 GL (GE Healthcare) equilibrated in 25 mM HEPES/NaOH, 200 mM NaCl, pH 8.0, to obtain homogenous protein samples; peak fractions containing the protein of interest were pooled and concentrated to 1 mg mL−1 and stored at −80 °C until usage.
Enzymatic activity measurements
Evaluation of glucosylgalactosyltransferase enzymatic activity was performed following the protocols described in Scietti et al.16 using gelatin as substrate. Recombinant PLOD1 or PLOD3 samples at 0.2 mg mL−1 were mixed with 4 mg mL−1 gelatin (solubilized in water through heating denaturation at 95 °C for 10 min), 50 μM MnCl2, and 100 μM UDP-Glc in a total volume of 5 μL, and let incubate for 1 hour at 37 °C. All reactions were stopped by heating at 95 °C for 2 min, prior to transfer into Proxiplate white 384-well plates (Perkin-Elmer), then 5 μL of the UDP-Glo luminescence detection reagent (Promega) were added and incubated 1 hour at 25 °C. For assessment of the lysyl hydroxylase activity, recombinant PLOD1 or PLOD3 samples at 0.2 mg mL−1 were mixed with 4 mg mL−1 gelatin (solubilized in water through heating denaturation at 95 °C for 10 min), 50 μM FeCl2, 100 μM 2-oxoglutarate, and 500 μM sodium ascorbate in a total volume of 5 μL, and let incubate for 1 hour at 37 °C. Reactions were stopped by heating at 95 °C for 2 min, prior to transfer into Proxiplate white 384-well plates (Perkin-Elmer), then 5 μL of the Succinate-Glo reagent I (Promega) were added and let incubate 1 hour at 25 °C, after that 10 μL of the Succinate-Glo reagent II (Promega) were added and let incubate 10 minutes at 25 °C.
The plates were then transferred into a GloMax plate reader (Promega) configured according to manufacturer’s instructions for luminescence detection. All experiments were performed in triplicates. Control experiments were performed using identical conditions by selectively removing PLOD1/3, donor or acceptor substrates. Data were analyzed and plotted using the GraphPad Prism 7 (Graphpad Software, USA).
Differential Scanning Fluorimetry (DSF)
DSF assays on PLOD1 and PLOD3 samples at a concentration of 1 mg mL−1 in a buffer composed of 25 mM HEPES, 100 mM NaCl, pH 8 were performed on using a Tycho NT.6 instrument (NanoTemper Technologies GmbH, Germany). Data were analyzed and plotted using the GraphPad Prism 7 (Graphpad Software, USA).
Statistical analysis
Data are represented as mean ± SD for continuous variables (one-way analysis of variance, ANOVA), as acquired in three or more independent experiments. Shapiro-Wilks criteria was used to evaluate normality of the data, and if non-normally distributed, Man-Whitney, Wilcoxon signed-rank and Kruskall-Wallis with Dunn’s multiple comparisons were used. Where appropriate, Chi-square was used to evaluate for differences in categorical variables. An α < 0.05 was defined as the cutoff for significance. JMP ® Pro, Version 12.2.0. SAS Institute Inc., Cary NC 1989–2019 was used to perform all statistical analyses, with exception of the enzymatic studies, which were analyzed and plotted using GraphPad Prism 7 (Graphpad Software, USA).
Results
Aortopathic Disease and Collagen Studies in a Family with FTAAD
We identified an incidental 50 mm ascending aortic aneurysm (sinus of valsalva) in an otherwise healthy, normotensive 39-year-old male. His family history was significant for a Type A aortic dissection that occurred in his half-brother at 36-years-old. This was treated emergently with placement of a 28 mm (valve-sparing) Hemashield graft. His brother had no known medical history prior to presentation of the dissection. The available extended family members underwent screening echocardiogram, and we reviewed this data to determine the prevalence of ascending aortic aneurysm in the family. (Fig 1, A) The proband (II-2, Fig1, A) was found to have a severely dilated aorta (cross-sectional-to-height (CSA/Ht) ratio >10.0), and his half-brother (II-6) had a moderate-sized aortic aneurysm prior to dissection (CSA/Ht 7.0 – 10.0). Mild aortic dilatation (CSA/Ht 5.0 – 6.9) was found in 3 other family members (I-2, II-5, III-1) who were asymptomatic. (Fig 1, B)
Figure 1. PLOD1-linked Aortopathy.
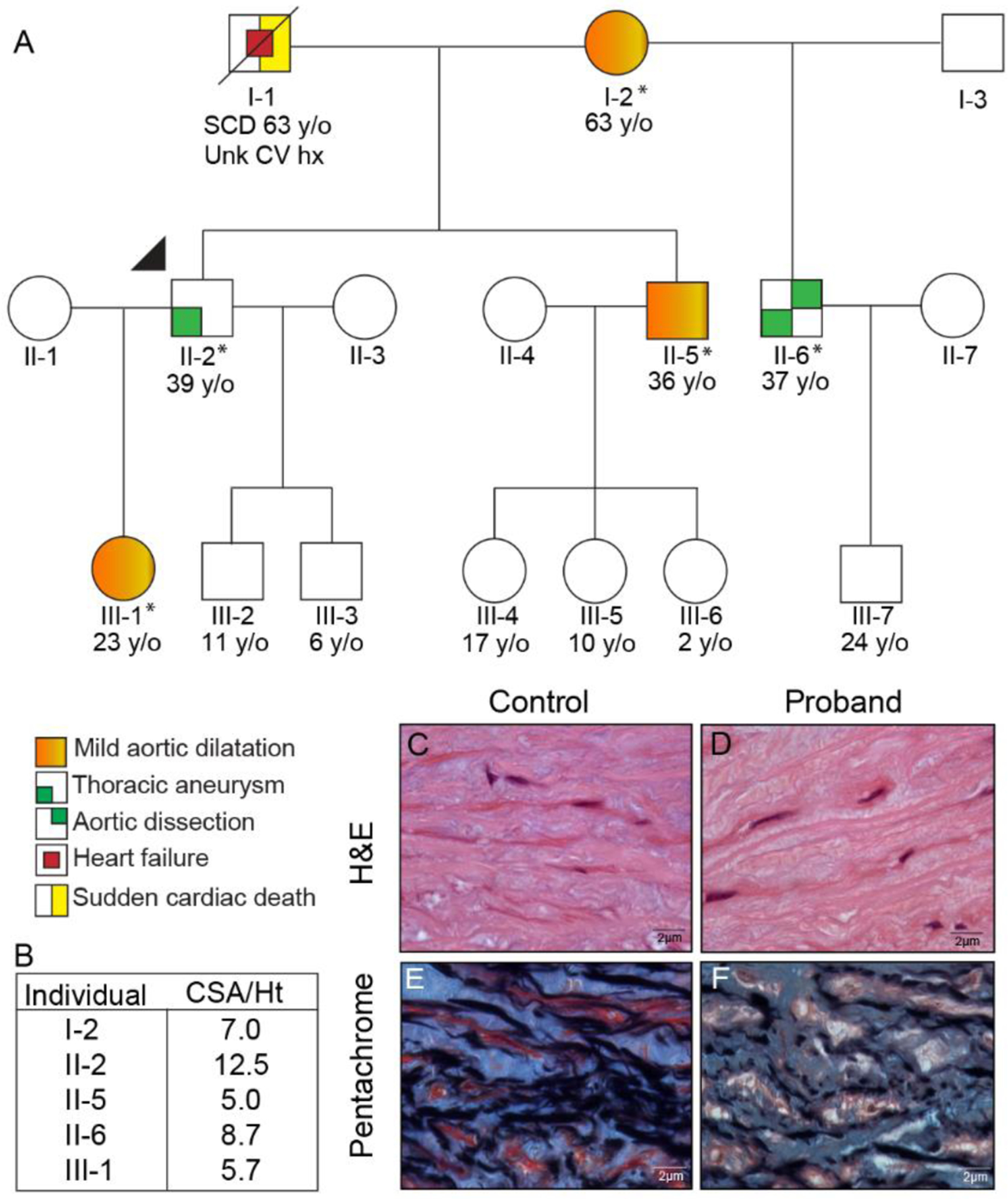
A 3-generation pedigree was compiled after a proband (II-2, arrow) with aortic aneurysm was identified (*Exome sequencing completed and PLOD1 variant detected). In total, 2 members of the family demonstrated aortic aneurysm and/or dissection, and 3 had mild aortic dilatation (A). Standard 2-dimensional echocardiographic imaging was reviewed to determine the Cross-sectional area / Height ratio (CSA/Ht) of affected family members, and identified 2 (II-2, II-6) with either a severely or moderately dilated aorta (CSA/Ht > 10.0 & 7.0, respectively) and 3 (I-2, II-5, III-1) with mild aortic dilatation (CSA/Ht 5.0–6.9) (B). Histologic sections obtained from the proband’s resected aneurysm demonstrated no pathologic alterations by Hematoxylin and Eosin stain. The Pentachrome stain however, shows closely packed elastic lamellae (similar to the control aorta); however, with small areas of separation and loss of lamellae along with increased glycosaminoglycans in the proband. (All original magnifications 100×) (C–F).
Clinical examination of the proband did not demonstrate any typical exam findings from the revised ghent nosology.26 Genetic testing was performed in the proband (II-2), but did not identify a causal variant in ACTA2, CBS, COL3A1, COL5A1, COL5A2, EFEMP2, FBN1, FBN2, FLNA, MAT2A, MED12, MYH11, MYLK, NOTCH1, PRKG1, SKI, SLC2A10, SMAD3, SMAD4, SMAD6, TGFB2, TGFB3, TGFBR1, TGFBR2. However, there was a variant of unknown significance (VUS) identified in the PLOD1 (c.534C>A p. (Ser178Arg), (MAF 7.9e−06) gene. The proband’s half-brother (II-6) who had sustained a prior aortic dissection also underwent standard clinical genetic testing, which revealed the same PLOD1 p. (Ser178Arg) variant in addition to a duplication VUS in MYH11 (Exons 2–43; 3 copies).
The proband was ultimately referred for elective aortic root replacement and pathologic specimens acquired from surgical resection were evaluated and compared to non-diseased sections of the ascending aorta available through Lifeline of Ohio. Histologic sections from the proband showed relatively preserved aortic medial structure with only mild focal separation and loss of elastic fibers and increased glycosaminoglycans. (Fig. 1 C–F, Fig. S1) This finding was attenuated compared to most FTAAD cases, which demonstrate significant medial necrosis.27
To qualitatively assess the impact of collagen in the proband, we first evaluated transmission electron microscopy (TEM) and grossly found no significant difference in the appearance of collagen fibrils. However when we assessed the size of 100 randomly selected fibrils, we found evidence of mild fibril narrowing in the proband (Proband: 25.7 ± 4.3 vs. Controls: 29.7 ± 5.8nm, p< 0.0001), which may reflect changes induced by the variant or the aneurysmal tissue itself. (Fig. 2 A–D, K) In addition, we performed Picrosirius red staining with polarized microscopy,28 which demonstrated that collagen in the proband was similar to control samples. (Fig. 2 E–G) This finding is dissimilar to what has been demonstrated in bicuspid aortic valve associated aortopathy,29 and together with the TEM data, suggests that the final collagen product grossly resembles mature tissue, however microscopically there is evidence of diminished girth in the proband, suggesting that post-translational modification of collagen is impacted.
Figure 2. Collagen Studies.
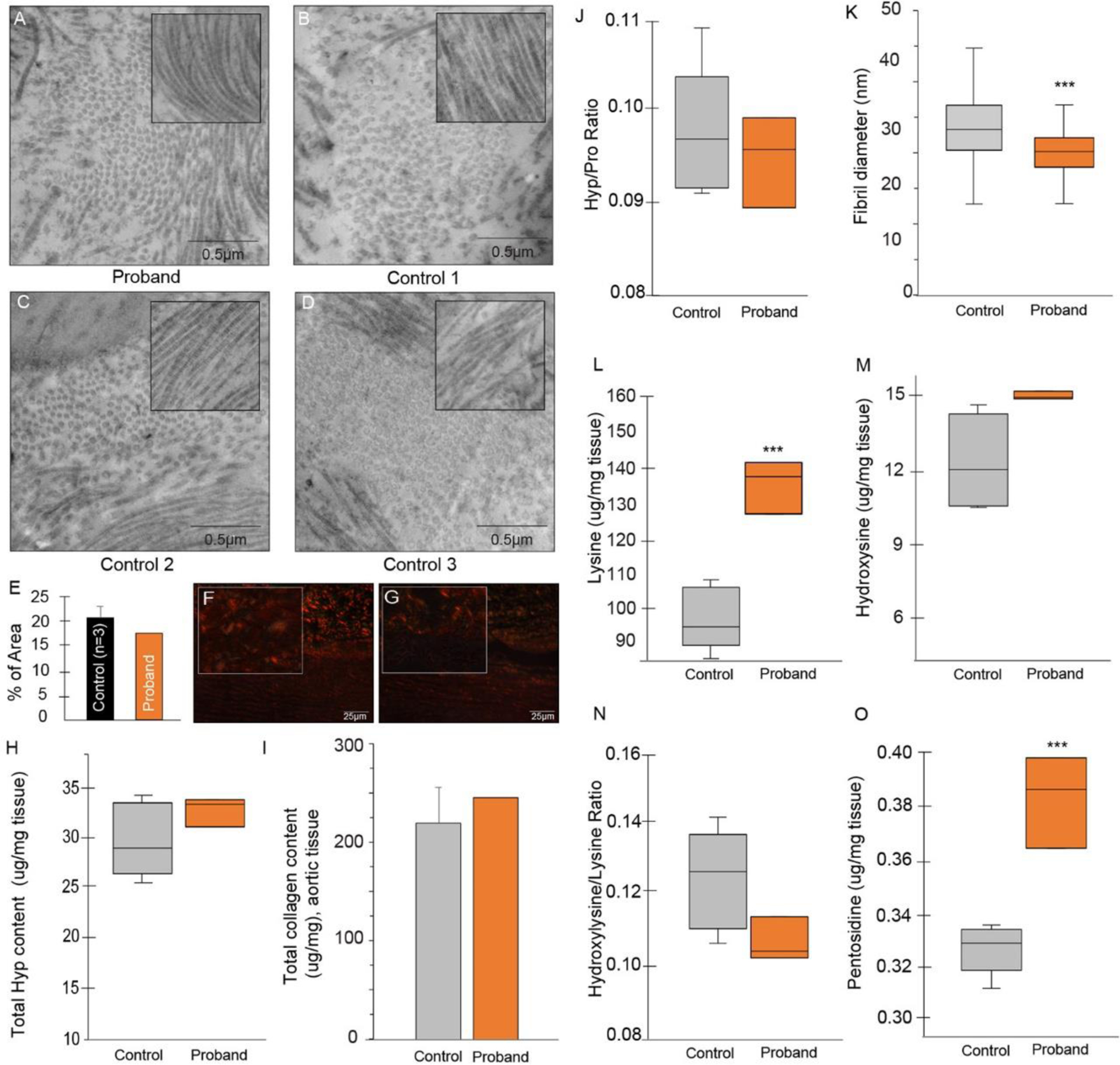
Transmission electron microscopy of the proband (A) and control samples (B-D) showing fibril size and organization pattern (inset: longitudinal fibril organization, same scale); 100 randomly selected fibers from each sample were subject fibril diameter measurements and fibril size was found to be ~ 3nm smaller in the proband (25.7 + 4.3 vs. 29.7 + 5.8 nm, p< 0.0001 (K). Picrosirius red staining (PSR, 10×) of representative control (F) and proband (G) tissue show a similar degree of mature collagen (red/orange) in the proband; magnifications are at 100x and quantification of PSR is shown in E. In collagen studies, the total hydroxyproline (Hyp) (H), collagen (I) and mean hydroxyproline-to-proline ratio was similar (control: 0.12 ± 0.03 vs. proband: 0.09 ± .001) in controls and the proband (J). However, the substrate for the PLOD1 enzyme, lysine, was significantly higher in the proband (140 ± 8 vs. 97 ± 9μg/mg aortic tissue, p = 0.0003) (L), yet the reaction product hydroxylysine was not different in controls versus proband (M) nor was the ratio of hydroxylysine to lysine (N). Finally, relative pentosidine level was assessed as a marker of collagen turnover, and found to be preserved (low collagen turnover) in the proband (0.043 ± 0.001 vs. 0.033 ± 0.000 μg/mg aortic tissue, p = 0.0005) (O). Unless otherwise specified, triplicate samples from 3 control patients and the proband were used for collagen analysis. *** p < 0.0001.
To quantitatively assess collagen production, we assessed the level of hydroxyproline and the hydroxyproline-to-proline ratio, both of which were preserved in the proband (Proband: 33.1 μg/mL vs. Controls: 29.6 ± 4.1 μg/mL and 0.32 ± 0.01 vs. 0.33 ± 0.02, p = 0.513) as was the total amount of collagen. (Fig. 2 H–J) The PLOD1 enzymatic substrate lysine was evaluated and found to be significantly higher in the proband (140 ± 8 vs. 97 ± 9 μg/mg aortic tissue, p = 0.0003), however the enzymatic product hydroxylysine was not different, nor was the hydroxylysine-to-lysine ratio, suggesting 1) preservation of lysyl hydroxylase activity of the PLOD1 enzyme, and 2) that increases in lysine may be compensatory for downstream changes in collagen production. (Fig. 2 L–N) Finally, we assessed the relative level of pentosidine, a known marker of collagen turnover, and found it to be significantly higher in the proband, consistent with preservation of, and/or lower collagen turnover (0.043 ± 0.001 vs. 0.033 ± 0.000 μg/mg aortic tissue, p = 0.0005). (Fig. 2O)
PLOD1 Variant c.534C>A (p. Ser178Arg) is Implicated in Human FTAAD
To determine if there were additional variants that may be responsible for the presentation in this pedigree, 4 family members (I-2, II-2, II-6, III-1) underwent whole exome sequencing. Putative causal variants were evaluated if 1) they were shared between affected family members, 2) the variant was rare with MAF ≤ 0.01% and 3) there was potential functional impact (search limited to: missense, nonsense, frameshift or splice site variants), particularly in the cardiovascular system. We identified 1,594 variants in coding regions, and after filtering for variants with known cardiovascular pathogenicity (n = 52) and demonstrable expression in cardiovascular tissue (n = 37), we hand-searched the list and identified 6 that met criteria (Table S1). Of the 6 potential genes, the only mutation implicated in connective tissue disease with co-segregation in affected members was PLOD1 p. (Ser178Arg). Aside from II-6, other members of the family did not have the MYH11 variant. Therefore, we concluded it was not likely the primary pathogenic variant (lack of co-segregation in all members with disease), however this duplication may have potentiated abnormal vascular pliability and fragility resulting in proclivity to aneurysm formation and dissection in II-6. We selectively evaluated the proband and control tissue for α-smooth muscle actin (αSMA), COLA3A1 and PLOD1 (IHC), and found that both PLOD1 and COL3A1 (PLOD1 >> COL3A1) were upregulated in the proband, whereas αSMA, was no different (Fig S1). To determine if this loss-of-function variant resulted in any change to the concentration of PLOD1 in human aortic tissue, we quantitatively evaluated the amount of PLOD1 in 4 control specimens of non-diseased human aorta and the proband. The proband demonstrated a 43% increase in PLOD1 (Band-to-Ponceau ratio: 0.33 vs. 0.188 ± 0.11), consistent with upregulation of PLOD1 (as assessed by quantification on Ponceau), likely in response to the loss-of-function induced by the variant. (Fig S2)
PLOD1 encodes for lysyl hydroxylase-1 (LH1, also known as PLOD1), an enzyme required to convert lysine to hydroxylysine post-translationally at the rough endoplasmic reticulum (rER). LH1 is essential for the formation of procollagen and the eventual triple helical structure of collagen, followed by crosslinking and maturation.29 Each of these steps is required for the development of a mature ECM, which provides the structure for VSMC stability. PLOD1 is located on the short arm of chromosome 1 (1p36.22) and is comprised of 41,338 base pairs which make up 727 amino acids (Fig 3, A, B). The PLOD1 p. (Ser178Arg) variant has not previously been reported, and real-time algorithms used to predict expected change in protein structure and function are lacking (designated VUS). We evaluated conservation of the Ser178 amino acid residue and pathogenicity of the p. (Ser178Arg) variant in multiple in silico analysis techniques (Table 1).30–36 This analysis demonstrates that the Ser178 amino acid residue is highly evolutionarily constrained, and that the surrounding amino acid residues are conserved between orthologs, suggesting that the functional effect of this missense variant is damaging (Fig 3, C).37
Figure 3. PLOD1 Amino Acid Conservation & Protein Structure/Function.
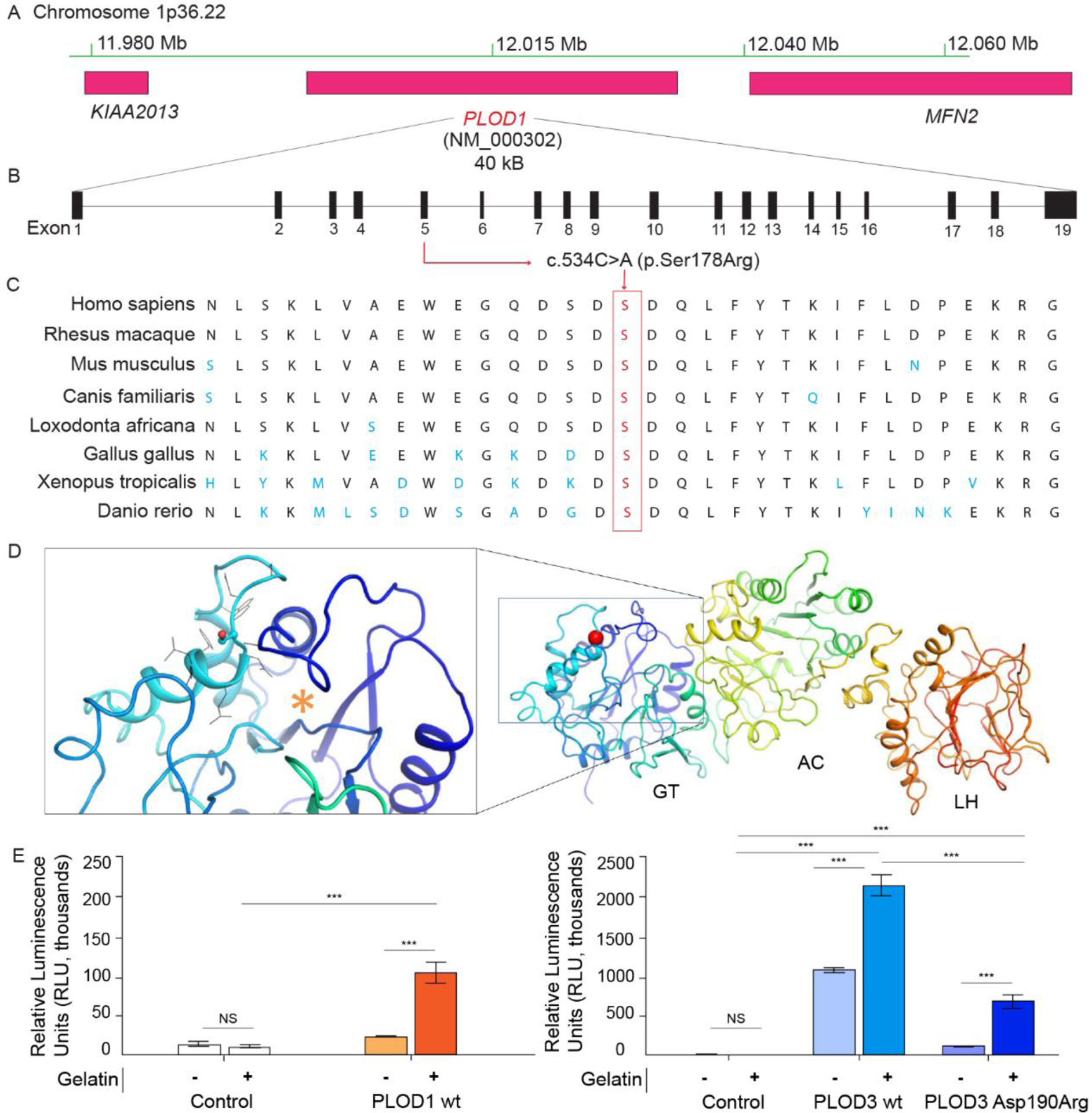
Sequencing identified a novel PLOD1 (c.534C>A (p. (Ser178Arg)), MAF 7.9e−06) mutation on chromosome 1p36.22 (A, B). Orthologous protein sequence alignment of PLOD1 from different species and outlined (red) amino acid proteins demonstrate that the novel mutation occurred in a highly conserved motif (C) of the glycosyltransferse (GT) domain (red sphere), close to the putative UDP-glycan donor substrate binding site (orange asterisk in the inset) (D). Activity assays identified PLOD1 glucosyltransferase (GlcT) activity on gelatin substrate (left) which is ten-fold lower if compared to PLOD3 (right) (E). Error bars represent standard deviations from average of triplicate independent experiments. Statistical evaluations based on pair sample comparisons between uncoupled and coupled assay values using Student’s t-test. NS, non-significant; ***, P-value < 0.001.
Table 1.
Conservation and Pathogenicity of PLOD1 p. (Ser178Arg)
| Method | Score | Prediction |
|---|---|---|
| PhastCons30, 36 | 0.98 | Conserved |
| GERP34 | 4.61 | Conserved |
| LRT33 | 0.00 | Deleterious |
| Mutation Taster35 | 0.99 | Disease-causing |
| SIFT31, 32 | 0.00 | Damaging |
| PROVEAN31, 32 | −2.72 | Damaging |
Conservation and pathogenicity of PLOD1 p. (Ser178Arg) missense variant via multiple in silico analyses
PLOD1-associated Enzyme Activity
To better assess the impact of the variant on PLOD1 enzymatic function specifically, computational modeling of the possible effects of the p. (Ser178Arg) variant on the overall PLOD1 structure was undertaken. Computational predictions consistently indicated that minor destabilization effects may occur, without significant impact on folding stability and/or quaternary structure assembly (Table S2). Therefore, we hypothesized that the p. (Ser178Arg) variant could result in a well-folded protein product, possibly affected by alterations in its enzymatic function. Ser178 is located in the N-terminal domain of PLOD1, corresponding to the glycosyltransferase (GT) catalytic site of the homologous multifunctional enzyme PLOD3 (Fig 3, D, Fig S3).8, 16 In the homolog, the corresponding residue Asp190 was identified using site-directed mutagenesis as critical for glycosyltransferase enzymatic activity,9 and here we used this site for comparative computational mutation analysis (Fig S3). To date, PLOD1 has not been described to possess glycosyltransferase activity in vivo or in vitro. To explore potential PLOD1 glycosyltransferase activities in vitro, we performed the same glycosyltransferase assays used to evaluate PLOD3 enzymatic activity,16 and observed that only in the presence of gelatin acceptor substrates, PLOD1 is capable of processing UDP-glycan donor substrates into free UDP, suggesting possible glucosylgalactosyltransferase activity (GlcT). (Fig 3E) Albeit ten-fold lower compared to multifunctional lysyl hydroxylase-glycosyltransferase PLOD3,16 such observed donor substrate processing appears exclusively specific for UDP-glucose, suggesting differences in UDP-sugar recognition in the PLOD1 GT catalytic site compared to its homolog. Attempts to obtain recombinant PLOD1 p. (Ser178Arg) resulted in extremely low enzyme yields, hampering subsequent biochemical characterizations. Conversely, we successfully produced PLOD3 p. (Asp190Arg) with yields and stability slightly lower than wild-type PLOD3, but clearly compatible with a folded protein product based on thermal stability, oligomeric state and LH activity (Fig 4). When probed in GlcT assays, this variant showed a strongly reduced activity compared to wild-type PLOD3 (Fig 3E). Taken together, these results suggest that the charge reversal introduced by the p. (Ser178Arg) mutation might affect the N-terminal GT domain of PLOD1, its interactions with collagen acceptor substrate molecules, and possibly, thus far uncharacterized GlcT activity of this enzyme. Glycosylation of collagen hydroxylysines is an extremely conserved post-translational modification with increasingly reported relevance on ECM integrity and stability.38, 39 In this respect, malfunctions in the newly identified GlcT activity of PLOD1 (as may be the case in this variant) may severely impact on mature collagen secretion and overall ECM organization.
Figure 4. Biochemical characterization of recombinant protein samples used in this study.
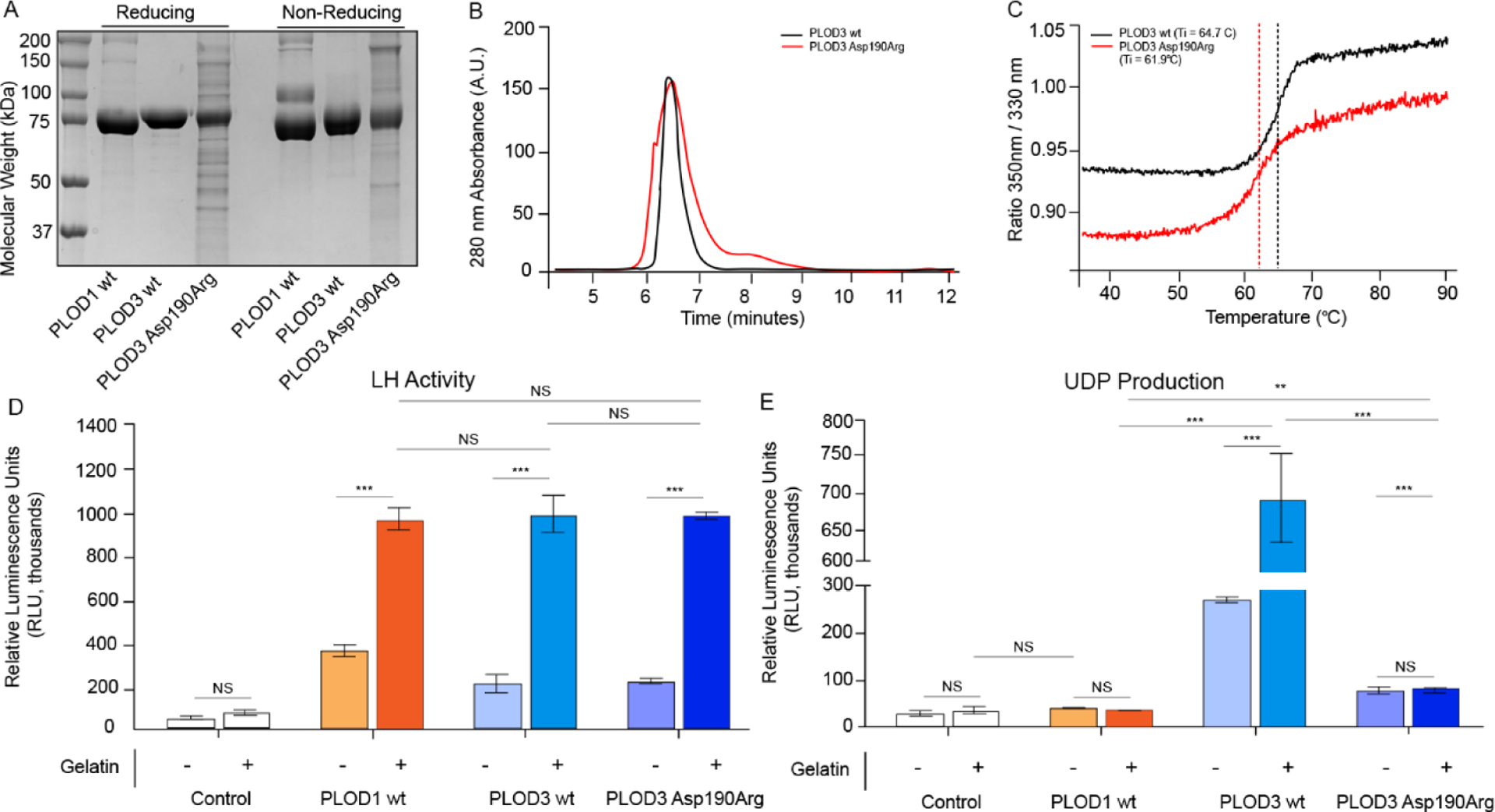
The folding stability and enzymatic activity of recombinantly-produced PLOD1 and PLOD3 enzymes were assessed using reducing and non-reducing SDS-PAGE (A) (M: molecular weight marker). Comparison between PLOD3 wild-type and Asp190Arg mutant using analytical gel filtration (B) and differential scanning fluorimetry (C) showed that both samples are in the correct folded state. This was further confirmed by LH activity assays (D) performed on all recombinant samples, consistently showing high activity on gelatin substrates. Galactosyltransferase activity assays monitoring UDP production (E) show that, contrary to promiscuous processing of UDP-Glucose and UDP-Galactose by wild-type PLOD3, wild-type PLOD1 substrate processing is specific for UDP-Glucose, and that the PLOD3 Arg190Asp mutation strongly affects this enzyme function. Error bars represent standard deviations from average of triplicate independent experiments. Statistical evaluations based on pair sample comparisons between uncoupled and coupled assay values using Student’s t-test. NS, non-significant; **, P-value < 0.01; ***, P-value < 0.001.
PLOD1 Deletion Induces Vascular Smooth Muscle Cell Contraction
Given our findings supporting new putative GT activity of PLOD1, and the potential for alteration of the ECM, we sought to investigate the impact of the variant at the ECU, where VSMCs play an integral role in contributing to maintenance of arterial integrity and vascular stability. To determine whether PLOD1 inhibition had any effect on VSMC contraction, we performed experiments on cultured human aortic smooth muscle cells and intentionally silenced (si-RNA) PLOD1 (achieving 91% silencing of the gene, Fig 5, A, Fig S4). Comparisons between control and si-PLOD1 knockdown human aortic VSMCs were performed to determine if PLOD1 impacted secretory, proliferative and/or contractile function.
Figure 5. PLOD1 pathogenicity in aortopathy is linked to vascular smooth muscle cell changes.
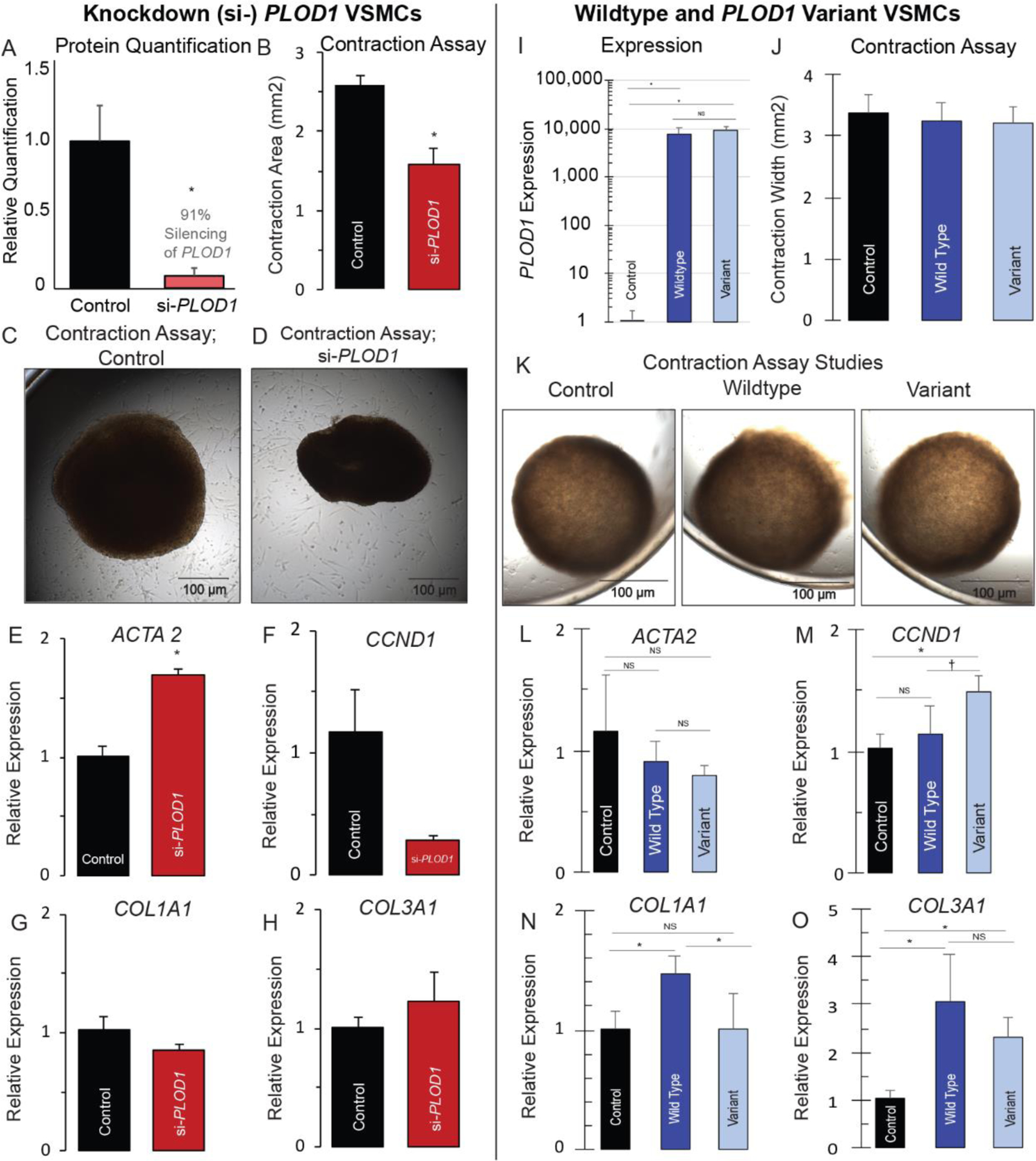
Left panel: Human vascular smooth muscle cells (VSMCs) were cultured and PLOD1 was intentionally silenced (91% si-RNA, *p< 0.05, A). Collagen gel-based assays were used to perform VSMC contraction studies, which showed that si-PLOD1 knockdown cells demonstrated hypercontractility (1.59 mm2±0.48 vs 2.59±0.312, p<0.01) (B–D). The same si-PLOD1 VSMCs demonstrated upregulation of contractile elements (ACTA2 (1.69±0.06 vs 1.01±0.09, p<0.01) (E) and MYH11 (Fig. S4), with insignificant reduction in the proliferative marker CCND1 (1.17+0.33 vs. 0.30+0.04, p=0.56) (F) and no significant difference in secretory elements COL1A1 and COL3A1 (G, H). Right panel: VSMCs were cultured and intentionally transfected with wildtype (WT, overexpression of PLOD1) or the p. (Ser178Arg) variant; WT and variant VSMCs demonstrated ~ 10,000-fold increase in PLOD1 and the variant, respectively (I). We compared contraction studies and genetic markers in control, wildtype and variant VSMCs. There was no difference in VSMC contractility in any of the cell lines (J, K). In WT cells there was no evidence of change in the contractile marker ACTA2 (L), however the proliferative marker CCND1 was increased in the variant, but not WT line (M). Finally, there was evidence of induction of a secretory phenotype in WT cells (increased COL1A1 and COL3A1), yet in the VSMCs transfected with the variant, this response was attenuated, consistent with a loss-of-function of PLOD1 (N, O). * p < 0.05.
Collagen gel-based assays were used to perform VSMC contraction studies and demonstrated hypercontractility of si-PLOD1 cells when compared to control cells (1.59 mm2 ± 0.48 vs 2.59 ± 0.31, p < 0.01) (Fig 5, B–D). To determine the potential etiology of si-PLOD1 hypercontractility, we performed qPCR to evaluate genetic markers of the secretory, proliferative and contractile phenotypes. The si-PLOD1 VSMCs displayed upregulation of the contractile genetic marker ACTA2 (1.69 ± 0.06 vs 1.01 ± 0.09, p < 0.01, Fig 5, E). Given the changes found in ACTA2, we assessed a second contractile marker, MYH11, and found that this was also upregulated in the si-PLOD1 VSMCs (4.71 ± 1.02 vs 1.03 ± 0.04, p < 0.05, Fig S4C). We also evaluated a proliferative marker (CCND1) and secretory markers (COL1A1 and COL3A1), but found no significant changes in these genes (Fig 5, F–H). Although statistically insignificant, CCND1 appeared to be reduced in the si-PLOD1 line, and to confirm that proliferation was not substantially altered, we assessed a second proliferative marker (CCND2) in this line, and confirmed there was no significant difference (Fig S4C). Taken together, this data suggests that VSMCs undergo phenotypic switching based upon the level of PLOD1 present. In the particular case where a secretory phenotype may be favorable, such as when there is low collagen substrate, attenuation of secretory function as was seen with the variant, could contribute to vascular pathogenicity. The VSMC data in combination with the newly identified enzymatic activity of PLOD1 hint that, similar to its homolog PLOD3, the enzyme is important for the development of mature collagen fibril formation through both lysine hydroxylation (LH activity) and their subsequent glycosylation (GlcT activity).
Novel c.534C>A (p. (Ser178Arg)) Variant Induces Loss of Function and VSMC Secretory Changes
To investigate the precise impact of the PLOD1 variant on functional changes in VSMCs, we cultured human aortic smooth muscle cells and intentionally transfected them with wildtype or the p. (Ser178Arg) variant (Fig 5, I). We evaluated contractile function with collagen gel-based assays and gene expression of contractile, proliferative and secretory markers comparing the PLOD1 p. (Ser178Arg) variant to both wild-type transfected and control VSMCs. There was no significant difference in contractile function via collagen contraction assay in the wildtype or variant PLOD1 VSMCs, and the contractile marker ACTA2 was no different in WT and variant cells (Fig 5, J–M). However, overexpression of wildtype PLOD1 resulted in increased expression of secretory markers (COL1A1 1.47 ± 0.16 vs 1.01 ± 0.15, p < 0.05 & COL3A1 3.06 ± 0.98 vs. 1.02 ± 0.19, p < 0.05), and was partially attenuated with overexpression of the p. (Ser178Arg) variant (COL1A1 1.00 ± 0.30 vs 1.47 ± 0.16, p = 0.07 & COL3A1 2.33 ± 0.38 vs. 3.06 ± 0.98, p < 0.29) (Fig 5, N, O). Taken together, our data provides evidence that the variant leads to a loss-of-function in PLOD1. In the presence of the variant, abnormal enzyme folding likely results in impaired collagen lysine glycosylation to support mature fibril formation and strengthen the integrity of the ECM. These changes likely result in compensatory phenotypic VSMC switching to increase the production of procollagen and the PLOD1 substrate lysine, a necessary adaptation that is attenuated in the presence of the variant.
Discussion
Here we demonstrate evidence of human aortopathic disease in a family with a novel PLOD1 (c.534C>A, p. (Ser178Arg)) variant. We demonstrated novel PLOD1 GT activity, describe a putative role for variant-induced misfolding in a homologous mutation, identify relatively mature collagen with mild fibril narrowing and report VSMC phenotypic switching contributing to PLOD1-mediated FTAAD. (Fig 6)
Figure 6. Collagen, enzymatic and vascular smooth muscle cell studies.
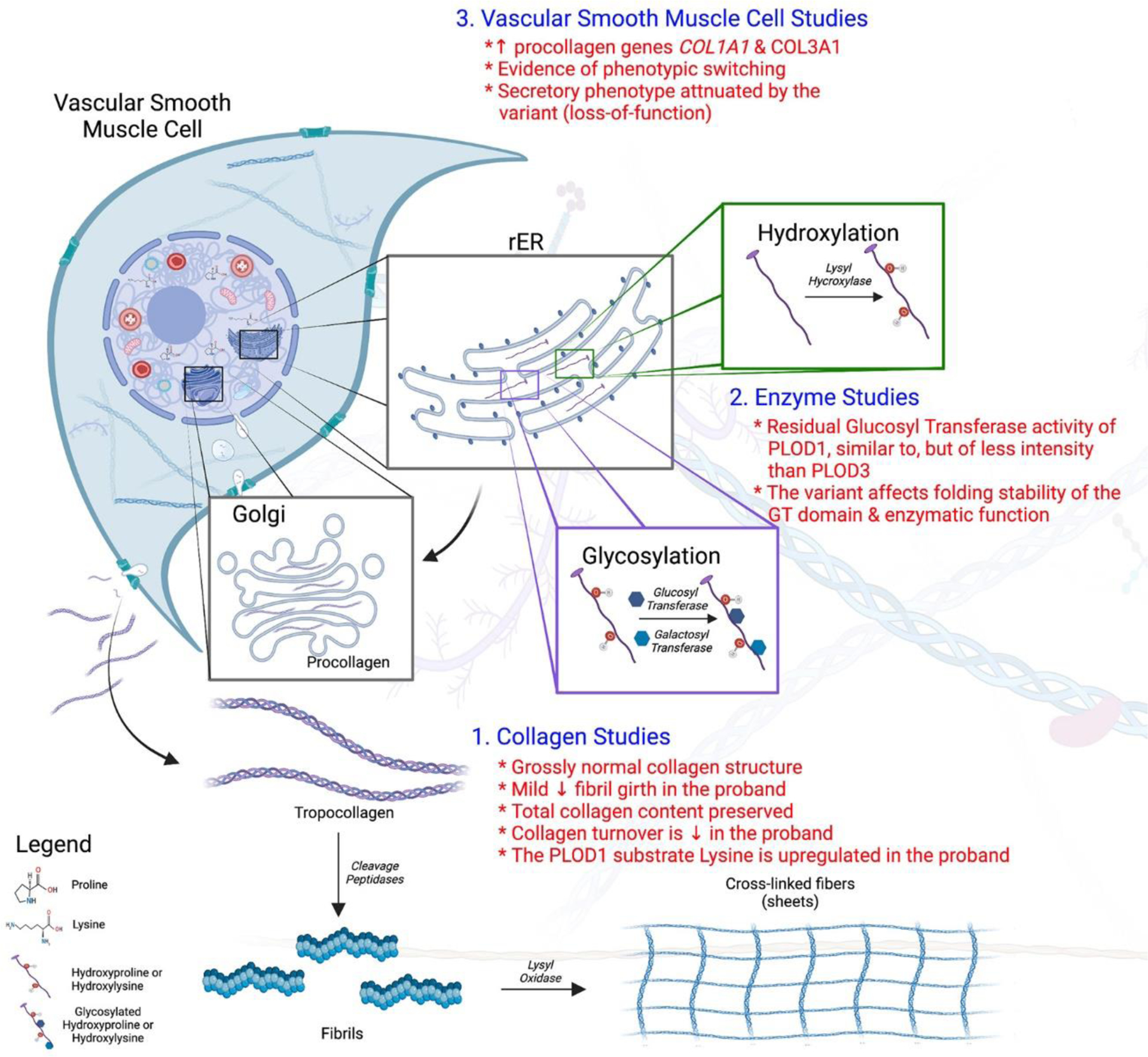
Collagen biosynthesis is a complex process involving hydroxylation, glycosylation, cross-link formation and tertiary structure integration. In this study we evaluated 1) collagen quantity and quality, 2) enzymatic function and 3) vascular smooth muscle cell function. Findings of this study include (in red): grossly normal appearing mature collagen and collagen content albeit with mild fibrillar narrowing, preservation of collagen turnover and likely compensatory upregulation of the PLOD1 substrate lysine, new GlcT functionality of the PLOD1 enzyme, possible aberrant enzyme folding in the p. (Ser178Arg) variant and vascular smooth muscle cell phenotypic switching with attenuation of the secretory state in the variant. Figure created with Biorender.com.
Historically, PLOD1 has been implicated in autosomal recessive non-vascular kyphoscoliotic EDS (formerly kEDS Type VI),39 yet here we provide evidence for vascular disease beyond kEDS. kEDS is typically characterized by hyperextensible joint and skin tissue, easy brusibility and kyphoscoliosis.40, 41 One of the first clinical series reported EDS Type VI findings in a pediatric population in 1989, where the diagnosis was made based upon clinical findings and reduced PLOD1 concentration in fibroblasts.42 In this neonatal cohort, where the genetic cause of PLOD1 deficiency was unknown (i.e. unknown genetic variant), 2 (17%) neonates presented with severe phenotypes (hypotonia, motor delay, weakness and joint dislocation) and demonstrated arterial fragility (vertebral and femoral artery ruptures) in the peripheral, but not central arterial vasculature. Based upon this early work, it had been postulated that variants in PLOD1 may induce vascular fragility, however this has not borne out in classic adolescent and adult kEDS disease outside of case reports, and there continues to be no direct evidence of clear association between HTAD/FTAAD and PLOD1 mutations.43 However, there have been a small number of dissections reported in medium-sized arteries from case reports44 and a systematic review, indicating that the type of PLOD1 variant present may be important to recognize sub-populations with elevated vascular risk.43 Additionally, there has been some suggestion that reduction in PLOD1, albeit with normal PLOD1 levels, is associated with the presence of aneurysms in bicuspid-valve associated aortopathy, however the mechanisms by which this occurs remain under study.45
In the human family presented, we have shown evidence of adult aortopathic disease (FTAAD) in the setting of a highly conserved and evolutionary constrained PLOD1 p. (Ser178Arg) variant, in absence of typical kEDS skeletal findings. This variant is likely important in the development of aortic dilatation demonstrated, however we postulate that the disease may be modified by other genetically-linked variants. In this family, the presence of an MYH11 duplication in the proband’s half-brother (II-6) was provocative, in that MYH11 variants have been shown to affect VSMC contraction and HTAD/FTAAD risk independently.6 However, this duplication was not necessary for additional members of the family to demonstrate aortic aneurysm formation, indicating that the underlying aortopathy was not strictly related to this single variant. Rather, we hypothesize that in this individual (II-6), MYH11 likely impacted predisposition to dissection at a smaller aortic diameter. Although it seems reasonable to conclude that the PLOD1 variant likely contributed to the development of aortic aneurysm in this family, it is unclear if this is sufficient to result in dissection. In the case of the family member (II-6) with dissection, it is likely that the MYH11 duplication contributed to a more aggressive phenotype, particularly now that we understand MYH11 is upregulated in the presence of the PLOD1 p. (Ser178Arg) variant.
To begin to appreciate the mechanisms by which seemingly unrelated functional enzymatic, collagen and VSMC changes occur, one must first recall the complex processes required for the biosynthesis of normal collagen. Vascular tissue (aorta) is commonly comprised of types I and III collagens, whose complex biosynthesis involves hydroxylation, glycosylation, cross-link formation and quaternary superstructure assembly. Related to this work, hydroxylation typically occurs on unfolded procollagens through the action of three enzymatic families (prolyl 4-hydroxylases/P4Hs, prolyl 3-hydroxylases/P3Hs and LHs) including PLOD1/LH1. Folding of the N- and C-propeptides precedes carbohydrate attachment (glycosylation), cross-linking and formation of triple helical structures, assisted by resident molecular chaperones and folding enzymes.46 In the proband we demonstrate grossly mature collagen with qualitative studies (TEM, Picrosirius red) albeit with mild fibril narrowing, which may be a result of the variant and/or simply an expected change in aneurysmal tissue itself. We also show that the absolute quantity of collagen was not different in the proband, suggesting that the variant doesn’t substantially affect the amount of collagen present. However, likely compensatory increases in procollagen precursor gene activity (COL1A1, COL3A1), lysine production and reduced collagen turnover were stimulated in an effort to preserve collagen production, presumably due to enzymatic and VSMC changes.
Broadly, the enzymatic family of lysyl hydroxylases function in the lysine hydroxylation of collagens, and when loss-of-function occurs, hyperextensibility and connective tissue disease result.40, 47–50 The structure and function of PLOD3 has been well described, and it is characterized by 3 domains, each with unique enzymatic function: glycosyltransferase (GT), accessory (AC) and lysyl hydroxylase LH domains.16 The identification of the p. (Ser178Arg) mutation in the N-terminal GT domain of the PLOD1 enzyme prompted us for investigating possible additional enzymatic activities resembling those known for homologous multifunctional PLOD3. The computational data indicated that the p. (Ser178Arg) variant likely has minimal impact upon the structure of PLOD1, which was further supported by demonstration of normal LH1 activity. Our analysis suggests that, at least in vitro, PLOD1 is capable of processing the glycosyltransferase donor substrate UDP-glucose in the presence of collagen-like acceptor substrates, hinting for possible glucosylgalactosyltransferase activity in this enzyme isoform, or for critical roles associated to PLOD1: collagen interactions in a non-catalytic site. One counterpoint for clinical significance however, is that this property is 10-fold less than what has been demonstrated in PLOD3. However, human proteomic data would support that PLOD1 is likely relevant for human vascular connective tissue disease, as it is actively expressed in VSMC tissue, unlike PLOD3.51 Nonetheless, further biochemical and in vivo functional investigations are required to verify the biological significance and provide a comprehensive understanding of the implications of this newly-observed PLOD1 function.
Finally, roles for PLOD1 in VSMC physiology have not been elucidated until now. Our data support that PLOD1 is important in maintaining balance between VSMC contractile and secretory function, although it is likely secondarily due to dysfunction first in the GlcT activity of the PLOD1 enzyme. In the complete knockdown line, contractile elements were upregulated and VSMCs demonstrated hypercontractility, whereas overexpression of PLOD1 resulted in upregulation of secretory elements, while preserving a normal contraction profile. This pattern provides in vivo support for VSMC phenotypic switching. VSMC overexpression of the human PLOD1 p. (Ser178Arg) variant led to attenuation of procollagen secretory marker upregulation, supporting pathogenicity via loss of normal PLOD1 function. Further, the possibility that novel GlcT activity of PLOD1 may be reduced or absent in the variant, would support this possibility, and future studies should focus on this aspect. We postulate that the effect of PLOD1 may be active in a “dose-dependent” manner, given that the variant induced a secretory phenotype with no significant contractile changes, yet the complete PLOD1 knockdown model (si-PLOD1) resulted in a hypercontractile phenotype. VSMCs are known to display phenotypic plasticity, allowing for phenotype switching into synthetic/secretory, proliferative and/or contractile phenotypes, depending upon what is needed in the current environment (i.e. injury).52 In cases of heterogenous aortopathic disease, changes in VSMC phenotype are central to the underlying pathophysiology of FTAAD/HTAD, caused in part by changes in expression of contractile proteins.53 VSMC function, and contractility in particular, are hypothesized to contribute to FTAAD via the interaction between cells and the ECM. More specifically, we postulate that aberrant PLOD1 GlcT activity may affect post-translational glycosylation and collagen strength despite only minimal changes on gross collagen structure. This may then trigger compensatory VSMC phenotypic switching to favor collagen precursor secretion, and in the case of the variant, perhaps pathologic attenuation of this compensatory response.
There are limitations inherent to genetic studies in single families such as this. Next generation whole exome sequencing data, although informative, does not provide information about sizable noncoding regions of the genome that may influence disease and/or phenotypes. Additionally, exome sequencing across tissue types may limit the applicability of results more broadly to vascular connective tissue disease. Continued analysis of aortopathic disease, genetic contribution and environmental factors will be required to fully understand the pathogenesis and contribution of new variants to heritable vascular disease and individual risk. Additional future investigation should assess the impact of the variant on: collagen function, tertiary structure, precursor signaling, and novel GlcT enzymatic activity. These studies will allow better understanding of the implications of PLOD1-mediated aortopathic disease. In summary, the identification of a PLOD1 variant in a human family with FTAAD, grossly preserved collagen content and architecture, aberrant PLOD1 putative enzymatic dysfunction and attenuation of compensatory VSMC phenotypic switching with upregulation of procollagen precursors, support roles for this enzyme in human large artery vascular disease. Future research in families with “genetic negative” FTAAD should focus on the molecular impact of genetic changes (i.e. VUS), which will be important to better understand the pathogenesis, and potential future therapeutic targets for vascular connective tissue disease.
Supplementary Material
Brief Commentary.
Background
Heritable thoracic aortic disease (HTAD) and familial thoracic aortic aneurysm and dissection (FTAAD) are important causes of human morbidity and mortality, most of which do not have a readily identifiable genetic cause. In new variants of unknown significance, functional data are important to provide evidence for association between genetic heritability and disease.
Translational Significance
In this study, we identified kindred with a new missense PLOD1 variant and human aortopathic disease. We provide in vitro and in vivo functional data that suggest PLOD1 may impact arterial integrity through newly described enzymatic roles and modulation of vascular smooth muscle cell function.
Acknowledgements
We thank the family presented in this work for participating in this study. We thank Brenda Lilly, PhD, Professor of Pediatrics, for the collagen gel-based contraction assay protocol (Nationwide Children’s Hospital, Columbus OH). Thank you to the Morphology Core at Nationwide Children’s Hospital for performing Picrosirius Red Staining. We thank The James Comprehensive Cancer Center and Office of Research at The Ohio State University for use of mass spectrometry and electron microscopy cores. We thank Dr. Antonella Chiapparino, Dr. Alberta Pinnola and Mr. Matteo De Marco for assistance in the production and characterization of recombinant PLOD1 and PLOD3 enzymes and their variants. All authors have read the journal’s authorship agreement and policy on conflicts of interest.
Funding Support
National Heart, Lung, and Blood Institute Grants: K08HL148701 (EAB), R35HL135754 (PMJ); Italian Association for cancer research MFAG Grant 20075 (FF); Giovanni Armenise-Harvard Foundation Grant CDA 2013 (FF); Mizutani Foundation for Glycoscience Grant 200039 (FF); University of Pavia InROAD program (LS); Italian Ministry for Education, University and Research (MIUR), Dipartimenti di Eccellenza 2018-2022 (FDG, LS, SF, FF).
Abbreviations
- CSA/Ht
Cross-sectional-area to Height ratio
- ECM
Extracellular matrix
- ECU
Elastin-contractile unit
- FTAAD
Familial thoracic aortic aneurysm and dissection
- GT
Glycosyltransferase
- HTAD
Heritable thoracic aortic disease
- kEDS
kyphoscoliotic Ehlers Danlos Syndrome
- PLOD1
Procollagen-lysine 2-oxoglutarate 5-dioxygenase 1
- VSMC (human aortic)
Vascular smooth muscle cell
- VUS
Variant of unknown significance
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Competing Interest
None related to this project, all authors (no relationships with industry). All authors have approved the final version of this article.
References
- 1.Centers for Disease Control and Prevention, National Center for Health Statistics. Underlying Cuase of Death, 1999–2017. Accessed 2019.
- 2.Biddinger A, Rocklin M, Coselli J and Milewicz DM. Familial thoracic aortic dilatations and dissections: a case control study. J Vasc Surg. 1997;25:506–11. [DOI] [PubMed] [Google Scholar]
- 3.Milewicz DM and Regalado ES. Use of genetics for personalized management of heritable thoracic aortic disease: how do we get there? J Thorac Cardiovasc Surg. 2015;149:S3–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Milewicz DM, Carlson AA and Regalado ES. Genetic testing in aortic aneurysm disease: PRO. Cardiol Clin. 2010;28:191–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davis EC. Smooth muscle cell to elastic lamina connections in developing mouse aorta. Role in aortic medial organization. Lab Invest. 1993;68:89–99. [PubMed] [Google Scholar]
- 6.Pinard A, Jones GT and Milewicz DM. Genetics of Thoracic and Abdominal Aortic Diseases. Circ Res. 2019;124:588–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meester JAN, Verstraeten A, Schepers D, Alaerts M, Van Laer L and Loeys BL. Differences in manifestations of Marfan syndrome, Ehlers-Danlos syndrome, and Loeys-Dietz syndrome. Ann Cardiothorac Surg. 2017;6:582–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scietti L, Campioni M and Forneris F. SiMPLOD, a Structure-Integrated Database of Collagen Lysyl Hydroxylase (LH/PLOD) Enzyme Variants. J Bone Miner Res. 2019;34:1376–1382. [DOI] [PubMed] [Google Scholar]
- 9.Wang C, Risteli M, Heikkinen J, Hussa AK, Uitto L and Myllyla R. Identification of amino acids important for the catalytic activity of the collagen glucosyltransferase associated with the multifunctional lysyl hydroxylase 3 (LH3). J Biol Chem. 2002;277:18568–73. [DOI] [PubMed] [Google Scholar]
- 10.Shrestha B and Dunn L. The Declaration of Helsinki on Medical Research involving Human Subjects: A Review of Seventh Revision. J Nepal Health Res Counc. 2020;17:548–552. [DOI] [PubMed] [Google Scholar]
- 11.Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, McGuire AL, Nussbaum RL, O’Daniel JM, Ormond KE, Rehm HL, Watson MS, Williams MS, Biesecker LG, American College of Medical G and Genomics. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15:565–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C and Chanda SK. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10:1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang H, Robinson PN and Wang K. Phenolyzer: phenotype-based prioritization of candidate genes for human diseases. Nat Methods. 2015;12:841–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Milani-Nejad N, Canan BD, Elnakish MT, Davis JP, Chung JH, Fedorov VV, Binkley PF, Higgins RS, Kilic A, Mohler PJ and Janssen PM. The Frank-Starling mechanism involves deceleration of cross-bridge kinetics and is preserved in failing human right ventricular myocardium. Am J Physiol Heart Circ Physiol. 2015;309:H2077–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morikawa Y, Shibata A, Sasajima Y, Suenami K, Sato K, Takekoshi Y, Endo S, Ikari A and Matsunaga T. Sibutramine facilitates apoptosis and contraction of aortic smooth muscle cells through elevating production of reactive oxygen species. Eur J Pharmacol. 2018;841:113–121. [DOI] [PubMed] [Google Scholar]
- 16.Scietti L, Chiapparino A, De Giorgi F, Fumagalli M, Khoriauli L, Nergadze S, Basu S, Olieric V, Cucca L, Banushi B, Profumo A, Giulotto E, Gissen P and Forneris F. Molecular architecture of the multifunctional collagen lysyl hydroxylase and glycosyltransferase LH3. Nat Commun. 2018;9:3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ittisoponpisan S, Islam SA, Khanna T, Alhuzimi E, David A and Sternberg MJE. Can Predicted Protein 3D Structures Provide Reliable Insights into whether Missense Variants Are Disease Associated? J Mol Biol. 2019;431:2197–2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rodrigues CH, Pires DE and Ascher DB. DynaMut: predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res. 2018;46:W350–W355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Laimer J, Hofer H, Fritz M, Wegenkittl S and Lackner P. MAESTRO--multi agent stability prediction upon point mutations. BMC Bioinformatics. 2015;16:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dehouck Y, Kwasigroch JM, Gilis D and Rooman M. PoPMuSiC 2.1: a web server for the estimation of protein stability changes upon mutation and sequence optimality. BMC Bioinformatics. 2011;12:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pires DE, Ascher DB and Blundell TL. mCSM: predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics. 2014;30:335–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng J, Randall A and Baldi P. Prediction of protein stability changes for single-site mutations using support vector machines. Proteins. 2006;62:1125–32. [DOI] [PubMed] [Google Scholar]
- 23.Pires DE, Ascher DB and Blundell TL. DUET: a server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res. 2014;42:W314–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Capriotti EFP, Casadio R. I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005;33:306–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Faravelli SCM, Palamini M, Cancianaj A, Chiapparino A, Forneris F. Optimized Recombinant Production of Secreted Proteins Using Human Embryonic Kidney (HEK293) Cells Grown in Suspension. Bio-protocol. 2021;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chandra A, Patel D, Aragon-Martin JA, Pinard A, Collod-Beroud G, Comeglio P, Boileau C, Faivre L, Charteris D, Child AH and Arno G. The revised ghent nosology; reclassifying isolated ectopia lentis. Clin Genet. 2015;87:284–7. [DOI] [PubMed] [Google Scholar]
- 27.Yuan SM and Jing H. Cystic medial necrosis: pathological findings and clinical implications. Rev Bras Cir Cardiovasc. 2011;26:107–15. [DOI] [PubMed] [Google Scholar]
- 28.Vogel B, Siebert H, Hofmann U and Frantz S. Determination of collagen content within picrosirius red stained paraffin-embedded tissue sections using fluorescence microscopy. MethodsX. 2015;2:124–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jover E, Silvente A, Marin F, Martinez-Gonzalez J, Orriols M, Martinez CM, Puche CM, Valdes M, Rodriguez C and Hernandez-Romero D. Inhibition of enzymes involved in collagen cross-linking reduces vascular smooth muscle cell calcification. FASEB J. 2018;32:4459–4469. [DOI] [PubMed] [Google Scholar]
- 30.Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, Weinstock GM, Wilson RK, Gibbs RA, Kent WJ, Miller W and Haussler D. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choi Y and Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31:2745–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choi Y, Sims GE, Murphy S, Miller JR and Chan AP. Predicting the functional effect of amino acid substitutions and indels. PLoS One. 2012;7:e46688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chun S and Fay JC. Identification of deleterious mutations within three human genomes. Genome Res. 2009;19:1553–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cooper GM, Stone EA, Asimenos G, Program NCS, Green ED, Batzoglou S and Sidow A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15:901–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwarz JM, Cooper DN, Schuelke M and Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11:361–2. [DOI] [PubMed] [Google Scholar]
- 36.Hubisz MJ, Pollard KS and Siepel A. PHAST and RPHAST: phylogenetic analysis with space/time models. Brief Bioinform. 2011;12:41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lamiable A, Thevenet P, Rey J, Vavrusa M, Derreumaux P and Tuffery P. PEP-FOLD3: faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res. 2016;44:W449–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.De Giorgi F, Fumagalli M, Scietti L and Forneris F. Collagen hydroxylysine glycosylation: non-conventional substrates for atypical glycosyltransferase enzymes. Biochem Soc Trans. 2021;49:855–866. [DOI] [PubMed] [Google Scholar]
- 39.Gardelli C, Russo L, Cipolla L, Moro M, Andriani F, Rondinone O, Nicotra F, Sozzi G, Bertolini G and Roz L. Differential glycosylation of collagen modulates lung cancer stem cell subsets through beta1 integrin-mediated interactions. Cancer Sci. 2021;112:217–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yeowell HN and Steinmann B. PLOD1-Related Kyphoscoliotic Ehlers-Danlos Syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K and Amemiya A, eds. GeneReviews((R)) Seattle (WA); 1993. [PubMed] [Google Scholar]
- 41.Yeowell HN and Pinnell SR. The Ehlers-Danlos syndromes. Semin Dermatol. 1993;12:22940. [PubMed] [Google Scholar]
- 42.Wenstrup RJ, Murad S and Pinnell SR. Ehlers-Danlos syndrome type VI: clinical manifestations of collagen lysyl hydroxylase deficiency. J Pediatr. 1989;115:405–9. [DOI] [PubMed] [Google Scholar]
- 43.D’Hondt S, Van Damme T and Malfait F. Vascular phenotypes in nonvascular subtypes of the Ehlers-Danlos syndrome: a systematic review. Genet Med. 2018;20:562–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Henneton P, Legrand A, Giunta C and Frank M. Arterial fragility in kyphoscoliotic Ehlers-Danlos syndrome. BMJ Case Rep. 2018;2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wagsater D, Paloschi V, Hanemaaijer R, Hultenby K, Bank RA, Franco-Cereceda A, Lindeman JH and Eriksson P. Impaired collagen biosynthesis and cross-linking in aorta of patients with bicuspid aortic valve. J Am Heart Assoc. 2013;2:e000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ishikawa Y and Bachinger HP. A molecular ensemble in the rER for procollagen maturation. Biochim Biophys Acta. 2013;1833:2479–91. [DOI] [PubMed] [Google Scholar]
- 47.Eyre D, Shao P, Weis MA and Steinmann B. The kyphoscoliotic type of Ehlers-Danlos syndrome (type VI): differential effects on the hydroxylation of lysine in collagens I and II revealed by analysis of cross-linked telopeptides from urine. Mol Genet Metab. 2002;76:211–6. [DOI] [PubMed] [Google Scholar]
- 48.Ewans LJ, Colley A, Gaston-Massuet C, Gualtieri A, Cowley MJ, McCabe MJ, Anand D, Lachke SA, Scietti L, Forneris F, Zhu Y, Ying K, Walsh C, Kirk EP, Miller D, Giunta C, Sillence D, Dinger M, Buckley M and Roscioli T. Pathogenic variants in PLOD3 result in a Stickler syndrome-like connective tissue disorder with vascular complications. J Med Genet. 2019;56:629–638. [DOI] [PubMed] [Google Scholar]
- 49.Gistelinck C, Weis M, Rai J, Schwarze U, Niyazov D, Song KM, Byers PH and Eyre DR. Abnormal Bone Collagen Cross-Linking in Osteogenesis Imperfecta/Bruck Syndrome Caused by Compound Heterozygous PLOD2 Mutations. JBMR Plus. 2021;5:e10454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Salo AM, Cox H, Farndon P, Moss C, Grindulis H, Risteli M, Robins SP and Myllyla R. A connective tissue disorder caused by mutations of the lysyl hydroxylase 3 gene. Am J Hum Genet. 2008;83:495–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thul PJ, Akesson L, Wiking M, Mahdessian D, Geladaki A, Ait Blal H, Alm T, Asplund A, Bjork L, Breckels LM, Backstrom A, Danielsson F, Fagerberg L, Fall J, Gatto L, Gnann C, Hober S, Hjelmare M, Johansson F, Lee S, Lindskog C, Mulder J, Mulvey CM, Nilsson P, Oksvold P, Rockberg J, Schutten R, Schwenk JM, Sivertsson A, Sjostedt E, Skogs M, Stadler C, Sullivan DP, Tegel H, Winsnes C, Zhang C, Zwahlen M, Mardinoglu A, Ponten F, von Feilitzen K, Lilley KS, Uhlen M and Lundberg E. A subcellular map of the human proteome. Science. 2017;356. [DOI] [PubMed] [Google Scholar]
- 52.Owens GK, Kumar MS and Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. [DOI] [PubMed] [Google Scholar]
- 53.Inamoto S, Kwartler CS, Lafont AL, Liang YY, Fadulu VT, Duraisamy S, Willing M, Estrera A, Safi H, Hannibal MC, Carey J, Wiktorowicz J, Tan FK, Feng XH, Pannu H and Milewicz DM. TGFBR2 mutations alter smooth muscle cell phenotype and predispose to thoracic aortic aneurysms and dissections. Cardiovasc Res. 2010;88:520–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.