

Abstract
Many microsatellite DNA sequences are able to form non-B form DNA secondary structures, such as hairpin loops, cruciforms, triplex DNA or G-quadruplexes. These DNA structures can form a significant impediment to DNA replication and repair, leading to DNA nicks, gaps, and breaks, which can be repaired by homologous recombination (HR). Recent work understanding HR at structure-forming repeats has focused on genetic requirements for replication fork restart, break induced replication (BIR) at broken forks, recombination during and after relocalization of breaks or stalled forks to the nuclear periphery, and how repair pathway choice and kinetics are navigated in the presence of a repeat tract. In this review, we summarize recent developments that illuminate the role of recombination in repairing DNA damage or causing tract length changes within repetitive DNA and its role in maintaining genome stability.
Keywords: microsatellites, recombination, replication fork restart, chromosome fragility, instability, DNA structure
INTRODUCTION
Eukaryotic genomes contain many repetitive DNA sequences that exhibit size instability. Expanded tracts of repetitive DNA sequences are the cause of over 30 genetic diseases and can consist of trinucleotide or larger repetitive units [1]. Some of these expandable repeats can form stable non-B-form DNA structures such as hairpin loops, cruciforms, G-quadruplexes or triplex structures [1]. These non-B form DNA structures can impede normal cellular processes like DNA replication and repair which can result in DNA breakage and inaccurate or failed repair [1,2]. Damage that results in DSBs can be repaired by various types of end-joining (EJ), by annealing of processed ends, or by recombination-based mechanisms using either a sister chromatid or homolog as the template [1,3]. Due to the challenges of aligning DNA across a repetitive sequence, gain or loss of repeat units can occur during both HR and EJ [3]. In addition, recombination is a primary mechanism used in restarting stalled or collapsed replication forks and in repairing gaps left behind the replication fork [4]. In this review we will summarize the important new findings in replication-associated recombination and recombinational repair at DSBs in the context of DNA microsatellites. This area is an emerging field which has implications for our understanding of expandable repeats in relation to genetic disease but also in cancer, as inaccurate repair at repetitive sequences can lead to deletions, genomic rearrangements and loss of heterozygosity.
Replication associated recombination within DNA repeats
DNA structures formed by repetitive DNA sequences are impediments for DNA synthesis and can cause fork stalling or gaps behind the replication fork if bypassed[1,2]. One consequence of impairment of the replisome is repeat instability [1,2]. Though it has long been recognized that mutation of polymerases and helicases leads to repeat instability by increasing stalling and slippage [5], recent work shows that destabilization of the replisome can have dramatic consequences at structure-forming repeats [6,7]. For example, absence of Mrc1 or Tof1 of the fork protection complex leads to fork breakage and frequent deletions of an expanded CAG tract [6], and mutation in Psf1, an essential component of the GINS complex associated with the eukaryotic replicative helicase, increased repeat instability of several different microsatellite sequences including (GAA)25, (TTC)25, (CTG)25, (G)18, (GT)49, (AACGCAATGCG)4 and (CAACGCAATGCGTTGGATCT)3 [7]. In the event of a fork stalling event, two pathways can be used to bypass the impediment, either synthesis by translesion polymerases or template switch. In the GINS mutant, microsatellite instability was dependent on Rad51 and Rad52 and independent of polymerase ζ (Zeta), implicating recombination as a mechanism for repeat instability at unstable replication forks [7]. In situations where the replisome is unstable, BIR may also be employed, as deletion of Pol32 or Pif1 in cells with a destabilized replisome resulted in decreased repeat instability at microsatellites [7].
BIR is a highly mutagenic form of HR where a DSB end invades a region of homology and replicates for several kilobases, potentially to the telomere end [8,9]. Replication during BIR is highly mutagenic as it proceeds via a migrating bubble of conservative DNA synthesis and long tracts of ssDNA accumulate, allowing for increased base damage [10,11]. The accessory subunit of DNA Polymerase δ, Pol32 in S. cerevisiae or POLD3 in mammals, is required for repair via BIR [12,13]. Previous work implicated BIR or broken fork repair (BFR) as a mechanism for repeat-associated large scale expansion at a (CAG)140 tract inserted into yeast [14]. Recent results indicate this mechanism is conserved. A mammalian reporter system with an expanded (CGG)n repeat inserted exhibited an increase in expansions, contractions, and reporter gene mutagenesis. Both instability and mutagenesis depended on genes POLD3, POLD4, RAD51, RAD52 and SMARCAL1, supporting BFR as a mechanism for repeat instability [15](*). In addition, replication stress at either a hairpin-forming (CAG)102 repeat or an expanded purine/pyrimidine (Pu/Py)88 mirror repeat that forms a triplex H-DNA and a G-quadruplex structure in human cells resulted in increased replication associated DSBs [16] (*). Breaks that occurred at expanded CAG tracts were dependent on the MUS81 nuclease, repaired via BIR, and resulted in increased PolD3-mediated mutagenesis [16] (*).
Interstitial telomeric sequences (ITSs) are fragile and prone to expansion, and breakage at an ITS can result in de novo telomere addition by telomerase [17,18]. Introduction of an ITS into two different BIR assay systems resulted in slowed repair synthesis and increased abortive repair at the site of the ITS [19,20] (**) (Figure 1C). Aborted BIR synthesis is ITS repeat length dependent, but shorter ITS repeats were still unstable and underwent tract length changes [20] (**). Aborted BIR on the recipient (invading) strand resulted in telomere addition either via telomerase or by recombination-mediated telomere elongation for a non-native ITS or in cells lacking telomerase [20] (**). However deletion of factors that promote D-loop dissociation, such as the helicase Mph1, resulted in decreased abortive BIR (Figure 1A) [19]. ITSs can form G-quadruplex structures but they can also be bound by proteins such as RAP1 or TRF2, resulting in a protein-mediated replication stall [17,21,22]. It’s likely that the aborted BIR at the ITS is due to a protein-mediated stall rather than G4 structures as deletion of Rrm3, which can unwind G4 structures, does not worsen the abortive synthesis [20] (**). Further, addition of a canonical G4 sequence, even in the presence of a G4 stabilizer, did not result in the same abortive synthesis [20] (**). Taken together, BIR is a repair pathway that can lead to repeat instability through polymerase slippage. However, repetitive sequences may also pose a barrier to BIR leading to unanticipated template switches, which could result in genomic rearrangement and LOH events.
Figure 1:
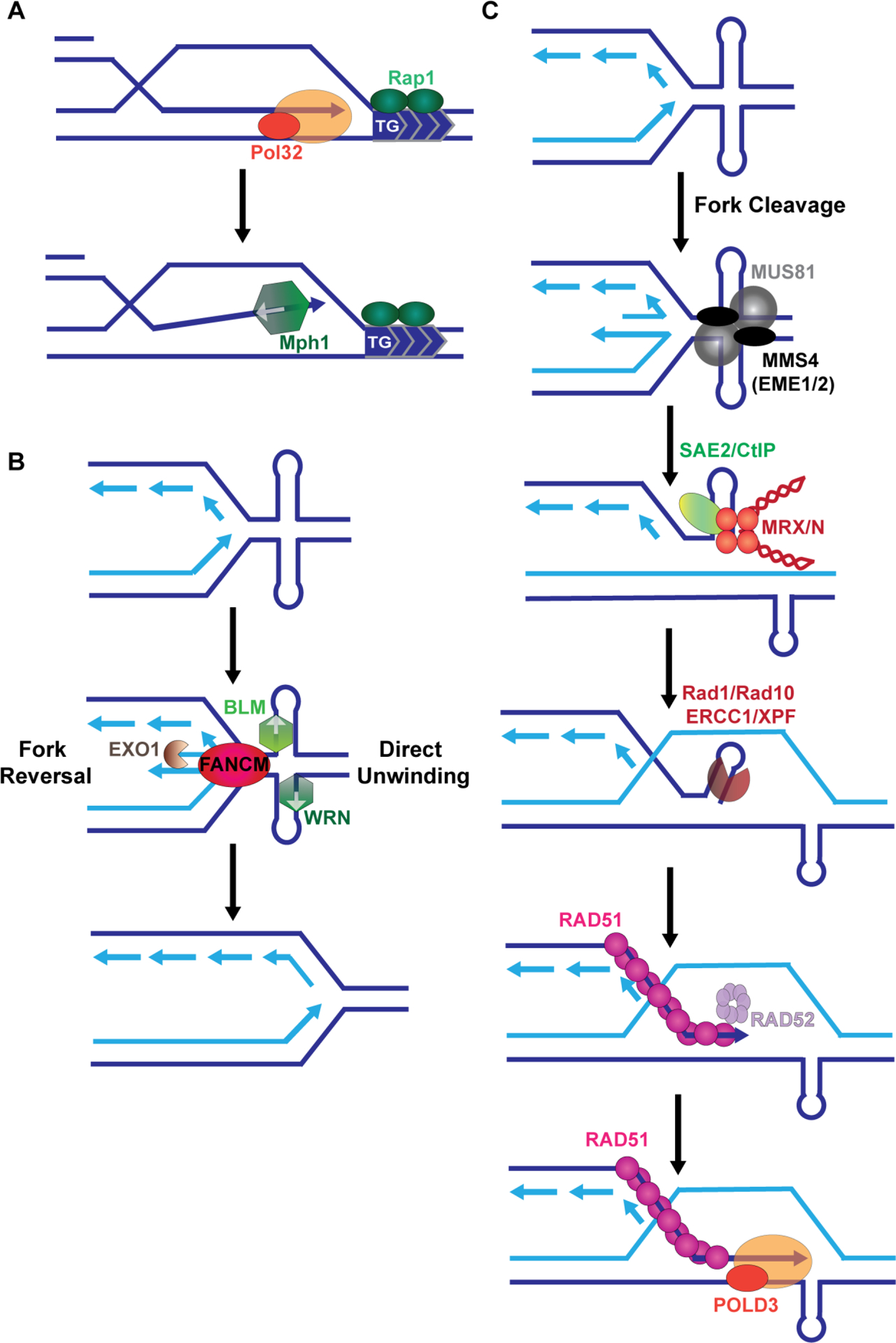
Models for replication fork restart and repair of collapsed forks at DNA structures. A. During BIR, if the replisome encounters a barrier, repair synthesis is more likely to abort. For internal telomeric sequences (ITS) native to that organism, de novo telomere addition on the recipient strand can lead to chromosome truncation. Abortive BIR due to the ITS is promoted by the helicase Mph1, which disassociates D-loops. B. DNA structures can stall replication, as illustrated for an AT repeat forming a cruciform structure. One mechanism for fork restart could be fork reversal which requires the translocase FANCM. Alternatively, helical unwinding of cruciform structures by the 3’−5’ helicases BLM or WRN can result in replication fork restart (note: other helicases are involved at G-rich hairpins or quadruplex structures, such as Srs2, Sgs1, RTEL and FANCJ [3]). C. In the event that fork restart fails, the replication fork may collapse. For some structures, breaks require cleavage by the MUS81-EME1 and/or the SLX1-SLX4 nucleases. The broken fork end with a hairpin is processed by MRX/N and Sae2/CtIP. Additional processing of the hairpin may occur by XPF/ERCC1 (Rad1/Rad10). Second end capture is mediated by RAD52 and repair synthesis is completed by Polδ with the accessory subunit POLD3.
Cleavage and processing of DNA structures is a prerequisite to recombinational repair
Common fragile sites (CFSs) are late replicating chromosomal regions that undergo breakage upon replication stress. One characteristic of CFSs is an enrichment of AT repeat sequences, which can form cruciform structures at a length around 24 perfect repeat units ([23] and references therein). A portion of the FRA16D CFS with a polymorphic AT dinucleotide (Flex1) showed increasing ability to stall replication in an AT-length dependent manner when inserted into either a yeast or human chromosome [24–26]. Stalled replication forks may be resolved by fork reversal or helical unwinding to promote fork restart [4]. Accordingly, several helicases have been shown to be important in preventing breaks at AT repeat structures, including FANCM [27] (*), which can reverse forks, and the BLM [28] and WRN helicases [29] (**) (Figure 1B). Wang et al showed that BLM is enriched at CFS undergoing replication stress [28]. In addition, loss of BLM or its helicase function resulted in increased Flex1 breakage, supporting a model where helical unwinding of DNA structures can help restart replication stalled at these sites [30]. Loss of both FANCM and BLM resulted in a synergistic increase in DSBs and decreased viability, suggesting they work in separate but overlapping pathways to prevent chromosomal breakage at CFSs (Figure 1B) [28].
Failure to resolve a stalled replication fork by fork remodeling and unwinding of the DNA structure may result in fork collapse, DNA breaks and recombinational repair (Figure 1C) [23]. In a yeast model, increasing chromosome fragility of the AT repeat found in Flex1 was correlated with the propensity of the sequence to form a cruciform structure and required both Mus81-Mms4 and the other nucleases associated with the Slx4 scaffold (Slx1 and Rad1-Rad10) [26] (Figure 1C). In humans, microsatellite instable (MSI) cancers require the WRN helicase for survival. Interestingly, WRN depletion in MSI cancer cells caused impaired proliferation and metaphase spreads showed ~35% of chromosomes were shattered [29](**). DSBs that accumulate in WRN deficient MSI cancer cells accumulate around sites of (TA)n repeats that were longer in the MSI cells compared to controls, including some located within CFSs. The DNA breaks in WRN deficient MSI cells are dependent on MUS81 and SLX4, which is consistent with the idea that nucleolytic cleavage drives fork collapse at cruciform structures (Figure 1C) [29](**). Inverted repeats can also form hairpin and cruciform structures. Palindromic sequences were found at breakpoints of chromosomal translocations that result in oncogene amplification and disease pathogenesis [31]. Ait Saada et al. showed that structures formed by palindromic sequences are targeted by different nucleases depending on the size of the loop and their transcriptional status [32]. For example, MRX and Sae2 initiated DSBs at hairpins with small loops that were likely occurring during lagging strand replication. Conversely, Mus81 initiated DSBs of perfect palindromic sequences at actively transcribed genes, suggesting that a structure generated during transcription is important for Mus81 recognition and cleavage. Altogether, the data support that nuclease cleavage is commonly used to resolve problems at cruciform structures in cells.
Once a cleavage occurs within a structure-forming sequence the ends must be processed for healing of the break to occur. The MRN/MRX complex is a conserved multimeric complex comprised of the nuclease Mre11 along with Rad50 and Nbs1 (Xrs2 in S. cerevisiae). The endonuclease CtIP/Sae2 functions with the MRN/X complex to promote endonuclease activities occurring at hairpin-capped ends [25,26,33]. Kaushal et al. showed that hairpin-forming sequences flanking the Flex1 AT cruciform could inhibit processing of the break by the Sae2-stimulated Mre11 nuclease, resulting in decreased healing [26]. This suggests that the MRN/X complex may not be able to efficiently process all structures. The ERCC1/XPF nuclease (yeast Rad1-Rad10) was found to be important for processing breaks within short inverted repeats and other structure-forming sequences [34,35]. ERCC1/XPF was also required for removing blocked ends at Flex1 and G4 structures [30]. Interestingly, loss of the helicase/translocase FANCM resulted in increased mitotic recombination at Flex1 which depended on Rad51 and more surprisingly Rad52. The authors suggest that Rad52 is important in mediating second end capture or in DSB repair resulting from a broken fork when one end is blocked (Figure 1C) [27](*).Taken together, these data suggest that if fork restart cannot occur, fork collapse leads to structure blocked broken ends that need processing by MRN/CtIP and/or ERCC1/XPF, and that Rad52 is needed to facilitate second end capture of these DSBs. Thus, CFSs can be regions that have both an increased propensity to break and a decreased ability to repair. It’s not currently known whether ERCC1/XPF and Rad52 work cooperatively in processing broken ends and second end capture during recombination at broken forks, which may be an interesting future avenue to investigate.
Mitotic DNA Synthesis (MiDAS) of repetitive sequences requires the BIR machinery
In cases where replication is not completed by the mitotic phase of the cell cycle, which often happens at CFS regions, the Hickson lab showed that DNA synthesis can occur during mitosis [36]. This mitotic DNA synthesis is termed MiDAS. The requirements for MiDAS that is induced by aphidicolin, an inhibitor of DNA polymerases, are MUS81-EME1, which cleaves DNA, and POLD3 which fills in the DNA gaps [36]. Supportive of the possibility that MiDAS is a derivative of BIR, Pif1 has recently been shown to have a role in promoting both BIR and MiDAS in mammalian cells [37](*). Loss of FANCM and PIF1 resulted in increased chromosome gaps and breaks upon replication stress, suggesting that PIF1 helps repair synthesis at broken forks. Deletion of Pif1 resulted in less MiDAS in replication stressed cells and shorter BIR tracts in a genetic assay that measures long-tract gene conversion. Interestingly, repair tracts were longer at forks stalled at the Flex1 AT repeat and cleaved by MUS81 than they were at an endonuclease-induced break, suggesting that BIR in mammalian cells is more processive when established at a naturally stalled fork [37](*).
The genetic requirements for mediating MiDAS may be context or structure dependent. The expanded CGG repeat associated with Fragile X syndrome is a Rare Fragile Site (RFS). Conditions of folate deprivation result in mis-segregation of the FRAXA locus, ssDNA anaphase bridges and increased formation of micronuclei that contain the FRAXA locus [38]. Folate deprivation in cells that contained an expanded CGG repeat at the FRAXA locus resulted in MiDAS on all chromosomes expressing the fragile site in a SLX1–4, RAD51, and POLD3 dependent manner [39](**). These observations are in contrast to MiDAS at CFSs, which require MUS81-EME1 and RAD52 [40], neither of which played a role at FRAXA. Thus, initiation of MiDAS may be different depending on the repeat structure or type of replication perturbation.
In addition, BIR resulting from a broken fork may be a consequence of other cellular processes gone awry. In the absence of RTEL1 there’s increased MiDAS at loci prone to forming G-quadruplex-associated R-loops, suggesting that it normally unwinds these structures to prevent breaks [41]. Stabilization of R-loops by other means also results in BIR as deletion of RNase H enzymes resulted in GAA repeat expansions that depended on Pol32 and Rad52. The authors propose that increased H-loop (RNA transcript stabilized H-DNA) formation stalls replication, resulting in replication fork collapse and repair via BIR, and that repeat expansions occur during this process [42].
Telomeres are sites of replication associated recombinational repair
Replication of telomeres can lead to replication fork stalling as they can be protein bound and are also comprised of G-rich repetitive sequences capable of forming G4 structures [43]. Stalled telomeric replication forks are substrates for the enzyme telomerase, which suggests that a mechanism to recover from stalled replication is telomere addition by telomerase [43]. Matmati et al. showed that there is a competition between Ku and telomerase binding to reversed fork ends, leading to the idea that Ku removal at collapsed telomeric forks allows telomerase to repair broken telomeres, thereby shielding telomeres from homologous recombination [44](*). In telomerase deficient fission yeast cells telomere replication is severely impaired, and replication intermediates that arise from stalled and collapsed replication forks accumulate [44](*). In S. pombe, loss of Rad51, the MRN complex, Exo1 and Ctp1/CtIP were essential for telomere maintenance, suggesting that fork processing and recombination is being utilized as a back-up mechanism to promote fork restart when telomerase is not available. Consistently, in mammalian cells, Stroik et al. showed that EXO1 depleted human cells are hypersensitive to G4 stabilizing agents, have telomere length defects, and replication at G4 structures is impaired [45]. Similarly, loss of endonucleolytic processing by CtIP resulted in accumulation of stalled telomeric replication forks and increased levels of telomere fusions, breaks and telomere loss [46]. Together, these results indicate that nucleolytic processing is especially important for replication fork restart within telomeric repeats.
The helicase RTEL1 can unwind G4 structures [47]. In the absence of RTEL1, there is increased telomeric fragility due to G4-associated fork collapse [47]. Margalef et al, showed that loss of RTEL1 resulted in aberrant recruitment of telomerase to the reversed fork and replication fork collapse. Importantly, deletion of telomerase or blocking fork reversal rescued telomere dysfunction in the Rtel1- cells suggesting that inappropriate recruitment of telomerase can impede restart and cause telomere fragility [48]. This result is somewhat contradictory to above results which showed that telomerase is important in promoting fork restart at telomeres [44](*). To help resolve this contradiction, the authors in Matmati et al postulate that one possible reason for the differential requirements for telomerase in fork restart could be due to differences between fission yeast and mammalian cells. Mammalian cells have very long telomeres, and in the absence of RTEL1, replication through the G-rich telomere would be exceptionally difficult, leading to more fork stalling and collapse. Matmati et al suggest that it’s possible that human telomerase is unable to complete synthesis of many kilobases of DNA synthesis, especially in cells lacking RTEL1 where replication stress is exacerbated. It is not currently known whether mammalian telomerase is able to heal telomeres when replication forks collapse in non-mutant conditions.
G4 sequences, at telomeres and genome-wide, are also targets of helical unwinding. Interestingly, in BLM deficient cells, sister chromatid exchange events occur frequently at G4 motifs, especially those present in transcribed genes [49]. The authors propose that, in the absence of BLM, G4 structures aren’t resolved on transcribed strands, leading to fork stalling and recombinational lesion bypass using template switch. Because of the dual role of BLM, the resulting double Holliday junction can’t be dissolved, leading to resolvase recruitment and sister chromatid exchange [49]. Recently, novel mutations in the 5’−3’ DNA helicase DDX11 were identified in Warsaw Breakage Syndrome patients [50]. Patient derived cell lines with siRNA knockdown of DDX11 showed impaired replication, increased DNA damage and cohesion defects upon treatment with G4 stabilizers. This phenotype was mirrored in DDX11 helicase domain mutants. The authors postulate that DDX11 may normally unwind G4 structures on the lagging strand to prevent fork collapse [50]. Interestingly, the Timeless component of the fork protection complex binds G4s, providing a mechanism for recruitment of DDX11 to G4-stalled forks [51].
Relocation to the nuclear periphery is an important step for HR-mediated repair of repetitive sequences.
Fork stalling at an expanded CAG repeat tract can result in the transient relocalization of chromosomes to the nuclear pore complex (NPC) during S-phase in a manner dependent on the SUMO-targeted ubiquitin ligase (StUbL) Slx5/8 [52]. Impaired relocalization of the stalled fork resulted in increased CAG repeat fragility and instability which was dependent on Rad52, suggesting that the NPC plays a role in restraining inaccurate recombination at collapsed forks. Continuation of this work identified that Mms21-mediated sumoylation of RPA, Rad52 and Rad59 drove stalled fork relocalization to the nuclear pore (Figure 2A) [53](*). Most intriguingly, Rad51 foci only co-localized with the CAG repeat tract after relocalization to the pore. Additionally, Rad51 loading onto the early collapsed fork is prevented by sumoylated RPA, suggesting that there are mechanisms in place to constrain recombination at a stalled fork until it is needed for restart. Relocation to the NPC is also a mechanism for regulating telomere recombination in the absence of telomerase. Mutations in the nuclear pore protein Nup1 result in reduced telomere and CAG repeat relocation to the pore (Figure 2A) [54]. In the absence of this relocation, short telomeres bypass senescence and utilize low fidelity Rad51-dependent sister chromatid recombination to maintain their length, and CAG repeat tracts have increased Rad52-dependent contractions. Together, these data indicate that recombination events can be controlled by pathways that depend on nuclear location, though the details of how that regulation occurs are not yet clear.
Figure 2:
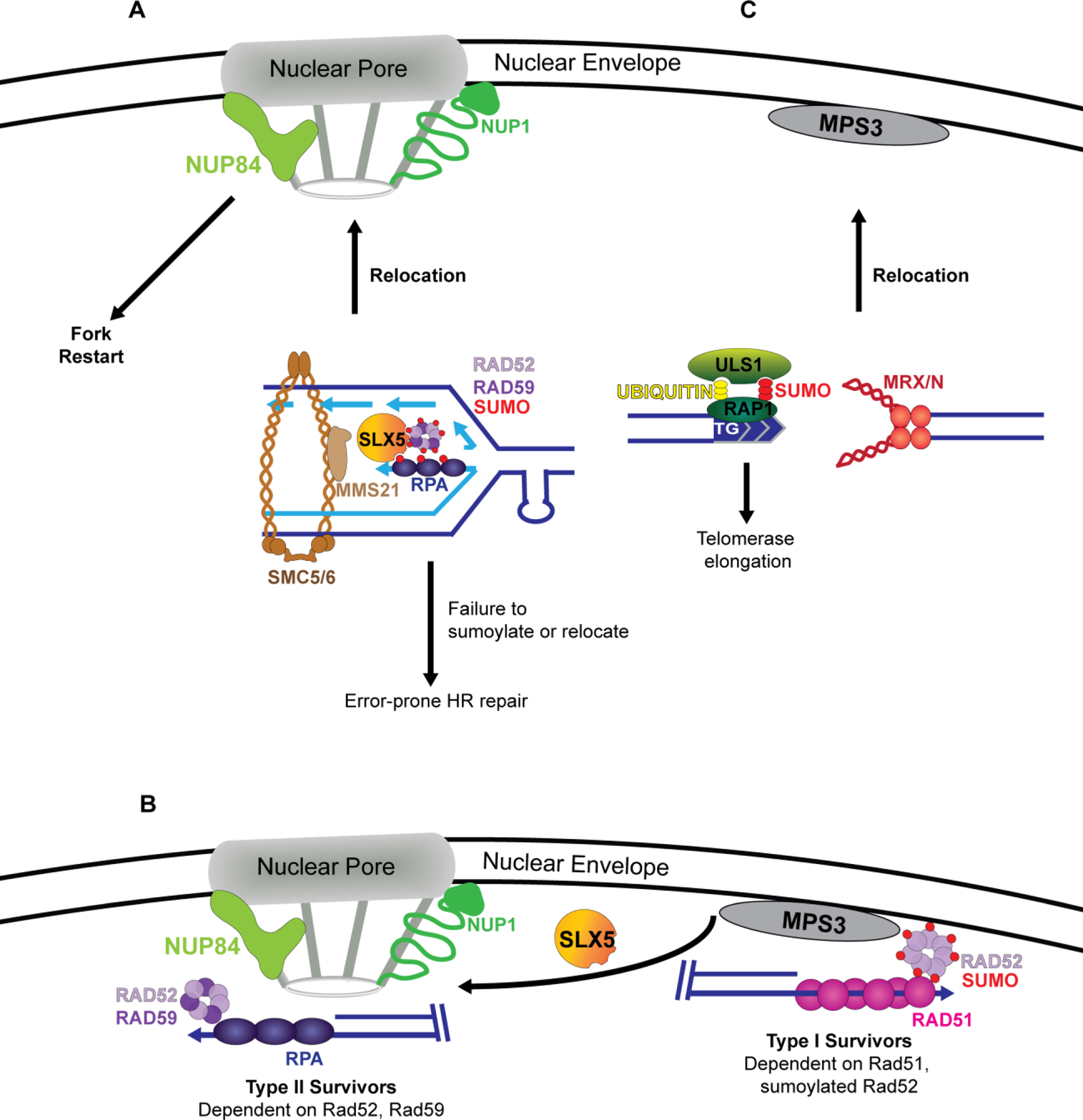
The role of nuclear location in controlling recombination and maintaining repetitive sequences. A. Replication forks stalled due to an expanded CAG repeat relocate to the nuclear pore complex (NPC) for repair and fork restart. Relocation is dependent on the StUbL Slx5/8 via Slx5 SIM domains and Mms21-mediated sumoylation of RPA, Rad52 and Rad59. Failure to sumoylate these proteins leads to impaired replication fork relocation, resulting in unconstrained recombination and repeat instability. B. Telomeres normally associate with the nuclear envelope. In the absence of telomerase, telomeres get progressively shorter until a crisis point, at which point they use recombination to extend the telomeric end. Eroded telomeres with sumoylated Rad52 bound result in type I survivors which depend on Rad51-mediated recombination. Further erosion results in relocation to the nuclear pore and the formation of type II survivors, which is dependent on Rad59 and de-sumoylation of Rad52. C. Targeted DSBs containing a TG repeat on one side of the break results in differential end processing. Broken ends that do not contain a telomeric seed are bound by MRX and relocate to the nuclear envelope. The end containing a telomeric seed is bound by the telomeric binding protein Rap1 which interacts with the StUbL Uls1, helping to signal telomerase recruitment to the TG sequence.
In both mammalian and yeast systems, telomeres are normally tethered to the nuclear envelope, where recombination is suppressed [55]. Eroded telomeres have more ssDNA bound by RPA and have been previously shown to relocate from the nuclear envelope to the nuclear pore complex [56]. In the absence of telomerase, telomeres progressively shorten each replicative cycle until a crisis point. During crisis, telomeres utilize recombination to elongate telomeres and survive. In yeast, telomerase-independent survival is mediated by two different genetic pathways, type I survivors utilize Rad52 and Rad51 while type II survivors require Rad52 and Rad59 [57]. Recent work has established that the sumoylation state of Rad52 influences this choice. Sumoylated Rad52 favors Rad51-dependent type I survivors. However, upon severe erosion, telomeres relocate from the nuclear envelope to the NPC in a Slx5- and Rad59-dependent manner, and relocation is associated with Rad52 de-sumoylation and formation of type II survivors. (Figure 2B). These observations suggest that sumoylation plays a key role in pathway choice that leads to survival in the absence of telomerase [58].
Quiescent cells are cells that are in G0 phase and not undergoing replication, though DNA damage and repair still occur. To determine the cellular response in G0 to eroded telomeres the Coulon group deleted the RNA subunit of telomerase and cultured cells to senescence, at which point they shifted cells into quiescence via nitrogen starvation. Eroded telomeres were found to undergo increased rearrangement at subtelomeric regions which depended on recombination and transcription of the telomeric RNA TERRA [59]. In quiescence, telomeres form a telomeric cluster at the nuclear envelope. However, if telomeres are critically short or interaction with the nuclear periphery is disrupted by deletion of Bqt4, telomeres detach from the nuclear periphery resulting in increased TERRA transcription and subtelomeric recombination. Telomere detachment resulted in the inability to properly exit quiescence. Thus, the nuclear periphery is a safe harbor for telomeres to prevent transcription and recombination [60].
One important step that drives recombinational repair choice is resection. It is not well understood how a repetitive sequence may influence resection and control repair choice. Typically, DSB broken ends relocate to Mps3, a SUN domain containing protein embedded in the nuclear membrane which has roles in nuclear organization [61]. The Gasser lab sought to understand how an internal TG tract would alter chromosome dynamics and DNA repair processing at an induced DSB. They found that the broken end with the TG repeat tract, but not the non-repetitive end, had impaired recruitment of the MRX complex and resection, which resulted in poor tethering between the broken ends and delayed chromosome relocation to the nuclear envelope (Figure 2C) [62]. They identified a requirement for the StUbL Uls1 in mediating telomerase recruitment to the TG sequence and suppressing NHEJ. Taken together, these studies support the importance of post-translational modification of repair proteins by sumoylation in mediating repair processes at repetitive sequences, whether it be at a stalled replication fork, an eroded telomere end, or a DSB.
DSB and gap repair in the context of repetitive DNA tracts
One goal in the field is to develop therapies that shorten disease-length microsatellite sequences to delay symptom onset or disease progression. One attractive strategy is to use gene editing technologies such as CrispR/Cas9 to target the repeat tract and force repair such that the repeat tract shrinks. Work in yeast using CrispR/Cas9 to shorten an expanded CAG repeat tract resulted in large chromosomal deletions near the repeat tract, which were mediated by both annealing and NHEJ pathways; in addition, CrispR/Cas9 mediated breaks at the CTG repeat resulted in increased cell death [63](*). In contrast, targeting the same repeat with a TALEN endonuclease induced repeat contractions primarily by Rad52-dependent annealing which were confined to the repeat tract and did not cause large deletions of surrounding DNA [64]. Mosbach et al. envision a variety of possible explanations for the differences in repair events at the repeat locus between TALEN and CrispR/Cas9 induced breaks including: the type of end created (4 nucleotide 5’ overhangs with TALENs vs. blunt end with CrispR/Cas9), differences in enzyme substrate or kinetics, the role of the gRNA in end-tethering after Cas9 cutting, or differences in checkpoint activation [63]. Using a GFP reporter system in human cells, it was shown that SpCas9 induced contractions as well as expansions of long CTG trinucleotide repeats, whereas the nickase mutant Cas9-D10A only induced contractions [65]. Subsequent studies have been able to successfully shrink an expanded CAG repeat tract in Huntington’s patient-derived fibroblasts by using paired CrispR/Cas9 nickases to make nicks on both sides of the repeat tract [66]. Taken together, DSB induction using CrispR/Cas9 does not result in limited contraction of the repeat tract but rather in expansion events or larger chromosomal deletions, but targeted use of TALENS or the CrispR/Cas9 nickase to the repeat tract could be effective strategies for future therapies.
Structure-forming DNA sequences are natural fragile sites that cause chromosomal DSBs. When a DSB occurs, one of the key steps that drives repair away from NHEJ to HR is the initiation of resection. Short-range resection is initiated by the MRX/N complex in conjunction with Sae2/CtIP. Long range resection is performed by both the 5’−3’ exonuclease Exo1 and coordinated Sgs1/BLM helicase unwinding along with Dna2 endonuclease cleavage in partially redundant roles [67]. Previously, it has not been understood how repetitive, structured sequences would alter resection kinetics. Insertion of a TG repeat tract at an inducible break resulted in impaired MRX recruitment and resection [62]. In addition, deletion of Sae2 resulted in impaired resection on the side of a Cas9-induced break within an expanded CAG repeat where a hairpin capped end was likely formed (Figure 3A) [63](*). Long range resection may also be impaired by DNA structures. Depletion of PIF1, which can unwind G4 structures, or treatment with G4 stabilizers resulted in reduced resection and impaired recombinational repair of an induced DSB, whereas overexpression of PIF1 rescued resection in cells treated with G4 stabilizers, suggesting that PIF1 is important for unwinding non-B form structures the resection machinery may encounter (Figure 3B) [68](*). These observations are consistent with data suggesting that loss of BRCA1, which facilitates resection, results in sensitivity to G4 stabilization [69,70]. Co-immunoprecipitation of PIF1 and BRCA1 was increased in the cells treated with G4 stabilizer, suggesting they function together to unwind G4 structures to facilitate resection [68](*).
Figure 3:
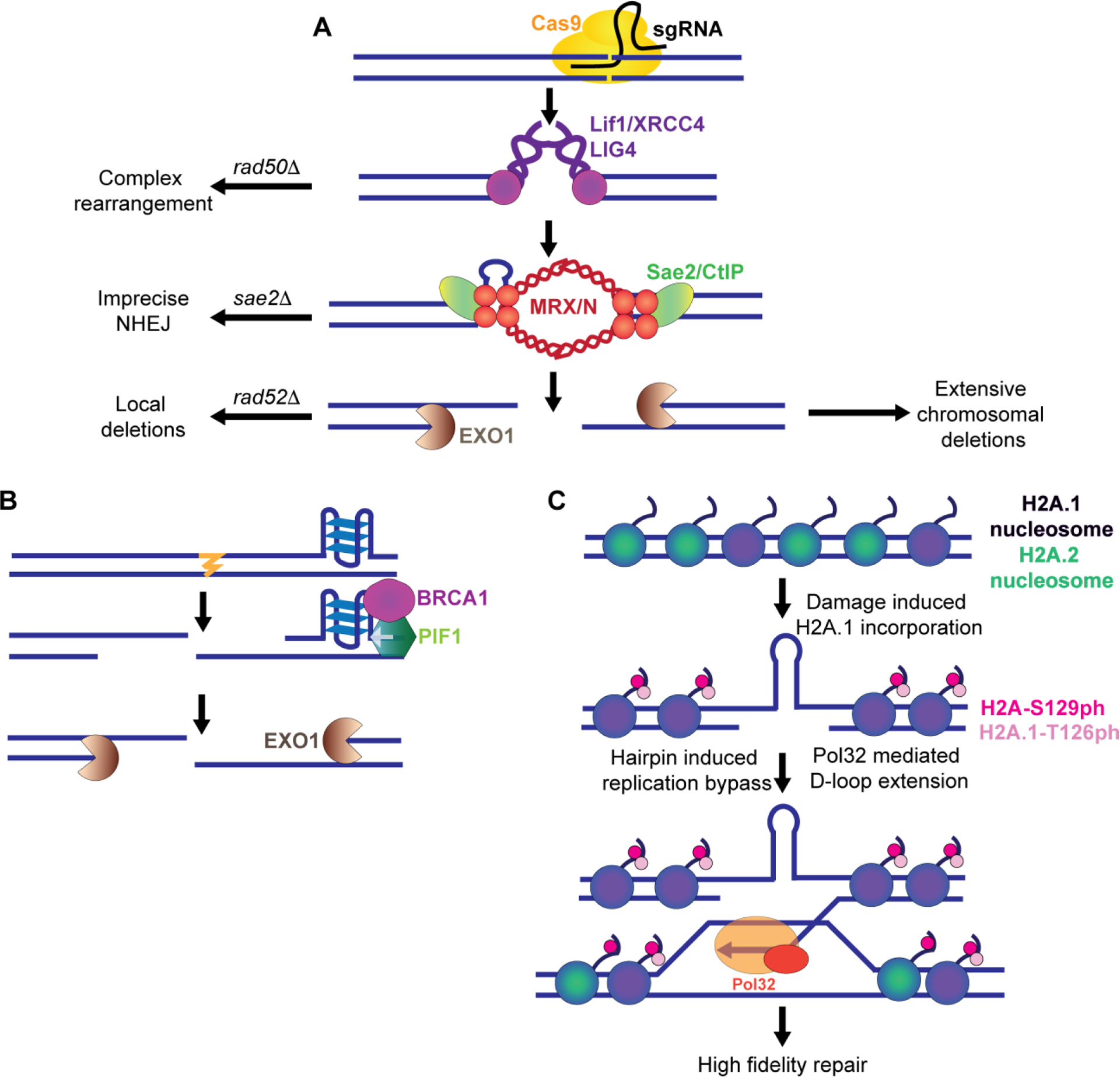
Navigation of resection and repair synthesis through microsatellites. A. At a CrispR/Cas9 break targeting the CTG repeat, repair results in various chromosomal changes. In NHEJ defective rad50Δ mutants, complex rearrangements can be found. In the absence of Sae2, which processes hairpin capped ends, repair occurs via imprecise end-joining. After long range resection by Exo1, repair can result in SSA mediated long range deletions while loss of RAD52 results in smaller local deletions. B. Resection can be impeded by DNA secondary structures. Pif1, which unwinds G4 structures, interacts with BRCA1 to unwind secondary structures that impede long range resection. C. Replication of bypassed DNA hairpins or DNA damage at an expanded CAG repeat tract results in incorporation of copy 1 of histone H2A (H2A.1) which can be phosphorylated at a threonine 126 on the histone tail. The incorporation of histone H2A.1 and phosphorylation of T126 promote efficient D-loop extension during sister chromatid recombination or other D-loop mediated repair, resulting in high fidelity repair and preventing repeat expansions.
The chromatin environment at gaps contributes to efficient repair by error-free template switch within repetitive DNA regions (reviewed in [71,72]). The absence of proper chromatin remodeling or impaired histone modification can result in recombination-dependent expansions that occur during template switching [73,74]. Histone variant deposition is also important for mediating repair fidelity as deletion of HTA1 (copy 1 of histone H2A) as well as a mutation of a phosphorylatable threonine (T126) in the S. cerevisiae Hta1 C-terminus showed a significant increase in (CAG)85 repeat expansions which depended on recombination (Figure 3C). The Hta1 and Hta2 proteins are nearly identical at the amino acid level except for a threonine switch at position 126 in Hta1. These data suggest that post-translational modification of histone H2A copy 1 can serve as a signal to help promote D-loop synthesis fidelity through a repetitive tract (Figure 3C) [75](*).
In repeat expansion diseases, the timing of symptom onset and disease progression is influenced by the level of age-related somatic instability of the repeat tract. In mouse models of triplet repeat expansion diseases, MutS and MutL complexes are required for somatic expansions, though the exact complex varies by disease (MutSβ/MLH1/MutLγ for HD and DM1, MutSα/MLH1 in Friederichs’s ataxia mouse models (see [76] for review). To explore the mechanisms of somatic expansion, yeast cells containing the Friederichs’s ataxia (GAA)n repeat tract were subjected to chronological aging. Expansions occurred during quiescence and required the mismatch repair complexes MutSβ and MutLα as well as the Rad1-Rad10 nuclease [77]. Interestingly, in quiescent yeast, large deletions and gene conversion events arose that were end-joining and recombination dependent. These were hypothesized to occur due to mismatch repair proteins initiating a break that was resected by Exo1 and then repaired via non-homologous end-joining or recombination [77]. More broadly, all the above studies suggest that recombinational repair at breaks or gaps that occur within or near a repeat tract must be negotiated by the repair machinery and impaired recombination could be a source of repeat instability or fragility.
Summary and conclusions
Many repetitive DNA sequences form secondary structures that serve as constant challenges to DNA replication and repair machineries, resulting in stalled forks, nicks, gaps, and DSBs. Recombination is an important pathway to repair these lesions and serves as a powerful guardian of the genome. Recombinational repair at repetitive sequences serves to preserve genome stability, though it can be a mutagenic process that leads to repeat length changes. Future work understanding genetic requirements in promoting accurate recombinational repair at microsatellites will be key in our understanding of repair and replication fork restart as well as harnessing genome-modifying technologies to develop therapies for genetic disease and cancer.
Funding sources:
American Cancer Society-Ellison Foundation Postdoctoral Fellowship PF-18-125-10-DMC to EJP. NIGMS (GM122880) and NSF (MCB1817499) to CHF.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- 1.Khristich AN, Mirkin SM: On the wrong DNA track: Molecular mechanisms of repeat-mediated genome instability. J Biol Chem 2020, 295:4134–4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brown RE, Freudenreich CH: Structure-forming repeats and their impact on genome stability. Curr Opin Genet Dev 2021, 67:41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Polleys EJ, House NCM, Freudenreich CH: Role of recombination and replication fork restart in repeat instability. DNA Repair (Amst) 2017, 56:156–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ait Saada A, Lambert SAE, Carr AM: Preserving replication fork integrity and competence via the homologous recombination pathway. DNA Repair (Amst) 2018, 71:135–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gadgil R, Barthelemy J, Lewis T, Leffak M: Replication stalling and DNA microsatellite instability. Biophys Chem 2017, 225:38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gellon L, Kaushal S, Cebrian J, Lahiri M, Mirkin SM, Freudenreich CH: Mrc1 and Tof1 prevent fragility and instability at long CAG repeats by their fork stabilizing function. Nucleic Acids Res 2019, 47:794–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jedrychowska M, Denkiewicz-Kruk M, Alabrudzinska M, Skoneczna A, Jonczyk P, Dmowski M, Fijalkowska IJ: Defects in the GINS complex increase the instability of repetitive sequences via a recombination-dependent mechanism. PLoS Genet 2019, 15:e1008494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anand RP, Lovett ST, Haber JE: Break-induced DNA replication. Cold Spring Harb Perspect Biol 2013, 5:a010397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Malkova A, Ira G: Break-induced replication: functions and molecular mechanism. Curr Opin Genet Dev 2013, 23:271–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saini N, Ramakrishnan S, Elango R, Ayyar S, Zhang Y, Deem A, Ira G, Haber JE, Lobachev KS, Malkova A: Migrating bubble during break-induced replication drives conservative DNA synthesis. Nature 2013, 502:389–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Donnianni RA, Symington LS: Break-induced replication occurs by conservative DNA synthesis. Proc Natl Acad Sci U S A 2013, 110:13475–13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lydeard JR, Jain S, Yamaguchi M, Haber JE: Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature 2007, 448:820–823. [DOI] [PubMed] [Google Scholar]
- 13.Costantino L, Sotiriou SK, Rantala JK, Magin S, Mladenov E, Helleday T, Haber JE, Iliakis G, Kallioniemi OP, Halazonetis TD: Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science 2014, 343:88–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim JC, Harris ST, Dinter T, Shah KA, Mirkin SM: The role of break-induced replication in large-scale expansions of (CAG)n/(CTG)n repeats. Nat Struct Mol Biol 2017, 24:55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kononenko AV, Ebersole T, Vasquez KM, Mirkin SM: Mechanisms of genetic instability caused by (CGG)n repeats in an experimental mammalian system. Nat Struct Mol Biol 2018, 25:669–676. [DOI] [PMC free article] [PubMed] [Google Scholar]; *This work builds off previous work suggesting large-scale repeat expansions occur during BIR. This work along with [30] show that BIR is a mechanism for repeat instability in mammalian cells. Specifically, the authors show (CGG)n repeat instability and reporter mutatgeneisis is driven by POLD3, POLD4, RAD51, RAD52, and SMARCAL1.
- 16.Gadgil RY, Romer EJ, Goodman CC, Rider SD Jr., Damewood FJ, Barthelemy JR, Shin-Ya K, Hanenberg H, Leffak M: Replication stress at microsatellites causes DNA double-strand breaks and break-induced replication. J Biol Chem 2020, 295:15378–15397. [DOI] [PMC free article] [PubMed] [Google Scholar]; *This work builds off previous work in yeast suggesting large-scale repeat expansions occur during BIR. This work along with [29] show that BIR is a mechanism for repeat instability in mammalian cells. The authors show that expanded CAG repeats and Pu/Py microsatellites result increased DSBs during replication. The DSBs at expanded CAG repeat were dependent on MUS81 and resulted in complex rearrangements and POLD3-mediated mutagenesis.
- 17.Aksenova AY, Greenwell PW, Dominska M, Shishkin AA, Kim JC, Petes TD, Mirkin SM: Genome rearrangements caused by interstitial telomeric sequences in yeast. Proc Natl Acad Sci U S A 2013, 110:19866–19871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aksenova AY, Han G, Shishkin AA, Volkov KV, Mirkin SM: Expansion of Interstitial Telomeric Sequences in Yeast. Cell Rep 2015, 13:1545–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stivison EA, Young KJ, Symington LS: Interstitial telomere sequences disrupt break-induced replication and drive formation of ectopic telomeres. Nucleic Acids Res 2020, 48:12697–12710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu L, Yan Z, Osia BA, Twarowski J, Sun L, Kramara J, Lee RS, Kumar S, Elango R, Li H, et al. : Tracking break-induced replication shows that it stalls at roadblocks. Nature 2021, 590:655–659. [DOI] [PMC free article] [PubMed] [Google Scholar]; **The authors in this work show that BIR synthesis is impaired if the replisome encounters a replication barrier such as an internal telomeric sequence (ITS). The ITS is bound by Rap1 which impedes BIR. In the event that BIR is aborted, the recipient chromosome becomes a substrate for de novo telomere addition or healing by recombination dependent telomere elongation. This work underscores that impediments to replication may also impair break repair synthesis.
- 21.Anand RP, Shah KA, Niu H, Sung P, Mirkin SM, Freudenreich CH: Overcoming natural replication barriers: differential helicase requirements. Nucleic Acids Res 2012, 40:1091–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Simonet T, Zaragosi LE, Philippe C, Lebrigand K, Schouteden C, Augereau A, Bauwens S, Ye J, Santagostino M, Giulotto E, et al. : The human TTAGGG repeat factors 1 and 2 bind to a subset of interstitial telomeric sequences and satellite repeats. Cell Res 2011, 21:1028–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaushal S, Freudenreich CH: The role of fork stalling and DNA structures in causing chromosome fragility. Genes Chromosomes Cancer 2019, 58:270–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang H, Freudenreich CH: An AT-rich sequence in human common fragile site FRA16D causes fork stalling and chromosome breakage in S. cerevisiae. Mol Cell 2007, 27:367–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang H, Li Y, Truong LN, Shi LZ, Hwang PY, He J, Do J, Cho MJ, Li H, Negrete A, et al. : CtIP maintains stability at common fragile sites and inverted repeats by end resection-independent endonuclease activity. Mol Cell 2014, 54:1012–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaushal S, Wollmuth CE, Das K, Hile SE, Regan SB, Barnes RP, Haouzi A, Lee SM, House NCM, Guyumdzhyan M, et al. : Sequence and Nuclease Requirements for Breakage and Healing of a Structure-Forming (AT)n Sequence within Fragile Site FRA16D. Cell Rep 2019, 27:1151–1164 e1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang H, Li S, Oaks J, Ren J, Li L, Wu X: The concerted roles of FANCM and Rad52 in the protection of common fragile sites. Nat Commun 2018, 9:2791. [DOI] [PMC free article] [PubMed] [Google Scholar]; *The translocase FANCM normally functions in the Fanconia Anemia pathway and is often mutated in breast cancers. The authors identified a novel role of FANCM in replication fork restart at CFSs. In addition, they showed that loss of RAD52 has a synthetic lethal phenotype in FANCM depleted cells, suggesting they function in different pathways at CFSs. The authors postulate that inhibition of Rad52 in FANCM deficient cancers could be a novel therapeutic target.
- 28.Wang H, Li S, Zhang H, Wang Y, Hao S, Wu X: BLM prevents instability of structure-forming DNA sequences at common fragile sites. PLoS Genet 2018, 14:e1007816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Wietmarschen N, Sridharan S, Nathan WJ, Tubbs A, Chan EM, Callen E, Wu W, Belinky F, Tripathi V, Wong N, et al. : Repeat expansions confer WRN dependence in microsatellite-unstable cancers. Nature 2020, 586:292–298. [DOI] [PMC free article] [PubMed] [Google Scholar]; **In MSI (microsatellite instability) cancers, the WRN (Werner syndrome) helicase is a synthetic lethal target. The authors identify that in the absence of WRN in MSI cancer cells, DNA breakpoints were often located at expanded (AT) dinucleotide sequences, which can form cruciform structures. In the absence of WRN, breaks at the AT repeats occur in a MUS81 and SLX4-dependent manner. These results suggest a model where repeat expansions accumulate in MSI cancer cells, making the cells dependent on WRN to unwind the resulting cruciform structures
- 30.Li S, Lu H, Wang Z, Hu Q, Wang H, Xiang R, Chiba T, Wu X: ERCC1/XPF Is Important for Repair of DNA Double-Strand Breaks Containing Secondary Structures. iScience 2019, 16:63–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Svetec Miklenic M, Svetec IK: Palindromes in DNA-A Risk for Genome Stability and Implications in Cancer. Int J Mol Sci 2021, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ait Saada A, Costa AB, Sheng Z, Guo W, Haber JE, Lobachev KS: Structural parameters of palindromic repeats determine the specificity of nuclease attack of secondary structures. Nucleic Acids Res 2021, 49:3932–3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lobachev KS, Rattray A, Narayanan V: Hairpin- and cruciform-mediated chromosome breakage: causes and consequences in eukaryotic cells. Front Biosci 2007, 12:4208–4220. [DOI] [PubMed] [Google Scholar]
- 34.Lu S, Wang G, Bacolla A, Zhao J, Spitser S, Vasquez KM: Short Inverted Repeats Are Hotspots for Genetic Instability: Relevance to Cancer Genomes. Cell Rep 2015, 10:1674–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McKinney JA, Wang G, Mukherjee A, Christensen L, Subramanian SHS, Zhao J, Vasquez KM: Distinct DNA repair pathways cause genomic instability at alternative DNA structures. Nat Commun 2020, 11:236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Minocherhomji S, Ying S, Bjerregaard VA, Bursomanno S, Aleliunaite A, Wu W, Mankouri HW, Shen H, Liu Y, Hickson ID: Replication stress activates DNA repair synthesis in mitosis. Nature 2015, 528:286–290. [DOI] [PubMed] [Google Scholar]
- 37.Li S, Wang H, Jehi S, Li J, Liu S, Wang Z, Truong L, Chiba T, Wang Z, Wu X: PIF1 helicase promotes break-induced replication in mammalian cells. EMBO J 2021, 40:e104509. [DOI] [PMC free article] [PubMed] [Google Scholar]; *The authors of this work make strides in connecting BIR to MiDAS and implicate the helicase PIF1 in both repair processes. Deletion of Pif1 resulted in less MiDAS in replication stressed cells and shorter BIR tracts in a genetic assay that measures long tract gene conversion. The authors also demonstrate that BIR repair synthesis may be longer when initiated from a naturally stalled fork, suggestting BIR may be more processive when established at a naturally stalled replication fork.
- 38.Bjerregaard VA, Garribba L, McMurray CT, Hickson ID, Liu Y: Folate deficiency drives mitotic missegregation of the human FRAXA locus. Proc Natl Acad Sci U S A 2018, 115:13003–13008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garribba L, Bjerregaard VA, Goncalves Dinis MM, Ozer O, Wu W, Sakellariou D, Pena-Diaz J, Hickson ID, Liu Y: Folate stress induces SLX1- and RAD51-dependent mitotic DNA synthesis at the fragile X locus in human cells. Proc Natl Acad Sci U S A 2020, 117:16527–16536. [DOI] [PMC free article] [PubMed] [Google Scholar]; **Previous work from this group showed increased anaphase bridges and missegregation at the expanded CGG repeat associated with fragile X syndrome. The authors expanded on these findings to show that MiDAS occurs at the CGG repeat when it expressed as a rare fragile site by folate deprivation, and MiDAS depended on SLX1, RAD51, and POLD3.
- 40.Ozer O, Hickson ID: Pathways for maintenance of telomeres and common fragile sites during DNA replication stress. Open Biol 2018, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu W, Bhowmick R, Vogel I, Ozer O, Ghisays F, Thakur RS, Sanchez de Leon E, Richter PH, Ren L, Petrini JH, et al. : RTEL1 suppresses G-quadruplex-associated R-loops at difficult-to-replicate loci in the human genome. Nat Struct Mol Biol 2020, 27:424–437. [DOI] [PubMed] [Google Scholar]
- 42.Neil AJ, Liang MU, Khristich AN, Shah KA, Mirkin SM: RNA-DNA hybrids promote the expansion of Friedreich’s ataxia (GAA)n repeats via break-induced replication. Nucleic Acids Res 2018, 46:3487–3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Simon M-N, Churikov D, Géli V: Replication stress as a source of telomere recombination during replicative senescence in Saccharomyces cerevisiae. FEMS Yeast Research 2016, 16. [DOI] [PubMed] [Google Scholar]
- 44.Matmati S, Lambert S, Geli V, Coulon S: Telomerase Repairs Collapsed Replication Forks at Telomeres. Cell Rep 2020, 30:3312–3322 e3313. [DOI] [PubMed] [Google Scholar]; * In this work, the authors investigate replication dynamics in telomerase negative cells. They show that telomerase repairs telomeric stalled replication forks. They additionally suggest that recombination is a back-up mechanism to promote fork restart at telomeric stalled replication forks.
- 45.Stroik S, Kurtz K, Lin K, Karachenets S, Myers CL, Bielinsky AK, Hendrickson EA: EXO1 resection at G-quadruplex structures facilitates resolution and replication. Nucleic Acids Res 2020, 48:4960–4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stroik S, Kurtz K, Hendrickson EA: CtIP is essential for telomere replication. Nucleic Acids Res 2019, 47:8927–8940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vannier JB, Pavicic-Kaltenbrunner V, Petalcorin MI, Ding H, Boulton SJ: RTEL1 dismantles T loops and counteracts telomeric G4-DNA to maintain telomere integrity. Cell 2012, 149:795–806. [DOI] [PubMed] [Google Scholar]
- 48.Margalef P, Kotsantis P, Borel V, Bellelli R, Panier S, Boulton SJ: Stabilization of Reversed Replication Forks by Telomerase Drives Telomere Catastrophe. Cell 2018, 172:439–453 e414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van Wietmarschen N, Merzouk S, Halsema N, Spierings DCJ, Guryev V, Lansdorp PM: BLM helicase suppresses recombination at G-quadruplex motifs in transcribed genes. Nat Commun 2018, 9:271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van Schie JJM, Faramarz A, Balk JA, Stewart GS, Cantelli E, Oostra AB, Rooimans MA, Parish JL, de Almeida Esteves C, Dumic K, et al. : Warsaw Breakage Syndrome associated DDX11 helicase resolves G-quadruplex structures to support sister chromatid cohesion. Nat Commun 2020, 11:4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lerner LK, Holzer S, Kilkenny ML, Svikovic S, Murat P, Schiavone D, Eldridge CB, Bittleston A, Maman JD, Branzei D, et al. : Timeless couples G-quadruplex detection with processing by DDX11 helicase during DNA replication. EMBO J 2020, 39:e104185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Su XA, Dion V, Gasser SM, Freudenreich CH: Regulation of recombination at yeast nuclear pores controls repair and triplet repeat stability. Genes Dev 2015, 29:1006–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Whalen JM, Dhingra N, Wei L, Zhao X, Freudenreich CH: Relocation of Collapsed Forks to the Nuclear Pore Complex Depends on Sumoylation of DNA Repair Proteins and Permits Rad51 Association. Cell Rep 2020, 31:107635. [DOI] [PMC free article] [PubMed] [Google Scholar]; *The authors of this paper show that Mms21 dependent sumoylation of RPA, Rad52 and Rad59 is a key step to promote fork relocation to the nuclear pore. If sumoylation of these repair factors is impaired, there is unconstrained Rad51 loading. This work emphasizes that proper location in the nucleus and sumoylation status of repair proteins are key elements that promote recombinational repair fidelity at collapsed forks.
- 54.Aguilera P, Whalen J, Minguet C, Churikov D, Freudenreich C, Simon MN, Geli V: The nuclear pore complex prevents sister chromatid recombination during replicative senescence. Nat Commun 2020, 11:160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Geli V, Lisby M: Recombinational DNA repair is regulated by compartmentalization of DNA lesions at the nuclear pore complex. Bioessays 2015, 37:1287–1292. [DOI] [PubMed] [Google Scholar]
- 56.Khadaroo B, Teixeira MT, Luciano P, Eckert-Boulet N, Germann SM, Simon MN, Gallina I, Abdallah P, Gilson E, Geli V, et al. : The DNA damage response at eroded telomeres and tethering to the nuclear pore complex. Nat Cell Biol 2009, 11:980–987. [DOI] [PubMed] [Google Scholar]
- 57.McEachern MJ, Haber JE: Break-induced replication and recombinational telomere elongation in yeast. Annu Rev Biochem 2006, 75:111–135. [DOI] [PubMed] [Google Scholar]
- 58.Charifi F, Churikov D, Eckert-Boulet N, Minguet C, Jourquin F, Hardy J, Lisby M, Simon MN, Geli V: Rad52 SUMOylation functions as a molecular switch that determines a balance between the Rad51- and Rad59-dependent survivors. iScience 2021, 24:102231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Maestroni L, Audry J, Matmati S, Arcangioli B, Geli V, Coulon S: Eroded telomeres are rearranged in quiescent fission yeast cells through duplications of subtelomeric sequences. Nat Commun 2017, 8:1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maestroni L, Reyes C, Vaurs M, Gachet Y, Tournier S, Geli V, Coulon S: Nuclear envelope attachment of telomeres limits TERRA and telomeric rearrangements in quiescent fission yeast cells. Nucleic Acids Res 2020, 48:3029–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sosa Ponce ML, Moradi-Fard S, Zaremberg V, Cobb JA: SUNny Ways: The Role of the SUN-Domain Protein Mps3 Bridging Yeast Nuclear Organization and Lipid Homeostasis. Front Genet 2020, 11:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Marcomini I, Shimada K, Delgoshaie N, Yamamoto I, Seeber A, Cheblal A, Horigome C, Naumann U, Gasser SM: Asymmetric Processing of DNA Ends at a Double-Strand Break Leads to Unconstrained Dynamics and Ectopic Translocation. Cell Rep 2018, 24:2614–2628 e2614. [DOI] [PubMed] [Google Scholar]
- 63.Mosbach V, Viterbo D, Descorps-Declere S, Poggi L, Vaysse-Zinkhofer W, Richard GF: Resection and repair of a Cas9 double-strand break at CTG trinucleotide repeats induces local and extensive chromosomal deletions. PLoS Genet 2020, 16:e1008924. [DOI] [PMC free article] [PubMed] [Google Scholar]; *The data in this paper provides a cautionary tale on whether CrispR/Cas9-induced breaks within a repeat tract could be a feasible therapy for repeat expansion diseases. They show that spCas9 cleavage of the CAG repeat tract results in increased cell death and large chromosomal deletions mediated by end-joining and annealing pathways. In constrast, previous data using TALENs [64] or Cas9 nickases [65] did lead to more targeted CAG deletions.
- 64.Mosbach V, Poggi L, Viterbo D, Charpentier M, Richard GF: TALEN-Induced Double-Strand Break Repair of CTG Trinucleotide Repeats. Cell Rep 2018, 22:2146–2159. [DOI] [PubMed] [Google Scholar]
- 65.Cinesi C, Aeschbach L, Yang B, Dion V: Contracting CAG/CTG repeats using the CRISPR-Cas9 nickase. Nat Commun 2016, 7:13272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dabrowska M, Juzwa W, Krzyzosiak WJ, Olejniczak M: Precise Excision of the CAG Tract from the Huntingtin Gene by Cas9 Nickases. Front Neurosci 2018, 12:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Symington LS: Mechanism and regulation of DNA end resection in eukaryotes. Crit Rev Biochem Mol Biol 2016, 51:195–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jimeno S, Camarillo R, Mejias-Navarro F, Fernandez-Avila MJ, Soria-Bretones I, Prados-Carvajal R, Huertas P: The Helicase PIF1 Facilitates Resection over Sequences Prone to Forming G4 Structures. Cell Rep 2018, 24:3262–3273 e3264. [DOI] [PubMed] [Google Scholar]; *In this work the authors show one way that resection is navigated in the presence of non-B form DNA structures. Loss of Pif1, which unwinds G4 structures, or stabilization of G4 structures results in impaired resection and altered repair outcome. This work identifies that Pif1 and BRCA1 may function together to negotiate resection of non-B form DNA.
- 69.Xu H, Di Antonio M, McKinney S, Mathew V, Ho B, O’Neil NJ, Santos ND, Silvester J, Wei V, Garcia J, et al. : CX-5461 is a DNA G-quadruplex stabilizer with selective lethality in BRCA1/2 deficient tumours. Nat Commun 2017, 8:14432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zimmer J, Tacconi EMC, Folio C, Badie S, Porru M, Klare K, Tumiati M, Markkanen E, Halder S, Ryan A, et al. : Targeting BRCA1 and BRCA2 Deficiencies with G-Quadruplex-Interacting Compounds. Mol Cell 2016, 61:449–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.House NC, Koch MR, Freudenreich CH: Chromatin modifications and DNA repair: beyond double-strand breaks. Front Genet 2014, 5:296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lahue RS, Frizzell A: Histone deacetylase complexes as caretakers of genome stability. Epigenetics 2012, 7:806–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Daee DL, Mertz T, Lahue RS: Postreplication repair inhibits CAG.CTG repeat expansions in Saccharomyces cerevisiae. Mol Cell Biol 2007, 27:102–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.House NC, Yang JH, Walsh SC, Moy JM, Freudenreich CH: NuA4 Initiates Dynamic Histone H4 Acetylation to Promote High-Fidelity Sister Chromatid Recombination at Postreplication Gaps. Mol Cell 2014, 55:818–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.House NC, Polleys EJ, Quasem I, De la Rosa Mejia M, Joyce CE, Takacsi-Nagy O, Krebs JE, Fuchs SM, Freudenreich CH: Distinct roles for S. cerevisiae H2A copies in recombination and repeat stability, with a role for H2A.1 threonine 126. eLife 2019, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]; *The authors in House, Polleys et al show that incorporation of certain histone variants can aid repair fidelity. Loss of copy 1 of histone H2A (Hta1) in S. cerevisiae results in increased recombination-dependent CAG repeat expansions. In addition, they identify that a phosphorylatable residue in Hta1, threonine 126, is essential for preventing repeat expansions.
- 76.Usdin K, House NC, Freudenreich CH: Repeat instability during DNA repair: Insights from model systems. Crit Rev Biochem Mol Biol 2015, 50:142–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Neil AJ, Hisey JA, Quasem I, McGinty RJ, Hitczenko M, Khristich AN, Mirkin SM: Replication-independent instability of Friedreich’s ataxia GAA repeats during chronological aging. Proc Natl Acad Sci U S A 2021, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]