

Abstract
RNA editing exerts critical impacts on numerous biological processes. While millions of RNA editings have been identified in humans, much more are expected to be discovered. In this work, we constructed Convolutional Neural Network (CNN) models to predict human RNA editing events in both Alu regions and non-Alu regions. With a validation dataset resulting from CRISPR/Cas9 knockout of the ADAR1 enzyme, the validation accuracies reached 99.5% and 93.6% for Alu and non-Alu regions, respectively. We ported our CNN models in a web service named EditPredict. EditPredict not only works on reference genome sequences but can also take into consideration single nucleotide variants in personal genomes. In addition to the human genome, EditPredict tackles other model organisms including bumblebee, fruitfly, mouse, and squid genomes. EditPredict can be used stand-alone to predict novel RNA editing and it can be used to assist in filtering for candidate RNA editing detected from RNA-Seq data.
INTRODUCTION
In humans, RNA editing is a common molecular process that causes nucleotide substitutions in RNA as compared to the corresponding DNA sequence. Of all 12 possible types of single-base substitutions, adenosine-to-inosine (A-to-I) RNA editing is the primary canonical RNA editing type, comprising over 95% of all known RNA editing events. Functional impacts of RNA editing are increasingly appreciated, as studies have shown that RNA editing can alter protein products [1], affect drug sensitivity [2], and be associated with prognosis [3]. Through high-throughput genomic research efforts during the last decade, a few million human RNA editing events have been curated in dedicated databases such as REDIportal [4] and DARNED [5]. Because RNA editing takes effect at the co-transcriptional or post-transcriptional level, the identity and quantity of RNA editing events in differential spatiotemporal contexts are different. The complete landscape of RNA editing in the human genome still is not fully characterized [6].
Mass identification of human RNA editing events began with the rapidly increasing use of high throughput sequencing (HTS) technologies [7]. Consequently, typical RNA editing identification strategies inevitably involve comparing sequencing data from paired RNA and DNA samples. Approaches have also been developed for when DNA sequencing data is absent, which heavily rely on post-processing filters [8]. Candidate RNA editing sites screened by these HTS dependent pipelines must go through gold standard validation by RT-PCR or Sanger Sequencing. Unfortunately, HTS data are subject to various types of bias and noise [8], which typically lead to substantial inflation of false-positive discoveries in the final candidates of RNA editing. For instance, controversies [8] surrounded the 2011 impressive report of “Widespread RNA and DNA sequence differences in the human transcriptome,” and many groups attributed non-canonical editing events exclusively to sequencing biases and bioinformatics flaws [9, 10].
Given the imperfect quality of sequencing-detected RNA editing, an RNA editing prediction algorithm independent of sequencing data may substantially reduce the false positive rate of a candidate list and alleviate the overall financial and labor cost of RNA editing studies. Like other genomic features, RNA editing sites are probably dependent on proximal DNA context to some extent; indeed, sporadic studies [11, 12] have leveraged flanking nucleotides to improve sequencing data-independent prediction of RNA-editing. A Convolutional Neural Network (CNN) was wrapped in a method DeepRed [13] to identify RNA editing events from RNA-Seq data, adding to successful Deep Learning applications on genomic sequence features such as splicing junctions and transcription factor binding targets [14–16].
In this study, we developed EditPredict, a Deep Learning solution of RNA editing prediction from genome DNA sequence. Specifically, we utilized CNN to model flanking DNA sequences of known A-to-I RNA editing events, with consideration of alternate flanking directions and variant sequence lengths. Other than working with reference genome sequences, EditPredict can take in individualized Single-Nucleotide Variants (SNVs) to make dynamic RNA-editing prediction with variable sequence context. EditPredict does not involve HTS data in either the training or the application process, thus rendering itself a completely parallel workflow in complement to sequencing-based pipelines. EditPredict empowers researchers to gain a good estimation of RNA-editing propensity at any adenosine position in the whole genome, prior to an actual HTS experiment that entails tedious sample collection, library preparation, sequencing, and HTS data processing. While being designed as a self-contained tool independent of HTS data, EditPredict produces results that can be checked against HTS output.
Our resultant models have been implemented as an online tool, EditPredict. The default application model facilitates users to comprehend the RNA-editing propensity of interested genomic sites in the human reference genome. When given an optional input in Variant Call Format (VCF) file, it makes personalized RNA-editing inferences for a particular subject characterized by individualized genomic variants. In addition to humans, EditPredict can also tackle genome sequences of bumblebee, fruit fly, mouse, and squid.
METHODS
Flanking sequences of RNA-editing sites and non-edited sites used for training CNN models
We utilized a vast positive dataset comprising all known RNA-editing sites (~4.67 million) curated in REDIportal [4]. Of the 4.67 million reported RNA editing sites, around 10% are non-Alu RNA editing. The point positions were converted to DNA sequences by extracting neighboring nucleotides in either or both directions. Precisely, three direction modes were considered: upstream, downstream, and bi-direction. If the RNA editing is on the reverse strain, sequence from the reverse strand was extracted. Eight sequence length values were tested: 10, 30, 50, 70, 90, 100, 150, and 200 nucleotides. Of note, for the bi-directional mode, the actual sequence lengths were magnified to 2*l+1, with l taken from the foresaid series. It is for narrative ease that we refer to the sequences under bi-directional mode with the corresponding length under a uni-directional mode.
Additionally, we randomly selected non-edited genomic sites from the human genome and extracted their flanking sequences to build negative datasets. The sequence number and length of the negative datasets were chosen to match those of the positive datasets with the same region type (e.g. Alu, non-Alu, coding, etc.) and were at least 200 base pairs away from known RNA editing sites. Millions of positive sequences and negative sequences were first converted to binary data vectors in four series through the One-Hot Encoding process [17], with each series corresponding to one of the four possible nucleotide variants (A/T/C/G). The value of 1 affirmed the identity of a particular nucleotide variant at the particular position, while a value of 0 negated that identity.
In addition to the primary focus of humans, we also trained CNN models for another four species. Mouse (8,823 A-to-I sites) and fruitfly (5,025 A-to-I sites) RNA editing data were downloaded from RADAR [18]. Bumblebee (65,534 A-to-I sites) RNA editing sites were obtained from the publication by Porath et al. [19]. Squid (62,250 A-to-I sites) RNA editing sites were obtained from the publication by Liscovitch-Brauer et al. [20]. Negative genomic sequences in these additional species were extracted similarly as we did in the human genome.
CNN architecture and parameters
In the preliminary design phase, we tested recurrent neural networks (RNN) for RNA editing prediction. It turned out that the performance of RNN models was inferior to CNN models. So, the final classifiers used deep CNN networks to infer RNA-editing from One-Hot-encoded input sequences (Figure 1). The CNN models consist of multiple convolutional layers following the input layer and two traditional fully connected layers right before the output layer. Pooling operations, batch normalization, and neuron dropout (rate=0.25) were implemented. For the activation function of hidden layers, we chose the rectified linear unit (ReLU), defined as ReLU(x) = max(0, x), with x being the input to a neuron. For the output layer, logistic regression for binary classification was applied to derive the prediction score , where x denoted the input to an output neuron and s(x) the prediction score. The loss function for iterative optimization was set as cross-entropy loss, defined the entropy between a true distribution p and the estimated class probabilities q, as . where x takes value among the possible class labels (in our case, 0 and 1).
Figure 1. EditPredict model schema. The two left panels with light grey shading correspond to the training (top) and application (bottom) processes, respectively.
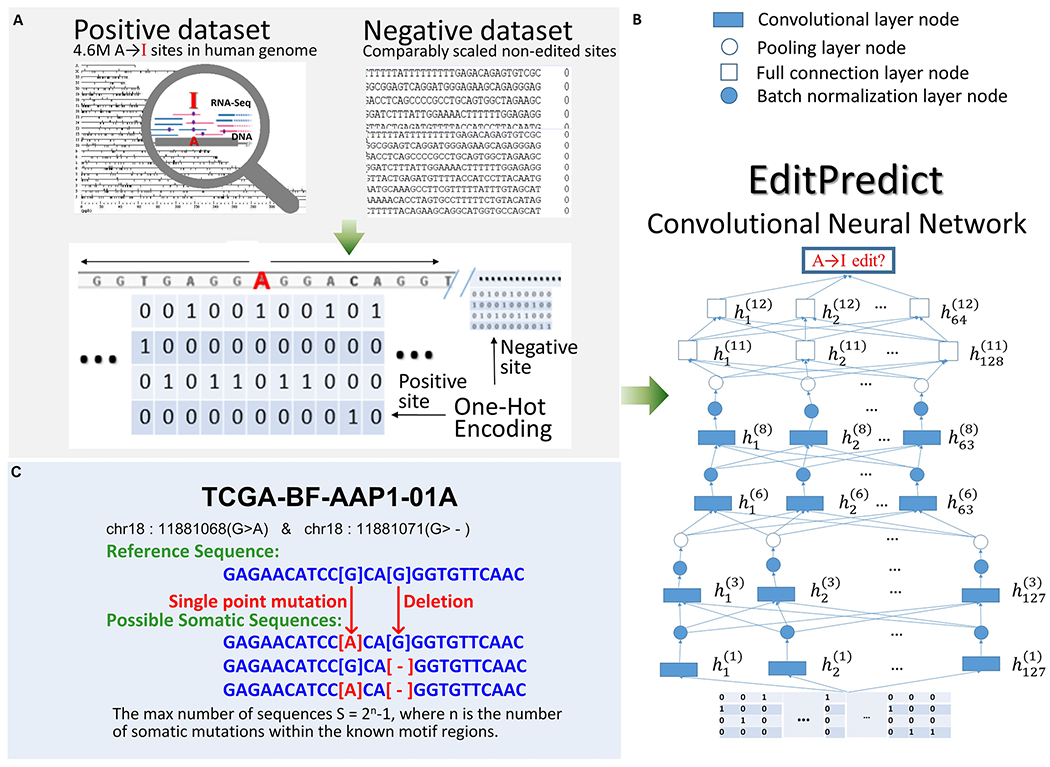
A. Approximately, flanking sequences of ~4.6 million known RNA editing sites were extracted as the positive set, and matched negative sites were constructed. One-hot encoding for a segment of 11 nucleotides was illustrated. B. The CNN architecture handles bi-directional long flanking sequences of potential A-to-I editing sites. The number of layers and the number of neurons per layer are depicted. The convolution process has been omitted for clarity. C. An example of a personalized genome approach. Multiple flanking sequences for a real subject may be generated based on mutation and INDELs.
To accommodate varied input sequence lengths, we designed variant CNN architectures with differences in kernel size, pooling size, and possibly other parameters. A representative CNN architecture that handles bi-directional 50-nucleotide flanking sequences (i.e., 101-nucleotide full length) is illustrated in Figure 1 (A and B), and its more technical details are provided in Supplementary Table 1.
CNN models were trained with the assistance of Python library Keras. We took advantage of GPU processing with an NVIDIA 1080TI. The most cost-effective model, entitled “EditPredict,” was put online through PHP scripts for user application. EditPredict is capable of inferring RNA-editing propensity for any user-interested positions in the reference genome, and can also parse an optional input of individualized genomic variant set to achieve personalized inference of RNA-editing propensity for a particular individual (Figure 1C).
Evaluation of CNN prediction performance
Four prediction metrics were calculated to measure the performance of the initially trained CNN model: Precision, Recall, F1 score, and Accuracy. These metrics are defined in Eq. 1–4, where TP, FP, TN, FN represent the numbers of true positive (correctly predicted RNA-editing sites), false positive (non-edited sites incorrectly predicted as editing sites), true negative (non-edited sites predicted as non-edited), and false negative (RNA-editing sites predicted as non-edited), respectively.
| (Equation 1) |
| (Equation 2) |
| (Equation 3) |
| (Equation 4) |
Validation of CNN Model
An independent validation experiment was conducted using RNA-Seq. CRISPR/Cas9 experiments were carried out to the knockout of the primary Adenosine Deaminase acting on RNA (ADAR1), the enzyme responsible for the A-to-I RNA editing reaction. Jurkat cells (4x106) were electroporated using the NEON transfection system at 1,350 volts for 10 ms and 3 pulses. Electroporation of cells introduced HiFi Cas9 (IDT 1081061) complexes with tracRNA-550 (IDT 1075927) and gsRNA targeting the PAM sequence “AGG” starting at chr1: 154,601,042 (HG38). Cells were harvested at 48 hours post electroporation for RNA isolation and collection. Total RNA was extracted from fresh frozen tissues (30mg) using the RNeasy Universal Kit (QIAGEN) according to the manufacturer’s instructions. Synthesis of cDNA and library preparation was performed using the SMARTer Universal Low Input RNA Kit for Sequencing (Clontech) and the Ion Plus Fragment Library Kit (ThermoFisher) as previously described [21–23]. Sequencing was performed using the Ion Proton S5/XL systems (Life Technologies) in the Analytical and Translational Genomics Shared Resource at the University of New Mexico Comprehensive Cancer Center.
To filter out potential false positives in our data sets, we included nucleotide sites that had >20 read coverage in both samples, including the replicates. Only sites occurring at a frequency of >0.10 were reported. Putative RNA-editing sites were identified by comparing the editing sites between wild-type Jurkat cells and ADAR1 knockout Jurkat cells. A second ADAR1 KO RNA-Seq data of HEK293T cells were downloaded from Short Read Archive (GSE99249). The RNA-Seq data were processed following GATK’s RNA-Seq variant calling best practice protocols to produce binary alignment map files (BAM). A-to-I RNA editings were inferred by comparing HEK293T cells BAMs with and without ADAR1, knockout using REDItools [24].
RESULTS
Model performance in cross-validation
A positive set of RNA editing sites (4.67 million, Alu and non-Alu combined) and a negative set (also 4.67 million) of non-edited sites were used for training the CNN model. The complete model design and CNN architecture are illustrated in Figure 1. The first set of models we trained were without distinguishing Alu and non-Alu RNA editing. We employed ten-fold cross-validation to evaluate the performance of the various CNN models dependent on alternate modes and length parameters. As shown in the result summary (Figure 2A), our CNN models achieved good classification performance, returning accuracy, precision, recall, and F1 score as high as 95-97%. For up to 50 nucleotides, the sequence length showed a positive effect on model performance: the elongation of sequence length from 10 nucleotides to 50 nucleotides boosted the accuracy by more than 10% for uni-directional models, and by 6% for the bi-directional model. The bi-directional model generally outperformed the counterpart uni-directional models by 2%~10% increased accuracy. The bi-directional model on flanking sequences of length 100-nucleotide (50 nucleotides in both downstream and upstream directions) returned an accuracy of 96.1%, which exceeded 100-nucleotide uni-directional models by 3% accuracy. Increasing sequence length beyond 100 nucleotides did not yield a substantial performance gain. The results for RNA editing CNN models for other non-human species are available in Supplementary Table 2.
Figure 2. Performance of various models of RNA editing prediction out of ten-fold cross-validation.
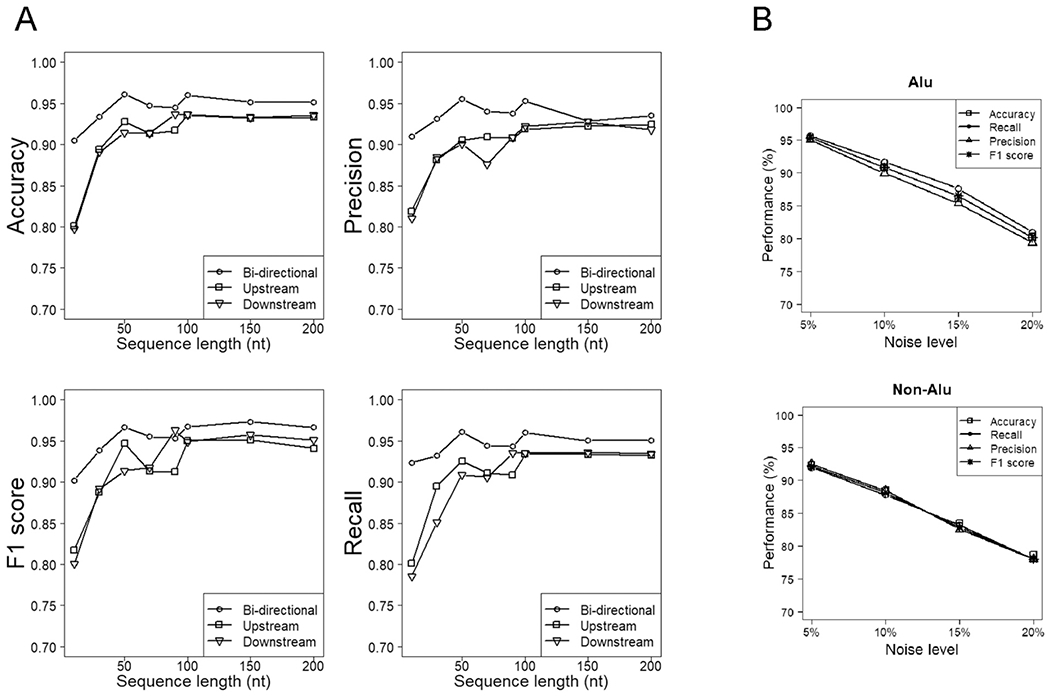
A, Performance of the overall models (with Alu and non-Alu RNA editing sites combined) of various directional modes and handling sequence lengths. “Upstream” and “downstream” refer to sequences adjoining the concerned sites in only one direction, while “both sides” refer to sequences extended from the central concerned sites bidirectionally. Compared to the uni-directional models, the corresponding bi-directional models deal with sequences of a roughly doubled length, although they are depicted with the same series of length parameters for illustrative symmetry. B, Performance of the Alu/non-Alu models trained on perturbed training datasets, where a percentage of positive datapoints were replaced with random negative datapoints.
Following a prior research example [25], we investigated the CNN models’ robustness against wrong training data. For this sake, we replaced a moderate portion (5%-20%) of the 4.67 RNA-editing sites with random genomic sites and built new models resulting from the perturbed data. With CNN models trained for Alu and non-Alu sites separately (details below), cross-validation accuracies decreased from 92~95% to 78~80% when the training data contained up to 20% false positive datapoints (Figure 2B). This label-perturbation experiment reassured us that a moderate noise level in training data does not impede the CNN performance significantly, which is consistent with the earlier investigation conclusion [25].
Alu vs. non-Alu RNA editing models
It is well known that A-to-I RNA editing occurs mostly in Alu elements [26]. The location of A-to-I RNA editing is also not random within the Alu elements, with noticeable peak patterns (Figure 3A). There are 1,131,306 Alu elements in the human genome, and 78.1% of Alu elements have a length between 250 nucleotides and 340 nucleotides. These Alu elements of more regular length were enrolled to reveal a noticeable spatial pattern of RNA editing occurrence (Figure 3A). The full length of each Alu element was divided into 100 bins, and we counted the number of RNA edits falling into each bin. The bin-wise occurrence of RNA edits across all Alu elements was divided by the number of analyzed Alu elements, thus giving rise to an RNA editing rate of each bin (Figure 3A).
Figure 3. A-to-I RNA editing associations with Alu elements.
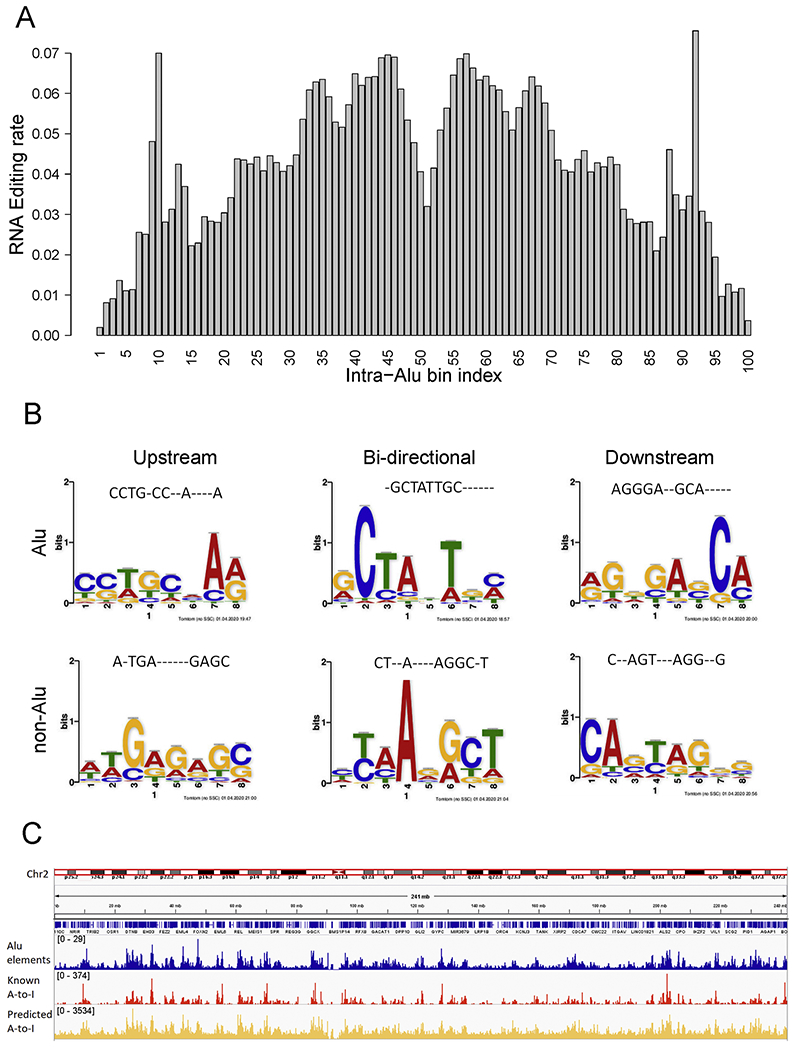
A) A-to-I RNA editing site distribution over the span of an Alu segment (normalized to 100 bins). Hotspot of A-to-I RNA editing can be observed near the boundary (10th & 92th bins), and the other peak is near the center point (45th & 57th bins). The precise boundary sites have the least likelihood of editing, and the absolute center has a moderate level of editing. This figure used data only from the positive strand perspective, thus producing a symmetry effect. B) Motifs from upstream, downstream and bi-directional models filters with highest weight. Each sequence logo is converted to an oligo nucleotide sequence by taking the nucleotide of the highest probability, and the alignment of the motif sequence with the best-hitting previous RNA editing motif is displayed on the top. C) Overlay of Alu elements, known A-to-I RNA editing sites, and predicted A-to-I RNA editing sites across chromosome 2. Similar peaks location can be observed.
The overwhelming enrichment of A-to-I RNA editing in Alu elements and the spatial pattern of A-to-I RNA editing within Alu elements suggest that our overall CNN model could be identifying a pattern specific to Alu elements. To remove the potential confounding effect of Alu/non-Alu distinction, we divided the training set into Alu and non-Alu set and retrained the models. The Alu RNA editing model achieved an accuracy of 99.6% and the non-Alu model achieved an accuracy of 94.1%. Applying the human Alu model on human non-Alu data yielded an accuracy of around 50%. Cross-species application of the human Alu model on mouse RNA editing data which is considered mostly Alu-free RNA editing sites also yielded an accuracy of around 50%. These results suggest that Alu model and non-Alu model captured distinct flanking sequence patterns of two disparate types of RNA-editing sites: The Alu model captured the Alu sequence patterns, and non-Alu model captured the sequence patterns for non-Alu RNA editing sites.
To learn the motif which led to the positive prediction of RNA editing, we tried to visualize filters and feature maps in our CNN models. Specifically, we inspected and visualized the two-dimensional filters and the activation maps output by convolutional layers to understand exact features for a given input sequence. We retrieved filter weights and feature maps for the first convolutional layer. Then, we backtracked to the input sequence which is corresponding to the feature maps. The size of the sequence motif was determined by the filter size of the CNN model. The top motifs, as ranked by the first-layer edge weight of the CNN model, are displayed in Figure 3B for upstream, downstream, and bi-directional models, respectively. We utilized R package Biostrings [27] to performed global Needleman-Wunsch alignment between the current six motifs and 13 previous motifs [12], and displayed the best alignment pattern for each current motif (Figure 3B). These six current motifs were congruent with three of the previous 13 motifs (#4, #6, and #7 in Table 1 of the previous study [12]), so our new results have helped to narrow down the valid RNA editing motifs.
We applied EditPredict to predict the RNA edit propensity for the entire chromosome 2 of 121,450,757 positions. Overall, 5% nucleotides of chromosome 2 were predicted to be potential RNA editing sites; this percentage far exceeds a baseline editing rate of 0.16% obtained by dividing the total number of known RNA-editing events (4.67 million) by nucleotide volume of the human reference genome (3 billion). By overlaying known Alu elements, known A-to-I RNA editing, and predicted A-to-I RNA editing sites, clear concordance can be observed (Figure 3C).
Validations
Adenosine Deaminases acting on RNA (ADAR) are responsible for A-to-I RNA editing, consisting of two major isoforms ADAR1 and ADAR2 in mammals. We conducted an independent validation experiment for the trained CNN models using RNA-Seq. Knockout of ADAR1 in Jurkat cells was performed with CRISPR/Cas9 (Figure 4A). The knockout of ADAR1 was verified by western blot (Figure 4B). RNA editing sites were identified by comparing RNA-Seq data pre- and post-ADAR1 knockout. By comparing RNA-Seq data pre and post ADAR1 knockout, 5,372 novel RNA-editing sites (5,076 Alu and 296 non-Alu) outside the scope of the training dataset were identified. These 5,372 novel RNA editing sites and the same amount of novel negative sites were used as the validation dataset for testing trained CNN models. Receiver operating characteristic curves show that the bidirectional models performed the best (Figure 4C). Alu bi-directional model achieved an overall accuracy of 99.4% and non-Alu bi-directional model achieved an overall accuracy of 95.2%. These two models also obtained respectable sensitivity and specificity (Figure 4D).
Figure 4.
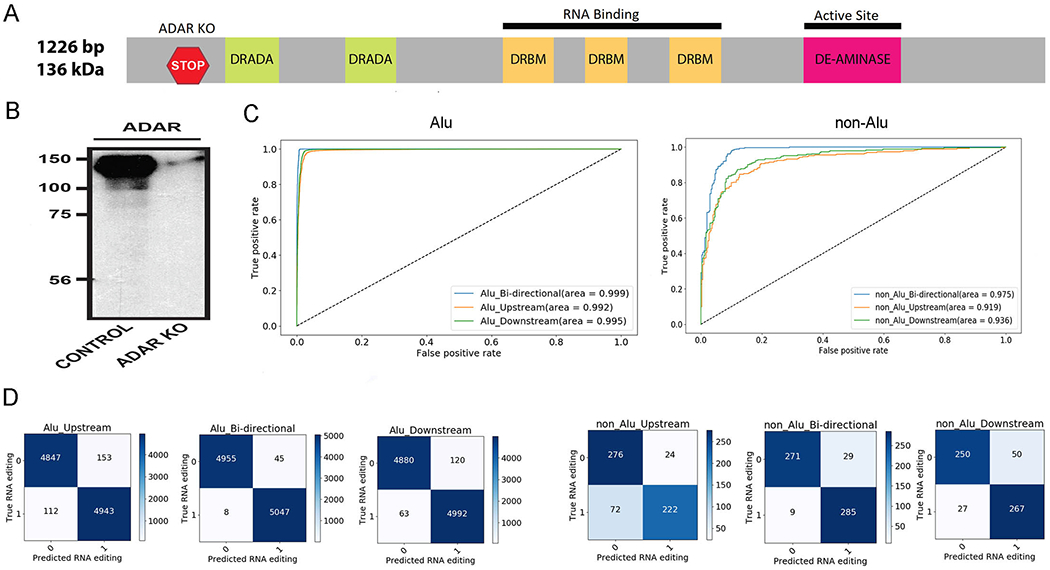
Validation results for CNN RNA editing models. A. Location of the ADAR1 KO with CRISPR/Cas9. B. Western blot shows the successful knockout of ADAR1. C. ROC curves suggest the excellent performance of CNN Alu and non-Alu models on the independent validation set of novel RNA editing sites. Models used 50-nucleotide flanking sequences for upstream, downstream, and bi-directional modes. D. Confusion tables display both true positive and true negative validation results for both Alu and non-Alu models.
Another independent novel RNA editing dataset was obtained by analyzing RNA-Seq data from HEK293T (GSE99249) cells pre and post ADKAR1 knockout which identified 889 novel Alu and 100 novel non-Alu RNA editing sites [28]. Validation on these data produced accuracy 99.0% and 91.4% for Alu and non-Alu CNN bidirectional models, respectively. All models tested in the validation phase were constructed from 101-nucleotide long sequences.
Comparison with Other Tools
Many other RNA editing detection tools have been developed previously, such as RNAeditor [29], REDItools [24], etc. These tools detect RNA editing from sequencing data and are fundamentally different from EditPredict which predicts the binary RNA editing outcome from the reference genome. EditPredict should be used as a supplement to existing RNA editing detection tools. After novel RNA editing sites are detected from sequencing data, EditPredict can be applied to select top candidate for further wetlab validation or downstream analysis. DeepRed [13] is another tool that uses flanking sequences to predict RNA editing. There is one major technical difference between EditPredict and DeepRed. DeepRed is a deep learning-based hybrid framework integrated with ensemble learning. It combined multiple ensemble Deep Neural Networks (DNNs) by using an averaging method. EditPredict is based on deep learning model using CNN which can take the advantage of inherent properties of nucleotide sequence. DNN does not see any order in their inputs. If the nucleotide sequence is cut into pieces and reorder, DNN may not be able to recognize it. On the other hand, CNN takes the advantage of local spatial coherence of the sequence and can reduce the number of operations needed to process input by using convolution on patches of adjacent pixels. CNN contains pooling layers, which downscale the input and thus allow faster computation.
In addition to the technical difference, EditPredict has several notable advantages. DeepRed applied a bootstrap resampling method to avoid class imbalance problem. EditPredict is trained with 4.6 million real RNA editing data. EditPredict also identifies different flanking patterns between Alu and non-Alu RNA editing and employs a personalized genome approach. Furthermore, DeepRed was developed with MATLAB commercial software. EditPredict is developed in Python and freely usable online without any complication due to installation and software dependencies. To compare the performance between EditPredict and DeepRed, we used RNA editing derived from U87 cell. The same data were used in the original DeepRed study. DeepRed achieved an accuracy of 93.23%, while EditPredict achieved an accuracy of 97.89%. The complete comparison result including precision, recall, and F1 statistics are available in Supplementary Table 3.
EditPredict Implementation
Based on the cross-validation and independent validation results, the bi-directional 100-nucleotide model achieved an optimal balance between accuracy and runtime. EditPredict is capable of inferring RNA-editing propensity for any user-interested positions in the human reference genome (GRCh38 and GRCh37), and can also parse an optional input of individualized genomic variant set to achieve personalized inference of RNA-editing propensity for a personal genome. EditPredict is also equipped with CNN models for bumblebee, fruity fly, mouse, and squid. The parameters of models populated in EditPredict for all five species are detailed in Supplementary Table 1. EditPredict is developed using a combination of Python, R, PHP, JavaScript, and HTML. CNN models were trained with the assistance of Python library Keras by taking advantage of GPU processing with an NVIDIA 1080TI. The online server can make predictions for up to 300 candidate genomic positions in less than two minutes, but it may take an appreciably longer time when the position total number goes beyond a thousand. If the user invokes the personalized inference with a custom VCF file, the computational time normally increases by 20% compared to the counterpart job session without a VCF input.
DISCUSSION
So far, there have been many successful applications of deep learning algorithms in genomic sequence analyses. Through this work, we developed yet another deep learning application EditPredict to add to this expanding research direction, which was proved to predict editable RNA-editing sites with an accuracy of ~95% in general. We implemented the validated cost-effective CNN model in a user-friendly online tool, where users can employ our server to predict RNA editing propensity for any adenosine sites within five genomes: human, bumblebee, fruit fly, mouse, and squid. Of note, for humans, both the reference genome or a particular personal genome defined by a set of individualized genomic variants.
Here, through ten-fold cross-validation of 4.67 million RNA editing sites and two independent tests of novel editing sites, we proved that sequence patterns hidden in RNA editing flanking sequences can be modeled by CNN and that the established model can reach an accuracy of ~95% in general. Our analyses also show that Alu and non-Alu RNA editing have different sequence patterns. The models constructed for Alu did not perform well for non-Alu RNA editing data, and vise versa. Also, models did not perform well when used cross-species, which suggests sequence pattern uniqueness within each species. In parallel to the study of RNA editing sites, we had tried predicting expression quantitative loci (eQTL) with a similar CNN approach. With 2.6 million sequences flanking GTEx eQTLs [30] as a positive dataset, a CNN model configured similarly to the one of EditPredict attained a 60% accuracy at best – not much better than a random coin-flip classifier. The unsuccessful trial with eQTL suggests that a sequence pattern permissive for accurate prediction is not intrinsic with all genomic features, but it must be embedded in RNA-editing sites’ flanking sequences. The CNN model from our tool EditPredict was capable of discerning the intricate sequence context pattern shared by general A-to-I RNA editing events and thereby precisely discriminating editable Adenosine sites from non-editable ones.
EditPredict achieves a comparable performance to the existing tool DeepRed [13], which is also built upon Deep Neural Networks. The major difference between EditPredict and DeepRed lies in the degree of independence of HTS data. DeepRed trained its classifiers on a set of RNA editing sites called ab initio from 64 RNA-Seq samples, and it is expected to distinguish probable RNA-editing events from SNVs, a typical output from general sequencing data analyses. Our strategy, however, is purely based on genomic sequences, which are always accessible through the public human reference genome. At the application interface, both DeepRed and EditPredict can accept a set of SNVs as input, but these SNVs were interpreted in fundamentally different ways. DeepRed takes these SNVs as candidate sites and, by analyzing the flanking sequences retrieved from the reference genome, sets out to assess RNA-editing likelihood for each candidate site independently. In contrast, EditPredict requires another mandatory input of candidate sites and takes the optional set of SNVs as personal alterations in relation to the human reference genome. In this way, the flanking sequences fed to EditPredict are tailored to reflect the individualized local context of candidate sites, thus allowing a personalized RNA-editing inference for an individual genome (Figure 1C).
While we investigated a wide range of possible length values for flanking sequences [15], the pursuit of the optimal tradeoff between performance and computation burden motivated us to settle on a full sequence length of 101-nucleotide for the bi-directional mode (50-nucleotide on each side of the central candidate site). Coincidentally, this sequence length is roughly the same length used by DeepBind [15], a well-known Deep Learning application in transcription factor binding analysis. The sequence length of 100-nucleotides is apparently longer than the corresponding parameter of an earlier logistic regression classifier [12] but is only half the size as required by DeepRed. Theoretically, EditPredict should compete favorably against DeepRed in terms of computation time.
In the compilation of the positive dataset for CNN training, we have decided to include all A-to-I RNA editing sites but none of the other RNA-editing subtypes. This is because A-to-I RNA editing events remarkably dominate an RNA editome with a percentage of over 95%, and this biological mechanism has been resolved to the most rigorous extent. Cytidine-to-uridine changes form another minor yet canonical class of RNA editing, but these editing events tend to harbor less functional importance than A-to-I RNA editing sites [31]. Given the quantitative and functional advantage of A-to-I RNA editing events, we decided to build a CNN model for this class of RNA editing events only. This restricts the application of EditPredict to A-to-I RNA editing events only. In the future, it may be feasible to extend EditPredict with additional models tailored towards other classes of RNA-editing events or other genomic features, as long as we obtain sufficient positive examples with satisfactory experimental evidence.
Our study has limitations. For example, we did not attempt to refine the enormous RNA-editing sites curated by REDIportal, but have included all 4.67 million RNA-editing sites as positive examples in our CNN model. It is possible that a certain number of false positives were present in our training data. Additionally, RNA editing has different efficiencies. Such efficiency is not captured by our model. We treated RNA editing as a binary event for simplicity. Furthermore, in our knockout experiment, we only knocked out ADAR1 instead of both ADAR1 and ADAR2. While ADAR1 and ADAR2 work similarly, they could have slightly different recognition sequence patterns. By only knocking out ADAR1, we didn’t independently validate ADAR2 RNA editing. However, the high performance of cross-validation and independent ADAR1 validation is a good indication of overall model performance which includes ADAR2 induced RNA editing. Our positive results of CNN predicting RNA editing sites add to the growing successes of Deep Learning applications on various genomic sequence features such as splicing junctions and transcription factor binding targets. Despite its optimistic performance estimation, EditPredict is a sequence-only, sequencing-independent tool, which is not purported to replace sequencing technology as the definitive method for detecting RNA editing events. EditPredict can be used stand-alone to predict novel RNA editing sites as most genomic prediction tools. However, it can be applied to enhance RNA editing detection by offering an independent layer of confidence filtering. For example, RNA editing sites detected by other RNA editing detection tools from RNA-Seq data, can be compared to EditPredict results and ranked by EditPredict’s editing probability to select most likely true RNA editing sites for downstream analysis.
Supplementary Material
Using Convolutional Neural Network, we trained a human RNA editing prediction model based on flanking sequence.
The model was validated in two independent datasets.
The same strategy was used to train four non-human species.
The models were implemented into an online application.
Funding
This work was supported by Cancer Center Support Grant from National Cancer Institute (P30CA118100) and also supported by Bioinformatics, Biostatistics, and Analytical & Translational Genomics Shared Resources at the University of New Mexico Comprehensive Cancer Center. YG was supported by National Cancer Institute Grant R01ES030993-01A1.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Availability of data and materials
EditPredict web application is freely accessible to the public at http://www.innovebioinfo.com/Sequencing_Analysis/RNAediting/RNA1.php. The programming code to implement EditPredict is publicized at GitHub at https://github.com/wjd198605/EditPredict.
The RNA-Seq data on ADAR1 KO with CRISPR/CAS9 has been submitted to NIH under BioProject number PRJNA604003.
Competing interests
Each co-author has no financial and non-financial competing interests.
Ethics approval
NA.
REFERENCES
- 1.Peng X, Xu X, Wang Y, Hawke DH, Yu S, Han L, Zhou Z, Mojumdar K, Jeong KJ, Labrie M, et al. A-to-I RNA Editing Contributes to Proteomic Diversity in Cancer. Cancer Cell 2018, 33:817–828 e817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Han L, Diao L, Yu S, Xu X, Li J, Zhang R, Yang Y, Werner HMJ, Eterovic AK, Yuan Y, et al. The Genomic Landscape and Clinical Relevance of A-to-I RNA Editing in Human Cancers. Cancer Cell 2015, 28:515–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paz-Yaacov N, Bazak L, Buchumenski L, Porath HT, Danan-Gotthold M, Knisbacher BA, Eisenberg E, Levanon EY: Elevated RNA Editing Activity Is a Major Contributor to Transcriptomic Diversity in Tumors. Cell Reports 2015, 13:267–276. [DOI] [PubMed] [Google Scholar]
- 4.Picardi E, D’Erchia AM, Lo Giudice C, Pesole G: REDIportal: a comprehensive database of A-to-I RNA editing events in humans. Nucleic Acids Research 2017, 45:D750–D757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kiran A, Baranov PV: DARNED: a DAtabase of RNa EDiting in humans. Bioinformatics 2010, 26:1772–1776. [DOI] [PubMed] [Google Scholar]
- 6.Bazak L, Haviv A, Barak M, Jacob-Hirsch J, Deng P, Zhang R, Isaacs FJ, Rechavi G, Li JB, Eisenberg E, Levanon EY: A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res 2014, 24:365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li JB, Levanon EY, Yoon JK, Aach J, Xie B, LeProust E, Zhang K, Gao Y, Church GM: Genome-Wide Identification of Human RNA Editing Sites by Parallel DNA Capturing and Sequencing. Science 2009, 324:1210–1213. [DOI] [PubMed] [Google Scholar]
- 8.Guo Y, Yu H, Samuels DC, Yue W, Ness S, Zhao YY: Single-nucleotide variants in human RNA: RNA editing and beyond. Brief Funct Genomics 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schrider DR, Gout JF, Hahn MW: Very Few RNA and DNA Sequence Differences in the Human Transcriptome. Plos One 2011, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Piskol R, Peng Z, Wang J, Li JB: Lack of evidence for existence of noncanonical RNA editing. Nat Biotechnol 2013, 31:19–20. [DOI] [PubMed] [Google Scholar]
- 11.Eggington JM, Greene T, Bass BL: Predicting sites of ADAR editing in double-stranded RNA. Nature Communications 2011, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nigita G, Alaimo S, Ferro A, Giugno R, Pulvirenti A: Knowledge in the Investigation of A-to-I RNA Editing Signals. Front Bioeng Biotechnol 2015, 3:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ouyang Z, Liu F, Zhao C, Ren C, An G, Mei C, Bo X, Shu W: Accurate identification of RNA editing sites from primitive sequence with deep neural networks. Sci Rep 2018, 8:6005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Quang D, Xie X: DanQ: a hybrid convolutional and recurrent deep neural network for quantifying the function of DNA sequences. Nucleic Acids Res 2016, 44:e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alipanahi B, Delong A, Weirauch MT, Frey BJ: Predicting the sequence specificities of DNA-and RNA-binding proteins by deep learning. Nat Biotechnol 2015, 33:831–838. [DOI] [PubMed] [Google Scholar]
- 16.Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, Darbandi SF, Knowles D, Li YI, Kosmicki JA, Arbelaez J, Cui W, Schwartz GB, et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176:535–548 e524. [DOI] [PubMed] [Google Scholar]
- 17.Choong ACH, Lee NK: Evaluation of convolutionary neural networks modeling of DNA sequences using ordinal versus one-hot encoding method. In International Conference on Computer and Drone Applications; November 9-11; Kuching, Malaysia. 2017 [Google Scholar]
- 18.Ramaswami G, Li JB: RADAR: a rigorously annotated database of A-to-I RNA editing. Nucleic Acids Res 2014, 42:D109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Porath HT, Hazan E, Shpigler H, Cohen M, Band M, Ben-Shahar Y, Levanon EY, Eisenberg E, Bloch G: RNA editing is abundant and correlates with task performance in a social bumblebee. Nature Communications 2019, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liscovitch-Brauer N, Alon S, Porath HT, Elstein B, Unger R, Ziv T, Admon A, Levanon EY, Rosenthal JJC, Eisenberg E: Trade-off between Transcriptome Plasticity and Genome Evolution in Cephalopods. Cell 2017, 169:191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brayer KJ, Frerich CA, Kang H, Ness SA: Recurrent Fusions in MYB and MYBL1 Define a Common, Transcription Factor-Driven Oncogenic Pathway in Salivary Gland Adenoid Cystic Carcinoma. Cancer Discov 2016, 6:176–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brown RB, Madrid NJ, Suzuki H, Ness SA: Optimized approach for Ion Proton RNA sequencing reveals details of RNA splicing and editing features of the transcriptome. PLoS One 2017, 12:e0176675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frerich CA, Brayer KJ, Painter BM, Kang H, Mitani Y, El-Naggar A, Ness SA: Transcriptomes define distinct subgroups of salivary gland adenoid cystic carcinoma with different driver mutations and outcomes. Oncotarget 2018, 9:7341–7358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Picardi E, Pesole G: REDItools: high-throughput RNA editing detection made easy. Bioinformatics 2013, 29:1813–1814. [DOI] [PubMed] [Google Scholar]
- 25.Yu H, Samuels DC, Zhao YY, Guo Y: Architectures and accuracy of artificial neural network for disease classification from omics data. Bmc Genomics 2019, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Athanasiadis A, Rich A, Maas S: Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol 2004, 2:e391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Biostrings: Efficient manipulation of biological strings [https://bioconductor.org/packages/Biostrings] [Google Scholar]
- 28.Chung H, Calis JJA, Wu X, Sun T, Yu Y, Sarbanes SL, Dao Thi VL, Shilvock AR, Hoffmann HH, Rosenberg BR, Rice CM: Human ADAR1 Prevents Endogenous RNA from Triggering Translational Shutdown. Cell 2018, 172:811–824 e814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.John D, Weirick T, Dimmeler S, Uchida S: RNAEditor: easy detection of RNA editing events and the introduction of editing islands. Brief Bioinform 2017, 18:993–1001. [DOI] [PubMed] [Google Scholar]
- 30.Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, S S, Consortium GT: The Genotype-Tissue Expression (GTEx) project. Nat Genet 2013, 45:580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Z, Zhang J: Human C-to-U Coding RNA Editing Is Largely Nonadaptive. Mol Biol Evol 2018, 35:963–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.