

Abstract
Objective
To investigate the role of PF‐06650833, a highly potent and selective small‐molecule inhibitor of interleukin‐1–associated kinase 4 (IRAK4), in autoimmune pathophysiology in vitro, in vivo, and in the clinical setting.
Methods
Rheumatoid arthritis (RA) inflammatory pathophysiology was modeled in vitro through 1) stimulation of primary human macrophages with anti–citrullinated protein antibody immune complexes (ICs), 2) RA fibroblast‐like synoviocyte (FLS) cultures stimulated with Toll‐like receptor (TLR) ligands, as well as 3) additional human primary cell cocultures exposed to inflammatory stimuli. Systemic lupus erythematosus (SLE) pathophysiology was simulated in human neutrophils, dendritic cells, B cells, and peripheral blood mononuclear cells stimulated with TLR ligands and SLE patient ICs. PF‐06650833 was evaluated in vivo in the rat collagen‐induced arthritis (CIA) model and the mouse pristane‐induced and MRL/lpr models of lupus. Finally, RNA sequencing data generated with whole blood samples from a phase I multiple‐ascending‐dose clinical trial of PF‐06650833 were used to test in vivo human pharmacology.
Results
In vitro, PF‐06650833 inhibited human primary cell inflammatory responses to physiologically relevant stimuli generated with RA and SLE patient plasma. In vivo, PF‐06650833 reduced circulating autoantibody levels in the pristane‐induced and MRL/lpr murine models of lupus and protected against CIA in rats. In a phase I clinical trial (NCT02485769), PF‐06650833 demonstrated in vivo pharmacologic action pertinent to SLE by reducing whole blood interferon gene signature expression in healthy volunteers.
Conclusion
These data demonstrate that inhibition of IRAK4 kinase activity can reduce levels of inflammation markers in humans and provide confidence in the rationale for clinical development of IRAK4 inhibitors for rheumatologic indications.
INTRODUCTION
Rheumatoid arthritis (RA) affects ~1% of the population, manifesting with joint pain and tissue destruction and characterized in seropositive cases by the presence of antibodies against posttranslationally modified proteins, as well as IgM (rheumatoid factor) (1, 2). Cells involved in RA inflammation include not only lymphocytes but also neutrophils, macrophages, osteoclasts, and synovial fibroblasts. Treatments for RA include small‐molecule disease‐modifying antirheumatic drugs as well as biologic agents (3). The rate of sustained remission over time in RA remains disappointingly low, highlighting the need for additional therapeutic mechanisms (4).
Systemic lupus erythematosus (SLE) is also a systemic disease mediated by autoantibodies, and is characterized by tissue inflammation and damage to multiple organ systems including joints, skin, and kidneys (5). Defects in the clearance of apoptotic and necrotic cells have been demonstrated, allowing access to nuclear antigens by autoantibodies (6). Resulting immune complexes activate numerous immune cell types, including dendritic cells (DCs) and B lymphocytes. Treatments for SLE include glucocorticoids and antimalarial agents, but efficacy is limited and long‐term use is associated with toxicity. The BAFF‐neutralizing antibody belimumab was approved in 2015, and while it provides benefit in some SLE patients, there remains a clear need for additional treatment modalities (5).
Interleukin‐1 receptor–activated kinase 4 (IRAK4) is a central regulator of the innate immune response. IRAK4 transmits signals from Toll‐like receptors (TLRs) and interleukin‐1 receptor (IL‐1R) by binding the adaptor protein myeloid differentiation factor 88 (MyD88) and inducing signaling through IRAK1 and IRAK2 (7, 8). The downstream result of myddosome assembly is activation of NF‐κB, interferon regulatory factor 5 (IRF‐5), and MAPK (9). Deletion of IRAK4 or inactivation of IRAK4 activity in mice prevents the development of inflammation in multiple models of inflammatory disease (10, 11, 12, 13, 14). Cells from IRAK4‐deficient humans also show no response to TLR or IL‐1R family ligands that signal through MyD88 (15).
Several recent studies have highlighted the antiinflammatory efficacy of IRAK4 inhibitors in human cells and preclinical models of inflammation (16, 17, 18, 19, 20). However, clinical development has been hampered by debate over the role of IRAK4 kinase activity in disease, as a significant role of kinase‐independent signaling by IRAK4 has been demonstrated (21, 22). We have shown that inhibition of the kinase activity of IRAK4 does not significantly affect IL‐1– or TLR‐induced NF‐κB or MAPK activation and only minimally suppresses IL‐1–induced cytokines, whereas NF‐κB or MAPK activation by IL‐1 or TLR and production of cytokines are not observed with IRAK4‐null cells (7, 23). Additionally, several in vitro studies have suggested that IRAK4 inhibitors, which inhibit inflammatory responses in rodent cells, are not equally efficacious for the same activity in human cells (22, 24, 25). It has also been speculated that inhibition of both IRAK4 and IRAK1 kinase activities may be required for efficacy in human cells (21, 22). A recent study using an IRAK4 inhibitor has confirmed the requirement for IRAK4 kinase activity in mouse and human DC and B cell activation as well as in several in vivo models of SLE (20). However, translation of in vitro antiinflammatory efficacy of IRAK4 inhibition to reduction of inflammation in humans is lacking.
We have recently developed potent and selective inhibitors of IRAK4 with little activity against IRAK1 (18). In the present study we demonstrated that PF‐06650833, a small‐molecule inhibitor of IRAK4, reduces responses to disease‐relevant stimuli in human cells and in animal models of RA and SLE. Importantly, we showed that administration of PF‐06650833 to humans resulted in the suppression of an interferon (IFN) gene signature in a phase I multiple‐ascending‐dose clinical trial (26). Our findings show that selective IRAK4 inhibitors reduce signals of inflammation in humans and are potential therapies for autoimmune disease.
MATERIALS AND METHODS
The identification of PF‐06650833 and methods used to define its pharmacology have been described previously (18). Details on materials and experimental procedures used in the present study are provided in Supplementary Materials, on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41953/abstract.
In vitro cellular assays
Human, mouse, and rat whole blood collected under institutional review board (IRB)– and institutional animal care and use committee (IACUC)–approved protocols and anticoagulated with heparin, or peripheral blood mononuclear cells (PBMCs) derived from these blood samples, were incubated with PF‐06650833, exposed to TLR ligands, and cytokines were assayed with Meso Scale Discovery assay kits. Occupancy of the IRAK4 ATP‐binding site was quantified via blockade of covalent labeling with a biotin‐labeled ATP probe, followed by immunoprecipitation and Western blotting for IRAK4.
Human macrophages were exposed to inhibitors and then incubated with anti–citrullinated protein antibody (ACPA) immune complexes (ICs), and cytokines in the supernatant were measured with Meso Scale Discovery kits. Broad antiinflammatory phenotypic profiling was performed at Eurofins Scientific (St. Charles, MO), using a BioMAP Diversity Plus Panel with protocols that have been described previously (27, 28).
Human RA fibroblast‐like synoviocytes (FLS) were incubated with inhibitors, and stimulatory ligands added. Supernatants were analyzed with a Meso Scale Discovery assay kit.
Neutrophils were isolated from healthy volunteers and SLE patients under IRB‐approved protocols. Neutrophils from healthy volunteers were primed with IFNα2β and stimulated with R837 alone or in combination with PF‐06650833. SLE patient neutrophils were cultured in media for 8–12 hours. Double‐stranded DNA (dsDNA) was quantified using a Quant‐iT PicoGreen kit.
PBMCs were isolated from healthy volunteers, incubated with PF‐06650833, and stimulated with R848 or SLE patient sera. Nuclear localization of IRF‐5 was measured using an Amnis imaging cytometer (7).
For B cell maturation assays, human B cells were isolated from Leukopaks, primed with IFNα, incubated with PF‐06650833, and stimulated with R848. Supernatants were analyzed for cytokines, and the cells were assessed for plasmablast differentiation.
The total DC fraction was obtained from healthy volunteer buffy coats, and plasmacytoid DCs (pDCs) were isolated by fluorescence‐activated cell sorting and cultured with 40% SLE neutrophil supernatants with or without PF‐06650833. IFNα levels were measured by enzyme‐linked immunosorbent assay (ELISA).
SLE ICs were generated from patient plasma and used to stimulate human PBMCs, with or without PF‐06650833. IFNα was measured by ELISA. IFN‐responsive gene expression was analyzed by quantitative reverse transcription–polymerase chain reaction.
In vivo disease models
To model RA, female Lewis rats were immunized with collagen. Subsequently they were administered PF‐06650833 or tofacitinib orally for 7 days. Paw volume and body weight were measured daily. All procedures were reviewed and approved by the Pfizer IACUC.
To model SLE, BALB/c mice were treated with pristane. Subsequently PF‐06650833 was administered in chow at weeks 8–20, while prednisone was administered orally once daily at weeks 1–20. Serum was collected at weeks, 4, 8, 12, and 20. Anti‐dsDNA, anti‐SSA, and anti‐RNP were quantified by ELISA. Kidney specimens were prepared as previously described (29) and evaluated by a pathologist who was blinded with regard to the treatment group.
Female lupus‐prone MRL/lpr mice were fed standard chow with or without added PF‐06650833 for 12–13 weeks. Body weights were recorded once a week, proteinuria was assessed every 2 weeks, and blood was collected every 4 weeks.
Clinical trial
The phase I studies of PF‐06650833 have been described previously (26). RNA was extracted from whole blood and subjected to RNA sequencing. To assess effects on the IFN gene signature, an equally weighted 21‐gene signature (30) was analyzed for percent change on day 14 compared to day 0.
RESULTS
Pharmacologic properties of PF‐06650833
PF‐06650833 was identified as described previously (18) (Figure 1A). The compound is selective as measured by its ability to compete with a covalent analog of ATP in monocyte cell lysates (assessed with an ActivX ATP occupancy assay) (31). Twelve kinases other than IRAK4 had 50% inhibition concentration (IC50) values of <1 μM (Figure 1B and Supplementary Table 1, on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41953/abstract), and PF‐06650833 was nearly 7,000 times more selective for IRAK4 than for IRAK1 (Figure 1B). In PBMCs stimulated with the TLR‐7/8 ligand R848, PF‐06650833 had an IC50 of 2.4 nM for inhibition of tumor necrosis factor (TNF) release and was potent in human whole blood, with an IC50 of 8.8 nM (Figure 1C). The free IC50 values for PF‐06650833 in rodent whole blood were determined to be 5–10‐fold less potent than in human whole blood, in contrast to previous reports indicating lack of IRAK4 inhibitor activity in human cells (24, 25).
Figure 1.
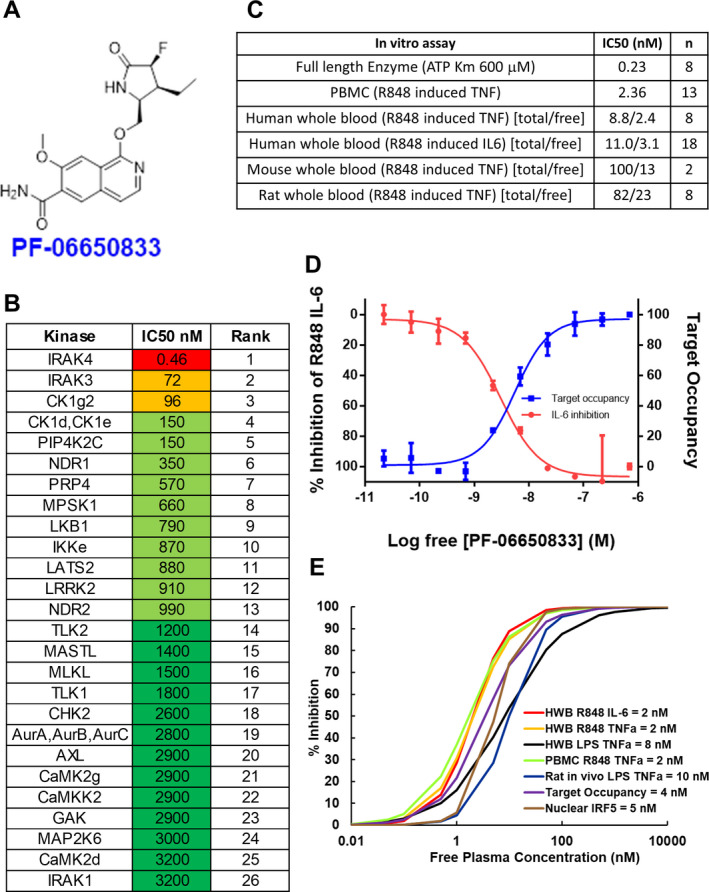
Pharmacologic properties of PF‐06650833, an interleukin‐1–associated kinase 4 (IRAK4) inhibitor. A, Structure of PF‐06650833. B, Selectivity of PF‐06650833 in an ActivX ATP occupancy assay using THP‐1 lysates. The top 26 of >200+ kinases are shown. Fifty percent inhibition concentrations (IC50) were determined using a 5‐point dose‐response curve (full data set shown in Supplementary Table 1, on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41953/abstract). C, Potency of PF‐06650833 in enzyme and cell‐based assays. IC50 values for enzyme and peripheral blood mononuclear cell (PBMC) assays are a single value, whereas whole blood (wb) assays for human, mouse, and rat are denoted as total concentration of compound over the non–protein‐bound (free [f]) concentration of compound, calculated as (wb IC50 × [fu/(B/P)] = IC50 free, where fraction unbound [fu] = 0.22 and blood/plasma ratio [B/P] = 0.91). D, Demonstration of the relationship between free drug concentration, IRAK4 ATP binding site occupancy, and inhibition of downstream biology by PF‐06650833 in a whole blood sample from a patient with systemic lupus erythematosus. Values are the mean ± SEM. E, Use of 50% maximum response concentration data from each preclinical assay to determine that 100 nM was the target free compound concentration required to maintain >90% IRAK4 inhibition across species and assays. CK1g2 = casein kinase 1 gamma 2; PIP4K2C = phosphatidylinositol‐5‐phosphate 4‐kinase 2C; NDR1 = nuclear Dbf2‐related kinase 1; PRP4 = pre–mRNA processing factor 4; MPSK1 = myristoylated and palmitoylated serine/threonine kinase 1; LKB1 = liver kinase B1; LATS2 = large tumor suppressor kinase 2; LRRK2 = leucine‐rich repeat kinase 2; TLK2 = tousled‐like kinase 2; MASTL = microtubule‐associated serine/threonine kinase–like; MLKL = mixed‐lineage kinase domain–like; CHK2 = checkpoint kinase 2; AurA = aurora A kinase; AXL = AXL receptor tyrosine kinase; CAMK2g = Ca2+/calmodulin‐dependent protein kinase 2g; GAK = cyclin G–associated kinase; TNF = tumor necrosis factor; IL‐6 = interleukin‐6; HWB = human whole blood; LPS = lipopolysaccharide; IRF‐5 = interferon regulatory factor 5.
Modeling of PF‐06650833 IRAK4 pharmacology to select efficacious dose
Pfizer has established the “Three Pillars of Survival” concept (32) to predict clinical success based on the ability to measure candidate pharmacology. For PF‐06650833, we established a system that captured all 3 pillars (free drug concentration [pillar I], target occupancy [pillar II], and downstream biologic effect [pillar III]) in an in vitro assay performed in SLE patient whole blood (Figure 1D). Increasing concentrations of PF‐06650833 (pillar I) resulted in increased target occupancy (pillar II), as measured by blockade of the IRAK4 binding site from covalent modification by a probe. With a 50% maximum response concentration (EC50) value very similar to the target occupancy value, R848‐induced IL‐6 release (pillar III) was inhibited by PF‐06650833. Similar results were obtained using blood from a healthy volunteer (Supplementary Figure 1, http://onlinelibrary.wiley.com/doi/10.1002/art.41953/abstract). Thus, target occupancy and cytokine inhibition were proportional and predictable from compound concentration, allowing us to infer pillar II (target occupancy) from pillar III assay results. Using experimentally generated EC50, Hill slope, and protein binding values, we then examined the relationship between free drug concentration and target inhibition in the preclinical assays (Figure 1E). The EC50 values for PF‐06650833 varied by only 5‐fold in these assays—from 2 nM to 10 nM—such that achieving >100 nM free drug concentration would be expected to inhibit >90% of any downstream biologic events dependent on IRAK4 kinase activity. Thus, the target efficacious concentration for preclinical experiments was set at 100 nM.
PF‐06650833–induced inhibition of pathophysiologic processes central to RA
In seropositive RA, ACPAs are observed prior to the clinical diagnosis of RA, are a specific marker of autoimmunity, and indicate more rapid progression of disease and poorer treatment response (33, 34). ACPA ICs activate macrophages to release TNF (35). Thus, we tested the ability of PF‐06650833 to block ACPA IC–induced TNF in samples from 11 donors. PF‐06650833 at 100 nM inhibited TNF release to a mean ± SEM of 57.5 ± 17.9% of that seen with DMSO treatment alone (P = 0.003 by paired t‐test) (Figure 2A). As a positive control, we included a Bruton’s tyrosine kinase (BTK) inhibitor, as this kinase has been previously implicated in macrophage TNF responses to both TLR and Fcγ receptor ligands (36). Results with this inhibitor were similar to those observed with IRAK inhibition.
Figure 2.
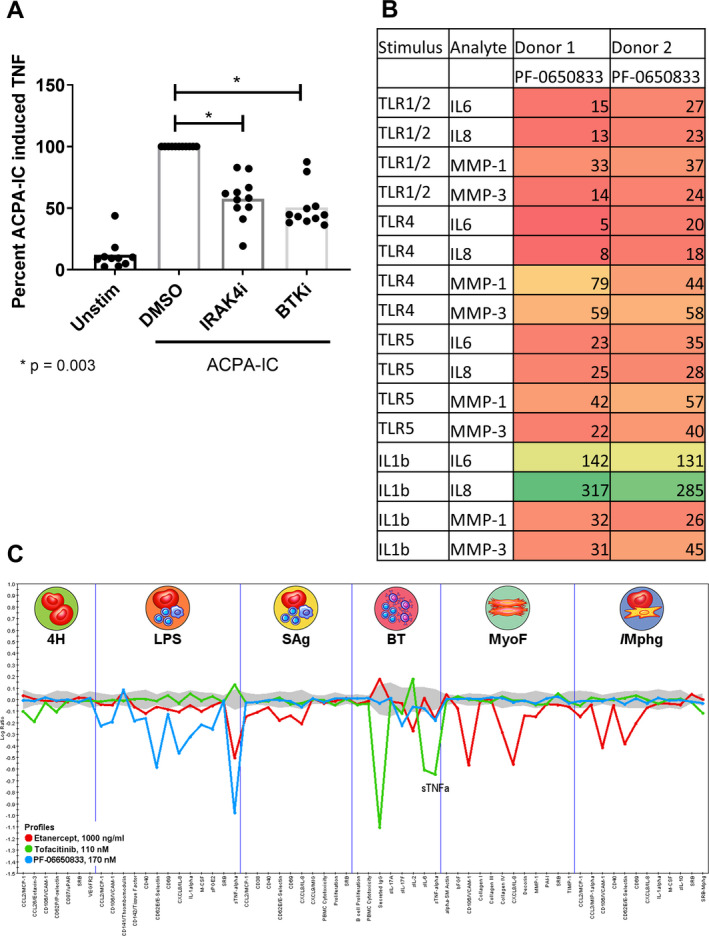
PF‐06650833 inhibits rheumatoid arthritis (RA) pathophysiology. A, Human macrophages were exposed to anti–citrullinated protein antibody (ACPA) immune complexes (ICs) formed with cyclic citrullinated peptide–positive RA sera in the presence or absence of 100 nM interleukin‐1–associated kinase 4 inhibitor (IRAK4i) (PF‐06650833) or 100 nM Bruton’s tyrosine kinase inhibitor (BTKi) (PF‐303). Supernatants were analyzed by enzyme‐linked immunosorbent assay for induction of tumor necrosis factor (TNF). Values are the percent in relation to DMSO treatment (set at 100). Symbols represent individual subjects (n = 11 per group); bars show the mean. B, Human RA fibroblast‐like synoviocytes were stimulated with 10 μg/ml Pam3Cys (Toll‐like receptor 1/2 [TLR‐1/2]), 10 ng/ml lipopolysaccharide (LPS) (TLR‐4), 100 ng/ml flagellin (TLR‐5), or 0.1 ng/ml interleukin‐1β (IL‐1β) in the presence or absence of 100 nM PF‐06650833. Cytokine and matrix metalloproteinase (MMP) content in supernatants was assessed by Meso Scale Discovery assay. Values are the percent of those observed with vehicle control. C, Compounds at the noted concentrations were profiled with DiscoverRx on the BioMAP platform. Data represent the log ratio of values in compound‐treated samples to controls, with negative values indicating inhibition and points outside of the gray shading demonstrating a statistically significant difference based on assay variability. Each point on the x‐axis represents the result of a different end point in each assay system: venular endothelial cells (4H), peripheral blood mononuclear cells (PBMCs) plus venular endothelial cells stimulated with LPS (LPS), PBMCs plus venular endothelial cells stimulated with superantigen (SAg), B cells plus PBMCs (BT), lung fibroblasts (MyoF), and macrophages plus venular endothelial cells (/Mphg). Unstim = unstimulated; MCP‐1 = monocyte chemotactic factor 1; VCAM‐1 = vascular cell adhesin molecule 1; uPAR = urokinase‐type plasminogen activator receptor; SRB = sulforhodamine B staining; VEGFR‐2 = vascular endothelial growth factor receptor 2; M‐CSF = macrophage colony‐stimulating factor; sPGE2 = soluble prostaglandin E2; MIG = monokine induced by interferon‐γ; bFGF = basic fibroblast growth factor; PAI‐1 = plasminogen activator inhibitor 1; TIMP‐1 = tissue inhibitor of metalloproteinases 1.
Synovial fibroblasts from RA patients display increased inflammatory responses to TLR ligands and IL‐1β (37, 38). We exposed RA FLS to inflammatory stimuli in the presence or absence of 100 nM PF‐06650833 and quantified their inflammatory response. As depicted in Figure 2B, PF‐06650833 substantially reduced cytokine and matrix metalloproteinase (MMP) release in response to all ligands profiled. However, it did not inhibit IL‐1–induced cytokines in RA FLS. This is similar to our previous findings in dermal fibroblasts stimulated with IL‐1β, in which cytokine release was only weakly affected by IRAK4 inhibition (8, 23). Similar results were also generated using FLS from non‐RA donors (n = 2), although the magnitude of inflammatory mediators released was lower (data not shown). Intriguingly, PF‐06650833 was able to block MMP induction by IL‐1β: the first response to IL‐1β in nonhematopoietic cells we have found to be IRAK4 kinase dependent.
We next tested the ability of PF‐06650833 to inhibit inflammatory processes in more physiologic tissue culture models involving multiple human primary cells, using the DiscoveRx BioMAP platform. The effect was compared to those of 2 approved RA therapies, the TNF inhibitor etanercept and the JAK inhibitor tofacitinib. As depicted in Figure 2C, PF‐06650833 showed the greatest inhibition of inflammatory readouts in assays of innate immunity, notably the lipopolysaccharide (LPS)–stimulated PBMC plus endothelium assay, with modest activity in a small number of end points in the B–T cell assay. In contrast and as expected, the JAK inhibitor had no impact on assays of innate immunity (LPS, lung myofibroblasts, macrophages plus venular endothelial cells), blocking only responses in the B–T cell assay. Etanercept exhibited yet a third pattern, with predominant activity in the myofibroblast‐ and macrophage‐containing assays, and a modest effect on some readouts in the LPS and bacterial superantigen–stimulated PBMC assays. These results indicate that an IRAK4 inhibitor would block multicellular inflammatory processes in RA in a pattern distinct from those of the approved RA treatments.
PF‐06650833–induced reduction of inflammation in the rat collagen‐induced arthritis (CIA) model
To profile PF‐06650833 in vivo, we used the rat CIA model. Starting on the first day of disease activity (day 11–14 after collagen boost), rats with CIA were administered vehicle, 10 mg/kg tofacitinib daily, or 3 mg/kg PF‐06650833 twice daily for 7 days. The kinetic disease activity data for the treatment days from a representative study (n = 10 animals per group) are presented in Figure 3, demonstrating that both PF‐06650833 and tofacitinib significantly inhibited paw volume compared to that observed in vehicle‐treated animals (mean ± SEM paw volume on day 7 0.786 ± 0.043 ml, 0.555 ± 0.034 ml, and 0.371 ± 0.044 ml with vehicle, PF‐06650833, and tofacitinib, respectively; P = 0.0005, PF‐06650833 versus vehicle and P < 0.0001, tofacitinib versus vehicle, by t‐test). These results indicate that PF‐06650833 is capable of inhibiting inflammation in vivo, even when administered weeks after the initial inflammatory insult.
Figure 3.
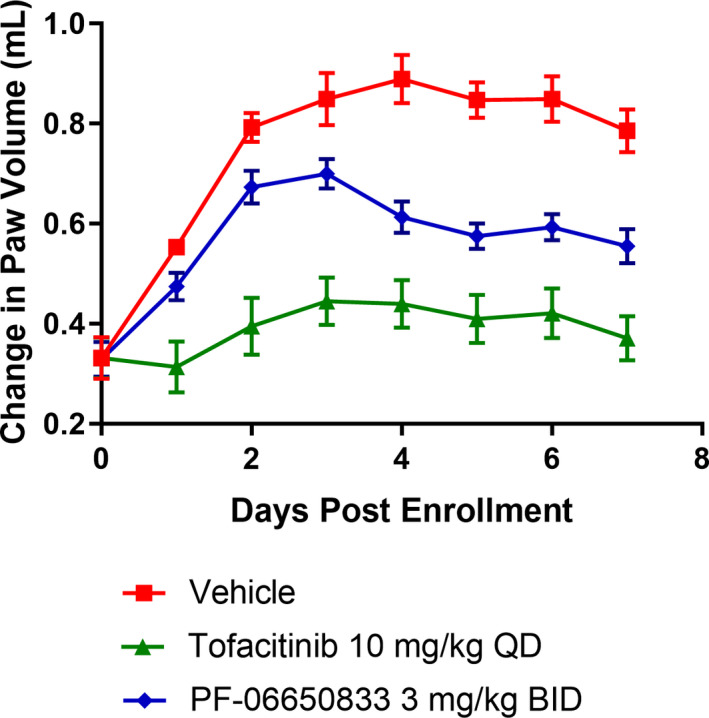
PF‐06650833 is efficacious in rat collagen‐induced arthritis (CIA). Rats with CIA were treated for 7 days with PF‐06650833 3 mg/kg twice daily (bid), tofacitinib 10 mg/kg daily (qd), or vehicle, and paw volume was measured daily. Data are from a representative experiment (of 3 experiments performed). Values are the mean ± SEM.
PF‐06650833–induced inhibition of pathophysiologic processes central to SLE
High titers of antinuclear antibodies are diagnostic for SLE, and formation of ICs contributes to the multiorgan inflammation characteristic of the disease (39). Exposure of the immune system to nuclear antigens results from impaired clearance of apoptotic cells and/or enhanced release from neutrophils via NETosis (40, 41, 42). Lupus ICs can then induce cytokine release from monocytes and T cell–independent B cell maturation and can activate pDCs to release type I IFNs, resulting in the characteristic IFN gene signature of SLE (43, 44). We stimulated DNA release from human neutrophils by application of the TLR‐7 agonist R837 for 15 hours. DNA content in the supernatants was significantly higher than that in neutrophils incubated in medium alone (mean ± SEM 113.5 ± 10.7 ng/ml versus 43.3 ± 9.6 ng/ml; P = 0.0001 by paired t‐test [n = 3 per group]), and was completely reversed by preincubation with PF‐06650833 (45.83 ± 9.7 ng/ml; P = 0.0017 versus R837 without PF‐06650833) (Figure 4A). Monocytes from SLE patients exhibit increased IRF‐5 protein expression and nuclear localization (45). After confirming the ability of PF‐06650833 to inhibit nuclear localization of IRF‐5 stimulated by R848 (23.0 ± 13.0% versus 6.8 ± 4.4%, respectively, without versus with PF‐06650833 exposure; P = 0.012 by two‐way analysis of variance [ANOVA] [n = 3]), we demonstrated its capacity to inhibit IRF‐5 nuclear localization in response to SLE patient sera (38.6 ± 18.4% versus 24.2 ± 13.0%, respectively; P < 0.0001 by two‐way ANOVA [n = 12]) (Figure 4B).
Figure 4.
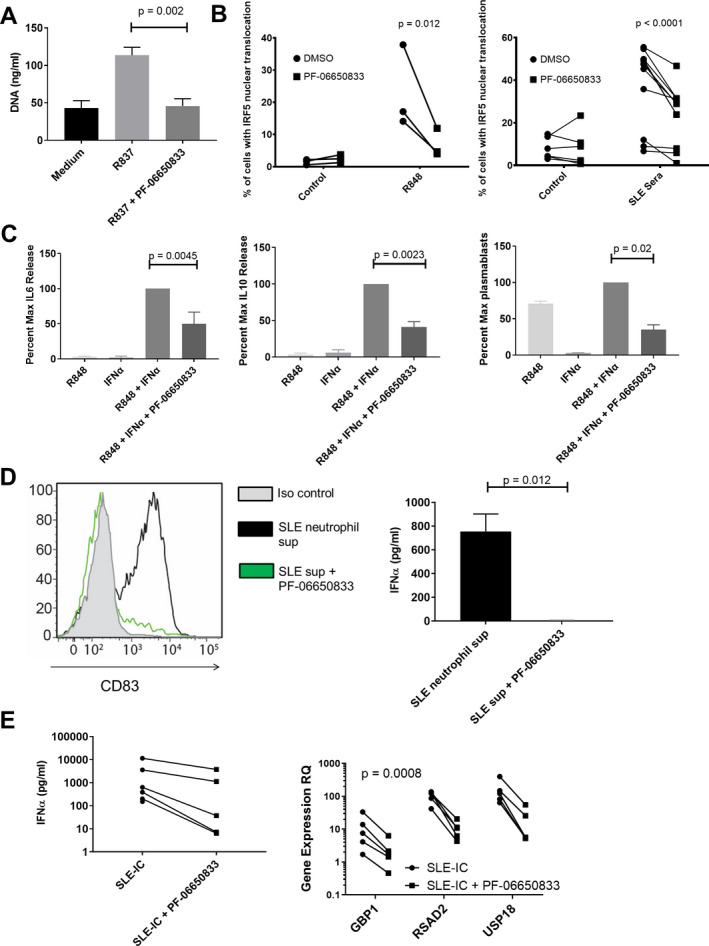
PF‐06650833 inhibits pathophysiology in human systemic lupus erythematosus (SLE). A, DNA release was measured in neutrophil supernatant (sup) following exposure to R837 in the presence or absence of 100 nM PF‐06650833. Values are the mean ± SEM (n = 3 donors). B, Peripheral blood mononuclear cells (PBMCs) were exposed to either R848 or SLE sera in the presence or absence of 100 nM PF‐06650833, incubated for 2 hours at 37°C, and then interferon regulatory factor 5 (IRF‐5) nuclear translocation was analyzed by Amnis imaging cytometry. C, B cells were exposed to interferon‐α (IFNα) plus R848 in the presence or absence of 100 nM PF‐06650833. Supernatants were harvested at 24 hours to assess interleukin‐6 (IL‐6) release and at 72 hours to assess IL‐10 release. Plasmablasts were quantified by flow cytometry after 6 days. D, Neutrophils were isolated from SLE patients and NETosis induced with R837. Supernatants were used to stimulate plasmacytoid dendritic cells for 24 hours in the presence or absence of 200 nM PF‐06650833 prior to quantification of CD83 induction by flow cytometry (n = 5) or IFNα release by enzyme‐linked immunosorbent assay (ELISA) (mean ± SEM; n = 3). Iso = isotype. E, PBMCs were exposed to SLE immune complexes (ICs) in the presence or absence of 100 nM PF‐06650833. After 24 hours, supernatant was analyzed by ELISA for IFNα, and IFN‐induced gene expression in cells was measured by quantitative reverse transcription–polymerase chain reaction. RQ = relative quantification.
Exposure to IFN and TLR‐7 ligands results in B cell cytokine release and T cell–independent B cell maturation into plasmablasts (46, 47). Incubation of human B cells with 100 nM PF‐06650833 prior to addition of IFNα and R848 inhibited B cell IL‐6 production at 24 hours to a mean ± SEM of 50.0 ± 16.7% of control (P = 0.0045 by paired t‐test [n = 6 donors]), IL‐10 production at 72 hours to 41.3 ± 7.1% of control (P = 0.0023 by paired t‐test [n = 6 donors]), and CD27+CD38+ plasmablast differentiation at 7 days to 35.1 ± 6.6 % of control (P = 0.02 by paired t‐test [n = 6 donors]) (Figure 4C). Although the potency of PF‐06650833 (IC50) for inhibiting these activities in primary human B cells (not shown) is comparable to that for inflammatory cytokine release by other cell types, inhibition is less complete than for inflammatory cytokine production by monocytes (48), suggesting IRAK4 kinase‐independent signaling.
Plasmacytoid DCs are a rare population in peripheral blood, but are responsible for the majority of IFNα release (44, 49). Neutrophil damage‐associated molecular patterns (DAMPs) induced pDCs to up‐regulate CD83, indicating maturation; this response was significantly inhibited by preincubation with 200 nM PF‐06650833, and IFNα release from pDCs was significantly inhibited by PF‐06650833 (mean ± SEM 5.7 ± 2.5 pg/ml versus 755 ± 147 pg/ml [n = 3]; P = 0.012 by paired t‐test) (Figure 4D). SLE ICs formed by purifying IgG from plasma from an anti‐RNP/anti‐dsDNA–positive SLE patient and mixing with apoptotic cells were used to stimulate human PBMCs. PF‐06650833 exposure inhibited IFNα release in all donors (n = 5), as well as the induction of 3 IFN‐responsive genes (RSAD2, USP18, and GBP1) (P = 0.0008 by 2‐way ANOVA) (Figure 4E).
Taken together, the above findings demonstrate that numerous SLE pathophysiologic processes, including neutrophil DAMP release, monocyte inflammatory cytokine response, B cell cytokine release and plasmablast differentiation, pDC maturation, and IFNα release as well as the resulting IFN gene signature, are dependent on IRAK4 kinase activity.
Therapeutic efficacy of PF‐06650833 in mouse models of SLE
The pristane‐induced model of SLE is dependent on TLR activation and results in an antinuclear antibody repertoire similar to that in human SLE (50). BALB/c mice were treated intraperitoneally with pristane to induce disease, and standard chow was replaced 8 weeks later with PF‐06650833–containing chow for the remaining 12 weeks of the study. Longitudinal quantitation of antibody levels is shown in Figure 5. Anti‐dsDNA titers were significantly increased in pristane‐treated animals versus controls beginning at 12 weeks (mean ± SEM 10,977 ± 1,503 units/ml versus 5,294 ± 678 units/ml [n = 10 animals per group]; P = 0.0023 by two‐way ANOVA), which persisted at week 20 (17,645 ± 3527 units/ml versus 5,103 ± 816 units/ml; P < 0.0001 by two‐way ANOVA). At week 20, therapeutic dosing with PF‐06650833 had significantly reduced anti‐dsDNA titers compared to control (8,159 ± 1282 units/ml versus 17,645 ± 3,527 units/ml; P < 0.0001 by Mann‐Whitney test), as had prednisone treatment (8,608 ± 1,637 units/ml; P < 0.0001). With PF‐06650833 treatment, there also were significant reductions at week 20 in anti‐SSA IgG (2,726 ± 494 units/ml versus 7,693 ± 1,241 units/ml; P < 0.004 by Mann‐Whitney test) and anti‐RNP IgG (3,730 ± 2,861 units/ml versus 18,127 ± 7,902 units/ml; P = 0.011 by Mann‐Whitney test).
Figure 5.
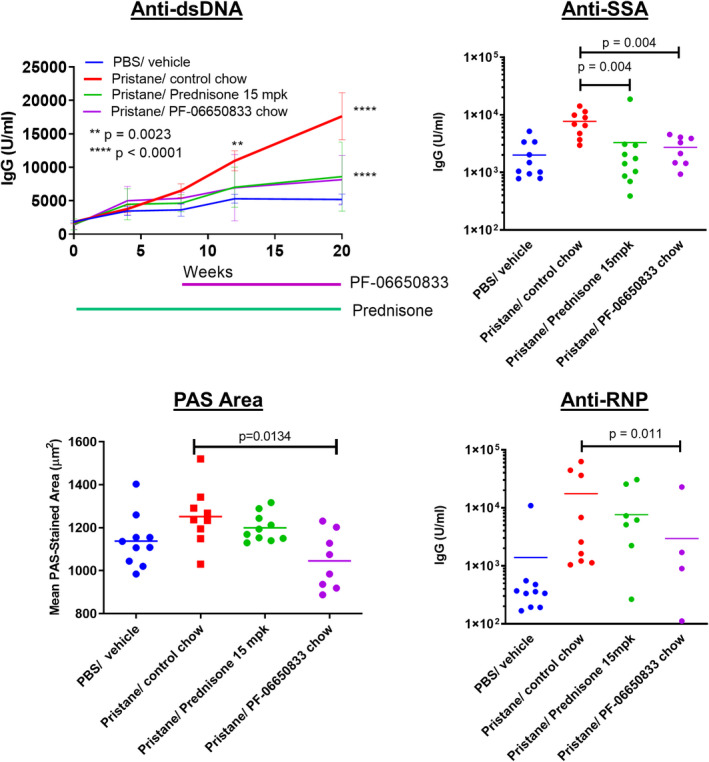
Effect of PF‐06650833 on pristane‐induced systemic lupus erythematosus. PF‐06650833 was administered to BALB/c mice by chow dosing during weeks 8–20 following pristane administration. Anti–double‐stranded DNA (anti‐dsDNA) was quantified by enzyme‐linked immunosorbent assay at weeks 4, 8, 12, and 20, and the mean ± SEM value at each of these time points is shown. Additional autoantibody titers (anti‐SSA, anti‐RNP) were determined, and immunohistochemical assessments (periodic acid–Schiff [PAS] staining) were performed, at week 20. Data are from a representative experiment (of 2 experiments performed). Symbols represent individual subjects; bars show the mean. PBS = phosphate buffered saline; mpk = mg/kg.
Kidney inflammation in the pristane‐induced lupus model was mild and did not result in proteinuria. As there was no increase in renal inflammation, tubular injury, or proteinuria/cast scores in response to pristane exposure, the effects of the IRAK4 inhibitor on these end points were not evaluated. Kidneys in all groups were also evaluated by quantitative image analysis, as described previously (51), for glomerular tuft area, periodic acid–Schiff (PAS) staining area per tuft, glomerular nuclear area, IgG immunohistochemistry scores, and C3 immunohistochemistry scores. Of these, only mean glomerular tuft area was significantly increased by pristane, and only mean PAS area per tuft was significantly reduced by PF‐06650833 (mean ± SEM 1,045 ± 47 μm2, versus 1,252 ± 45 μm2 with pristane treatment alone [n = 8 animals per group]; P = 0.013 by Kruskal‐Wallis test) (Figure 5).
Treatment with PF‐06650833 was also explored in the MRL/lpr mouse model of SLE. In 2 studies, PF‐06650833 significantly reduced lymphadenopathy, anti–histone IgG, histologic kidney inflammation, glomerular nephropathy, and C3 and IgG deposition by immunohistochemistry, and glomerular tuft area and glomerular nuclear area by image analysis (Supplementary Figures 2 and 3, on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41953/abstract), while showing trends toward reductions in splenomegaly, proteinuria, anti‐dsDNA, anti‐SSA, and tubular injury. As BTK inhibitors have previously shown efficacy in this model (52, 53, 54) and we previously showed in vivo efficacy of the BTK inhibitor PF‐06250112 in the NZB/NZW mouse model of lupus (29), it was included as a positive control. PF‐06250112 significantly reduced proteinuria, autoantibody titers, and all renal histologic pathology end points other than PAS area per tuft (Supplementary Figures 2 and 3). It may be that in the pristane‐induced SLE model, in which autoantibodies are induced via TLR activation, IRAK4 kinase activity is essential, while in the MRL/lpr model, in which autoantibody production is initiated by faulty B cell apoptosis, IRAK4 kinase activity is more important for the resulting inflammation than for the production of autoantibodies themselves.
Reduction of IFN signature genes in healthy volunteers by selective IRAK4 inhibition
We have completed 2 randomized, double‐blind, sponsor‐open phase I studies of the safety, pharmacokinetics, and pharmacodynamics of single‐ and multiple‐ascending doses of PF‐06650833 (26). Since the 300‐mg dose of a modified‐release formulation showed maximum pharmacologic effect in reducing C‐reactive protein (CRP) levels on day 14 of dosing, we evaluated the effect of PF‐06650833 on an IFN signature (comprising the normalized expression of 21 genes [30]) as a biomarker of systemic inflammation relevant to SLE pathogenesis at this time point, in comparison to the respective gene signature for each volunteer on day 0, prior to dosing. As shown in Figure 6, the IFN gene signature in the placebo group had changed positively by a median of 9.1%, while that in the PF‐06650833–treated trial participants was reduced by a median of 28.8% (difference −37.9%; P = 0.015 by Wilcoxon test). The paired gene score values for each volunteer on day 0 and day 14 are also plotted in Supplementary Figure 4 (http://onlinelibrary.wiley.com/doi/10.1002/art.41953/abstract), showing minimal regulation of gene scores in placebo‐treated subjects (positive change in 6 of 11 and negative change in 5 of 11), whereas the magnitude of change between day 0 and day 14 was consistently higher, and the change was negative in 6 of 7 cases, among PF‐06650833–treated subjects.
Figure 6.
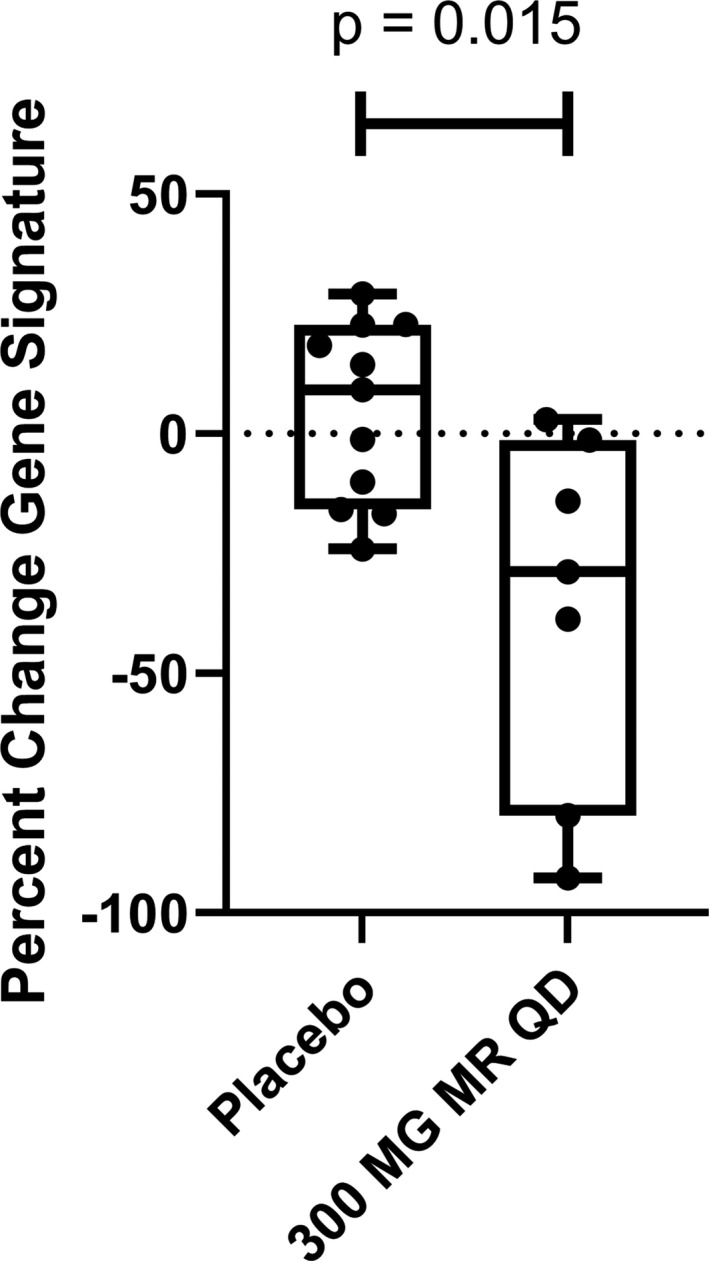
PF‐06650833 inhibits type I interferon signature in vivo in humans. Moderate‐release (MR) PF‐06650833 (300 mg/day [qd]) (n = 7) or placebo (n = 11) was administered for 14 days in a phase I multiple‐ascending‐dose trial in healthy human volunteer subjects. Whole blood was collected in a PAXgene tube on day 0 prior to administration of the first dose and on day 14 prior to administration of the last dose, RNA extracted, and a composite gene signature calculated. The percent change in the composite gene signature for each individual participant between the 2 time points is shown. Each box represents the 25th to 75th percentiles. Lines inside the boxes represent the median. Lines outside the boxes represent the full range of values. Symbols represent individual patients.
Thus, we have demonstrated that in humans, an IRAK4 inhibitor reduces biomarkers of inflammation that are relevant to RA and SLE pathophysiology.
DISCUSSION
Inhibitors of innate immune signaling pathways are potential targets for treatment of autoimmune diseases (55, 56). In this study, we demonstrated that PF‐06650833 effectively inhibits cytokines produced by macrophages activated by ACPA ICs from RA patients as well as pDCs activated by SLE ICs. We also showed that PF‐06650833 effectively inhibits cytokines induced by TLR ligands on RA FLS but is not effective against IL‐1β–induced cytokines. However, PF‐06650833 is effective at blocking MMPs induced by IL‐1β on these cells. The reason for this difference is not known but may result from the differential effects of IRAK4 inhibition on early versus late myddosome formation and/or differential effects on transcription factor activation. Previously, investigators in our group have shown that inhibition of IRAK4 kinase activity affects IL‐1β signaling by stabilizing the early form of the myddosome that signals primarily through IRAK1 and NF‐κB (8, 57). It is possible that IL‐6/8 secretion is induced primarily by signaling via the early myddosome, whereas activation of MMPs might be mediated by the late myddosome, which signals through IRAK2 (58). Regarding transcription factors, inhibition of IRAK4 kinase activity is known to block activation of IRF‐5, but not NF‐κB or MAPK (7), and these effects are cell type specific (38). It is possible that in RA FLS, chromatin remodeling makes the MMP promoters more dependent on IRF‐5 or other IRAK4 kinase‐dependent transcription factors.
We also demonstrated that PF‐06650833 is efficacious in a rat model of arthritis that did not include use of Freund’s complete adjuvant (to ensure it was not driven by TLR). While it is encouraging that PF‐06650833 significantly reduces severity in the rat CIA model, it is notable that the degree of inhibition was not as great as that observed with tofacitinib. It is possible that the rapid clearance of PF‐06650833 from rodent species (rat T½ = 0.6 hours [18]) resulted in transient loss of IRAK4 target inhibition in rat CIA. It has been shown previously that the efficacy of tofacitinib is driven by average target occupancy (59), while the evidence thus far suggests that minimum target occupancy is more relevant for IRAK4‐mediated efficacy. Thus, the respective efficacies of PF‐06650833 and tofacitinib in rat CIA may be related to target occupancy, mechanism of inhibition, or the respective contribution of each kinase to disease pathophysiology. Of greater relevance to the treatment of human RA, our group recently identified significant effects on disease activity in a phase II study of the efficacy and safety of PF‐06650833 in patients with active RA and inadequate response to methotrexate (60). The rate of clinical response at 12 weeks in that study was consistent with those reported for tofacitinib, and the study itself included a cohort treated with tofacitinib. Thus, the respective efficacy of the 2 molecules will be revealed by the publication of data from this clinical trial.
We have shown here that PF‐06650833 can block type I IFN induced by SLE serum and by neutrophil DAMPs in primary human pDCs. We also demonstrated that it can block plasmablast differentiation and B cell activation induced by TLR ligands and activation of the transcription factor IRF‐5 by SLE serum in monocytes (61, 62). Therapeutic dosing of PF‐06650833 was efficacious in reducing the induction of all antinuclear antibodies tested in the pristane model of SLE, while significantly reducing only 1 of 3 reactivities assayed in the MRL/lpr model. Interestingly, PF‐06650833 did demonstrate robust inhibition of kidney inflammatory histology in the MRL/lpr mouse model. It is unknown whether this difference is due to the different mechanisms of disease initiation in the 2 models or differences in ability to cover the target, as PF‐06650833 is rapidly cleared from mouse circulation.
Despite lower potency in rodent cells and rapid clearance from rodent circulation, the in vitro and in vivo results generated with PF‐06650833 presented here provide important insights with regard to some of the controversies concerning the relative importance of IRAK1 and IRAK4 kinase activity in various cell types in mice and humans (16, 19, 21, 22, 24, 25). IRAK4 kinase activity is necessary for DAMP‐induced inflammatory signaling in rodent and human leukocytes, and IRAK1 kinase activity does not provide a sufficient substitute. Findings of a recent study (20) are consistent with our data, confirming the importance of IRAK4 activity to lupus pathophysiologic signaling in DCs and B cells, and in in vivo models using an unrelated small molecule inhibitor of IRAK4. Further work is needed in the effort to understand the role of IRAK4 kinase activity in stromal cells, as well as in IL‐1β–induced inflammation. We have shown that IL‐1β–induced cytokines from human fibroblasts treated with an IRAK4 inhibitor were only weakly inhibited even though IRAK4 autophosphorylation was completely inhibited (8, 63), while inhibition of TLR‐7/8 (R848)–induced cytokine production by IRAK4 inhibition coincided with the inhibition of IRAK4 autophosphorylation in primary human monocytes (7, 63). Likewise, in the results presented here, IL‐1β–induced cytokines in RA FLS were not inhibited by PF‐06650833, in contrast to MMPs, which were inhibited.
To demonstrate proof of pharmacologic effect in humans, we assessed markers of inflammation in a phase I clinical trial in healthy volunteers. These markers were IFN signature (increased in SLE) (44) and CRP levels (increased in RA) (64, 65). Basal levels of these markers were present in healthy volunteers, and notably, with modified‐release PF‐06650833 at a daily dosage of 300 mg, we observed statistically significant reductions in both end points, demonstrating that an IRAK4 inhibitor reduces markers of inflammation in humans. To our knowledge, this is the first demonstration of modulation of IFN‐regulated genes in healthy volunteers. Together with the CRP data (26), the present results represent the first proof of pharmacologic effect of a selective IRAK4 inhibitor on inflammatory signaling pathways in humans. Taken together, these data strongly support the clinical utility of IRAK4 inhibitors for the treatment of multiple human inflammatory autoimmune diseases.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Winkler had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Winkler, Sun, De, Sharif, Athale, Jacobson, Ramsey, Dower, Hegen, Homer, Brodfuehrer, Tilley, Danto, Beebe, Barnes, Pascual, Lin, Kilty, Fleming, Rao.
Acquisition of data
Winkler, Sun, De, Jiao, Sharif, Symanowicz, Athale, Shin, Wang, Jacobson, Ramsey, Dower, Andreyeva, Liu, Homer, Danto, Beebe, Rao.
Analysis and interpretation of data
Winkler, Sun, De, Jiao, Sharif, Symanowicz, Athale, Shin, Wang, Jacobson, Ramsey, Dower, Andreyeva, Liu, Hegen, Homer, Brodfuehrer, Tilley, Gilbert, Danto, Beebe, Barnes, Pascual, Lin, Kilty, Fleming, Rao.
ROLE OF THE STUDY SPONSOR
Pfizer facilitated the study design, provided writing assistance for the manuscript, and reviewed and approved the manuscript prior to submission. The authors independently collected the data, interpreted the results, and had the final decision to submit the manuscript for publication. Publication of this article was contingent upon approval by Pfizer.
Supporting information
Supplementary Material
Fig S1
Fig S2
Fig S3
Fig S4
Table S1
Supplementary Material
Supplementary Material
ACKNOWLEDGMENTS
The authors wish to thank Tatyana Souza and Varenka Rodriguez for optimizing the plasmablast differentiation assay, Jill Wright and Yanyu Zhang for human IgG purification from plasma, Alison O’Mahoney (Eurofins) for providing the DiscoverRx figure, the scientists at ActivX for performing kinase selectivity assays and at Washington Biotechnology for performing the pristane SLE model, our clinical collaborators in the phase I multiple‐ascending‐dose study, and especially all of the human volunteers who contributed to every phase of this work.
ClinicalTrials.gov identifier: NCT02485769.
Supported by Pfizer. Dr. Barnes’ work was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases, NIH (grant 1R21‐AR‐065959‐01).
Author disclosures are available at https://onlinelibrary.wiley.com/action/downloadSupplement?doi=10.1002%2Fart.41953&file=art41953-sup-0001-Disclosure form.pdf
REFERENCES
- 1. Malmström V, Catrina AI, Klareskog L. The immunopathogenesis of seropositive rheumatoid arthritis: from triggering to targeting [review]. Nat Rev Immunol 2016;17:60–75. [DOI] [PubMed] [Google Scholar]
- 2. Smolen JS, Aletaha D, Barton A, Burmester GR, Emery P, Firestein GS, et al. Rheumatoid arthritis [review]. Nat Rev Dis Primers 2018;4:18001. [DOI] [PubMed] [Google Scholar]
- 3. Aletaha D, Smolen JS. Diagnosis and management of rheumatoid arthritis: a review. JAMA 2018;320:1360–72. [DOI] [PubMed] [Google Scholar]
- 4. Smolen JS, Aletaha D. Rheumatoid arthritis therapy reappraisal: strategies, opportunities and challenges [review]. Nat Rev Rheumatol 2015;11:276–89. [DOI] [PubMed] [Google Scholar]
- 5. Dörner T, Furie R. Novel paradigms in systemic lupus erythematosus [review]. Lancet 2019;393:2344–58. [DOI] [PubMed] [Google Scholar]
- 6. Tsokos GC, Lo MS, Reis PC, Sullivan KE. New insights into the immunopathogenesis of systemic lupus erythematosus [review]. Nat Rev Rheumatol 2016;12:716–30. [DOI] [PubMed] [Google Scholar]
- 7. Cushing L, Winkler A, Jelinsky SA, Lee K, Korver W, Hawtin R, et al. IRAK4 kinase activity controls Toll‐like receptor–induced inflammation through the transcription factor IRF5 in primary human monocytes. J Biol Chem 2017;292:18689–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. De S, Karim F, Kiessu E, Cushing L, Lin LL, Ghandil P, et al. Mechanism of dysfunction of human variants of the IRAK4 kinase and a role for its kinase activity in interleukin‐1 receptor signaling. J Biol Chem 2018;293:15208–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Balka KR, De Nardo D. Understanding early TLR signaling through the Myddosome [review]. J Leukoc Biol 2019;105:339–51. [DOI] [PubMed] [Google Scholar]
- 10. Suzuki N, Suzuki S, Duncan GS, Millar DG, Wada T, Mirtsos C, et al. Severe impairment of interleukin‐1 and Toll‐like receptor signalling in mice lacking IRAK‐4. Nature 2002;416:750–6. [DOI] [PubMed] [Google Scholar]
- 11. Kim TW, Staschke K, Bulek K, Yao J, Peters K, Oh KH, et al. A critical role for IRAK4 kinase activity in Toll‐like receptor‐mediated innate immunity. J Exp Med 2007;204:1025–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Koziczak‐Holbro M, Littlewood‐Evans A, Pöllinger B, Kovarik J, Dawson J, Zenke G, et al. The critical role of kinase activity of interleukin‐1 receptor–associated kinase 4 in animal models of joint inflammation. Arthritis Rheum 2009;60:1661–71. [DOI] [PubMed] [Google Scholar]
- 13. Nanda SK, Lopez‐Pelaez M, Arthur JS, Marchesi F, Cohen P. Suppression of IRAK1 or IRAK4 catalytic activity, but not type 1 IFN signaling, prevents lupus nephritis in mice expressing a ubiquitin binding‐defective mutant of ABIN1. J Immunol 2016;197:4266–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kawagoe T, Sato S, Jung A, Yamamoto M, Matsui K, Kato H, et al. Essential role of IRAK‐4 protein and its kinase activity in Toll‐like receptor‐mediated immune responses but not in TCR signaling. J Exp Medicine 2007;204:1013–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Picard C, Puel A, Bonnet M, Ku CL, Bustamante J, Yang K, et al. Pyogenic bacterial infections in humans with IRAK‐4 deficiency. Science 2003;299:2076–9. [DOI] [PubMed] [Google Scholar]
- 16. Kelly PN, Romero DL, Yang Y, Shaffer AL III, Chaudhary D, Robinson S, et al. Selective interleukin‐1 receptor‐associated kinase 4 inhibitors for the treatment of autoimmune disorders and lymphoid malignancy. J Exp Med 2015;212:2189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chaudhary D, Robinson S, Romero DL. Recent advances in the discovery of small molecule inhibitors of interleukin‐1 receptor‐associated kinase 4 (IRAK4) as a therapeutic target for inflammation and oncology disorders [review]. J Med Chem 2015;58:96–110. [DOI] [PubMed] [Google Scholar]
- 18. Lee KL, Ambler CM, Anderson DR, Boscoe BP, Bree AG, Brodfuehrer JI, et al. Discovery of clinical candidate 1‐{[(2S,3S,4S)‐3‐Ethyl‐4‐fluoro‐5‐oxopyrrolidin‐2‐yl]methoxy}‐7‐methoxyisoquinoline‐6‐carboxamide (PF‐06650833), a potent, selective inhibitor of interleukin‐1 receptor associated kinase 4 (IRAK4), by fragment‐based drug design. J Med Chem 2017;60:5521–42. [DOI] [PubMed] [Google Scholar]
- 19. Dudhgaonkar S, Ranade S, Nagar J, Subramani S, Prasad DS, Karunanithi P, et al. Selective IRAK4 inhibition attenuates disease in murine lupus models and demonstrates steroid sparing activity. J Immunol 2017;198:1308–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Corzo CA, Varfolomeev E, Setiadi AF, Francis R, Klabunde S, Senger K, et al. The kinase IRAK4 promotes endosomal TLR and immune complex signaling in B cells and plasmacytoid dendritic cells. Sci Signal 2020;13:eeaz1053. [DOI] [PubMed] [Google Scholar]
- 21. Qin J, Jiang Z, Qian Y, Casanova JL, Li X. IRAK4 kinase activity is redundant for interleukin‐1 (IL‐1) receptor‐associated kinase phosphorylation and IL‐1 responsiveness. J Biol Chem 2004;279:26748–53. [DOI] [PubMed] [Google Scholar]
- 22. Song KW, Talamas FX, Suttmann RT, Olson PS, Barnett JW, Lee SW, et al. The kinase activities of interleukin‐1 receptor associated kinase (IRAK)‐1 and 4 are redundant in the control of inflammatory cytokine expression in human cells. Mol Immunol 2009;46:1458–66. [DOI] [PubMed] [Google Scholar]
- 23. Cushing L, Stochaj W, Siegel M, Czerwinski R, Dower K, Wright Q, et al. Interleukin 1/Toll‐like receptor‐induced autophosphorylation activates interleukin 1 receptor‐associated kinase 4 and controls cytokine induction in a cell type‐specific manner. J Biol Chem 2014;289:10865–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chiang EY, Yu X, Grogan JL. Immune complex‐mediated cell activation from systemic lupus erythematosus and rheumatoid arthritis patients elaborate different requirements for IRAK1/4 kinase activity across human cell types. J Immunol 2011;186:1279–88. [DOI] [PubMed] [Google Scholar]
- 25. Sun J, Li N, Oh KS, Dutta B, Vayttaden SJ, Lin B, et al. Comprehensive RNAi‐based screening of human and mouse TLR pathways identifies species‐specific preferences in signaling protein use. Sci Signal 2016;9:ra3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Danto SI, Shojaee N, Singh RS, Li C, Gilbert SA, Manukyan Z, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of PF‐06650833, a selective interleukin‐1 receptor‐associated kinase 4 (IRAK4) inhibitor, in single and multiple ascending dose randomized phase 1 studies in healthy subjects. Arthritis Res Ther 2019;21:269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. O'Mahony A, John MR, Cho H, Hashizume M, Choy EH. Discriminating phenotypic signatures identified for tocilizumab, adalimumab, and tofacitinib monotherapy and their combinations with methotrexate. J Transl Med 2018;16:156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shah F, Stepan AF, O'Mahony A, Velichko S, Folias AE, Houle C, et al. Mechanisms of skin toxicity associated with metabotropic glutamate receptor 5 negative allosteric modulators. Cell Chem Biol 2017;24:858–69. [DOI] [PubMed] [Google Scholar]
- 29. Rankin AL, Seth N, Keegan S, Andreyeva T, Cook TA, Edmonds J, et al. Selective inhibition of BTK prevents murine lupus and antibody‐mediated glomerulonephritis. J Immunol 2013;191:4540–50. [DOI] [PubMed] [Google Scholar]
- 30. Yao Y, Higgs BW, Richman L, White B, Jallal B. Use of type I interferon‐inducible mRNAs as pharmacodynamic markers and potential diagnostic markers in trials with sifalimumab, an anti‐IFNα antibody, in systemic lupus erythematosus [review]. Arthritis Res Ther 2010;12 Suppl 1:S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Patricelli MP, Nomanbhoy TK, Wu J, Brown H, Zhou D, Zhang J, et al. In situ kinase profiling reveals functionally relevant properties of native kinases. Chem Biol 2011;18:699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Morgan P, Van Der Graaf PH, Arrowsmith J, Feltner DE, Drummond KS, Wegner CD, et al. Can the flow of medicines be improved? Fundamental pharmacokinetic and pharmacological principles toward improving Phase II survival. Drug Discov Today 2012;17:419–24. [DOI] [PubMed] [Google Scholar]
- 33. Castelar‐Pinheiro GR, Xavier RM. The spectrum and clinical significance of autoantibodies in rheumatoid arthritis. Front Immunol 2015;6:320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Catrina AI, Svensson CI, Malmström V, Schett G, Klareskog L. Mechanisms leading from systemic autoimmunity to joint‐specific disease in rheumatoid arthritis [review]. Nat Rev Rheumatol 2016;13:79–86. [DOI] [PubMed] [Google Scholar]
- 35. Sokolove J, Zhao X, Chandra PE, Robinson WH. Immune complexes containing citrullinated fibrinogen costimulate macrophages via Toll‐like receptor 4 and Fcγ receptor. Arthritis Rheum 2011;63:53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Horwood NJ, Page TH, McDaid JP, Palmer CD, Campbell J, Mahon T, et al. Bruton's tyrosine kinase is required for TLR2 and TLR4‐induced TNF, but not IL‐6, production. J Immunol 2006;176:3635–41. [DOI] [PubMed] [Google Scholar]
- 37. Falconer J, Murphy AN, Young SP, Clark AR, Tiziani S, Guma M, et al. Synovial cell metabolism and chronic inflammation in rheumatoid arthritis [review]. Arthritis Rheumatol 2018;70:984–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Doody KM, Bottini N, Firestein GS. Epigenetic alterations in rheumatoid arthritis fibroblast‐like synoviocytes [review]. Epigenomics 2017;9:479–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pisetsky DS. Anti‐DNA antibodies: quintessential biomarkers of SLE [review]. Nat Rev Rheumatol 2015;12:102–10. [DOI] [PubMed] [Google Scholar]
- 40. Lee KH, Kronbichler A, Park DD, Park Y, Moon H, Kim H, et al. Neutrophil extracellular traps (NETs) in autoimmune diseases: a comprehensive review. Autoimmun Rev 2017;16:1160–73. [DOI] [PubMed] [Google Scholar]
- 41. Bouts YM, Wolthuis DF, Dirkx MF, Pieterse E, Simons EM, van Boekel AM, et al. Apoptosis and NET formation in the pathogenesis of SLE [review]. Autoimmunity 2012;45:597–601. [DOI] [PubMed] [Google Scholar]
- 42. Mahajan A, Herrmann M, Muñoz LE. Clearance deficiency and cell death pathways: a model for the pathogenesis of SLE [review]. Front Immunol 2016;7:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Muñoz LE, Janko C, Schulze C, Schorn C, Sarter K, Schett G, et al. Autoimmunity and chronic inflammation: two clearance‐related steps in the etiopathogenesis of SLE. Autoimmun Rev 2010;10:38–42. [DOI] [PubMed] [Google Scholar]
- 44. Crow MK. Type I interferon in the pathogenesis of lupus [review]. J Immunol 2014;192:5459–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Stone RC, Feng D, Deng J, Singh S, Yang L, Fitzgerald‐Bocarsly P, et al. Interferon regulatory factor 5 activation in monocytes of systemic lupus erythematosus patients is triggered by circulating autoantigens independent of type I interferons. Arthritis Rheum 2012;64:788–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kiefer K, Oropallo MA, Cancro MP, Marshak‐Rothstein A. Role of type I interferons in the activation of autoreactive B cells [review]. Immunol Cell Biol 2012;90:498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Douagi I, Gujer C, Sundling C, Adams WC, Smed‐Sörensen A, Seder RA, et al. Human B cell responses to TLR ligands are differentially modulated by myeloid and plasmacytoid dendritic cells. J Immunol 2009;182:1991–2001. [DOI] [PubMed] [Google Scholar]
- 48. Cushing L, Winkler A, Jelinsky SA, Lee K, Korver W, Hawtin R, et al. IRAK4 kinase activity controls Toll‐like receptor‐induced inflammation through the transcription factor IRF5 in primary human monocytes. J Biol Chem 2017;292:18689–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Colonna M, Trinchieri G, Liu YJ. Plasmacytoid dendritic cells in immunity [review]. Nat Immunol 2004;5:1219–26. [DOI] [PubMed] [Google Scholar]
- 50. Zhuang H, Szeto C, Han S, Yang L, Reeves WH. Animal models of interferon signature positive lupus [review]. Front Immunol 2015;6:291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Homer BL, Dower K. 41‐week study of progressive diabetic nephropathy in the ZSF1 fa/faCP rat model. Toxicol Pathol 2018;46:976–7. [DOI] [PubMed] [Google Scholar]
- 52. Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, et al. The Bruton tyrosine kinase inhibitor PCI‐32765 blocks B‐cell activation and is efficacious in models of autoimmune disease and B‐cell malignancy. Proc Nat Acad Sci U S A 2010;107:13075–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kim YY, Park KT, Jang SY, Lee KH, Byun JY, Suh KH, et al. HM71224, a selective Bruton's tyrosine kinase inhibitor, attenuates the development of murine lupus. Arthritis Res Ther 2017;19:211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chalmers SA, Wen J, Doerner J, Stock A, Cuda CM, Makinde HM, et al. Highly selective inhibition of Bruton's tyrosine kinase attenuates skin and brain disease in murine lupus. Arthritis Res Ther 2018;20:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shi FD, Ljunggren HG, Sarvetnick N. Innate immunity and autoimmunity: from self‐protection to self‐destruction [review]. Trends Immunol 2001;22:97–101. [DOI] [PubMed] [Google Scholar]
- 56. Waldner H. The role of innate immune responses in autoimmune disease development [review]. Autoimmun Rev 2009;8:400–4. [DOI] [PubMed] [Google Scholar]
- 57. De Nardo D, Balka KR, Gloria YC, Rao VR, Latz E, Masters SL. Interleukin‐1 receptor–associated kinase 4 (IRAK4) plays a dual role in myddosome formation and Toll‐like receptor signaling. J Biol Chem 2018;293:15195–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pauls E, Nanda SK, Smith H, Toth R, Arthur JS, Cohen P. Two phases of inflammatory mediator production defined by the study of IRAK2 and IRAK1 knock‐in mice. J Immunol 2013;191:2717–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lamba M, Hutmacher MM, Furst DE, Dikranian A, Dowty ME, Conrado D, et al. Model‐informed development and registration of a once‐daily regimen of extended‐release tofacitinib. Clin Pharmacol Ther 2017;101:745–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Danto SI, Shojaee N, Singh RS, Manukyan Z, Mancuso J, Peeva E, et al. Efficacy and safety of the selective interleukin‐1 receptor associated kinase 4 inhibitor, PF‐06650833, in patients with active rheumatoid arthritis and inadequate response to methotrexate [abstract]. Arthritis Rheumatol 2019;71 Suppl 10. URL: https://acrabstracts.org/abstract/efficacy‐and‐safety‐of‐the‐selective‐interleukin‐1‐receptor‐associated‐kinase‐4‐inhibitor‐pf‐06650833‐in‐patients‐with‐active‐rheumatoid‐arthritis‐and‐inadequate‐response‐to‐methotrexate/. [Google Scholar]
- 61. Lazzari E, Jefferies CA. IRF5‐mediated signaling and implications for SLE [review]. Clin Immunol 2014;153:343–52. [DOI] [PubMed] [Google Scholar]
- 62. Ban T, Sato GR, Tamura T. Regulation and role of the transcription factor IRF5 in innate immune responses and systemic lupus erythematosus [review]. Int Immunol 2018;30:529–36. [DOI] [PubMed] [Google Scholar]
- 63. Cushing L, Stochaj W, Siegel M, Czerwinski R, Dower K, Wright Q, et al. Interleukin 1/Toll‐like receptor‐induced autophosphorylation activates interleukin 1 receptor‐associated kinase 4 and controls cytokine induction in a cell type‐specific manner. J Biol Chem 2014;289:10865–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Otterness IG. The value of C‐reactive protein measurement in rheumatoid arthritis [review]. Semin Arthritis Rheum 1994;24:91–104. [DOI] [PubMed] [Google Scholar]
- 65. Tishler M, Caspi D, Yaron M. C‐reactive protein levels in patients with rheumatoid arthritis: the impact of therapy. Clin Rheumatol 1985;4:321–4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Fig S1
Fig S2
Fig S3
Fig S4
Table S1
Supplementary Material
Supplementary Material