

Abstract
Background:
The most prominent risk factor for atrial fibrillation (AF) is chronological age, however underlying mechanisms are unexplained. Algorithms using epigenetic modifications to the human genome effectively predict chronological age. Chronological and epigenetic predicted ages may diverge, a phenomenon termed epigenetic age acceleration (EAA), which may reflect accelerated biological aging. We sought to evaluate for associations between epigenetic age measures and incident AF.
Methods:
Measures for 4 epigenetic clocks (Horvath, Hannum, DNAm PhenoAge, and DNAm GrimAge) and an epigenetic predictor of PAI-1 levels (DNAm PAI-1) were determined for study participants from 3 population-based cohort studies. Cox models evaluated for associations with incident AF and results were combined via random-effects meta-analysis. Two-sample summary-level Mendelian randomization analyses evaluated for associations between genetic instruments of the EAA measures and AF.
Results:
Among 5,600 individuals (mean age: 65.5 years; 60.1% female; 50.7% black), there were 905 incident AF cases during a mean follow-up of 12.9 years. Unadjusted analyses revealed all 4 epigenetic clocks and the DNAm PAI-1 predictor were associated with statistically significant higher hazards of incident AF, though the magnitudes of their point estimates were smaller relative to the associations observed for chronological age. The pooled EAA estimates for each epigenetic measure, with the exception of Horvath EAA, were associated with incident AF in models adjusted for chronological age, race, sex, and smoking variables. Following multivariable adjustment for additional known AF risk factors that could also potentially function as mediators, pooled EAA measures for 2 clocks remained statistically significant. Five year increases in EAA measures for DNAm GrimAge and DNAm PhenoAge were associated with 19% (adjusted hazard ratio [HR]: 1.19; 95% confidence intervals [CI]: 1.09–1.31; p<0.01) and 15% (adjusted HR: 1.15; 95% CI: 1.05–1.25; p<0.01) higher hazards of incident AF, respectively. Mendelian randomization analyses for the 5 EAA measures did not reveal statistically significant associations with AF.
Conclusions:
Our study identified adjusted associations between EAA measures and incident AF, suggesting biological aging plays an important role independent of chronological age, though a potential underlying causal relationship remains unclear. These aging processes may be modifiable and not constrained by the immutable factor of time.
Keywords: atrial fibrillation, aging, genetics, epigenetics
INTRODUCTION
Insight into the pathophysiology of atrial fibrillation (AF), the most common sustained cardiac arrhythmia, remains limited and the efficacy of treatment strategies is modest.1 The most prominent risk factor for AF is chronological age, however underlying mechanisms remain unexplained.2 Although the rate of chronological aging is uniform and fixed, the rate of biological aging varies widely.3 Insight into this variability may provide clues into the drivers of aging, potentially leading to treatments that render the risk of age-related disease modifiable and perhaps even preventable.4
Although the intricate details governing aging have yet to be unravelled, significant progress has been made towards identifying biological markers of aging.5 Telomeres are repetitive DNA sequences at chromosomal ends that shorten with repeated somatic cell division.6 Though a robust marker and perhaps mediator of cellular aging, we and others have failed to find evidence that telomere length associates with incident AF.7,8 More recently, mathematical algorithms involving epigenetic modifications, specifically 5-cytosine methylations within CpG dinucleotides, have been shown to predict chronological age with remarkable accuracy, particularly those developed by Horvath and Hannum, leading to their being referred to as epigenetic clocks.9,10 Subsequent epigenetic clocks tailored towards various aspects of clinical phenotype have also been shown to be predictive of disease development.11,12 Although alterations in the methylation status of relevant CpG dinucleotides may lead to changes in gene expression that underlie the observed associations between epigenetic modifications and aging, this remains speculative.5
Consistent with the notion that biological aging is variable, the age predicted by epigenetic clocks often diverges from chronological age, a phenomenon termed epigenetic age acceleration (EAA).5 Individuals whose epigenetic age is older than their chronological age are considered to have positive EAA, whereas negative EAA indicates an epigenetic age younger than the corresponding chronological age. As each clock represents a continuum across a spectrum, everyone exhibits a relative biological age for these epigenetic markers. In addition to being impacted by a heritable genetic predisposition, epigenetic predicted age also theoretically takes into account cumulative life insults impacting biological aging.13
EAA has been shown to associate with disease development, potentially identifying a novel treatment target to prevent and treat age-related diseases, though the phenomenon has yet to be evaluated for cardiac arrhythmias.5 Through a collaboration involving the AFGen Consortium, we sought to evaluate associations between epigenetic age measures and incident AF.14
METHODS
Transparency and Openness Promotion
Anonymized data and materials for the Framingham Heart Study [FHS], Atherosclerosis Risk in Communities [ARIC], and Cardiovascular Health Study [CHS] have been made publicly available at BIOLINCC and can be accessed at https://biolincc.nhlbi.nih.gov/home/.
Study Cohorts
Our study was conducted using 3 population-based cohort studies that had systematically ascertained incident AF and had performed genome-wide DNA methylation analysis on a subgroup of study participants. The design, recruitment, baseline characterization, outcome ascertainment procedures, and approach to DNA methylation analyses for FHS, ARIC, and CHS have been previously described in detail; brief cohort summaries are provided below and remaining details are contained in the Online Supplement.15–24 The institutional review committees for each cohort approved the study methodology and all study participants provided informed consent for their clinical and genetic data to be used for research.
Framingham Heart Study (FHS)
The FHS, initiated in 1948, is a 3-generational community-based prospective cohort study. Study participants for the current analysis were from a subgroup of 5,124 individuals recruited between 2005–2008 comprising the FHS Offspring cohort (offspring and spouses of the Original cohort recruited in 1971).15 Methods describing ascertainment of baseline clinical features have been previously reported.15 Prevalent AF was documented through cardiovascular history performed at enrollment and baseline ECG.18 Ascertainment of incident AF was pursued through biennial surveillance interviews, cardiovascular disease-related hospitalizations, and clinician visits. ECGs and other electrocardiographic data performed for clinical reasons were reviewed by at least 2 cardiologists to adjudicate AF.19
Atherosclerosis Risk in Communities Study (ARIC)
ARIC enrolled 15,792 adults aged 45–64 years between 1987 and 1989 from 4 US communities: the northwest suburbs of Minneapolis, MN; Washington County, MD; Jackson, MS; and Forsyth County, NC.16 Comprehensive baseline evaluations were performed followed by annual or semi-annual phone interviews and up to 6 follow-up examinations through 2017. Methods for ascertaining baseline co-morbidities have been documented previously.16 Prevalent AF was identified from the baseline ECG, and incident AF was identified from study visit ECGs (performed at each visit), hospital discharge diagnoses, and death certificates.20 Prior work has revealed this method of AF ascertainment is associated with a sensitivity of 84% and specificity of 98%.20
Cardiovascular Health Study (CHS)
CHS enrolled 5,201 participants aged 65 years and older that were recruited in 1989–1990 from Medicare eligibility lists in four US communities: Forsyth County, North Carolina; Washington County, Maryland; Sacramento County, California; and Pittsburgh, Pennsylvania. An additional 687 participants, almost all Black, were recruited between 1992–1993. Participants underwent comprehensive examinations at study entry to document baseline demographics and medical co-morbidities.21 Subsequent follow-up was performed with alternating clinic visits and phone calls every six months until 1999 and semi-annual phone calls thereafter. Resting 12-lead ECGs were performed at each visit. Prevalent AF at baseline was documented using the baseline ECG, while incident AF was ascertained on the basis of clinic visit ECGs and hospital discharge diagnosis codes that were supplemented with Medicare inpatient and outpatient claims data.22
Epigenetic Measures
Four separate epigenetic clocks (1) Horvath, 2) Hannum, 3) DNAm PhenoAge, and 4) DNAm GrimAge), and an epigenetic predictor of plasminogen activator inhibitor-1 (PAI-1) levels (DNAm PAI-1) were evaluated. These measures were chosen because they had been previously shown to provide the most accurate estimates of chronological age (Horvath and Hannum) or had exhibited the strongest associations with aspects of clinical phenotype and disease (DNAm PhenoAge, DNAm GrimAge, and DNAm PAI-1).9–12 Details of each epigenetic measure have been previously reported.9–12 Briefly, the Horvath clock, considered pan-tissue, is composed of 353 CpG sites.9 The Hannum clock involves 71 CpG sites and, similar to the remaining clocks, was strictly derived from blood leukocytes.10 Both the Horvath and Hannum clocks were designed to calibrate with chronological age. The DNAm PhenoAge algorithm involves 513 CpG sites and was calibrated using a weighted average of chronological age and 9 clinical laboratory values, whereas DNAm PAI-1 uses 211 CpGs that were predictive of plasma PAI-1 levels.11,12 DNAm GrimAge was developed by regressing time to death on a composite of 7 DNA methylation scores calibrated for plasma clinical biomarkers associated with mortality, including PAI-1, a methylation score for pack years of smoking, and chronological age at the time of blood draw.12 Composed of 1,030 CpGs, DNAmGrimAge was calibrated to predict mortality.
Clock values for all cohorts and estimates for cell type proportions for FHS and ARIC were determined using an online calculator (https://dnamage.genetics.ucla.edu/new).
Two-Sample Summary-Level Mendelian Randomization
Genetic instruments for the 5 EAA measures were derived from a recent genome-wide association study.13 This prior study revealed that the maximum average percentage of variance explained by each genetic instrument (composed of independent single nucleotide polymorphisms [SNPs] that had been identified to associate with the EAA measure at genome-wide significance [p<5×10−8]) among individuals of European ancestry was: Horvath (2.37%), Hannum (1.13%), DNAm PhenoAge (1.38%), DNAm PAI-1 (1.62%), and DNAm GrimAge (1.07%).13 Genetic effect sizes for DNAm PAI-1 were standardized against the mean standard deviation of DNAm PAI-1 (3055 pg/mL) across all cohorts contributing to the genome-wide association study. Analyses evaluating the associations between the genetic instruments and AF were performed using a publicly available dataset from a recent AF genome-wide association study involving 60,620 AF cases and 970,216 controls of European ancestry.25 Notably, overlap was present between the cohorts used in the EAA and AF genome-wide association studies, which was unavoidable due to the use of summary level data.
Statistical Analysis
Normally distributed continuous variables are presented as means + standard deviation. Time-to-event analyses using Cox proportional hazards models were employed to evaluate for associations between epigenetic age measures and incident AF. Study participants with prevalent AF were excluded. FHS utilized mixed effects Cox models to account for familial aggregation. All models possessing an epigenetic age measure, including those unadjusted for clinical variables, involved covariables for cell counts and technical factors, which included sample batch, laboratory site, and follow-up visit. The EAA measures, consistent with prior work, were defined as the residuals obtained after regressing the measure of epigenetic age over chronological age.11,12
Models containing epigenetic measures unadjusted for chronological age were used to enable comparisons of the magnitudes of the measures of association between the epigenetic measures and incident AF relative to those for chronological age. Models containing both chronological and epigenetic predicted age or PAI-1 levels, along with sex and race, assessed for residual associations between chronological age and incident AF following adjustment for the epigenetic measure. Subsequent models (intermediate adjusted and multivariable) were used to evaluate for associations between EAA measures and incident AF. Acknowledging that multiple potential covariables could function as both confounders and mediators, the intermediate adjusted model was restricted to covariables not considered potential mediators (chronological age, sex, race, smoking status [categorized as current, former, and never and analyzed using never as the reference category], and pack year smoking history; Table I in the Supplement). The multivariable model additionally included body mass index, systolic and diastolic blood pressure, anti-hypertensive medication, diabetes, and history of myocardial infarction and heart failure (Table II in the Supplement). Analyses were additionally stratified by study participants with positive and negative EAA and potential differences in the measures of association for the EAA measures on the risk of incident AF were evaluated through an interaction term in both the intermediate and multivariable Cox regression models. Analysis of Schoenfeld residuals for the various models indicated no evidence of violation of the proportional hazards assumption.
Analyses accounting for the competing risk of death were also performed using the competing risk regression model described by Fine and Gray.26 Given our primary goal of evaluating EAA as a risk modifier of incident AF, rather than a risk predictor, the competing risk regression models served as sensitivity analyses.27
Assessment of the ability of the EAA measures to predict incident AF was evaluated using time-dependent C-statistics. Time-dependent C-statistics were initially determined for each EAA measure in models that contained only the EAA measure and technical factors. The change in the predictive capacity of the intermediate and multivariable models for incident AF following addition of each EAA measure was subsequently assessed.
All baseline values for variables used in the regression models corresponded to those ascertained at the time of the blood draw used for DNA methylation analysis. The Horvath, Hannum, DNAm PhenoAge, and DNAm GrimAge clocks are expressed in years and their corresponding measures of associations were reported per 5-year increment, whereas the measures of association for DNAm PAI-1 are per standard deviation due to its being predictive of plasma PAI-1 levels.
Analyses to clarify if the additional covariables included in the multivariable model could function as mediators in the relationships between the EAA measures and incident AF were pursued. Baseline associations between the EAA measures and 5 of the covariables were assessed using linear (body mass index and systolic and diastolic blood pressure) and logistic (use of an anti-hypertensive medication and diabetes) regression, as appropriate. Cox regression analyses assessed for associations between the EAA measures and incident heart failure and myocardial infarction. The regression analyses were adjusted for technical factors, chronological age, sex, and race and the measures of association were reported per 5-year increment of the epigenetic clock and the measures of association for DNAm PAI-1 were per standard deviation.
The proportion of treatment effect (PTE) method was used to assess for statistical evidence that the covariables considered potential mediators mediated the association between the EAA measures and AF. The PTE evaluates the proportional reduction in the regression coefficient of the predictor of interest when the putative mediator is included in the Cox regression model.28 PTE values were determined for models containing each covariable considered a potential mediator in isolation and a PTE value was also determined for a model containing all of these covariables. Confidence intervals were obtained using bootstrap resampling with 1,000 repetitions, with the standard errors calculated as the standard deviation of the bootstrapped estimates after omitting the smallest and largest ten estimates.
The DNAm GrimAge clock, calibrated for all-cause mortality, was derived using a training set of 1,731 study participants from FHS that were also evaluated in the current study. Given the previously documented association between AF and mortality in FHS, sensitivity analyses adjusting for the subgroup of individuals used for clock derivation were performed to screen for potential bias and no differences were observed for the DNAm GrimAge EAA associations with incident AF (Table III in the Supplement).29
Random-effects meta-analysis was used to estimate pooled measures of association. The Q and I-squared statistics were used to assess between-cohort heterogeneity.
Mendelian randomization estimates for individual SNPs used for the genetic instruments of the 5 EAA measures were determined and their collective measures of association were assessed through inverse variance weighted fixed effects meta-analysis. Sensitivity analyses assessing for directional pleiotropy were performed using MR-Egger and MR-RAPS and heterogeneity was evaluated using the Q-statistic.30,31 Each instrument was assessed for weak instrument bias using the F-statistic (Horvath EAA [1,010], Hannum EAA [170], DNAm PhenoAge [98], DNAm PAI-1 [196], DNAm GrimAge [3]). Statistical power calculations revealed 80% power to detect the following odds ratios (OR) (per standard deviation change in the genetic instrument) for the associations between EAA genetic instruments and AF: Horvath EAA (1.07), Hannum EAA (1.16), DNAm PhenoAge (1.22), DNAm PAI-1 (1.15), DNAm GrimAge (3.23).
Two-tailed p-values <0.05 were considered statistically significant. Statistical analyses were performed using Stata version 16 (College Station, TX, USA), SAS version 9.4 (Cary, NC, USA), and R versions 3.6.0 and 3.6.2. The 2-sample Mendelian randomization analyses were conducted using the TwoSampleMR package.32
RESULTS
Study Participants
FHS
A total of 2,362 individuals without prevalent AF from the FHS Offspring cohort underwent methylation analyses and were included (Table 1). All FHS study participants were White, 1,313 (55.6%) were female, and the mean age at baseline was 65.8 ± 8.8 years. Baseline epigenetic measures are provided in Table 2. Predicted mean epigenetic ages ranged from 58.6 ± 10.2 years (DNAm PhenoAge) to 67.1 ± 9.3 years (Hannum). During a median follow-up period of 11.8 years, 278 individuals were diagnosed with incident AF.
Table 1:
Baseline Clinical Characteristics of FHS, ARIC, and CHS Study Participants
| FHS n = 2,362 | ARIC n = 2,519 | CHS n = 719 | |
|---|---|---|---|
|
| |||
| Chronological Age, years | 65.8 ± 8.8 | 56.6 ± 5.8 | 74.1 ± 5.2 |
| Female | 1,313 (55.6) | 1,609 (63.9) | 443 (61.6) |
| BMI (kg/m 2 ) | 28.2± 5.3 | 30.2 ± 6.3 | 27.7 ± 5.3 |
| Black Race | 0 (0) | 2,519 (100) | 321 (44.6) |
| Cigarette Smoking | |||
| Current | 192 (8.1) | 625 (24.8) | 95 (13.2) |
| Past | 1,304 (55.2) | 753 (29.9) | 319 (44.4) |
| Never | 866 (36.7) | 1,141 (45.3) | 305 (42.4) |
| Pack Years | 15.2 ± 21.2 | 11.2 ± 19.2 | 15 ± 22 |
| Systolic BP (mm Hg) | 128 ± 17 | 128 ± 21 | 138 ± 21 |
| Diastolic BP (mm Hg) | 74 ± 10 | 75 ± 11 | 74 ± 11 |
| Hypertensive Medication | 1,112 (47.1) | 1,225 (48.6) | 335 (46.6) |
| Diabetes Mellitus | 304 (12.9) | 673 (26.7) | 145 (20.2) |
| Prior Heart Failure | 23 (1.0) | 196 (7.8) | 5 (0.7) |
| Prior Myocardial Infarction | 79 (3.3) | 117 (4.6) | 3 (0.4) |
| Incident AF Cases | 278 (11.8) | 351 (13.9) | 276 (38.4) |
| Median FU Time, years | 9.6 | 21.8 | 10.2 |
| Baseline Years | 2005–2008 | 1990–95 | 1992–93 |
| End of FU | 2016 | 2016 | 2014 |
Data are n (%) or mean ± standard deviation. FHS = Framingham Heart Study, ARIC = Atherosclerosis Risk In Communities, CHS = Cardiovascular Health Study, BMI = body mass index, kg/m2 = kilograms/meters2, BP = blood pressure, mm Hg = millimeters of mercury, AF = atrial fibrillation, FU = follow-up.
Table 2:
Baseline Epigenetic Age Measures of FHS, ARIC, and CHS Study Participants
| FHS n = 2,362 | ARIC n = 2,519 | CHS n = 719 | |
|---|---|---|---|
|
| |||
| Horvath Predicted Age, years | 65.0 ± 8.6 | 57.1 ± 9.4 | 70.2 ± 7.7 |
| Horvath Predicted EAA, years | 0.0 ± 5.2 | 0.0 ± 7.3 | 0.0 ± 6.4 |
| Hannum Predicted Age, years | 67.1 ± 9.3 | 57.1 ± 8.7 | 72.2 ± 7.1 |
| Hannum Predicted EAA, years | 0.0 ± 5.1 | 0.0 ± 6.4 | 0.0 ± 5.4 |
| DNAm PhenoAge, years | 58.6 ± 10.2 | 52.8 ± 9.1 | 64.2 ± 8.3 |
| DNAm PhenoAge EAA, years | 0.0 ± 6.4 | 0.0 ± 7.4 | 0.0 ± 6.8 |
| DNAm PAI-1* | 19,696.4 ± 3201.6 | 21,467.0 ± 2825.0 | 24,382.0 ± 2,994.0 |
| DNAm PAI-1* EAA | 0.0 ± 3,153.9 | 0.0 ± 2,800.0 | 0.0 ± 2,993.0 |
| DNAm GrimAge, years | 59.2 ± 8.7 | 57.6 ± 7.2 | 71.7 ± 5.7 |
| DNAm GrimAge EAA, years | 0.0 ± 4.8 | 0.0 ± 5.8 | 0.0 ± 4.4 |
Data are n (%) or mean ± standard deviation.
DNAm PAI-1 is an epigenetic predictor of plasma PAI-1 levels (picograms/milliliter). FHS = Framingham Heart Study, ARIC = Atherosclerosis Risk In Communities, CHS = Cardiovascular Health Study, EAA = epigenetic age acceleration.
ARIC
Within the ARIC cohort, methylation analysis was performed on 2,519 Black individuals without prevalent AF (Table 1). Baseline mean epigenetic predicted ages (Table 2) ranged from 52.8 ± 9.1 years (DNAm PhenoAge) to 57.6 ± 7.2 years (DNAm GrimAge). The baseline mean age of study participants was 56.6 ± 5.8 years and 1,609 (63.9%) were female. Incident AF was diagnosed in 351 study participants during a median follow-up period of 21.8 years.
CHS
Genome-wide methylation analysis was conducted on 719 individuals from the CHS cohort without prevalent AF (Table 1). A total of 398 (55.4%) individuals were White and the remainder were Black. Baseline mean epigenetic predicted ages and EAA values for each epigenetic clock are provided in Table 2. A majority of the study participants were female (443/719; 61.6%) and their mean age at baseline was 74.1 ± 5.2 years. Among the CHS cohort, 276 developed incident AF during a median follow up period of 10.2 years.
The remaining baseline clinical characteristics and epigenetic measures for the 3 cohorts are summarized in Tables 1 and 2.
Chronological Age, Epigenetic Predicted Age, and Incident AF
Chronological age exhibited statistically significant associations with AF in all 3 cohorts in unadjusted analyses (Table IV in the Supplement). Combined analysis revealed that a 5-year increase in baseline chronological age was associated with a 1.44-fold (95% CI: 1.34–1.54; p<0.0001) higher hazard of developing incident AF. Each of the 5 epigenetic age measures, in models adjusted only for technical factors and unadjusted for chronological age or other clinical factors, was associated with a statistically significant higher hazard of incident AF in pooled analyses involving the 3 cohorts (Table IV in the Supplement). Relative to chronological age, the calculated point estimates for each epigenetic measure were smaller in magnitude for the hazard of incident AF.
Models that included both chronological and epigenetic measures revealed an attenuation of the associations observed between chronological age and incident AF (Figure I in the Supplement). Following adjustment for measures of epigenetic predicted age or PAI-1 levels, the magnitude of the point estimates identified on pooled analysis per 5-year increase in chronological age ranged from 1.26 (95% CI: 1.09–1.47, p=0.0020; DNAm PhenoAge clock adjustment [Figure ID in the Supplement]) to 1.43 (95% CI: 1.32–1.54, p<0.0001; DNAm PAI-1 adjustment [Figure IE in the Supplement]). Similar findings were also observed for the multivariable model (Figure II in the Supplement).
Epigenetic Age Acceleration and Incident AF
The meta-analyzed estimates for EAA, adjusted for chronological age, sex, race, and smoking variables exhibited statistically significant associations for incident AF for all EAA measures with the exception of Horvath EAA (Figure 1A–E). The meta-analyzed point estimate for DNAm GrimAge EAA had the largest magnitude association (hazard ratio [HR]: 1.25; 95% CI: 1.15–1.37; p<0.0001) (Figure 1E) and the smallest magnitude statistically significant association was observed for Hannum EAA (HR: 1.12; 95% CI: 1.02–1.24; p=0.0193); both per 5-year increment (Figure 1B). Similar results were observed for the competing risk regression models (Figure III in the Supplement).
Figure 1: Meta-Analyses Evaluating Associations Between 5 Epigenetic Age Acceleration Measures and Incident Atrial Fibrillation.
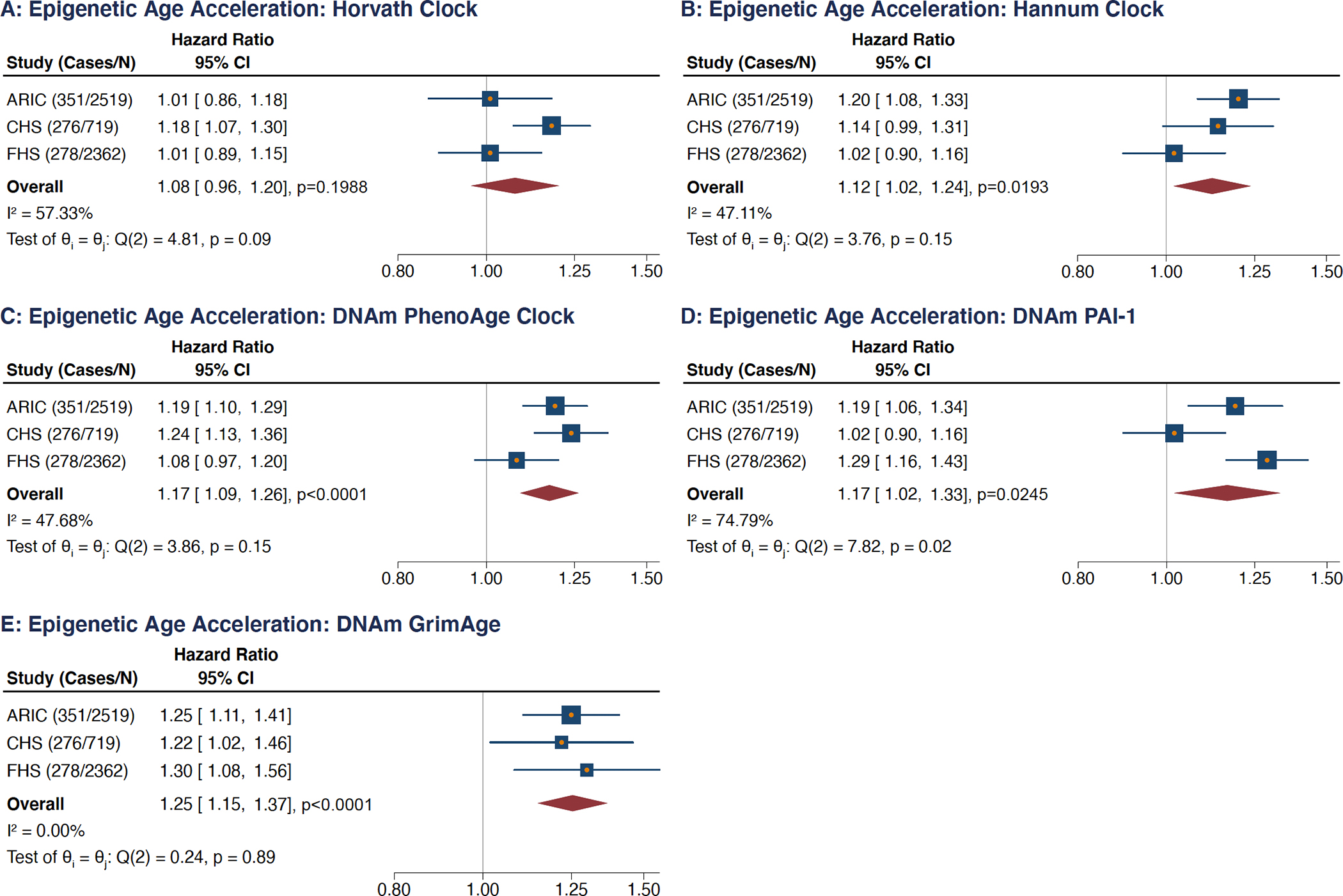
Analyses adjusted for chronological age, sex, race, smoking variables, and technical factors. (A) Horvath clock, (B) Hannum clock, (C) DNAm PhenoAge clock, (D) DNAm PAI-1 epigenetic predictor, (E) DNAm GrimAge clock.
HRs are per 5-year increment in each age measure, per standard deviation increment for DNAm PAI-1.
Technical factors include cell types, sample batch, laboratory site, and follow-up visit.
CI = confidence intervals, FHS = Framingham Heart Study, ARIC = Atherosclerosis Risk in Communities, CHS = Cardiovascular Health Study.
Tests for overall effect heterogeneity performed with the Q- and I-squared statistics.
Multivariable Adjustment
Following adjustment for the 7 additional AF risk factors present in the multivariable model, statistical significance persisted for EAA measures for 2 clocks, namely DNAm GrimAge (HR: 1.19; 95% CI: 1.09–1.31; p=0.0002) and DNAm PhenoAge (HR: 1.15; 95% CI: 1.05–1.25; p=0.0017); both per 5-year increment (Figure 2). Although the meta-analyzed measures of association for EAA measures for the other clocks and DNAm PAI-1 were not statistically significant following multivariable adjustment, their directionality remained consistent relative to the associations observed on unadjusted analysis. Overall similar results were obtained on competing risk regression analyses, though statistical significance for DNA GrimAge EAA was no longer observed (subdistribution HR: 1.08; 95% CI: 0.99–1.19; p=0.0790; Figure IV in the Supplement).
Figure 2: Meta-Analyses of Multivariable Models Evaluating Associations Between 5 Epigenetic Age Acceleration Measures and Incident Atrial Fibrillation.
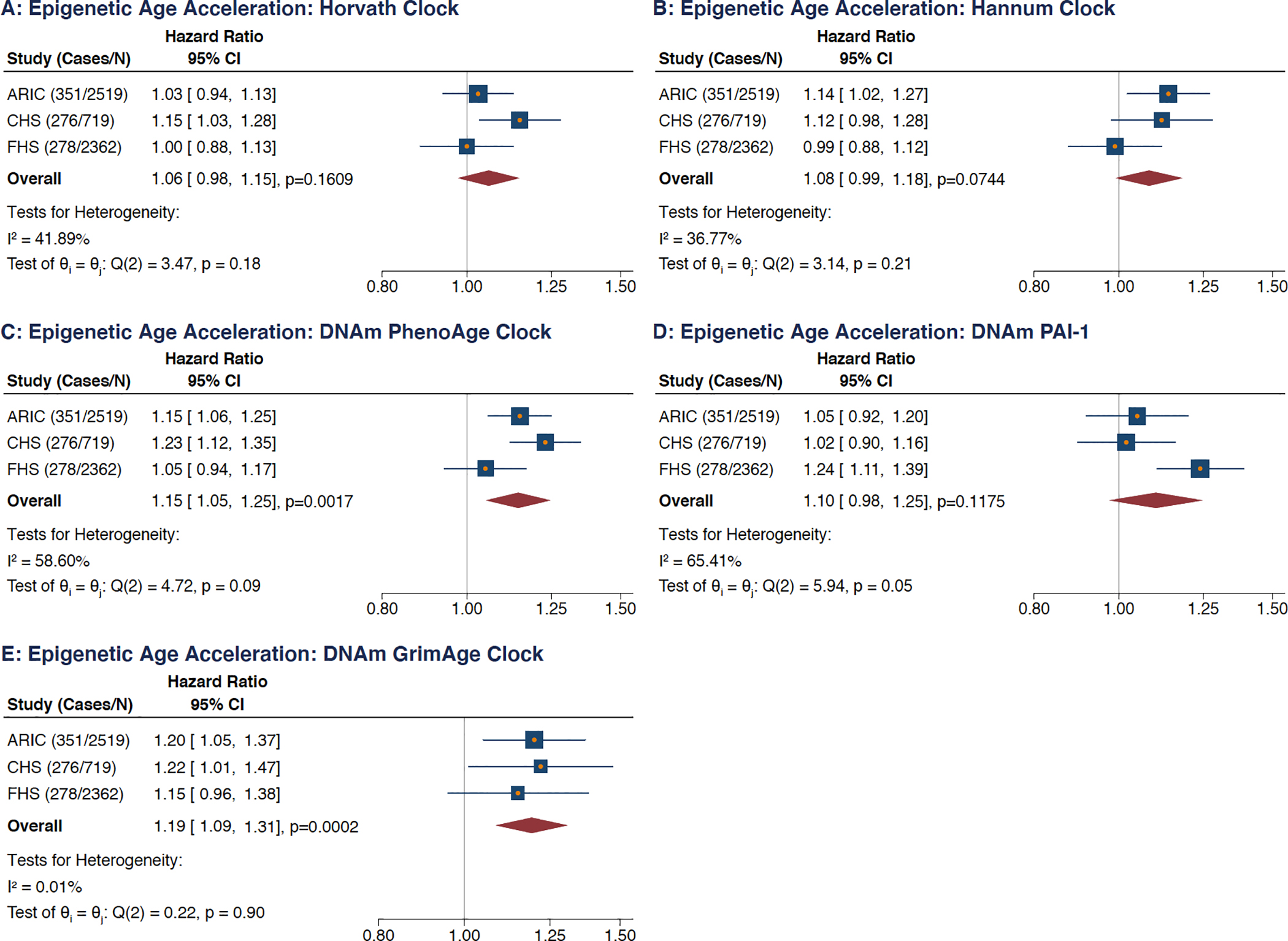
(A) Horvath clock, (B) Hannum clock, (C) DNAm PhenoAge clock, (D) DNAm PAI-1 epigenetic predictor, (E) DNAm GrimAge clock.
HRs are per 5-year increment in each age measure, per standard deviation increment for DNAm PAI-1.
Analyses adjusted for technical factors (cell types, sample batch, laboratory site, and follow-up visit), chronological age, sex, race, body mass index, smoking status, smoking pack year, systolic blood pressure, diastolic blood pressure, hypertensive medication, diabetes, history of congestive heart failure, history of myocardial infarction.
CI = confidence intervals, FHS = Framingham Heart Study, ARIC = Atherosclerosis Risk in Communities, CHS = Cardiovascular Health Study.
Tests for overall effect heterogeneity performed with the Q- and I-squared statistics.
Evaluation of time-dependent C-statistics in models containing only the EAA measure and technical factors revealed comparable predictive capacity for each EAA measure with C-statistic values ranging from 0.62 to 0.63 (Figure V in the Supplement). Addition of the EAA measures to the intermediate and multivariable adjusted models did not result in statistically significant improvements in the time-dependent C-statistics (Figures VI–IX in the Supplement).
Screening for Different EAA Measures of Association Among Individuals with Positive and Negative EAA
Although our findings suggest that increasing EAA is associated with a higher risk of AF, we additionally sought to clarify if this higher risk (and magnitude of risk) was consistent among individuals with positive and negative EAA. Analyses stratified by positive and negative EAA status for each EAA measure in both the intermediate and multivariable models did not identify significantly different hazards for incident AF when evaluated statistically for interaction for 4 of the EAA measures; the sole exception being DNAm PAI-1 EAA (Figures X and XI in the Supplement).
Mediation Analysis
EAA measures were found to exhibit positive associations with the covariables considered as potential mediators with the exception of baseline diastolic blood pressure (Figures XII–IIXX in the Supplement). Each EAA measure was associated with statistically significant increased baseline BMI and systolic blood pressure, with isolated exceptions (DNAm GrimAge EAA for BMI [Figure XIIE in the Supplement] and Horvath EAA for systolic blood pressure [Figure XIIIA in the Supplement]). Similarly statistically significant results were observed for the EAA measures and baseline prevalent diabetes and treatment with an anti-hypertensive medication (Figures XV and XVI in the Supplement). Each EAA measure exhibited a statistically significant association with incident congestive heart failure (Figure XVII in the Supplement), and all but one of the EAA measures were associated with significantly heightened risks of incident myocardial infarction (Figure XVIII in the Supplement).
None of the covariables considered as potential mediators, either in isolation (Figures IXX–XXV in the Supplement) or in combination (Figure 3) was found to be a statistically significant mediator for the associations between the EAA measures and incident AF when evaluated using the PTE. PTE values for models containing all of these covariables were found to range from 11% for DNAm PhenoAge EAA (95% CI: −0.11 to 0.34; Figure 3C) to 27% for Hannum EAA (95% CI: −0.13 to 0.66; Figure 3B). When evaluated in isolation, none of the covariables had a PTE point estimate greater than 10% (Figures IXX–XXV in the Supplement).
Figure 3: Combined Proportion of Treatment Effect (PTE) of Potential Mediators on the Association Between 5 Epigenetic Age Acceleration Measures and Incident Atrial Fibrillation.
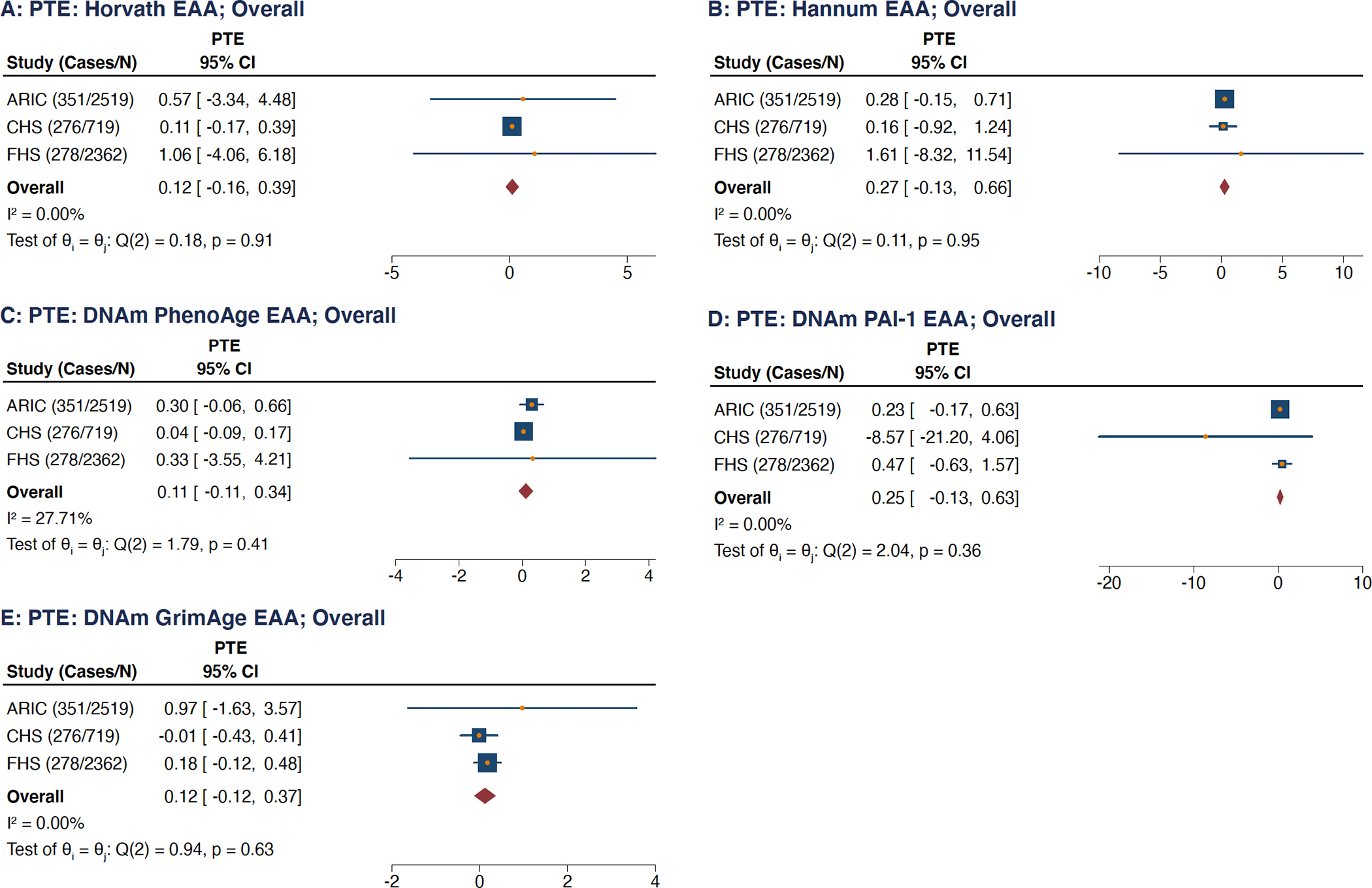
(A) Horvath clock, (B) Hannum clock, (C) DNAm PhenoAge clock, (D) DNAm PAI-1 epigenetic predictor, (E) DNAm GrimAge clock.
PTE = Proportion of Treatment Effect, CI = confidence intervals, FHS = Framingham Heart Study, ARIC = Atherosclerosis Risk in Communities, CHS = Cardiovascular Health Study.
Tests for overall effect heterogeneity performed with the Q- and I-squared statistics.
Two-Sample Summary-Level Mendelian Randomization
In order to investigate for a potential causal relationship underlying the associations between the EAA measures and incident AF, 2-sample Mendelian randomization was pursued. None of the genetic instruments for the 5 EAA measures associated with a higher odds of the arrhythmia, including the 4 EAA measures that associated with incident AF following adjustment for potential confounders in the intermediate model (Figure 4 and Figure XXVI in the Supplement). Inverse variance weighted meta-analysis of the genetic instruments for the 4 epigenetic clocks revealed ORs that ranged from 1.02 (Horvath EAA, OR: 1.02, 95% CI: 0.95–1.10, p=0.57 and DNAm PhenoAge, OR: 1.02, 95% CI: p=0.53) to 1.12 (DNAm GrimAge; 95% CI: 0.93–1.35, p=0.24), whereas the genetic instrument for EAA of DNAm PAI-1 levels was 0.90 (95% CI: 0.75–1.09) (Figure 4). Sensitivity analyses revealed no evidence for directional pleiotropy or heterogeneity for any of the genetic instruments and AF (Table V in the Supplement).
Figure 4: Two-Sample Mendelian Randomization Analysis of Epigenetic Age Acceleration (EAA) Measures and Atrial Fibrillation (AF).
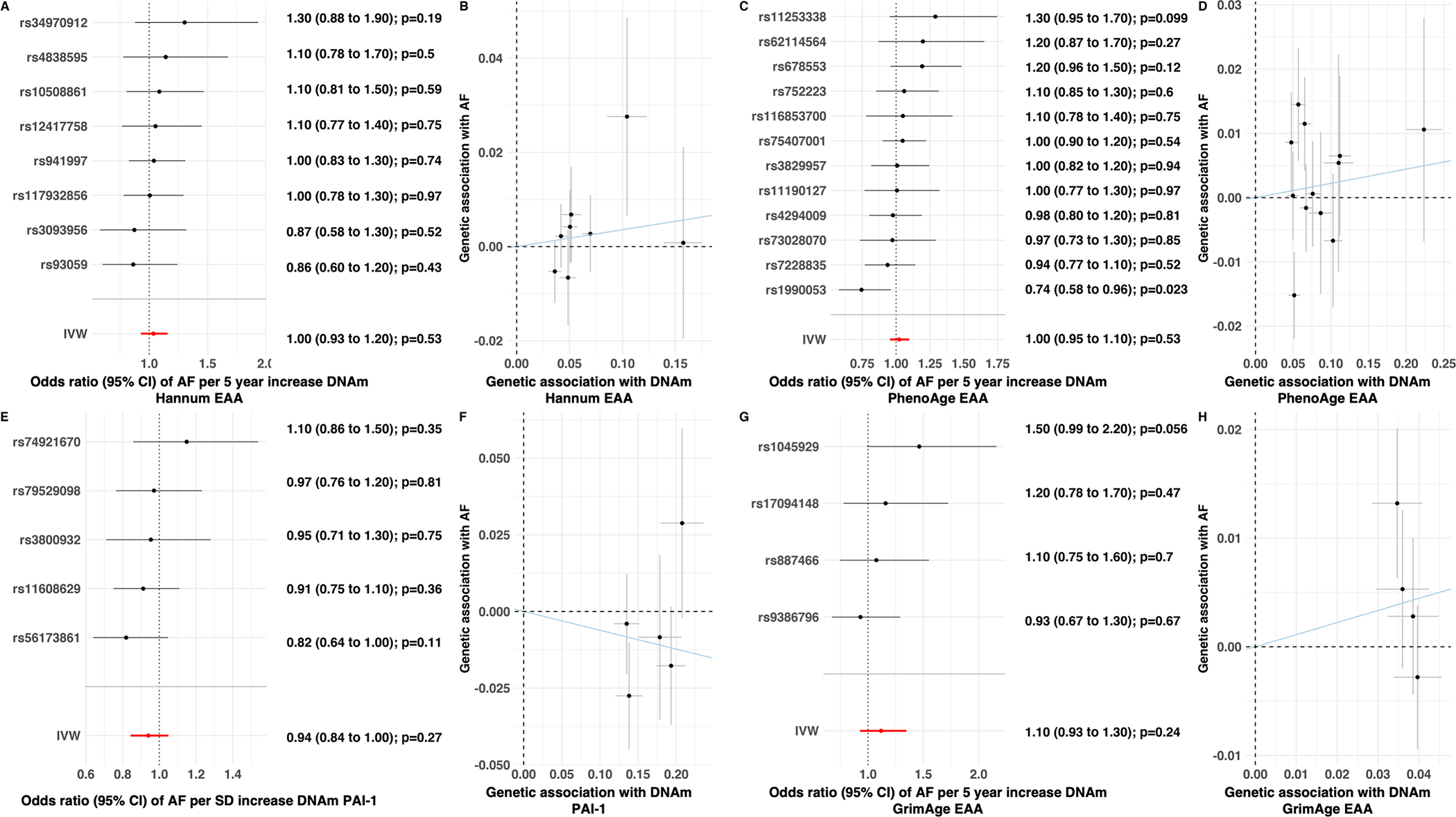
Forest plots demonstrating collective inverse variance weighted effect estimates and individual measures of association for each genetic variant associated with the Hannum (A), DNAm PhenoAge (C), DNAm PAI-1 (E), and DNAm GrimAge (G) EAA measures and atrial fibrillation. Scatter plots of associations for each Hannum (B), DNAm PhenoAge (D), DNAm PAI-1 (F), and DNAm GrimAge (H) EAA genetic variant with atrial fibrillation (y-axis) and associated EAA measure (x-axis) with line representing inverse variance weighted estimate.
Odds ratios are per 5-year increment in each age measure and per SD increment in DNAm PAI
EAA = epigenetic age acceleration, AF = atrial fibrillation
DISCUSSION
Our study involving 5,600 individuals from 3 population-based cohorts identified associations between methylation-based predictors of biological aging and incident AF. Among 5 different EAA measures, measures of accelerated biological aging, 4 exhibited associations with incident AF following adjustment for chronological age, sex, race, and smoking variables on pooled analysis. Multivariable adjustment with 7 additional clinical traits considered risk factors for AF yielded persistently significant associations for 2 clocks, DNAm PhenoAge and DNAm GrimAge, suggesting their potential role as novel AF risk factors. Notably, these additional clinical traits may also potentially function as mediators, and hence their inclusion in models may inappropriately mask or attenuate associations following adjustment, though statistically significant mediation was not observed. Despite adjustment with epigenetic measures, the association between chronological aging and incident AF persisted indicating that epigenetic measured biological aging only partially accounts for the overall association underlying AF and chronological aging.
Epigenetic clocks incorporate DNA methylation status at CpG dinucleotides into mathematical algorithms to predict chronological age and various aspects of clinical phenotype.5 DNA methylation and nucleosome occupancy, the probability that a region of DNA is occupied by a histone octamer, are hypothesized to impact gene expression through altered accessibility of transcription machinery to DNA binding sites.33 Although a large body of work has established that methylation patterns of CpG dinucleotides are tightly correlated with aging, debate remains regarding potential causal relationships underlying CpG methylation status, nucleosome formation, and gene expression.34–40 The initial two epigenetic clocks to gain widespread acceptance were those from Horvath and Hannum and focused on estimating chronological age in isolation.9,10 Subsequent clocks were developed to predict additional phenotypic features and include the DNAm PhenoAge clock, designed to optimally predict clinical phenotype, and the DNAm GrimAge clock, which has been shown to be the strongest predictor of all-cause mortality.11,12
Though 4 EAA measures evaluated in our study exhibited statistically significant associations with incident AF following adjustment for chronological age, sex, race, and smoking variables in isolation, only the measures derived from DNAm PhenoAge and DNAm GrimAge remained associated with the arrhythmia upon multivariable adjustment for additional clinical factors. In addition to chronological age, DNAmPhenoAge also incorporates 9 values gleaned from blood testing, namely, albumin, creatinine, glucose, C-reactive protein, lymphocyte percentage, mean cell volume, red blood cell distribution width, alkaline phosphatase and white blood cell count. Notably, multiple of these laboratory measures relate to various clinical features, such as inflammation, kidney disease, and diabetes, previously linked to AF.2 In a similar manner, the 7 plasma proteins used to derive DNAm GrimAge (adrenomedullin, β−2-microglobulin, cystatin-C, growth differentiation factor-15, leptin, PAI-1, and tissue inhibitor metalloproteinases-1) are also involved in biological pathways implicated in AF pathogenesis.2 Given that both clocks involve features previously linked to AF, it is perhaps not surprising that an associated measure meant to be reflective of accelerated biological aging predicted a higher risk of the arrhythmia. Consistent with this notion, it is conceivable that either mediation or residual confounding involving these blood features or plasma proteins could potentially account for the observed EAA associations with incident AF, however this could not be further evaluated due to their values at the time of the blood draw used for methylation analysis being generally unavailable.
It should also be noted that the risk factors included in the multivariable models assumes their roles as confounders, however it is conceivable that many of these clinical features may function as a mediators in the relationship between epigenetic age and incident AF. For example, accelerated biological aging ascertained on the basis of increased epigenetic age may increase the risk of developing hypertension, which would be anticipated to lead to a corresponding increase in the risk of developing AF.41 Consistent with this concept, with the exception of diastolic blood pressure, we identified statistically significant positive associations between EAA measures and the 6 other covariables considered to be potential mediators. Although we did not identify statistically significant mediation when evaluated through the PTE method, which may have been secondary to inadequate statistical power, it is important to consider the possibility that these potential mediators may have attenuated the true magnitude of association between the EAA measures and incident AF. In this context, it is conceivable that the measures of association identified through the intermediate adjusted model may be more reflective of the actual measures of association for EAA measures and AF risk.
The lack of association identified for the EAA genetic instruments and incident AF on Mendelian randomization analysis may suggest that the observed associations on Cox regression analyses are not due to an underlying causal link. Given the multiple potential factors that may contribute to EAA measures, it is conceivable that observed associations may be secondary to residual confounding or other forms of bias. Although conceivable, as highlighted above, all 5 genetic instruments accounted for very modest variability of the EAA measures, ranging from 1.07% (DNAm GrimAge) to 2.37% (Horvath), and hence it is also possible that these particular analyses failed to yield sufficient power to detect true associations.13 Notably, DNAm GrimAge EAA exhibited the largest magnitude association with incident AF in Cox regression analyses, however due to the limited strength of its genetic instrument, the related Mendelian randomization analysis had only 80% statistical power to detect an OR of 3.23 or greater (per standard deviation increase in its genetic instrument). As future genome-wide association studies further unravel the genetic contributors to EAA and enable the development of more powerful genetic instruments, it is conceivable that significant associations may yet emerge.
Although the EAA measures evaluated appear unlikely to improve AF risk prediction beyond current multivariable models, it is conceivable that epigenetic measures of aging may eventually help guide development of improved prevention and treatment strategies for the arrhythmia. It should be noted that, while chronological aging is inevitable, methylation of CpG dinucleotides is reversible and evidence suggests that epigenetic aging may be modifiable.42 Treatment of somatic cells with Yamanaka factors, which converts them to a pluripotent state, remarkably resets their epigenetic clocks to zero.9 Human trials evaluating potential therapies to reverse biological aging, as measured by epigenetic clocks, have begun to emerge and have shown intriguing preliminary results.43 Additionally, lifestyle factors such as diet and exercise have been suggested to be associated with reduced epigenetic predicted biological aging.42 In this context, it is appealing to consider that lifestyle interventions and treatments that slow or potentially reverse epigenetic measured biological aging could serve as effective upstream preventive therapies for the arrhythmia. Though intriguing, it is crucial to emphasize that causality should not be inferred from the associations identified in our observational study, a concept highlighted by the lack of associations observed in the Mendelian randomization analyses. Future work will be necessary to both confirm and decipher the significance of the links observed between epigenetic age measures and incident AF.
Limitations
Although our analysis involved 3 large population-based cohorts and 905 incident AF cases, our study size is modest and future studies with improved statistical power may further inform insights into the relationship between epigenetic age measures and incident AF. Despite inclusion of both White and Black individuals, our study lacked statistical power to compare the measures of associations between epigenetic age measures and incident AF observed for different races. Although the 3 cohorts evaluated are population-based cohorts, differences in their demographic and clinical features may have potentially contributed to differences in the magnitudes of the observed measures of association, however the consistent directions of effect noted in all 3 cohorts appear to reinforce the generalizability of our findings, strengthening their relevance. It is important to also acknowledge the possibility that some participants may have experienced undetected AF, however given that ascertainment of AF is not anticipated to vary significantly by EAA, the corresponding likelihood of spurious false positive associations is considered low. The potential significance of the different measures of association observed for DNAm PAI-1 EAA on the risk of AF among individuals with positive and negative EAA is uncertain; future studies will be necessary to confirm this finding. We acknowledge that our study is observational; we cannot exclude residual confounding, and cannot establish causal relations. Equally important, owing to the modest statistical power stemming from the limited strength of the genetic instruments, the lack of associations observed in the Mendelian randomization analyses should not be viewed as strong evidence to refute potential causality. Furthermore, as epigenetic phenomena by their nature may be more related to environmental exposures rather than fixed genetic determinants, these findings may highlight an inherent limitation of Mendelian randomization in such circumstances.
Conclusions
Our meta-analysis involving 3 large population-based cohort studies identified associations between epigenetic age measures and incident AF. Measures of accelerated biological aging remained associated with incident AF following adjustment for possible confounders, revealing a potential role for prevention and treatment strategies to mitigate age-related AF risk that otherwise may be perceived as inexorable.
Supplementary Material
Clinical Perspective.
What is new?
DNA-based markers of accelerated biological aging, termed epigenetic age acceleration, independently associate with a higher risk of incident atrial fibrillation.
Epigenetic measured biological aging is suggested to partially account for the association between chronological aging and atrial fibrillation, though underlying mechanisms remain to be clarified.
What are the clinical implications?
Although chronological aging is a presumed non-modifiable risk factor for atrial fibrillation, the notion that biological aging may be slowed or potentially reversible is an area of intensive investigation.
Should accelerated biological aging have an underlying causal relationship with atrial fibrillation, future treatments and lifestyle modifications that successfully slow or reverse biological aging may serve as effective forms of preventive therapy for the arrhythmia.
Acknowledgements:
The authors thank the staff and participants of the ARIC, FHS, and CHS studies for their invaluable contributions.
Disclosures:
UC Regents (the employer of Drs. Horvath and Lu) has filed patents surrounding several epigenetic biomarkers of aging (including GrimAge), which list Drs. Horvath and Lu as inventors. Dr. Lubitz receives sponsored research support from Bristol Myers Squibb / Pfizer, Bayer AG, Boehringer Ingelheim, and Fitbit, and has consulted for Bristol Myers Squibb / Pfizer and Bayer AG, and participates in a research collaboration with IBM. Dr. Ellinor is supported by a grant from Bayer AG to the Broad Institute focused on the genetics and therapeutics of cardiovascular diseases, and he has served on advisory boards or consulted for Bayer AG, Quest Diagnostics, MyoKardia and Novartis. Dr. Benjamin serves as an uncompensated member for the MyHeartLab Steering Committee. The MyHeartLab Study is a PI-initiated study from the University of California San Francisco: PI, Jeffrey Olgin, MD, through a research grant to UCSF from Samsung. The remaining authors have no relevant disclosures.
Sources of Funding:
ARIC: The Atherosclerosis Risk in Communities study has been funded in whole or in part with Federal funds from the National Heart, Lung, and Blood Institute, National Institutes of Health, Department of Health and Human Services (contract numbers HHSN268201700001I, HHSN268201700002I, HHSN268201700003I, HHSN268201700004I and HHSN268201700005I). Funding was also provided by 5RC2HL102419, R01NS087541, K24HL148521, and American Heart Association grant 16EIA26410001.
FHS: The Framingham Heart Study is funded by NIH, NHLBI, 75N92019D00031; HHSN268201500001I. Funding also provided by NHLBI: R01HL128914; 2R01 HL092577; American Heart Association, 18SFRN34110082; The laboratory work for this investigation was funded by the Division of Intramural Research, National Heart, Lung, and Blood Institute, National Institutes of Health, and by a Director’s Challenge Award, National Institutes of Health.
CHS: Infrastructure for the CHARGE Consortium is supported in part by the National Heart, Lung, and Blood Institute grant R01HL105756. The CHS research was supported by NHLBI contracts HHSN268201200036C, HHSN268200800007C, HHSN268201800001C, N01HC55222, N01HC85079, N01HC85080, N01HC85081, N01HC85082, N01HC85083, N01HC85086; and NHLBI grants U01HL080295, U01HL130114, K08HL116640, R01HL087652, R01HL092111, R01HL103612, R01HL105756, R01HL103612, R01HL111089, R01HL116747 and R01HL120393 with additional contribution from the National Institute of Neurological Disorders and Stroke (NINDS). Additional support was provided through R01AG023629 from the National Institute on Aging (NIA), Merck Foundation / Society of Epidemiologic Research as well as Laughlin Family, Alpha Phi Foundation, and Locke Charitable Foundation. A full list of principal CHS investigators and institutions can be found at CHS-NHLBI.org. The provision of genotyping data was supported in part by the National Center for Advancing Translational Sciences, CTSI grant UL1TR000124, and the National Institute of Diabetes and Digestive and Kidney Disease Diabetes Research Center (DRC) grant DK063491 to the Southern California Diabetes Endocrinology Research Center.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Drs. Lu and Horvath are supported by a National Institute of Health U01 grant, U01AG060908 - 01. Dr. Kornej received funding from the Marie Sklodowska-Curie Actions under the European Union’s Horizon 2020 research and innovation programme (grant agreement No 838259). Dr. Lubitz is supported by NIH grant 1R01HL139731 and American Heart Association grant 18SFRN34250007. Dr. Sotoodehnia is supported by R01HL141989 from the NHLBI.
Abbreviations
- AF
atrial fibrillation
- EAA
epigenetic age acceleration
- FHS
Framingham Heart Study
- ARIC
Atherosclerosis Risk in Communities
- CHS
Cardiovascular Health Study
- PAI-1
plasminogen activator inhibitor-1
- SNP
single nucleotide polymorphism
- PTE
proportion of treatment effect
- OR
odds ratio
- HR
hazard ratio
Footnotes
References
- 1.Heijman J, Guichard J-B, Dobrev D, Nattel S. Translational Challenges in Atrial Fibrillation. Circ Res. 2018;122:752–773. [DOI] [PubMed] [Google Scholar]
- 2.Staerk L, Sherer JA, Ko D, Benjamin EJ, Helm RH. Atrial Fibrillation: Epidemiology, Pathophysiology, and Clinical Outcomes. Circ Res. 2017;120:1501–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hamczyk MR, Nevado RM, Barettino A, Fuster V, Andrés V. Biological Versus Chronological Aging: JACC Focus Seminar. J Am Coll Cardiol. 2020;75:919–930. [DOI] [PubMed] [Google Scholar]
- 4.Longo VD, Antebi A, Bartke A, Barzilai N, Brown-Borg HM, Caruso C, Curiel TJ, de Cabo R, Franceschi C, Gems D, et al. Interventions to Slow Aging in Humans: Are We Ready? Aging Cell. 2015;14:497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Horvath S, Raj K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat Rev Genet. 2018;19:371–384. [DOI] [PubMed] [Google Scholar]
- 6.Blackburn EH, Epel ES, Lin J. Human telomere biology: A contributory and interactive factor in aging, disease risks, and protection. Science. 2015;350:1193–1198. [DOI] [PubMed] [Google Scholar]
- 7.Roberts JD, Dewland TA, Longoria J, Fitzpatrick AL, Ziv E, Hu D, Lin J, Glidden DV, Psaty BM, Burchard EG, et al. Telomere length and the risk of atrial fibrillation: insights into the role of biological versus chronological aging. Circ Arrhythm Electrophysiol. 2014;7:1026–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Staerk L, Wang B, Lunetta KL, Helm RH, Ko D, Sherer JA, Ellinor PT, Lubitz SA, McManus DD, Vasan RS, et al. Association Between Leukocyte Telomere Length and the Risk of Incident Atrial Fibrillation: The Framingham Heart Study. J Am Heart Assoc. 2017;6:e006541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Horvath S DNA methylation age of human tissues and cell types. Genome Biol. 2013;14:R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, Klotzle B, Bibikova M, Fan J-B, Gao Y, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013;49:359–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levine ME, Lu AT, Quach A, Chen BH, Assimes TL, Bandinelli S, Hou L, Baccarelli AA, Stewart JD, Li Y, et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging. 2018;10:573–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu AT, Quach A, Wilson JG, Reiner AP, Aviv A, Raj K, Hou L, Baccarelli AA, Li Y, Stewart JD, et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging. 2019;11:303–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCartney DL, Min JL, Richmond RC, Lu AT, Sobczyk MK, Davies G, Broer L, Guo X, Jeong A, Jung J, et al. Genome-wide association studies identify 137 loci for DNA methylation biomarkers of ageing. bioRxiv. 2020;2020.06.29.133702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roselli C, Chaffin MD, Weng L-C, Aeschbacher S, Ahlberg G, Albert CM, Almgren P, Alonso A, Anderson CD, Aragam KG, et al. Multi-ethnic genome-wide association study for atrial fibrillation. Nat Genet. 2018;50:1225–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kannel WB, Feinleib M, McNamara PM, Garrison RJ, Castelli WP. An investigation of coronary heart disease in families. The Framingham offspring study. Am J Epidemiol. 1979;110:281–290. [DOI] [PubMed] [Google Scholar]
- 16.The Atherosclerosis Risk in Communities (ARIC) Study: design and objectives. The ARIC investigators. Am J Epidemiol. 1989;129:687–702. [PubMed] [Google Scholar]
- 17.Fried LP, Borhani NO, Enright P, Furberg CD, Gardin JM, Kronmal RA, Kuller LH, Manolio TA, Mittelmark MB, Newman A. The Cardiovascular Health Study: design and rationale. Ann Epidemiol. 1991;1:263–276. [DOI] [PubMed] [Google Scholar]
- 18.Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham Study. Stroke. 1991;22:983–988. [DOI] [PubMed] [Google Scholar]
- 19.Piccini JP, Hammill BG, Sinner MF, Jensen PN, Hernandez AF, Heckbert SR, Benjamin EJ, Curtis LH. Incidence and prevalence of atrial fibrillation and associated mortality among Medicare beneficiaries, 1993–2007. Circ Cardiovasc Qual Outcomes. 2012;5:85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alonso A, Agarwal SK, Soliman EZ, Ambrose M, Chamberlain AM, Prineas RJ, Folsom AR. Incidence of atrial fibrillation in whites and African-Americans: the Atherosclerosis Risk in Communities (ARIC) study. Am Heart J. 2009;158:111–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tell GS, Fried LP, Hermanson B, Manolio TA, Newman AB, Borhani NO. Recruitment of adults 65 years and older as participants in the Cardiovascular Health Study. Ann Epidemiol. 1993;3:358–366. [DOI] [PubMed] [Google Scholar]
- 22.Wallace ER, Siscovick DS, Sitlani CM, Dublin S, Mitchell PH, Odden MC, Hirsch CH, Thielke S, Heckbert SR. Incident Atrial Fibrillation and Disability-Free Survival in the Cardiovascular Health Study. J Am Geriatr Soc. 2016;64:838–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maksimovic J, Gordon L, Oshlack A. SWAN: Subset-quantile within array normalization for illumina infinium HumanMethylation450 BeadChips. Genome Biol. 2012;13:R44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, Wiencke JK, Kelsey KT. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nielsen JB, Thorolfsdottir RB, Fritsche LG, Zhou W, Skov MW, Graham SE, Herron TJ, McCarthy S, Schmidt EM, Sveinbjornsson G, et al. Biobank-driven genomic discovery yields new insight into atrial fibrillation biology. Nat Genet. 2018;50:1234–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fine JP, Gray RJ. A Proportional Hazards Model for the Subdistribution of a Competing Risk. J Am Stat Assoc. 1999;94:496–509. [Google Scholar]
- 27.Austin PC, Lee DS, Fine JP. Introduction to the Analysis of Survival Data in the Presence of Competing Risks. Circulation. 2016;133:601–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin DY, Fleming TR, De Gruttola V. Estimating the proportion of treatment effect explained by a surrogate marker. Stat Med. 1997;16:1515–1527. [DOI] [PubMed] [Google Scholar]
- 29.Benjamin EJ, Wolf PA, D’Agostino RB, Silbershatz H, Kannel WB, Levy D. Impact of atrial fibrillation on the risk of death: the Framingham Heart Study. Circulation. 1998;98:946–952. [DOI] [PubMed] [Google Scholar]
- 30.Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44:512–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao Q, Wang J, Hemani G, Bowden J, Small DS. Statistical inference in two-sample summary-data Mendelian randomization using robust adjusted profile score. ArXiv180109652 Math Stat [Internet]. 2019. [cited 2020 Dec 14];Available from: http://arxiv.org/abs/1801.09652 [Google Scholar]
- 32.Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, Laurin C, Burgess S, Bowden J, Langdon R, et al. The MR-Base platform supports systematic causal inference across the human phenome. eLife. 2018;7:e34408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luo C, Hajkova P, Ecker JR. Dynamic DNA methylation: In the right place at the right time. Science. 2018;361:1336–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berdyshev GD, Korotaev GK, Boiarskikh GV, Vaniushin BF. [Nucleotide composition of DNA and RNA from somatic tissues of humpback and its changes during spawning]. Biokhimiia Mosc Russ. 1967;32:988–993. [PubMed] [Google Scholar]
- 35.Wilson VL, Jones PA. DNA methylation decreases in aging but not in immortal cells. Science. 1983;220:1055–1057. [DOI] [PubMed] [Google Scholar]
- 36.Bjornsson HT, Sigurdsson MI, Fallin MD, Irizarry RA, Aspelund T, Cui H, Yu W, Rongione MA, Ekström TJ, Harris TB, et al. Intra-individual change over time in DNA methylation with familial clustering. JAMA. 2008;299:2877–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bell JT, Tsai P-C, Yang T-P, Pidsley R, Nisbet J, Glass D, Mangino M, Zhai G, Zhang F, Valdes A, et al. Epigenome-wide scans identify differentially methylated regions for age and age-related phenotypes in a healthy ageing population. PLoS Genet. 2012;8:e1002629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Portela A, Liz J, Nogales V, Setién F, Villanueva A, Esteller M. DNA methylation determines nucleosome occupancy in the 5′-CpG islands of tumor suppressor genes. Oncogene. 2013;32:5421–5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Collings CK, Anderson JN. Links between DNA methylation and nucleosome occupancy in the human genome. Epigenetics Chromatin. 2017;10:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lövkvist C, Sneppen K, Haerter JO. Exploring the Link between Nucleosome Occupancy and DNA Methylation. Front Genet. 2017;8:232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huxley RR, Lopez FL, Folsom AR, Agarwal SK, Loehr LR, Soliman EZ, Maclehose R, Konety S, Alonso A. Absolute and attributable risks of atrial fibrillation in relation to optimal and borderline risk factors: the Atherosclerosis Risk in Communities (ARIC) study. Circulation. 2011;123:1501–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Quach A, Levine ME, Tanaka T, Lu AT, Chen BH, Ferrucci L, Ritz B, Bandinelli S, Neuhouser ML, Beasley JM, et al. Epigenetic clock analysis of diet, exercise, education, and lifestyle factors. Aging. 2017;9:419–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fahy GM, Brooke RT, Watson JP, Good Z, Vasanawala SS, Maecker H, Leipold MD, Lin DTS, Kobor MS, Horvath S. Reversal of epigenetic aging and immunosenescent trends in humans. Aging Cell. 2019;18:e13028. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.