

SUMMARY
Chromosome loops shift dynamically during development, homeostasis, and disease. CTCF is known to anchor loops and construct 3D genomes, but how anchor sites are selected is not yet understood. Here we unveil Jpx RNA as a determinant of anchor selectivity. Jpx RNA targets thousands of genomic sites, preferentially binding promoters of active genes. Depleting Jpx RNA causes ectopic CTCF binding, massive shifts in chromosome looping, and downregulation of >700 Jpx target genes. Without Jpx, thousands of lost loops are replaced by de novo loops anchored by ectopic CTCF sites. Although Jpx controls CTCF binding on a genome-wide basis, it acts selectively at the subset of developmentally sensitive CTCF sites. Specifically, Jpx targets low-affinity CTCF motifs and displaces CTCF protein through competitive inhibition. We conclude that Jpx acts as a CTCF release factor and shapes the 3D genome by regulating anchor site usage.
Graphical Abstract
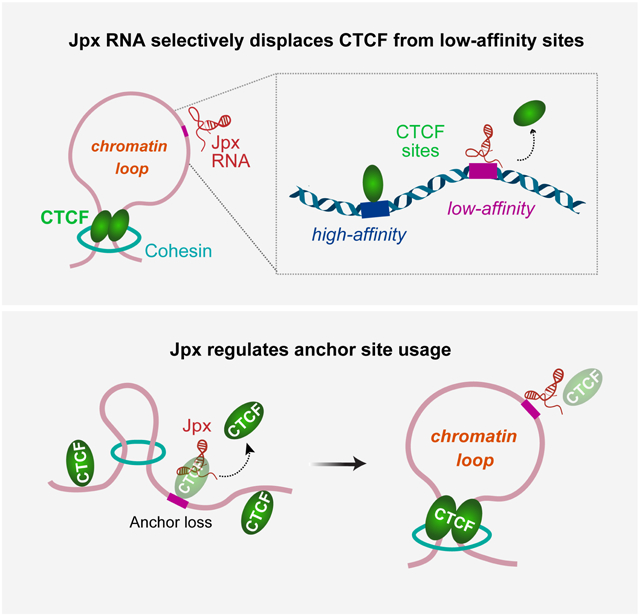
The Jpx noncoding RNA controls where CTCF binds across autosomes, determining the location of chromosome loops and genome architecture.
INTRODUCTION
The mammalian genome displays many levels of architectural organization, from chromatin loops between enhancers and promoters to higher-order structures such as “compartments” in which active chromatin self-associates and is partitioned away from inactive chromatin (Dekker and Mirny, 2016; Dixon et al., 2012; Lieberman-Aiden et al., 2009; Mirny et al., 2019; Rowley and Corces, 2018). These structures are highly dynamic, changing significantly during development, regulation of homeostasis, and response to physiological stress. The CCCTC-binding factor, CTCF, has long been known to construct 3D chromatin and direct long-range chromatin contacts by promoting specific loops and blocking ectopic enhancer-promoter contacts (Handoko et al., 2011; Kurukuti et al., 2006; Phillips-Cremins and Corces, 2013; Splinter et al., 2006). Although CTCF was originally described as a transcription factor (Klenova et al., 1993), many of its effects can be attributed to its architectural properties, enabling transcriptional repression in one context and gene activation in another, as exemplified by allele-specific regulation (Bell and Felsenfeld, 2000; Chao et al., 2002; Hark et al., 2000; Holmgren et al., 2001). More recently, CTCF has been implicated in formation of borders around topological domains (Dixon et al., 2012; Nora et al., 2012; Phillips-Cremins et al., 2013; Rao et al., 2014; Tang et al., 2015). In prevailing loop-extrusion models, cohesins form a “ring”, capture a chromatin loop, and extrude chromatin through the loop until they encounter a pair of convergent or tandem CTCF sites (Alipour and Marko, 2012; Davidson et al., 2019; Fudenberg et al., 2016; Kim et al., 2019; Sanborn et al., 2015). The binding of CTCF to key sites therefore underlies the 3D organization of the mammalian genome, but how specific “anchor sites” are selected for loop creation and how anchors shift to form new loops in a physiologically relevant manner are not fully understood.
Notably, although CTCF has been shown to favor binding to a 20-bp consensus motif (Bell and Felsenfeld, 2000; Kim et al., 2007), these sites are numerous in the mammalian genome. How then does CTCF determine its sites of occupancy? DNA methylation, post-translational modifications, and interactions with protein partners have all been proposed to play a role (Del Rosario et al., 2019; Donohoe et al., 2007; Klenova et al., 2001; MacPherson et al., 2009; Wang et al., 2012). Combinatorial usage of its 11-Zn fingers can also be a factor (Nakahashi et al., 2013; Ohlsson et al., 2001). Nevertheless, these features are insufficient to explain the full range of specificity that underlies differential binding. In recent years, CTCF has been shown to bind RNA, raising the possibility of co-regulation by RNA. In the first instance, Jpx RNA was shown to regulate initiation of X-chromosome inactivation (XCI) by titrating away CTCF from the Xist promoter (Sun et al., 2013). Thousands of mammalian transcripts have now been shown to bind CTCF, but only a few among them have been implicated in functions, such as chromosome pairing and counting (Kung et al., 2015; Saldana-Meyer et al., 2014; Sun et al., 2013; Xu et al., 2007; Yang et al., 2015). The idea of an RNA cofactor is especially attractive because RNAs can provide a missing link in the site-specific recruitment of regulatory proteins (Lee, 2012). Two recent studies reported that mutating a domain within CTCF with putative RNA-binding activity affects formation of topological domains (Hansen et al., 2019; Saldana-Meyer et al., 2019). However, it is unclear whether the observed effects were due to loss of RNA binding or due instead to secondary consequences of altering CTCF protein conformation.
Here we identify Jpx as a noncoding RNA that regulates CTCF’s architectural function on a genome-wide scale. Jpx is an X-linked transcript (Chow et al., 2003; Chureau et al., 2002; Johnston et al., 2002) known to be obligatory for Xist induction during X-chromosome inactivation (Carmona et al., 2018; Karner et al., 2019; Sun et al., 2013; Tian et al., 2010). Because of its well-characterized X-linked activities, Jpx’s function is currently thought to be restricted to X-inactivation (da Rocha and Heard, 2017; Disteche, 2016; Jegu et al., 2017; Lee, 2011; Starmer and Magnuson, 2009). On the other hand, Jpx RNA has also been shown to be diffusible, capable of migrating between X chromosomes and autosomes (Tian et al., 2010), suggesting the possibility of non-X-linked roles. Here, by mapping Jpx binding sites using epigenomic techniques, we discover that Jpx targets hundreds of autosomal genes and reveal a genome-wide role in the selection of CTCF anchor sites for chromosome looping.
RESULTS
Jpx RNA binds thousands of chromatin sites and associates with active genes
To map Jpx binding sites in the mouse genome, we performed CHART-seq (Capture Hybridization Analysis of RNA Targets), an epigenomic method that maps RNA-binding sites by pulling down chromatin with capture probes for an RNA of interest (Chu et al., 2017; Simon et al., 2013; Simon et al., 2011). We designed a cocktail of 22~25 nt biotinylated DNA probes that bind three proximal Jpx exons (i.e. Jpx CHART probes; Fig. S1A). In parallel, we performed two CHART controls to exclude artifacts — first, a Jpx “antisense (AS)” CHART (using reverse complement probes) to control for strand-specificity and rule out hybridization to genomic DNA; and second, a Jpx CHART elution without RNase H (“no-RNase H”) to control for non-specific, RNase H-independent elution. qPCR confirmed that Jpx CHART captured Jpx RNA specifically, whereas the reverse complement did not (Fig. S1B). We performed time-course analysis in female mouse embryonic stem (ES) cells at differentiation days (d), d0, d3 and d7 in 2–3 biological replicates of each timepoint. Approximately 60–90 million paired-end reads were obtained for each library and only reads uniquely mapping to the mouse genome were used (Fig. S1C). As expected, the Jpx locus showed a major peak correlating with its nascent transcription (Fig. 1A, left; Fig. S1D).
Figure 1. Jpx RNA binds thousands of genomic sites and targets active promoters.
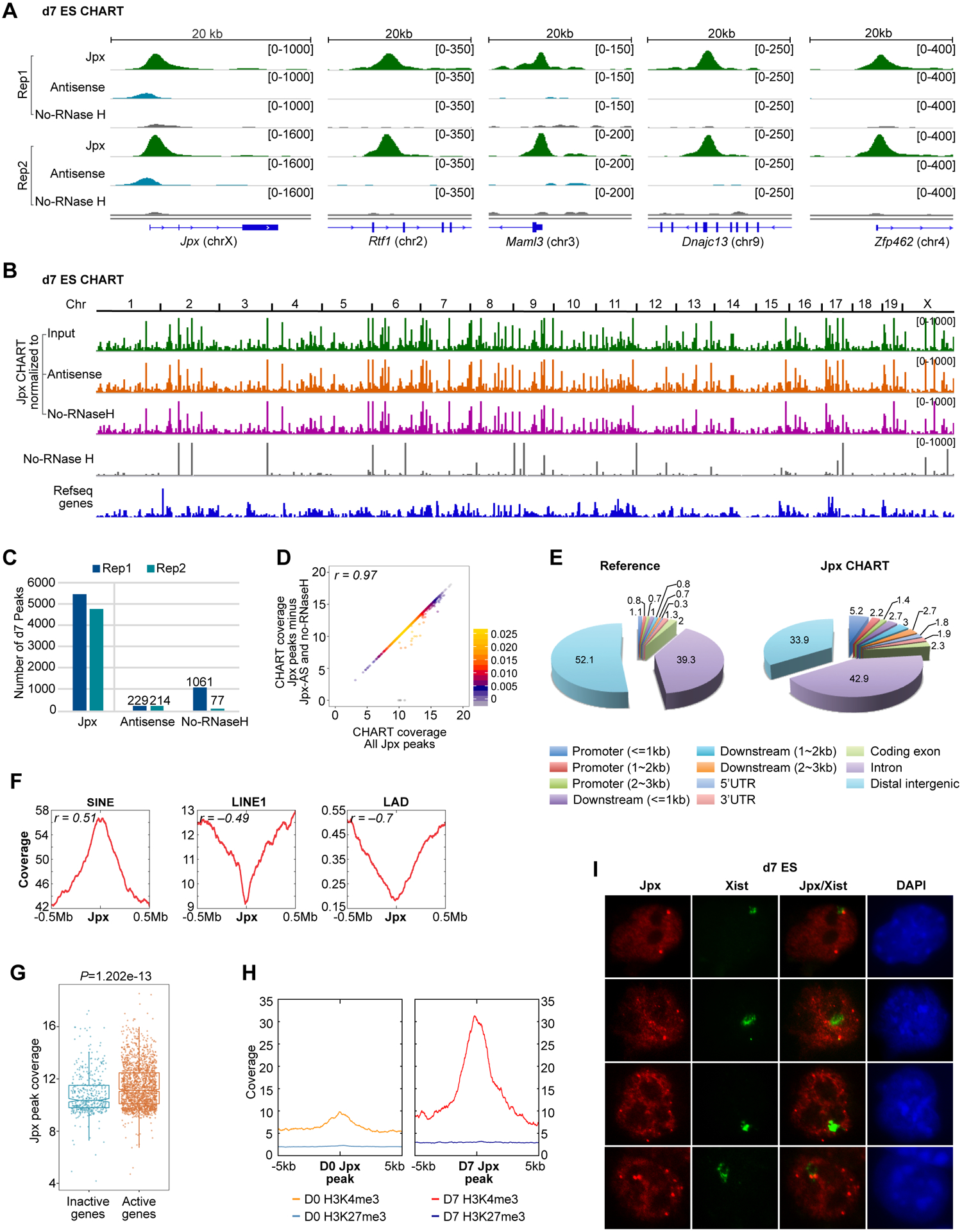
(A) Input-subtracted coverage for Jpx CHART in d7 ES cells. Two biological replicates shown (Rep1, Rep2). Negative controls: antisense (AS) and no-RNase H.
(B) Jpx CHART coverage for the whole genome, normalized to input, AS, or no-RNase H controls. Input-subtracted coverage for no-RNase H CHART also shown.
(C) Number of significant Jpx peaks in two d7 CHART replicates, after subtraction of AS and no-RNase H background peaks.
(D) 2D Kernel density scatterplot showing Jpx peak coverage (log2 scale) between unfiltered peaks and bona fide peaks described in (C). Bins, 10 kb. Pearson’s correlation coefficient (r) indicated.
(E) CEAS analysis: Pie charts show percentage of Jpx peaks with indicated genomic features compared to the reference genome.
(F) Plots representing SINE, LINE1, and LAD coverages within ±500 kb of Jpx peaks. Bins, 50 kb. Pearson’s correlation coefficient (r) indicated.
(G) Boxplot comparing log2 Jpx peak coverage between active (FPKM ≥ 0.5, n=12,907) or inactive (FPKM < 0.5, n=10,636) genes in d7 ES cells. P-value, Wilcoxon ranked sum test.
(H) Significant H3K4me3 enrichment around Jpx peaks in d7 ES cells compared to undifferentiated (d0) ES cells. No H3K27me3 enrichment was detected.
(I) Representative Xist and Jpx RNA-FISH of d7 ES cells. Nuclei were stained with DAPI (blue).
Intriguingly, the X-inactivation center was not the only site of enrichment (Fig. 1A,B; S1E,F). Additional CHART signals were observed across the entire genome, whether normalized to input, Jpx-AS, or no-RNaseH control (Fig. 1B, top 3 tracks; Fig. S1F). Thousands of statistically significant peaks for each sample were identified using MACS software in d7 ES cells, whereas few were seen in the Jpx-AS and no-RNaseH controls (Fig. 1C; Tables S1–S3). To obtain bona fide Jpx binding sites, we filtered away peaks that were present in Jpx-AS and no-RNase H CHART and observed ~5000 specific peaks (Fig. 1C,D). Whereas Jpx CHART replicates showed excellent correlation (Fig. S1G, top), there was poor correlation with control CHART samples (Fig. S1G, bottom). Thousands of Jpx binding sites were also called in two biological replicates (Rep) of d0 and d3 ES cells (Fig. S2A). Interestingly, whereas the correlation between biological replicates for all 3 timepoints was high, the correlation between timepoints was more modest (Fig. S1G, top vs. Fig.S2B), hinting at a dynamic regulation of Jpx localization during cell differentiation in light of a 10-fold increase in Jpx levels (Tian et al., 2010).
Jpx preferentially bound promoters, proximal regions of the transcription start site (TSS) or the transcription termination site (TTS), exons and introns, relative to their representation in the genome (Fig. 1E). There were a positive correlation with SINE-rich, gene-rich regions (r=0.51) and negative correlations with LINE1-rich, gene-poor regions (r=–0.49) and lamina-associated domains (r=–0.70)(Fig. 1F; S2C,D). Significantly, Jpx peak coverage was higher within actively transcribed genes (Fig. 1G). H3K4me3, a chromatin mark that typifies active promoters, was also strongly enriched over Jpx peaks, especially on d7 when Jpx RNA is upregulated. In contrast, no such relationship was observed for the repressive histone mark, H3K27me3 (Fig. 1H). To determine whether Jpx signals were visible outside of the X-inactivation center, we performed RNA fluorescence in situ hybridization (FISH) and observed multiple Jpx RNA clusters in a majority of d7 cells (54%, n=425; Fig. 1I). Additionally, a diffuse haze of Jpx signals was evident in nearly all d7 ES cells, which may account, at least in part, for the genome-wide CHART peaks. These signals were abolished by knockdown of Jpx (Fig. S2E), arguing for specificity of the Jpx signals. These data reveal that Jpx RNA binds to thousands of genomic sites and preferentially associate with active genes.
Acute depletion of Jpx dramatically downregulates >700 target genes
To investigate function, we acutely depleted Jpx in differentiating ES cells using locked nucleic acids (LNA) gapmers that degrade target transcripts using an RNase H-mediated mechanism. Two independent LNA gapmers (LNA#1, LNA#2), a scrambled (Scr) gapmer control, and two biological replicates of each were employed to enhance specificity and exclude off-target effects (Fig. S3A,B). Both Jpx LNAs and replicates yielded >95% depletion in differentiating ES cells after 8 hours (Fig. 2A). RNA-seq analysis identified 900 differentially expressed genes (DEGs; FDR < 0.05) between control and Jpx-depleted cells (Table S4). Jpx-depleted cells showed a strong overall downregulation (738 of 900 genes, 82%; Fig. 2B,C) and two independent LNAs showed similar trends for down- or up-regulation of genes (Fig. 2D; S3B). Downregulated DEGs were significantly associated with Jpx peaks (Fig. S3C,D). Upregulated DEGs did not show a significant enrichment relative to non-DEGs or randomized gene sets of 900 genes (Fig. S3C,D).
Figure 2. Acute Jpx depletion results in downregulation of >700 genes.
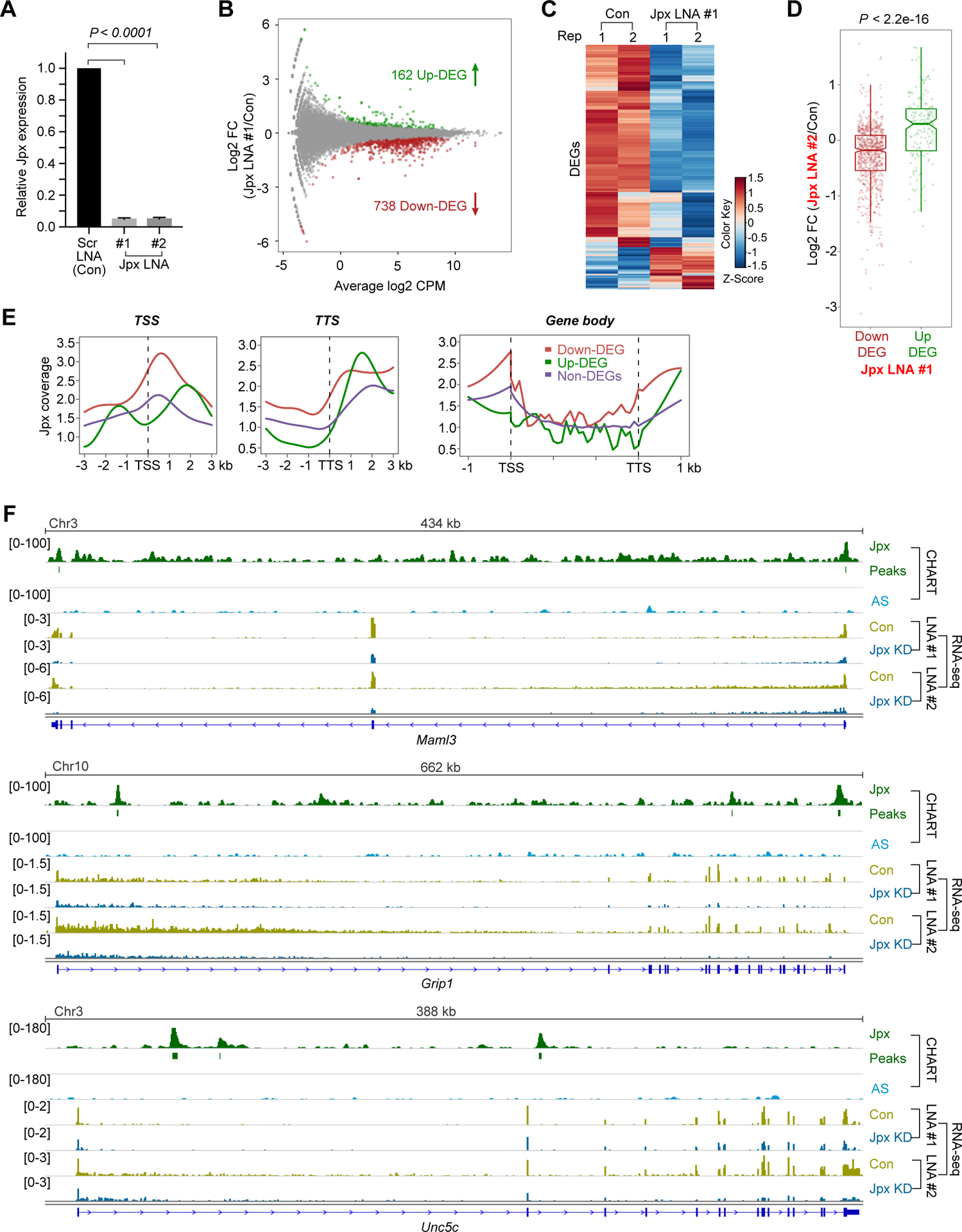
(A) Relative Jpx expression (normalized to Gapdh) in d7 ES cells transfected with scrambled (con) or two distinct Jpx LNA#1 or LNA#2 for 8 hr. Means ± SEM from three independent experiments shown. P-value, Student’s t-test.
(B) MA Bland–Altman plot showing log2 fold-change (FC) and average log2 counts per million (CPM) mapped reads for each gene after Jpx LNA#1 vs. scrambled (con) LNA treatment in d7 ES cells. Two biological replicates shown. Green, upregulated DEGs. Red, downregulated DEGs.
(C) Heatmap of normalized expression values (row-based Z-scores) for 900 DEGs in two biological replicates.
(D) Boxplot of transcriptomic changes between two Jpx LNAs (#1, #2). 738 downregulated DEGs and 162 upregulated DEGs from LNA#1 compared to Log2 FC in CPM for LNA#2.
(E) Metagene analysis of d7 Jpx coverage around TSS and TTS for downregulated DEGs, upregulated DEGs, and non-DEGs.
(F) Genome browser (IGV) views of d7 Jpx CHART-seq and RNA-seq data for representative DEGs following Jpx LNA treatment (#1, #2). Significant enrichment peaks as noted. Antisense CHART background serves as control.
Metagene analysis revealed that downregulated DEGs showed the greatest Jpx coverage at the TSS (Fig. 2E), though binding could also be seen cross the gene body and near the TTS. The localization patterns were exemplified by Maml3, Grip1, and Unc5c, which showed high expression when Jpx was bound and significant downregulation following Jpx depletion by both Jpx-specific LNAs (Fig. 2F). As the effects were evident after only 8 hours of LNA treatment, Jpx binding may be continually required to maintain gene activity. Gene Ontology (GO) analysis (Table S4) demonstrated that 349 DEGs — predominantly the downregulated genes — were related to general differentiation and development (Fig. S3E), with 161 DEGs specifically related to neural differentiation and development (Fig. S3F). These data argue that Jpx RNA localizes to and activates hundreds of target genes.
Jpx RNA antagonizes CTCF binding to target genes
De novo motif analysis of Jpx peaks identified 18 significant underlying motifs, several of which demonstrated similarity to motifs for known transcription factors, including PRDM1, FOXB1, ZNF263, JUN, POU3F2, CTCF and CTCFL (Fig. S4). Among them, CTCF stood out, as this motif bore a striking resemblance to CTCF’s established motif (Fig. 3A) and Jpx was previously implicated in evicting CTCF from the Xist promoter (Sun et al., 2013). We then asked if Jpx binding affected CTCF localization on a genome-wide scale by performing CTCF ChIP-seq in d7 ES cells depleted of Jpx RNA (LNA#1). As expected, Jpx depletion resulted in increased CTCF binding and downregulation of Xist (Fig. S5A), consistent with published data (Sun et al., 2013). Intriguingly, autosomal targets — specifically the downregulated DEGs — also demonstrated significantly increased CTCF binding after Jpx depletion (Fig. 3B), as indicated by the right shift in cumulative distribution plots (CDPs)(Fig. 3C, left panel). The gain in CTCF peak coverage was large when compared to unaffected genes (non-DEG; Fig. 3D). By contrast, upregulated DEGs did not change in CTCF binding (Fig. 3C, right panel). These observations hinted that CTCF binding may be antagonized by Jpx.
Figure 3. Jpx loss causes a massive global displacement of CTCF.
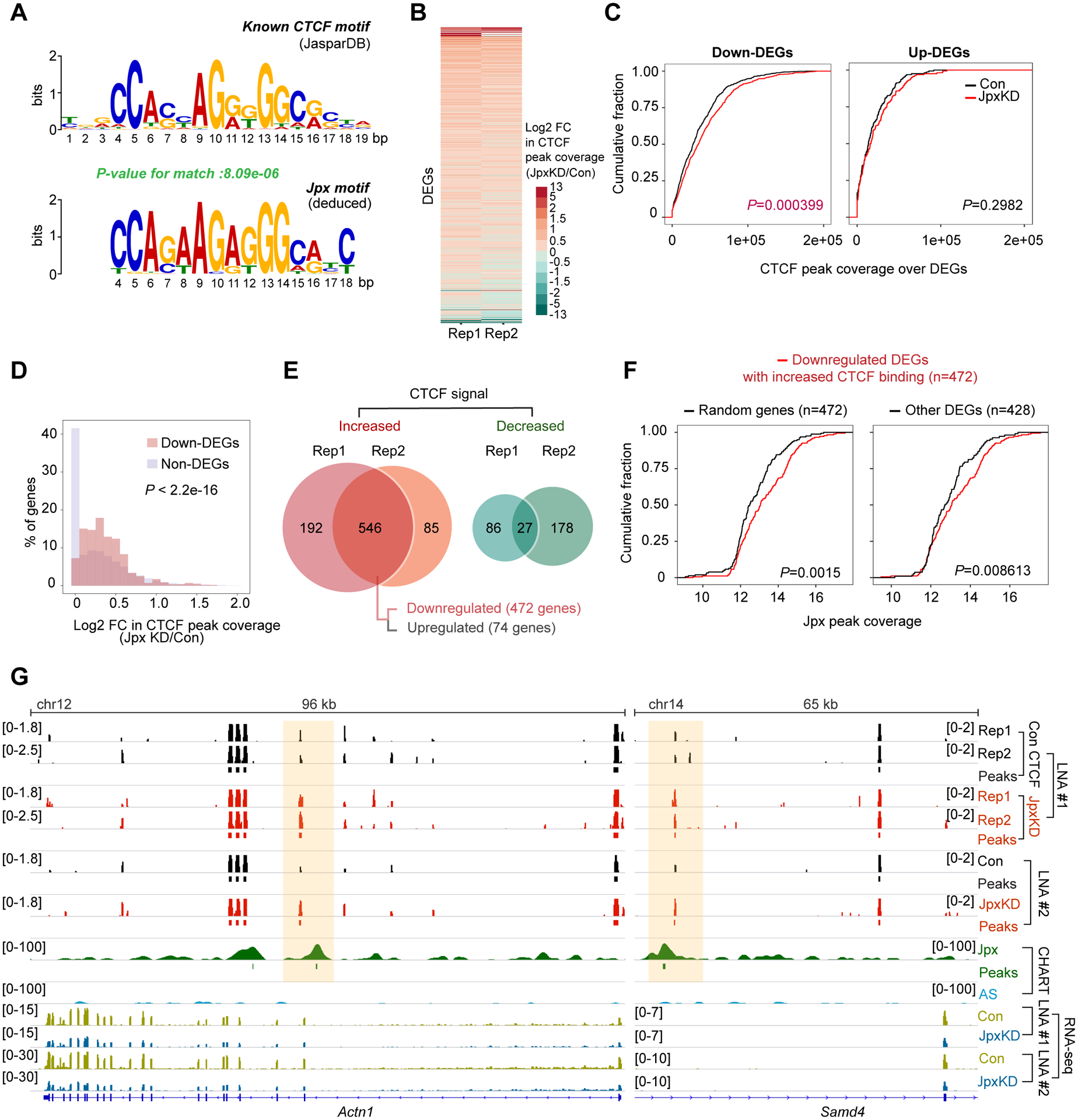
(A) Logograms reveal sequence similarities between CTCF motif (JASPAR database) and Jpx motif.
(B) Heatmap depicting log2 FC in CTCF peak coverage for DEGs between control and Jpx-depleted d7 ES cells in two biological replicates.
(C) Cumulative distribution plots (CDP) comparing CTCF peak coverage between control and Jpx-depleted cells for downregulated (left) or upregulated (right) DEGs. P determined by Kolmogorov-Smirnov (KS) test.
(D) Histogram showing the percentage of down-DEGs vs. non-DEGs with the indicated log2 FC in CTCF peak coverage. P-value, KS test.
(E) Venn diagrams showing the overlap of DEGs with increased or decreased CTCF peak coverage across two biological replicate experiments. DEGs exhibiting the reproducible increase in CTCF peak coverage were further categorized into downregulated (n=472) or upregulated (n=74) genes.
(F) CDP of log2 Jpx peak coverage for downregulated 472 DEGs with increased CTCF binding compared to 472 genes randomly selected from non-DEGs (left) or remaining 428 DEGs (right). Randomizing gene subset was generated by random selection from non-DEG (n=22,824). P-values, KS test.
(G) Genome browser views of d7 Jpx CHART-seq, CTCF ChIP-seq and RNA-seq data for the indicated down-DEGs. See also Fig. S5C. CTCF coverage, log2 fold-enrichment estimates relative to input. Significant CTCF and Jpx peaks shown as bars. Two biological replicates and two distinct Jpx LNAs shown.
Indeed, analysis of two biological replicates indicated that 546 DEGs showed increased CTCF peak coverage when Jpx was depleted (Fig. 3E). In wildtype cells, the 546 DEGs had greater Jpx binding relative to a randomized gene set (Fig. S5B). Among the 546 DEGs with increased CTCF coverage, 472 were downregulated after Jpx depletion (Fig. 3E). The 472 genes also had high initial Jpx peak coverage, as compared to a randomized gene set and to other DEGs (Fig. 3F). For instance, Jpx targets, Actn1, Samd4, and Tenm4, showed significant increases in CTCF binding in multiple biological replicates and regardless of whether LNA#1 or LNA#2 was used to deplete Jpx (Fig. 3G, S5C). Interestingly, the change in CTCF binding was not binary (all-or-none binding). Rather, Jpx modulates the degree of binding. We conclude that Jpx antagonizes and fine-tunes CTCF binding at target sites across the genome.
Jpx regulates low-occupancy, developmentally sensitive CTCF sites
Among tens of thousands CTCF sites in the mammalian genome, which are regulated by Jpx? Unperturbed d7 ES cells demonstrated 60,944 CTCF binding sites (Fig. 4A). When Jpx was depleted, the vast majority (58,325 sites) remained unchanged. However, 8,595 new CTCF peaks were gained and 2,619 were lost (Fig. 4A; Table S5). What distinguished the 8,595 from other CTCF sites? Notably, two types of CTCF binding motifs had been identified previously (Plasschaert et al., 2014)— (i) Low Occupancy (LowOc) sites that have lower affinity for CTCF and are more likely to be developmentally regulated; versus (ii) High Occupancy (HighOc) sites that have higher affinity for CTCF and bind CTCF constitutively (Fig. 4B). HighOc sites have greater similarity to the CTCF consensus sequence. In particularly, the 7th, 9th and 18th nucleotides (asterisks) were shown to be major determinants of LowOc versus HighOc binding, with the presence of C or G at the 18th position being especially critical for stable binding.
Figure 4. Jpx selectively controls a subset of developmentally sensitive CTCF motifs.
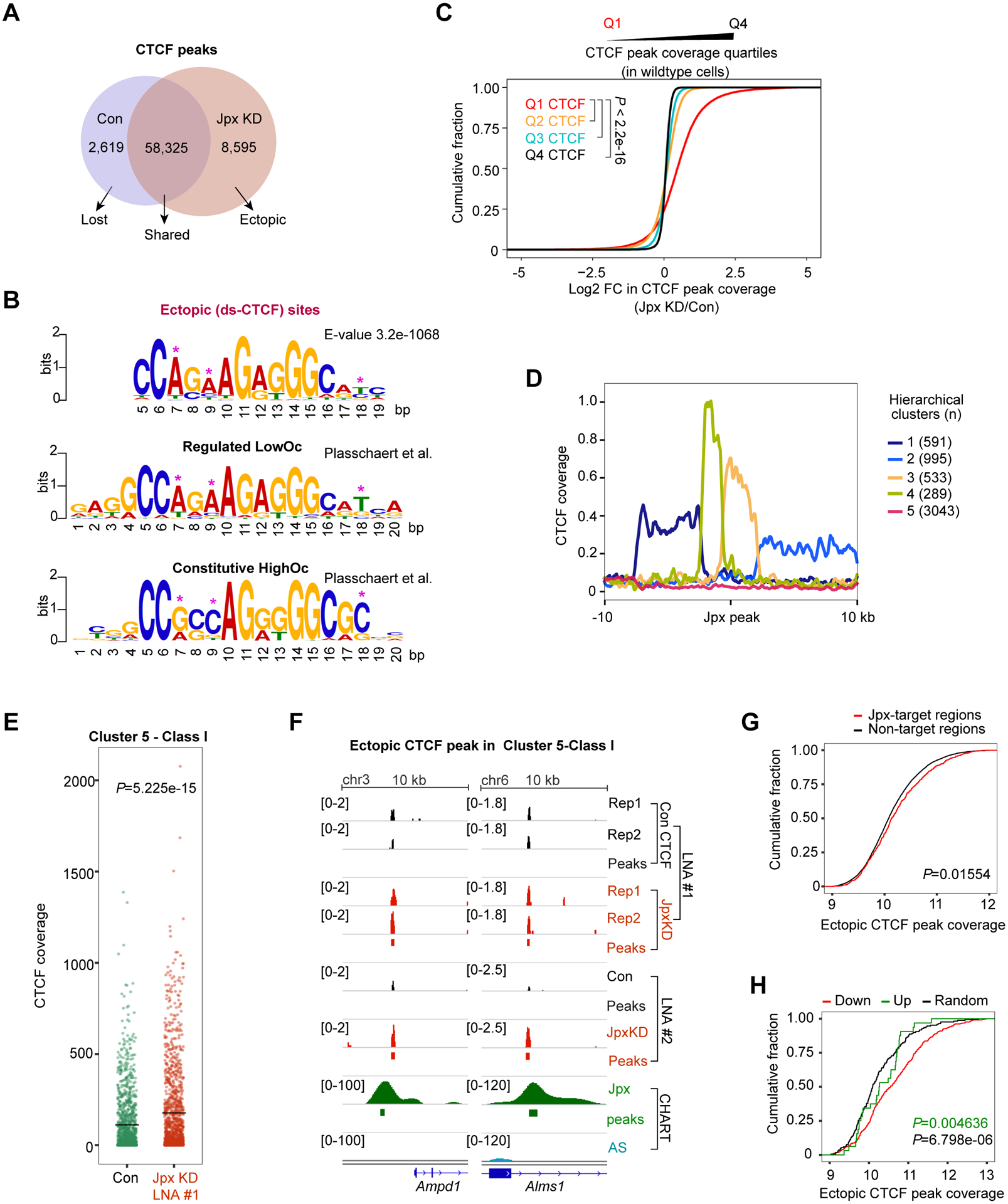
(A) Venn diagram representing the number of overlapping or exclusive CTCF sites in control and Jpx-depleted d7 ES cells. CTCF peaks common to two biological replicates were used.
(B) Logograms of sequence motifs for 8,595 ectopic CTCF peaks described in (A). LowOc and HighOc sites as defined by Plasschaert et al.
(C) CDP comparing log2 FC in CTCF peak coverage over Q1, Q2, Q3 and Q4 CTCF quartiles. CTCF sites were divided into quartiles based on peak coverage. P-values, the KS test.
(D) Five hierarchical clusters defined by CTCF’s relationship to Jpx peaks in d7 ES cells. See also Fig. S6A.
(E) CTCF coverage values calculated over Cluster 5-Class I genes in control vs. Jpx LNA#1 cells. See also Fig. S6E. Black crossbar, mean. P-value, Wilcoxon ranked sum test.
(F) Cluster 5-Class I: Increased CTCF coverages following Jpx depletion.
(G) CDP comparing log2 coverage of ectopic CTCF peaks between Jpx-targets and non-targets (20 kb bins). P-value, KS test.
(H) CDP showing log2 coverage of ectopic CTCF peaks for down-DEGs (n=738), up-DEGs (n=162) and randomly selected non-DEGs (n=900). P-values, KS test: Green, down vs. up. Black, down vs. random.
Here we derived a consensus motif for the subset of 8,595 sites that gained CTCF binding. Strikingly, these sites exhibited a high similarity to the LowOc sites, especially at the 7th, 9th, and 18th nucleotide positions (Fig. 4B). We divided CTCF peak coverages (in wildtype cells) into four quartiles (Q1-Q4) and asked how depleting Jpx changed CTCF profiles in each quartile (Fig. 4C). Significantly, the lowest CTCF quartile (Q1) displayed the greatest increase in CTCF binding following Jpx depletion. Thus, Jpx controls developmentally sensitive CTCF (ds-CTCF) binding with lowest in vivo affinity.
To understand CTCF binding profiles around Jpx sites, we performed hierarchical clustering analysis of CTCF coverage over ±10 kb of Jpx peak centers (Fig. 4D, S6A). Five clusters (subtypes) of Jpx sites were revealed. Cluster 1 (n=591 sites) and Cluster 2 (n=995) exhibited broad CTCF enrichment > ~2 kb upstream and downstream of Jpx peak centers, respectively, whereas Cluster 3 (n=533) localized over the Jpx peak center and Cluster 4 (n=289) localized just upstream. Clusters 1–4 accounted for ~44% of Jpx sites (n=2,408). Cluster 5 accounted for the remaining 3,043 Jpx sites (~56%). Within Cluster 5, Class I (n=1,838; Fig. S6B) had no CTCF peaks in wildtype cells but gained CTCF binding after Jpx depletion (Fig. 4E,F). Class II (n=1,205; Fig. S6B) showed low-level binding with peaks called in wildtype cells and increased in CTCF binding after Jpx depletion (Fig. S6C,D). These results were consistent between replicates and for LNA#1 versus LNA#2. For the 8,595 ectopic CTCF peaks (Fig. 4A), CDP analysis showed a right shift (increase) in CTCF binding over Jpx-target sites (Fig. 4G). Conversely, for the 2,619 lost CTCF peaks (Fig. 4A), CDP analysis showed a left shift (decrease) in CTCF binding (Fig. S6F). Furthermore, metagene profiles affirmed that CTCF gains were greatest over the initially highest Jpx quartile (Q4)(Fig. S6G). Lastly, the subgroup of downregulated DEGs showed the most dramatic increase in CTCF peak coverage (Fig. 4H). Altogether, these data argue that Jpx RNA regulates the developmental binding of CTCF and specifically antagonizes CTCF binding at low-affinity CTCF sites.
Jpx RNA is a CTCF release factor
To understand mechanism, we performed electrophoretic mobility shift assay (EMSA) with purified CTCF protein and asked whether Jpx differentially affects CTCF binding at LowOc versus HighOc sites in vitro. The HighOc site from Mettl21a retained strong CTCF binding in Jpx-depleted cells, whereas the LowOc site from Cald1 only gained a CTCF site after Jpx depletion (Fig. 5A). Mettl21a and Cald1 sites differed at the 7th, 9th, and 18th positions of the CTCF consensus (pink shading). Notably, while Jpx RNA-CTCF interactions were of extremely high affinity with a dissociation constant (Kd) of <1.0 nM (Kung et al., 2015; Sun et al., 2013), the HighOc (Mettl21a) DNA site showed a relatively low affinity of 18.5 nM and the LowOc (Cald1) showed an even lower affinity of 29.9 nM (Fig. 5B–D). Additional HighOc sites (Narfl and Kcna7) and LowOc sites (E330021D16Rik and Arhgap1) revealed similar differences in Kd between HighOc and LowOc probes (Fig. 5A,E,F). Indeed, the average affinity of HighOc probes (Mettl21a, Narfl, Kcna7) significantly exceeded that of LowOc probes (Cald1, E330021D16Rik, Arhgap1)(Fig.5G). Thus, coverages observed by ChIP-seq analysis (Fig. 5A) reflect biochemical affinities of CTCF for underlying motifs.
Figure 5. Jpx RNA is a CTCF release factor.
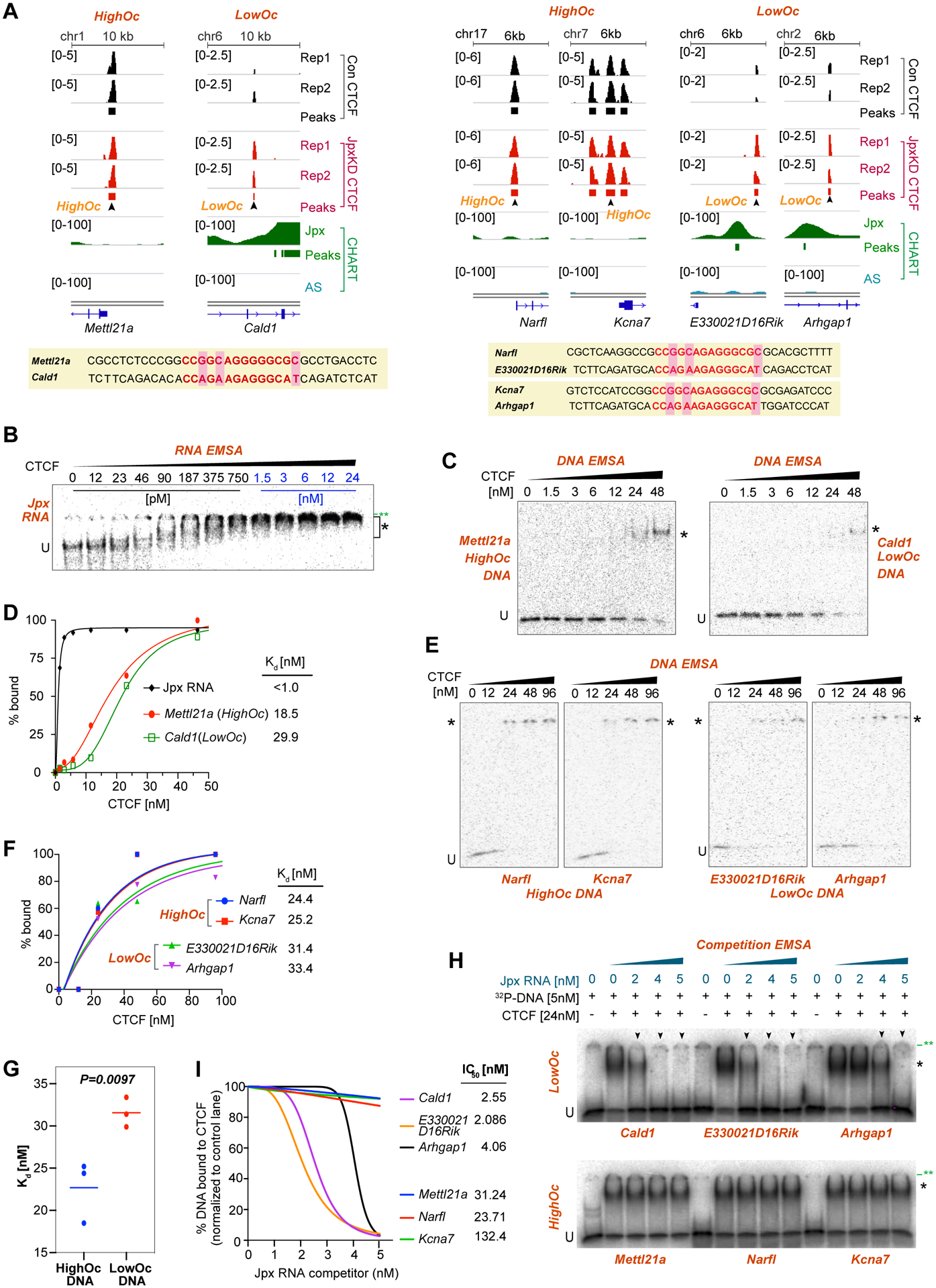
(A) Representative HighOc and LowOc CTCF sites used in EMSA experiments. d7 Jpx CHART-seq and CTCF ChIP-seq data shown. For Kcna7 DNA probe, middle site (marked by arrowhead) chosen. Bottom: Probe sequences. Red bases highlight CTCF core binding motifs. Pink shading marks critical bases.
(B) RNA EMSA with 5pM Jpx RNA (E1-E3, 383nt) with increasing CTCF amounts as indicated. U, unbound probe. *, CTCF-probe shift. **, well position.
(C) DNA EMSA using 5 pM Mettl21a or Cald1 DNA probe with increasing amounts of CTCF.
(D) Relative Kd values for indicated probes, as determined by EMSAs in (B) and (C).
(E) DNA EMSA with 5 pM of HighOc (Narfl and Kcna7) or LowOc (E330021D16Rik and Arhgap1) DNA probes with increasing amounts of CTCF.
(F) Relative Kd values for the indicated probes, as determined by EMSAs shown in (E).
(G) Differential affinities (Kd) of HighOc vs. LowOc DNA probes for CTCF. Crossbar, mean. P-value, Student’s t test.
(H) Competition EMSA. 32P-labeled LowOc or HighOc DNA probes were mixed with increasing amounts of cold Jpx RNA (E1-E3, 383 nt) competitor. U, unbound probe., *, CTCF-probe shift. **, well also indicated. Arrowheads, Jpx-mediated competition.
(I) IC50 for Jpx RNA inhibiting CTCF binding to LowOc and HighOc sites. %DNA bound was determined from EMSA in (H).
Notably, none of the DNA affinities approached Jpx RNA’s superior affinity (Fig. 5D,F). Whereas CTCF did not appreciably shift either Mettl21a or Cald1 until the protein concentration exceeded 24 nM, CTCF shifted Jpx RNA in concentrations as low as 23 pM. With Kd differences of 1–2 log10, we hypothesized that Jpx might antagonize CTCF binding at LowOc sites through competitive inhibition. To test this hypothesis, we performed competition experiments by mixing DNA probe and RNA competitor together prior to adding CTCF. HighOc DNA sites robustly bound CTCF in the absence of Jpx RNA, and addition of 2–5 nM Jpx RNA (0.4–1.0x molar excess) minimally competed away CTCF binding (Fig. 5H bottom). By contrast, although LowOc DNA sites bound CTCF well in the absence of Jpx, addition of sub-stoichiometric (2–4 nM) Jpx concentrations partially titrated away the binding (Fig. 5H top). At 5 nM of Jpx, no DNA binding was evident for LowOc sites. To quantitate differences, we calculated an IC50, the concentration of Jpx at which 50% of CTCF-DNA binding is inhibited. Whereas 23–132 nM of Jpx RNA was required to inhibit CTCF binding to HighOc sites, only 2–4 nM was necessary for LowOc sites (Fig. 5I). We conclude that Jpx RNA is a CTCF release factor — specifically at low-affinity CTCF sites (Fig. 6A). HighOc sites are immune to Jpx titration due to their higher affinity for CTCF.
Figure 6. Jpx controls chromosomal looping on a genome-wide scale.
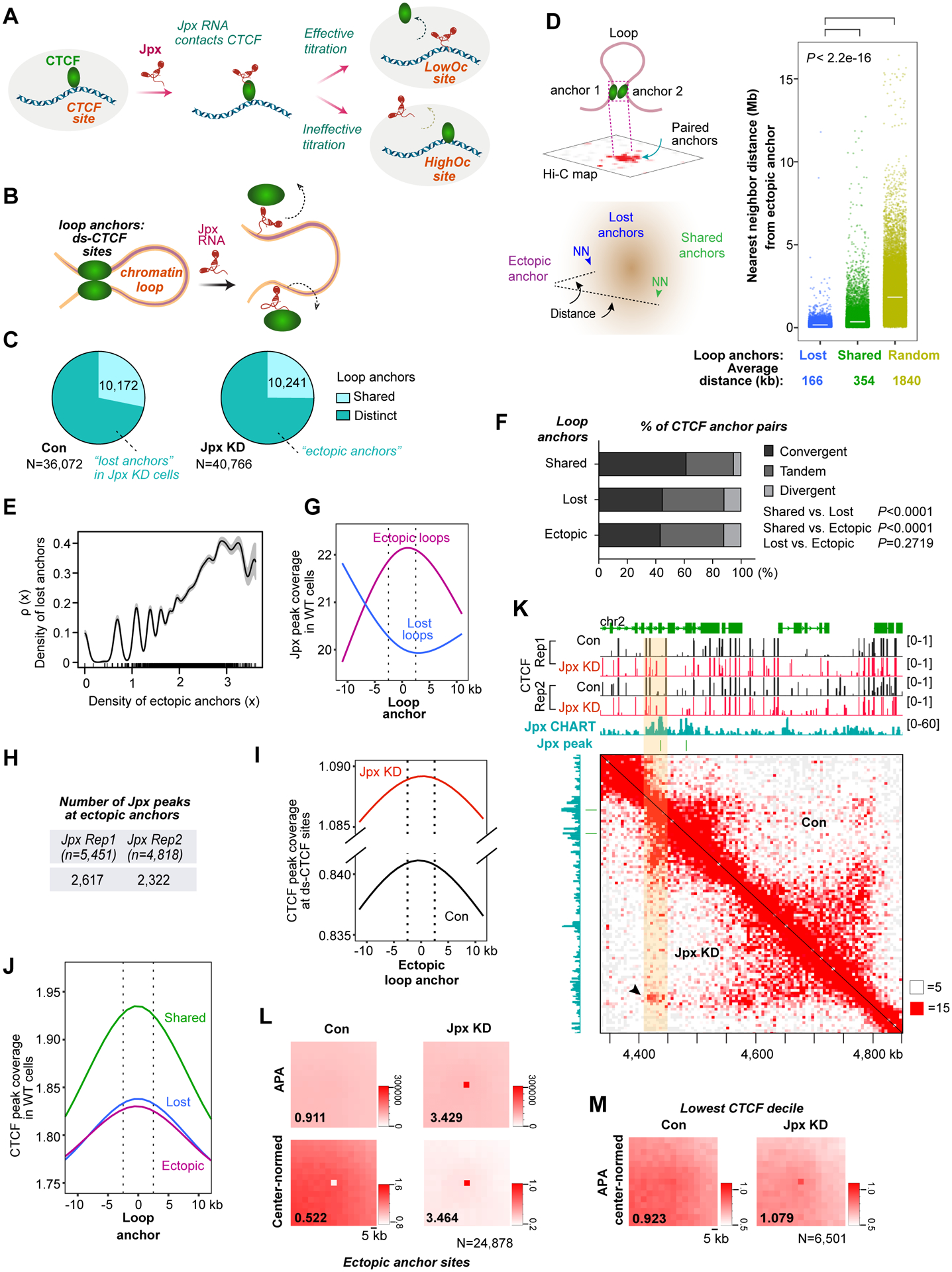
(A) Model: Jpx RNA operates as a CTCF release factor for ds-CTCF sites. Jpx transiently contacts CTCF on chromatin and evicts CTCF from LowOc sites through competitive inhibition. HighOc sites are resistant.
(B) Hypothesis: Jpx controls chromosome looping by controlling CTCF anchor site selection.
(C) Venn diagram showing the number of loop anchors in control and Jpx-depleted d7 ES cells at 5 kb resolution. N, total number of loop anchors. Anchors are considered ‘shared’ between control and Jpx-depleted cells if anchors occur within a 40 kb window.
(D) Left: Nearest neighbor (NN) analysis to determine distance between an ectopic anchor and the closest lost or shared anchor. Right: Dot plot of measured distances for each category as shown. Crossbar, mean. P-values, Wilcoxon ranked sum test.
(E) Covariation of density of ectopic loop anchors (x) with density of lost loop anchors ρ(x). Grey-shaded region indicates 95% confidence interval.
(F) Percentage of CTCF anchor pairs in convergent, tandem, and divergent orientations in shared, lost, or ectopic categories. P-values, Chi-square test.
(G) Plot comparing Jpx peak coverage in d7 ES cells over anchors associated with ectopic vs. lost loops. Vertical dash lines, 5 kb anchor region.
(H) Number of Jpx binding sites (Jpx peaks ±10 kb in rep1, 2) overlapping ectopic anchors. N = total number of Jpx peaks.
(I) Plot comparing CTCF peak coverage at ds-CTCF sites over ectopic anchors in control vs. Jpx-depleted cells. Vertical dash lines, 5 kb anchor region.
(J) Plot showing CTCF peak coverage normally found in WT cells over ectopic, lost, and shared anchors. Vertical dash lines, 5 kb anchor region.
(K) Hi-C contact matrix at 5 kb resolution showing a significant de novo interaction (arrow) at the region bound by Jpx and increased CTCF (shaded) in Jpx-depleted cells. White and Red squares, minimum and maximum intensity, respectively.
(L) Top: APA showing aggregate strength of looping interactions between paired CTCF sites. Bottom: Center-normed APA. P2LL ratio shown. N, number of the ectopic anchor sites after filtering for distance threshold relative to diagonal.
(M) Center-normed APA showing the normalized-aggregate strength of paired anchor interactions at ds-CTCF anchor sites of the lowest CTCF decile. P2LL ratio and N (6,501 loop anchors) shown.
Jpx controls chromosome looping by shifting anchor sites
We hypothesized that Jpx could promote gene activation by releasing CTCF and thereby reorganizing chromosome loops (Fig. 6B). To obtain high-resolution contact maps, we performed in situ Hi-C in control and Jpx-depleted d7 ES cells, with two biological replicates yielding ~1 billion read pairs and ~900 M of valid contacts for each sample (Table S6). We called loops using HiCCUPS (Rao et al., 2014) at 5 kb resolution and identified 36,072 loop anchors from control cells (Fig. 6C). When Jpx was depleted, total loop anchors increased 13% to 40,766. The apparently small increase belied major shifts in looping patterns. Indeed, only a quarter of total loop anchors was actually shared between control and Jpx-depleted cells (Fig. 6C). The vast majority (72–75%) was distinct. A large number of anchors was lost in Jpx-depleted cells (“lost anchors”). Simultaneously, a large number (30,525) of anchors was gained (“ectopic anchors”). The characteristic loss and gain of loops could be visualized in Hi-C contact heat maps (Fig. S7A). Similar results were obtained when loops were called at 20 kb resolution, which yielded 22,921 and 23,722 loop anchors from control and Jpx-depleted cells, respectively — among which were 16,972 lost and 17,751 ectopic anchors. Therefore, consistent with conclusions made at 5 kb resolution, the vast majority (74–75%) of loop anchors were distinct (ectopic or lost). Henceforth, all analyses are described at 5 kb resolution. These data demonstrate that Jpx RNA dramatically shifts anchor site usage.
In keeping with loop-extrusion models, we surmised that loss of a CTCF anchor would force cohesin to progress to a neighboring CTCF site. Conversely, gaining a CTCF site within the original loop would “short-circuit” loop extrusion and yield a smaller loop. In either scenario, we would expect lost anchors to reside near new anchors. To test this idea, we performed a “nearest neighbor” analysis to quantify the physical distance from an ectopic anchor to the nearest lost anchor (Fig. 6D). On average, the nearest-neighbor (NN) distance between them was 166 kb. Significantly, this distance was much shorter than the average distance (354 kb) to the nearest shared anchor site — i.e., sites that were not affected by Jpx depletion (Fig. 6D). This distance was also significantly shorter than the average distance (1,840 kb) for a randomized model in which loop anchor coordinates were scrambled by randomly shuffling paired anchor coordinates. These findings demonstrate a tight linkage between where anchors are lost and where new ones appear to create a loop. Furthermore, the density of gained loop anchors (x) covaried with the density of lost loop anchors [ρ(x)](Fig. 6E), indicating a dependence between the new, tightly linked loop anchor and the original. Thus, Jpx depletion causes a shift in anchor site usage to the nearest neighbor.
Interestingly, among shared CTCF anchor pairs, a majority (61.5%) was in the convergent orientation (Rao et al., 2014; Tang et al., 2015), while 33.3% was in the tandem orientation (Fig. 6F), an orientation associated with more dynamic regulation (Tang et al., 2015). Among ectopic CTCF anchor pairs, tandem (44.6%) and convergent pairs (43.3%) became nearly equal. Among lost pairs, tandem (43.3%) and convergent (44.8%) were similarly equalized. By contrast, divergently oriented pairs did not change in usage. These data support the idea that tandem loops are more dynamically and locally regulated (Tang et al., 2015).
About half (4,187 of 8,595) of ds-CTCF peaks that appeared in Jpx-depleted cells became ectopic loop anchors. We therefore predicted that anchor site usage would shift to CTCF sites previously occupied by Jpx. To test this, we quantified Jpx coverage (in wildtype cells) over gained versus lost anchor sites. Whereas Jpx was not enriched at lost anchors, ectopic anchors occurred where there was once high Jpx levels (Fig. 6G), indicating that Jpx depletion enabled a new anchor to form. Consistent with this, ~48 % of Jpx peaks called in CHART (2,617 of 5,451 for Rep1; 2,322 of 4,818 for Rep2) overlapped with new anchors in Jpx-depleted cells (Fig. 6H). Thus, de novo anchors formed at almost half of sites where Jpx formerly bound. These new anchors were associated with significantly increased CTCF peak coverage at ds-CTCF sites (Fig. 6I). The findings were again similar when analyzed at 20 kb resolution: Jpx was enriched at ectopic anchor sites identified at 20 kb resolution (Fig. 6G versus S7B) and CTCF peak coverages increased at ds-CTCF sites after Jpx knockdown (Fig. 6I versus S7C). We conclude that Jpx loss drives a massive shift in anchor site usage, resulting in the formation of ectopic loops associated with ds-CTCF sites.
A key corollary of our hypothesis is that shared anchors would be high-affinity CTCF sites that are unaffected by Jpx. Approximately 25% of CTCF sites did not change following Jpx depletion (Fig. 6C). To determine whether these are HighOc sites, we examined CTCF coverage at shared anchors versus lost and gained anchors. Indeed, shared loops showed the highest CTCF coverage (Fig. 6J) and the greatest enrichment of HighOc motifs relative to lost or ectopic loops (Fig. S7D). Jpx therefore indeed had little effect on anchor usage of high-affinity CTCF sites.
We next examined Jpx’s effect in the context of the paired loop anchors by measuring the strength of the CTCF pair. In a Hi-C matrix, paired loop anchors appear as “dots” representing juxtaposition of distant CTCF pairs and creation of looping of intervening chromatin (Fig. 6D diagram). On chr2, for example, Jpx depletion resulted in gain of CTCF binding and creation of an ectopic loop (Fig. 6K, yellow highlight). We then performed meta-loop analysis using Aggregation Peak Analysis (APA)(Durand et al., 2016b; Rao et al., 2014) to quantitate the aggregate strength of all paired loop anchors (dots) across the genome. APA and center-normed APA affirmed that aggregate strength of looping interactions was significantly enhanced over 24, 878 anchor sites in Jpx-depleted cells relative to controls (P2LL, 3.429 vs. 0.911) (Fig. 6L). When APA analysis was restricted to loop anchors with lowest occupancy ds-CTCF sites, aggregate peak strength became significant after Jpx deficiency (Fig. 6M). Thus, loss of Jpx strengthens ectopic looping interactions across the genome.
For a specific example, we turned to the Ftx-Xist domain involved in X-chromosome inactivation, where multiple CTCF sites occur in the Xist promoter region (P2, Xist5’) and Ftx (Fig. S7E) and robust contact could be seen between Ftx and the Xist promoter (Fig. S7E,F)(van Bemmel et al., 2019). It is known that Jpx evicts CTCF binding at the Xist P2 promoter to induce Xist expression (Fig. S5A)(Sun et al., 2013). Here we confirmed CTCF binding at Xist P2 using P2-mutated cells (Fig. S7G). Using the 3C assay, we observed increased P2-Ftx interactions when Jpx was depleted, and this dependence on Jpx was lost when P2 was mutated (Fig. S7H). There was a concurrent decrease in Xist5’-Ftx interactions when looping shifted to Xist P2, and this shift was also dependent on the P2 motif (Fig. S7I). The shift was not evident in the Hi-C matrix (Fig. S7F), as the Xist P2 and 5’ sites are too close to each other (~3kb), and was only observed by using higher resolution of 3C assay (Fig. S7H,I). These specific examples further support the notion that Jpx regulates loop formation by selecting anchor site usage.
Given the proposed requirement for cohesin to stabilize CTCF anchors (Li et al., 2020; Pugacheva et al., 2020), we asked if Jpx depletion affected localization of the cohesin subunit, RAD21. Two ChIP-seq biological replicates showed a total of 51,481 RAD21-binding sites in wildtype d7 ES cells. While the vast majority (44,871) was not affected by Jpx depletion, 6,216 sites were gained and 6,610 sites were lost (Fig. S7J) — reminiscent of CTCF dynamics following Jpx depletion (Fig. 4). In control cells, 40,512 RAD21 peaks (78.7%) overlapped CTCF peaks (Fig. S7K). Reciprocally, 41,048 CTCF sites (67.4%) colocalized with RAD21 (Fig. S7L). Although Jpx depletion did not dramatically shift colocalization percentages, the bulk analysis again belied underlying patterns. When RAD21 colocalization was examined among various CTCF quartiles in wildtype cells, we learned that RAD21 occupancy was highly correlated with CTCF peak coverage, with the highest CTCF quartile showing >98% colocalization with RAD21 and the lowest quartile only 29% (Fig. 7A, S7M). Because low-affinity CTCF sites are more dynamic, cohesin may have reduced residence times at these sites. Indeed, RAD21 peak coverage was more dynamic in the lower CTCF quartiles (Fig. 7B, S7N). When Jpx was depleted and CTCF bound ds-CTCF sites, a significant increase in RAD21 peak coverage at ds-CTCF sites was observed (Fig. 7C,D) — suggesting that ectopic CTCF sites became stabilized by RAD21. Thus, a majority of CTCF and cohesin sites (corresponding to high-affinity sites) are unaffected by Jpx, but Jpx loss results in stabilization of cohesin binding to ectopic loops gained at low-affinity CTCF sites.
Figure 7. Jpx controls looping and gene expression by shifting anchor site usage.
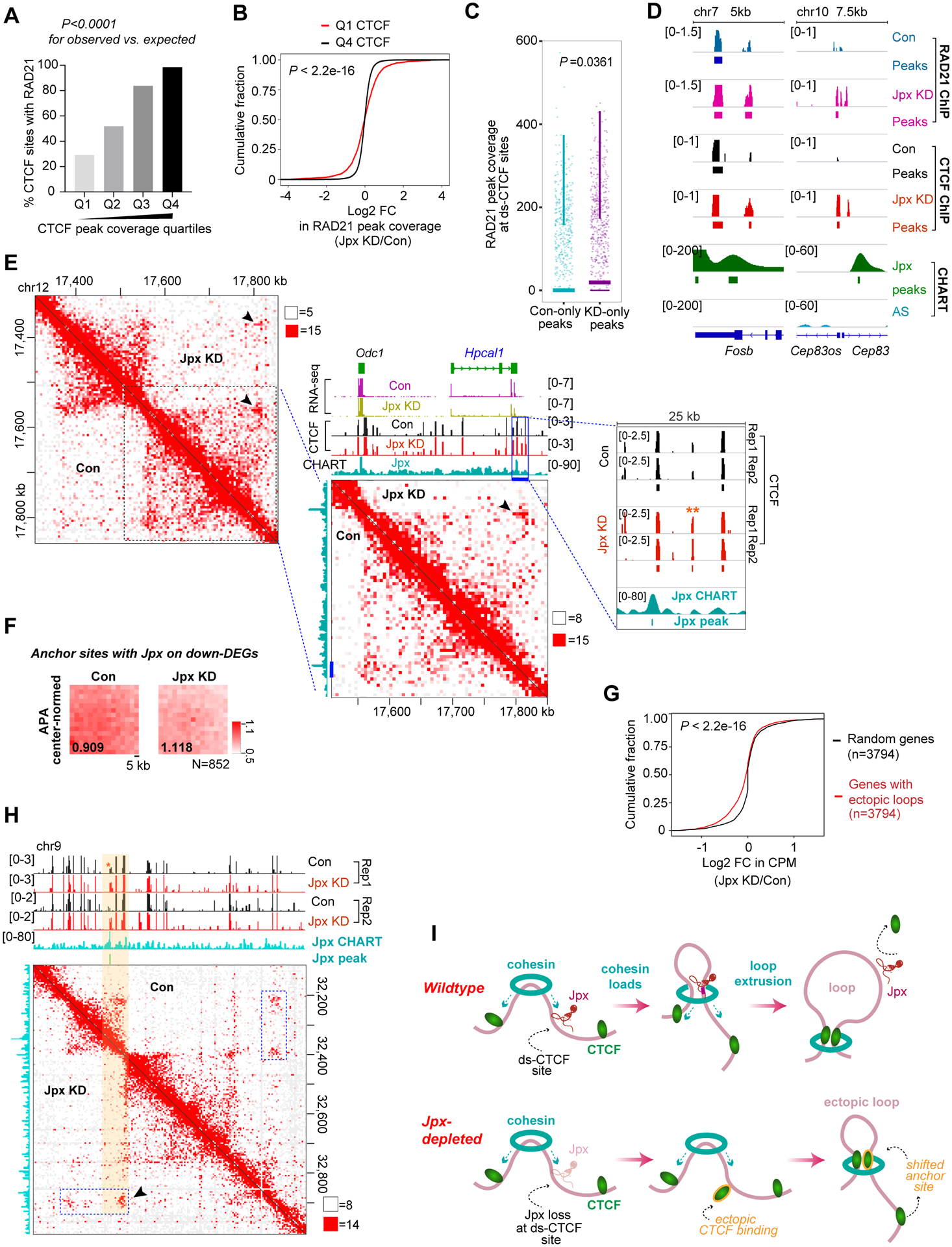
(A) RAD21 colocalizes with CTCF sites, with greatest overall coverage in higher CTCF peak quartiles in d7 ES cells. P-values, Chi-square test for observed vs. expected values.
(B) CDP comparing log2 FC in RAD21 peak coverages over Q1 vs. Q4 CTCF sites. P-value, KS test. See also Fig.S7N.
(C) Box and whisker plot for coverages of RAD21 peaks (control-only vs. KD-only shown in Fig. S7J) at ds-CTCF sites in control vs. Jpx-depleted d7 ES cells. P-value, Wilcoxon ranked sum test.
(D) Two representative loci showing colocalization of Jpx, CTCF and RAD21 peaks. Jpx depletion results in increased CTCF and RAD21 coverages over ectopic CTCF sites.
(E) Hi-C matrix at 5 kb resolution for Hpcal1 showing the relationship between increased CTCF binding, Jpx binding, ectopic loop formation, and gene downregulation. Arrows, changes in looping interactions coincide with Jpx binding sites (in wildtype state) and increased CTCF upon Jpx KD. **, ectopic, ds-CTCF peak. RNA-seq, CTCF ChIP-seq, Jpx CHART tracks, with significant peaks shown in magnified views.
(F) Center-normed APA showing the normalized-aggregate strength of paired anchor interactions at Jpx binding sites overlapping down-DEGs. P2LL ratio and N (852 loop anchors) shown.
(G) Log2 FC in CPM values for Jpx target genes with ectopic loops anchors (n=3,794). P (KS test) compares genes with ectopic loops to 3,794 randomized genes.
(G) Shifting loops caused by Jpx depletion, as shown by Hi-C matrix at 5 kb resolution. Blue box, shifting loop. Arrow, strengthened loop following Jpx depletion at the expense of upstream loop.
(I) Shifting Loops Model. See Discussion for description.
Finally, we assessed phenotypic consequences by asking whether transcriptomic changes (Fig. 2,3) occurred around shifted loops. Hi-C matrix showed that Jpx loss and CTCF enrichment at Hpcal1 were accompanied by formation of a de novo loop anchor where Jpx once bound (arrowhead, Fig. 7E). These changes corresponded to a downregulation of Hpcal1 (Fig. 7E, RNA-seq). On a transcriptome-wide scale, for the 852 Jpx-associated loop anchors over downregulated DEGs, APA revealed strengthened interactions after Jpx depletion (Fig. 7F). To examine gene expression changes in aggregate across 3,794 genes with ectopic loop anchors, we calculated log2 fold-changes in Jpx-depleted versus control cells and found significantly decreased gene expression in Jpx-depleted cells (Fig. 7G). We conclude that ectopic loops caused by Jpx loss are accompanied by suppression of gene expression.
DISCUSSION
We have identified an RNA coregulator of 3D genome architecture. Once thought to only control X-inactivation, our study demonstrates that Jpx RNA actually controls CTCF binding on a genome-wide scale. Jpx determines anchor site selection and specifically affects low-affinity CTCF sites that are associated with developmental regulation. In normal cells, Jpx binds thousands of genomic sites, preferentially engages promoters of active genes, modulates the looping landscape, and thereby regulates gene activation. Without Jpx, 72–75% of chromosome loops are displaced, thousands of new loops appear, and >700 genes are downregulated in expression. In our “shifting anchors” model (Fig. 7H,I), Jpx binding precludes CTCF binding at low-affinity sites, while having no effect on cohesin’s movement across chromatin. Cohesin progresses past Jpx and stops when it encounters a CTCF anchor pair. When Jpx is depleted, CTCF binds ectopically and “short-circuits” the advancing cohesin ring, causing an anchor shift and formation of a smaller loop. Notably, this mechanism could yield larger or smaller loops. If the ectopic CTCF site is proximal to the original loop anchor (e.g., Fig. 7I), a shorter loop would result. If the ectopic CTCF site is distal, a larger loop would be created.
We propose that Jpx RNA acts as a CTCF release factor — just as WAPL serves as a release factor for cohesin (Haarhuis et al., 2017). Through its sub-nanomolar affinity for CTCF (Kd of <1.0 nM, Fig. 5), Jpx outcompetes CTCF-DNA interactions at low-affinity CTCF motifs (eg. Cald1). On the other hand, Jpx cannot do so at higher-affinity CTCF sites (e.g., Mettl21a). Thus, its release action is highly specific and only affects a subset of ds-CTCF anchors. CTCF is known to have much shorter residence times on chromatin than cohesin. Whereas CTCF associates and dissociates on a timescale of ~1–2 min, cohesin does so on a timescale of ~22 min (Hansen et al., 2018; Hansen et al., 2017). We presume that CTCF occupancy times could be further stratified by their LowOc and HighOc status, with the LowOc sites — i.e., the Jpx-sensitive sites — having even shorter residence times. CTCF’s fast on/off rates would enable rapid binding to LowOc anchors when Jpx detaches from chromatin (e.g., by 8 hours of Jpx depletion). Similarly, its favorable on/off rates would enable CTCF to extricate from chromatin when Jpx levels rise dramatically during cell differentiation (Tian et al., 2010). Notably, CTCF occupancy has been correlated with extensive alternative promoter usage, which is in turn associated with tissue- or lineage-specific gene expression (Davuluri et al., 2008; Kim et al., 2007). These tissue-specific CTCF sites significantly overlap with enhancers, in line with CTCF’s function in developmental regulation of enhancer-promoter interactions (Phillips-Cremins and Corces, 2013; Shen et al., 2012). Supporting this notion, when loops shift in response to Jpx depletion, >700 genes are downregulated, with an enrichment for development- and differentiation-associated loci.
We predict that Jpx will not be the only RNA cofactor for CTCF’s architectural function, as indeed CTCF has a large family of interacting transcripts (Kung et al., 2015; Saldana-Meyer et al., 2014). Conversely, we also predict that Jpx’s action may not be limited to CTCF, as our study identified multiple other DNA motifs enriched within Jpx binding sites (Fig. S4). Intriguingly, some motifs have unique patterns of enrichment within the five clusters of Jpx peaks (Fig. 4D; Table S7). For example, while motifs 1 and 2 show significant enrichment in all clusters, motif 16 (SPDEF, ZNF410, YY2) shows highest enrichment in Cluster 3 where CTCF overlaps Jpx peaks, Motifs 3 (ESR2) and 4 (Zfp652_DBD) show high enrichment in Cluster 1, and Motif 13 (NF1A, RFX2/3/4, MEIS3) was specifically enriched in Cluster 5. Our study thereby points the way for future investigation into potential regulators of Jpx-CTCF interactions and other RNA determinants of 3D architecture.
LIMITATIONS OF THE STUDY
Specific experiments and controls were designed to minimize off-target hybridization to CHART capture probes, including use of (1) Jpx-AS capture probes to control for strand-specificity of pulldown and to rule out probe hybridization to genomic DNA rather than RNA target, as well as (2) a no-RNase H elution to control for RNase H-independent elution. Despite these precautions, we cannot exclude the possibility of off-target hybridization, as this is a known limitation of CHART (Simon et al., 2011). We also note that, for the Jpx perturbation studies, we chose an 8 hr timepoint to focus on direct effects and minimize secondary effects. It is possible that the overall transcriptomic profile could differ at longer timepoints, both due to secondary effects and to long-term cellular adaptation to Jpx loss.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for reagents should be directed to and will be fulfilled by the Lead Contact, Jeannie T. Lee (lee@molbio.mgh.harvard.edu).
Material Availability
Requests for materials generated in this study should be directed to and will be fulfilled by the lead contact upon completion of a Material Transfer Agreement.
Data and Code Availability
All raw and processed high-throughput sequencing data generated in this study have been deposited to GEO with accession number: GSE144056. Data are publicly available as of the date of publication. This paper also analyzed existing, publicly available data. All accession numbers are listed in the Key Resource Table.
This study does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key Resource Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-CTCF | Cell Signaling Technology | Cat#2899S; RRID:AB_2086794 |
| Rabbit polyclonal anti-RAD21 | Abcam | Cat#ab992; RRID:AB_2176601 |
| Chemicals, peptides, and recombinant proteins | ||
| Formaldehyde solution | Sigma-Aldrich | Cat#F8775 |
| Recombinant mouse LIF | Sigma-Aldrich | Cat#ESG1107 |
| Protector RNase Inhibitor | Sigma-Aldrich | Cat#3335402001 |
| cOmplete EDTA-free Protease Inhibitor Cocktail | Sigma-Aldrich | Cat#11873580001 |
| Protease Inhibitor Cocktail | Sigma-Aldrich | Cat#P8340 |
| Proteinase K | Sigma-Aldrich | Cat#03115844001 |
| RNase A | Thermo Fisher Scientific | Cat#12091021 |
| RNase H | New England Biolabs | Cat#M0297L |
| Superscript III Reverse Transcriptase | Thermo Fisher Scientific | Cat#18080085 |
| TRIzol | Thermo Fisher Scientific | Cat#15596018 |
| Turbo DNase | Thermo Fisher Scientific | Cat#AM2238 |
| Ribonucleoside vanadyl complex | New England Biolabs | Cat#S1402S |
| Critical commercial assays | ||
| Agencourt AMPure XP | Beckman Coulter | Cat#A63881 |
| Mouse ES Cell Nucleofector Kit | Lonza-Walkersville | Cat#VPH-1001 |
| NEBNext ChIP-Seq Library Prep Master Mix Set for Illumina | New England Biolabs | Cat#E6240S |
| NEBNext Ultra II directional RNA Second Strand Synthesis Module | New England Biolabs | Cat#E7550S |
| NEBNext Multiplex Oligos for Illumina | New England Biolabs | Cat#E7335S (Index Primers Set1) |
| NEBNext Multiplex Oligos for Illumina | New England Biolabs | Cat#E7500S (Index Primers Set2) |
| NEBNext Ultra II DNA Library Prep Kit for Illumina | New England Biolabs | Cat#E7645S |
| Ribominus Eukaryote Kit v2 | Thermo Fisher Scientific | Cat#A15020 |
| RNeasy MinElute Cleanup kit | QIAGEN | Cat#74204 |
| Deposited data | ||
| Jpx CHART-seq in d0 ES cells | This study | GEO: GSE144056 |
| Jpx CHART-seq in d3 differentiating ES cells | This study | GEO: GSE144056 |
| Jpx CHART-seq in d7 differentiating ES cells | This study | GEO: GSE144056 |
| H3K4me3 ChIP-seq in d0 ES cells | Pinter et al., 2012 | GEO: GSE36905 |
| H3K4me3 ChIP-seq in d7 differentiating ES cells | Pinter et al., 2012 | GEO: GSE36905 |
| H3K27me3 ChIP-seq in d0 ES cells | Pinter et al., 2012 | GEO: GSE36905 |
| H3K27me3 ChIP-seq in d7 differentiating ES cells | Pinter et al., 2012 | GEO: GSE36905 |
| LAD (Lamina-Associated Domain) regions | Peric-Hupkes et al., 2010 | GEO: GSE17051 |
| RNA-seq in d7 differentiating ES cells transfected with scrambled LNA (control for Jpx LNA #1) | This study | GEO: GSE144056 |
| Jpx KD RNA-seq in d7 differentiating ES cells transfected with Jpx LNA #1 | This study | GEO: GSE144056 |
| RNA-seq in d7 differentiating ES cells transfected with scrambled LNA (control for Jpx LNA #2) | This study | GEO: GSE144056 |
| Jpx KD RNA-seq in d7 differentiating ES cells transfected with Jpx LNA #2 | This study | GEO: GSE144056 |
| CTCF ChIP-seq in d7 differentiating ES cells transfected with scrambled LNA (control for Jpx LNA #1) | This study | GEO: GSE144056 |
| Jpx KD CTCF ChIP-seq in d7 differentiating ES cells transfected with Jpx LNA #1 | This study | GEO: GSE144056 |
| CTCF ChIP-seq in d7 differentiating ES cells transfected with scrambled LNA (control for Jpx LNA #2) | This study | GEO: GSE144056 |
| Jpx KD CTCF ChIP-seq in d7 differentiating ES cells transfected with Jpx LNA #2 | This study | GEO: GSE144056 |
| RAD21 ChIP-seq in d7 differentiating ES cells transfected with scrambled LNA (control for Jpx LNA #1) | This study | GEO: GSE144056 |
| Jpx KD RAD21 ChIP-seq in d7 differentiating ES cells transfected with Jpx LNA #1 | This study | GEO: GSE144056 |
| in situ Hi-C in d7 differentiating ES cells transfected with scrambled LNA (control for Jpx LNA #1) | This study | GEO: GSE144056 |
| Jpx KD in situ Hi-C in d7 differentiating ES cells transfected with Jpx LNA #1 | This study | GEO: GSE144056 |
| Experimental models: Cell lines | ||
| Mouse ES cells (female, 16.7 TsixTST/+) Strain: M. musculus/M. castaneus | Ogawa et al., 2008 | N/A |
| Xist P2-mutant mouse ES cells (female, 16.7 TsixTST/+) | This study | N/A |
| Oligonucleotides | ||
| 3′ biotin-TEG Jpx CAHRT probes, see Table S8 | Integrated DNA Technologies | N/A |
| 3′ biotin-TEG Jpx AS CHART (negative control) probes, see Table S8 | Integrated DNA Technologies | N/A |
| Antisense LNA GapmeR Control, see Table S8 | QIAGEN | Cat#339515 |
| Antisense LNA GapmeR Jpx LNAs, see Table S8 | QIAGEN | Sequences designed in this study; Cat#339517 |
| Primers used for RT-qPCR, see Table S8 | Integrated DNA Technologies | N/A |
| DNA EMSA probes, see Table S8 | Integrated DNA Technologies | N/A |
| Primers used to amplify Jpx (383 nt) for EMSA, see Table S8 | Integrated DNA Technologies | N/A |
| Xist P2 CTCF sgRNAs used to generate the P2-mutant cell line, see Table S8 | Integrated DNA Technologies | N/A |
| PCR screening primers used to generate the P2-mutant cell line, see Table S8 | Integrated DNA Technologies | N/A |
| Primers for 3C assay, see Table S8 | Integrated DNA Technologies | N/A |
| Recombinant DNA | ||
| pSpCas9-(BB)-2A-GFP (PX458) | Ran et al., 2013 | Addgene Cat#48138 |
| Software and algorithms | ||
| BEDTools v2.25.0 | Quinlan and Hall, 2010 | https://bedtools.readthedocs.io/en/latest/ |
| CEAS v1.0.2 | Shin et al., 2009 | N/A |
| Cutadapt v1.2.1 | Martin, 2011 | https://cutadapt.readthedocs.io/en/stable/# |
| Cutadapt v1.8.1 | Martin, 2011 | https://cutadapt.readthedocs.io/en/stable/# |
| Cufflinks v2.2.1 | Trapnell et al., 2012 | http://cole-trapnell-lab.github.io/cufflinks/ |
| deepTools v3.1.2 | Ramírez et al., 2016 | https://deeptools.readthedocs.io/en/develop/ |
| Homer v4.8 | Heinz et al., 2010 | http://homer.ucsd.edu/homer/ngs/ |
| Homer v4.10 | Heinz et al., 2010 | http://homer.ucsd.edu/homer/ngs/ |
| ImageJ v1.53a | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Juicebox v1.9.8 | Durand et al., 2016a | https://github.com/aidenlab/Juicebox |
| Juicer v1.5.3 | Durand et al., 2016b | https://github.com/aidenlab/juicer |
| Juicer v1.7.6 for HiCCUPS (GPU version) | Durand et al., 2016b | https://github.com/aidenlab/juicer |
| MACS2 v2.1.1.2016309 | Zhang et al., 2008 | https://pypi.org/project/MACS2/ |
| MEME suite v4.10.1 | Machanick and Bailey, 2011 | https://meme-suite.org/index.html |
| MEME suite v5.3.0 | Machanick and Bailey, 2011 | https://meme-suite.org/index.html |
| NovoAlign v3.00.02 | Novocraft | http://www.novocraft.com/products/novoalign/ |
| NovoAlign v4.02.01 | Novocraft | http://www.novocraft.com/products/novoalign/ |
| Pgltools | Greenwald et al., 2017 | https://github.com/billgreenwald/pgltools |
| SAMtools v0.1.19 | Li et al., 2009 | http://samtools.sourceforge.net/ |
| SAMtools v1.4.1 | Li et al., 2009 | http://samtools.sourceforge.net/ |
| SPP v1.11 | Kharchenko et al., 2008 | http://compbio.med.harvard.edu/Supplements/ChIP-seq/ |
| TopHat2 v2.0.10 | Kim et al., 2013 | https://ccb.jhu.edu/software/tophat/index.shtml |
| Trim Galore! v0.4.1 | Babraham Bioinformatics | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ |
EXPERIMENTAL MODEL and SUBJECT DETAILS
Mouse ES cell culture and differentiation
Mouse embryonic stem cells (female M. musculus/M. castaneus hybrid 16.7, its TsixTST/+ clone with predesignated Xa and Xi by truncated Tsix expression (Ogawa et al., 2008)) were cultured and differentiated as previously described (Lee and Lu, 1999). Specifically, ES cells were grown on the irradiated MEF feeders in the complete DMEM medium [DMEM (high glucose, GlutaMAX, pyruvate, Thermo Fisher Scientific, 10569044) supplemented with 15% Fetal Bovine Serum (HyClone), 25 mM HEPES (Thermo Fisher Scientific, 15630130), 1X MEM NEAA (Thermo Fisher Scientific, 11140-076), 1X PEN/STREP (Thermo Fisher Scientific, 15140163), 0.1 mM ß-mercaptoethanol (Thermo Fisher Scientific, 21985023) and 500 U/mL of LIF (Sigma, ESG1107)].
For differentiation of ES cells, ES cells and feeders were trypsinized, separated into single cells and incubated on the plate in the media at 37°C for approximately 40 min. After confirming ES cells floating in the media while most of feeders were attached, ES cells were separated and re-plated on non-gelatinized petri dishes in the complete DMEM medium without LIF. Embryonic bodies (EB) formed from ES cells were grown in suspension, transferred to the gelatinized cell culture plates on day 4 of differentiation and further differentiated until day 7.
Generation of the P2-mutant cell line
P2 mESCs were generated by transfecting TsixTST/+ mESCs with a plasmid (pSpCas9-(BB)-2A-GFP; Addgene, 48138) (Ran et al., 2013) expressing Cas9 with an sgRNA targeted to the P2 CTCF motif. Specifically, 100,000 TST mESCs were transfected using Lipofectamine LTX reagent (Thermo Fisher Scientific, 15338100) and 500ng of the Cas9-sgRNA plasmid. Transfected cells were GFP sorted after 24 hours and the entire GFP positive pool plated. After 6 days, the pool was split and plated at low density and, after 5 more days clones were picked from the low-density plate into a 96-well plate. Clones were grown to confluency, then triplicate plated in 96-well plates. Clones were screened via PCR followed by BfaI restriction digestion in order to determine clones carrying an indel in the P2 CTCF motif. Clone A4 was thawed from one of the triplicate plates, expanded, and re-screened by PCR and Sanger sequencing to confirm a 12 bp deletion in the P2 CTCF motif (deletion of AAACCACTAGAG in the P2 motif, AAACCACTAGAGGGCAGGT).
SgRNA oligos (Xist P2 CTCF sgRNA top, Xist P2 CTCF sgRNA bottom) and PCR screening primers (Xist P2 CTCF flanking F1, Xist P2 CTCF flanking R1) are listed in Table S8.
METHOD DETAILS
RNA FISH
Differentiating d7 ES Cells were trypsinized, cytospun on slides, permeabilized with CSKT buffer (100 mM NaCl, 300 mM sucrose, 10 mM PIPES, 3 mM MgCl2, 0.5% Triton X-100, 2 mM ribonucleoside vanadyl complex (New England Biolabs, S1402S), pH 6.8) and fixed with 4 % formaldehyde. Cells were hybridized with 24 ng of Jpx probes (a 90kb BAC 399K20 subclone) (Sun et al., 2013) and 12 ng of Xist probes (pSx9-3 plasmid) (Ogawa et al., 2008), which were prepared by nick-translation, in hybridization buffer (50% formamide, 2X SSC, 10% dextran sulfate, 240ng mouse Cot-1 DNA (Thermo Fisher Scientific, 18440016), 2mM ribonucleoside vanadyl complex) overnight at 37°C. Slides were washed with 2X SSC/50% formamide at 37°C for 5 min three times, washed with 2X SSC at 37°C for 5 min three times, and mounted with Vectashield mounting media containing DAPI (Thermo Fisher Scientific, H-1200).
LNA-mediated Jpx knockdown
5 million ES cells on day 7 of differentiation were transfected with 2 μM of scrambled LNA (AACACGTCTATACGC; Antisense LNA GapmeR Control; Qiagen, 339515), Jpx-targeting LNA #1 (Jpx LNA #1; GGACGCCGCCATTTTA; Antisense LNA GapmeR; Qiagen, 339517) or LNA #2 (Jpx LNA #2; CAGTTTCTCCACTCTC; Antisense LNA GapmeR; Qiagen, 339517) (Table S8) using Mouse ES cell Nucleofector Kit (Lonza, VVPH-1001) per manufacturer’s instruction. Differentiating ES cells transfected with LNA were immediately transferred to the gelatinized plates, incubated at 37°C for 8 hr for successful depletion of Jpx, and then subjected to the following experiments including RT-qPCR, RNA-seq, ChIP-seq, 3C assay and Hi-C.
RT-qPCR
Total RNA was extracted using Trizol (Thermo Fisher Scientific, 15596018), depleted of DNA with Turbo DNA-free Kit (Thermo Fisher Scientific, AM1907), and subjected to cDNA synthesis with random primers using SuperScript III Reverse Transcriptase (Thermo Fisher Scientific, 18080085). RNA expression was normalized to the level of Gapdh expression. Primers for Jpx and Gapdh expression are listed in Table S8.
EMSA
DNA EMSAs were performed according to the previous report (Sun et al., 2013) with modifications. The HighOc and LowOc probe oligos (Table S8) were ordered from Integrated DNA Technologies (IDT) in duplex form. Double-stranded DNA probes were end-labeled with ATP[Double-stranded DNA probes were end-labeled with ATP[γ-32P] and unincorporated ATP was removed with a microspin G-50 columns (GE Healthcare). Recombinant FLAG-CTCF-6xHis protein was purified from Rosetta-Gami B cells (Novagen), as previous described (Sun et al., 2013). Recombinant CTCF was incubated at room temperature (RT) with 5 pM DNA probes in CTCF binding buffer (50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 5 mM MgCl2, 0.1 mM ZnSO4, 10% glycerol, and 0.05% Tween-20). Samples were resolved at RT in a 1x TBE-5% PAGE gel. The gel was dried prior to image capture with an Amersham Typhoon imager using a phosphor-screen. RNA EMSAs were performed as described previously (Sun et al., 2013) with modifications. Briefly, RNA probes were in vitro transcribed with a Lucigen T7 transcription kit from PCR-amplified cDNA templates created using T7_Jpx_ex1F, TAATACGACTCACTATAGACGGCACCACCAGGCTTCT; and Jpx_ex3R, GAGTTTATTTGGGCTTACAGTTC (Sun et al., 2013) (Table S8). In-vitro-transcribed RNA was purified in by size-exclusion chromatography in an Akta Pure system (Chillon et al., 2015), treated with fast calf intestinal phosphatase (New England Biolabs; NEB), end-labeled with ATP[γ−32P], and purified through a microspin G-50 column. The resulting RNA probes were denatured at 95°C for 2 min, incubated at 70°C for 5 min, 37°C for 15 min, 20° for 15 min, then cooled down to 4°C, and maintained in folding buffer (50 mM NaCl, 2 mM MgCl2) on ice prior to the binding reaction. Recombinant CTCF was incubated at room temperature with 5 pM RNA probes for 30 min in CTCF binding buffer plus 8 U RNase Inhibitor (Roche, 03335399001) per reaction. Samples were resolved at RT by 1x TBE-5% PAGE gel. During competition experiments, 5 nM DNA probes and the corresponding pre-folded RNA probes were mixed before addition of protein. IC50 measurements were performed using ImageJ (v1.53a)(Schneider et al., 2012).
CHART-seq
Design of CHART probes
Using computational tools (https://www.idtdna.com/calc/analyzer, BLAST, BLAT(https://genome.ucsc.edu/cgi-bin/hgBlat)), four CHART probes were designed to target three exons common to all Jpx variants (Fig. S1A). All probes meet the following criteria.
BLAST was used to screen off-target transcripts with mouse genomic plus transcripts database. E-values for the match with the other transcripts are > 1, indicating no significance.
BLAT was used to screen off-target genomic DNA sites with mm9 and mm10 database. No other match was found.
Probes have 21–25 nucleotides in length with similar GC content (40–52.4%) and melting temperature (54.4–56.8°C)
We also checked the specificity of probes using other search engines (http://ggrna.dbcls.jp/help.html for RNAs, and https://gggenome.dbcls.jp/help.html for genomic sites). No other match was found.
The 3’ biotin-TEG Jpx CHART probes and the reverse complement probes that are able to capture the antisense strand of Jpx are listed in Table S8.
Technical improvement in CHART protocol
While optimizing our Jpx CHART protocol we made several key protocol improvements to obtain higher signal-to-noise ratios than previously. Our CHART protocol is based on the published ChIRP (Chu et al., 2011) or CHART (Simon et al., 2013) protocols with the following modifications: (i) To make the elution step compatible with RNase H activity, we used NP-40 detergent instead of SDS or N-lauryl sarcosine. (ii) We improved the signal-to-noise ratio by using formamide during hybridization and wash steps. (iii) We lowered the background signal by maintaining the same salt concentration (250 mM NaCl) during elution as was used in the prior wash steps. Reference protocols used lower salt concentrations (e.g. 150 mM NaCl). Because high salt concentrations reduce RNase H activity by as much as ~50% at 300 nM (Berkower et al., 1973), we used excess RNase H to compensate and improved recovery of samples during elution.
CHART Protocol detail
Undifferentiated (d0) and differentiating (d3 and d7) ES cells were harvested for CHART-seq. Approximately 16 million cells were crosslinked with 1% formaldehyde (Sigma, F8775) in PBS at room temperature (RT) for 10 min and the reaction was quenched with 0.125 M glycine at RT at 5 min. The cells were washed with cold PBS three times, snap-frozen in liquid nitrogen and stored at −80°C. The cell pellet was resuspended in 2 mL sucrose buffer [0.3 M sucrose, 1% Triton X-100, 100 mM HEPES pH 7.5, 100 mM KOAc, 0.1 mM EGTA, 0.5 mM Spermidine, 0.15 mM Spermine, 1 mM DTT, 1X Complete EDTA-free Protease Inhibitor Cocktail (Roche, 11873580001), 50 U/ml RNase inhibitor (Roche, 3335399001), 0.5 mM PMSF], ruptured with 10 ~ 15 strokes of an ice-cold dounce homogenizer, and incubated for 10 min on ice. 2 mL of glycerol buffer (25% glycerol, 10 mM HEPES pH 7.5, 1 mM EDTA, 0.1 mM EGTA, 100 mM KOAc, 0.5 mM Spermidine, 0.15 mM Spermine, 1 mM DTT, 1X PIC (Sigma, 11873580001), 50 U/ml RNase inhibitor, 0.5 mM PMSF) was added to the Dounce homogenizer, and then the subsequent mixture was loaded slowly on the top of 2 mL of glycerol buffer and centrifugated at 1500 g for 10 min at 4°C to collect the nuclei. The nuclei pellet was washed once with cold PBS and further crosslinked with 3% formaldehyde in 6 mL of PBS for 30 min at RT. After crosslinking, the nuclei were washed twice with cold PBS, resuspended in 600 μL of 250 mM NaCl nuclei resuspension buffer [50 mM HEPES pH 7.5, 250 mM NaCl, 0.1 mM EGTA, 0.5% N-laurylsarcosine, 0.1% Sodium deoxycholate, 5 mM DTT, 1X Protease Inhibitor Cocktail (Sigma, P8340), 100 U/mL RNase inhibitor] and incubated on ice for 10 min. The nuclei were collected by centrifugation at 400 g for 5 min at 4°C, resuspended in 300 ul of 75 mM NaCl nuclei resuspension buffer [50 mM HEPES pH 7.5, 75 mM NaCl, 0.1 mM EGTA, 0.5% N-laurylsarcosine, 0.1% Sodium deoxycholate, 5 mM DTT, 1X Protease Inhibitor Cocktail (Sigma, P8340), 100 U/ml RNase inhibitor], aliquoted into microtubes and sonicated using a Covaris E220 for 5 min (10 % duty cycle, 105 peak incident power and 200 cycles/burst). After centrifugation at 16,100 g for 15 min at 4°C, 2X volume of hybridization buffer [25 mM HEPES pH 7.5, 1.175 M NaCl, 7.5 mM EDTA, 1.25 mM DTT, 0.5% SDS, 7.5X Denhardt’s solution, 15% formamide, 1X Protease Inhibitor Cocktail (Sigma, P8340), 100U/mL RNase inhibitor, 0.5 mM PMSF] were added to the supernatant. The resulting CHART extracts were pre-cleared by incubation with Dynabeads MyOne Streptavidin C1 (33 μL of beads per 100 μL of extracts; Thermo Fisher Scientific, 65001) for 1.5 hr at RT. The precleared extracts were incubated with the probe sets for Jpx or its antisense (10 pmol of probes per 100 μL of extracts) overnight at RT. After overnight hybridization, the beads (66 μL of beads per 100 μL of extracts) were added to the hybridized sample, incubated with rotation for 2 hr at RT and captured on a magnet rack. The beads were washed by incubation in pre-warmed wash buffer I (30 mM HEPES pH 7.5, 240 mM NaCl, 15% formamide, 1.5 mM EDTA, 0.75 mM EGTA, 0.65% SDS, 0.75% N-laurylsarcosine, 1X Protease Inhibitor Cocktail (Sigma, 11873580001), 0.5 mM PMSF) once and wash buffer II (10 mM HEPES pH 7.5, 250 mM NaCl, 2 mM EDTA, 1 mM EGTA, 0.2% SDS, 0.1% N-laurylsarcosine, 1X Protease Inhibitor Cocktail (Sigma, 11873580001), 0.5 mM PMSF) four times with rotation for 5 min at 37 °C. After additional two brief washes at RT with wash buffer III (50 mM HEPES pH 7.5, 250 mM NaCl, 0.1% NP-40), DNA was eluted twice with 75 U of RNase H (NEB, M0297L) in 150 ul of RNase H elution buffer (50 mM Tris-Cl pH 7.5, 250 mM NaCl, 3 mM MgCl2, 10 mM DTT, 0.5% NP-40) for 20 min at RT (RNase H was not added to the sample for No-RNase H CHART). The eluant DNA was treated with RNase A (Thermo Fisher Scientific, 12091021) at a concentration of 200 μg/μL for 1 hr at 37°C, incubated with Proteinase K (Sigma, 03115844001) at a concentration of 1 μg/μL for 1hr at 55°C, and kept at 65 °C overnight. The phenol/chloroform extraction followed by ethanol precipitation was used for CHART DNA purification.
The CHART eluant in 10 mM Tris-Cl pH 8.0 buffer was further sheared in microtube with a Covaris E220 for 4 min (5 % duty cycle, 175 peak incident power and 200 cycles/burst) and subjected to library preparation. Input and CHART-seq libraries were prepared per manufacturer’s instructions, using NEBNext ChIP-seq library Prep Master Mix Set for Illumina (NEB, E6240S), NEBNext Multiplex Oligos for Illumina (index primers set 1, NEB, E7335S; index primers set 2, NEB, E7500S), and AMPure beads (Beckman Coulter, A63881) for double-size selection (0.6X-1.2X). NEBNext Ultra II DNA library prep kit for Illumina (NEB, E7645) was used after NEBNext ChIP-seq library Prep Master Mix Set for Illumina (NEB, E6240S) was discontinued. Libraries were generated in two biological replicates for d0 and d3, and three biological replicates for d7 ES cells.
CHART-seq data processing
We initially used the older Illumina HiSeq 2000 and 2500 platforms, which have paired-end 50 bp (PE50) reads. As the technology developed, our later sequencing was performed on the newer HiSeq 4000 using paired-end 150 bp reads (PE150). Rep1 and Rep2 were derived from PE50 reads, whereas Rep3 was from PE150 reads. Approximately 57–78 million, 61–69 million and 66–89 million paired-end reads per sample were generated for rep1, rep2 and rep3, respectively. The data were consistent across all three replicates, irrespective of the platform used.
CHART-seq reads were subjected to the trimming of adaptor sequence and removal of PCR duplicates using the software Trim Galore! (v0.4.1) (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) with the parameters “--dont_gzip --stringency 12 --phred33 -e 0.2 --paired --retain_unpaired -a GATCGGAAGAGC -a2 GATCGGAAGAGC”, which in turn called the software Cutadapt (v1.2.1 for rep1 and rep2; v1.18 for rep3) (Martin, 2011). Because we used M. musculus/M. castaneus hybrid cell line, reads were mapped to CAST/Eih (cas) and 129S1/SvJm (mus) genomes using Novoalign (v3.00.02 for rep1 and rep2; v4.02.01 for rep3) with the parameters “i300 100 -F STDFQ -t180 -rRandom -h180 180 -v180” for both versions (Pinter et al., 2012). From the independent mapping to cas and mus reads, we generated composite (sum of neutral, cas-specific, and mus-specific) reads mapped back to the NCBI37/mm9 genome using a previously published pipeline (Minajigi et al., 2015; Pinter et al., 2012). Composite bam files having only uniquely aligned reads were used for the following analyses.
To obtain the mm9 chromosomal coverage of these data sets, input-subtracted coverages for each CHART (Jpx, Jpx AS, no-RNase H) were generated using the software SPP (v1.11) (Kharchenko et al., 2008), and visualized using Integrative Genomics Viewer (IGV). In detail, SPP is a library (R package) of routines that uses read auto-correlation to identify the places of the RNA/Protein/DNA complex binding. The function “get.smoothed.tag.density” was utilized for generating smoothed Jpx CHART coverage [get.smoothed.tag.density(CHART data, input data, bandwidth=500, step=100, tagshift), here bandwidth is the window size, step is the step size, and tagshift shifts the peaks to the center of the binding footprint].
To ascertain high signal-to-noise ratio (Fig.1B), Jpx CHART coverages were normalized (subtraction method) to negative control CHARTs (Jpx AS or no-Rnase H) using SPP (500bp of window size and 100bp of step size) in addition to normalization to input. Jpx CHART coverage profiles were consistent regardless of whether Jpx CHART was normalized to input or negative controls.
To determine where Jpx RNA was binding DNA with statistical significance and compare across experiments, we called enriched peaks using the function “callpeak” of the software MACS2 (v2.1.1.2016309) (Zhang et al., 2008) with composite CHART bam file (Jpx, Jpx AS, no-RNAse H CHARTs) and the composite input bam file. Default parameters were used (band width=300, model fold = [5,50], q-value cutoff = 0.05). Additionally, in order to remove non-specific signals and thus ensure the use of Jpx CHART peaks with high significance, we excluded Jpx AS and no-RNase H peaks from Jpx CHART peaks. This was accomplished by subtracting Jpx AS and no-Rnase H MACS2 peaks from Jpx CHART MACS2 peaks using the function “subtract” of the software BEDTools (v2.25.0)(Quinlan and Hall, 2010), generating filtered Jpx peaks (henceforth Jpx peaks).
To perform correlation analysis between replicates and different conditions (Fig. S1G,S2B), the smoothed SPP-generated Jpx CHART coverages (wig) were averaged over 10 kb windows tiled across the genome. Then we created a scatterplot in the software R after coordinating respective experiment windows.
To identify genomic distribution of Jpx peaks and perform metagene analysis (Fig. 1E and 2E), we used the software CEAS (Cis-regulatory Element Annotation System)(v1.0.2) (Shin et al., 2009), which required a region (bed) file and a coverage (wig) file. We used Jpx peaks for the bed file and SPP-generated Jpx CHART coverages for the wig file.
Quantification of peak signals
To quantify the CHART and ChIP signals, we used two distinct statistics-based, peak-centered methods to derive “peak coverages”. Method 1 obtains a monotonic value for each MACS2-generated peak and then sums up over a region of interest. Method 2 obtains the binned coverage generated by SPP around peaks. Method 1 was used to compare and contrast peak signals between experimental units (e.g. box plot, Fig. 1D; CDP, Fig. 3B). Method 2 was used to obtain plots of peak coverage around meta-loops anchors (e.g. meta-loop analysis using deepTools, Fig. 6J). The CHART or ChIP signal quantified by both methods reflects the frequency of peaks in a region of interest:
Method 1: Both width and height of peaks are taken into account for peak signal. SignalValue (measurement of overall enrichment obtained from MACS2 output narrowPeak) per peak was used. In detail, “BEDTools intersect -loj” command was first used to intersect two bed files by left outer joining. The -a bed file contains the peak regions with signalValues. The -b bed file contains the regions of interest. To maintain enrichment structure of the peak, we used integrated values of enrichment. This was obtained by multiplying the width of peak by its height (signalValue). These integrated values were summed over their intersected regions, and these sum values along with the regions of interest, including gene bodies ± 3kb, were compared and plotted (Fig. 1D, 1G, 3B–F, 4G–H, 7B–C, S5B, S6F–G, S7N).
Method 2: The binned coverages obtained using SPP were generated around the MACS2 peaks. By doing so, the structure of original signals that generated the MACS2 peaks is maintained. This signal representation is based on smoothed counts and is not spatially limited to the exact extent of a peak.
First, to define regions around peaks we tiled the mm9 genome with windows (BEDTools makewindows) then intersected these windows with MACS2 generated peaks (BEDTools intersect). Then we merged the windows (BEDTools merge) to generate regions surrounding peaks. We then intersected SPP-generated coverage bins with these regions to generate a bedGraph file of SPP-generated coverage values spatially overlapping the extent of the peaks. The resulting bedGraph file was converted to the bigWig format of the peak-centric Jpx or CTCF signal. We used the function “computeMatrix” of the software deepTools (v3.1.2) (Ramirez et al., 2016) with the reference point as the center of loop anchors and the bigWig file to generate a coverage matrix (computeMatrix reference-point –skipZeros). This matrix was then imported into R to plot coverage around meta-loop anchors.
For Jpx peak coverage around meta-loop anchors, we used 3 kb windows with step size 300 for generating the extent around MAC2-generated Jpx peaks and -bs=300 for deepTools (Fig. 6G, S7B). For CTCF peak coverage around meta-loop anchors, we used a 500 bp window and step size 250 for generating the extent around either MACS2-generated CTCF peaks (Fig. 6J) or the subset of CTCF peaks (Fig. 6I, S7C) and -bs=1000 for deepTools.
Enrichment profiles around Jpx peaks
H3K4me3 and H3K27me3 ChIP-seq data in d0 and d7 mouse ES cells were obtained from the previous study (Pinter et al., 2012) (GEO: GSE36905) and coverages (wig) were generated using the pipeline described in the study (Pinter et al., 2012). These data were derived using the same mouse ES cell line as this study. SINE and LINE1 element regions were obtained using RepeatMasker from UCSC genome browser (http://genome.ucsc.edu/cgi-bin/hgTables). LAD (lamina-associated domain) regions were obtained from the previous report (Peric-Hupkes et al., 2010). From these region (bed) files, SINE, LINE1 and LAD coverages were obtained using the function “coverage” of BEDTools over 50 kb windows. The windows were generated by tiling the mm9 genome using the function “makewindows” of BEDTools. The resulting coverage files were used to generate the enrichment profiles around Jpx peaks using deepTools with the reference point as the center of Jpx peaks (computeMatrix reference-point --skipZeros, histone marks -bs=10, rest -bs=100; plotProfile). Pearson’s r values are reported in the figures (Fig. 1F,H).
Strand-specific Total RNA-seq
Differentiating ES cells (d7) were transfected as described in LNA-mediated knockdown. From these cells, 4 ug of total RNA extracted using TRIzol (Thermo Fisher Scientific, 15596018) was depleted of ribosomal RNA using RiboMinus Eukaryote Kit v2 (Thermo Fisher Scientific, A15020), depleted of DNA with TURBO DNase (Thermo Fisher Scientific, AM2238) for 15 min at 37°C, and purified by RNeasy MinElute Cleanup kit (Qiagen, 74204). Ribosomal RNA depletion was confirmed by Bioanalyzer (Agilent RNA 6000 Pico Kit). The resulting rRNA-depleted RNA was fragmented by incubation in First-strand Buffer including magnesium supplied with SuperScript III Reverse Transcriptase (Thermo Fisher Scientific, 18080093) for 10 min at 94°C, incubated with random primer and dNTP for 5 min at 65°C, and subjected to RT reaction in the presence of 5mM DTT, 40 U of RNase inhibitor (Roche, 03335399001), 0.5 ug of actinomycin D (Sigma, A1410) and SuperScript III Reverse Transcriptase for 50 min at 50°C. The subsequent first-strand cDNA was subjected to second strand cDNA synthesis using NEBNext Ultra Directional RNA Second Strand Synthesis Module (NEB, E7550S) containing dUTP mix. Library preparation was performed as described in CHART-seq. Two biological replicates were generated for each set of RNA-seq (scrambled and Jpx LNA #1; scrambled and Jpx LNA #2).
RNA-seq data processing
RNA-seq libraries were sequenced on the Illumina HiSeq 2000 (PE50), generating 134–151 million paired-end reads per sample (scrambled and Jpx LNA #1) or the Illumina HiSeq 4000 (PE150), generating 87–107 million paired-end reads per sample (scrambled and Jpx LNA #2). As noted above, we initially used the older Illumina HiSeq 2000 platform, which has paired-end 50 bp (PE50) reads. As the technology developed, our later sequencing was performed on the newer HiSeq 4000 using paired-end 150 bp reads (PE150). Two biological replicates from Jpx LNA #1 dataset were derived from PE50 reads, whereas two biological replicates from Jpx LNA #2 dataset were from PE150 reads. The data were consistent across all datasets, irrespective of the platform used.
RNA-seq reads were adaptor-trimmed using Trim Galore! (v0.4.1) and Cutadapt (v1.2.1 for Jpx LNA #1 dataset; v1.18 for Jpx LNA #2 dataset) with the same parameters described in CHART-seq data processing, and then aligned to the mouse genome (NCBI37/mm9) using the alignment software TopHat2 (v2.0.10)(Kim et al., 2013) with the parameters “--mate-inner-dist 100 --mate-std-dev 100 --library-type fr-firststrand”. We obtained 76~94 % concordant pair alignment rate.
Only uniquely and concordantly mapped reads were selected without removal of duplicates (Parekh et al., 2016) and converted into “Tag directory” using the function “makeTagDirectory” in the software package HOMER (v4.8) analysis pipeline (http://homer.ucsd.edu/homer/ngs/) (Heinz et al., 2010), Then, DE analysis was performed using the software edgeR (utility getDiffExpression.pl) in the HOMER analysis pipeline. Counts per million (CPM) mapped reads for each gene and log2 fold-change in gene expression were determined by edgeR. An FDR (adjusted P-value) of < 0.05 was used to identify significantly differentially expressed genes (DEGs).
For display purposes, strand-specific FPM (fragments per million)-normalized coverage files (bigWig) were generated using a custom script (using linux utilities sort, sed, and awk (coreutils/8.27)) and the software SAMtools (v.0.1.19)(Li et al., 2009) as described previously (Kung et al., 2015), and then visualized using IGV. In detail, starting with uniquely aligned reads without duplicates removed i) Samflags were fixed using the function “fixmate” of SAMtools to accommodate drop outs during adaptor-trimming ii) Fragment counts used as the scaling factor were derived from the function “flagstats” of SAMtools as: “with itself and mate mapped”/2 + “singletons.” iii) Watson (+) and Crick (−) strands were identified using the sam tag XS:A. iv) Wig coverage files were obtain using samtools depth. v) Finally, FPM counts were derived by dividing by fragments scaled by 1000000.
To determine transcripts of constitutively active genes and those less expressed in d7 ES cells (Fig. 1G), active genes were defined as having an FPKM (Fragments Per Kilobase of transcript per Million mapped reads) >= 0.5. FPKM values were obtained by the software Cufflinks (v2.2.1) (Trapnell et al., 2012) as outlined in the cufflinks suite of tools work flow (http://cole-trapnell-lab.github.io/cufflinks/manual/). Both upper-quartile-norm and compatible-hits-norm were chosen as the normalization method used.
Enrichment of Jpx peaks over down-DEGs
To determine DEGs overlapping with Jpx peaks (Fig. S3C,D), DEGs (genes bodies ±3 kb) or non-DEGs (gene bodies ±3 kb) were intersected with rep1 or rep2 of Jpx peak regions (extended ± 10kb from Jpx peaks, considering a 3 kb resolution of CHART) using the function “intersect” of BEDTools. The numbers of down-DEGs, up-DEGs or non-DEGs having rep1 Jpx (rep1), rep2 Jpx (rep2), either rep1 or rep2 Jpx (union of rep1+2), or both rep1 and rep2 Jpx (intersect of rep1+2) were indicated (Fig. S3C). Percentages of genes with Jpx peak regions out of total down-DEGs (738), up-DEGs (162), or non-DEGs (22,824) were also indicated (Fig. S3D). To determine whether Jpx peak regions were enriched over down-DEGs with statistical significance, we randomly sampled 900 non-DEGs out of total 22,824 non-DEGs 100 times. The number of random genes with Jpx peak regions (Fig. S3C) is the averaged value from 100 repeated random samplings. Using these randomized gene sets of 900 genes, p-values were determined by Wilcoxon ranked sum test (one-sided) with null hypothesis that the enrichment of Jpx peak regions over down-DEGs occurs by random chance. We used an alpha of 0.05 as the rejection value.
Gene Ontology (GO) analysis
GO analysis was performed using the R package goseq (v1.26.0) (Young et al., 2010). Specifically, DEGs determined by edgeR were reformatted into a vector suitable for goseq. The length bias present in the data was corrected using the Probability Weighting Function (PWF). Significantly enriched GO terms (Biological Process; BP) were determined using Wallenius approximation method with a cut-off of FDR (adjusted P-value) < 0.05 (adjustment method: Benjamini-Hochberg (BH)). Among total 86 significantly enriched GO terms (Table S4), we selected 16 GO terms related to general differentiation and developments (Fig. S3E) and 18 GO terms related to neural development and differentiation (Fig. S3F) with a higher significance (P-value < 0.0001). Then, these GO categories were mapped to DEGs, and the resulting matrix table with the log2 fold-change value for each DEG was used to generate heatmap shown in Fig. S3E and S3F.
ChIP-seq of CTCF and RAD21
Differentiating ES cells (d7) were transfected as described in LNA-mediated knockdown. CTCF and RAD21 ChIPs were performed as described previously (Jeon and Lee, 2011; Kung et al., 2015; Wang et al., 2018). Specifically, 10 million cells were cross-linked with 1% formaldehyde in 10 mL of complete medium, quenched with 0.125 M glycine, washed with cold PBS and stored at −80°C with snap-freezing in LN2. The nuclei pellet was collected by the sequential incubations in Buffer I [50 mM HEPES pH 7.5, 1 mM EDTA pH 8.0, 150 mM NaCl, 0.5% NP-40, 0.25% Triton X-100, 1X Protease Inhibitor Cocktail (Sigma, P8340)] and Buffer II (10 mM Tris pH 8.0, 200 mM NaCl, 5 mM EDTA pH 8.0, 2.5 mM EGTA pH 8.0, 1X Protease Inhibitor Cocktail), and centrifugation at 1700 g for 5 min at 4°C after each incubation for 10 min at 4°C. The subsequent nuclei were incubated in Buffer III (10 mM Tris pH 8.0, 5 mM EDTA pH 8.0, 2.5 mM EGTA pH 8.0, 1X Protease Inhibitor Cocktail) with 500 ug of RNase A (Termo Fisher Scientific, 12091021) for 20 min at 37 °C, supplied with N-lauroyl sarcosine at a concentration of 0.5%, further incubated with rotation for 15 min at 4°C, and then sonicated. Sonication was performed using a Covaris E220 for 18 min with the parameters of 10% duty cycle, 140 peak incident power, 200 cycles/burst (CTCF ChIP-seq using Jpx LNA #1), or a Qsonica (Q800R) for 6 min (on time) with the parameters of 40% amplitude and 30 s on/30 s off (CTCF ChIP using Jpx LNA #2, RAD21 ChIP using Jpx LNA #1). After centrifugation at maximum speed, the same volume of IP buffer (2% Triton X-100, 300 mM NaCl, 30 mM Tris pH 8.0 and 1X Protease Inhibitor Cocktail) was added to the supernatant. The resulting lysates were incubated with CTCF antibody (Cell Signaling Technology, 2899S), RAD21 antibody (Abcam, ab992) or normal rabbit IgG (Cell signaling Technology, 2729) on the rotator at 4°C overnight (1 μL of antibody/100 μL of lysates) and then incubated with equilibrated Dynabeads protein G (Thermo Fisher Scientific, 1003D) for 2 hr (7 μL of Dynabeads / 100 μL of lysates) at 4°C. Beads were collected on magnet, washed with Wash buffer I (50 mM HEPES pH 7.5, 10 mM EDTA pH 8.0, 0.5% Sodium Deoxycholate, 1% NP-40, 500 mM NaCl and 1X Roche protease inhibitor cocktail) three times, Wash buffer II [50 mM HEPES pH 7.5, 10 mM EDTA pH 8.0, 0.5% Sodium Deoxycholate, 1% NP-40, 250 mM NaCl and 1X Complete EDTA-free Protease Inhibitor Cocktail (Roche, 11873580001)] three times, and Last wash buffer (10 mM Tris pH 8.0, 1mM EDTA pH 8.0, 0.1% NP-40 and 50 mM NaCl) once. DNA was eluted by incubation of beads in Elution buffer (50 mM Tris pH 8.0, 10 mM EDTA pH 8.0, 1% SDS) at 65 °C for 20 min twice. The DNA eluants were treated with Proteinase K (0.5 μg/μL, Sigma, 03115844001) at 55°C for 1 hr, incubated at 65°C overnight, and purified by phenol/chloroform extraction and ethanol precipitation. Input DNA and ChIP DNA were further subjected to library construction steps using NEBNext ChIP-seq library Prep Master Mix Set for Illumina (NEB, E6240S) for Jpx LNA #1 CTCF ChIP-seq or NEBNext Ultra II DNA library prep kit for Illumina (NEB, E7645) for Jpx LNA #2 CTCF ChIP-seq and RAD21 ChIP-seq, as described in CHART-seq. Libraries were generated in two biological replicates and one experiment for CTCF ChIP-seq using Jpx LNA #1 (scrambled vs. Jpx LNA #1) and Jpx LNA #2 (scrambled vs. Jpx LNA #2), respectively. Libraries for RAD21 ChIP-seq using Jpx LNA #1 were generated in two biological replicates.
ChIP-seq data processing
CTCF ChIP-seq libraries were sequenced on the Illumina HiSeq 2000 (PE50) for CTCF ChIP-seq using Jpx LNA #1, or 4000 (PE150) platform for CTCF ChIP-seq using Jpx LNA #2. Approximately 89–113 million and 116–152 million paired-end reads per sample were generated for Jpx LNA #1 ChIP-seq dataset and Jpx LNA #2 ChIP-seq dataset, respectively.
CTCF ChIP-seq reads were subjected to the trimming of adaptor sequence and removal of PCR duplicates using the software Trim Galore! (v0.4.1) and the software Cutadapt (v1.2.1 for Jpx LNA #1 dataset; v1.18 for Jpx LNA #2 dataset) with the same parameters described in CHART-seq analysis. As described in CHART-seq data processing, adaptor-trimmed reads without duplicates were mapped to CAST/Eih (cas) and 129S1/SvJm (mus) genomes (mm9) using Novoalign (v3.00.02 for CTCF ChIP-seq using Jpx LNA #1; v4.02.01 for CTCF ChIP-seq using Jpx LNA #2) with the parameter “i300 100 -F STDFQ -t180 -rRandom -h180 180 -v180” for both versions. The composite (sum of neutral, cas-specific and mus-specific) bam files having uniquely mapped reads were generated for each library and then used for the following analyses.
CTCF ChIP coverage was derived using the function “get.conservative.fold.enrichment.profile” of SPP (v1.11)[get.conservative.fold.enrichment.profile (ChIP data, input.data, fws=150, step=25, alpha=0.01), here fws is the window size, step is the step size, and alpha is the cutoff, which generated log2 fold-enrichment estimates (ratio of signal tag density (IP) over background tag density (Input)) with statistical significance (alpha=0.01) under the assumption of Poisson distribution]. Coverage profiles were visualized with IGV (Fig. 3G,4G, S5C, S6A, S6D). FPM (fragments per million)-normalized CTCF coverages were averaged from all three CTCF ChIP-seq datasets (two biological replicates using Jpx LNA #1 one using Jpx LNA #2 ChIP-seq dataset), and then visualized with IGV (Fig. S5A, S7C).
Enriched CTCF peaks were identified in each replicate of Jpx LNA #1-associated control and Jpx KD samples. MACS2 (v2.1.1.2016309) was used to call the significant peaks with the default parameters (callpeak; band width=300, model fold = [5,50], q-value cutoff = 0.05). We only consider the regions where both biological replicates have a peak called in either control or Jpx KD, using the function “intersect” of BEDTools (-a rep1 bed file -b rep2 bed file). This resulted in two bed files, one for control and one for Jpx KD. We defined three categories of differential binding peaks (lost, shared and ectopic peaks) (Fig.4A). Lost and ectopic peaks were derived using intersect complement (BEDTools intersect -v). Shared peaks were derived using union of intersects (BEDTools intersect -wa; BEDTools intersect -wb; BEDTools merge).
RAD21 ChIP-seq libraries using Jpx LNA #1 were sequenced on the Illumina HiSeq 4000 (PE150) platform, generating 62–106 million paired-end reads per sample. Adaptor-trimming, removal of PCR duplicates and alignment using Novoalign (v4.02.01) were performed exactly as CTCF ChIP-seq data processing above. The resulting composite bam files having unique reads were merged from two biological replicated in order to increase the sequencing depth, and then used to generate coverage and call peaks. Coverages (log2 fold-enrichment estimate) were generated using SPP (v1.11) with the same command and parameters as CTCF. Enriched RAD21 peaks were called using MACS2 (v2.1.1.2016309) with the default parameters.
Analysis of peak coverage quartiles
To group Jpx or CTCF peak regions into quartiles based on the degree of their signals in wildtype (control) cells (Fig. 4C, 7A, 7B, S6G, S7M, S7N), we used “Method 1” described above. We made one modification to “Method 1” for CTCF peak quartiles: All regions with CTCF peaks in either control or Jpx KD cells (i.e. lost, shared and ectopic peak regions; See Fig. 4A) were taken into account. This required us to use a non-peak based signalValue because signalValues are absent (null) for ectopic peak regions in control cells. The signalValues used were log2 fold-enrichment estimates over input, generated by SPP.
Hierarchical cluster analysis
For hierarchical cluster analysis of CTCF signal around Jpx peaks (Fig. 4E), we used the non-peak centric SPP-generated CTCF signal instead of the CTCF peak coverages. This was done so that we could identify all clusters including those with little CTCF signal. Jpx peaks were extended ±10 kb from the center of Jpx peaks (Jpx peak regions). Cluster analysis was performed using deepTools with the reference point as the center of Jpx peaks (computeMatrix reference-point -bs=50; plotProfile --hclust 5). For further analysis of the clusters (Fig. 4F, S6C, S6E), control or Jpx KD CTCF coverages generated by SPP were integrated by breadth of their bins, and then summed up over the indicated Jpx peak regions (cluster 5, class I or II), as described above.
De novo motif analysis
Ectopic CTCF motifs (Fig. 4B) were analyzed using the software MEME-ChIP (v4.10.1) from the MEME Suite (http://meme-suite.org/index.html) (Machanick and Bailey, 2011). De novo motif analysis for Jpx binding sites (Fig. 3A, S4) were performed MEME-ChIP (v5.3.0) with the threshold of E-value (adjusted p-value) < 0.05. Jpx peaks longer than 8 bp were used as input with the parameters “-minw 6 -maxw 20 -meme-mod anr”.
For each Jpx binding motif, the similarity to the binding motifs of the known vertebrate transcription factors (JASPAR database, Uniprobe database and the previous report (Jolma et al., 2013) were assessed by Tomtom software (v5.3.0) (https://meme-suite.org/meme/tools/tomtom) with the parameters “-min-overlap 5 -dist pearson”. We included up to three non-redundant motifs matched with each Jpx motif (p-value for match < 0.05) in Fig. S4.
To investigate the relationship between de novo Jpx motifs (Fig. S4) and Jpx peak clusters (Fig. 4E), we determined which motifs were enriched in each cluster (Table S7). Significant enrichment was determined using a cut-off of p-value < 0.001. P-values were calculated by the software AME (Analysis of Motif Enrichment) (https://meme-suite.org/meme/tools/ame) from MEME suite (v4.10.1) with the parameters “ame --pvalue-report-threshold 1 --verbose 1 --scoring avg --method fisher”.
In situ Hi-C
In situ Hi-C was performed essentially as described (Rao et al., 2014; Wang et al., 2018) with minor modifications. Specifically, differentiating (d7) ES cells were transfected with scrambled or Jpx-targeting LNA #1 using the transfection method described in LNA-mediated knockdown. At 8hr after transfection, 3 million cells for each sample were crosslinked with 1 % formaldehyde in complete medium (1 million cells/ 1 mL) for 10 min at RT, quenched with 0.125 M glycine for 5 min at RT, washed with cold PBS and stored at −80°C with snap-freezing in LN2. Cells were lysed in 250 μl of Hi-C lysis buffer (10 mM Tris-Cl pH 8.0, 10 mM NaCl, 0.2% NP-40) supplemented with 50 ul of PIC (Sigma, P8340) for 30 min on ice. The nuclei pellet was collected by centrifugation at 2500 g for 5 min, washed once with 500 μl of ice-cold Hi-C lysis buffer, incubated in 50 μl of 0.5% SDS buffer (0.5% SDS, 10mM Tris-Cl pH8.0) at 62°C for 10 min, and then further incubated at 37°C for 15 min after 145 μl of DW and 25 μl of 10% TritonX-100 were added. To digest the nuclei, 25.3 μl of 10X NEBuffer2 and 200 U (8 ul) of MboI restriction enzyme (25 U/μl; NEB, R0147M) were added to the resuspended nuclei. After incubation on thermomixer with 900 rpm at 37°C overnight, MboI was inactivated at 62°C for 20 min. The resulting nuclei in the digestion buffer was subjected to the fill-in reaction with biotin-14-dATP (19524016, Thermo Fisher Scientific) by adding 50 μl of fill-in master mix (15 mM of bioin-14-dATP, 15 mM of dCTP, 15 mM of dGTP, 15 mM of dTTP, 40 U of DNA Polymerase I, Large (Klenow) Fragment (NEB, M0210S)). After incubation with rotation at 37°C for 1 hr, ligation was performed by adding 900 ul of ligation master mix (120 μl of 10X NEB T4 DNA ligase buffer, 100 μl of 10% Triton X-100, 6 μl of 20mg/ml BSA (NEB, B9000S), 2000 U of T4 DNA ligase (NEB, M0202S), up to 900ul of water) at RT for 4 hr with rotation. The nuclei pellet was collected by centrifugation at 4000 rpm for 5 min, incubated in TES buffer (50 mM Tris pH 8.0, 10 mM EDTA pH 8.0, 1% SDS) with Proteinase K at a concentration of 1 μg/μl at 55°C for 1 hr, supplied with NaCl at a concentration of 0.3 M, and then subjected to decrosslinking at 65°C overnight. DNA was purified by phenol/chloroform extraction and ethanol precipitation, treated with RNase A (Thermo Fisher Scientific, 12091021) at a concentration of 200 ng/μL for 1 hr at 37°C, and purified again phenol/chloroform and ethanol precipitation. Purified DNA was sheared to 300–500 bp in microtube with a Covaris E220 for 110 s (10% duty cycle, 140W peak incident power, 200 cycles/burst), and subjected to double-size selection using AMPure beads (0.55X-0.8X).
Pull-down of biotinylated ligation fragments and library preparation were performed exactly as Rao et al. (Rao et al., 2014) performed. The exceptions are i) 30 μL of Dynabeads MyOne Streptavidin C1 beads was used per sample, ii) After adaptor-ligation with 3 μl of 15 μM NEBNext adaptor for Illumina (included in NEBNext Multiplex Oligos for Illumina; NEB, E7335S) at RT for 15 min, the C1 beads were separated o n a magnet, washed with 600 μl of 1X TWB (5mM Tris-HCl, pH 7.5, 0.5mM EDTA, 1M NaCl, 0.05% Tween 20) twice at 55°C for 2 min and washed briefly with 100 μl of 1X NEBuffer 2.1 at RT. The resulting C1 beads were resuspended in 50 μl of 1X NEBuffer 2.1 with 3 μl USER enzyme (included in NEBNext Multiplex Oligos for Illumina; NEB, E7335S) and incubated at 37°C for 20 min with rotation. After the buffer was discarded, the beads were resuspended in 100 μl of 1X PCR master mix [50 μl of Phusion High-Fidelity PCR Master Mix with HF Buffer (NEB, 0531S), 2.5 μl of universal primer (NEBNext Multiplex Oligos for Illumina; NEB, E7335S), 2.5 μl of index primer (NEBNext Multiplex Oligos for Illumina; NEB, E7335S, 45 μl of water]. In situ Hi-C libraries were directly amplified from the beads with 12 cycles. Libraries were generated in two biological replicates.
In situ Hi-C data processing
In situ Hi-C libraries were sequenced on the Illumina HiSeq 4000 (PE150) platforms. See Table S6 for Hi-C statistics. Illumina adaptor-trimmed reads were further trimmed with the options “-a GATCGATC (MboI ligation junction) and -m 20”. Individual reads of each pair were separately mapped to the mus and cas genomes (mm9) using Novoalign (v4.02.01) with the parameters “-F STDFQ -r None”. Composite reads having only uniquely aligned reads were generated as described in CHART-seq data processing, and then merged into Hi-C summary files and filtered using HOMER (v4.10) with the parameters “makeTagDirectory -tbp1; makeTagDirectory -update -removePEbg -restrictionSite GATC -both -removeSelfLigation” as previously described (Kriz et al., 2021; Minajigi et al., 2015). To increase sequencing depth, HOMER tag directories from the two biological replicates were combined. To obtain the same number of valid contacts for control and Jpx-depleted cells, down-sampling of the larger HOMER tag file (control sample) was performed as follows. First, the linux command shuf (coreutils/8.27) was used to randomize tags. Then, because there are paired-tags in HOMER directory, both paired-tags were removed until the number of tags were reduced to the same size of the smaller tag file. As a result, we used the exact same number (898,009,206) of valid contacts for both control and Jpx KD Hi-C. Hi-C contact maps in “hic” format were generated using the command “Juicer.sh pre mm9” of Juicer tools (v1.5.3)(Durand et al., 2016b), and visualized with the “Balanced (KR)” normalization option using the Juicebox tool (v1.9.8) (Durand et al., 2016a).
In situ Hi-C Loop analysis
HiCCUPS (GPU version from Juicer (v1.7.6)) was used to call loops with the following parameters: “-m 500 -k KR -r 5000 -f 0.1 -p 4 -i 7 -d 20000” for 5 kb resolution and “-m 500 -k KR -r 20000 -f 0.1 -p 2 -i 5 -d 40000” for 20 kb resolution (Durand et al., 2016b). Then we determined shared and distinct (ectopic or lost) loops after calling loops from control and Jpx-depleted cells. Because looping interactions appear as a punctate signal spanning several pixels, the loops were considered shared loops when the anchors of control loops and Jpx KD loops are <40kb apart. Ectopic and lost loops were defined as Jpx KD loops and control loops excluding shared loops, respectively. Shared loops for control or Jpx KD were derived using the function “intersect” of the software pgltools (Greenwald et al., 2017). Then, lost and ectopic loops were derived using intersect complement (pgltools intersect -v).
As a meta-loop analysis, APA was performed with parameters “-n 30 -w 10 -r 5000 -q 3 -k KR” for the loops called at 5 kb resolution using Juicer (v1.5.3) (Durand et al., 2016b). Center-normed APA is generated by dividing all entries of each submatrix by the mean value of the central pixel (https://github.com/aidenlab/juicer/wiki/APA). Therefore, a high contrast ratio to the nearby regions is important to interpret the strength of enrichment at the center. APA and Center-Normed APA matrix values were imported and presented as heat map using the R package qplots (v3.0.1). Nearest-neighbor (NN) search was conducted using the function “closest1D” of pgltools. To generate random loop anchors, individual coordinates of each NN lost loop anchors were randomly shuffled. The NN distance between them was determined by calculating the shortest distance (the hypotenuse of the triangle). In order to examine the dependence of the loss of loops on the gain of loops, the R package spatstat (v1.54–0)(https://spatstat.org/about.html) was used. A plot of the density of ectopic loop anchors (x) versus density of lost loop anchors ρ(x) was generated with the assumption that x=Z(u) where Z=covariate and u=locus. Rho (ρ) was computed by the ratio method.
To examine whether HighOc CTCF motif is enriched in shared loops (Fig. S7D), we first generated the Position Weight Matrices (PWM) for constitutive HighOc and regulated LowOc CTCF sites with top 10 % scores as previously described (Plasschaert et al., 2014) by using MEME (v5.3.0) with the parameters “ -mod zoops -minw 6 -maxw 50 -objfun classic -revcomp -markov_order 0”. Using this PWM, the software AME (Analysis of Motif Enrichment) (https://meme-suite.org/meme/tools/ame)(v5.3.0) was employed to test the relative enrichment of constitutive HighOc motif in shared loops to that in in lost or ectopic loops.
To investigate the directionality of CTCF pairs in loop anchor sites (Fig. 6F), we adapted the method described in (Rao et al., 2014) with modification. To assign a single motif to each loop anchor, the strongest CTCF peak among multiple CTCF peak candidates was chosen for each loop anchor (±10 kb) and matched to the CTCF consensus motif (shown in Fig.3A) with significance (p-value < 1e-5). CTCF motif search was conducted using PWMscan (https://ccg.epfl.ch/pwmtools/pwmscan.php) and CTCF PWM from JasparDB 2020 vertebrate (MA0139) with default parameters (p-value < 1e-5) (Ambrosini et al., 2018). CTCF peaks with no significance (p-value > 1e-5) for motif match were not considered. In this manner, we only selected a pair of CTCF peaks each of which has a motif with significant confidence in order to ensure directions. As a result, we obtained total 8,589 of CTCF pairs for shared loop anchors, a total of 6,007 of CTCF pairs for ectopic loop anchors, and a total 5,609 of CTCF pairs for lost loop anchors. Among total CTCF pairs for shared, ectopic, lost loop anchors, the directions of CTCF motif pairs were classified as convergent, divergent, or tandem orientation.
3C assay
Differentiating (d7) ES cells (WT P2 or mutant P2) were transfected with scrambled or Jpx LNA #1 as described in LNA-mediated knockdown. At 8hr after transfection, 3 million cells for each sample were crosslinked with 1% formaldehyde in complete medium (one million cells/ 1 mL) for 10 min at RT and quenched with 0.125 M glycine for 5 min at RT. To construct in situ 3C library, we essentially performed in situ Hi-C (Rao et al., 2014) described above, omitting the biotin fill-in step. Briefly, nuclei collected from 3 million cells for each condition were digested with 200 U MboI (NEB, R0147M) at 37°C overnight. After heat inactivation of enzyme next day, ligation was performed with 10,000 U T4 DNA ligase (NEB, M0202L) for 4 hr at RT, followed by Proteinase K treatment at 55°C for 1 hr and overnight decrosslinking at 68°C. DNA was purified by phenol/chloroform extraction and ethanol precipitation, treated with RNase A at a concentration of 200 ng/μL for 1 hr at 37°C, and purified again by phenol/chloroform and ethanol precipitation. In order to generate the positive control for comparison of size on agarose gel, 10 μg of the BAC 399K20 (Augui et al., 2007) clone was digested with 100 U of Sau3AI (NEB, R0169S) and randomly ligated with 20 U of T4 DNA ligase (Promega, M1794), as previously described (Naumova et al., 2012). 3C-PCR primers were unidirectionally designed to target the region 40~60 bp away from the restriction site (Hagege et al., 2007; Naumova et al., 2012). Because there are 6 MboI sites between the P2 and Xist 5’, we were able to distinguish the interactions mediated by the P2 vs. Xist 5’ CTCF. 3C primers (shown in Fig. S7E) are listed in Table S8. The primers targeting a Gapdh region (Table S8) (Li et al., 2014) were used to control the amount of input DNA and normalize the interactions.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification methods, statistical parameters and the exact values of p and Pearson’s r are described in figures, figure legends, associated method details, or main texts. Randomized controls were generated to compare the same number of data points. Statistical tests were performed using R or GraphPad Prism software. A one-sided alternative hypothesis was used for the KS test, Student’s t test and Wilcoxon ranked sum test. Statistical significance was determined by the value of p or adjusted p < 0.05 unless otherwise indicated.
Supplementary Material
Figure S1. Characterization of Jpx CHART-seq.
Related to Figure 1
(A) Schematic illustration of Jpx showing alternative splice forms. RefSeq for Jpx shows three exons, but splice variants also occur (Johnston et al., 2002). Red bars, Jpx CHART probes for Jpx exon sequences common to all variants.
(B) RT-qPCR measuring Jpx and the negative control Gapdh RNAs enriched by Jpx CHART probes or the antisense CHART probes in d7 ES cells. Enrichment level is presented as the percentage relative to input RNA. Jpx CHART shows strand-specific enrichment of Jpx RNA and clean background. Data are means ± standard error of the mean (SEM) from three independent experiments.
(C) Statistics and sequencing depth for CHART-seq analysis in three independent biological replicates, as indicated by the numbers of mapped reads.
(D) Input-subtracted d0, d3 and d7 Jpx CHART coverages for the Jpx locus in ES cells.
(E) Rep3 of the Jpx CHART shown in Figure 1A.
(F) Input-subtracted coverage tracks across the whole genome for the indicated CHART experiments in undifferentiated (d0) ES cells. Comparison of Jpx CHART to either antisense or no-RNase H CHART validates the specificity of Jpx CHART.
(G) Correlation analyses of input-subtracted Jpx CHART coverages between 3 independent biological replicates (top), compared to the negative control experiments (antisense or no-RNaseH CHART) (bottom), as indicated. Bin, 10 kb. Pearson’s correlation coefficients (r) for all dots (black) and red dots within the range of 0–100 linear scale (red) are indicated in each plot.
Figure S2. Jpx RNA localization is not restricted to the X chromosome and occurs at thousands of genomic sites.
Related to Figure 1
(A) Number of d0, d3 and d7 Jpx peaks called from various Jpx, AS, and no-RNase H replicates in CHART experiments. Note three biological replicates were performed for d7 ES cells
(B) Correlation analyses of Jpx CHART coverages between different differentiation timepoints.
(C) Input-subtracted d7 Jpx coverage on the entire genome are shown with the indicated genomic features. SINE, LINE1 and LAD coverage values were calculated over 200 kb bins. Jpx peaks (d7) described in Fig.1C are also displayed. The mean option for IGV windowing function was chosen to display correlation versus anti-correlation.
(D) Related to (C). Input-subtracted d7 Jpx coverage along X chromosome is shown with indicated genomic features. Jpx CHART coverage zoomed on X chromosome clearly reveals that Jpx CHART is enriched in the regions associated with SINE, but not LINE1 or LAD. SINE, LINE1 and LAD coverages were computed over 200 kb bins. IGV windowing function is mean, as described in (C).
(E) Examples of Jpx RNA-FISH images obtained from d7 ES cells transfected with LNA targeting Jpx or harboring control/scrambled sequence for 8 hr. Nuclei were stained with DAPI.
Figure S3. The effect of Jpx depletion on global gene expression.
Related to Figure 2
(A) 2D Kernel density scatterplots comparing log2 counts per million (CPM) mapped reads for each gene (dots) between two biological replicates, as determined by RNA-seq analysis using Jpx LNA#1 (left) and LNA#2 (right). Two biological replicates for control or Jpx KD cells shown. Pearson’s correlation coefficients (r) indicated.
(B) 2D Kernel density scatterplots showing good correlation between RNA-seq results using two distinct Jpx LNAs. Log2 CPM for each gene between Jpx LNA#1-versus Jpx LNA#2-treated cells shown. Pearson’s correlation coefficients (r) indicated.
(C) Number of genes (gene bodies ±3 kb) with Jpx peaks shown for each CHART replicate, or union of two replicates (merged datasets), or the intersect (overlapping) between two replicates. Number for random genes is the averaged value from 100 random samplings from non-DEGs.
(D) Percentage of genes with Jpx peak regions. P-values, Wilcoxon ranked sum test.
(E) Gene Ontology (Biological Process) analysis showing 349 DEGs functionally categorized into general differentiation and development with a cut-off of FDR (adjusted p-value) <0.05, as determined by the Benjamini-Hochberg (BH) method. P-values for each GO term indicated. Heatmap representing log2 FC in expression for individual DEG co-clustered into the indicated GO terms. See also Table S4.
(F) Gene Ontology analysis showing 161 DEGs further subcategorized into neural development and differentiation with a cut-off of FDR <0.05, as determined by the Benjamini-Hochberg (BH) method. P-values for each GO term indicated. Heatmap shown, as described in Fig. 3SE. See also Table S4.
Figure S4. De novo motif analysis for Jpx-binding sites.
Related Figure 3
Using MEME-ChIP with a cut-off threshold of E-value < 0.05, 18 de novo motifs (column 2) were deduced under Jpx peaks. Using Tomtom software, significant matches (column 5) were identified for each de novo motif. Known motifs from Uniprobe, JASPAR, or Jolma 2013 databases were evaluated using Tomtom, with P-values for the match shown (column 6).
Figure S5. Jpx depletion results in increased CTCF binding over downregulated DEGs.
Related to Figure 3
(A) Increased CTCF coverage at the Xist P2 site (*) following Jpx depletion. Fragments per million (FPM)-normalized CTCF ChIP-seq combining data from Jpx LNA#1 and LNA#2 versus control cells.
(B) Cumulative distribution plots of Jpx peak coverage (log2 scale) for 546 DEGs with increased CTCF binding indicated in Fig. 3F against 546 genes randomly selected from non-DEGs. P-value, the KS test.
(C) Related to Fig. 3G. Genome browser view of d7 Jpx CHART-seq, CTCF ChIP-seq and RNA-seq data for for DEG, Tenm4. Significant increase in CTCF binding (shaded) and Tenm4 downregulation are reproducible between Jpx LNA#1 and #2.
Figure S6. Relationship between Jpx localization and CTCF binding.
Related to Figure 4
(A) Representative view for control CTCF ChIP coverage with enriched peaks relative to Jpx CHART peaks in each cluster shown in Fig. 4D.
(B) Class I and Class II subgroups within Cluster 5, with their respective numbers of Jpx peaks (±10 kb of peak center) and associated CTCF binding.
(C) Comparison of CTCF signal between control and Jpx KD CTCF ChIP for Class II of Cluster 5. Plotted are CTCF coverage values calculated over ±10 kb from the center of Jpx peaks grouped into Class II of Cluster 5. Black crossbar, mean. P-value determined by Wilcoxon ranked sum test.
(D) Representative IGV view for overlapping CTCF peaks from control and Jpx KD CTCF ChIP data in Class II of Cluster 5. Significant increase in the level of CTCF binding following Jpx depletion is shown compared to control CTCF coverage in two replicates.
(E) Related to Fig. 4E. Dotplot of CTCF coverages over Cluster 5-Class I peaks using independent Jpx LNA #2. Black crossbar, mean. P-value, Wilcoxon ranked sum test. Results are similar to those for Jpx LNA#1 (Fig. 4E).
(F) Cumulative distribution plot comparing peak coverage (log2 scale) of 2,619 CTCF peaks that were lost upon Jpx depletion between Jpx-target regions (±10 kb from Jpx peak’s center) and non-target regions (20 kb bins). P-values, KS test.
(G) Metagene plot showing coverage profiles of ectopic CTCF peaks around Jpx peak sites in a given quartile. Jpx peaks were divided into quartiles based on the degree of peak coverage in d7 wildtype ES cells. Q1, lowest quartile. Q4, highest quartile. Average ectopic CTCF peak coverages were calculated over 3 kb bins within ± 15 kb from the center of Jpx peaks. P-value, the KS test.
Figure S7. Shifting loops and cohesin (RAD21) binding.
(A) Representative Hi-C contact matrices displaying significant changes (indicated by arrows) in looping interactions, as shown by comparing control and Jpx-depleted cells. Anchor loci for lost and ectopic loops are indicated by blue and green squares, respectively. Hi-C resolution, 5kb. White and Red squares, minimum and maximum intensity, respectively.
(B) Comparing analysis at 5 kb vs. 20 kb resolution. Plot comparing Jpx peak coverage in d7 WT cells over anchors associated with ectopic loops vs. lost loops. Vertical dash lines indicate the 20 kb anchor region. See Fig. 6G for comparison to analysis at 5 kb resolution.
(C) Plot comparing CTCF coverage at ds-CTCF sites over anchors associated with ectopic loops in control vs. Jpx-depleted cells when loops were called at 20 kb resolution. Vertical dash lines indicate the 20 kb anchor region. See Fig. 6I for comparison to analysis at 5 kb resolution.
(D) Plot showing relative enrichment of HighOc motifs in shared loop anchors when compared to lost or ectopic loop anchors. −Log (adjusted p-values) were determined using AME software.
(E) Map of the X-inactivation center showing known interactions between Ftx and Xist (van Bemmel et al., 2019).
(F) Hi-C contact map showing looping interactions between Ftx and Xist promoter in both control and Jpx-depleted cells.
(G) ChIP-qPCR showing that CTCF binding (relative ratio shown) is lost when Xist P2 site is mutated. Mean ± SEM from three independent experiments shown. P-value, Student’s t test.
(H) 3C-qPCR showing increased P2-Ftx interactions when Jpx was depleted using Jpx LNA#1, and this dependence on Jpx was lost when the Xist P2 motif was mutated. Mean ± SEM from three independent experiments shown. P-value, Student’s t test. Interaction frequencies were normalized to Gapdh.
(I) 3C-qPCR showing a concomitant decrease in Xist5-Ftx interactions when the loop shifted to Xist P2. This shift was also dependent on the P2 motif. Mean ± SEM from three independent experiments shown. P-value, Student’s t test.
(J) Venn diagram representing RAD21 bindings sites that are shared between control and Jpx-depleted cells, or present in only control or Jpx-depleted cells. RAD21 peaks were called from two biological replicates that were merged.
(K) Number of RAD21 sites with or without colocalizing CTCF in each indicated cells. N, total number of RAD21 sites. P-value, Fisher’s exact test.
(L) Number of CTCF sites with or without colocalizing RAD21 in each indicated cells. N, total number of CTCF sites. P-value, Fisher’s exact test.
(M) Colocalization of RAD21 and CTCF. All CTCF peak sites (lost, shared, and ectopic peak sites; See Fig. 4A) were divided into quartiles based on peak coverage. Number of colocalizing RAD21 peaks is shown for each quartile.
(N) Related to Fig.7B. Cumulative distribution plot showing log2 FC in RAD21 peak coverage over Q1, Q2, Q3 and Q4 CTCF sites. P-value, the KS test.
Table S3. Day 7 Jpx CHART peaks.
Related to Figure 1.
Table S1–S3 Jpx peak regions in d0, d3 and d7 ES cells were annotated in the mm9 genome using the utility ‘annotatePeaks.pl’ in the software HOMER (v4.10).
Table S4. DEGs and enriched GO categories.
Related to Figure 2. Lists of significantly downregulated DEGs, upregulated DEGs and enriched GO categories, as identified with a cut-off of FDR <0.05.
Table S5. Control and Jpx KD CTCF ChIP peaks.
Related to Figure 4. Regions of differential binding peaks (lost, shared and ectopic peaks). See also Figure 4A.
Table S6. Hi-C statistics.
Related to Figures 6 and 7. Hi-C data processing statistics for Control and Jpx KD samples.
Table S7. De novo Jpx motifs enriched in Jpx peak clusters.
Related to Figure 4. Enrichment of Jpx motifs (Fig. S4) in Jpx peak clusters (Fig. 4D), as determined using the software AME from MEME suite (v4.10.1). A cut-off of p-value < 0.001 was used to determine significant enrichment.
Table S8. List of oligonucleotides.
Related to STAR Methods.
HIGHLIGHT.
Jpx RNA binds and activates hundreds of mammalian genes in the mouse genome.
Jpx selectively evicts CTCF bound to low-affinity, developmentally sensitive sites.
Jpx loss causes genome-wide shifts in CTCF binding and chromosome looping.
Jpx determines anchor site usage by serving as a CTCF release factor.
Jpx RNA regulates chromatin architecture via CTCF on autosomes
ACKNOWLEDGMENTS
We thank R. Blum for help with bioinformatic analyses, Y. Jeon for experimental advice, and all lab members for support and stimulating discussions. This work was supported by the National Research Foundation of Korea (NRF) grant to H.J.O. and a grant from the National Institutes of Health (R37-GM58839) and Howard Hughes Medical Institute to J.T.L.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
J.T.L. is a cofounder of Fulcrum Therapeutics and is an advisor to Skyhawk Therapeutics.
REFERENCES
- Alipour E, and Marko JF (2012). Self-organization of domain structures by DNA-loop-extruding enzymes. Nucleic Acids Research 40, 11202–11212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrosini G, Groux R, and Bucher P (2018). PWMScan: a fast tool for scanning entire genomes with a position-specific weight matrix. Bioinformatics 34, 2483–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augui S, Filion GJ, Huart S, Nora E, Guggiari M, Maresca M, Stewart AF, and Heard E (2007). Sensing X Chromosome Pairs Before X Inactivation via a Novel X-Pairing Region of the Xic. Science 318, 1632–1636. [DOI] [PubMed] [Google Scholar]
- Bell AC, and Felsenfeld G (2000). Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 405, 482–485. [DOI] [PubMed] [Google Scholar]
- Berkower I, Leis J, and Hurwitz J (1973). Isolation and characterization of an endonuclease from Escherichia coli specific for ribonucleic acid in ribonucleic acid-deoxyribonucleic acid hybrid structures. J Biol Chem 248, 5914–5921. [PubMed] [Google Scholar]
- Carmona S, Lin B, Chou T, Arroyo K, and Sun S (2018). LncRNA Jpx induces Xist expression in mice using both trans and cis mechanisms. PLoS Genet 14, e1007378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao W, Huynh KD, Spencer RJ, Davidow LS, and Lee JT (2002). CTCF, a candidate trans-acting factor for X-inactivation choice. Science 295, 345–347. [DOI] [PubMed] [Google Scholar]
- Chillon I, Marcia M, Legiewicz M, Liu F, Somarowthu S, and Pyle AM (2015). Native Purification and Analysis of Long RNAs. Methods Enzymol 558, 3–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow JC, Hall LL, Clemson CM, Lawrence JB, and Brown CJ (2003). Characterization of expression at the human XIST locus in somatic, embryonal carcinoma, and transgenic cell lines. Genomics 82, 309–322. [DOI] [PubMed] [Google Scholar]
- Chu C, Qu K, Zhong FL, Artandi SE, and Chang HY (2011). Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol Cell 44, 667–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu HP, Cifuentes-Rojas C, Kesner B, Aeby E, Lee HG, Wei C, Oh HJ, Boukhali M, Haas W, and Lee JT (2017). TERRA RNA Antagonizes ATRX and Protects Telomeres. Cell 170, 86–101.e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chureau C, Prissette M, Bourdet A, Barbe V, Cattolico L, Jones L, Eggen A, Avner P, and Duret L (2002). Comparative sequence analysis of the X-inactivation center region in mouse, human, and bovine. Genome Res 12, 894–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Rocha ST, and Heard E (2017). Novel players in X inactivation: insights into Xist-mediated gene silencing and chromosome conformation. Nat Struct Mol Biol 24, 197–204. [DOI] [PubMed] [Google Scholar]
- Davidson IF, Bauer B, Goetz D, Tang W, Wutz G, and Peters JM (2019). DNA loop extrusion by human cohesin. Science. [DOI] [PubMed] [Google Scholar]
- Davuluri RV, Suzuki Y, Sugano S, Plass C, and Huang TH (2008). The functional consequences of alternative promoter use in mammalian genomes. Trends Genet 24, 167–177. [DOI] [PubMed] [Google Scholar]
- Dekker J, and Mirny L (2016). The 3D Genome as Moderator of Chromosomal Communication. Cell 164, 1110–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Rosario BC, Kriz AJ, Del Rosario AM, Anselmo A, Fry CJ, White FM, Sadreyev RI, and Lee JT (2019). Exploration of CTCF post-translation modifications uncovers Serine-224 phosphorylation by PLK1 at pericentric regions during the G2/M transition. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disteche CM (2016). Dosage compensation of the sex chromosomes and autosomes. Semin Cell Dev Biol 56, 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, and Ren B (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donohoe ME, Zhang LF, Xu N, Shi Y, and Lee JT (2007). Identification of a Ctcf cofactor, Yy1, for the X chromosome binary switch. Mol Cell 25, 43–56. [DOI] [PubMed] [Google Scholar]
- Durand NC, Robinson JT, Shamim MS, Machol I, Mesirov JP, Lander ES, and Aiden EL (2016a). Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst 3, 99–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand NC, Shamim MS, Machol I, Rao SS, Huntley MH, Lander ES, and Aiden EL (2016b). Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst 3, 95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fudenberg G, Imakaev M, Lu C, Goloborodko A, Abdennur N, and Mirny LA (2016). Formation of Chromosomal Domains by Loop Extrusion. Cell Rep 15, 2038–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwald WW, Li H, Smith EN, Benaglio P, Nariai N, and Frazer KA (2017). Pgltools: a genomic arithmetic tool suite for manipulation of Hi-C peak and other chromatin interaction data. BMC Bioinformatics 18, 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haarhuis JHI, van der Weide RH, Blomen VA, Yanez-Cuna JO, Amendola M, van Ruiten MS, Krijger PHL, Teunissen H, Medema RH, van Steensel B, et al. (2017). The Cohesin Release Factor WAPL Restricts Chromatin Loop Extension. Cell 169, 693–707 e614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagege H, Klous P, Braem C, Splinter E, Dekker J, Cathala G, de Laat W, and Forne T (2007). Quantitative analysis of chromosome conformation capture assays (3C-qPCR). Nat Protoc 2, 1722–1733. [DOI] [PubMed] [Google Scholar]
- Handoko L, Xu H, Li G, Ngan CY, Chew E, Schnapp M, Lee CW, Ye C, Ping JL, Mulawadi F, et al. (2011). CTCF-mediated functional chromatin interactome in pluripotent cells. Nat Genet 43, 630–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen AS, Cattoglio C, Darzacq X, and Tjian R (2018). Recent evidence that TADs and chromatin loops are dynamic structures. Nucleus 9, 20–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen AS, Hsieh T-HS, Cattoglio C, Pustova I, Saldaña-Meyer R, Reinberg D, Darzacq X, and Tjian R (2019). Distinct Classes of Chromatin Loops Revealed by Deletion of an RNA-Binding Region in CTCF. Molecular Cell 76, 395–411.e313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen AS, Pustova I, Cattoglio C, Tjian R, and Darzacq X (2017). CTCF and cohesin regulate chromatin loop stability with distinct dynamics. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hark AT, Schoenherr CJ, Katz DJ, Ingram RS, Levorse JM, and Tilghman SM (2000). CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature 405, 486–489. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmgren C, Kanduri C, Dell G, Ward A, Mukhopadhya R, Kanduri M, Lobanenkov V, and Ohlsson R (2001). CpG methylation regulates the Igf2/H19 insulator. Curr Biol 11, 1128–1130. [DOI] [PubMed] [Google Scholar]
- Jegu T, Aeby E, and Lee JT (2017). The X chromosome in space. Nat Rev Genet 18, 377–389. [DOI] [PubMed] [Google Scholar]
- Jeon Y, and Lee JT (2011). YY1 tethers Xist RNA to the inactive X nucleation center. Cell 146, 119–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston CM, Newall AE, Brockdorff N, and Nesterova TB (2002). Enox, a novel gene that maps 10 kb upstream of Xist and partially escapes X inactivation. Genomics 80, 236–244. [DOI] [PubMed] [Google Scholar]
- Jolma A, Yan J, Whitington T, Toivonen J, Nitta KR, Rastas P, Morgunova E, Enge M, Taipale M, Wei G, et al. (2013). DNA-binding specificities of human transcription factors. Cell 152, 327–339. [DOI] [PubMed] [Google Scholar]
- Karner HM, Webb CH, Carmona S, Liu Y, Lin B, Erhard M, Chan D, Baldi P, Spitale RC, and Sun S (2019). Functional Conservation of lncRNA JPX despite Sequence and Structural Divergence. J Mol Biol. [DOI] [PubMed] [Google Scholar]
- Kharchenko PV, Tolstorukov MY, and Park PJ (2008). Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nat Biotechnol 26, 1351–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TH, Abdullaev ZK, Smith AD, Ching KA, Loukinov DI, Green Roland D., Zhang MQ, Lobanenkov VV, and Ren B (2007). Analysis of the Vertebrate Insulator Protein CTCF-Binding Sites in the Human Genome. Cell 128, 1231–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Shi Z, Zhang H, Finkelstein IJ, and Yu H (2019). Human cohesin compacts DNA by loop extrusion. Science 366, 1345–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klenova EM, Chernukhin IV, El-Kady A, Lee RE, Pugacheva EM, Loukinov DI, Goodwin GH, Delgado D, Filippova GN, Leon J, et al. (2001). Functional phosphorylation sites in the C-terminal region of the multivalent multifunctional transcriptional factor CTCF. Mol Cell Biol 21, 2221–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klenova EM, Nicolas RH, Paterson HF, Carne AF, Heath CM, Goodwin GH, Neiman PE, and Lobanenkov VV (1993). CTCF, a conserved nuclear factor required for optimal transcriptional activity of the chicken c-myc gene, is an 11-Zn-finger protein differentially expressed in multiple forms. Mol Cell Biol 13, 7612–7624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriz AJ, Colognori D, Sunwoo H, Nabet B, and Lee JT (2021). Balancing cohesin eviction and retention prevents aberrant chromosomal interactions, Polycomb-mediated repression, and X-inactivation. Mol Cell 81, 1970–1987.e1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung JT, Kesner B, An JY, Ahn JY, Cifuentes-Rojas C, Colognori D, Jeon Y, Szanto A, del Rosario BC, Pinter SF, et al. (2015). Locus-specific targeting to the X chromosome revealed by the RNA interactome of CTCF. Mol Cell 57, 361–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurukuti S, Tiwari VK, Tavoosidana G, Pugacheva E, Murrell A, Zhao Z, Lobanenkov V, Reik W, and Ohlsson R (2006). CTCF binding at the H19 imprinting control region mediates maternally inherited higher-order chromatin conformation to restrict enhancer access to Igf2. Proc Natl Acad Sci U S A 103, 10684–10689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JT (2011). Gracefully ageing at 50, X-chromosome inactivation becomes a paradigm for RNA and chromatin control. Nat Rev Mol Cell Biol 12, 815–826. [DOI] [PubMed] [Google Scholar]
- Lee JT (2012). Epigenetic regulation by long noncoding RNAs. Science 338, 1435–1439. [DOI] [PubMed] [Google Scholar]
- Lee JT, and Lu N (1999). Targeted mutagenesis of Tsix leads to nonrandom X inactivation. Cell 99, 47–57. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Liang Y, LeBlanc M, Benner C, and Zheng Y (2014). Function of a Foxp3 cis - Element in Protecting Regulatory T Cell Identity. Cell 158, 734–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Haarhuis JHI, Sedeño Cacciatore Á, Oldenkamp R, van Ruiten MS, Willems L, Teunissen H, Muir KW, de Wit E, Rowland BD, et al. (2020). The structural basis for cohesin-CTCF-anchored loops. Nature 578, 472–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machanick P, and Bailey TL (2011). MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics 27, 1696–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacPherson MJ, Beatty LG, Zhou W, Du M, and Sadowski PD (2009). The CTCF insulator protein is posttranslationally modified by SUMO. Mol Cell Biol 29, 714–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. 2011 17, 3. [Google Scholar]
- Minajigi A, Froberg J, Wei C, Sunwoo H, Kesner B, Colognori D, Lessing D, Payer B, Boukhali M, Haas W, et al. (2015). Chromosomes. A comprehensive Xist interactome reveals cohesin repulsion and an RNA-directed chromosome conformation. Science 349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirny LA, Imakaev M, and Abdennur N (2019). Two major mechanisms of chromosome organization. Curr Opin Cell Biol 58, 142–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahashi H, Kieffer Kwon KR, Resch W, Vian L, Dose M, Stavreva D, Hakim O, Pruett N, Nelson S, Yamane A, et al. (2013). A genome-wide map of CTCF multivalency redefines the CTCF code. Cell Rep 3, 1678–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumova N, Smith EM, Zhan Y, and Dekker J (2012). Analysis of long-range chromatin interactions using Chromosome Conformation Capture. Methods 58, 192–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, et al. (2012). Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485, 381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa Y, Sun BK, and Lee JT (2008). Intersection of the RNA interference and X-inactivation pathways. Science 320, 1336–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlsson R, Renkawitz R, and Lobanenkov V (2001). CTCF is a uniquely versatile transcription regulator linked to epigenetics and disease. Trends Genet 17, 520–527. [DOI] [PubMed] [Google Scholar]
- Parekh S, Ziegenhain C, Vieth B, Enard W, and Hellmann I (2016). The impact of amplification on differential expression analyses by RNA-seq. Sci Rep 6, 25533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peric-Hupkes D, Meuleman W, Pagie L, Bruggeman SW, Solovei I, Brugman W, Graf S, Flicek P, Kerkhoven RM, van Lohuizen M, et al. (2010). Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol Cell 38, 603–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips-Cremins JE, and Corces VG (2013). Chromatin insulators: linking genome organization to cellular function. Mol Cell 50, 461–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips-Cremins JE, Sauria ME, Sanyal A, Gerasimova TI, Lajoie BR, Bell JS, Ong CT, Hookway TA, Guo C, Sun Y, et al. (2013). Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell 153, 1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinter SF, Sadreyev RI, Yildirim E, Jeon Y, Ohsumi TK, Borowsky M, and Lee JT (2012). Spreading of X chromosome inactivation via a hierarchy of defined Polycomb stations. Genome Res 22, 1864–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plasschaert RN, Vigneau S, Tempera I, Gupta R, Maksimoska J, Everett L, Davuluri R, Mamorstein R, Lieberman PM, Schultz D, et al. (2014). CTCF binding site sequence differences are associated with unique regulatory and functional trends during embryonic stem cell differentiation. Nucleic Acids Res 42, 774–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugacheva EM, Kubo N, Loukinov D, Tajmul M, Kang S, Kovalchuk AL, Strunnikov AV, Zentner GE, Ren B, and Lobanenkov VV (2020). CTCF mediates chromatin looping via N-terminal domain-dependent cohesin retention. Proc Natl Acad Sci U S A 117, 2020–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowley MJ, and Corces VG (2018). Organizational principles of 3D genome architecture. Nat Rev Genet 19, 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldana-Meyer R, Gonzalez-Buendia E, Guerrero G, Narendra V, Bonasio R, Recillas-Targa F, and Reinberg D (2014). CTCF regulates the human p53 gene through direct interaction with its natural antisense transcript, Wrap53. Genes Dev 28, 723–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldana-Meyer R, Rodriguez-Hernaez J, Escobar T, Nishana M, Jacome-Lopez K, Nora EP, Bruneau BG, Tsirigos A, Furlan-Magaril M, Skok J, et al. (2019). RNA Interactions Are Essential for CTCF-Mediated Genome Organization. Mol Cell 76, 412–422.e415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanborn AL, Rao SS, Huang SC, Durand NC, Huntley MH, Jewett AI, Bochkov ID, Chinnappan D, Cutkosky A, Li J, et al. (2015). Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc Natl Acad Sci U S A 112, E6456–6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Yue F, McCleary DF, Ye Z, Edsall L, Kuan S, Wagner U, Dixon J, Lee L, Lobanenkov VV, et al. (2012). A map of the cis-regulatory sequences in the mouse genome. Nature 488, 116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin H, Liu T, Manrai AK, and Liu XS (2009). CEAS: cis-regulatory element annotation system. Bioinformatics 25, 2605–2606. [DOI] [PubMed] [Google Scholar]
- Simon MD, Pinter SF, Fang R, Sarma K, Rutenberg-Schoenberg M, Bowman SK, Kesner BA, Maier VK, Kingston RE, and Lee JT (2013). High-resolution Xist binding maps reveal two-step spreading during X-chromosome inactivation. Nature 504, 465–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon MD, Wang CI, Kharchenko PV, West JA, Chapman BA, Alekseyenko AA, Borowsky ML, Kuroda MI, and Kingston RE (2011). The genomic binding sites of a noncoding RNA. Proc Natl Acad Sci U S A 108, 20497–20502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splinter E, Heath H, Kooren J, Palstra RJ, Klous P, Grosveld F, Galjart N, and de Laat W (2006). CTCF mediates long-range chromatin looping and local histone modification in the beta-globin locus. Genes Dev 20, 2349–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starmer J, and Magnuson T (2009). A new model for random X chromosome inactivation. Development 136, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Del Rosario BC, Szanto A, Ogawa Y, Jeon Y, and Lee JT (2013). Jpx RNA activates Xist by evicting CTCF. Cell 153, 1537–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Luo OJ, Li X, Zheng M, Zhu JJ, Szalaj P, Trzaskoma P, Magalska A, Wlodarczyk J, Ruszczycki B, et al. (2015). CTCF-Mediated Human 3D Genome Architecture Reveals Chromatin Topology for Transcription. Cell 163, 1611–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian D, Sun S, and Lee JT (2010). The long noncoding RNA, Jpx, is a molecular switch for X chromosome inactivation. Cell 143, 390–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, and Pachter L (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bemmel JG, Galupa R, Gard C, Servant N, Picard C, Davies J, Szempruch AJ, Zhan Y, Zylicz JJ, Nora EP, et al. (2019). The bipartite TAD organization of the X-inactivation center ensures opposing developmental regulation of Tsix and Xist. Nat Genet 51, 1024–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CY, Jegu T, Chu HP, Oh HJ, and Lee JT (2018). SMCHD1 Merges Chromosome Compartments and Assists Formation of Super-Structures on the Inactive X. Cell 174, 406–421.e425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Maurano MT, Qu H, Varley KE, Gertz J, Pauli F, Lee K, Canfield T, Weaver M, Sandstrom R, et al. (2012). Widespread plasticity in CTCF occupancy linked to DNA methylation. Genome Res 22, 1680–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu N, Donohoe ME, Silva SS, and Lee JT (2007). Evidence that homologous X-chromosome pairing requires transcription and Ctcf protein. Nat Genet 39, 1390–1396. [DOI] [PubMed] [Google Scholar]
- Yang F, Deng X, Ma W, Berletch JB, Rabaia N, Wei G, Moore JM, Filippova GN, Xu J, Liu Y, et al. (2015). The lncRNA Firre anchors the inactive X chromosome to the nucleolus by binding CTCF and maintains H3K27me3 methylation. Genome Biol 16, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young MD, Wakefield MJ, Smyth GK, and Oshlack A (2010). Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol 11, R14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Characterization of Jpx CHART-seq.
Related to Figure 1
(A) Schematic illustration of Jpx showing alternative splice forms. RefSeq for Jpx shows three exons, but splice variants also occur (Johnston et al., 2002). Red bars, Jpx CHART probes for Jpx exon sequences common to all variants.
(B) RT-qPCR measuring Jpx and the negative control Gapdh RNAs enriched by Jpx CHART probes or the antisense CHART probes in d7 ES cells. Enrichment level is presented as the percentage relative to input RNA. Jpx CHART shows strand-specific enrichment of Jpx RNA and clean background. Data are means ± standard error of the mean (SEM) from three independent experiments.
(C) Statistics and sequencing depth for CHART-seq analysis in three independent biological replicates, as indicated by the numbers of mapped reads.
(D) Input-subtracted d0, d3 and d7 Jpx CHART coverages for the Jpx locus in ES cells.
(E) Rep3 of the Jpx CHART shown in Figure 1A.
(F) Input-subtracted coverage tracks across the whole genome for the indicated CHART experiments in undifferentiated (d0) ES cells. Comparison of Jpx CHART to either antisense or no-RNase H CHART validates the specificity of Jpx CHART.
(G) Correlation analyses of input-subtracted Jpx CHART coverages between 3 independent biological replicates (top), compared to the negative control experiments (antisense or no-RNaseH CHART) (bottom), as indicated. Bin, 10 kb. Pearson’s correlation coefficients (r) for all dots (black) and red dots within the range of 0–100 linear scale (red) are indicated in each plot.
Figure S2. Jpx RNA localization is not restricted to the X chromosome and occurs at thousands of genomic sites.
Related to Figure 1
(A) Number of d0, d3 and d7 Jpx peaks called from various Jpx, AS, and no-RNase H replicates in CHART experiments. Note three biological replicates were performed for d7 ES cells
(B) Correlation analyses of Jpx CHART coverages between different differentiation timepoints.
(C) Input-subtracted d7 Jpx coverage on the entire genome are shown with the indicated genomic features. SINE, LINE1 and LAD coverage values were calculated over 200 kb bins. Jpx peaks (d7) described in Fig.1C are also displayed. The mean option for IGV windowing function was chosen to display correlation versus anti-correlation.
(D) Related to (C). Input-subtracted d7 Jpx coverage along X chromosome is shown with indicated genomic features. Jpx CHART coverage zoomed on X chromosome clearly reveals that Jpx CHART is enriched in the regions associated with SINE, but not LINE1 or LAD. SINE, LINE1 and LAD coverages were computed over 200 kb bins. IGV windowing function is mean, as described in (C).
(E) Examples of Jpx RNA-FISH images obtained from d7 ES cells transfected with LNA targeting Jpx or harboring control/scrambled sequence for 8 hr. Nuclei were stained with DAPI.
Figure S3. The effect of Jpx depletion on global gene expression.
Related to Figure 2
(A) 2D Kernel density scatterplots comparing log2 counts per million (CPM) mapped reads for each gene (dots) between two biological replicates, as determined by RNA-seq analysis using Jpx LNA#1 (left) and LNA#2 (right). Two biological replicates for control or Jpx KD cells shown. Pearson’s correlation coefficients (r) indicated.
(B) 2D Kernel density scatterplots showing good correlation between RNA-seq results using two distinct Jpx LNAs. Log2 CPM for each gene between Jpx LNA#1-versus Jpx LNA#2-treated cells shown. Pearson’s correlation coefficients (r) indicated.
(C) Number of genes (gene bodies ±3 kb) with Jpx peaks shown for each CHART replicate, or union of two replicates (merged datasets), or the intersect (overlapping) between two replicates. Number for random genes is the averaged value from 100 random samplings from non-DEGs.
(D) Percentage of genes with Jpx peak regions. P-values, Wilcoxon ranked sum test.
(E) Gene Ontology (Biological Process) analysis showing 349 DEGs functionally categorized into general differentiation and development with a cut-off of FDR (adjusted p-value) <0.05, as determined by the Benjamini-Hochberg (BH) method. P-values for each GO term indicated. Heatmap representing log2 FC in expression for individual DEG co-clustered into the indicated GO terms. See also Table S4.
(F) Gene Ontology analysis showing 161 DEGs further subcategorized into neural development and differentiation with a cut-off of FDR <0.05, as determined by the Benjamini-Hochberg (BH) method. P-values for each GO term indicated. Heatmap shown, as described in Fig. 3SE. See also Table S4.
Figure S4. De novo motif analysis for Jpx-binding sites.
Related Figure 3
Using MEME-ChIP with a cut-off threshold of E-value < 0.05, 18 de novo motifs (column 2) were deduced under Jpx peaks. Using Tomtom software, significant matches (column 5) were identified for each de novo motif. Known motifs from Uniprobe, JASPAR, or Jolma 2013 databases were evaluated using Tomtom, with P-values for the match shown (column 6).
Figure S5. Jpx depletion results in increased CTCF binding over downregulated DEGs.
Related to Figure 3
(A) Increased CTCF coverage at the Xist P2 site (*) following Jpx depletion. Fragments per million (FPM)-normalized CTCF ChIP-seq combining data from Jpx LNA#1 and LNA#2 versus control cells.
(B) Cumulative distribution plots of Jpx peak coverage (log2 scale) for 546 DEGs with increased CTCF binding indicated in Fig. 3F against 546 genes randomly selected from non-DEGs. P-value, the KS test.
(C) Related to Fig. 3G. Genome browser view of d7 Jpx CHART-seq, CTCF ChIP-seq and RNA-seq data for for DEG, Tenm4. Significant increase in CTCF binding (shaded) and Tenm4 downregulation are reproducible between Jpx LNA#1 and #2.
Figure S6. Relationship between Jpx localization and CTCF binding.
Related to Figure 4
(A) Representative view for control CTCF ChIP coverage with enriched peaks relative to Jpx CHART peaks in each cluster shown in Fig. 4D.
(B) Class I and Class II subgroups within Cluster 5, with their respective numbers of Jpx peaks (±10 kb of peak center) and associated CTCF binding.
(C) Comparison of CTCF signal between control and Jpx KD CTCF ChIP for Class II of Cluster 5. Plotted are CTCF coverage values calculated over ±10 kb from the center of Jpx peaks grouped into Class II of Cluster 5. Black crossbar, mean. P-value determined by Wilcoxon ranked sum test.
(D) Representative IGV view for overlapping CTCF peaks from control and Jpx KD CTCF ChIP data in Class II of Cluster 5. Significant increase in the level of CTCF binding following Jpx depletion is shown compared to control CTCF coverage in two replicates.
(E) Related to Fig. 4E. Dotplot of CTCF coverages over Cluster 5-Class I peaks using independent Jpx LNA #2. Black crossbar, mean. P-value, Wilcoxon ranked sum test. Results are similar to those for Jpx LNA#1 (Fig. 4E).
(F) Cumulative distribution plot comparing peak coverage (log2 scale) of 2,619 CTCF peaks that were lost upon Jpx depletion between Jpx-target regions (±10 kb from Jpx peak’s center) and non-target regions (20 kb bins). P-values, KS test.
(G) Metagene plot showing coverage profiles of ectopic CTCF peaks around Jpx peak sites in a given quartile. Jpx peaks were divided into quartiles based on the degree of peak coverage in d7 wildtype ES cells. Q1, lowest quartile. Q4, highest quartile. Average ectopic CTCF peak coverages were calculated over 3 kb bins within ± 15 kb from the center of Jpx peaks. P-value, the KS test.
Figure S7. Shifting loops and cohesin (RAD21) binding.
(A) Representative Hi-C contact matrices displaying significant changes (indicated by arrows) in looping interactions, as shown by comparing control and Jpx-depleted cells. Anchor loci for lost and ectopic loops are indicated by blue and green squares, respectively. Hi-C resolution, 5kb. White and Red squares, minimum and maximum intensity, respectively.
(B) Comparing analysis at 5 kb vs. 20 kb resolution. Plot comparing Jpx peak coverage in d7 WT cells over anchors associated with ectopic loops vs. lost loops. Vertical dash lines indicate the 20 kb anchor region. See Fig. 6G for comparison to analysis at 5 kb resolution.
(C) Plot comparing CTCF coverage at ds-CTCF sites over anchors associated with ectopic loops in control vs. Jpx-depleted cells when loops were called at 20 kb resolution. Vertical dash lines indicate the 20 kb anchor region. See Fig. 6I for comparison to analysis at 5 kb resolution.
(D) Plot showing relative enrichment of HighOc motifs in shared loop anchors when compared to lost or ectopic loop anchors. −Log (adjusted p-values) were determined using AME software.
(E) Map of the X-inactivation center showing known interactions between Ftx and Xist (van Bemmel et al., 2019).
(F) Hi-C contact map showing looping interactions between Ftx and Xist promoter in both control and Jpx-depleted cells.
(G) ChIP-qPCR showing that CTCF binding (relative ratio shown) is lost when Xist P2 site is mutated. Mean ± SEM from three independent experiments shown. P-value, Student’s t test.
(H) 3C-qPCR showing increased P2-Ftx interactions when Jpx was depleted using Jpx LNA#1, and this dependence on Jpx was lost when the Xist P2 motif was mutated. Mean ± SEM from three independent experiments shown. P-value, Student’s t test. Interaction frequencies were normalized to Gapdh.
(I) 3C-qPCR showing a concomitant decrease in Xist5-Ftx interactions when the loop shifted to Xist P2. This shift was also dependent on the P2 motif. Mean ± SEM from three independent experiments shown. P-value, Student’s t test.
(J) Venn diagram representing RAD21 bindings sites that are shared between control and Jpx-depleted cells, or present in only control or Jpx-depleted cells. RAD21 peaks were called from two biological replicates that were merged.
(K) Number of RAD21 sites with or without colocalizing CTCF in each indicated cells. N, total number of RAD21 sites. P-value, Fisher’s exact test.
(L) Number of CTCF sites with or without colocalizing RAD21 in each indicated cells. N, total number of CTCF sites. P-value, Fisher’s exact test.
(M) Colocalization of RAD21 and CTCF. All CTCF peak sites (lost, shared, and ectopic peak sites; See Fig. 4A) were divided into quartiles based on peak coverage. Number of colocalizing RAD21 peaks is shown for each quartile.
(N) Related to Fig.7B. Cumulative distribution plot showing log2 FC in RAD21 peak coverage over Q1, Q2, Q3 and Q4 CTCF sites. P-value, the KS test.
Table S3. Day 7 Jpx CHART peaks.
Related to Figure 1.
Table S1–S3 Jpx peak regions in d0, d3 and d7 ES cells were annotated in the mm9 genome using the utility ‘annotatePeaks.pl’ in the software HOMER (v4.10).
Table S4. DEGs and enriched GO categories.
Related to Figure 2. Lists of significantly downregulated DEGs, upregulated DEGs and enriched GO categories, as identified with a cut-off of FDR <0.05.
Table S5. Control and Jpx KD CTCF ChIP peaks.
Related to Figure 4. Regions of differential binding peaks (lost, shared and ectopic peaks). See also Figure 4A.
Table S6. Hi-C statistics.
Related to Figures 6 and 7. Hi-C data processing statistics for Control and Jpx KD samples.
Table S7. De novo Jpx motifs enriched in Jpx peak clusters.
Related to Figure 4. Enrichment of Jpx motifs (Fig. S4) in Jpx peak clusters (Fig. 4D), as determined using the software AME from MEME suite (v4.10.1). A cut-off of p-value < 0.001 was used to determine significant enrichment.
Table S8. List of oligonucleotides.
Related to STAR Methods.
Data Availability Statement
All raw and processed high-throughput sequencing data generated in this study have been deposited to GEO with accession number: GSE144056. Data are publicly available as of the date of publication. This paper also analyzed existing, publicly available data. All accession numbers are listed in the Key Resource Table.
This study does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key Resource Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-CTCF | Cell Signaling Technology | Cat#2899S; RRID:AB_2086794 |
| Rabbit polyclonal anti-RAD21 | Abcam | Cat#ab992; RRID:AB_2176601 |
| Chemicals, peptides, and recombinant proteins | ||
| Formaldehyde solution | Sigma-Aldrich | Cat#F8775 |
| Recombinant mouse LIF | Sigma-Aldrich | Cat#ESG1107 |
| Protector RNase Inhibitor | Sigma-Aldrich | Cat#3335402001 |
| cOmplete EDTA-free Protease Inhibitor Cocktail | Sigma-Aldrich | Cat#11873580001 |
| Protease Inhibitor Cocktail | Sigma-Aldrich | Cat#P8340 |
| Proteinase K | Sigma-Aldrich | Cat#03115844001 |
| RNase A | Thermo Fisher Scientific | Cat#12091021 |
| RNase H | New England Biolabs | Cat#M0297L |
| Superscript III Reverse Transcriptase | Thermo Fisher Scientific | Cat#18080085 |
| TRIzol | Thermo Fisher Scientific | Cat#15596018 |
| Turbo DNase | Thermo Fisher Scientific | Cat#AM2238 |
| Ribonucleoside vanadyl complex | New England Biolabs | Cat#S1402S |
| Critical commercial assays | ||
| Agencourt AMPure XP | Beckman Coulter | Cat#A63881 |
| Mouse ES Cell Nucleofector Kit | Lonza-Walkersville | Cat#VPH-1001 |
| NEBNext ChIP-Seq Library Prep Master Mix Set for Illumina | New England Biolabs | Cat#E6240S |
| NEBNext Ultra II directional RNA Second Strand Synthesis Module | New England Biolabs | Cat#E7550S |
| NEBNext Multiplex Oligos for Illumina | New England Biolabs | Cat#E7335S (Index Primers Set1) |
| NEBNext Multiplex Oligos for Illumina | New England Biolabs | Cat#E7500S (Index Primers Set2) |
| NEBNext Ultra II DNA Library Prep Kit for Illumina | New England Biolabs | Cat#E7645S |
| Ribominus Eukaryote Kit v2 | Thermo Fisher Scientific | Cat#A15020 |
| RNeasy MinElute Cleanup kit | QIAGEN | Cat#74204 |
| Deposited data | ||
| Jpx CHART-seq in d0 ES cells | This study | GEO: GSE144056 |
| Jpx CHART-seq in d3 differentiating ES cells | This study | GEO: GSE144056 |
| Jpx CHART-seq in d7 differentiating ES cells | This study | GEO: GSE144056 |
| H3K4me3 ChIP-seq in d0 ES cells | Pinter et al., 2012 | GEO: GSE36905 |
| H3K4me3 ChIP-seq in d7 differentiating ES cells | Pinter et al., 2012 | GEO: GSE36905 |
| H3K27me3 ChIP-seq in d0 ES cells | Pinter et al., 2012 | GEO: GSE36905 |
| H3K27me3 ChIP-seq in d7 differentiating ES cells | Pinter et al., 2012 | GEO: GSE36905 |
| LAD (Lamina-Associated Domain) regions | Peric-Hupkes et al., 2010 | GEO: GSE17051 |
| RNA-seq in d7 differentiating ES cells transfected with scrambled LNA (control for Jpx LNA #1) | This study | GEO: GSE144056 |
| Jpx KD RNA-seq in d7 differentiating ES cells transfected with Jpx LNA #1 | This study | GEO: GSE144056 |
| RNA-seq in d7 differentiating ES cells transfected with scrambled LNA (control for Jpx LNA #2) | This study | GEO: GSE144056 |
| Jpx KD RNA-seq in d7 differentiating ES cells transfected with Jpx LNA #2 | This study | GEO: GSE144056 |
| CTCF ChIP-seq in d7 differentiating ES cells transfected with scrambled LNA (control for Jpx LNA #1) | This study | GEO: GSE144056 |
| Jpx KD CTCF ChIP-seq in d7 differentiating ES cells transfected with Jpx LNA #1 | This study | GEO: GSE144056 |
| CTCF ChIP-seq in d7 differentiating ES cells transfected with scrambled LNA (control for Jpx LNA #2) | This study | GEO: GSE144056 |
| Jpx KD CTCF ChIP-seq in d7 differentiating ES cells transfected with Jpx LNA #2 | This study | GEO: GSE144056 |
| RAD21 ChIP-seq in d7 differentiating ES cells transfected with scrambled LNA (control for Jpx LNA #1) | This study | GEO: GSE144056 |
| Jpx KD RAD21 ChIP-seq in d7 differentiating ES cells transfected with Jpx LNA #1 | This study | GEO: GSE144056 |
| in situ Hi-C in d7 differentiating ES cells transfected with scrambled LNA (control for Jpx LNA #1) | This study | GEO: GSE144056 |
| Jpx KD in situ Hi-C in d7 differentiating ES cells transfected with Jpx LNA #1 | This study | GEO: GSE144056 |
| Experimental models: Cell lines | ||
| Mouse ES cells (female, 16.7 TsixTST/+) Strain: M. musculus/M. castaneus | Ogawa et al., 2008 | N/A |
| Xist P2-mutant mouse ES cells (female, 16.7 TsixTST/+) | This study | N/A |
| Oligonucleotides | ||
| 3′ biotin-TEG Jpx CAHRT probes, see Table S8 | Integrated DNA Technologies | N/A |
| 3′ biotin-TEG Jpx AS CHART (negative control) probes, see Table S8 | Integrated DNA Technologies | N/A |
| Antisense LNA GapmeR Control, see Table S8 | QIAGEN | Cat#339515 |
| Antisense LNA GapmeR Jpx LNAs, see Table S8 | QIAGEN | Sequences designed in this study; Cat#339517 |
| Primers used for RT-qPCR, see Table S8 | Integrated DNA Technologies | N/A |
| DNA EMSA probes, see Table S8 | Integrated DNA Technologies | N/A |
| Primers used to amplify Jpx (383 nt) for EMSA, see Table S8 | Integrated DNA Technologies | N/A |
| Xist P2 CTCF sgRNAs used to generate the P2-mutant cell line, see Table S8 | Integrated DNA Technologies | N/A |
| PCR screening primers used to generate the P2-mutant cell line, see Table S8 | Integrated DNA Technologies | N/A |
| Primers for 3C assay, see Table S8 | Integrated DNA Technologies | N/A |
| Recombinant DNA | ||
| pSpCas9-(BB)-2A-GFP (PX458) | Ran et al., 2013 | Addgene Cat#48138 |
| Software and algorithms | ||
| BEDTools v2.25.0 | Quinlan and Hall, 2010 | https://bedtools.readthedocs.io/en/latest/ |
| CEAS v1.0.2 | Shin et al., 2009 | N/A |
| Cutadapt v1.2.1 | Martin, 2011 | https://cutadapt.readthedocs.io/en/stable/# |
| Cutadapt v1.8.1 | Martin, 2011 | https://cutadapt.readthedocs.io/en/stable/# |
| Cufflinks v2.2.1 | Trapnell et al., 2012 | http://cole-trapnell-lab.github.io/cufflinks/ |
| deepTools v3.1.2 | Ramírez et al., 2016 | https://deeptools.readthedocs.io/en/develop/ |
| Homer v4.8 | Heinz et al., 2010 | http://homer.ucsd.edu/homer/ngs/ |
| Homer v4.10 | Heinz et al., 2010 | http://homer.ucsd.edu/homer/ngs/ |
| ImageJ v1.53a | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Juicebox v1.9.8 | Durand et al., 2016a | https://github.com/aidenlab/Juicebox |
| Juicer v1.5.3 | Durand et al., 2016b | https://github.com/aidenlab/juicer |
| Juicer v1.7.6 for HiCCUPS (GPU version) | Durand et al., 2016b | https://github.com/aidenlab/juicer |
| MACS2 v2.1.1.2016309 | Zhang et al., 2008 | https://pypi.org/project/MACS2/ |
| MEME suite v4.10.1 | Machanick and Bailey, 2011 | https://meme-suite.org/index.html |
| MEME suite v5.3.0 | Machanick and Bailey, 2011 | https://meme-suite.org/index.html |
| NovoAlign v3.00.02 | Novocraft | http://www.novocraft.com/products/novoalign/ |
| NovoAlign v4.02.01 | Novocraft | http://www.novocraft.com/products/novoalign/ |
| Pgltools | Greenwald et al., 2017 | https://github.com/billgreenwald/pgltools |
| SAMtools v0.1.19 | Li et al., 2009 | http://samtools.sourceforge.net/ |
| SAMtools v1.4.1 | Li et al., 2009 | http://samtools.sourceforge.net/ |
| SPP v1.11 | Kharchenko et al., 2008 | http://compbio.med.harvard.edu/Supplements/ChIP-seq/ |
| TopHat2 v2.0.10 | Kim et al., 2013 | https://ccb.jhu.edu/software/tophat/index.shtml |
| Trim Galore! v0.4.1 | Babraham Bioinformatics | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ |