

Abstract
Two-photon fluorescence lifetime microscopy (2P-FLIM) is a non-invasive optical technique that can obtain cellular metabolism information based on the intrinsic autofluorescence lifetimes of free and enzyme-bound NAD(P)H, which reflect the metabolic state of single cells within the native microenvironment of the living tissue. NAD(P)H 2P-FLIM was initially performed in bone marrow stromal cell (BMSC) cultures established from Col (I) 2.3GFP or OSX-mCherry mouse models, in which osteoblastic lineage cells were labeled with green or red fluorescence protein, respectively. Measurement of the mean NAD(P)H lifetime, τM, demonstrated that osteoblasts in osteogenic media had a progressively increased τM compared to cells in regular media, suggesting that osteoblasts undergoing mineralization had higher NAD+/NAD(P)H ratio and may utilize more oxidative phosphorylation (OxPhos). In vivo NAD(P)H 2P-FLIM was conducted in conjunction with two-photon phosphorescence lifetime microscopy (2P-PLIM) to evaluate cellular metabolism of GFP+ osteoblasts as well as bone tissue oxygen at different locations of the native cranial bone in Col (I) 2.3GFP mice. Our data showed that osteocytes dwelling within lacunae had higher τM than osteoblasts at the bone edge of suture and marrow space. Measurement of pO2 showed poor correlation of pO2 and τM in native bone. However, when NAD(P)H 2P-FLIM was used to examine osteoblast cellular metabolism at the leading edge of the cranial defects during repair in Col (I) 2.3GFP mouse model, a significantly lower τM was recorded, which was associated with lower pO2 at an early stage of healing, indicating an impact of hypoxia on energy metabolism during bone tissue repair. Taken together, our current study demonstrates the feasibility of using non-invasive optical NAD(P)H 2P-FLIM technique to examine cellular energy metabolism at single cell resolution in living animals. Our data further support that both glycolysis and OxPhos are being used in the osteoblasts, with more mature osteoblasts exhibiting higher ratio of NAD+/NAD(P)H, indicating a potential change of energy mode during differentiation. Further experiments utilizing animals with genetic modification of cellular metabolism could enhance our understanding of energy metabolism in various cell types in living bone microenvironment.
Keywords: Energy metabolism, 2P-FLIM, 2P-PLIM, osteoblasts, glycolysis, oxidative phosphorylation, 2-photon microscopy, bone repair, intravital imaging
1. Introduction
Energy metabolism plays an important role in osteoblast differentiation and bone mineralization. While a series of studies have shown that aerobic glycolysis is the key mode of energy metabolism in primary calvarial osteoblasts [1, 2], studies measuring energy consumption and mitochondrial metabolism show that osteoblast differentiation coincides with increased oxygen consumption and the propagation of the mitochondria network [3, 4]. Oxidative phosphorylation (OxPhos) increases during differentiation and mineralization in response to ascorbic acid and β-glycerophosphate [4, 5], and altered mitochondria function can impact osteoblast differentiation and bone formation [3, 6–9]. It is unclear which energy pathway is most utilized by osteoblasts at different differentiation stages in vivo and how the oxygen microenvironment could affect the metabolic state of osteoblasts which in turn impact differentiation and mineralization of bone forming cells. It is important to note that in vitro culture does not well represent in vivo conditions, particularly when considering hypoxic niches in bone tissue. So far, no studies have been reported which attempt to define bone cell bioenergetics in vivo.
Nicotinamide adenine dinucleotide (NADH), flavin adenine dinucleotide (FADH2), and their oxidized forms (NAD+ and FAD2+) are essential enzymatic cofactors in the cellular respiration process. NADH and NAD+ reside in the cytosol and the mitochondria, and aid in shuttling of electrons during glycolysis, the TCA cycle, and OxPhos. In comparison, FAD and FADH2 are primarily located within mitochondria and participate in the TCA cycle and OxPhos pathways. Reduced and oxidized forms of NADH and FAD are used as biomarkers to determine the relative cellular energy metabolism in various cell types including those in tumors [10], the brain [11], and cardiac tissues [12]. However, chemical extraction and analysis of these molecules are often terminal, precluding dynamic and longitudinal assessment of cellular metabolism in vitro and in vivo.
A noninvasive optical technique based upon intrinsic fluorescence has been developed to obtain information on cellular metabolism and to differentiate different metabolic states of cells and tissues [13–18]. Since NADH and FAD are intrinsically autofluorescent, the intensities of each cofactor can be analyzed at single-cell level non-invasively and non-destructively with current microscopy techniques. While NADH and FAD are separable by excitation and emission wavelength, NADH and the phosphorylated form, NAD(P)H, have similar excitation and emission, and therefore are combined when collecting intensity measurements as NAD(P)H. The redox partners of NADH and FAD, namely, NAD+ and FADH2 are not autofluorescent. Based upon the unique optical properties of these molecules, a “redox ratio” of NADH and FAD has been used to assess cellular energy metabolism in various cell cultures [15, 17, 19, 20]. Since the ratio is dependent on signal from FAD, which resides primarily within the mitochondria and is utilized within the TCA cycle and OxPhos, a decrease in the redox ratio corresponds to increased glycolysis. The redox ratio of NADH and FAD has been used extensively to determine energy metabolism of cancer cells [21, 22], neurons [23] and skin cells [17, 20].
Intensity based measurements such as the one described above can be challenging to implement in the highly scattering in vivo environment, therefore fluorescence lifetime-dependent measurements have been developed via two-photon microscopy (2P-FLIM) to analyze contributions of free and bound NAD(P)H and FAD to ascertain energy metabolism [24–26]. This method exploits the intrinsic autofluorescence lifetimes of free and bound NADH and FAD, which reflect the metabolic state of single cells within the native microenvironment of the living tissue. Since fluorescence lifetime is determined by how long an excited molecule stays in the excited state before transitioning to the ground state, its measurements are independent of molecular concentration as well as the scattering properties of tissues. The free and enzyme bound forms of NAD(P)H and FAD have different and separable lifetimes. For NAD(P)H, when bound to an enzyme, its lifetime is markedly increased as compared to the free form. In contrast, when FAD is bound to an enzyme, its lifetime decreases as compared to the free form. By evaluating fluorescence lifetime, one can infer the relative amounts of free and bound molecule and hence determine cellular energy metabolism information. 2P-FLIM has been performed in cultures [27, 28], tissue sections [17], as well as in vivo [29], where a decrease of the bound form of NAD(P)H has been shown to correspond to increased glycolysis [13, 28] whereas an increase of the bound form of NAD(P)H correlates with increased OxPhos usage. Additionally, a phasor approach can be used to assign the lifetime of each fluorescent molecular source to its unique location on the phasor plot, creating fingerprints of different intrinsic fluorescence signals from living tissues [30–34].
The goal of our current study is to utilize 2P-FLIM, in conjunction with two-photon phosphorescence lifetime microscopy (2P-PLIM) and an established oxygen reporting nanoprobe, PtP-C343, to understand osteoblastic cellular metabolism at high spatial resolution in vivo in the context of normal and regenerative microenvironments. To our knowledge, our study is the first to conduct such high-resolution analysis of cellular energy metabolism in bone. Our study demonstrates that cellular metabolism is associated with differentiation stage of osteoblasts and can be further influenced by the in vivo microenvironment during bone healing and regeneration.
2. Materials and Methods
2.1. Materials.
For cell culture, α-MEM, ascorbic acid (AA), β-glycerophosphate (BGP), d-glucose, and alkaline phosphatase detection kit were purchased from Millipore Sigma (St. Louis, MO, USA). Tetramethlyrhodamine (TMR; 2,000,000 MW) was also purchased from Millipore Sigma. Platinum porphyrin Coumarin-343 (PtP-C343) was generously provided by Sergei Vinogradov from the University of Pennsylvania (Philadelphia, PA).
2.2. Experimental Animals.
B6.Cg-Tg(Col1a1*2.3-GFP) as well as Tg(Sp7/mCherry) reporter mice, also known as Col (I) 2.3GFP [35, 36] and OSX-mCherry mice [37], respectively, were purchased from Jackson Laboratories (Bar Harbor, ME). All in vivo experiments were performed using adult mice housed in pathogen-free, temperature and humidity-controlled facilities with a 12h day-night cycle in the vivarium at the University of Rochester Medical Center. All mouse housing cages contained wood shavings and a plastic house for environmental enrichment as well as general water and food with frequent changes. All experimental procedures were reviewed and approved by the University Committee on Animal Resources. General anesthesia and analgesia procedures were performed based on the mouse formulary provided by the University Committee on Animal Resources. The health status of the animals was monitored throughout the experiments by experienced veterinarians according to the Guide for the Care and Use of Laboratory Animals outlined by the National Institute of Health.
2.3. MC3T3-e1 Cell Culture.
Cryopreserved MC3T3-e1 cells (ATCC, clone 4) were thawed and seeded in wells of a 6-well plate. Base media consisting of α-MEM and 15% fetal bovine serum (FBS, Millipore-Sigma, St. Louis, MO) were supplemented to the cells. Media was exchanged every other day and after 7 days, once the cells had reached >60% confluence, osteogenic differentiation media consisting of α-MEM, 50μg/mL ascorbic acid, 5 mM β-glycerophosphate, and 10% FBS was added to some wells containing cells. The remaining wells had α-MEM with 10% FBS supplemented as a regular media group. Media was changed every two days.
2.4. Bone Marrow Stromal Cell Isolation and Culture.
Bone marrow stromal cells (BMSCs) were isolated from 8–15 week-old transgenic mice as previously described [38–40]. Briefly, femurs and tibiae were removed from mice. A syringe filled with α-MEM and a 23-gauge needle was inserted into the marrow cavity and flushed. The resulting marrow suspension was aspirated gently to evenly disperse the cells. The cell suspension was filtered via 70μm cell strainer (Falcon) to remove bone debris. Cells were centrifuged, resuspended, counted, and seeded at 5.0×105 cells per well of a 6-well plate. The cells were initially cultured in α-MEM and 15% FBS for 10 days with media changes every two days. Osteogenic differentiation media with 10% FBS, 50μg/mL ascorbic acid, 5 mM β-glycerophosphate was supplemented at day 10 to some wells with media changes every two days. For glucose experiment, BMSCs from Col (I) 2.3GFP mice were harvested and split into 6-well plate containing α-MEM (A10490-01, GIBICO) and 15%FBS with a final glucose concentration of 5.5mM or 10mM. The corresponding media was changed every 2 days. To suppress glycolysis, BMSCs were also treated with 2-DG (2-Deoxy-D-glucose) at 4mM for 30 min before performing 2P-FLIM.
2.5. Two-photon Laser Scanning Microscopy (MPLSM).
The 2-photon microscope was equipped with a tunable Mai Tai laser (100fs, 80MHz; Spectra-physics, Santa Clara, CA) used for excitation with an attached modified FluoView confocal 300 unit for beam scanning. For general in vitro imaging, an XLPlan N 25X/1.05 NA Olympus water immersion objective lens was used. In vitro imaging was conducted to collect signal from NAD(P)H (740nm excitation, 447/60nm emission), FAD (880nm excitation, 526/37nm emission), mCherry (740nm excitation, 605/55nm emission), or GFP (850nm excitation, 526/37nm emission). Fluorescence emission was collected into photomultiplier tubes (PMTs, H7422P-40, Hamamatsu, Japan). Images were taken at 256×256 pixels.
In vivo imaging was performed with a LUMPlan fI/IR 20X/0.95NA Olympus water immersion objective lens where bone collagen was imaged via Second Harmonic Generation (SHG), blood vessels via red fluorescence from perfused TMR as well as osteoblasts via green fluorescence (GFP) from the Col (I) 2.3GFP+ cells. An excitation wavelength of 880nm was chosen. Emission was collected with 448/20nm, 605/55nm, and 534/30nm bandpass filters (Semrock, Rochester, NY) for SHG, TMR, and GFP collection, respectively. Images at 1024×1024 pixels were acquired at a rate corresponding to a pixel dwell time of 0.2ms.
2.6. Two-photon Imaging and Optical Redox Ratio Determination.
BMSCs were extracted from OSX-mCherry mice and cultured in a standard 6-well plate as previously described. At designated time intervals, the dish containing cells was removed from the cell incubator and moved to the multiphoton microscope. MPLSM was conducted in regions where heterogeneous cells were located. NAD(P)H and FAD images were collected. Using ImageJ, mean NAD(P)H and FAD signal from each mCherry+ osteoblast was measured. A redox ratio for each cell was calculated based on the intensity of NAD(P)H and FAD as shown in Equation 1.
| Eq. 1 |
2.7. Two-photon NAD(P)H Fluorescence Lifetime Imaging (2P-FLIM) and Cellular Energy Metabolism Index Determination.
Two-photon excitation of NAD(P)H was performed at 740nm. NAD(P)H emission was filtered via a 447/60nm bandpass filter to minimize GFP bleed-through into the channel. The filtered signal was directed to a PMT (Hamamatsu H7422P-40) connected to a photon counting board (ISS A320 FastFLIM). Time-domain NAD(P)H images were collected at 256×256 pixels with a pixel dwell time of 20μs and 100 repetition rate, in other words producing a fluorescence decay curve for each pixel in the image. The resulting NAD(P)H fluorescence decay curves were transformed to the Fourier-domain via Fourier Transform within the ISS VistaVision software to produce the phasor plot for the region of interest. From the clustered data shown on the phasor plot, using the ISS Vista Vision software, individual cells were separately identified to produce a cluster of data within the phasor space corresponding to that cell. The G and S coordinates of the centroid of the data cluster was recorded. Using G and S coordinates as well as the laser modulation frequency ω, a mean lifetime, τM, for each cell was calculated (Eq. 2). To note, phasor data that appeared to be similar to the background or to other autofluorescent molecules such as collagen were removed from analyses based on known locations on the phasor plot as described in the Discussion Section. See schematic in Figure 1B.
Fig. 1. Schematic of Energy Metabolism Index Determination.
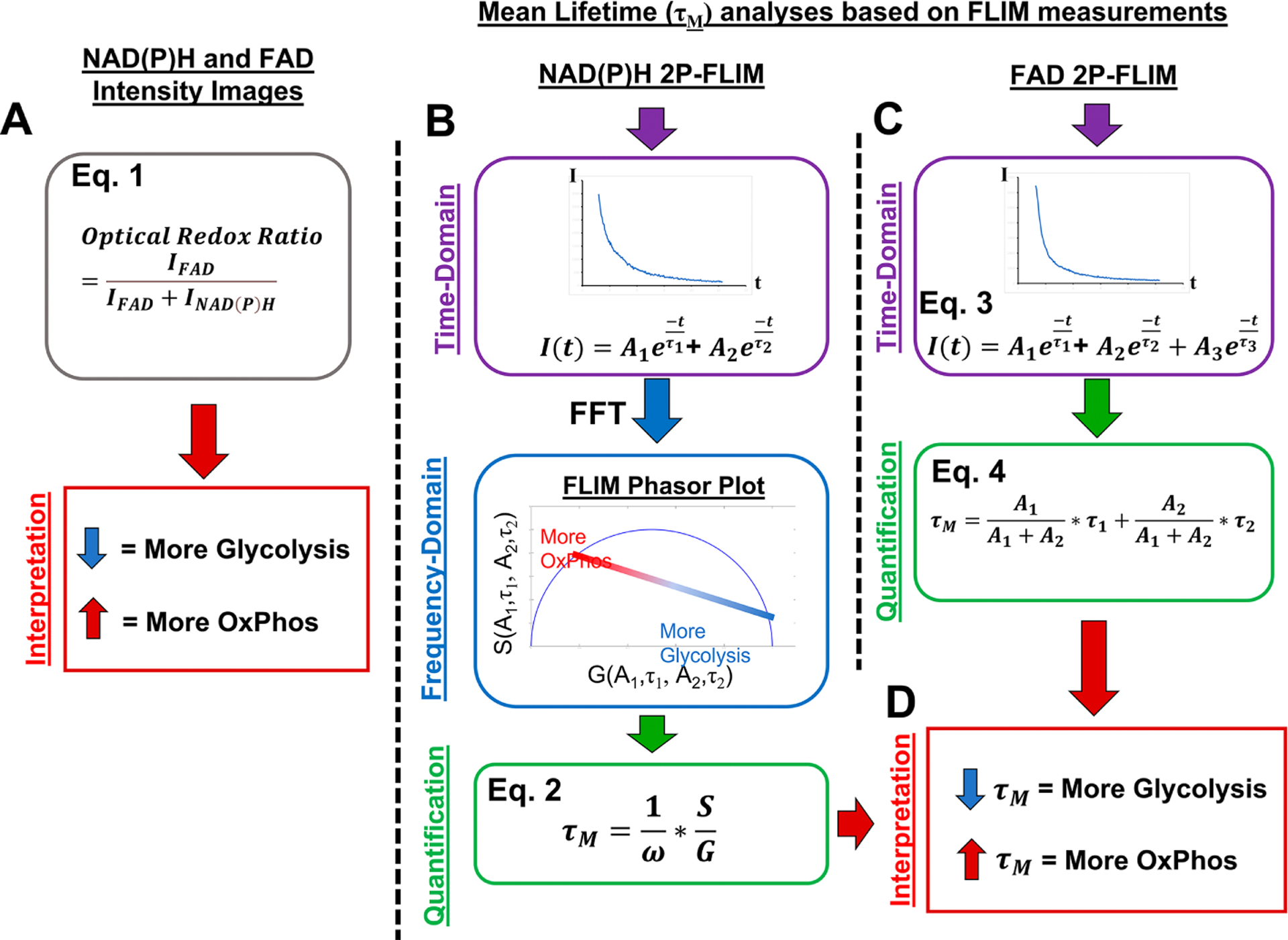
NAD(P)H and FAD intensity images as well as lifetime images of cells were collected within the same region. Cells which had both FAD and NAD(P)H intensity images collected were analyzed via the optical ratio where the intensity of FAD was divided by the sum of NAD(P)H and FAD intensities on a pixel-by-pixel basis. Single cells were evaluated such that a lower redox ratio corresponding to more glycolysis and higher corresponding to more OxPhos (A). NAD(P)H lifetime images collected from cells in vitro and in vivo were processed via Fourier Transform (FFT) to enter the frequency-domain. This resulted in time-domain data converting into the FLIM phasor plot where individual pixels in time correspond to points in phasor space. Single cells were selected on the phasor plot, the G and S coordinates of median location of the point cluster were determined, and mean lifetime, τM, was calculated (B). FAD lifetime images were collected from cells in vitro and each pixel was fit to a triple exponential function. A τM was calculated from the first two amplitudes (A1 and A2) as well as lifetimes (τ1 and τ2) (C). The calculated τM from either NAD(P)H or FAD analyses were interpreted similarly where lower τM corresponds to increased utilization of glycolysis and in contrast, higher τM corresponds to increased OxPhos (D).
| Eq. 2 |
2.8. FAD FLIM and Cellular Energy Metabolism Index Determination.
Excitation of FAD was performed at 880nm. Emission light was filtered via 526/37nm bandpass filter and directed to a PMT (Hamamatsu H7422P-40) connected to the photon counting board. Utilizing the ISS VistaVision software, individual cells were selected from the resulting time-domain images and a decay profile for each cell was generated. The fluorescence decay profile was extracted from the ISS VistaVision software and analyzed via an in-house MATLAB program where a 3-componenet exponential function, with offset, was fit via linear least squares fitting method (Eq. 3). A resulting Chi-squared test value of 1 ± 0.1 from the exponential fit was determined acceptable and used for data calculation. As previously described, the third component of the fit which corresponded to mitochondrial flavin mononucleotide was ignored and a τM was calculated with the remaining two amplitudes and lifetimes of the curves (Eq. 4) [41]. See schematic in Figure 1C.
| Eq. 3 |
| Eq. 4 |
2.9. In vitro Fluorescence Lifetime Imaging.
Cells were cultured in 6-well plates as previously described. At designated time intervals, the cell culture plate was removed from the incubator and taken to the microscope. A XLPLN-MP 25X/1.05NA Olympus water immersion objective lens was immersed into the media containing cells. NAD(P)H and/or FAD 2P-FLIM was performed in various regions of the plate. To ensure minimal interference from eGFP or collagen which can be excited at 740nm [42–45], we chose a bandpass filter of 447/60 nm to eliminate eGFP emission bleed-through into the NAD(P)H channel while providing a large bandwidth to collect the NAD(P)H emission. To verify that GFP contamination was eliminated, measurements of NAD(P)H τM from mCherry labelled osteoblasts using this 447/60nm filter showed a similar τM to the eGFP labelled osteoblasts (Supplemental Fig. 1). To separate collagen autofluorescence from NAD(P)H when performing 2P-FLIM, we utilized the phasor plot as a fit-free approach to depict lifetime distributions from the lifetime images [45–47]. As fibrillar collagen produces a lifetime that can be readily shown in the phasor plot [45], we are able to highlight pixels in cells versus pixels pertaining to extracellular collagen, allowing τM to be determined from individual osteoblasts using the software provided by ISS.
2.10. In vivo Cellular Energy Metabolism Index Measurements in Intact Cranial Bone.
To image the osteoblasts within native intact bone, Col (I) 2.3GFP mice were subjected to a cranial window model which has previously been established [48, 49]. Briefly, the skin above the skull was removed and a plastic ring was fixed to the skull surface. Media was then added to the space around the ring, and it was sealed with a glass window.
When performing NAD(P)H 2P-FLIM of the intact bone, mice were imaged 3 days after surgery. Prior to imaging, a retro-orbital injection of a mixture of TMR and PtP-C343 was performed to label vasculature within the bone and measure intra- and extra-vascular pO2, respectively. The native bone surrounding the suture was initially imaged via MPLSM to determine a location where GFP+ osteoblasts were found in the suture, marrow, and lacunae regions. These three separate regions were selected to perform NAD(P)H 2P-FLIM analyses on a single cell resolution.
2.11. Cranial Window Defect Chamber Model.
To measure the osteoblast energy metabolism within a healing defect, the cranial window defect chamber model was established where a 1-mm full thickness defect was created in the parietal bones of Col (I) 2.3GFP mice using a 0.86 mm diameter inverted Busch burr bore (Armstrong Tool & Supply Company, Livonia, MI). The dura remained intact during the surgery[48, 49]. NAD(P)H 2P-FLIM was performed 18 days post-surgery within regions of the healing bone where osteoblasts were present. Additionally, NAD(P)H FLIM was performed on the opposite, un-injured, side of the skull where GFP+ osteoblasts were located.
2.12. Phosphorescence Lifetime Imaging and Oxygen Tension Determination.
2P-PLIM was performed as previously described [38]. Briefly, excitation of PtP-C343 was performed at 900nm and gated in 10 μs intervals over a 1.3 ms total interval. PtP-C343 phosphorescence was filtered via two-706/167nm bandpass filters and directed to a PMT (Hamamatsu R10699, Shizuoka, Japan). Detected photons were counted via SR 400 (Stanford Research Systems, Sunnyvale, CA) and binned into 320 ns-long bins. Typically, 10,000 excitation-collection cycles were averaged to achieve adequate signal-to-noise ratios (SNR>1.5). Data acquisition was controlled using a custom-written program in LabView (National Instruments). The resulting raw decay curves were fit to a single-exponential function where the decay time constant τ was calculated. Using an independently created calibration curve, pO2 was determined. 2P-PLIM was conducted by making point measurements within labelled vessels and surrounding extravascular space where the GFP+ osteoblasts resided.
2.13. Statistical Analyses.
All in vivo data is expressed as mean ± standard error of the mean and all in vitro data is reported as mean ± standard deviation due to the large number of samples analyzed in this study. Statistical analyses were conducted via one-way or two-way ANOVA with a Bonferroni post-hoc analyses of individual groups unless specified otherwise. A p value of less than 0.05 was considered statistically significant.
3. Results
3.1. Longitudinal Measurements of Optical Redox Ratio at Single Cell Resolution in BMSC Cultures.
The optical redox ratio is a well-established method to determine the metabolic state of cells in vitro [17, 50]. NAD(P)H and FAD intensity images were collected from bone marrow stromal cell culture derived from OSX-mCherry mice in which osteoblasts are labeled with RFPmcherry. Cells were cultured in either regular or osteogenic media for a total of 28 days. As shown in Fig. 2A, there was adequate fluorescence signal collected from the mCherry+ osteoblasts in culture, which was used to calculate the redox ratio at single cell resolution (Fig. 2B) at week 2, 3 and 4-week post seeding. Quantitative analyses showed that cells cultured in regular media had a mean redox ratio of ~0.36 with no significant change over 28-day period. In contrast, cells in osteogenic media had a progressively increasing redox ratio from 0.44 ± 0.08 at day 14 to 0.54 ± 0.11 at day 28 post-seeding (Fig. 2C, p<0.05). These data indicate a shift towards increased production of FAD and OxPhos as the more mature mCherry+ osteoblastsutilize mitochondria and more OxPhos as their primary energy source.
Fig. 2. In vitro NAD(P)H and FAD fluorescence intensity analyses of OSX-mCherry BMSCs in regular or osteogenic media.
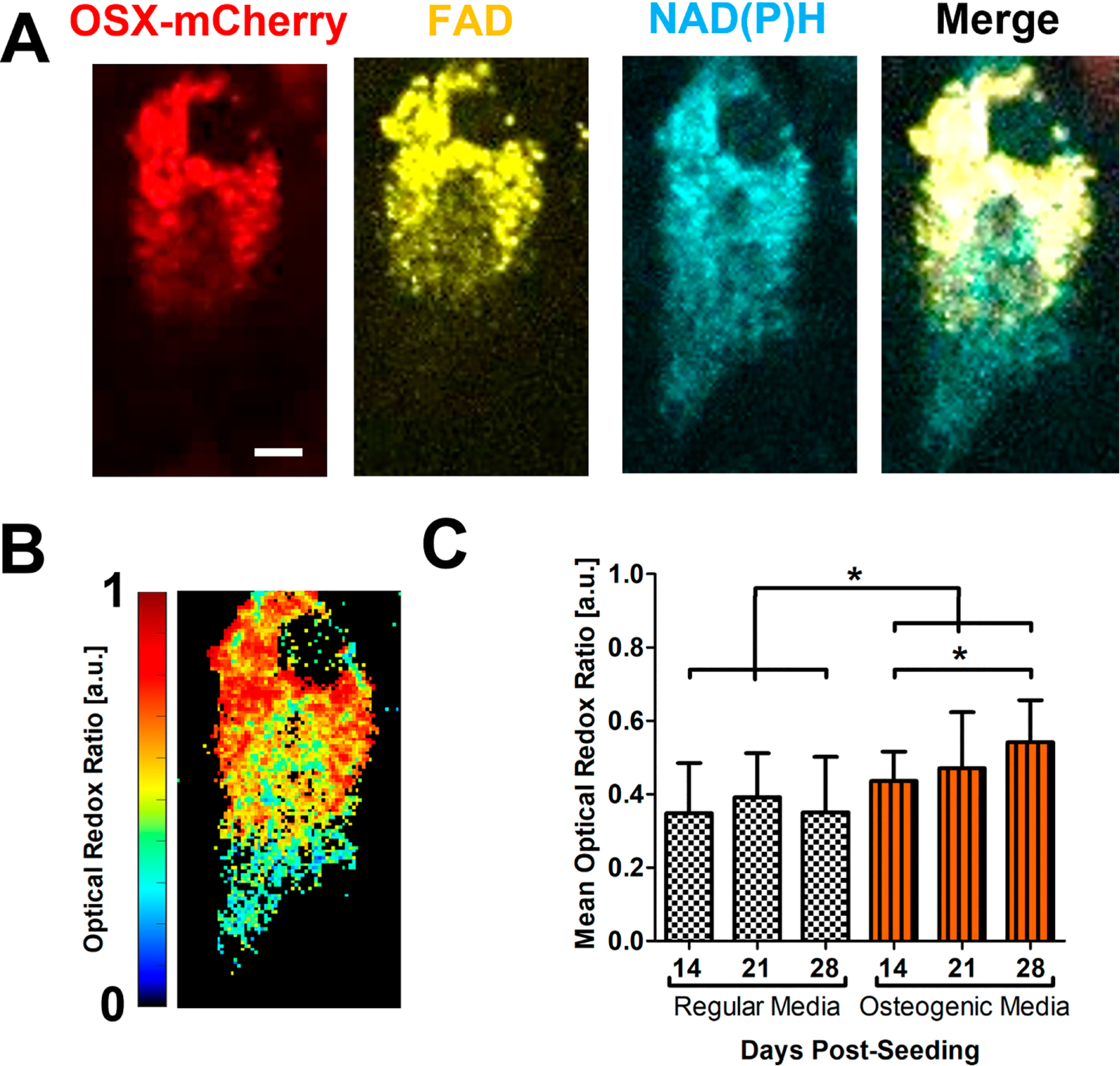
Representative fluorescence intensity images of OSX-cherry(red), FAD(yellow), NAD(P)H (cyan) (A) of an osteoblast in BMSC culture derived from OSX-mCherry mice. The redox ratio was calculated at each pixel to create a heatmap at single cell resolution as shown (B) [scale bar = 3 μm]. The mean optical redox ratio of mCherry+ osteoblasts in regular or osteogenic media at days 14, 21, and 28 post-seeding are shown (C). [n≈18 cells in each of 3 parallel experiments per group, *, p<0.05, One-Way ANOVA]
3.2. Longitudinal Measurements of τM of NAD(P)H and FAD via 2P-FLIM at Single Cell Resolution in BMSC Cultures.
NAD(P)H 2P-FLIM (Figs. 3B1&2) and FAD 2P-FLIM (Fig. 3C1–3) were performed on BMSC cultures derived from OSX-mCherry mice as described in the Methods section. Longitudinal measurements of FAD τM from mCherry+ osteoblasts showed that osteoblasts in osteogenic media had an increasing τM over time with the highest value of 1.07±0.16 recorded at 28 days post-seeding, which was significantly higher than that in regular media (Fig. 3D, p<0.05). Similarly, measurements of τM for NAD(P)H showed a trend towards higher value over 28 day-period in the presence of osteogenic media (Fig. 3E, p<0.05).
Fig. 3. In vitro NAD(P)H and FAD fluorescence lifetime analyses of OSX-mCherry BMSCs in regular or osteogenic media.
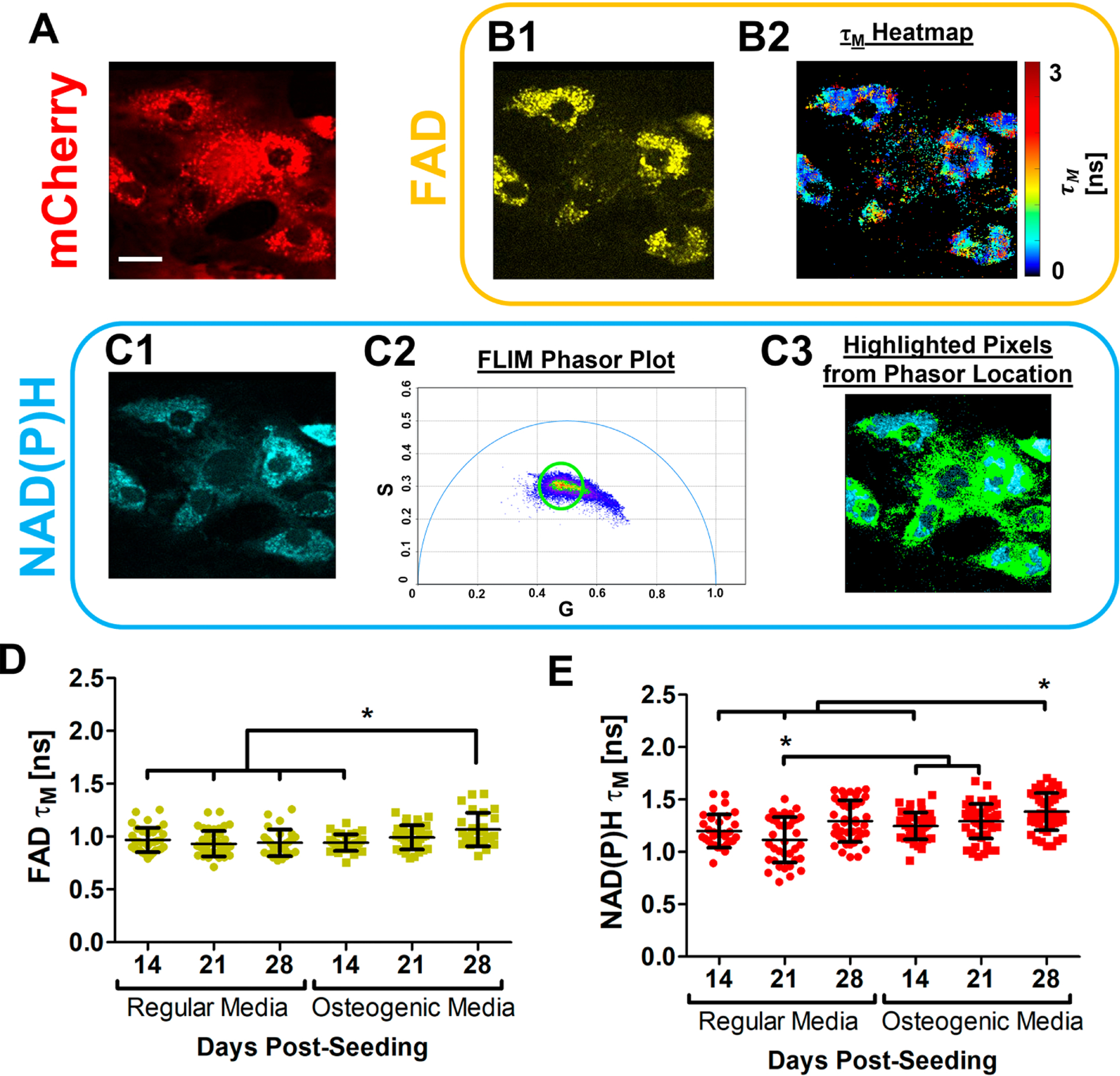
Representative fluorescence intensity images of OSX-mCherry+ osteoblasts (red) (A) at day 21 post-seeding in regular media. FAD 2P-FLIM was conducted on the mCherry+ osteoblasts. Representative FAD intensity image (B1, yellow) was collected and a corresponding τM intensity distribution heatmap image (B2) was calculated. NAD(P)H 2P-FLIM (C1) was also performed where the NAD(P)H lifetime was converted into the phasor plot (C2), through which the circled lifetime species (green) were assigned to the corresponding regions within the cells (C3). The mean FAD τM (D) and NAD(P)H τM (E) calculated for OSX-cherry+ osteoblasts in regular or osteogenic media at days 14, 21, and 28 post-seeding are shown, respectively. [scale bar = 5 μm]. [N≈17 cells in each of 3 parallel experiments per group, *, p<0.05, One-Way ANOVA]
Similarly, NAD(P)H 2P-FLIM was performed in BMSC cultures established from bone marrow harvested from Col (I) 2.3GFP mice. The cellular metabolism of GFP+ osteoblasts and GFP− stromal cells was examined at single cell resolution (Figs. 4A1–3) via phasor plot analyses to determine NAD(P)H τM (Fig. 4B1 & B2). GFP+ and GFP− cells were separately evaluated at weeks 2, 3 and 4 post-seeding with or without osteogenic media. Longitudinal τM of NAD(P)H again showed progressively increased τM in GFP+ osteoblasts treated with osteogenic media (Fig. 4C, p<0.05) with highest value recorded at day 28 (mean = 1.612 ± 0.19 ns), while NAD(P)H τM in GFP+ osteoblasts cultured in regular media exhibited no significant change over 28-day period. The GFP− stromal cells had a significantly lower τM than GFP+ osteoblasts in osteogenic media over 4-week culture period (mean = 1.12 ± 0.15 ns, p<0.05). NAD(P)H τM in GFP− stromal cell remained unchanged in the presence or absence of osteogenic media (Fig. 4C, p>0.05). Taken together, our data suggest that osteoblasts treated with osteogenic media utilized more OxPhos than those treated with regular media.
Fig. 4. NAD(P)H FLIM of Col (I) 2.3GFP BMSCs.
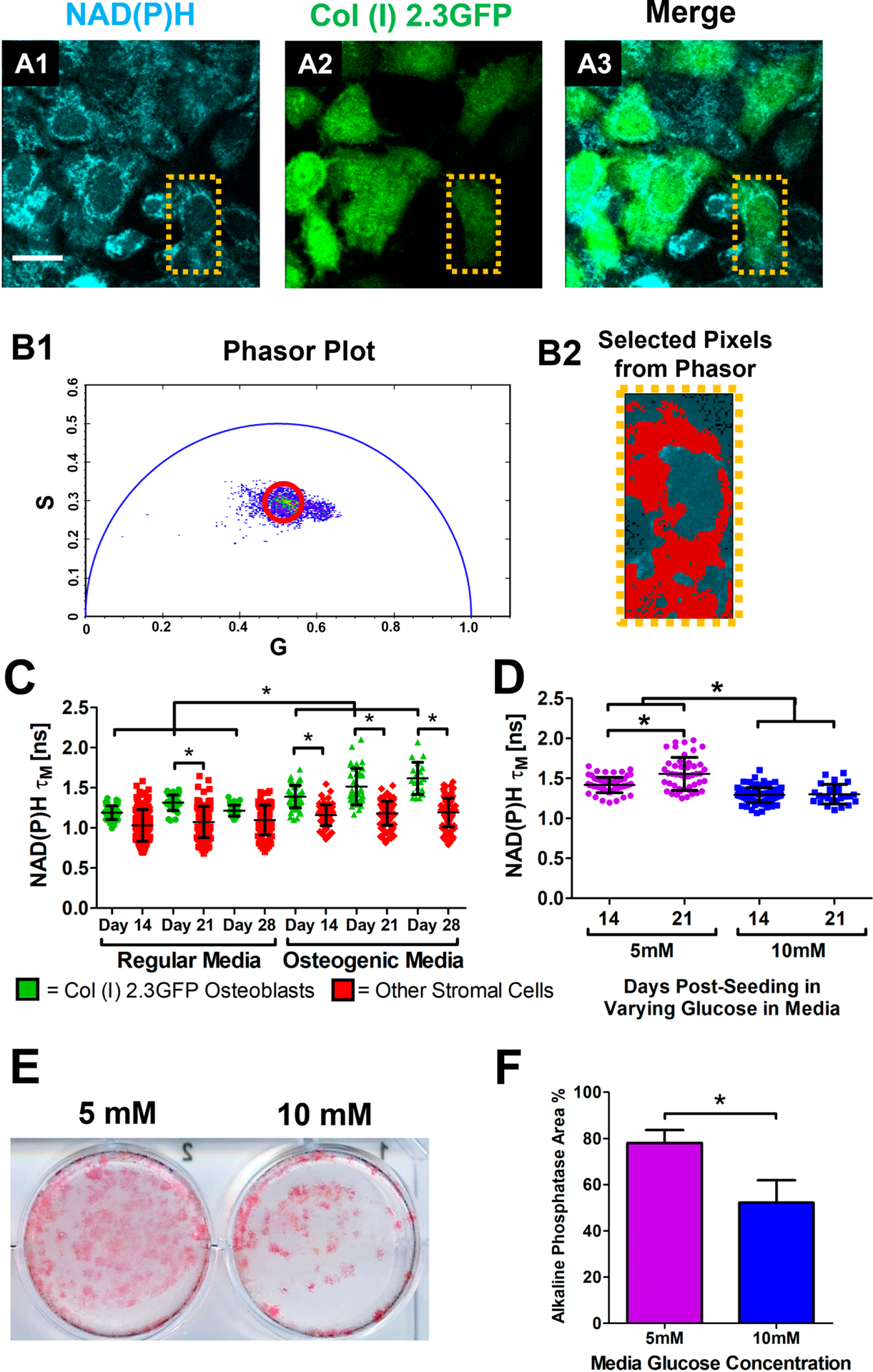
Representative images of bone marrow stromal cells (BMSCs) at day 14 in regular media from Col (I) 2.3GFP mice where NAD(P)H from all cells is shown in cyan and GFP from osteoblasts is shown in green (A1–3). Calculated phasor plot (B1) of the image with a highlighted region in red corresponding to pixels that contain that lifetime information (B2). Measurements of NAD(P)H τM from GFP+ osteoblasts (green dots) as well as the GFP− stromal cells (red dots) in regular or osteogenic media at days 14, 21, and 28 post-seeding (C). Measurements of NAD(P)H τM from GFP+ osteoblasts in media containing either 5mM or 10mM glucose at days 14 and 21 post-seeding (D). Representative alkaline phosphatase staining of osteoblasts in varying glucose media after 28 days in culture (E). Percentage of alkaline phosphatase staining coverage within the culture dishes (F). [n≈14 GFP+ and 40 GFP− cells in each of 3 parallel experiments per group for media treatment experiment; n≈19 cells in each of 3 parallel experiments per group for glucose treatment; *, p<0.05; One-Way ANOVA]
Previous studies show glucose concentration in culture media can affect osteoblast proliferation and differentiation [51–53]. To determine the effect of glucose concentration on osteoblasts, BMSCs extracted from Col (I) 2.3GFP mice were placed in α-MEM media containing 5.5mM (standard) or 10mM glucose. NAD(P)H 2P-FLIM was conducted on GFP+ osteoblasts at 14 and 21-days post-seeding, and demonstrated that osteoblasts cultured in 5.5mM glucose had a higher τM than those in 10mM glucose (mean = 1.42 ± 0.09 ns, Fig. 4D, p<0.05). Additionally, there was an increase in τM from osteoblasts after 21 day-culture in 5.5 mM glucose media upon osteogenic media treatment (mean = 1.56 ± 0.21 ns, Fig. 4D, p<0.05). When BMSCs were stained with alkaline phosphatase on day 21 days, there was an observed increase in stained area in 5.5mM glucose media (mean = 78.1 ± 5.57%, Fig. 4E & 4F), indicating that high glucose inhibits the enhanced OxPhos at a later stage of differentiation and the osteogenic differentiation of BMSC cultures. The inhibitory effects of high glucose on osteogenic differentiation has been previously reported [53].
Longitudinal NAD(P)H 2P-FLIM was performed in an osteoblastic cell line, MC3T3-e1, with or without osteogenic media in multiple regions and wells at days 10, 14, and 21 post-seeding. Abundant NAD(P)H autofluorescence signal was observed via 2P-FLIM (Fig. 5A) Measurement of NAD(P)H τM showed that cells in osteogenic media had increasing τM longitudinally from 1.45 ± 0.09 ns at day 10 to 1.48 ± 0.09 ns at day 14 and 1.7 ± 0.12 ns at day 21 post-seeding whereas cells in regular media had a consistently similar τM of ~1.4 ns among all time points. Further 1-way ANOVA tests showed that τM from cells in osteogenic media at days 14 and 21 were different from cells in regular media (Fig. 5B, p<0.05). Increased alkaline phosphatase staining was confirmed following 21-day culture in osteogenic media (Fig. 5C), indicating that more matured osteoblasts had increased OxPhos compared to less mature osteoblasts.
Fig. 5. NAD(P)H FLIM of MC3T3-e1 cells in regular or osteogenic media cultures.
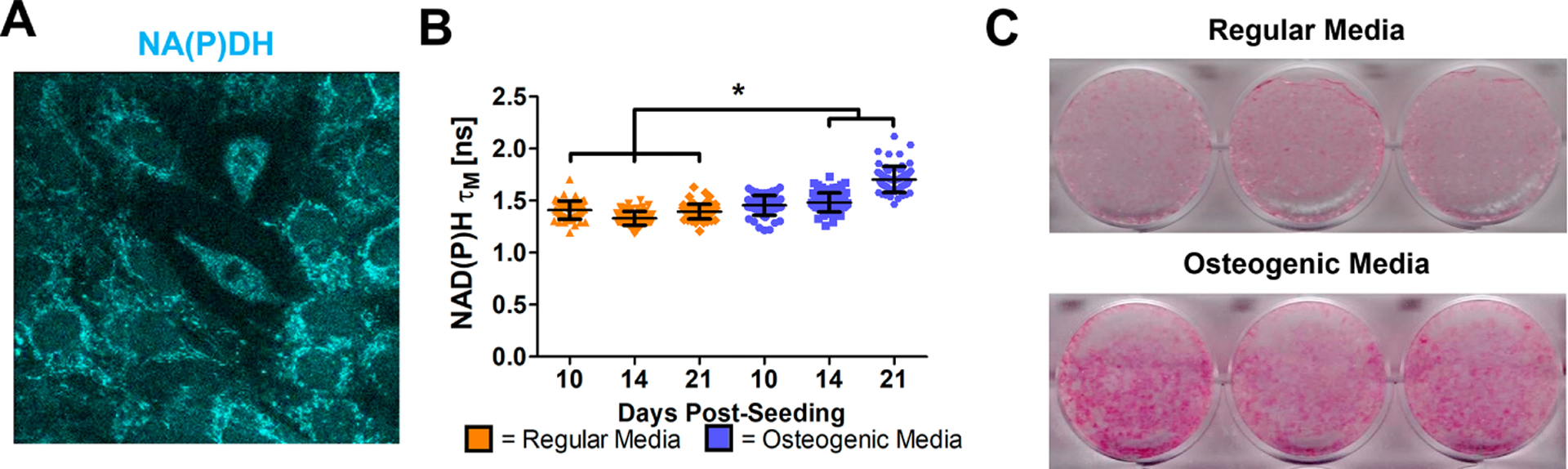
Representative NAD(P)H intensity image of MC3T3-e1 cells after 10 days of culture (A). NAD(P)H τM was calculated on MC3T3-e1 cells cultured in regular or osteogenic media at days 10, 14, and 21 post-seeding (B). Alkaline phosphatase staining of MC3T3-e1 cells after 21 days of culture (C). [n≈23 cells in each of 3 parallel experiments per group; *, p<0.05; One-Way ANOVA]
3.3. In vivo Measurements of τM of NAD(P)H via 2P-FLIM at Single Cell Resolution in the Cranial Bone.
Cranial windows were established on 3-month old Col (I) 2.3GFP mice. NAD(P)H 2P-FLIM was performed on GFP+ osteoblasts within the native intact bone at single cell resolution. Although collagen autofluorescence has a similar excitation and emission as NAD(P)H, it has a separable and different lifetime from both bound and free NAD(P)H [45]. As shown in Figs. 6A1–3&B1–3, where 2P-FLIM was conducted at two representative regions (marrow space and lacunae), populations of lifetime species from collagen in bone (orange) and NAD(P)H (green) within GFP+ cells can be separated via phasor plot, demonstrating that measurements of NAD(P)H τM in vivo is feasible.
Fig. 6. NAD(P)H FLIM of GFP+ osteoblasts in native bone of Col (I) 2.3GFP mice.
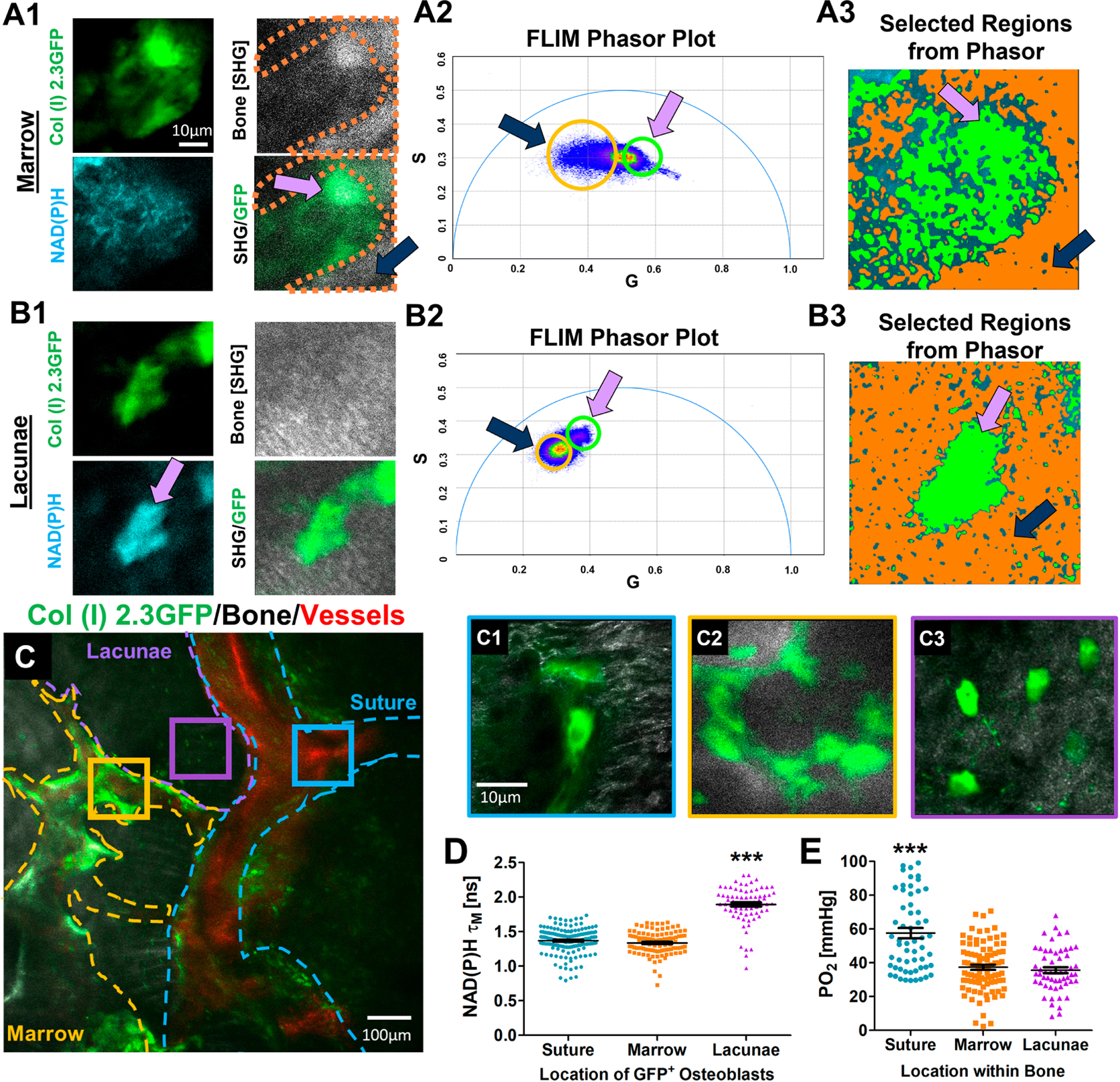
Representative intensity images of regions where NAD(P)H FLIM was performed illustrating osteoblasts (green), bone via SHG (white), and NAD(P)H signal (cyan) within marrow space (A1) and lacunae (panel B1). Phasor plot analyses of NAD(P)H FLIM (A2 and B2) within the corresponding regions. Selected regions within the phasor plot reveals separable lifetime species of collagen in bone (highlighted in orange) and NAD(P)H in cells (highlighted in green). Pink arrows show the selected lifetime species where GFP+ osteoblasts reside, and the black arrows show selected lifetime species from bone tissue (A3 & B3). Representative MPLSM image of native bone in Col (I) 2.3GFP mice (C). Bone/SHG (white), osteoblasts (green), and vasculature (red) with highlighted regions where NAD(P)H FLIM was performed on individual cells in the suture (C1), marrow space (C2), and lacunae space (C3). Measurements of NAD(P)H τM of GFP+ osteoblasts within the different regions of the native bone (D). Measurements of oxygen tension (pO2) at the similar regions where NAD(P)H FLIM was performed (E). [N≈23 cells for NAD(P)H τM measurements and N≈13 pO2 point measurements in each of 5 mice; ***, p<0.001; One-Way ANOVA]
The GFP+ osteoblasts were analyzed individually and categorized into 3 separate regions: the suture (blue), bone marrow (orange), and the lacuna-canalicular (purple) spaces (Fig. 6C and C1–3). Measurements of NAD(P)H τM for osteoblasts within the respective region showed that GFP+ osteoblasts/osteocytes residing in the lacunae had an average τM of 1.8 ± 0.21 ns, which was significantly higher than both τM measurements made in the suture and bone marrow spaces (Fig. 6D, p<0.05, n = 5 mice). There were no differences in τM among osteoblasts aligned along the bone surface of marrow space and suture (mean = 1.3 ± 0.17 ns). Oxygen tension measurements were made using 2P-PLIM via vascular perfusion of oxygen probe PtP-C343. Oxygen tension pO2 was recorded in the vasculature as well as the surrounding extravascular space where NAD(P)H 2P-FLIM was conducted. As shown in Fig. 6E, the suture region had a higher mean pO2 (mean= 57.6 ± 23.2 mmHg, p<0.05, n = 5 mice) than the marrow regions and the lacunae spaces. Poor correlation between pO2 and NAD(P)H τM of osteoblasts were noted in the intact native cranial bone (Supplemental Fig. 2).
3.4. In vivo NAD(P)H 2P-FLIM of Osteoblasts during Cranial Defect Healing in Mice.
2PFLIM was conducted 18-days post-surgery on Col I (2.3) GFP+-labelled osteoblasts within the defect as well as on the contralateral side on the intact bone. As shown in Fig. 7A, significant number of GFP+ osteoblasts can be seen around the leading edge of bone defect. Measurements of NAD(P)H τM from GFP+ osteoblasts showed that the osteoblasts at the leading edge of the defect had significantly lower τM than those within the contralateral native bone (mean = 1.28 ± 0.15 ns; Fig. 7B, p<0.05, N = 4 mice). Further measurements of pO2 in the same region showed lower value associated with expanding osteoblasts at the leading edge of bone defect (Fig. 7C, p<0.05, n=3), suggesting a change of the mode of energy metabolism to a more glycolytic state during healing where the hypoxic environment could exert an impact.
Fig. 7. NAD(P)H FLIM of osteoblasts within healing defect or nearby native bone of Col (I) 2.3GFP mice.
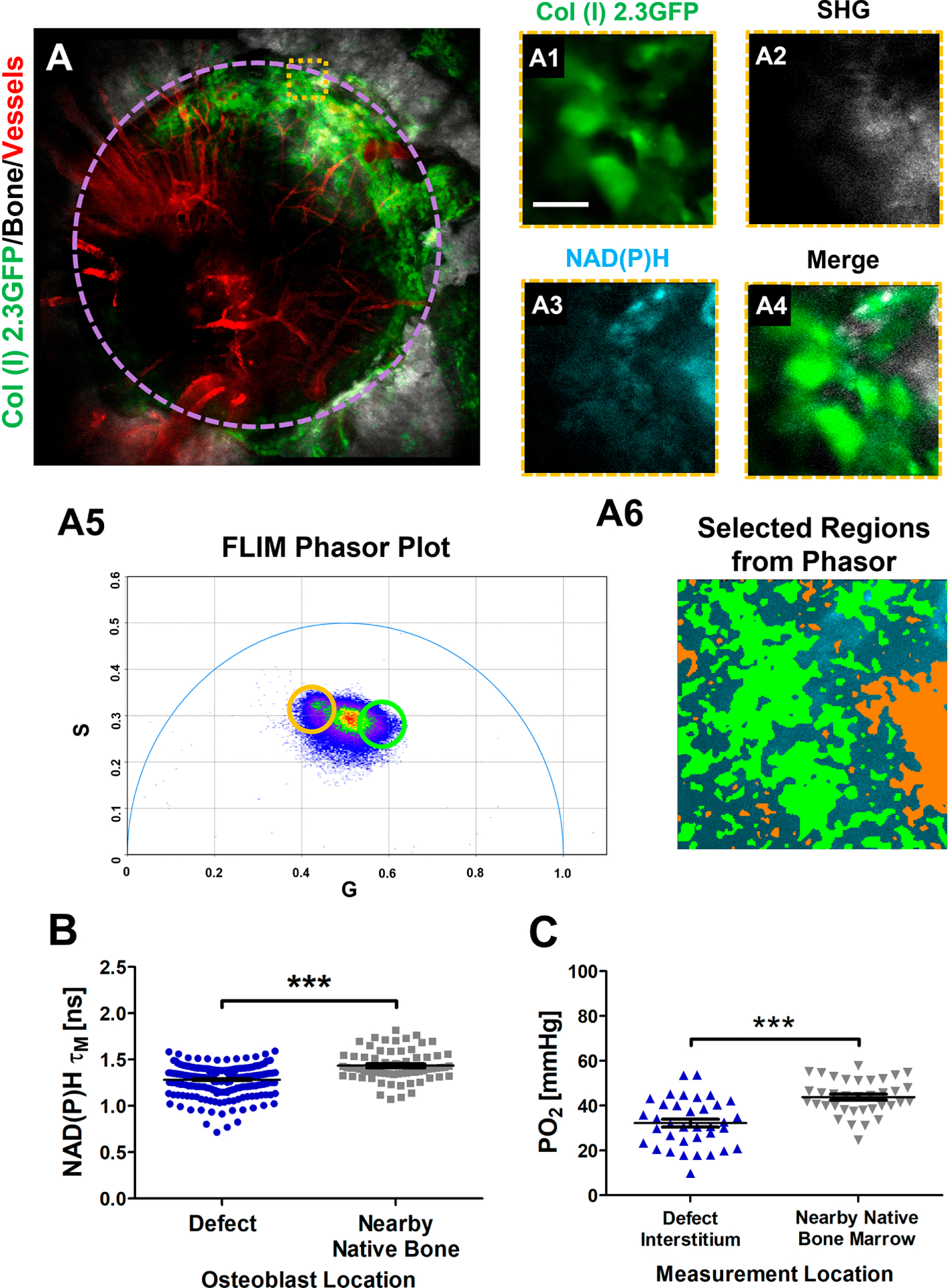
MPLSM of a 1 mm defect performed 18 days after surgery (A). Representative images of native bone illustrate bone/SHG (white), osteoblasts (green), and vasculature (red) with highlighted region where NAD(P)H FLIM was performed on individual cells (A1–4) [scale bar = 10 μm]. Corresponding phasor plot of the region shows a distribution of points (A5) where selected regions from the phasor demonstrate separable species among collagen (highlighted orange) and GFP+ osteoblasts (highlighted green) (A6). Calculated NAD(P)H τM of GFP+ osteoblasts within the healing defect as well as the nearby uninjured native bone (B) as well as pO2 within measured regions (C). [N≈37 cells for NAD(P)H τM measurements and N≈9 pO2 point measurements in each of 4 mice; ***, p<0.001; Student’s t-Test]
4. Discussion
A series of studies have demonstrated that aerobic glycolysis is a key component of energy metabolism in osteoblasts [2, 54, 55]. In primary osteoblast culture, most glucose carbons are found to be incorporated into lactate in the presence of abundant oxygen [57]. Stimulation of aerobic glycolysis through stabilization of HIF-1α in preosteoblasts markedly increases osteoblast production and bone formation in the mouse [5]. Additionally, intermittent use of anabolic parathyroid hormone PTH is shown to promote bone formation by stimulating aerobic glycolysis via insulin growth factor signaling pathway [58]. A more recent study further demonstrates that aerobic glycolysis accounts for approximately 80% of the ATP production in calvarial derived mature osteoblasts [59]. However, despite the evidence that points to an essential role of glycolysis in osteoblastic bone formation [56], genetic deletion of Glut1 via Prx1Cre showed a mild phenotype in osteoblast differentiation and mineralization [60]. Similarly, mice following deletion of Glut4 via osteocalcin-cre also show normal bone architecture [61], suggesting the redundant role of glucose transporter as well as the strong ability of osteoblasts to adapt to other energy sources. In fact, the extensive mitochondrial network formed in mature osteoblasts and the enhanced oxygen consumption rate (OCAR) upon treatment of osteogenic differentiation media [4, 62] or anabolic PTH [58, 63] suggest the potential of osteoblasts to use other alternative substrates such as glutamine and other amino acids to fuel mitochondrial OxPhos during osteoblastic bone formation [64, 65].
Utilizing 2P-FLIM analyses at single cell resolution, in our current study, we examined the optical redox ratio of NAD(P)H and FAD, as well as the mean lifetime τM of NAD(P)H and FAD longitudinally during osteoblast differentiation in BMSC cultures. In contrast to other studies, in which only a short duration of time was examined in culture during osteoblast differentiation [59, 66], we tracked the cellular metabolism in Col 1 (2.3) GFP+ or OSX-cherry+ osteoblasts over a 4-week period in the presence and absence of osteogenic media via 2P-FLIM. While similar redox ratio and mean τM of FAD and NAD(P)H were recorded over time in osteoblasts in the absence of osteogenic media, we found a significant and progressively increased NAD(P)H and FAD τM, as well as the redox ratio over time upon the addition of osteogenic media starting at day 10 post-seeding, indicating a shift of metabolism to supply more energy for enhanced collagen production and mineralization in culture. This data is consistent with previous studies that show a shift from glycolysis to OxPhos in osteoblast culture following addition of ascorbic acid and β-glycerophosphate[4, 66]. Further in vitro experiments via inhibitor of glycolysis (2DG) (supplemental Fig. 3) and manipulation of glucose concentration (Fig. 4E&F) demonstrate the sensitivity of our current imaging approach to changes of energy metabolism and allow us to perform in vivo experiments to determine cellular metabolism via 2P-FLIM.
To determine cellular metabolism in living animals, NAD(P)H 2P-FLIM was performed in Col 1 (2.3) GFP transgenic animals. Combining MPLSM and 2P-FLIM, we examined the energy metabolism in osteoblasts at various locations in adult injured or non-injured bone. Our data demonstrated that osteoblasts aligned along the bone surface of the suture and marrow spaces had a similar NAD(P)H τM as those obtained in culture at 21-days following treatment of osteogenic media, suggesting a similar metabolic profile of osteoblasts both in vivo and in vitro. Interestingly, when we examined metabolism in isolated Col (I) 2.3GFP+ osteoblasts residing in bone lacunae, we identified a significantly enhanced mean NAD(P)H τM, in these cells, suggesting that osteocytes may utilize more oxidative phosphorylation than mature osteoblasts.
The energy metabolism of osteocytes remains unclear and has emerged as an area of intense research interest. Since osteocytes cannot maintain their phenotype in culture, very limited in vitro work has been performed so far to assess energy metabolism. However, by utilizing excised bone, it has been shown that osteocytes have increased mitochondria, suggesting enhanced mitochondrial activities and perhaps more usage of OxPhos [67]. Additionally, Sánchez-de-Diego et al. demonstrated that decreasing glucose in culture media lead to increased expression of DMP1 in osteoblasts, an osteocyte marker. They proposed that these osteocytes minimize consumption of glucose but perhaps utilize alternative metabolites such as glutamine as a source to fuel OxPhos and produce energy [68]. So far, no available data can be found regarding the energy sources of osteocytes in living bone although it is suggested that mitochondrial metabolism has a role in osteocyte bioenergetics [69].
Local oxygen availability in bone tissue could affect the mode and pathway of energy metabolism in osteoblasts [16, 70]. To determine the potential correlation of pO2 and energy metabolism in vivo, we utilized high resolution 2P-PLIM to interrogate localized pO2 via the oxygen sensitive probe PtP-343 delivered through the circulation [38]. Our data show that the normal bone environment exhibited a wide range of pO2 within the suture, marrow spaces and lacunae with mean pO2 around 40–60 mmHg, e.g., 5–7% of atmosphere oxygen. At this range of oxygen level, we found poor correlation between pO2 and NAD(P)H τM in osteoblasts in suture and marrow space, suggesting that in normal bone pO2 may have a smaller effect on cell metabolism. In contrast to normal bone, measurements of pO2 and NAD(P)H τM during healing showed a significant drop of pO2 surrounding clusters of osteoblasts in the defect (Fig.7D). Corresponding to this drop of pO2, a significant shift of NAD(P)H τM to glycolysis was recorded. A comparison of NAD(P)H τM from osteoblasts within the healing bone and the three regions of the intact bone demonstrated that osteoblasts in the defect had the lowest NAD(P)H τM, likely due activation of the hypoxia pathway. Future analyses to characterize the dynamic changes of oxygen environment and metabolic changes/programming of osteoblasts and other cell types during healing could provide further insights into the control of cellular metabolism in vivo during repair and regeneration.
It is important to note that our current optical approach does not measure the absolute usage of glycolysis or OxPhos in a cell. Instead, this approach takes into account both energy pathways and provides a snapshot of the relative ratio of the usage of these pathways in a single cell. Therefore, this method is very useful in longitudinal analyses to compare energy metabolism during cell differentiation. It could also be very effective in vivo or in vitro when comparison is made among multiple cell types present in a culture or in a tissue. It is also important to note that NAD(P)H has many uses in addition to energetic metabolism including glutathione disulfide reduction, redox regulation, oxidative protein folding, amino acid and fatty acid biosynthesis etc. Thus, analyses using methods such as metabolic tracing could provide more precise measurement of energy metabolism in bone cells during normal bone homeostasis and during repair.
5. Conclusion
A minimally-invasive 2-photon FLIM-based optical technique was utilized to assess cellular metabolism in real-time at a single cell resolution in cell culture and in living animals that labeled osteoblastic cell populations with fluorescent proteins. We demonstrate that mature osteoblasts shifted to use more OxPhos as their energy source upon treatment of osteogenic media. By measuring cell metabolism and oxygen tension in un-injured and injured bone in living animals, we demonstrate that cellular metabolism could be dependent upon locations and the differentiation status of the osteoblasts. The cellular metabolism in osteoblasts could be further affected by oxygen availability during bone repair and regeneration. Using a minimally-invasive optical technique to assess cellular metabolism and tissue oxygenation will provide a deeper understanding of energy metabolism and hypoxia niche in normal and injured bone microenvironment, offering new insights into the dynamics of bone tissue oxygenation on bone cell differentiation and function.
Supplementary Material
Highlights:
NAD(P)H lifetime imaging enables single cell analyses of osteoblast metabolism in vitro and in vivo.
Higher NAD(P)H τM and OxPhos is associated with more mature osteoblasts.
Osteoblast metabolism could be dependent upon its location and differentiation status.
Osteoblast metabolism could be further affected by oxygen availability during bone repair.
ACKNOWLEDGEMENTS:
This study is supported by grants from the National Institutes of Health R01AR067859, R01DE019902, R21DE026256, R01DE029790, R21AR076056, P30AR069655, and the Department of Defense BCRP (W81XWH-17-1-0011).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author credit statement
Kevin Schilling: performing all experiments and analyses, original draft preparation.
Edward Brown: Supervision, reviewing and editing
Xinping Zhang: Conceptualization, Methodology, supervision, reviewing and editing.
REFERENCES:
- [1].Motyl KJ, Guntur AR, Carvalho AL, Rosen CJ. Energy Metabolism of Bone. Toxicol Pathol 2017;45: 887–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lee WC, Guntur AR, Long F, Rosen CJ. Energy Metabolism of the Osteoblast: Implications for Osteoporosis. Endocr Rev 2017;38: 255–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Shares BH, Busch M, White N, Shum L, Eliseev RA. Active mitochondria support osteogenic differentiation by stimulating beta-catenin acetylation. J Biol Chem 2018;293: 16019–16027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Guntur AR, Le PT, Farber CR, Rosen CJ. Bioenergetics during calvarial osteoblast differentiation reflect strain differences in bone mass. Endocrinology 2014;155: 1589–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Regan JN, Lim J, Shi Y, Joeng KS, Arbeit JM, Shohet RV, Long F. Up-regulation of glycolytic metabolism is required for HIF1alpha-driven bone formation. Proc Natl Acad Sci U S A 2014;111: 8673–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Meleshina AV, Dudenkova VV, Bystrova AS, Kuznetsova DS, Shirmanova MV, Zagaynova EV. Two-photon FLIM of NAD(P)H and FAD in mesenchymal stem cells undergoing either osteogenic or chondrogenic differentiation. Stem Cell Res Ther 2017;8: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Shum LC, White NS, Mills BN, Bentley KL, Eliseev RA. Energy Metabolism in Mesenchymal Stem Cells During Osteogenic Differentiation. Stem Cells Dev 2016;25: 114–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Shum LC, White NS, Nadtochiy SM, Bentley KL, Brookes PS, Jonason JH, Eliseev RA. Cyclophilin D Knock-Out Mice Show Enhanced Resistance to Osteoporosis and to Metabolic Changes Observed in Aging Bone. PLoS One 2016;11: e0155709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Dobson PF, Dennis EP, Hipps D, Reeve A, Laude A, Bradshaw C, Stamp C, Smith A, Deehan DJ, Turnbull DM, Greaves LC. Mitochondrial dysfunction impairs osteogenesis, increases osteoclast activity, and accelerates age related bone loss. Sci Rep 2020;10: 11643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Moreira JD, Hamraz M, Abolhassani M, Bigan E, Peres S, Pauleve L, Nogueira ML, Steyaert JM, Schwartz L. The Redox Status of Cancer Cells Supports Mechanisms behind the Warburg Effect. Metabolites 2016;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhu XH, Lu M, Lee BY, Ugurbil K, Chen W. In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc Natl Acad Sci U S A 2015;112: 2876–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Berthiaume JM, Kurdys JG, Muntean DM, Rosca MG. Mitochondrial NAD(+)/NADH Redox State and Diabetic Cardiomyopathy. Antioxidants & Redox Signaling 2019;30: 375–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kolenc OI, Quinn KP. Evaluating Cell Metabolism Through Autofluorescence Imaging of NAD(P)H and FAD. Antioxid Redox Signal 2019;30: 875–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Le Marois A, Suhling K. Quantitative Live Cell FLIM Imaging in Three Dimensions. Adv Exp Med Biol 2017;1035: 31–48. [DOI] [PubMed] [Google Scholar]
- [15].Blacker TS, Duchen MR. Investigating mitochondrial redox state using NADH and NADPH autofluorescence. Free Radic Biol Med 2016;100: 53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Penjweini R, Roarke B, Alspaugh G, Gevorgyan A, Andreoni A, Pasut A, Sackett DL, Knutson JR. Single cell-based fluorescence lifetime imaging of intracellular oxygenation and metabolism. 2020. [DOI] [PMC free article] [PubMed]
- [17].Skala MC, Riching KM, Bird DK, Gendron-Fitzpatrick A, Eickhoff J, Eliceiri KW, Keely PJ, Ramanujam N. In vivo multiphoton fluorescence lifetime imaging of protein-bound and free nicotinamide adenine dinucleotide in normal and precancerous epithelia. J Biomed Opt 2007;12: 024014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bird DK, Yan L, Vrotsos KM, Eliceiri KW, Vaughan EM, Keely PJ, White JG, Ramanujam N. Metabolic mapping of MCF10A human breast cells via multiphoton fluorescence lifetime imaging of the coenzyme NADH. Cancer Res 2005;65: 8766–73. [DOI] [PubMed] [Google Scholar]
- [19].Cannon Tm Fau - Shah AT, Shah At Fau - Walsh AJ, Walsh Aj Fau - Skala MC, Skala MC. High-throughput measurements of the optical redox ratio using a commercial microplate reader. 2015. [DOI] [PMC free article] [PubMed]
- [20].Varone A, Xylas J, Quinn KP, Pouli D, Sridharan G, McLaughlin-Drubin ME, Alonzo C, Lee K, Münger K, Georgakoudi I. Endogenous two-photon fluorescence imaging elucidates metabolic changes related to enhanced glycolysis and glutamine consumption in precancerous epithelial tissues. 2014. [DOI] [PMC free article] [PubMed]
- [21].Alhallak K, Rebello LG, Muldoon TJ, Quinn KP, Rajaram N. Optical redox ratio identifies metastatic potential-dependent changes in breast cancer cell metabolism. Biomedical Optics Express 2016;7: 4364–4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ostrander JH, McMahon CM, Lem S, Millon SR, Brown JQ, Seewaldt VL, Ramanujam N. Optical redox ratio differentiates breast cancer cell lines based on estrogen receptor status. Cancer Res 2010;70: 4759–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Stuntz E, Gong Y, Sood D, Liaudanskaya V, Pouli D, Quinn KP, Alonzo C, Liu Z, Kaplan DL, Georgakoudi I. Endogenous Two-Photon Excited Fluorescence Imaging Characterizes Neuron and Astrocyte Metabolic Responses to Manganese Toxicity. Sci Rep 2017;7: 1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Yaseen MA, Sutin J, Wu W, Fu B, Uhlirova H, Devor A, Boas DA, Sakadžić S. Fluorescence lifetime microscopy of NADH distinguishes alterations in cerebral metabolism in vivo. Biomedical Optics Express 2017;8: 2368–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kolenc OI, Quinn KP. Evaluating Cell Metabolism Through Autofluorescence Imaging of NAD(P)H and FAD. 2018. [DOI] [PMC free article] [PubMed]
- [26].Datta R, Heaster T, Sharick J, Gillette A, Skala M. Fluorescence lifetime imaging microscopy: fundamentals and advances in instrumentation, analysis, and applications. Journal of Biomedical Optics 2020;25: 071203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sharick JT, Favreau PF, Gillette AA, Sdao SM, Merrins MJ, Skala MC. Protein-bound NAD(P) H Lifetime is Sensitive to Multiple Fates of Glucose Carbon. Scientific Reports 2018;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Chacko JV, Eliceiri KW. Autofluorescence lifetime imaging of cellular metabolism: Sensitivity toward cell density, pH, intracellular, and intercellular heterogeneity. Cytometry A 2019;95: 56–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Castro PR, Barbosa AS, Pereira JM, Ranfley H, Felipetto M, Goncalves CAX, Paiva IR, Berg BB, Barcelos LS. Cellular and Molecular Heterogeneity Associated with Vessel Formation Processes. Biomed Res Int 2018;2018: 6740408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Stringari C, Cinquin A, Cinquin O, Digman MA, Donovan PJ, Gratton E. Phasor approach to fluorescence lifetime microscopy distinguishes different metabolic states of germ cells in a live tissue. Proc Natl Acad Sci U S A 2011;108: 13582–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ma N, Digman MA, Malacrida L, Gratton E. Measurements of absolute concentrations of NADH in cells using the phasor FLIM method. Biomed Opt Express 2016;7: 2441–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Stringari C, Edwards RA, Pate KT, Waterman ML, Donovan PJ, Gratton E. Metabolic trajectory of cellular differentiation in small intestine by Phasor Fluorescence Lifetime Microscopy of NADH. Sci Rep 2012;2: 568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Stringari C, Wang H, Geyfman M, Crosignani V, Kumar V, Takahashi JS, Andersen B, Gratton E. In vivo single-cell detection of metabolic oscillations in stem cells. Cell Rep 2015;10: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wright BK, Andrews LM, Markham J, Jones MR, Stringari C, Digman MA, Gratton E. NADH distribution in live progenitor stem cells by phasor-fluorescence lifetime image microscopy. Biophys J 2012;103: L7–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kalajzic I, Kalajzic Z, Kaliterna M, Gronowicz G, Clark SH, Lichtler AC, Rowe D. Use of type I collagen green fluorescent protein transgenes to identify subpopulations of cells at different stages of the osteoblast lineage. J Bone Miner Res 2002;17: 15–25. [DOI] [PubMed] [Google Scholar]
- [36].Kalajzic Z, Liu P, Kalajzic I, Du Z, Braut A, Mina M, Canalis E, Rowe DW. Directing the expression of a green fluorescent protein transgene in differentiated osteoblasts: comparison between rat type I collagen and rat osteocalcin promoters. Bone 2002;31: 654–60. [DOI] [PubMed] [Google Scholar]
- [37].Strecker S, Fu Y Fau - Liu Y, Liu Y Fau - Maye P, Maye P. Generation and characterization of Osterix-Cherry reporter mice. 2013. [DOI] [PMC free article] [PubMed]
- [38].Schilling K, El Khatib M, Plunkett S, Xue J, Xia Y, Vinogradov SA, Brown E, Zhang X. Electrospun Fiber Mesh for High-Resolution Measurements of Oxygen Tension in Cranial Bone Defect Repair. ACS Appl Mater Interfaces 2019;11: 33548–33558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Wang T, Zhai Y, Nuzzo M, Yang X, Yang Y, Zhang X. Layer-by-layer nanofiber-enabled engineering of biomimetic periosteum for bone repair and reconstruction. Biomaterials 2018;182: 279–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lyu S, Huang C, Yang H, Zhang X. Electrospun fibers as a scaffolding platform for bone tissue repair. J Orthop Res 2013;31: 1382–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kalinina S, Freymueller C, Naskar N, von Einem B, Reess K, Sroka R, Rueck A. Bioenergetic Alterations of Metabolic Redox Coenzymes as NADH, FAD and FMN by Means of Fluorescence Lifetime Imaging Techniques. International Journal of Molecular Sciences 2021;22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Mamontova AV, Solovyev ID, Savitsky AP, Shakhov Acapital Em C, Lukyanov KA, Bogdanov AM. Bright GFP with subnanosecond fluorescence lifetime. Sci Rep 2018;8: 13224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Datta R, Heaster TM, Sharick JT, Gillette AA, Skala MC. Fluorescence lifetime imaging microscopy: fundamentals and advances in instrumentation, analysis, and applications. J Biomed Opt 2020;25: 1–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Conklin MW, Provenzano PP, Eliceiri KW, Sullivan R, Keely PJ. Fluorescence lifetime imaging of endogenous fluorophores in histopathology sections reveals differences between normal and tumor epithelium in carcinoma in situ of the breast. Cell Biochem Biophys 2009;53: 145–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ranjit S, Dvornikov A, Stakic M, Hong SH, Levi M, Evans RM, Gratton E. Imaging Fibrosis and Separating Collagens using Second Harmonic Generation and Phasor Approach to Fluorescence Lifetime Imaging. Sci Rep 2015;5: 13378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ranjit S, Datta R, Dvornikov A, Gratton E. Multicomponent Analysis of Phasor Plot in a Single Pixel to Calculate Changes of Metabolic Trajectory in Biological Systems. The Journal of Physical Chemistry A 2019;123: 9865–9873. [DOI] [PubMed] [Google Scholar]
- [47].Ranjit S, Malacrida L, Jameson DM, Gratton E. Fit-free analysis of fluorescence lifetime imaging data using the phasor approach. Nature Protocols 2018;13: 1979–2004. [DOI] [PubMed] [Google Scholar]
- [48].Zhang X Intravital Imaging to Understand Spatiotemporal Regulation of Osteogenesis and Angiogenesis in Cranial Defect Repair and Regeneration. Methods Mol Biol 2018;1842: 229–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Huang C, Ness VP, Yang X, Chen H, Luo J, Brown EB, Zhang X. Spatiotemporal Analyses of Osteogenesis and Angiogenesis via Intravital Imaging in Cranial Bone Defect Repair. J Bone Miner Res 2015;30: 1217–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Walsh TG, Metharom P, Berndt MC. The functional role of platelets in the regulation of angiogenesis. Platelets 2015;26: 199–211. [DOI] [PubMed] [Google Scholar]
- [51].Liu JM, Rosen CJ, Ducy P, Kousteni S, Karsenty G. Regulation of Glucose Handling by the Skeleton: Insights From Mouse and Human Studies. Diabetes 2016;65: 3225–3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Fang Y, Wang ZY, Mao Y, Xin HT, Ren GL, Bai XF. Effects of insulin-like growth factor I on the development of osteoblasts in hyperglycemia. Diabetes Res Clin Pract 2006;73: 95–7. [DOI] [PubMed] [Google Scholar]
- [53].Wu M, Ai W, Chen L, Zhao S, Liu E. Bradykinin receptors and EphB2/EphrinB2 pathway in response to high glucose-induced osteoblast dysfunction and hyperglycemia-induced bone deterioration in mice. Int J Mol Med 2016;37: 565–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Karner CM, Long F. Glucose metabolism in bone. Bone 2018;115: 2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Esen E, Long F. Aerobic glycolysis in osteoblasts. Curr Osteoporos Rep 2014;12: 433–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Wei J, Shimazu J, Makinistoglu MP, Maurizi A, Kajimura D, Zong H, Takarada T, Lezaki T, Pessin JE, Hinoi E, Karsenty G. Glucose Uptake and Runx2 Synergize to Orchestrate Osteoblast Differentiation and Bone Formation. Cell 2015;161: 1576–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Borle AB, Nichols N, Nichols G, Jr. Metabolic studies of bone in vitro. I. Normal bone. J Biol Chem 1960;235: 1206–10. [PubMed] [Google Scholar]
- [58].Esen E, Lee SY, Wice BM, Long F. PTH Promotes Bone Anabolism by Stimulating Aerobic Glycolysis via IGF Signaling. J Bone Miner Res 2015;30: 2137. [DOI] [PubMed] [Google Scholar]
- [59].Lee WC, Ji X, Nissim I, Long F. Malic Enzyme Couples Mitochondria with Aerobic Glycolysis in Osteoblasts. Cell Rep 2020;32: 108108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Lee SY, Abel ED, Long F. Glucose metabolism induced by Bmp signaling is essential for murine skeletal development. Nat Commun 2018;9: 4831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Li Z, Frey JL, Wong GW, Faugere MC, Wolfgang MJ, Kim JK, Riddle RC, Clemens TL. Glucose Transporter-4 Facilitates Insulin-Stimulated Glucose Uptake in Osteoblasts. Endocrinology 2016;157: 4094–4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Gao J, Feng Z, Wang X, Zeng M, Liu J, Han S, Xu J, Chen L, Cao K, Long J, Li Z, Shen W, Liu J. SIRT3/SOD2 maintains osteoblast differentiation and bone formation by regulating mitochondrial stress. Cell death and differentiation 2018;25: 229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Riddle RC. Parathyroid Hormone Reprograms Osteoblast Metabolism. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 2015;30: 1956–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Stegen S, Devignes CS, Torrekens S, Van Looveren R, Carmeliet P, Carmeliet G. Glutamine Metabolism in Osteoprogenitors Is Required for Bone Mass Accrual and PTH-Induced Bone Anabolism in Male Mice. Journal of bone and mineral research 2021;36: 604–616. [DOI] [PubMed] [Google Scholar]
- [65].Yu Y, Newman H, Shen L, Sharma D, Hu G, Mirando AJ, Zhang H, Knudsen E, Zhang G-F, Hilton MJ, Karner CM. Glutamine Metabolism Regulates Proliferation and Lineage Allocation in Skeletal Stem Cells. Cell Metabolism 2019;29: 966–978.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Komarova SV, Ataullakhanov FI, Globus RK. Bioenergetics and mitochondrial transmembrane potential during differentiation of cultured osteoblasts. American Journal of Physiology-Cell Physiology 2000;279: C1220–C1229. [DOI] [PubMed] [Google Scholar]
- [67].Frikha-Benayed D, Basta-Pljakic J, Majeska RJ, Schaffler MB. Regional differences in oxidative metabolism and mitochondrial activity among cortical bone osteocytes. Bone 2016;90: 15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Sanchez-de-Diego C, Artigas N, Pimenta-Lopes C, Valer JA, Torrejon B, Gama-Perez P, Villena JA, Garcia-Roves PM, Rosa JL, Ventura F. Glucose Restriction Promotes Osteocyte Specification by Activating a PGC-1alpha-Dependent Transcriptional Program. iScience 2019;15: 79–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Karthik V, Guntur AR. Energy Metabolism of Osteocytes. Curr Osteoporos Rep 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Tuncay OC, Ho D, Barker MK. Oxygen tension regulates osteoblast function. American Journal of Orthodontics and Dentofacial Orthopedics 1994;105: 457–463. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.