

Abstract
Uterine PEComas often present a diagnostic challenge as they share morphological and immunohistochemical features with smooth muscle tumors. Herein we evaluated a series of 19 uterine PEComas to compare the degree of melanocytic marker expression with their molecular profile. Patients ranged from 32 to 77 (median 48) years, with six tumors classified as malignant based on the modified gynecologic-specific prognostic algorithm. All patients with malignant PEComas were alive with disease or dead from disease at last follow-up, while all those of uncertain malignant potential were alive and well (median follow-up, 47 months).
Seventeen of 19 (89%) PEComas harbored either a TSC1 or TSC2 alteration. One of the two remaining tumors showed a TFE3 rearrangement, but the other lacked alterations in all genes evaluated. All showed at least focal (usually strong) positivity for HMB-45, with 15/19 (79%) having > 50% expression, while the tumor lacking TSC or TFE3 alterations was strongly positive in 10% of cells. Melan-A and MiTF were each positive in 15/19 (79%) tumors, but staining extent and intensity was much more variable than HMB-45. Five of six (83%) malignant PEComas also harbored alterations in TP53, ATRX, or RB1, findings not identified in any tumors of uncertain malignant potential. One malignant PEComa was microsatellite-unstable/mismatch repair protein-deficient.
In summary, TSC alterations/TFE3 fusions and diffuse (> 50%) HMB-45 expression are characteristic of uterine PEComas. In morphologically ambiguous mesenchymal neoplasms with myomelanocytic differentiation, especially those with metastatic or recurrent disease, next-generation sequencing is recommended to evaluate for TSC alterations, as such patients can be eligible for targeted therapy.
Keywords: PEComa, Perivascular Epithelioid Cell Tumor, Uterus, TSC, TFE3, HMB-45
INTRODUCTION
Uterine perivascular epithelioid cell tumors (PEComas) are diagnostically challenging mesenchymal neoplasms as their morphology often overlaps with smooth muscle tumors1–5. PEComas are characterized by the coexpression of melanocytic and smooth muscle markers, but a small subset of smooth muscle tumors may also be positive for the former, in particular, HMB-456–8. Novel immunostains such as cathepsin K and PNL2 were originally speculated to aid in this diagnostic issue, but have since been found to be positive in smooth muscle tumors2,3,9. Recently, a study on uterine sarcomas with myomelanocytic differentiation identified four subgroups of tumors based on integration of morphological, immunohistochemical, and molecular data—malignant PEComa, sarcoma with PEComa-like features, myogenic sarcoma, and sarcoma NOS4. TSC2 alterations or TFE3 fusions were detected in 6/7 (86%) primary or recurrent tumors in the first two groups, and absent in the latter two categories (n=8). However, one malignant PEComa, as well as one of the myogenic sarcomas, lacked TSC2 or TFE3 alterations, but showed ≥ 50% expression for HMB-45 and positivity for MiTF; thus, it is difficult to explain their classification within the respective categories. Herein we correlate the molecular findings and degree of melanocytic marker expression in a series of 19 uterine tumors previously diagnosed as PEComa3.
MATERIALS AND METHODS
Next-Generation Sequencing/TFE3 Fusion Status:
Formalin-fixed paraffin-embedded sections (FFPE) (n=22) or sequencing data (n=1) were available for 23/32 (72%) tumors from the prior study3. For the former, genomic DNA was isolated from macro-dissected sections using the QIAamp DNA FFPE Tissue Kit (Qiagen, Valencia, CA) according to manufacturer’s instructions. Extraction was successful in 18/22 (82%) tumors while the remaining four failed to meet quality control measures and were excluded from analysis. Next-generation sequencing was performed using the targeted, hybrid capture 1,213-gene OncoPlus panel at the University of Chicago, as previously described10,11. Somatic mutation calling was performed across all 1,213 genes using a custom in-house bioinformatics pipeline as previously described10. Variant review was performed by two authors with specific expertise in this area (Z.O., L.R.) and included filters based on population variant frequencies (Exome Aggregation Consortium, http://exac.broadinstitute.org/), variant frequencies in cancer databases (COSMIC: catalogue of somatic mutations in cancer https://cancer.sanger.ac.uk/cosmic and cBioPortal https://www.cbioportal.org/), and coding effects. Somatic variant calls were inspected using Integrated Genomics Viewer (IGV; Broad Institute, MIT Harvard, Cambridge, MA). TFE3 fusions were previously assessed by fluorescence in-situ hybridization3.
Melanocytic Marker Expression:
Immunohistochemistry for HMB-45, Melan-A, and MiTF was previously performed in tumors from our study3. Tumors were scored for both extent (percentage of positive cells) and intensity (1+ weak, 2+ moderate, 3+ strong) of each melanocytic marker. Stains were considered positive if nuclear (MiTF) or cytoplasmic (HMB-45, Melan-A) expression was noted.
Microsatellite Instability/Mismatch Repair Protein Immunohistochemical Testing:
As part of the OncoPlus panel, a microsatellite instability (MSI) detection module using data from 336 incidentally captured homopolymers across the 1,213 captured genes was utilized as previously described12. Due to the impact of pre-analytic factors on the MSI-calling module and the inclusion of many FFPE blocks in this study that were > 5 years old, we required that > 90% of microsatellite loci reach the minimum sequencing depth threshold (50X coverage) for quality control and accurate MSI calling. Tumors with < 9% unstable loci were classified as microsatellite-stable, 9 to 15% as indeterminate, and > 15% as unstable. Immunohistochemistry for MSH6 and PMS2 was performed on all tumors classified as indeterminate or unstable, as well as in those without available microsatellite information. PEComas were considered mismatch repair protein-deficient if there was absence of nuclear staining in tumor cells for either MSH6 (rabbit monoclonal EPR3945, dilution 1:200; Abcam, Cambridge, MA) or PMS2 (rabbit monoclonal EPR3945, dilution 1:50; Abcam, Cambridge, MA). For all other staining patterns, the tumor was considered mismatch repair protein-proficient.
RESULTS
Clinical and Morphological Features:
Patients ranged from 32 to 77 (mean 50, median 48) years and tumors from 1 to 17 (mean 7, median 6; unknown in two) cm. Two patients had a clinical diagnosis of tuberous sclerosis. Two other patients had a history of renal angiomyolipoma or pulmonary lymphangioleiomyomatosis but never underwent further clinical or genetic evaluation. The remaining patients either had no other signs suggestive of tuberous sclerosis (n=13) or additional clinical information was not available (n=2). All tumors showed morphological features of PEComas (Supplemental File 1) and were classified as such in our prior study3. Based on the modified gynecologic-specific prognostic algorithm where at least three atypical features (size ≥ 5 cm, necrosis, high nuclear grade, mitoses > 1 per 50 high-power fields, vascular invasion) are required for a diagnosis of malignancy2,3,13, 13/19 (68%) PEComas were classified as uncertain malignant potential and 6/19 (32%) as malignant (Table 1). Recurrences occurred in 6/18 (33%; unknown in one) patients with three experiencing multiple recurrences (two malignant, one uncertain malignant potential). Follow-up was available for all patients, ranging from 5 to 175 (mean 51, median 47) months, with 13/19 (68%) alive and well, 3/19 (16%) alive with disease, and 3/19 (16%) dead of disease. All patients with a diagnosis of malignant PEComa were either alive with disease or dead of disease at last follow-up, while all with PEComas of uncertain malignant potential were alive and well; however, one patient in the latter category experienced multiple recurrences.
Table 1:
Clinical and Morphological Features of Uterine PEComas
| Case Number | Age (years) | Tuberous Sclerosis | ≥ 5 cm | Necrosis | High-Nuclear Grade | Mitoses > 1 / 50 HPFs | Vascular Invasion | # of Atypical Features | Classification | Recurrences | Follow-Up |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 63 | N/A | + | - | - | - | - | 1 | UMP | Multiple sites | NED, 175 months |
| 2 | 35 | - | + | - | - | + | - | 2 | UMP | - | NED, 57 months |
| 3 | 63 | - | + | + | + | + | - | 4 | Malignant | N/A | DOD, 5 months |
| 4 | 67 | N/A | + | + | + | + | + | 5 | Malignant | Lung | DOD, 20 months |
| 5 | 36 | + | - | - | - | - | - | 0 | UMP | - | NED, 11 months |
| 6 | 35 | - | - | - | - | + | - | 1 | UMP | - | NED, 55 months |
| 7 | 77 | - | - | - | - | - | - | 0 | UMP | - | NED, 15 months |
| 8 | 44 | + | - | - | - | - | - | 0 | UMP | - | NED, 47 months |
| 9 | 62 | - | + | + | + | + | - | 4 | Malignant | Lung, liver | AWD, 66 months |
| 10 | 32 | - | + | - | - | - | - | 1 | UMP | - | NED, 49 months |
| 11 | 48 | - | N/A | - | - | - | - | 0 | UMP | - | NED, 117 months |
| 12 | 50 | - | + | + | + | + | + | 5 | Malignant | Mediastinum | AWD, 7 months |
| 13 | 55 | - | - | - | - | - | - | 0 | UMP | - | NED, 88 months |
| 14 | 43 | - | - | - | - | + | - | 1 | UMP | - | NED, 19 months |
| 15 | 36 | - | N/A | - | - | + | - | 1 | UMP | - | NED, 47 months |
| 16 | 48 | - | + | + | + | + | - | 4 | Malignant | Vagina | DOD, 18 months |
| 17 | 50 | Suspicious | + | - | - | + | - | 2 | UMP | - | NED, 40 months |
| 18 | 39 | Suspicious | + | - | - | - | + | 2 | UMP | - | NED, 118 months |
| 19 | 64 | - | + | + | + | + | - | 4 | Malignant | Pelvis, liver | AWD, 24 months |
HPF=high-power field, N/A=not available, + = present, - = absent, UMP=uncertain malignant potential, NED=no evidence of disease, DOD=dead of disease, AWD=alive with disease
Next-Generation Sequencing:
Seventeen of 19 (89%) PEComas harbored either a TSC1 or TSC2 alteration (Figure 1a). TSC1 alterations were detected in 9/19 (47%) tumors and included pathogenic/likely pathogenic mutations in eight and a rearrangement predicted to cause loss of function in one, with three of them also showing copy number losses. TSC2 alterations were noted in 8/19 (42%) PEComas and included pathogenic/likely pathogenic mutations in four (with one also showing an additional variant of uncertain significance), variants of uncertain clinical significance in two, a rearrangement predicted to cause loss of function in one (also with additional copy number loss), and only copy number loss in one. For the remaining two tumors, one had a TFE3 fusion and the other did not harbor alterations in any genes examined. TSC1, TSC2, and TFE3 alterations were mutually exclusive.
Figure 1.

Pathogenic/likely pathogenic mutations (A) and copy number alterations (B) detected in uterine PEComas.
ATRX and/or TP53 mutations were detected in 4/6 (67%) malignant PEComas but were absent in all those classified as uncertain malignant potential. Additional non-recurrent pathogenic/likely pathogenic mutations included TERT (1/18; 6%), ADGVR1 (1/18; 6%), FZR1 (1/18; 6%), KMT2D (1/19; 5%), and SUFU (1/19; 5%). All variants are summarized in supplemental file 2.
Copy number losses were noted in 7/19 (37%) tumors (Figure 1b). Recurrent ones included the previously mentioned losses in TSC1 (9q34.13) (3/19; 16%) and TSC2 (16p13.3) (2/19; 11%), as well as FANCA (16q24.3) (2/19; 11%), FGFR3 (4p16.3) (2/19; 11%), NOTCH1 (9q34.3) (2/19; 11%), and MYCN (2p24.3) (2/19; 11%). Similar to the ATRX and TP53 mutations described above, deletion of TP53 (17p13.1) (case 9b) and RB1 (13q14.2) (case 16) were only observed in malignant PEComas. Overall, 5/6 (83%) malignant PEComas harbored alterations in TP53, ATRX, or RB1.
Correlation Between Melanocytic Marker Expression and Molecular Results:
All tumors, regardless of their TSC alteration status, showed at least focal positivity (≥ 10%) for HMB-45. HMB-45 expression ranged from 10 to 100% (mean 72%, median 75%), with most (17/19; 89%) showing strong intensity. In PEComas with TSC alterations, HMB-45 was positive in 15 to 100% (mean 74%, median 75%) of cells, with > 50% staining in 14/17 (82%). Melan-A was positive in 15/19 (79%) tumors, ranging from 0 to 100% (mean 19%, median 5%), with most (8/15; 53%) displaying strong intensity. In PEComas with TSC alterations, Melan-A was positive in 14/17 (82%), ranging from < 1 to 100% (mean 25%, median 10%), with > 50% expression seen in 4/17 (24%). MiTF was expressed in 15/19 (79%) tumors, ranging from 0 to 100% (mean 20%, median 5%) of cells, with only 2/15 (13%) showing strong intensity. In PEComas with TSC alterations, MiTF was expressed in 14/17 (82%), ranging from < 1 to 100% (mean 21%, median 10%), with > 50% expression in 3/17 (18%) tumors. Immunohistochemical results are summarized in Table 2.
Table 2:
Immunohistochemical and Molecular Features of Uterine PEComas
| Case # | Classification | HMB-45 (Extent, Intensity) | Melan-A (Extent, Intensity) | MiTF (Extent, Intensity) | TFE3 Fusion | Mutations | Copy Number Losses | MSI (% unstable loci) | MMR |
|---|---|---|---|---|---|---|---|---|---|
| 1 | UMP | 100%, 3+ | 60%, 2+ | 10%, 1+ | - | TERT promoter | TSC2 | F | P |
| 2 | UMP | 10%, 3+ | 10%, 2+ | 80%, 3+ | - | - | - | 6.6 | NP |
| 3 | Malignant | 100%, 3+ | < 5%, 3+ | 0 | - | TSC2 (VUS), TP53 | EP300, WT1 | F | P |
| 4 | Malignant | 60%, 3+ | < 1%, 1+ | < 5% 1+ | - | TSC1 | TSC1, NOTCH1 | F | P |
| 5 | UMP | 60%, 3+ | 0 | < 5%. 1+ | - | TSC1 | - | 5.4 | NP |
| 6 | UMP | 75%, 2+ | 20%, 2+ | 30%, 2+ | - | TSC1 | ERCC3 | 8.4 | NP |
| 7 | UMP | 55%, 2+ | 5%, 1+ | 10%, 1+ | - | TSC2 (VUS x2) | - | 12.3 | P |
| 8 | UMP | 100%, 3+ | 0 | 5%, 1+ | - | TSC1 | - | F | P |
| 9a | Malignant | 100%, 3+ | 60%, 2+ | 0 | - | TSC2, ATRX, ADGRV1 | - | F | P |
| 9b | < 1%, 3+ | 0 | 0 | - | ATRX, ADGRV1 | TSC1, CDH1, CTCF, FANCA, FGFR3, TP53, NOTCH1, POLE, PTEN | F | P | |
| 10 | UMP | 100%, 3+ | 25%, 3+ | 100%, 3+ | - | TSC1 | - | F | P |
| 11 | UMP | 15%, 3+ | < 5%, 3+ | 5%, 1+ | - | TSC1 | - | 10.2 | P |
| 12 | Malignant | 40%, 3+ | < 1%, 3+ | < 5%, 1+ | - | TSC1, ATRX, TP53, SUFU, FZR1 | - | 18.0 | D |
| 13 | UMP | 35%, 3+ | 55%, 3+ | 70%, 2+ | - | TSC2 | - | F | P |
| 14 | UMP | 70%, 3+ | < 1%, 3+ | 10%, 1+ | - | TSC1 rearrangement | TSC1, MYCN | 8.0 | NP |
| 15 | UMP | 100%, 3+ | 0 | 0 | PSF-TFE3 | - | - | 7.6 | NP |
| 16 | Malignant | 80%, 3+ | 15%, 3+ | 0 | - | TSC2 rearrangement | TSC2, CEBPA, RB1 | 6.0 | NP |
| 17 | UMP | 90%, 3+ | < 1%, 2+ | 30%, 2+ | - | TSC2 | FANCA, CTCF, IDH1, ERBB4, FGFR3, MYCN, DNMT3A, ALK, MSH2, MTOR, SDHB, ARID1A, CSF3R, MPL | 5.9 | NP |
| 18 | UMP | 70%, 3+ | 0 | 10%, 1+ | - | TSC2, TSC2 (VUS x1) | - | 4.5 | NP |
| 19 | Malignant | 100%, 3+ | 100%, 3+ | < 1%, 1+ | - | TSC2, TP53, KMT2D | - | NP | P |
MSI=microsatellite instability testing, MMR=mismatch repair protein, UMP=uncertain malignant potential, - = absent, F=failed, NP=not performed, VUS=variant of uncertain clinical significance, P=proficient, D=deficient
Two tumors did not show any TSC alterations. One of them (case 15) was composed of an alveolar to nested growth of epithelioid cells with clear cytoplasm. It harbored a PSF-TFE3 fusion and was strongly and diffusely (100%) HMB-45 positive, but negative for Melan-A and MiTF. The other one (case 2; Figure 2) was comprised of sheets and nests of predominantly epithelioid cells (70%), with cytoplasm ranging from clear to eosinophilic and granular to rhabdoid, as well as a perivascular/radial distribution of tumor cells. It showed strong, but focal (10%) expression for HMB-45 and Melan-A, and strong and diffuse (80%) MiTF staining.
Figure 2.
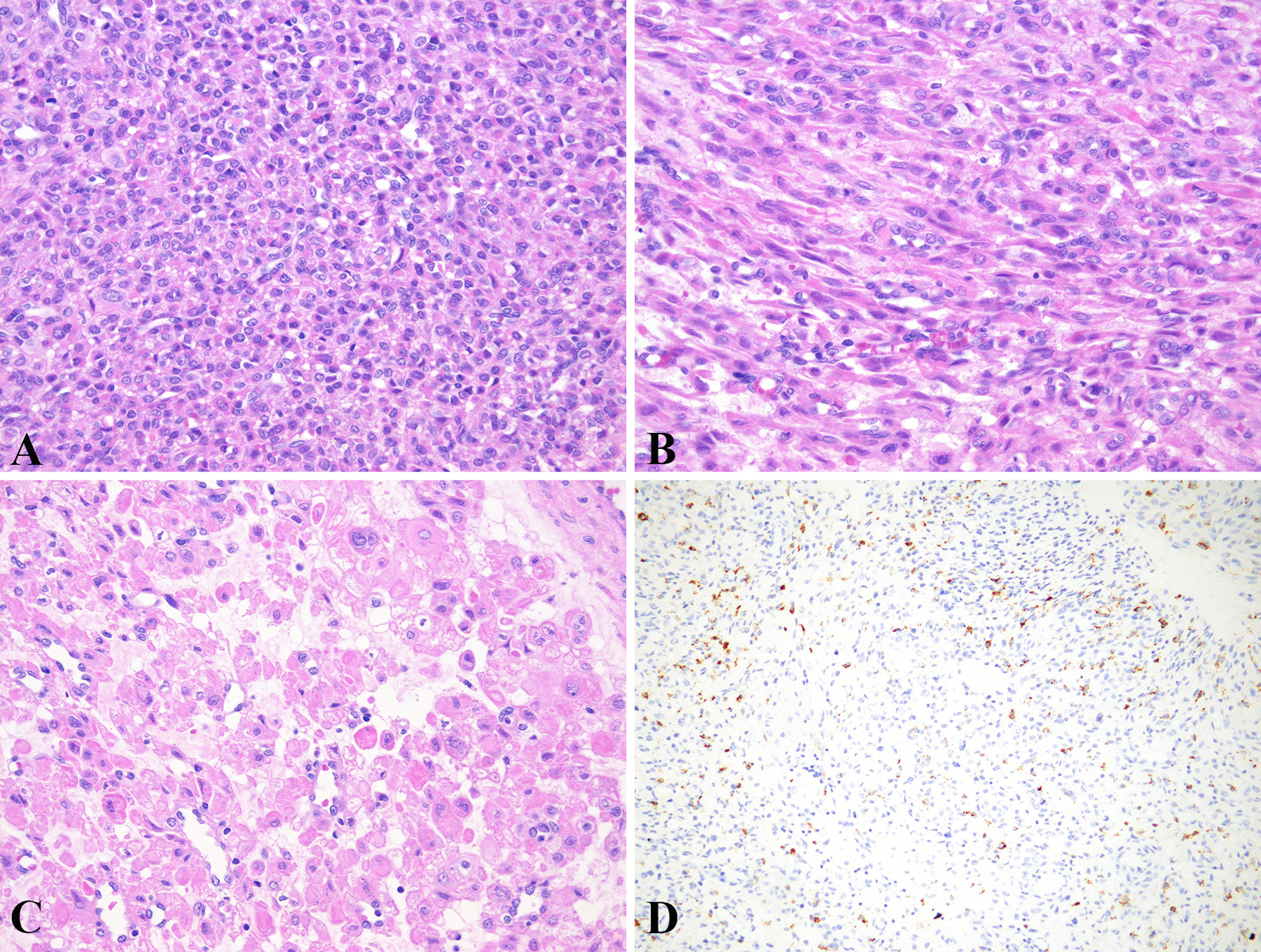
Sheets of epithelioid cells (A) and fascicles of spindle cells (B). Non-cohesive epithelioid cells with clear to eosinophilic and granular cytoplasm. Note scattered cells with a rhabdoid appearance (C). Strong, but focal HMB-45 expression (D).
One tumor (case 9; Figure 3) was biphasic, being comprised of two morphologically and immunohistochemically distinct areas. Approximately 40% of the tumor (case 9a) cells grew in sheets, nests, and trabeculae and were markedly pleomorphic, associated with brisk mitoses and tumor cell necrosis. This component was strongly and diffusely (100%) positive for HMB-45, moderately and diffusely (60%) positive for Melan-A, and negative for MiTF and all myogenic markers (data not shown). The remaining 60% (case 9b) was composed of fascicles of moderately atypical spindle cells with appreciable mitoses and tumor cell necrosis. It showed strong, but very rare (< 1%) HMB-45 expression, was negative for Melan-A and MiTF, and strongly and diffusely positive for smooth muscle actin, desmin, and caldesmon (data not shown). Both components harbored the same ATRX and ADGRV1 mutations, but a TSC1 mutation was detected only in the epithelioid component, and multiple copy number losses (including TSC1 and TP53, among others) only in the spindled component.
Figure 3.
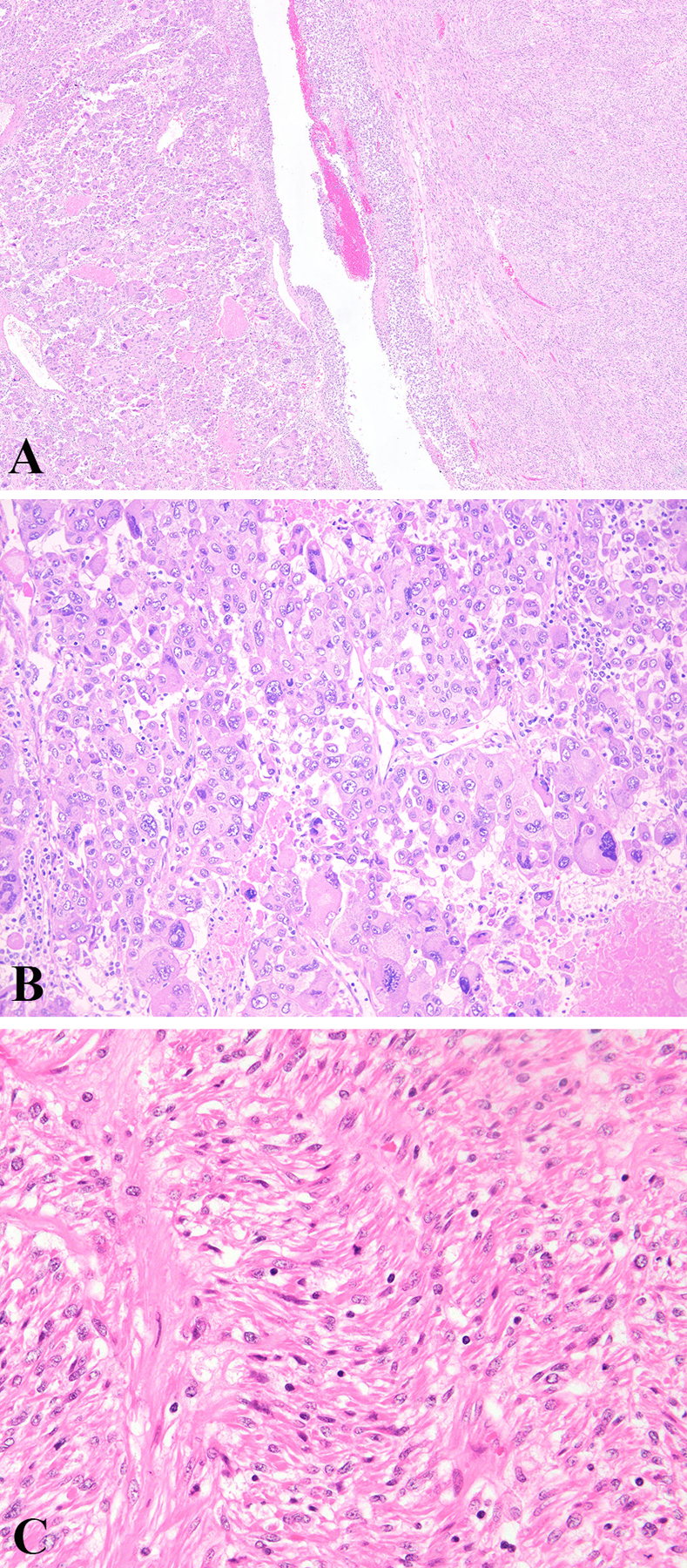
Biphasic neoplasm with epithelioid (left) and spindled (right) components. Non-cohesive epithelioid cells forming vague nests with marked atypia, brisk mitoses, and tumor cell necrosis (B). Fascicles of spindle cells with moderate atypia and scattered mitoses (C).
Microsatellite Instability/Mismatch Repair Protein Immunohistochemical Testing:
One PEComa (case 12) was microsatellite-unstable by next-generation sequencing, while the remaining tumors analyzed by sequencing were either stable (n=8), indeterminate (n=1), or unable to be determined based on insufficient sequencing coverage of microsatellite loci (< 90% of loci at 50X coverage) (n=8). The microsatellite-indeterminate PEComa and those without microsatellite information (n=9) were mismatch repair protein-proficient, while the one microsatellite-unstable tumor was mismatch repair protein-deficient (loss of MLH1 and PMS2, retained MSH2 and MSH6).
DISCUSSION
Even before the first series on uterine PEComas in 20021, there has been speculation to whether PEComas are a distinct entity or related to smooth muscle tumors, likely an epithelioid variant with melanocytic differentiation6–8,14–16. It is generally accepted that in the appropriate morphological setting, the presence of any expression for at least two melanocytic markers (or one melanocytic marker and cathepsin K), a diagnosis of PEComa can be rendered2. However, as there is often morphological overlap between PEComas and smooth muscle tumors, the question arises whether the extent of melanocytic marker expression is imperative in differentiating between the two entities. Cathepsin K was originally used in the differential diagnosis of renal cell carcinoma and epithelioid angiomyolipoma17, but its use has major limitations in the distinction between PEComa and smooth muscle tumors3,9. Herein, we further explored this issue by comparing the genomic findings with the degree of melanocytic marker expression in a series of 19 PEComas previously published3.
All tumors in this cohort fulfilled the immunohistochemical criteria used for PEComas (18 with positivity for at least two melanocytic markers and one positive for HMB-45 and cathepsin K). Since the latter PEComa (case 15) harbored a PSF-TFE3 fusion, it is not surprising that both Melan-A and MiTF were negative, as has been described in TFE3 translocation-associated PEComas18,19. Of the three melanocytic markers, only HMB-45 was strongly positive in all but two tumors, with just four showing < 50% expression. One of these four neoplasms (case 2) showed 10% HMB-45 expression and did not harbor a TSC/TFE3 alteration, while the remaining three with 15%, 35%, and 40% HMB-45 expression showed TSC alterations. We did not find Melan-A or MiTF to be particularly helpful as no correlation was obvious between TSC alterations and their expression pattern.
This naturally leads to the question as to whether case 2 is truly a PEComa versus a smooth muscle tumor with aberrant melanocytic marker expression. Several potential interpretations can be postulated. The presence of a different TSC dysregulation mechanism (epigenetic modification, miRNA, etc), a structural TSC alteration not detected by the assay, or alterations in a mTOR pathway-associated gene not covered by the assay may argue for this tumor still being a true PEComa. For instance, two TSC wild-type PEComas have recently been shown to harbor mutations in FLCN, a gene encoding for a protein involved in the mTOR pathway20. Although this gene was covered by our platform, it is still possible that there might be another mTOR pathway-associated gene that was not covered by our assay. Finally, although the morphology and immunoprofile was not typical of a TFE3 translocation-associated PEComa, it is possible that this tumor could harbor a TFE3 fusion that was not detected by FISH or a novel fusion in uterine PEComas yet to be identified. On the other hand, it may be postulated that this tumor may not be a PEComa, but perhaps an epithelioid smooth muscle neoplasm with PEComa-like morphological features and focal melanocytic marker expression. Myogenic markers, including desmin, caldesmon, and smooth muscle actin, were strongly and diffusely positive (data not shown), but this finding is also observed in PEComas1–3. We cannot further classify this mesenchymal tumor with certainty, but would favor it to be at most of uncertain malignant potential based on its morphology and wild-type status for genes commonly altered in malignancy (i.e. TP53, CDKN2A/2B, RB1, ATRX, etc). Thus, one could argue that due to the expression of three melanocytic markers, this tumor could still be part of the PEComa family.
One tumor (case 9) was quite unusual in that it was clearly biphasic with morphologically and immunophenotypically distinct spindled and epithelioid areas. Macro-dissection of these components revealed identical ATRX and ADGRV1 mutations in both, with a TSC1 mutation detected in the epithelioid areas and copy number losses in multiple genes in the spindled areas. Based on molecular data, we can infer this is a clonally-related neoplasm rather than a collision tumor, with two pathways of clonal evolution. One pathway where a TSC1 mutation was acquired likely resulted in a PEComa-like morphology and immunoprofile, and the second, with chromosomal instability with multiple copy number losses, led to a leiomyosarcoma-like morphology and immunophenotype. This neoplasm offers insight into whether PEComas and smooth muscle tumors are separate or related entities as it highlights that this tumor likely arose from the same progenitor cell and underwent divergent differentiation. Whether this concept is universal for all PEComas merits further study, especially those investigating epigenetic modifications/cell of origin.
Our study demonstrated several similarities with a recent series integrating morphological, immunohistochemical, and molecular features of 15 uterine mesenchymal tumors with myomelanocytic differentiation4. In that series, histologically ambivalent tumors were examined, while we typified neoplasms morphologically and immunohistochemically consistent with PEComa. Furthermore, they only sequenced 8/15 (53%) primary tumors (with the remainder studied being recurrences) whereas we only evaluated primary tumors. They identified a TSC2 alteration (n=5) or TFE3 fusion (n=1) in 6/7 (86%) malignant PEComas and PEComa-like sarcomas, that parallels our findings with 18/19 (95%) tumors having TSC or TFE3 alterations. Of note, none of their 15 tumors harbored TSC1 alterations, a finding we observed in 9/19 (47%) tumors, and has been previously been described in a subset of PEComas20–22. Those with characteristic PEComa genomic alterations were all positive for HMB-45 (two “positive”, “focal”, 15% strong, 20% strong, 100% strong; percentages and intensity not provided in tumors characterized as “positive” or “focal”), which aligns with our findings. One of their tumors (UMT01) classified as lung recurrence of a malignant PEComa showed moderate to strong HMB-45 expression in 70% of cells, lacked TSC or TFE3 alterations, but harbored mutations in TP53, RB1, and BRD44. As previously reported20,23 and also confirmed in our study (discussed in detail below), TSC-altered PEComas may show concurrent TP53 mutations. However, in contrast to our series, UMT01 lacked TSC alterations, but showed > 50% HMB-45 staining. The several interpretations speculated for our case 2 can be applied to this tumor. Another sarcoma and its recurrence from their study (UMT06 and UMT06-R) showed 5% and 20% strong HMB-45 expression, respectively4. The primary tumor harbored mutations in TP53 and ATRX, whereas the recurrence had the same ATRX mutation as well as novel TSC2 and TERT mutations. The change in the HMB-45 staining pattern is similar to a HMB-45 negative uterine leiomyosarcoma reported by Silva et al. that acquired variable positivity for HMB-45 in the recurrences7. Thus, it can be speculated that as only 5% of cells were HMB-45 positive in the primary tumor, a TSC2-mutated subclone was likely present that became the predominant component in the recurrence. This tumor might be analogous to our case 9 with both PEComa and LMS-like components, with the exception that its PEComa-like clone was a smaller interspersed component rather than the two morphologically and immunohistochemically distinct foci we observed.
None of the sarcomas from the other two groups described by Selenica et al. (myogenic sarcoma and sarcoma NOS) harbored TSC or TFE3 alterations, and all except one (UMT02) showed limited (≤ 15%) HMB-45 expression. However, in UMT02 tumor, 50% of cells were strongly HMB-45 positive, with alterations noted in TP53 and MED12. As MED12 mutations have been described in 11% to 21% of leiomyosarcomas24,25, and to our knowledge not in PEComas, this tumor likely represents a leiomyosarcoma with significant HMB-45 expression. As 5/6 (83%) of their tumors in the myogenic sarcoma category showed ≤ 15% HMB-45 expression, it is highly plausible that case 2 in this series represents a leiomyoma with aberrant (10%) HMB-45 expression. Of note, MED12 is covered by our platform and was not altered in this tumor.
The question of whether TSC alterations are specific to PEComas or if they may occur in morphological mimickers must be considered. Previously, TSC2 alterations in uterine mesenchymal neoplasms have been reported in a leiomyoma with bizarre nuclei26, two leiomyosarcomas27,28, and two “high-grade non-leiomyosarcoma sarcomas”24. Scanned slides without immunohistochemical data are available for the tumor reported as uterine leiomyosarcoma by The Cancer Genome Atlas, but in our opinion does not morphologically resemble leiomyosarcoma28. None of the other tumors with reported TSC2 alterations have any morphological or immunohistochemical information available for review. Although it is entirely plausible that TSC alterations may be detected in rare smooth muscle tumors, it appears that they are most prevalent in PEComas, and hence can help establish the diagnosis in morphologically and immunohistochemically ambiguous neoplasms. Currently, this distinction is largely academic and thus an extensive molecular evaluation is not necessary in most situations. However, it is important to note that in a patient with metastatic or recurrent disease if a TSC alteration is identified, she may be eligible for targeted therapy with MTOR inhibitors29–32. A proposed algorithm for the molecular work-up of a uterine mesenchymal neoplasm with immunohistochemical evidence of myomelanocytic differentiation is provided (Figure 4).
Figure 4.
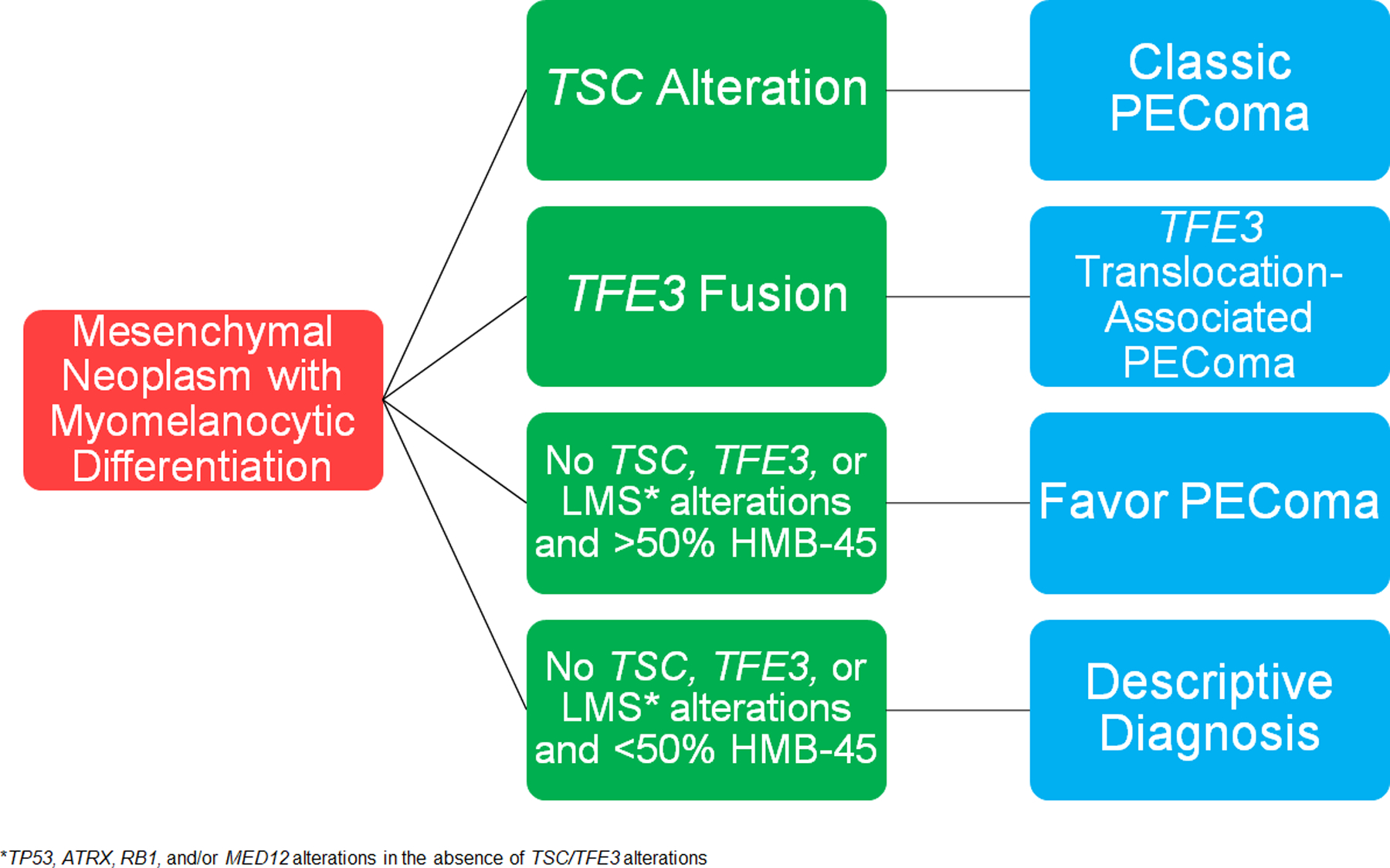
Proposed algorithm for evaluation of a uterine tumor with myomelanocytic differentiation by immunohistochemistry.
Aside from TSC alterations, only two other recurrent genetic aberrations were identified in this study, TP53 (3/19; 16%) and ATRX (2/19; 11%). TP53 and/or ATRX mutations were only detected in malignant PEComas (4/6; 67%), while another malignant PEComa showed a RB1 loss. The tumor that harbored both TP53 and ATRX mutations strongly expressed HMB-45 in 40% of cells, whereas the two with TP53 mutations (including one with a TSC2 variant of uncertain clinical significance), the one with an ATRX mutation, and the one with RB1 loss showed strong and diffuse HMB-45 positivity. Given the presence of a TSC variant and strong HMB-45 expression in ≥ 40% of cells, we believe that the diagnosis of malignant PEComa, as opposed to leiomyosarcoma with aberrant melanocytic marker expression is justified. Similarly, TP53, ATRX, and RB1 alterations have been described in a small subset of TSC-altered PEComas from diverse sites, most of which were classified as malignant, and analogous to our PEComas, showed variable amounts of HMB-45 expression4,20,23. Although the number of PEComas evaluated by next-generation sequencing is limited, the presence (or lack thereof) of an alteration in one or more of these genes might be a helpful adjunct to the morphological-based algorithm in predicting behavior; thus, additional studies are needed to corroborate this finding.
Brief discussion is warranted regarding the patient who suffered from multiple recurrences of her uncertain malignant potential PEComa. This tumor lacked a TSC mutation or rearrangement, but instead harbored a TSC2 deletion as well as a TERT promoter mutation. TERT promoter mutations are recurrent in multiple cancer types33. It is well recognized that they likely contribute to tumor biology and aggressive clinical behavior, including those in the gynecologic tract34–38. Whether this mutation is also characteristic for PEComas of uncertain malignant potential that recur merits further investigation.
By next-generation sequencing microsatellite testing, one tumor (case 12) was microsatellite-unstable and showed loss of MLH1 and PMS2. As there were no alterations in MLH1 or PMS2 by next-generation sequencing, we favor this abnormality to be secondary to MLH1 promoter methylation. To our knowledge, this is the second mismatch repair protein-deficient/microsatellite unstable uterine PEComa39, a finding which is important to note and warrants further study, as such patients can be eligible for targeted therapy with PD-L1 inhibitors40.
In summary, we identified TSC or TFE3 alterations in most (18/19; 95%) uterine tumors morphologically and immunohistochemically compatible with PEComa, and 14/17 (82%) had > 50% HMB-45 expression. We also recognized that most malignant PEComas harbor alterations in TP53, ATRX, or RB1, and rare PEComas are mismatch repair protein deficient/microsatellite unstable. Finally, detection of identical mutations (ATRX, ADGRV1) in a morphologically and immunohistochemically biphasic tumor that subsequently acquired different alterations (TSC1 mutation versus single copy deletions) in the different components, concludes this is a clonal neoplasm that underwent two pathways of evolution. Further studies are necessary to explore this finding and ultimately determine the relationship between PEComas and smooth muscle tumors.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Drs. Kristine Cornejo (Massachusetts General Hospital, Boston, MA, USA), Tomas Slavik (Ampath Private Pathology Laboratories and Department of Anatomical Pathology, University of Pretoria, Pretoria, South Africa), Trudy Jonges (University Medical Center Utrecht, Utrecht, Netherlands), Yukihiro Imai (Kobe City Medical Center General Hospital, Kobe, Japan), Jean-Francois Egger (Viollier Weintraub SA, Geneva, Switzerland), Carmen Tornos (Stony Brook University Hospital, Stony Brook, NY, USA), Carla Bartosch (Instituto Portugues de Oncologia, Porto, Portugual), and Takako Kiyokawa (The Jikei University School of Medicine, Tokyo, Japan) for providing follow-up information. They would also like to thank the University of Chicago Human Tissue Resource Center for slide preparation and the Molecular Diagnostic Laboratories for performing next-generation sequencing.
This work was presented in part as a platform presentation at the 110th United States and Canadian Academy of Pathology (USCAP) Annual Meeting.
FUNDING STATEMENT
This study was supported in part by P50-CA140146 (CRA) and P30-CA008748 (CRA).
Footnotes
ETHICS APPROVAL/CONSENT TO PARTICIPATE
This study was approved by the Institutional Review Boards at the individual institutions.
DATA AVAILABILITY STATEMENT
All data generated or analyzed during this study are included in this published article and its supplementary information files.
CONFLICT OF INTEREST STATEMENT
The authors have disclosed that they have no significant relationships with, or financial interest in, any commercial companies pertaining to this article.
REFERENCES
- 1.Vang R & Kempson RL Perivascular epithelioid cell tumor (‘PEComa’) of the uterus: a subset of HMB-45-positive epithelioid mesenchymal neoplasms with an uncertain relationship to pure smooth muscle tumors. Am J Surg Pathol 26, 1–13 (2002). [DOI] [PubMed] [Google Scholar]
- 2.Schoolmeester JK, Howitt BE, Hirsch MS, Dal Cin P, Quade BJ & Nucci MR Perivascular epithelioid cell neoplasm (PEComa) of the gynecologic tract: clinicopathologic and immunohistochemical characterization of 16 cases. Am J Surg Pathol 38, 176–188 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Bennett JA, Braga AC, Pinto A, Van de Vijver K, Cornejo K, Pesci A et al. Uterine PEComas: A Morphologic, Immunohistochemical, and Molecular Analysis of 32 Tumors. Am J Surg Pathol 42, 1370–1383 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Selenica P, Conlon N, Gonzalez C, Frosina D, Jungbluth AA, Beets-Tan RGH et al. Genomic Profiling Aids Classification of Diagnostically Challenging Uterine Mesenchymal Tumors With Myomelanocytic Differentiation. Am J Surg Pathol 10.1097/PAS.0000000000001572 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bennett JA & Oliva E Perivascular epithelioid cell tumors (PEComa) of the gynecologic tract. Genes Chromosomes Cancer 10.1002/gcc.22908 (2020). [DOI] [PubMed] [Google Scholar]
- 6.Silva EG, Deavers MT, Bodurka DC & Malpica A Uterine epithelioid leiomyosarcomas with clear cells: reactivity with HMB-45 and the concept of PEComa. Am J Surg Pathol 28, 244–249 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Silva EG, Bodurka DC, Scouros MA & Ayala A A uterine leiomyosarcoma that became positive for HMB45 in the metastasis. Ann Diagn Pathol 9, 43–45 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Simpson KW & Albores-Saavedra J HMB-45 reactivity in conventional uterine leiomyosarcomas. Am J Surg Pathol 31, 95–98 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Valencia-Guerrero A, Pinto A, Anderson WJ, Trevisan G, Nucci MR & Hirsch MS PNL2: A Useful Adjunct Biomarker to HMB45 in the Diagnosis of Uterine Perivascular Epithelioid Cell Tumor (PEComa). Int J Gynecol Pathol 10.1097/PGP.0000000000000653 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Kadri S, Long BC, Mujacic I, Zhen CJ, Wurst MN, Sharma S et al. Clinical Validation of a Next-Generation Sequencing Genomic Oncology Panel via Cross-Platform Benchmarking against Established Amplicon Sequencing Assays. J Mol Diagn 19, 43–56 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Bennett JA, Ritterhouse LL, Furtado LV, Lastra RR, Pesci A, Newell JM et al. Female adnexal tumors of probable Wolffian origin: morphological, immunohistochemical, and molecular analysis of 15 cases. Mod Pathol 33, 734–747 (2020). [DOI] [PubMed] [Google Scholar]
- 12.Chapel DB, Patil SA, Plagov A, Puranik R, Mendybaeva A, Steinhardt G et al. Quantitative next-generation sequencing-based analysis indicates progressive accumulation of microsatellite instability between atypical hyperplasia/endometrial intraepithelial neoplasia and paired endometrioid endometrial carcinoma. Mod Pathol 32, 1508–1520 (2019). [DOI] [PubMed] [Google Scholar]
- 13.WHO Classification of Tumours Female Genital Tumors (World Health Organization: Lyon, 2020). [Google Scholar]
- 14.Michal M & Zamecnik M Hyalinized Uterine Mesenchymal Neoplasms with HMB-45-Positive Epithelioid Cells: Epithelioid Leiomyomas or Angiomyolipomas? Report of Four Cases. Int J Surg Pathol 8, 323–328 (2000). [DOI] [PubMed] [Google Scholar]
- 15.Zamecnik M & Michal M HMB45+ hyalinized epithelioid tumor of the uterus is linked to epithelioid leiomyoma rather than to PEC-omas. Int J Surg Pathol 9, 341–343 (2001). [DOI] [PubMed] [Google Scholar]
- 16.Hurrell DP & McCluggage WG Uterine leiomyosarcoma with HMB45+ clear cell areas: report of two cases. Histopathology 47, 540–542 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Martignoni G, Pea M, Gobbo S, Brunelli M, Bonetti F, Segala D et al. Cathepsin-K immunoreactivity distinguishes MiTF/TFE family renal translocation carcinomas from other renal carcinomas. Mod Pathol 22, 1016–1022 (2009). [DOI] [PubMed] [Google Scholar]
- 18.Argani P, Aulmann S, Illei PB, Netto GJ, Ro J, Cho HY et al. A distinctive subset of PEComas harbors TFE3 gene fusions. Am J Surg Pathol 34, 1395–1406 (2010). [DOI] [PubMed] [Google Scholar]
- 19.Schoolmeester JK, Dao LN, Sukov WR, Wang L, Park KJ, Murali R et al. TFE3 translocation-associated perivascular epithelioid cell neoplasm (PEComa) of the gynecologic tract: morphology, immunophenotype, differential diagnosis. Am J Surg Pathol 39, 394–404 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Akumalla S, Madison R, Lin DI, Schrock AB, Yakirevich E, Rosenzweig M et al. Characterization of Clinical Cases of Malignant PEComa via Comprehensive Genomic Profiling of DNA and RNA. Oncology 98, 905–912 (2020). [DOI] [PubMed] [Google Scholar]
- 21.Schmiester M, Dolnik A, Kornak U, Pfitzner B, Hummel M, Treue D et al. TFE3 activation in a TSC1-altered malignant PEComa: challenging the dichotomy of the underlying pathogenic mechanisms. J Pathol Clin Res 7, 3–9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wagner AJ, Malinowska-Kolodziej I, Morgan JA, Qin W, Fletcher CD, Vena N et al. Clinical activity of mTOR inhibition with sirolimus in malignant perivascular epithelioid cell tumors: targeting the pathogenic activation of mTORC1 in tumors. J Clin Oncol 28, 835–840 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Agaram NP, Sung YS, Zhang L, Chen CL, Chen HW, Singer S et al. Dichotomy of Genetic Abnormalities in PEComas With Therapeutic Implications. Am J Surg Pathol 39, 813–825 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hensley ML, Chavan SS, Solit DB, Murali R, Soslow R, Chiang S et al. Genomic Landscape of Uterine Sarcomas Defined Through Prospective Clinical Sequencing. Clin Cancer Res 26, 3881–3888 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Makinen N, Aavikko M, Heikkinen T, Taipale M, Taipale J, Koivisto-Korander R et al. Exome Sequencing of Uterine Leiomyosarcomas Identifies Frequent Mutations in TP53, ATRX, and MED12. PLoS Genet 12, e1005850 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bennett JA, Chiang S, Chen Y, Bialik A, Aryee M, Young RH et al. Leiomyoma With Bizarre Nuclei: Correlation Between Morphology and Fumarate Hydratase/S-(2-Succino)-Cysteine Expression. Mod Pathol 28, 276A (2015). [Google Scholar]
- 27.Chudasama P, Mughal SS, Sanders MA, Hubschmann D, Chung I, Deeg KI et al. Integrative genomic and transcriptomic analysis of leiomyosarcoma. Nat Commun 9, 144 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cancer Genome Atlas Research Network. Electronic address, e. d. s. c. & Cancer Genome Atlas Research, N. Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas. Cell 171, 950–965 e928 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sanfilippo R, Jones RL, Blay JY, Le Cesne A, Provenzano S, Antoniou G et al. Role of Chemotherapy, VEGFR Inhibitors, and mTOR Inhibitors in Advanced Perivascular Epithelioid Cell Tumors (PEComas). Clin Cancer Res 25, 5295–5300 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Sanfilippo R, Fabbroni C, Fuca G, Fumagalli E, Morosi C, Sbaraglia M et al. Addition of Antiestrogen Treatment in Patients with Malignant PEComa Progressing to mTOR Inhibitors. Clin Cancer Res 10.1158/1078-0432.CCR-20-1191 (2020). [DOI] [PubMed] [Google Scholar]
- 31.Wagner AJ, Ravi V, Ganjoo KN, Van Tine BA, Riedel RF, Chugh R et al. ABI-009 (nab-sirolimus) in advanced malignant perivascular epithelioid cell tumors (PEComa): Preliminary efficacy, safety, and mutational status from AMPECT, an open label phase II registration trial. J. Clin. Oncol 37(15s), 11005 (2019). [Google Scholar]
- 32.Wagner AJ, Ravi V, Riedel RF, Ganjoo KN, Van Tine BA, Chugh R et al. Long‐term follow‐up for duration of response (DoR) after weekly nab‐sirolimus in patients with advanced malignant perivascular epithelioid cell tumors (PEComa): results from a registrational open‐label phase II trial, AMPECT. J Clin Oncol 38(15s), 11516 (2020). [Google Scholar]
- 33.Vinagre J, Almeida A, Populo H, Batista R, Lyra J, Pinto V et al. Frequency of TERT promoter mutations in human cancers. Nat Commun 4, 2185 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Wu RC, Ayhan A, Maeda D, Kim KR, Clarke BA, Shaw P et al. Frequent somatic mutations of the telomerase reverse transcriptase promoter in ovarian clear cell carcinoma but not in other major types of gynaecological malignancy. J Pathol 232, 473–481 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pilsworth JA, Cochrane DR, Xia Z, Aubert G, Farkkila AEM, Horlings HM et al. TERT promoter mutation in adult granulosa cell tumor of the ovary. Mod Pathol 31, 1107–1115 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Alexiadis M, Rowley SM, Chu S, Leung DTH, Stewart CJR, Amarasinghe KC et al. Mutational Landscape of Ovarian Adult Granulosa Cell Tumors from Whole Exome and Targeted TERT Promoter Sequencing. Mol Cancer Res 17, 177–185 (2019). [DOI] [PubMed] [Google Scholar]
- 37.Da Cruz Paula A, da Silva EM, Segura SE, Pareja F, Bi R, Selenica P et al. Genomic profiling of primary and recurrent adult granulosa cell tumors of the ovary. Mod Pathol 10.1038/s41379-020-0514-3 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang HN, Chiang YC, Cheng WF, Chen CA, Lin MC & Kuo KT Molecular alterations in endometrial and ovarian clear cell carcinomas: clinical impacts of telomerase reverse transcriptase promoter mutation. Mod Pathol 28, 303–311 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Doyle LA, Nowak JA, Nathenson MJ, Thornton K, Wagner AJ, Johnson JM et al. Characteristics of mismatch repair deficiency in sarcomas. Mod Pathol 32, 977–987 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357, 409–413 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.