

Abstract
Macadamia, a recently domesticated expanding nut crop in the tropical and subtropical regions of the world, is one of the most economically important genera in the diverse and widely adapted Proteaceae family. All four species of Macadamia are rare in the wild with the most recently discovered, M. jansenii, being endangered. The M. jansenii genome has been used as a model for testing sequencing methods using a wide range of long read sequencing techniques. Here, we report a chromosome level genome assembly, generated using a combination of Pacific Biosciences sequencing and Hi‐C, comprising 14 pseudo‐molecules, with a N50 of 52 Mb and a total genome assembly size of 758 Mb of which 56% is repetitive. Completeness assessment revealed that the assembly covered −97.1% of the conserved single copy genes. Annotation predicted 31,591 protein coding genes and allowed the characterization of genes encoding biosynthesis of cyanogenic glycosides, fatty acid metabolism, and anti‐microbial proteins. Re‐sequencing of seven other genotypes confirmed low diversity and low heterozygosity within this endangered species. Important morphological characteristics of this species such as small tree size and high kernel recovery suggest that M. jansenii is an important source of these commercial traits for breeding. As a member of a small group of families that are sister to the core eudicots, this high‐quality genome also provides a key resource for evolutionary and comparative genomics studies.
Keywords: endangered species, genome assembly, genome diversity, genome sequencing, Proteaceae, wild species
1. INTRODUCTION
Macadamia is a recent domesticate with a complex domestication history (Peace, 2005). The four currently recognized Macadamia species are endemic to the central coast of eastern Australia (Mast et al., 2008). However, macadamia was domesticated in Hawaii around 100 years ago, with most of the global production based upon the Hawaiian domesticated germplasm (Hardner, 2016). Macadamia is a member of the Proteaceae family, one of a group of families that are sister to the core eudicots (Christenhusz & Byng, 2016; Gross & Weston, 1992). Macadamia was the first Australian native plant to be widely grown as a food plant (Peace et al., 2013). All the Hawaiian macadamia cultivars have been reported to be based upon only a few or possibly even a single tree from Australia (Nock et al., 2019). The resulting narrow gene pool makes it susceptible to disease and climate change, whereas the unexploited wild macadamia germplasm of Australia provides an opportunity for great improvement of this newly domesticated crop. Despite a rapid international increase in macadamia production, breeding is restricted because of lack of genomic information (Topp et al., 2019).
Macadamia remains the most widely grown Australian native food crop (Peace et al., 2013). Macadamia production was valued at USD 1.17 billion in 2019, and production is expected to grow at a rate of 9.2% from 2020 to 2027 (https://www.grandviewresearch.com/industry-analysis/macadamia-nut-market). Among the M acadamia species, M. integrifolia, the species from which most of the domesticated gene pool is derived (Hardner, 2016), was the first genome to be sequenced (Nock et al., 2016). This genome, of cultivar HAES 741, has supported initial efforts at genome based breeding (O'Connor et al., 2018) and has recently been upgraded to chromosome level with a contig N50 of 413 Kb (Nock et al., 2020). The other species that has been a contributor to domesticated germplasm, M. tetraphylla, has also been sequenced with contig N50 of 1.18 Mb (Niu et al., 2020).
All species are rare in the wild, but M. jansenii is endangered and is only found in a limited area to the north‐west of Bundaberg, Queensland (Hayward et al., 2021; Shapcott & Powell, 2011). Macadamia jansenii is endangered under the Australian (EPBC) Act and critically endangered under the Queensland (Qld Nature Conservation Act) legislation (Gross & Weston, 1992). Due to the expected low heterozygosity associated with the extremely small population size, this species has been used as a model to compare available genome sequencing technologies (Murigneux et al., 2020; Sharma et al., 2021). Macadamia jansenii has been sequenced (Murigneux et al., 2020), using three long read sequencing technologies, Oxford Nanopore (PromethION), PacBio (Sequel I), and BGI (Single‐tube Long Fragment Read). The genome was recently updated by sequencing using the PacBio HiFi sequencing (Sharma et al., 2021). Here, we report chromosome level assembly of the same genotype using Hi‐C and annotation of the genome. This provides a platform that allows analysis of key genes of importance in macadamia breeding, a reference genome in this group of angiosperms, and insights into the impact of rarity on plant genomes.
This high‐quality reference genome also provides a platform for analysis of three unique attributes of macadamia, the high levels of unusual fatty acids (Hu et al., 2019), high cyanogenic glucoside content (Nock et al., 2016), and the presence of a novel anti‐microbial peptide (Marcus et al., 1999). The fatty acid, palmitoleic acid (16:1), is found in large amounts in macadamia and has been considered to have potential human health benefits (Solà Marsiñach & Cuenca, 2019; Song et al., 2018). Cyanogenic glycosides in plants are part of their defense against herbivores. However, the highly bitter nuts of M. jansenii are not edible, and the use of this species in macadamia breeding will require selection to ensure high levels of cyanogenic glycosides are avoided. Identification of the associated genes could assist by providing molecular tools for use in breeding selection. A novel antimicrobial protein was reported in the kernels of M. integrifolia (Marcus et al., 1999). These small antimicrobial proteins were found to be produced by processing of a larger pre‐cursor protein. As fungal infection and insect herbivores are major hurdles in macadamia production (Dahler et al., 1995; Marcus et al., 1999; Nock et al., 2016), retention of the antimicrobial protein and cyanogenesis in some parts of the plant may be important. Analysis of candidate genes for these traits may assist in understanding and manipulating them in macadamia breeding. The assembly and annotation of the genome presented here will allow characterization of the role of these novel genes and facilitate their use in breeding.
2. RESULTS
2.1. Genome sequencing and assembly
A pseudo‐molecule level genome assembly of PacBio contigs (Murigneux et al., 2020) was produced using Hi‐C. The estimated genome size of M. jansenii has been reported to be 780 Mb (Murigneux et al., 2020), and the size of the final Hi‐C assembly was 758 Mb, comprised of 219 scaffolds with an N50 of 52 Mb (Table 1). Of this, 97% was anchored to the 14 largest scaffolds representing the 14 chromosomes (Figure S1; Table S1). The statistical summary of self‐interacting genomic region analysis or topologically associating domain (TAD) analysis at three different resolutions is given in Tables S2 and S3. Comparison of the PacBio assembly with the Hi‐C chromosome assembly shows the number of scaffolds decreased from 762 to 219 and the length of the longest scaffold increased sixfold (Table 1). The L50 reduced from 135 to 7 scaffolds, and the N50 was improved from 1.58 to 52 Mb.
TABLE 1.
Macadamia jansenii genome sequencing and assembly statistics
| PacBio | Dovetail Chicago | Dovetail Hi‐C assembly | |
|---|---|---|---|
| Library statistics | 3,170,206 reads | 213 M read pairs; 2 × 150 bp | 156 M read pairs; 2× 150 bp |
| Coverage | 84X | 88X | 3,601X |
| Genome assembly | |||
| Total length | 758.28 Mb | 758.30 Mb | 758.43 Mb |
| L50/N50 a | 135 scaffolds; 1.58 Mb | 199 scaffolds; 1.0 Mb | 7 scaffolds; 52.1 Mb |
| L90/N90 a | 457 scaffolds; .51 Mb | 767 scaffolds; .23 Mb | 13 scaffolds; 45.61 Mb |
| Longest scaffold | 10,537,631 bp | 8,434,305 bp | 67,682,215 bp |
| Number of scaffolds | 762 | 1,529 | 219 |
| BUSCO results a | |||
| Complete genes (single+ duplicate) | 96.70% | 97.20% | 97.1% |
| Single genes | 79.10% | 80.10% | 80.80% |
| Duplicated genes | 17.60% | 17.10% | 16.30% |
| Fragmented genes | 0.90% | 1.00% | 1.00% |
| Missing genes | 2.00% | 2.00% | 2.10% |
Eudicots_odb10 dataset, Number of BUSCOs = 2,326.
2.2. Assembly completeness and repeat element analysis
The completeness of the M. jansenii assembly was assessed by Benchmarking Universal Single‐Copy orthologs (BUSCO) analysis (Simão et al., 2015). This analysis revealed 97.1% complete genes (single and duplicated) in the Hi‐C assembly (Table 1). A total of 423.6 Mb, representing 55.9% of the Hi‐C assembly, was identified as repetitive (Table 2). Class I TE (Transposable Elements) repeats were the most abundant repetitive elements representing 30% of the genome, including LTRs (24%), LINE (5.67%), and SINE (0%), and Class II TE repeats were 1.56%.
TABLE 2.
Annotation of repeat sequences in the M. jansenii genome
| Hi‐C assembly | |
|---|---|
| Total repetitive content | 55.9% |
| Class I TEs repeats | 29.9% |
| LTRs | 24% |
| LINE | 5.67% |
| SINE | 0% |
| Class II TEs repeats | 1.56% |
| Low complexity repeats | 0.33% |
| Simple repeats | 1.35% |
2.3. Structural and functional annotation
A total of 31,591 genes were identified in the repeat‐masked Hi‐C M. jansenii genome using a homology‐based and RNA assisted approach. The average length of the genes was 1,368 bp (Table 3). Of a total of 31,591 transcripts, only 22,500 sequences (71%) were annotated by BLAST2GO (Figure S2). Then, the transcripts were functionally annotated using Gene Ontology (GO) terms to assess the potential role of the genes in the M. jansenii genome. The most abundant M. jansenii specific gene families were organic cyclic and heterocyclic compounds among the molecular function; organic and cellular metabolic among the biological process; and protein‐containing binding membrane and intracellular organelle among the cellular component (Figure S3). The annotation edit distance (AED) score plot, which calculates how well the predicted genes agrees with the external evidence, is given in Figure S4, where zero represents the perfect evidence match and one represents no support for the predicted genes. The comparison of the three Macadamia genomes, assembled so far, showed M. jansenii has the highly continuous assembly with highest number of BUSCO genes (Table 4).
TABLE 3.
Genes predicted in the M. jansenii genome
| Gene prediction | |
|---|---|
| Total number of genes | 31,591 |
| Total coding region | 43,235,907 bp |
| Average length of genes | 1,368 bp |
| Number of single‐exon genes | 2,458 |
| Number of genes with annotation | 22,500 |
TABLE 4.
Comparison of genome assemblies of three Macadamia species
| M. integrifolia (V1) | M. integrifolia (V2) | M. tetraphylla | M. jansenii | |
|---|---|---|---|---|
| Assembly length (Mb) | 518.49 | 744.64 | 750.53 | 758.43 |
| N50 (Mb) | 4.7 kb | 413 kb | 1.2 Mb | 52.1 Mb |
| No. of contigs/scaffolds | 193,493 | 4094 | 4,335 | 219 |
| Repeats | 37.00% | 55.00% | 61.42% | 55.90% |
| Complete BUSCO | 77.40% | 90.20% | 89.72% | 97.1% |
| No. of coding genes | 35,337 | 34,274 | 31,571 | 31,591 |
2.4. Anti‐microbial genes
Blast analysis identified homologs of the macadamia antimicrobial protein in the M. jansenii genome (Figure S5A,B). The ANN01396 transcript from M jansenii also showed four repeat segments of cysteine motifs with the same structure as found in MiAMP‐2 (Figure 1a). Comparison of the translated protein sequences indicated a high level of homology with only 28 differences in the 665 amino acid sequence (Figure 1b). The M. jansenii sequence provides the first genomic sequence for this novel anti‐microbial gene and reveals the presence of an intron in the 5′ UTR (Figure S6).
FIGURE 1.
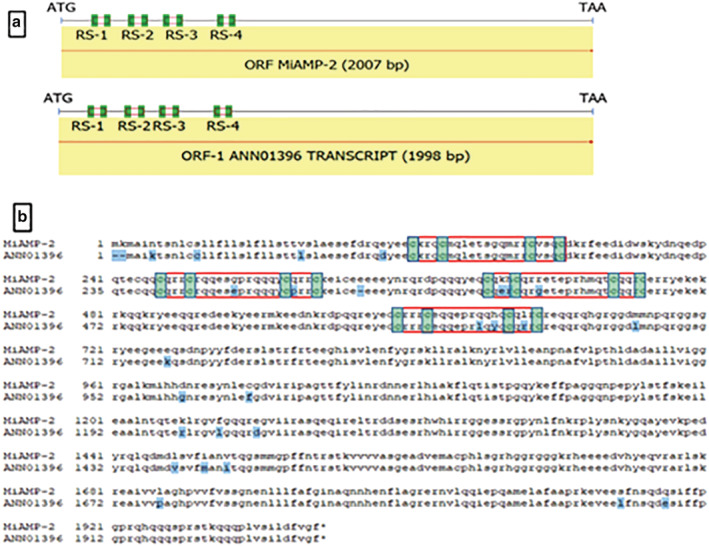
Anti‐microbial peptide structure. (a) The cDNA sequence of anti‐microbial gene of M. integrifolia with four repeat segments (RS), shown in red open boxes and cysteine residues in green filled boxes aligned with M. jansenii transcript sequence ANN01396, showing same pattern. (b) The alignment of the anti‐microbial peptide sequence from the M. integrifolia and M. jansenii. The first half of the sequence shows the repeat segments within red boxes with green highlighted cysteine residues. Differences in amino acid sequence throughout the alignment as shown in blue highlighted text
2.5. Cyanogenic glycoside genes
Analysis of genes of cyanogenic glycoside metabolism detected a total of 11 putative genes with high homology in the M. jansenii genome. These genes were distributed throughout the genome. The largest number of these genes (five) is encoded by CYP79 which is involved in the first step of the cyanogenesis, responsible for conversion of amino acids to oxime. In contrast, only one gene of CYP71 was found which is responsible for conversion of oxime to hydroxynitrile (Table S4).
2.6. Fatty acid metabolism genes
This study identified the key enzymes involved in fatty acid biosynthesis: elongases (e.g., KAS, FATA, and FATB) and desaturases (e.g., SAD). A total of 12 of these genes were found in the M. jansenii genome. Stearoyl‐ACP desaturases (SAD) which convert 18:0 to 18:1 were found to be abundant with five genes present. Ketoacyl‐ACP‐synthase (KAS), which is responsible for elongation of butyryl‐ACP from a 4 to 14 carbon chain, was found to be two in number (Table S5).
2.7. Heterozygosity and genetic diversity
To study the genetic diversity within the species, resequencing was undertaken for eight accessions including the one for which the genome assembly was generated. The trimmed read sequence data yield for the genotypes used in this study ranged from 19.7 to 22.8 Gb corresponding to 25X to 29X of the M. jansenii genome (assembled size 780 Mb).
The eight accessions analyzed had between 5.4 and 7.0 million variants relative to the reference genome (Tables 5 and S6). Most of these were SNPs with less than 600,000 indels in all genotypes. Most SNPs were heterozygous with approximately 1 million or less homozygous SNP variants in each individual (Table S6). The level of SNP heterozygosity for the eight genotypes (including the reference) was found to be in the range of 0.26% to 0.34% with an average of 0.31% (Table 5). The statistical polymorphic sites in up to seven of the eight M. jansenii accessions is given in Table S7. The genotypes varied in their divergence from the reference with most unique variants being heterozygous and only 762 to 44,014 unique homozygous SNPs being found in an individual and not present in the other seven genotypes (Table S8).
TABLE 5.
Heterozygosity and genetic variation in eight M. jansenii accessions
| AccessionID | Polymorphic variants | Heterozygosity analysis | Unique polymorphic SNP sites a | |||||
|---|---|---|---|---|---|---|---|---|
| Total b | Indels | SNP | Heterozygous SNP sites | Heterozygosity c | Heterozygous | Homozygous | Total | |
| 1005 d | 5,393,188 | 486,846 | 4,739,937 | 2,428,956 | 0.31 | 585,053 | 762.00 e | 585,815.00 |
| 1161004 | 5,311,865 | 377,580 | 4,797,864 | 2,038,553 | 0.26 | 187,441 | 8,615.00 | 196,056.00 |
| 1161003 | 6,649,485 | 555,641 | 5,898,975 | 2,465,089 | 0.32 | 521,184 | 44,014.00 | 565,198.00 |
| 1161005 | 6,109,728 | 531,550 | 5,393,088 | 2,347,362 | 0.3 | 608,485 | 40,629.00 | 649,114.00 |
| 1161001a | 6,868,915 | 574,625 | 6,087,318 | 2,649,035 | 0.34 | 95,089 | 8,155.00 | 103,244.00 |
| 1003 | 6,944,903 | 586,001 | 6,148,269 | 2,672,103 | 0.34 | 99,715 | 9,878.00 | 109,593.00 |
| 1002 | 6,642,855 | 586,334 | 5,857,020 | 2,447,418 | 0.31 | 219,933 | 27,408.00 | 247,341.00 |
| 1161001b | 6,594,383 | 548,292 | 5,852,433 | 2,556,695 | 0.33 | 83,662 | 4,843.00 | 88,505.00 |
Variant sites only found in this individual and not in any of the other seven genotypes.
Comprised of replacements, multi nucleotide variants (MNVs), Indels, and SNPs.
Calculated as a percentage of heterozygous SNP sites compared to the total genome size in bases.
Nuclear genome (780 Mb) used as reference for genetic variation and heterozygosity analysis.
Homozygous sites identified possibly due to errors in the reference genome.
2.8. Genome duplication
The MCScanX tool identified 6,303 genes (~20%) of the total genes as WGD/segmental genes. Among all the types of duplicated genes, dispersed genes (15,875 genes) were found to be highest in number, followed by WGD, singleton (3,183 genes) and tandem (2,966 genes). The proximal gene pairs which are separated by only a few genes on the chromosomes were found to be lowest in number, as 2,573 (Table S9). The distribution of the duplicated genes is shown in Figure 2, where pseudo‐molecules 3 and 10 represents a relationship based upon duplicated genes. The whole genome duplication (WGD) tool computed Ks distribution graph with a peak at 0.3 (Figure S7). This shows that M. jansenii genome has undergone a single whole genome duplication event about 20 Million years ago.
FIGURE 2.
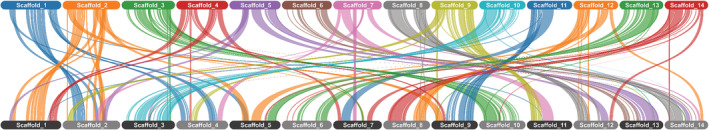
Chromosomal distribution of duplicated genes of M. jansenii, generated using SynVisio. The parallel horizontal lines represent the 14 pseudo‐molecules of M. jansenii genome, with connected ribbons representing the duplicated genes
3. DISCUSSION
A major constraint to the use of M. jansenii for commercial breeding is the risk of an inedible kernel due to high levels of toxic cyanogenic glycosides. Cyanogenic glycosides have been observed in all the four species of Macadamia. However, the concentration varies at different developmental stages (Castada et al., 2020). Even the edible cultivars derived from M. integrifolia have genes involved in the cyanogenic glycoside pathway (Nock et al., 2016). However, cyanogenic glycoside levels are extremely low in the kernel of the commercially important species M. integrifolia and M. tetraphylla (Dahler et al., 1995). The high level of bitterness in the seeds of M. jansenii may be associated with high concentrations of cyanogenic glycosides. Knowledge of these genes will support efforts to avoid their transfer to domesticated Macadamia when using M. jansenii as a source of other desirable genes.
Plants may produce antimicrobial proteins as part of their defense against microbial attack. Macadamia seed might have antimicrobial proteins that protect them against attack when germinating in the warm and moist rainforest environment. A new family of antimicrobial peptides, MiAMP‐2, was discovered in the seeds of M. integrifolia (Marcus et al., 1999). In addition to antimicrobial properties, these seed storage proteins are homologous to vicilin 7S globulins and have been identified as putative allergens (Rost et al., 2016, 2020). A cDNA sequence, from M. integrifolia, encoding these proteins, MiAMP‐2, has been reported to contain four repeat segments, with each segment comprised of cysteine rich motifs (C‐X‐X‐X‐C‐[10 to12] X‐C‐X‐X‐X‐C), where X is any other amino acid residue. Although only a single gene was found in the M. jansenii genome, it encoded a protein with four domains that correspond to the previously reported antimicrobial peptides, suggesting that four copies of the peptide could be derived from each translation of this gene. This is the first report of a gene structure for the macadamia anti‐microbial peptide with a single intron. This gene has potential for wide use as an antimicrobial protein in plant defense.
Macadamia oil has a unique composition being 75% fat, 80% of which is monounsaturated, for exampe, oleic oil (C18:1) 55–67%, followed by palmitoleic acid (C16:1) 15–22% (Aquino‐Bolaños et al., 2016; Curb et al., 2000; Hu et al., 2019). The results of analysis of the genes of lipid metabolism in the M. jansenii genome are consistent with this fatty acid profile. The number of SAD genes which are responsible for conversion of stearoyl‐ACP (18:0) to oleate (18:1) was found to be higher in number than the other genes in these pathways and may explain the desirable high oleic content of macadamias. Retention of these genes will be important in breeding. This species may provide a source of genes for manipulation of lipids in other food crops.
This rare species has a very small population size explaining the low heterozygosity (Ceballos et al., 2018). The heterozygosity was less than one third that of the more widespread, M. integrifolia, reported to have a heterozygosity of 0.98% (Nock et al., 2020; Topp et al., 2019). This analysis indicates the importance of conserving the diversity of this endangered species and retaining the unique alleles that may be useful in breeding. M. jansenii is a small tree with a high kernel recovery, and both of these traits are key for macadamia improvement. Sustainable intensification of production will be facilitated by the breeding of smaller trees, and improved kernel recovery is central to kernel yield. Genome level analysis will support field studies for the conservation of this species (Shapcott & Powell, 2011) and molecular analysis of diversity in support of breeding (Mai et al., 2020).
The use of M. jansenii as a model in testing genome sequencing and assembly methods (Murigneux et al., 2020; Sharma et al., 2021) is further enhanced by the chromosome level assembly presented here. This is currently the most complete genome sequence available for a macadamia and any member of the more than 1,660 Proteaceae species (Christenhusz & Byng, 2016) making a useful contribution to the goal of sequencing plant biodiversity (Lewin et al., 2018). The Proteaceae belongs to the basal eudicot order Proteales, a sister group to core eudicots (Chanderbali et al., 2016; Drinnan et al., 1994). Among the basal eudicots there are few well characterized genomes. Available genomes include Aquilegia coerulea (Ranuncules) (Filiault et al., 2018), Papaver somniferum (Ranuncules) (Pei et al., 2021), Nelumbo nucifera (Proteales) (Ming et al., 2013), Trochodendron aralioides (Trochodendrales) (Strijk et al., 2019), and Tetracentron sinense (Trochodendrales) (Liu et al., 2020). The M. jansenii genome provides a valuable contribution to comparative genomics in this group of flowering plants. The chromosome level assembly with an N50 scaffold length of 52 Mb and 97.1% of complete BUSCO genes compares favorably with those available for other endangered species, for example, Acer yangbiense with N50 45 Mb and 90.5% of complete BUSCO genes (Giordano et al., 2017), Ostrya rehderiana N50 2.31 Mb (Yang et al., 2018), and Nyssa yunnanensis with N50 of 985 Kb and BUSCO score of 90.5% (Weixue et al., 2020).
4. EXPERIMENTAL PROCEDURES
4.1. Plant material
Fresh leaf tissue of M. jansenii was collected from ex situ collections of trees at Nambour and Tiaro (three accessions were from the Maroochy Research Facility, Department of Agriculture and Fisheries, Nambour, Queensland, Australia, Accessions 1005, 1003, and 1002, and five from Tiaro, Queensland, Australia, Accession 1161003, 1161005, 1161001a, 1161001b, and 1161004). Fresh leaf tissue (fully expanded young flush) was collected and immediately frozen by placing under dry ice and stored at −80°C until further processed for DNA and RNA extraction.
4.2. DNA and RNA isolation
Leaf tissue was coarsely ground under liquid nitrogen using a mortar and pestle and further ground under cryogenic conditions into a fine powder using a Tissue Lyser (MM400, Retsch, Germany). All accessions were used for DNA isolation. DNA was extracted as per an established method (Furtado, 2014) with minor modification where phenol was excluded from the extraction method. DNA was extracted from 2–3 gm of leaf tissue and dissolved in up to 400 μl of TE buffer.
Accession 10051 was used for RNA isolation. RNA was extracted as per established methods (Furtado, 2014; Rubio‐Piña & Zapata‐Pérez, 2011). RNA was extracted from 2–3 gm of tissue and treated with extraction buffer, chloroform, and phenol/chloroform (1:1) in different steps, followed by further purification using DNase treatment from the Qiagen's RNeasy Mini kit. RNA quality and quantity were determined using A260/280 and A260/230 absorbance ratio (Nanodrop, Invitrogen USA) and RNA integrity measurements (Bioanalyser, Agilent technology, USA).
4.3. Chromosome level assembly
4.3.1. Chicago library sequencing and sequencing
DNA was isolated as per an established method (Furtado, 2014). Then, the library was prepared as described in Putnam et al. (2016). Briefly, ~500 ng of HMW gDNA was reconstituted into chromatin in vitro and fixed with formaldehyde. Fixed chromatin was digested with DpnII, the 5′ overhangs filled in with biotinylated nucleotides, and then, free blunt ends were ligated. After ligation, crosslinks were reversed, and the DNA was purified from protein. Purified DNA was treated to remove biotin that was not internal to ligated fragments. The DNA was then sheared to ~350 bp mean fragment size, and sequencing libraries were generated using NEBNext Ultra enzymes and Illumina‐compatible adapters. Biotin‐containing fragments were isolated using streptavidin beads before PCR enrichment of each library. The libraries were sequenced on an Illumina HiSeqX platform to produce 213 million 2 × 150 bp paired end reads, which provided 88.11 × physical coverage of the genome (1–100 kb pairs).
4.3.2. Dovetail Hi‐C library preparation and sequencing
A Dovetail Hi‐C library was prepared in a similar manner as described previously (Lieberman‐Aiden et al., 2009). Briefly, for each library, chromatin was fixed in place with formaldehyde in the nucleus and then extracted. Fixed chromatin was digested with DpnII, the 5′ overhangs filled in with biotinylated nucleotides, and then free blunt ends were ligated. After ligation, crosslinks were reversed, and the DNA purified from protein. Purified DNA was treated to remove biotin that was not internal to ligated fragments. The DNA was then sheared to ~350 bp mean fragment size, and sequencing libraries were generated using NEBNext Ultra enzymes and Illumina‐compatible adapters. Biotin‐containing fragments were isolated using streptavidin beads before PCR enrichment of each library. The libraries were sequenced on an Illumina HiSeqX platform to produce 156 million 2 × 150 bp paired end reads, which provided 3,601.74 × physical coverage of the genome (10–10,000 kb pairs).
4.3.3. Scaffolding the assembly with HiRise
The input de novo assembly, shotgun reads, Chicago library reads, and Dovetail Hi‐C library reads were used as input data for HiRise, a software pipeline designed specifically for using proximity ligation data to scaffold genome assemblies (Putnam et al., 2016). An iterative analysis was conducted. First, Shotgun and Chicago library sequences were aligned to the draft input assembly using a modified SNAP read mapper (http://snap.cs.berkeley.edu). The separations of Chicago read pairs mapped within draft scaffolds were analyzed by HiRise to produce a likelihood model for genomic distance between read pairs, and the model was used to identify and break putative misjoins, to score prospective joins, and to make joins above a threshold. After aligning and scaffolding Chicago data, Dovetail Hi‐C library sequences were aligned and scaffolded following the same method. After scaffolding, shotgun sequences were used to close gaps between contigs.
4.4. Re‐sequencing
To study the genetic diversity within the species, re‐sequencing of the eight different genotypes was performed on the DNBseq platform (Drmanac et al., 2010). The seven Macadamia jansenii samples were selected randomly to represent diversity in the population. A DNBseq library was prepared as follows. Briefly, genomic DNA (1 μg) was randomly fragmented using a Covaris; magnetic beads were used to select fragments with an average size of 300–400 bp, and DNA was quantified using a Qubit fluorometer. The fragments were subjected to end‐repair and 3′ adenylated; adaptors were ligated to the ends of these 3′ adenylated fragments. Then, the double stranded products were heat denatured and circularized by the splint oligo sequence; the single strand circle DNA (ssCir DNA) was formatted as the final library. The final library was then amplified to make DNA nanoball (DNB) which had more than 300 copies of each molecule, and the DNBs were loaded into the patterned nanoarray. Finally, pair‐end 150 bases reads were generated by combinatorial Probe‐Anchor Synthesis (cPAS) (MGISEQ‐2000).
4.5. RNA‐sequencing
RNA sequencing was undertaken by Macrogen, South Korea. Total RNA was subjected to ribosomal RNA depletion (Ribo zero plant) and then sequenced on Illumina platform using TruSeq stranded total RNA LT sample prep kit (plant) to obtain 154 M paired end reads of 23.3 Gb read length.
4.6. Genome assembly quality evaluation and repetitive element evaluation
The completeness of the genome assembly was evaluated by checking the integrity of the protein coding genes in the Hi‐C assembly using Benchmarking Universal Single‐Copy Orthologs (BUSCO) (version v5.0.0) analysis with eudicot odb10 dataset with 2,326 genes.
Repetitive elements in the Hi‐C assembly were identified de novo and classified using RepeatModeler (version 2.0.1). The repeat library obtained from RepeatModeler was used to identify and mask the repeats in the Hi‐C assembly file using RepeatMasker (Version 4.1.0).
4.7. Structural annotation and functional annotation
The prediction of the protein coding genes in the repeat masked genome was carried out using ab initio and evidence‐based approach. For ab initio prediction, Dovetail staff used Augustus (version 2.5.5) (Stanke et al., 2006) and SNAP (version 2006‐07‐28) (Johnson et al., 2008). For evidence‐based approach, MAKER (Cantarel et al., 2008) was used. For training the ab initio model for M. jansenii, coding sequences from Malus domestica, Prunus persica, and Arabidopsis thaliana were used using AUGUSTUS and SNAP. Six rounds of prediction optimization were done with the package provided by AUGUSTUS. To generate the peptide evidence in Maker pipeline, Swiss‐Prot peptide sequences from the UniProt database were downloaded and used in combination with the protein sequences from M. domestica, P. persica, and A. thaliana. To assess the quality of the gene prediction, AED scores were generated for each of the predicted genes as part of MAKER pipeline. Only those genes which were predicted by both SNAP and AUGUSTUS were retained in the final gene set. To generate the intron hints, a bam file was generated by aligning the RNAseq reads to the genome using the STAR aligner software (version 2.7), and then, bam2hints tool was used within the AUGUSTUS. The predicted genes were further characterized for their putative function by performing a BLASTx search against nr protein database (all non‐redundant GenBank CDS translations + PDB + SwissProt + PIR + PRF), as part of annotations undertaken by Dovetail and also by using OmicsBox Ver 1.3.11 (BioBam Bioinformatics, Spain).
4.8. Gene families
To identify the anti‐microbial genes in the genome, BLAST homology search was performed to identity transcripts similar to the M. integrifolia antimicrobial cDNA (MiAMP2, GenBank: AF161884.1) (Marcus et al., 1999). Then, sequence alignment was undertaken using Clone Manager ver 9.0 (SciEd, USA). Multiple Alignment was undertaken using a reference sequence as indicated in the results and alignment parameter scoring matrix of Mismatch (2), Open Gap (4), and Extension‐Gap (1). Genes involved in the metabolism of cyanogenic glycosides and fatty acids were identified in M. jansenii genome using BLASTp (1E‐10) and percentage identity >60%, and then, top hits were identified from the list based upon sequence similarity. For cyanogenic genes, the reference sequence was taken from the M. integrifolia genome (Nock et al., 2016), whereas for fatty acids reference sequences were used from A. thaliana.
4.9. Heterozygosity and genetic diversity analysis
All analysis including basic variant analysis (BVA) to determine polymorphic positions for heterozygosity analysis was performed using Qiagen CLC Genomics Workbench 21.0.4 (CLC bio, Aarhus, Denmark). BGI short read sequences of six genotypes (1003, 1002, 1161003, 1161005, 1161001a, and 1161001b) and Illumina reads of two genotypes (1005 and 1161004) of M. jansenii were first trimmed at quality limit of 0.01 (Phred score of 20 and above). The trimmed reads were then mapped to the M. jansenii Dovetail Hi‐C derived reference genome (Accession 1005). Mapping of short reads was conducted at settings as follows; Masking Mode (No), Match score (1), Mismatch score (2), Insertion cost (3), deletion cost (3), Length fraction (1), Similarity fraction (0.95), Global alignment (No), and Non‐specific match handling (Map randomly). To identify variants, the mapping file was subjected to the “Basic Variant Analysis” tool at the following settings: minimum coverage (10), minimum count (3), and minimum frequency (25%). Homozygous SNP variants were those filtered at 100% variant frequency. Heterozygous SNP variants were those filtered for a frequency range of between 25% to 75% for the alternate allele. The number of homozygous SNP variants also represented the number of homozygous sites. The number of heterozygous SNP sites were identified when the variant table was sorted for “Reference allele” as “Yes.” The heterozygosity for an accession was determined by representing the accession‐specific heterozygous SNPs sites as a percentage of the M. jansenii genome size (780 Mb).
Accession‐specific polymorphic SNP sites present in up to seven of the eight accessions were determined as outlined below. Accession‐specific SNP sites (homozygous and heterozygous), identified by filtering “sample Count” for “≤7,” were represented as a percentage of the M. jansenii genome size (780 Mb). Accession‐specific unique polymorphic SNP sites, defined as those variant sites present only once in any of the eight accessions, were identified by filtering the “sample Count” for “≤1.”
4.10. Genome duplication
MCScanX (Wang et al., 2012) was used to identify whole‐genome duplication (WGD)/segmental along with tandem, dispersed, singleton, and proximal duplication on the M. jansenii genome. An all‐versus‐all BLASTP was performed (E value: 1e−10, max target sequences: 5 and m6 format output), for the M. jansenii whole‐genome protein sequences. The duplicate gene classifier and MCScanX program was executed using the M. jansenii genome annotation (gff file) along with the BLASTP output file using the default parameters. The collinearity file (MCScanX output file) and the genome annotation file were used to generate the synteny plot using SynVisio toolkit (Bandi & Gutwin, 2020). The Ks distribution graph was generated using WGD tools (Zwaenepoel and van de Peer, 2018). Timing of WGD was estimated as described by Magallón et al. (2015).
AUTHOR CONTRIBUTIONS
Contributions of authors were as follows: designed the study and supervised the project: RJH, AF, BT, and MA; collected sample: MA, BT, AF, and PS; management of germplasm: MA and BT; DNA and RNA isolation: PS and AF; data analysis and prepared the figures: PS and AF; bioinformatics analysis: PS, AF, VM, JH, and AM; drafted the manuscript: PS, AF, JH, and WT; and data deposition: PS. All authors edited and approved the final manuscript.
CONFLICT OF INTEREST
The Authors did not report any conflict of interest.
Supporting information
Table S1: Size of each scaffold and number of genes per scaffold
Table S2: Topologically Associated Domains (TADs) analysis summary
Table S3: TAD statistics at different resolutions
Table S4: Location of cyanogenic genes on pseudo‐molecules
Table S5: Location of fatty acid genes on pseudo‐molecules
Table S6: SNP heterozygosity statistics in eight Macadamia jansenii accessions
Table S7: Polymorphic sites in up to seven of the eight Macadamia jansenii accessions
Table S8: Genotype‐specific unique polymorphic SNP sites
Table S9: List of duplicated genes in M. jansenii genome, using MCScanX tool
Figure S1: Linkage density histogram of Hi‐C assembly of M. jansenii genome
Figure S2: BLAST2GO sequence similarity search
Figure S3: Gene ontology (GO) analysis by BLAST2GO
Figure S4: Frequency graph of AED scores
Figure S5: Alignment of the vicilin‐like antimicrobial‐peptide transcript from M. integrifolia and M. jansenii
Figure S6: Alignment of anti‐microbial CDS sequence of M. integrifolia against the M. jansenii transcript sequence
Figure S7: Ks plot of M. jansenii was generated by Wgd tool.
Table S4: Location of cyanogenic genes on pseudo‐molecule (based on blastp results)
Table S5: Location of fatty acid genes on pseudo‐molecules (based on blastp results)
ACKNOWLEDGMENTS
This project was funded by the Hort Frontiers Advanced Production Systems Fund as part of the Hort Frontiers strategic partnership initiative developed by Hort Innovation, with co‐investment from the University of Queensland and contributions from the Australian Government. We thank the Research Computing Centre (RCC), University of Queensland, for support and providing high performance computing resources.
Sharma, P. , Murigneux, V. , Haimovitz, J. , Nock, C. J. , Tian, W. , Kharabian Masouleh, A. , Topp, B. , Alam, M. , Furtado, A. , & Henry, R. J. (2021). The genome of the endangered Macadamia jansenii displays little diversity but represents an important genetic resource for plant breeding. Plant Direct, 5(12), e364. 10.1002/pld3.364
DATA AVAILABILITY STATEMENT
The genome sequence reads, transcriptome sequences, and genome assembly of M. jansenii have been deposited under NCBI bioproject PRJNA694456.
REFERENCES
- Aquino‐Bolaños, E. N. , Mapel‐Velazco, L. , Martín‐del‐Campo, S. T. , Chávez‐Servia, J. L. , Martínez, A. J. , & Verdalet‐Guzmán, I. (2016). Fatty acids profile of oil from nine varieties of Macadamia nut. International Journal of Food Properties, 20(6), 1262–1269. [Google Scholar]
- Bandi, V. , & Gutwin, C. (2020). Interactive exploration of genomic conservation. In Proceedings of the 46th Graphics Interface Conference on Proceedings of Graphics Interface 2020 (GI'20). Canadian Human‐Computer Communications Society, Waterloo, CAN.
- Cantarel, B. L. , Korf, I. , Robb, S. M. , Parra, G. , Ross, E. , Moore, B. , Holt, C. , Sanchez Alvarado, A. , & Yandell, M. (2008). MAKER: An easy‐to‐use annotation pipeline designed for emerging model organism genomes. Genome Research, 18(1), 188–196. 10.1101/gr.6743907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castada, H. Z. , Liu, J. , Ann Barringer, S. , & Huang, X. (2020). Cyanogenesis in Macadamia and direct analysis of hydrogen cyanide in Macadamia flowers, leaves, husks, and nuts using selected ion flow tube‐mass spectrometry. Food, 2020(9), 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceballos, F. C. , Joshi, P. K. , Clark, D. W. , Ramsay, M. , & Wilson, J. F. (2018). Runs of homozygosity: Windows into population history and trait architecture. Nature Reviews Genetics, 19(4), 220–234. 10.1038/nrg.2017.109 [DOI] [PubMed] [Google Scholar]
- Chanderbali, A. S. , Berger, B. A. , Howarth, D. G. , Soltis, P. S. , & Soltis, D. E. (2016). Evolving ideas on the origin and evolution of flowers: New perspectives in the genomic era. Genetics, 202(4), 1255–1265. 10.1534/genetics.115.182964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christenhusz, M. J. M. , & Byng, J. W. (2016). The number of known plants species in the world and its annual increase. Phytotaxa, 261(3), 201–217. 10.11646/phytotaxa.261.3.1 [DOI] [Google Scholar]
- Curb, J. D. , Wergowske, G. , Dobbs, J. C. , Abbott, R. D. , & Huang, B. (2000). Serum lipid effects of a high–monounsaturated fat diet based on Macadamia nuts. Archives of Internal Medicine, 160(8), 1154–1158. 10.1001/archinte.160.8.1154 [DOI] [PubMed] [Google Scholar]
- Dahler, J. M. , McConchie, C. , & Turnbull, C. G. N. (1995). Quantification of cyanogenic glycosides in seedlings of three Macadamia (Proteaceae) species. Australian Journal of Botany, 43(6), 619–628. 10.1071/BT9950619 [DOI] [Google Scholar]
- Drinnan, A. N. , Crane, P. R. , & Hoot, S. B. (1994). Patterns of floral evolution in the early diversification of non‐magnoliid dicotyledons (eudicots). Paper presented at the Early Evolution of Flowers, Vienna.
- Drmanac, R. , Sparks, A. B. , Callow, M. J. , Halpern, A. L. , Burns, N. L. , Kermani, B. G. , Carnevali, P. , Nazarenko, I. , Nilsen, G. B. , Yeung, G. , Dahl, F. , Fernandez, A. , Staker, B. , Pant, K. P. , Baccash, J. , Borcherding, A. P. , Brownley, A. , Cedeno, R. , Chen, L. , … Reid, C. A. (2010). Human genome sequencing using unchained base reads on self‐assembling DNA nanoarrays. Science, 327(5961), 78–81. 10.1126/science.1181498 [DOI] [PubMed] [Google Scholar]
- Filiault, D. L. , Ballerini, E. S. , Mandáková, T. , Aköz, G. , Derieg, N. J. , Schmutz, J. , Jenkins, J. , Grimwood, J. , Shu, S. , Hayes, R. D. , Hellsten, U. , Barry, K. , Yan, J. , Mihaltcheva, S. , Karafiátová, M. , Nizhynska, V. , Kramer, E. M. , Lysak, M. A. , Hodges, S. A. , & Nordborg, M. (2018). The Aquilegia genome provides insight into adaptive radiation and reveals an extraordinarily polymorphic chromosome with a unique history. eLife, 7, e36426. 10.7554/eLife.36426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furtado, A. (2014). DNA extraction from vegetative tissue for next‐generation sequencing. In Cereal genomics (pp. 1–5). New York: Humana Press. [DOI] [PubMed] [Google Scholar]
- Giordano, F. , Aigrain, L. , Quail, M. A. , Coupland, P. , Bonfield, J. K. , Davies, R. M. , Tischler, G. , Jackson, D. K. , Keane, T. M. , Li, J. J. S. , Yue, J.‐X. , Liti, G. , Durbin, R. , & Ning, Z. (2017). De novo yeast genome assemblies from MinION, PacBio and MiSeq platforms. Scientific Reports, 7(1), 1–10. 10.1038/s41598-017-03996-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross, C. , & Weston, P. H. (1992). Macadamia jansenii (Proteaceae), a new species from central Queensland. Australian Systematic Botany, 5(6), 725–728. 10.1071/SB9920725 [DOI] [Google Scholar]
- Hardner, C. (2016). Macadamia domestication in Hawai'i. Genetic Resources and Crop Evolution, 63(8), 1411–1430. 10.1007/s10722-015-0328-1 [DOI] [Google Scholar]
- Hayward, G. , Nock, C. , Shimizu, Y. , & Shapcott, A. (2021). A comprehensive approach to assessing the future persistence of the endangered rainforest tree, Macadamia jansenii (Proteaceae) and the impact of fire. Australian Journal of Botany, 69, 285–300. 10.1071/BT20160 [DOI] [Google Scholar]
- Hu, W. , Fitzgerald, M. , Topp, B. , Alam, M. , & O'Hare, T. J. (2019). A review of biological functions, health benefits, and possible de novo biosynthetic pathway of palmitoleic acid in macadamia nuts. Journal of Functional Foods, 62, 103520. 10.1016/j.jff.2019.103520 [DOI] [Google Scholar]
- Johnson, A. D. , Handsaker, R. E. , Pulit, S. L. , Nizzari, M. M. , O'Donnell, C. J. , & de Bakker, P. I. (2008). SNAP: A web‐based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics, 24(24), 2938–2939. 10.1093/bioinformatics/btn564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewin, H. A. , Robinson, G. E. , Kress, W. J. , Baker, W. J. , Coddington, J. , Crandall, K. A. , Durbin, R. , Edwards, S. V. , Forest, F. , Gilbert, M. T. P. , Goldstein, M. M. , Grigoriev, I. V. , Hackett, K. J. , Haussler, D. , Jarvis, E. D. , Johnson, W. E. , Patrinos, A. , Richards, S. , Castilla‐Rubio, J. C. , … Zhang, G. (2018). Earth BioGenome Project: Sequencing life for the future of life. Proceedings of the National Academy of Sciences, 115(17), 4325–4333. 10.1038/nrg.2017.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman‐Aiden, E. , van Berkum, N. L. , Williams, L. , Imakaev, M. , Ragoczy, T. , Telling, A. , Amit, I. , Lajoie, B. R. , Sabo, P. J. , Dorschner, M. O. J. , Sandstrom, R. , Bernstein, B. , Bender, M. A. , Groudine, M. , Gnirke, A. , Stamatoyannopoulos, J. , Mirny, L. A. , Lander, E. S. , & Dekker, J. (2009). Comprehensive mapping of long‐range interactions reveals folding principles of the human genome. Science, 326(5950), 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, P.‐L. , Zhang, X. , Mao, J.‐F. , Hong, Y.‐M. , Zhang, R.‐G. , Yilan, E. , Nie, S. , Jia, K. , Jiang, C.‐K. , He, J. , Shen, W. , He, Q. , Zheng, W. , Abbas, S. , Jewaria, P. K. , Tian, X. , Liu, C.‐J. , Jiang, X. , Yin, Y. , … Lin, J. (2020). The Tetracentron genome provides insight into the early evolution of eudicots and the formation of vessel elements. Genome Biology, 21(1), 291. 10.1186/s13059-020-02198-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai, T. , Alam, M. , Hardner, C. , Henry, R. , & Topp, B. J. P. (2020). Genetic structure of wild germplasm of Macadamia: Species assignment. Diversity and Phylogeographic Relationships., 9(6), 714. 10.3390/plants9060714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magallón, S. , Gómez‐Acevedo, S. , Sánchez‐Reyes, L. L. , & Hernández‐Hernández, T. J. N. P. (2015). A metacalibrated time‐tree documents the early rise of flowering plant phylogenetic diversity. New Phytologist, 207(2), 437–453. 10.1111/nph.13264 [DOI] [PubMed] [Google Scholar]
- Marcus, J. P. , Green, J. L. , Goulter, K. C. , & Manners, J. M. (1999). A family of antimicrobial peptides is produced by processing of a 7S globulin protein in Macadamia integrifolia kernels. The Plant Journal, 19(6), 699–710. [DOI] [PubMed] [Google Scholar]
- Mast, A. R. , Willis, C. L. , Jones, E. H. , Downs, K. M. , & Weston, P. H. (2008). A smaller Macadamia from a more vagile tribe: Inference of phylogenetic relationships, divergence times, and diaspore evolution in Macadamia and relatives (tribe Macadamieae; Proteaceae). American Journal of Botany, 95(7), 843–870. 10.3732/ajb.0700006 [DOI] [PubMed] [Google Scholar]
- Ming, R. , van Buren, R. , Liu, Y. , Yang, M. , Han, Y. , Li, L.‐T. , Zhang, Q. , Kim, M.‐J. , Schatz, M. C. , Campbell, M. , Li, J. , Bowers, J. E. , Tang, H. , Lyons, E. , Ferguson, A. A. , Narzisi, G. , Nelson, D. R. , Blaby‐Haas, C. E. , Gschwend, A. R. , … Shen‐Miller, J. (2013). Genome of the long‐living sacred lotus (Nelumbo nucifera Gaertn.). Genome Biology, 14(5), R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murigneux, V. , Rai, S. K. , Furtado, A. , Bruxner, T. J. C. , Tian, W. , Ye, Q. , Wei, H. , Yang, B. , Harliwong, I. , Anderson, E. , Mao, Q. , Drmanac, R. , Wang, O. , Peters, B. A. , Xu, M. , Wu, P. , Topp, B. , Coin, L. J. M. , & Henry, R. J. (2020). Comparison of long read methods for sequencing and assembly of a plant genome. GigaScience, 9(12), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu, Y.‐F. , Li, G.‐H. , Ni, S.‐B. , He, X.‐Y. , Zheng, C. , Liu, Z.‐Y. , Gong, L.‐D. , Kong, G.‐H. , & Liu, J. (2020). Genome assembly and annotation of Macadamia tetraphylla . bioRxiv, 1–17. 10.1101/2020.03.11.987057 [DOI] [Google Scholar]
- Nock, C. J. , Baten, A. , Barkla, B. J. , Furtado, A. , Henry, R. J. , & King, G. J. (2016). Genome and transcriptome sequencing characterises the gene space of Macadamia integrifolia (Proteaceae). BMC Genomics, 17(1), 937. 10.1186/s12864-016-3272-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nock, C. J. , Baten, A. , Mauleon, R. , Langdon, K. S. , Topp, B. , Hardner, C. , Furtado, A. , Henry, R. J. , & King, G. J. (2020). Chromosome‐scale assembly and annotation of the Macadamia genome G3. Genes|Genomes|Genetics, 10(10), 3497–3504. 10.1534/g3.120.401326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nock, C. J. , Hardner, C. M. , Montenegro, J. D. , Ahmad Termizi, A. A. , Hayashi, S. , Playford, J. , Edwards, D. , & Batley, J. (2019). Wild origins of Macadamia domestication identified through intraspecific chloroplast genome sequencing. Frontiers in Plant Science, 10(334), 1–15. 10.3389/fpls.2019.00334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor, K. , Hayes, B. , & Topp, B. (2018). Prospects for increasing yield in macadamia using component traits and genomics. Tree Genetics & Genomes, 14(1), 7. 10.1007/s11295-017-1221-1 [DOI] [Google Scholar]
- Peace, C. P. (2005). Genetic characterisation of Macadamia with DNA markers. PhD thesis, The University of Queensland, Brisbane.
- Peace, C. P. , Allan, P. , Vithanage, V. , Turnbull, C. N. , & Carroll, B. J. (2013). Genetic relationships amongst macadamia varieties grown in South Africa as assessed by RAF markers. South African Journal of Plant and Soil, 22(2), 71–75. [Google Scholar]
- Pei, L. , Wang, B. , Ye, J. , Hu, X. , Fu, L. , Li, K. , Ni, Z. , Wang, Z. , Wei, Y. , Shi, L. , Zhang, Y. , Bai, X. , Jiang, M. , Wang, S. , Ma, C. , Li, S. , Liu, K. , Li, W. , & Cong, B. (2021). Genome and transcriptome of Papaver somniferum Chinese landrace CHM indicates that massive genome expansion contributes to high benzylisoquinoline alkaloid biosynthesis. Horticulture Research, 8(1), 5. 10.1038/s41438-020-00435-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putnam, N. H. , O'Connell, B. L. , Stites, J. C. , Rice, B. J. , Blanchette, M. , Calef, R. , Troll, C. J. , Fields, A. , Hartley, P. D. , Sugnet, C. W. , Haussler, D. , Rokhsar, D. S. , & Green, R. E. (2016). Chromosome‐scale shotgun assembly using an in vitro method for long‐range linkage. Genome Research, 26(3), 342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rost, J. , Muralidharan, S. , Campbell, D. , Mehr, S. , Nock, C. , & Lee, N. A. (2016). ASCIA‐P19: Discovery of 7s and 11s globulins as putative allergens in macadamia nut by combining allergenomics and patient serum ige binding. Internal Medicine Journal, 46(S4), 10–10. 10.1111/imj.19_13197 [DOI] [Google Scholar]
- Rost, J. , Muralidharan, S. , & Lee, N. A. (2020). A label‐free shotgun proteomics analysis of macadamia nut. Food Research International, 129, 108838. 10.1016/j.foodres.2019.108838 [DOI] [PubMed] [Google Scholar]
- Rubio‐Piña, J. A. , & Zapata‐Pérez, O. (2011). Isolation of total rna from tissues rich in polyphenols and polysaccharides of mangrove plants. Electronic Journal of Biotechnology, 14(5), 11–11. [Google Scholar]
- Shapcott, A. , & Powell, M. (2011). Demographic structure, genetic diversity and habitat distribution of the endangered, Australian rainforest tree Macadamia jansenii help facilitate an introduction program. Australian Journal of Botany, 59(3), 215–225. 10.1071/BT10132 [DOI] [Google Scholar]
- Sharma, P. , Aldossary, O. , Alsubaie, B. , Al‐Mssallem, I. , Nath, O. , Mitter, N. , Alves Margarido, G. R. , Topp, B. , Murigneux, V. , Masouleh, A. K. , Furtado, A. , & Henry, R. J. (2021). Improvements in the sequencing and assembly of plant genomes. Gigabyte, 1, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simão, F. A. , Waterhouse, R. M. , Ioannidis, P. , Kriventseva, E. V. , & Zdobnov, E. M. (2015). BUSCO: Assessing genome assembly and annotation completeness with single‐copy orthologs. Bioinformatics, 31(19), 3210–3212. 10.1093/bioinformatics/btv351 [DOI] [PubMed] [Google Scholar]
- Solà Marsiñach, M. , & Cuenca, A. P. (2019). The impact of sea buckthorn oil fatty acids on human health. Lipids in Health and Disease, 18(1), 145. 10.1186/s12944-019-1065-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, I.‐B. , Gu, H. , Han, H.‐J. , Lee, N.‐Y. , Cha, J.‐Y. , Son, Y.‐K. , & Kwon, J. (2018). Omega‐7 inhibits inflammation and promotes collagen synthesis through SIRT1 activation. Applied Biological Chemistry, 61(4), 433–439. 10.1007/s13765-018-0377-1 [DOI] [Google Scholar]
- Stanke, M. , Keller, O. , Gunduz, I. , Hayes, A. , Waack, S. , & Morgenstern, B. (2006). AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Research, 34(suppl_2), W435–W439. 10.1093/nar/gkl200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strijk, J. S. , Hinsinger, D. D. , Zhang, F. , & Cao, K. (2019). Trochodendron aralioides, the first chromosome‐level draft genome in Trochodendrales and a valuable resource for basal eudicot research. GigaScience, 8(11), 1–9. 10.1093/gigascience/giz136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topp, B. L. , Nock, C. J. , Hardner, C. M. , Alam, M. , & O'Connor, K. M. (2019). Macadamia (Macadamia spp.) Breeding. In Advances in plant breeding strategies: Nut and beverage crops (Vol. 4, pp. 221–251). Springer International Publishing. 10.1007/978-3-030-23112-5_7 [DOI] [Google Scholar]
- Wang, Y. , Tang, H. , Debarry, J. D. , Tan, X. , Li, J. , Wang, X. , Lee, T.‐H. , Jin, H. , Marler, B. , Guo, H. , Kissinger, J. C. , & Paterson, A. H. (2012). MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Research, 40(7), e49–e49. 10.1093/nar/gkr1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weixue, M. , Jinpu, W. , Ting, Y. , Yannan, F. , Le, C. , Jinlong, Y. , Ranchang, M. , Jie, L. , Jianming, Z. , Weibang, S. , Xun, X. , Xin, L. , Radoje, D. , & Huan, L. (2020). The draft genome assembly of the critically endangered Nyssa yunnanensis, a plant species with extremely small populations endemic to Yunnan Province, China. Gigabyte, 1–11. 10.46471/gigabyte.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y. , Ma, T. , Wang, Z. , Lu, Z. , Li, Y. , Fu, C. , Chen, X. , Zhao, M. , Olson, M. S. , & Liu, J. (2018). Genomic effects of population collapse in a critically endangered ironwood tree Ostrya rehderiana. Nature Communications, 9(1), 5449. 10.1038/s41467-018-07913-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwaenepoel, A. , & van de Peer, Y. (2018). wgd—Simple command line tools for the analysis of ancient whole‐genome duplications. Bioinformatics, 35(12), 2153–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Size of each scaffold and number of genes per scaffold
Table S2: Topologically Associated Domains (TADs) analysis summary
Table S3: TAD statistics at different resolutions
Table S4: Location of cyanogenic genes on pseudo‐molecules
Table S5: Location of fatty acid genes on pseudo‐molecules
Table S6: SNP heterozygosity statistics in eight Macadamia jansenii accessions
Table S7: Polymorphic sites in up to seven of the eight Macadamia jansenii accessions
Table S8: Genotype‐specific unique polymorphic SNP sites
Table S9: List of duplicated genes in M. jansenii genome, using MCScanX tool
Figure S1: Linkage density histogram of Hi‐C assembly of M. jansenii genome
Figure S2: BLAST2GO sequence similarity search
Figure S3: Gene ontology (GO) analysis by BLAST2GO
Figure S4: Frequency graph of AED scores
Figure S5: Alignment of the vicilin‐like antimicrobial‐peptide transcript from M. integrifolia and M. jansenii
Figure S6: Alignment of anti‐microbial CDS sequence of M. integrifolia against the M. jansenii transcript sequence
Figure S7: Ks plot of M. jansenii was generated by Wgd tool.
Table S4: Location of cyanogenic genes on pseudo‐molecule (based on blastp results)
Table S5: Location of fatty acid genes on pseudo‐molecules (based on blastp results)
Data Availability Statement
The genome sequence reads, transcriptome sequences, and genome assembly of M. jansenii have been deposited under NCBI bioproject PRJNA694456.