

Summary
Background
Lysosomal storage disorders (LSD) are a family of genetic diseases that have a devastating impact on the patient and family with a concomitant health burden. Although considered rare disorders, improved diagnostic capabilities, newborn screening programs and public awareness has witnessed the frequency of many LSD increase considerably over recent years. To quantify their footprint, the number of LSD diagnosed in the multicultural Australian population in a 12-year period was determined. The principle objective was to yield contemporary prevalence figures to inform public health policies.
Methods
From the national referral laboratory for LSD diagnoses in Australia, retrospective data from patient referrals and prenatal testing for the period January 1 2009 to December 31 2020 were collated. Diagnosis was established biochemically by enzyme activity and/or metabolite determinations, as well as molecular genetic testing. The incidence of each disorder was determined by dividing the number of postnatal diagnoses by the number of births with prevalence including prenatal diagnoses.
Findings
During this 12-year period 766 diagnosis of LSD were confirmed inclusive of 32 prenatal outcomes representing 38 individual disorders. Total diagnosis per 100,000 live births averaged 21 per year (range 16 – 26) with Fabry disease the most prevalent representing 34% of all diagnoses in the current (up to 2020) report.
Interpretation
The combined prevalence of LSD for this study period at 1 per 4,800 live births is considerably higher than 1 per 7,700 reported for a 17-year period up to 1996. Additionally, more adults were diagnosed than children, implying that LSD are more common in adulthood than childhood. These data highlight the requirements for physicians to consider LSD in symptomatic adults and should refigure public health policies steering newborn screening programs in the direction of adult-onset conditions.
Funding
No funding was received for this study.
Research in context.
Evidence before this study
Lysosomal storage disorders (LSD) are considered rare disorders with the majority occurring randomly in the general population irrespective of ethnicity and geographical location. Case ascertainment is difficult without universal population testing, and although newborn screening provides a conduit, it is also complicit with significant social, emotional and economic penalties. In order to assess the impact of LSD in the population with the primary objective of steering public health directions, PubMed was searched using the terms, lysosomal storage disorders in combination with incidence or prevalence or frequency, yielding only a handful of outdated reports that did not reflect heterogenous populations.
Added value of this study
Due to the absence of data from countries with similar ancestry to Australia, coupled with improved diagnostic capabilities for LSD, we sought to determine contemporary prevalence data in order to ascertain the burden of LSD to guide population health practice. Although newborn screening programs for LSD typically yield higher numbers of diagnoses, our Australian data reports that the current prevalence of LSD in the Australian population (without LSD newborn screening) was greater than anticipated; 1.6-fold higher than in the previous 17-year reporting period to 1996. The prevalence of the sum of 766 individual LSD was 1 in 4,800 live births, inclusive of an additional 11 individual disorders due mostly to improved laboratory diagnostic capabilities. In addition to the higher prevalence, our study also shows that the age of symptom onset for the majority of diagnoses was in the fourth decade of life.
Implications of all the available evidence
Data from our study, combined with newborn screening programs and pilot studies in other countries, reveal three seminal concepts regarding LSD. Firstly, the prevalence of LSD is on the rise and LSD are more common than historically contemplated. Secondly, a higher proportion of patients have adult-onset phenotypes, questioning the notion that LSD are childhood disorders and highlighting the clinical need to include LSD in the differential of symptomatic adults. Thirdly, these data have significant implications for LSD newborn screening programs, underscoring the quandary of screening neonates for conditions that will not manifest until much later in life.
Alt-text: Unlabelled box
Introduction
Lysosomal storage disorders (LSD) are rare genetic entities characterized by the dysfunction of lysosomal proteins. At least 70 distinct LSD have been reported and the majority adhere to autosomal inheritance with Danon disease, Fabry disease and mucopolysaccharidosis type II the exceptions as X-linked conditions. These disorders arise from pathogenic variants in genes typically encoding for lysosomal hydrolases and less commonly transmembrane proteins, modifiers or activators essential for proper lysosomal function. The net result is persistent lysosomal substrate deposition from inefficiently recycled cellular material, leading to the progressive deterioration of cells, tissues and organ systems, culminating in a complex phenotype complicit with the LSD.1 Often a cardinal clinical feature, progressive neurodegeneration is a source of significant distress for the patient and family, manifesting typically after an interval of apparent normality following birth. The personal and societal burden is immeasurable. This is often confounded by the relentless trajectory to attain an accurate LSD diagnosis, as more common causes of the disease picture are explored.
With the move to early recognition of LSD for the mitigation of misdiagnosis, and to enable targeted therapy for the handful of diseases for which treatment is commercially available, the addition of LSD to public health newborn screening programs seems reasonable. Disease-specific treatment is available for Pompe disease2 and mucopolysaccharidosis type I,3 and as the window of opportunity for effectiveness is significantly ameliorated if patients are diagnosed after symptom onset, both LSD are included in the US Secretary of Health and Human Services’ Recommended Uniform Screening Panel (RUSP).4 However, newborn screening for LSD is not straightforward from both an analytical and psychosocial standpoint. Results obtained thus far show 1) more false than true positives causing unnecessary stress to families, 2) more LSD newborns identified with a late-onset rather than an early-onset phenotype (in infancy/childhood) who may spend the majority of their life asymptomatic, and 3) the dilemma of screening for untreatable diseases, which includes the vast majority of LSD, particularly those with neurological symptoms.5, 6, 7
As debate continues over the feasibility, beneficiaries and economics of newborn screening for LSD, there is an unmet need to quantify the magnitude of the LSD burden in modern-day, real-life populations so public health policies are information rather than perception driven.5 There is a paucity of epidemiological data for LSD that is either outdated and/or confined to relatively homogenous demographics.8, 9, 10, 11, 12, 13, 14 This provided the impetus to determine contemporary prevalence data for LSD in the multicultural Australian population from January 2009 to December 2020 in a nation with no newborn screening for LSD.
Methods
This study compiled retrospective data on LSD diagnoses from both patient referrals and prenatal diagnoses for the 12-year period, January 1 2009 to December 31 2020 within Australia. All patient samples were submitted to our laboratory for LSD testing as part of routine patient care and their use was approved by the Institutional Human Research Ethics Committee (HREC/15/WCHN/69). The laboratory diagnosis of LSD was made by a combination of biochemical assessments (deficient enzyme and/or elevated substrate biomarkers) and genetic testing that identified pathogenic variants in the requisite genes.15
The number of live births was obtained from the Australian Bureau of Statistics.16 The prevalence of each LSD was calculated by dividing the total number of postnatal and prenatal diagnoses during the study period by the number of live births during the study period and incidence rates used the same approach with the exception of prenatal diagnoses. Carrier frequency was determined by dividing the prevalence value by 4 and finding the square root. For the X-linked disorders, the carrier frequency was equal to the prevalence rate assuming that a de novo event did not occur. Calculations and assumptions regarding the data have been detailed previously.9
Role of the funding source:
No funding was received for this study.
Results
In the Australian population during the 12-year period, January 2009 through to December 2020, there were 766 LSD diagnoses in the Australian population, inclusive of 32 positive prenatal outcomes (Table 1). Across the study period, there was a yearly average of 21 diagnoses per 100,000 births ranging from 16 to 26 with no significant difference over the 12 years (Figure 1). The entities spanned 38 individual disorders with Fabry disease clearly the most commonly diagnosed at 258 cases, accounting for 34% of all LSD diagnoses. This was followed by Pompe disease (81 cases, 11%), Gaucher disease (50 cases, 6.5%), metachromatic leukodystrophy (38 cases, 5%), Niemann Pick type C (33 cases, 4%) and MPS type I and IIIA each at 27 cases (3.5%) of the total number of diagnosed LSD. Of the neuronal ceroid lipofuscinosis (NCL), type 2 was the most frequently diagnosed with 23 cases representing 3% of the LSD cohort. There were no diagnoses recorded in the study period for Farber disease or β-mannosidosis. Collectively, the prevalence of LSD in the Australian population was determined to be 1 in 4,800 live births, with the two major subgroups being the sphingolipidoses at 1 in 8,000 and the mucopolysaccharidoses at 1 in 30,000.
Table 1.
Diagnosis of lysosomal storage disorders in Australia from January 2009 to December 2020
| Total live births 2009-2020 |
3,693,759 |
DIAGNOSES |
INCIDENCE |
PREVALENCE |
|||||
|---|---|---|---|---|---|---|---|---|---|
| DISORDER | Postnatal: median age at diagnosis (range), years |
Prenatal | Postnatal | Total | Per 100 000 | 1 per 1000 live births | Per 100 000 | 1 per 1000 live births | Carrier Frequency |
| SPHINGOLIPIDOSES (n=11) | 14 | 443 | 457 | 11.99 | 8 | 12.37 | 8 | 45 | |
| Acid lipase deficiency | 5.9 (3.4-30.5) | 5 | 5 | 0.14 | 739 | 0.14 | 739 | 430 | |
| Fabry diseasea | 44.6 (0.04-96.9) | 1 | 257 | 258 | 6.96 | 14 | 6.98 | 14 | 14 |
| Maleb | 46.1 (0.04-77.1) | 1 | 101 | 102 | 2.73 | 37 | 2.76 | 36 | 36 |
| Female | 42.8 (2.1-96.9) | 156 | 156 | 4.22 | 24 | 4.22 | 24 | 24 | |
| Gaucher disease | 16.4 (0.01-75.3) | 1 | 49 | 50 | 1.33 | 75 | 1.35 | 74 | 136 |
| GM1 gangliosidosis | 1.3 (0.02-40.5) | 3 | 16 | 19 | 0.43 | 231 | 0.51 | 194 | 220 |
| GM2 gangliosidosis type 1 | 1.1 (0.9-22.8) | 13 | 13 | 0.35 | 284 | 0.35 | 284 | 267 | |
| (Tay-Sachs disease) | |||||||||
| GM2 gangliosidosis type 2 | 1.1 (0.02-2.0) | 1 | 8 | 9 | 0.22 | 462 | 0.24 | 410 | 320 |
| (Sandhoff disease) | |||||||||
| Krabbe disease | 0.94 (0.5-67.2) | 2 | 16 | 18 | 0.43 | 231 | 0.49 | 205 | 226 |
| Metachromatic leukodystrophy | 4.5 (0.02-50.8) | 1 | 37 | 38 | 1.00 | 100 | 1.03 | 97 | 156 |
| Multiple sulphatase deficiency | 1.2 (0.4-2.0) | 1 | 2 | 3 | 0.05 | 1,847 | 0.08 | 1,231 | 555 |
| Niemann-Pick type A/B | 15 (0.4-75.3) | 1 | 10 | 11 | 0.27 | 369 | 0.30 | 336 | 290 |
| Niemann-Pick type C | 17.8 (0.02-72.2) | 3 | 30 | 33 | 0.81 | 123 | 0.89 | 112 | 167 |
| OLIGOSACCHARIDOSES (n=10) | 2 | 39 | 41 | 1.06 | 95 | 1.11 | 90 | 150 | |
| Aspartylglucosaminuria | 6.5 (4.8-56.2) | 6 | 6 | 0.16 | 616 | 0.16 | 616 | 392 | |
| Fucosidosis c | 2.5 | 1 | 1 | 2 | 0.03 | 3,694 | 0.05 | 1,847 | 679 |
| Galactosialidosis c | 0.07 (0.04-0.1) | 2 | 2 | 0.05 | 1,847 | 0.05 | 1,847 | 679 | |
| Galactosialidosis or Sialidosis d | 0.005 | 1 | 1 | 0.03 | 3,694 | 0.03 | 3,694 | 961 | |
| Infantile sialic acid storage disorder | 0.5 (0.08-0.8) | 2 | 2 | 0.05 | 1,847 | 0.05 | 1,847 | 679 | |
| α-Mannosidosis | 1.3 (1.2-8.6) | 3 | 3 | 0.08 | 1,231 | 0.08 | 1,231 | 555 | |
| Mucolipidosis type II/III | 0.2 (0.01-19.1) | 15 | 15 | 0.41 | 246 | 0.41 | 246 | 248 | |
| Mucolipidosis type IV c,e | 11.7 (1.7-21.8) | 2 | 2 | 0.05 | 1,847 | 0.05 | 1,847 | 679 | |
| Schindler disease c | 65.1 (64.0-66.1) | 2 | 2 | 0.05 | 1,847 | 0.05 | 1,847 | 679 | |
| Sialidosis | 0.9 (0.02-28.7) | 1 | 5 | 6 | 0.14 | 739 | 0.16 | 616 | 392 |
| Sialuria | 1.3 | 1 | 1 | 0.03 | 3,694 | 0.03 | 3,694 | 961 | |
| GLYCOGENOSES (GSD) (n=2) | 2 | 87 | 89 | 2.36 | 42 | 2.41 | 42 | 102 | |
| Pompe disease (GSD II) | 36 (0.2-69.7) | 2 | 79 | 81 | 2.14 | 47 | 2.19 | 46 | 107 |
| Cori (GSD III-debrancher deficiency) c | 1.6 (0.6-50.6) | 8 | 8 | 0.22 | 462 | 0.22 | 462 | 340 | |
| MUCOPOLYSACCHARIDOSES (MPS) (n=9) | 11 | 113 | 124 | 3.06 | 33 | 3.36 | 30 | 86 | |
| MPS type I (Hurler) | 1.3 (0.01-54.7) | 27 | 27 | 0.73 | 137 | 0.73 | 137 | 185 | |
| MPS type II (Hunter)f | 3.5 (0.04-12.3) | 2 | 21 | 23 | 0.57 | 176 | 0.62 | 161 | 161 |
| MPS type IIIA (Sanfillipo A) | 4.0 (0.3-39.7) | 2 | 25 | 27 | 0.68 | 148 | 0.73 | 137 | 185 |
| MPS type IIIB (Sanfillipo B) | 4.5 (1.4-39.6) | 10 | 10 | 0.27 | 369 | 0.27 | 369 | 304 | |
| MPS type IIIC (Sanfillipo C) | 2.3 (0.7-8.0) | 6 | 6 | 0.16 | 616 | 0.16 | 616 | 392 | |
| MPS type IVA (Morquio A) | 1.6 (1.0-45.5) | 2 | 17 | 19 | 0.46 | 217 | 0.51 | 194 | 220 |
| MPS type IVB (Morquio B) c | 1.8 (1.2-2.4) | 2 | 2 | 0.05 | 1,847 | 0.05 | 1,847 | 679 | |
| MPS type VI (Maroteaux-Lamy) | 4.9 (1.6-9.0) | 4 | 4 | 0.11 | 923 | 0.11 | 923 | 480 | |
| MPS type VII (Sly) | 13.1 | 5 | 1 | 6 | 0.03 | 3,694 | 0.16 | 616 | 392 |
| NEURONAL CEROID LIPOFUSCINOSES (NCL) (n=5) | 2 | 44 | 46 | 1.19 | 84 | 1.25 | 80 | 142 | |
| NCL 2 (late-infantile, | 4.1 (0.28-12.3) | 2 | 21 | 23 | 0.57 | 176 | 0.62 | 161 | 200 |
| Jansky-Bielschowsky) c | |||||||||
| NCL 3 (juvenile, Spielmeyer-Vogt) c,e | 11.9 (6.4-18.1) | 17 | 17 | 0.46 | 217 | 0.46 | 217 | 233 | |
| NCL 5 (Finnish variant, late-infantile) c,e | 14.5 (8.1-20.8) | 4 | 4 | 0.11 | 923 | 0.11 | 923 | 480 | |
| NCL 6 (Kufs variant, late-infantile) c,e | 7.9 | 1 | 1 | 0.03 | 3,694 | 0.03 | 3,694 | 961 | |
| NCL 7 (late-infantile) c,e | 4.7 | 1 | 1 | 0.03 | 3,694 | 0.03 | 3,694 | 961 | |
| MISCELLANEOUS (n=1) | 1 | 7 | 8 | 0.19 | 528 | 0.22 | 462 | 340 | |
| Cystinosis | 28.5 (0.67-54.5) | 1 | 7 | 8 | 0.19 | 528 | 0.22 | 462 | 340 |
| TOTAL LSD | 32 | 734 | 766 | 19.87 | 5 | 20.74 | 4.8 | 35 | |
Male and female combined.
One male with Fabry disease also had Klinefelter syndrome.
LSD not detected in Meikle and colleagues9 (MPS IIID was not seen in 2009-2020).
Specific diagnosis dependent on secondary testing.
Disorder by molecular diagnosis only.
All diagnoses were male.
Total live births n= 3,693,759: 2009-2019 ABS; 2020 calculated from: Australian population as of 06/01/2021.16
Figure 1.

Total LSD diagnosis and birth rate during the 12-year period of 2009 to 2020. The sum of all LSD diagnosis in each year is shown (filled squares) along with the number of live births (open circles) per year.
Although for many of the LSD the number of diagnoses was too small to comment (<10 diagnoses), Table 1 shows that there was broad heterogeneity in the age at diagnosis. These typically ranged from infancy to late adulthood. The median age of the patients at diagnosis was in adulthood for the two most prevalent LSD, Fabry disease and Pompe disease, at 45 and 36 years of age, respectively (Figure 2). Males with Fabry disease had a slightly higher median at 46.1 years of age (cf 42.8 for females) with the youngest patient diagnosed at 2 weeks of age and the oldest patient at 77.1. For females with Fabry disease the youngest patient was at 2.1 years of age with the eldest at 96.9. Pompe disease had a similar wide range of diagnoses from as early as 10 weeks of age to late adulthood (69.7 years). Gaucher disease, the third most prevalent, had a median age of diagnosis in the teenage years, and although out of the top six most prevalent LSD, metachromatic leukodystrophy, MPS I and IIIA revealed a median age in childhood, patients were diagnosed as late as the 4th and 5th decades of life (Figure 2).
Figure 2.
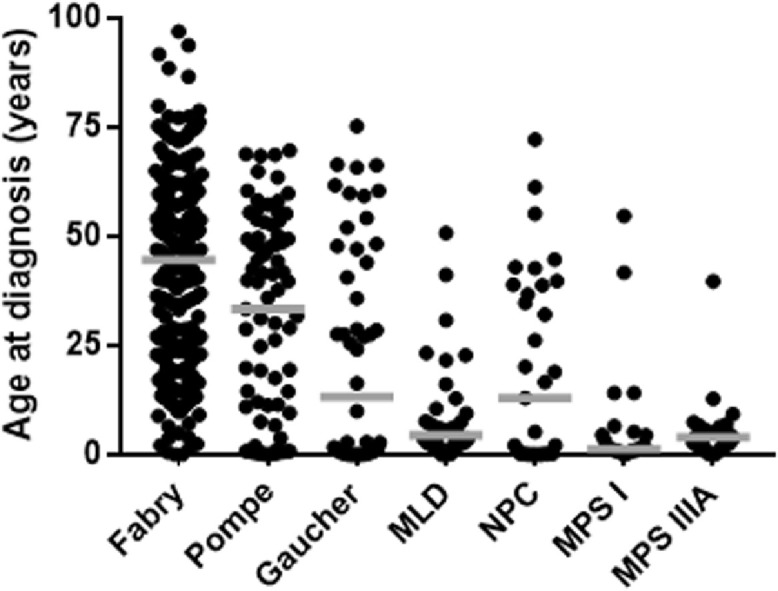
Diagnoses for the six most prevalent LSD. The number of individuals diagnosed with Fabry disease, Pompe disease, Gaucher disease, metachromatic leukodystrophy (MLD), Niemann Pick C (NPC), mucopolysaccharidosis type I (MPS I) and MPS IIIA are shown in order of prevalence from left to right, with MPS I and IIIA equal sixth. The midline marks the median age of diagnosis for each disorder.
Discussion
The prevalence in the Australian population is 1.6 times higher than the 1 in 7,700 live births previously reported in 1996.9 The occurrence of individual disorders has also changed with Fabry disease now replacing Gaucher disease as the most common LSD (Table 2). This is principally due to an almost 10-fold increase in the diagnosis of Fabry disease (from 1 in 117,000 to 1 in 14,000 live births). Gaucher disease now placed third but interestingly the prevalence reduced from 1 in 57,000 to 1 in 74,000 live births. The frequency of MPS I was lower, previously ranked as the second most prevalent LSD now moves to equal 6th along with MPS IIIA (Table 2). The 7th most common LSD was neuronal ceroid lipofuscinosis (NCL) 2 - belonging to the NCL family - and was not included in the previous report as laboratory testing for this LSD subgroup was not available. An additional 11 different LSD were diagnosed in this 12-year period from 2009 to 2020 compared with the 17-year period ending in 1996 (Table 1), primarily due to improved diagnostic capabilities that has refashioned testing for LSD.15
Table 2.
Comparison of current (2009-2020) and previous (1980-1996) six most prevalent LSD
| Disorder (2009-2020) | Prevalence (in 1000) | Disorder (1980-1996)* | Prevalence* (in 1000) |
|---|---|---|---|
| Fabry disease | 14 | Gaucher disease | 57 |
| Pompe disease | 46 | MPS type I | 88 |
| Gaucher disease | 74 | metachromatic leukodystrophy | 92 |
| metachromatic leukodystrophy | 97 | MPS type IIIA | 114 |
| Niemann-Pick type C | 112 | Fabry disease | 117 |
| MPS types I and IIIA | 137 | Krabbe disease | 141 |
data taken from Meikle and colleagues.9
A further aspect is the demographics of the Australian population. In the 1999 report, citizens were largely of British descent, with permanent migration from other European nations another major source, and Asian countries contributing to a smaller extent.15 The composition of the Australian population is progressively changing, with China surpassing the United Kingdom in 2010-2011, and India now a predominant contributor to Australian migration.17 Although cultural associations were not investigated in this report, higher frequencies of LSD do occur in particular populations, usually due to the phenomena of genetic drift exacerbated by geographical isolation.18 Comparing Australia with other nations is difficult due to the paucity of prevalence data and difficulties with ascertainment in the absence of universal population testing. Data from China reports MPS II to be the most common LSD, followed by Pompe disease,8 the latter also the second prevalent LSD in Australia. In European states, Sweden and the Czech Republic, both report Krabbe as the most common LSD, which ranked 12th in Australia.12,13 However, it is clear that the prevalence of LSD worldwide is increasing, and although newborn screening has undoubtedly improved ascertainment, case numbers are rising in its absence.
In addition to the increase in prevalence and change in ranking of LSD, is the noticeable older age at which patients are diagnosed. Traditionally considered childhood disorders, the average age of diagnosis for the two most common LSD (Fabry disease and Pompe disease) was in adulthood. Notwithstanding the importance of considering these rare disorders in the differential diagnoses of symptomatic adults, it challenges the fundamental premise of newborn screening, noting that Pompe disease is one of two LSD on the RUSP. Indeed, the majority of infants identified with an LSD via RUSP are predicted to have later-onset phenotypes.19,20 This requires remediation, particularly with regard to ongoing debate over expanding RUSP to include additional LSD, when screening newborns for adult-onset conditions is not recommended.21
Limitations of our study are acknowledged and have been recognized previously,9 additionally noting that other laboratories within Australia, typically due to readily available genetic testing, may have identified additional LSD patients that have not been confirmed by the national laboratory. It is predicted that the future will bring further escalations in LSD diagnosis emanating from increased awareness of these disorders and advances in technology that have grown the number and type of LSD that can be reliably diagnosed. The importance of accurate and contemporary epidemiological data is underscored by the ongoing drive to improve healthcare, which can only be achieved by assessing the societal burden of LSD so that public health policies can be appropriately targeted. With increasing pressure on expanding newborn screening programs, given technology is available for many LSD, incidence data becomes critical to ensure the objectives of newborn screening are fulfilled and not contraindicated.
Contributors
SJC compiled the data and SJC and MF analysed and interpreted it. MF and SJC wrote the manuscript.
Declaration of Interests
We declare that we have no conflicts of interest.
Acknowledgements
The authors are most grateful to Nicole Cadzow for her assistance with collating, verifying and preparing the numbers of positive diagnoses and to Dr Jennifer Saville for data handling and critical review.
References
- 1.Platt EF, d'Azzo A, Davidson BL, Neufeld EF, Tifft CJ. Lysosomal storage diseases. Nat Rev Dis Primers. 2018;4:27. doi: 10.1038/s41572-018-0025-4. [DOI] [PubMed] [Google Scholar]
- 2.Chien Y-H, Lee N-C, Thurberg BL, et al. Pompe disease in infants: Improving the prognosis by newborn screening and early treatment. Pediatrics. 2009;124:e116–e125. doi: 10.1542/peds.2008-3667. [DOI] [PubMed] [Google Scholar]
- 3.Clarke LA, Atherton AM, Burton BK. Mucopolysaccharidosis type I newborn screening: Best practices for diagnosis and management. J Pediatr. 2017;182:363–370. doi: 10.1016/j.jpeds.2016.11.036. [DOI] [PubMed] [Google Scholar]
- 4.Advisory Committee on Heritable Disorders in Newborns and Children. Available online: http://www.hrsa.gov/advisorycommittees/mchbadvisory/heritabledisorders/index/.html (accessed on 10th June 2021).
- 5.Lisi EC, Ali N. Opinions of adults affected with later-onset lysosomal storage diseases regarding newborn screening: A qualitative study. J Genet Couns. 2021;00:1–15. doi: 10.1002/jgc4.1421. [DOI] [PubMed] [Google Scholar]
- 6.Peck DS, Lacey JM, White AL, et al. Incorporation of second-tier biomarker testing improves the specificity of newborn screening for mucopolysaccharidosis type I. Int J Neonatal Screen. 2020;6:10. doi: 10.3390/ijns6010010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klug TL, Swartz LB, Washburn J, Brannen C, Kiesling JL. Lessons learned from Pompe disease newborn screening and follow-up. Int J Neonatal Screen. 2020;6:11. doi: 10.3390/ijns6010011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen X, Qiu W, Ye J, Han L, Gu X, Zhang H. Demographic characteristics and distribution of lysosomal storage disorder subtypes in Eastern China. J Hum Genet. 2016;61:345–349. doi: 10.1038/jhg.2015.155. [DOI] [PubMed] [Google Scholar]
- 9.Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA. 1999;281:249–254. doi: 10.1001/jama.281.3.249. [DOI] [PubMed] [Google Scholar]
- 10.Poorthuis BJ, Wevers RA, Kleijer WJ, et al. The frequency of lysosomal storage diseases in The Netherlands. Hum Genet. 1999;105:151–156. doi: 10.1007/s004399900075. [DOI] [PubMed] [Google Scholar]
- 11.Pinto R, Caseiro C, Lemos M, et al. Prevalence of lysosomal storage diseases in Portugal. Eur J Hum Genet. 2004;12:87–92. doi: 10.1038/sj.ejhg.5201044. [DOI] [PubMed] [Google Scholar]
- 12.Poupētová H, Ledvinová J, Berná L, Dvoráková L, Kozich V, Elleder M. The birth prevalence of lysosomal storage disorders in the Czech Republic: Comparison with data in different populations. J Inherit Metab Dis. 2010;33:387–396. doi: 10.1007/s10545-010-9093-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hult M, Darin N, von Döbeln U, Månsson JE. Epidemiology of lysosomal storage disorders in Sweden. Acta Paediatr. 2014;103:1258–1263. doi: 10.1111/apa.12807. [DOI] [PubMed] [Google Scholar]
- 14.Al-Jasmi FA, Tawfig N, Berniah A, et al. Prevalence and novel mutations of lysosomal storage disorders in United Arab Emirates: LSD in UAE. JIMD Rep. 2013;10:1–9. doi: 10.1007/8904_2012_182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fuller M. Laboratory diagnosis of lysosomal diseases: Newborn screening to treatment. Clin Biochem Rev. 2020;41:53–66. doi: 10.33176/AACB-19-00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.https://worldpopulationreview.com/countries/australia-population; https://www.macrotrends.net/countries/AUS/australia/birth-rate (accessed on 20th March 2021).
- 17.https://www.aph.gov.au/About_Parliament/Parliamentary_Departments/Parliamentary_Library/pubs/rp/rp1617/Quick_Guides/MigrationStatistics (accessed June 22nd).
- 18.Zimran A, Gelbert T, Westwood B, Grabowski GA, Beutler E. High frequency of the Gaucher disease mutation at nucleotide 1226 among Ashkenazi Jews. Am J Hum Genet. 1991;49:855–859. [PMC free article] [PubMed] [Google Scholar]
- 19.Burton BK, Charrow J, Hoganson GE, et al. Newborn screening for lysosomal storage disorders in Illinois: The initial 15-month experience. J Pediatr. 2017;190:130–135. doi: 10.1016/j.jpeds.2017.06.048. [DOI] [PubMed] [Google Scholar]
- 20.Wasserstein MP, Caggana M, Bailey SM, et al. The New York pilot newborn screening program for lysosomal storage diseases: Report of the first 65,000 infants. Genet Med. 2019;21:631–640. doi: 10.1038/s41436-018-0129-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duncan RE, Savulescu J, Gillam L, Williamson R, Delatycki MB. An international survey of predictive genetic testing in children for adult onset conditions. Genet Med. 2005;7:390–396. doi: 10.1097/01.gim.0000170775.39092.44. [DOI] [PubMed] [Google Scholar]