

Summary
A key facet of epithelial differentiation is the assembly of actin-based protrusions known as microvilli, which amplify apical membrane surface area for various cell functions. To probe mechanisms of microvillus assembly, we developed a protocol using spinning disk confocal microscopy to directly visualize microvillus biogenesis on the surface of cultured porcine kidney epithelial cell monolayers engineered to express fluorescent proteins. This protocol offers access to the molecular details of individual protrusion growth events at high spatiotemporal resolution.
For complete details on the use and execution of this protocol, please refer to Gaeta et al. (2021).
Subject areas: Cell Biology, Cell culture, Cell-based Assays, Microscopy
Graphical abstract
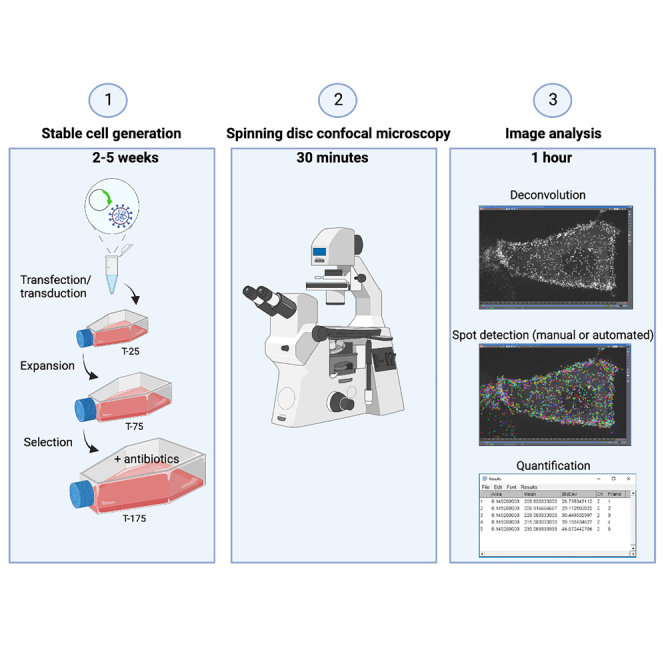
Highlights
-
•
Directly visualize microvillus growth events using spinning disc confocal microscopy
-
•
Procedure for stable cell line generation in LLC-PK1-CL4 cells
-
•
Analysis of microvillus growth dynamics and tip targeted proteins
A key facet of epithelial differentiation is the assembly of actin-based protrusions known as microvilli, which amplify apical membrane surface area for various cell functions. To probe mechanisms of microvillus assembly, we developed a protocol using spinning disk confocal microscopy to directly visualize microvillus biogenesis on the surface of cultured porcine kidney epithelial cell monolayers engineered to express fluorescent proteins. This protocol offers access to the molecular details of individual protrusion growth events at high spatiotemporal resolution.
Before you begin
This protocol for direct visualization of microvilli biogenesis requires the expression of fluorescent protein-tagged molecules of interest in the porcine kidney epithelial LLC-PK1-CL4 (CL4) cell line. CL4 cells are a genetically tractable polarized epithelial cell culture model that forms a mature brush border over ∼4–5 days post confluency (DPC) (Tyska and Mooseker, 2002). This cell line is well suited to this assay as individual protrusions can be visualized en face, with microvillus growth events occurring spontaneously at early stages of cellular differentiation. The timeline of differentiation and microvillus orientation offer advantages over other polarized cell culture models such as Caco2BBE, which form mature brush borders over ∼3 weeks (Peterson and Mooseker, 1992), and Ls174TW4 and MDCK cysts, which build microvilli that extend parallel to the xy plane (Baas et al., 2004; Elia and Lippincott-Schwartz, 2009). Thus, the short timeline of differentiation and en face view of the apical surface in combination with spinning disc confocal microscopy enables the study of unitary microvillus growth events with high spatiotemporal resolution. When visualizing two or more fluorescent proteins, creating a stably transfected cell line is strongly recommended to increase the pool of cells suitable for imaging, characterized by moderate expression levels without dramatic overexpression artifacts. To visualize microvilli growth and movement in live cells we also need to label the microvillar actin filaments with a fluorescent tag. Several options are available including labeled β-actin, labeled actin-binding domains such as Utrophin or LifeAct, or labeled actin bundling proteins such as Espin (ESPN). All of these methods are effective in labeling microvillar actin but have distinct advantages and disadvantages (Figure 1A) (Melak et al., 2017). We chose ESPN to label microvillar actin bundles because of its superior signal to noise ratio (SNR) compared to other markers such as β-actin or LifeAct (Figure 1B). This protocol outlines a two-step method for creating epithelial cells stably expressing fluorescent proteins (referred to herein as ‘stable cells‘) (Figure 2). In our example protocol given below, cells are selected for two unique selectable markers, G418 and puromycin, introduced using plasmid and lentiviral vectors respectively. Similar results can be achieved using two plasmids or two lentiviruses, as CL4 cells are amenable to both plasmid transfection and viral transduction, but the key is to have two distinct selectable markers. Alternatively, stable expression can be established via endogenously tagged proteins using CRISPR/Cas9 technology, or transient plasmid transfection if expression of only one fluorescent protein is required. An example protocol for creating double stable cells expressing EGFP-EPS8 and mCherry-ESPN is outlined below.
Figure 1.

Examples of CL4 cells expressing markers for the actin cytoskeleton
(A) Spinning disk confocal images of cells expressing mNEON-green-β-actin (left), EGFP-LifeAct (middle) and mCherry-ESPN (right).
(B) Microvilli signal to noise ratio (SNR) plots corresponding to images shown in A. N = 20 microvilli. SNR is defined as μ/σ, with μ = signal mean and σ = standard deviation of the noise.
Figure 2.

Workflow for creation of stable cells via plasmid transfection and viral transduction
Generation of CL4 stable cells by transfection
Timing:45 min, then 2–5 weeks
Note: The order of transfection/transduction for creating a double stable cell line is not critical.
Note: Refer to “Materials and equipment” for a complete list of solution/medium recipes.
-
1.Transfection of CL4 cells with plasmid DNA.
-
a.Start culture of CL4 cells.
-
i.Thaw vial of frozen cells (1mL) and immediately add to a T-75 flask containing 11 mL CL4 media (DMEM + 10% FBS, + 1% L-glutamine). CL4 cells may be used until a very high passage number (p∼100) without influencing the formation of microvilli.
-
i.
-
b.Split cells once in preparation for transfection. The following steps i-iv. outlines standard splitting procedure.
-
i.Rinse cells once with Dulbecco’s phosphate buffered saline (DPBS).
-
ii.Add 2 mL 0.05% trypsin, incubate at 37°C–3 min.Note: If necessary, tap flask to detach cells.
 CRITICAL: Do not let cells reach confluency as they become difficult to detach.
CRITICAL: Do not let cells reach confluency as they become difficult to detach. -
iii.Resuspend cells with 8 mL CL4 media to make a 10 mL total cell suspension.
-
iv.Split cells 1:10 by adding 1 mL cells to 11 mL CL4 media in a T-75 flask.
-
i.
-
c.Once cells reach 70%–80% confluency, split into a T-25 flask for transfection.
-
i.Rinse cells once with DPBS.
-
ii.Add 2 mL 0.05% trypsin, incubate at 37°C–3 min
-
iii.Resuspend cells with 8 mL CL4 media.
-
iv.Add 500 μL cells (∼3.5 × 105 cells) to 4.5 mL CL4 media in T-25 flask.
-
i.
-
d.Grow CL4 cells to 70%–80% confluency in a T-25 flask (∼16 h after splitting).
-
e.Transfection of CL4 cells using plasmid DNA (EGFP-EPS8) and FuGENE 6.
-
i.Bring Opti-MEM and FuGENE 6 to room temperature (RT; 20°C–22°C).
-
ii.Calculate volumes of Opti-MEM, FuGENE 6, and plasmid DNA to be added to a 1.5 mL Eppendorf tube, guided by manufacturer’s instructions. A 1:3 ratio of DNA:FuGENE 6 is recommended as a starting point (e.g., 4 μg DNA:12 μL FuGENE 6). Calculate volume of Opti-MEM needed to bring total volume to 200 μL. For example, assuming DNA stock concentration of 4 μg/μL, add 1 μL DNA, 12 μL FuGENE, and 187 μL Opti-MEM).Note: It is recommended to use endotoxin free DNA throughout this protocol to maximize transfection efficiency.CRITICAL: Add FuGENE 6 directly to media as this reagent can adhere to plastic.
-
iii.Combine FuGENE 6 and Opti-MEM in 1.5 mL Eppendorf tube. Tap the tube to mix.
-
iv.Incubate 5 min at RT.
-
v.Add plasmid DNA to FuGENE 6 and Opti-MEM mixture. Tap tube to mix and then incubate 15–30 min at RT.
-
i.
-
a.
Add total volume of transfection reaction (200 μL) dropwise to a T-25 flask.
Alternatives: Transfection of CL4 cells can also be accomplished using Lipofectamine 2000. If using lipofectamine 2000, do not incubate with reagent longer than 4 h.
-
2.Expansion and selection of CL4 stable cells.
-
a.The next day, expand cells into a T-75 flask.Note: Expansion of cells to a larger volume flask is performed to maintain the pool of transfected cells going into selection.
-
i.Rinse cells once with DPBS.
-
ii.Add 1 mL 0.05% trypsin, incubate at 37°C–3 min.
-
iii.Resuspend cells with 4 mL CL4 media.
-
iv.Add total cell suspension (5 mL) to 7 mL CL4 media in a T-75 flask.
-
i.
-
b.Once cells reach 70%–80% confluency (typically 24–48 h post-split), expand into a T-175 flask.Note: Expansion of cells to a larger volume flask is performed to maintain the pool of transfected cells going into selection.
-
i.Rinse cells once with DPBS.
-
ii.Add 2 mL 0.05% trypsin, incubate at 37°C–3 min.
-
iii.Resuspend cells with 8 mL CL4 media.
-
iv.Add total suspension (10 mL) to 20 mL CL4 media in T-175 flask, for a total of 30 mL.
-
v.Add 600 μL of 50 mg/mL stock solution of G-418 antibiotic (1:50 dilution, 1 mg/mL final concentration) to the flask.
-
vi.Monitor cells and replace media every 1–3 days as needed, maintaining antibiotic selection.Note: Selection antibiotic is typically added 48–72 h post transfection.Optional: If expressing a fluorescent protein, monitor expression of probe by epifluorescence. The pool of expressing cells should increase with more time in selection antibiotic.
-
i.
-
c.Once cells reach 70%–80% confluency, split cells at 2:10 dilution in T-175 flask, maintaining antibiotic selection.
-
i.Rinse cells once with DPBS.
-
ii.Add 4 mL 0.05% trypsin, incubate at 37°C–3 min.
-
iii.Resuspend cells with 6 mL CL4 media.
-
iv.Add 2 mL of total suspension to 28 mL CL4 media in T-175 flask.
-
v.Add 600 μL of 50 mg/mL stock solution of G-418 antibiotic (1:50 dilution; 1 mg/mL final concentration).Note: This split ratio may need to be optimized depending on transfection efficiency. We recommend 2:10 dilution as a good starting point. Lower dilutions typically lead to more stringent selection and a more robust population of stable cells.Note: When selecting with G-418 antibiotic at a 1 mg/mL concentration, a bulk of the cell death will occur after this step. Only discard culture if there are no surviving (attached) cells. CL4 cells are robust and the culture can recover from a handful of surviving cells. See troubleshooting 1 if recovery is slow.
-
i.
-
d.Repeat step 2c and freeze cells in 10% DMSO, 30% FBS to make back-up stocks.Note: Longer selection may be necessary. Cells are ready to freeze when media is clear of floating dead cells.Optional: Perform fluorescence activated cell sorting (FACS) to enrich for a more uniform population of EGFP expressing cells, see troubleshooting 2.
-
a.
Generation of CL4 double stable cells by lentiviral transduction
-
3.Preparation of 293FT cells:
-
a.Start culture of 293FT cells.
-
i.Thaw vial of frozen cells (1mL) and immediately add to a T-75 flask containing 11 mL 293FT media.CRITICAL: It is recommended to use cells with low passage number (<15) for efficient virus production.
-
i.
-
b.Split cells 1–2× in preparation for transfection.
-
i.Rinse cells once with DPBS.
-
ii.Add 2 mL 0.05% trypsin, incubate at 37°C–1 min.
-
iii.Resuspend cells with 8 mL HEK-239FT media to make a 10 mL total suspension.
-
iv.Split cells 1:10 by adding 1 mL cells to 11 mL 239FT media in a T-75 flask.
-
i.
-
a.
-
4.Transfection of 293FT cells with lentiviral DNA.
-
a.Grow 293FT cells to 70%–80% confluency in a T-75 flask in 293FT medium.
-
b.Add 40 μL FuGENE 6–760 μL Opti-MEM and tap the tube several times to mix.
-
c.Incubate 5 min at RT.CRITICAL: Add FuGENE6 directly to the media as this reagent can adhere to plastic.
-
d.Add the following plasmids to the tube with FuGENE 6 and Opti-MEM:
-
i.6 μg plasmid of interest (e.g., pLVX-mCherry-ESPN)
-
ii.4 μg psPAX2 (Addgene #12260)
-
iii.0.8 μg pMD2.G (Addgene #12259)
-
iv.Tap tube to mix and incubate DNA:FuGENE 6 mixture 15–30 min at RT.
-
i.
-
e.Add the mixture drop wise to cells.
-
f.Change media with 12 mL 293FT media 16 h after transfection.
-
g.Incubate ∼24 h.
-
h.Harvest the virus by spinning media at 500 x g for 10 min at 4°C. At this point any 293FT cells in the media will be found in the pellet. Transfer 10.5 mL of supernatant to a 15 mL conical and add 3.5 mL Lenti-X concentrator (Clontech) (1:3 concentrator:virus).
-
i.Incubate at 4°C packed on ice for 16 h.Optional: Filter virus through a 0.45μm PES filter after the 500 x g spin. This further ensures exclusion of 293FT cells from viral supernatant. See troubleshooting 3.Optional: Add 7 mL 293FT medium to flask to harvest more virus the next day. Follow 3 e-h, except concentrate virus using 2.3 mL Lenti-X concentrator.
-
j.Spin the virus/Lenti-X concentrator mixture 1500 x g for 45 min.
-
k.Resuspend the pellet in 2 mL serum free DMEM.
-
a.
Pause Point: Use virus immediately or freeze in 250 μl aliquots at −80°C. Virus is generally stable for 6 months - 1 year.
-
5.Transduction of target cells (e.g., CL4 cells expressing EGFP-EPS8).
-
a.Grow target cells to 70%–80% confluency in a T-25 flask.
-
b.Add 250 μL virus and 10 μg/mL polybrene to cells.
-
c.The next day split target cells up to a T-75 flask.Note: Expansion of cells to a larger volume flask is performed to maintain the pool of transfected cells going into the second selection.
-
i.Rinse cells once with DPBS.
-
ii.Add 1 mL 0.05 % trypsin, incubate at 37°C–3 min.
-
iii.Add 4 mL media to the flask.
-
iv.Add the 5 mL total suspension to 7 mL media in a T-75 flask.
-
v.Maintain selection for EGFP-EPS8 with G-418 antibiotic by adding 240 μL G-418 (1 mg/mL final concentration).
-
i.
-
d.Once cells are 70%–80% confluent (∼1–2 days), expand into a T-175 flask.Note: Expansion of cells to a larger volume flask is performed to maintain the pool of transfected cells going into the second selection.
-
i.Rinse cells once with DPBS.
-
ii.Add 2 mL 0.05% trypsin, incubate at 37°C–3 min.
-
iii.Resuspend cells with 8 mL CL4 media.
-
iv.Add total suspension (10 mL) to 20 mL CL4 media in T-175 flask, for a total of 30 mL.
-
v.Add 30 μL of 10 mg/mL stock solution of puromycin antibiotic (1:1000 dilution, 10 μg/mL final concentration) to the flask. Add 1 mg/mL G-418 antibiotic.
-
i.
-
a.
Monitor cells and replace media every 1–3 days as needed, maintaining dual antibiotic selection. Cells are ready to freeze after ∼2–4 weeks, when the amount of floating dead cells has significantly decreased. See step 2d.
Note: Constructing a kill curve will facilitate selecting the optimal concentration of antibiotics. In our case, we select and maintain stable cells in 1 mg/mL G-418 and 10 ug/mL puromycin.
Optional: If expressing a fluorescent protein, monitor expression of probe by epifluorescence. Check for co-expression of the transfected/transduced probes. If it is difficult to find co-expressing cells, see troubleshooting 2.
Optional: Perform fluorescence activated cell sorting (FACS) to enrich for a population of EGFP and mCherry co-expressing cells.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| DMEM, high glucose | Corning | Cat#10-013-CV |
| FluoroBrite DMEM | Gibco | Cat#A18967-01 |
| Dulbecco’s Phosphate Buffered Saline (DPBS) | Corning | Cat#21-031-CV |
| Fetal Bovine Serum | R&D Systems | Cat#S11550 |
| 200 mM L-glutamine | Corning | Cat#25-005-cl |
| 0.05% trypsin | Gibco | Cat#25300054 |
| Opti-MEM | Gibco | Cat#31985-070 |
| FuGENE 6 | Promega | Cat# E2691 |
| Puromycin dihydrochloride | Sigma-Aldrich | Cat# P8833 |
| G418 sulfate | GoldBio | Cat# G-418-25 |
| Polybrene infection reagent | Sigma-Aldrich | Cat# TR-1003-G |
| Lenti-X concentrator | Clontech | Cat# 631231 |
| Experimental models: Cell lines | ||
| LLC-PK1-CL4 (CL4) | Gift from Dr. Carolyn Slayman (Yale University) | N/A |
| 293FT | Thermo Fisher Scientific | Cat# R70007 |
| Recombinant DNA | ||
| psPAX2 | Dr. Didier Trono | Addgene #12260 |
| pMD2.G envelope plasmid | Dr. Didier Trono | Addgene #12259 |
| EGFP-EPS8 | (Gaeta et al., 2021; Meenderink et al., 2019; Postema et al., 2018) | N/A |
| pLVX-mCherry-Espin | (Gaeta et al., 2021) | N/A |
| Software and algorithms | ||
| NIS Elements AR Analysis | Nikon Instruments | https://www.nikoninstruments.com/Products/Software |
| FIJI/ ImageJ | Open source software | http://imagej.net/Fiji/Downloads |
| Prism10 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Other | ||
| 35 mm glass bottom dishes | Cellvis | D35-20-1.5-N |
| 0.22 um filter | GenClone | Cat#25-240 |
| 10 mL syringe | BD | Cat#305482 |
| T-25 flask | Greiner Bio-One | Cat# 690175 |
| T-75 flask | Greiner Bio-One | Cat# 658175 |
| T-175 flask | Greiner Bio-One | Cat# 661175 |
| Basic Plasma Cleaner PDC-32G | Harrick Scientific Products | Cat# PDC-32G |
| Nikon Ti2-E | Nikon | MEA54000 |
| CSU-X1 Confocal Scanner Unit | Yokogawa | 99582 |
| Prime 95B sCMOS | Teledyne Photometrics | N/A |
| OrCA-Fusion BT Digital CMOS camera | Hamamatsu | Cat#C11440-36U |
| 488, 561,647 LASERS | Nikon | N/A |
| STX Series Stage Top Incubator System | Tokai-Hit | Cat#TOKAI-HIT-STXG |
Materials and equipment
| CL4 / 293FT medium | ||
|---|---|---|
| Reagent | Final concentration | Amount |
| DMEM | n/a | 500 mL |
| Fetal bovine serum | 10% | 50 mL |
| L-glutamine | 1% | 5.5 mL |
| Total | n/a | 555.5 mL |
Store CL4/293FT medium at 4°C for 4–6 weeks. Prewarm media to 37°C before use.
Imaging medium
| Reagent | Final concentration | Amount |
|---|---|---|
| FluoroBrite DMEM | n/a | 500 mL |
| Fetal bovine serum | 10% | 50 mL |
| L-glutamine | 1% | 5.5 mL |
| Total | n/a | 555.5 mL |
Store complete FluoroBrite DMEM at 4°C for 4–6 weeks. Prewarm media to 37°C before use.
| Reagent | Final concentration | Amount |
|---|---|---|
| G-418 | 50 mg/mL | 500 mg |
| MiliQ H20 | n/a | 10 mL |
| Total | n/a | 10 mL |
Filter sterilize using a 10 mL syringe equipped with 0.22 μm filter, store at −20°C for up to one year.
| Reagent | Final concentration | Amount |
|---|---|---|
| Puromycin dihydrochloride | 10 mg/mL | 25 mg |
| MiliQ H20 | n/a | 2.5mL |
| Total | n/a | 2.5 mL |
Filter sterilize using a 10 mL syringe equipped with 0.22 μm filter, aliquot and store at −20°C for up to two years. Keep working stock at 4°C for up to one year.
Step-by-step method details
Plating CL4 cells for imaging
-
1.When stable cells reach 70–80% confluency, split cells from T-75 flask.
-
a.Rinse cells once in DPBS.
-
b.Add 2 mL 0.05% trypsin. Incubate cells at 37°C, ∼3 min.
-
c.Resuspend cells with 8 mL CL4 media.
-
d.In a 35 mm glass bottom dish (CellVis), add 300 μL (∼2 × 105 cells) to 1.7 mL CL4 media with appropriate selection antibiotics.
-
e.Incubate at 37°C for 16h and use for imaging once cells grow to ∼50–80% confluency.
-
a.
Optional: Use a Plasma Cleaner to prepare the surface of 35 mm glass bottom dishes before plating to ensure efficient cell spreading and adherence. See manufacturer’s guide for instructions. It is not necessary to coat with fibronectin or laminin unless cell lifting during acquisition becomes an issue. See troubleshooting 4.
Figure 3.

Example of optimal cell density for capturing microvillus growth events
Cells are growing in sub-confluent islands.
Optional: Plate multiple glass bottom dishes of cells at different dilutions to ensure that the optimal density for imaging is covered. See troubleshooting 5.
Spinning disk confocal imaging
We perform spinning disk confocal imaging using a Nikon Ti2 inverted light microscope equipped with a Yokogawa CSU-X1 spinning disk head, a Photometrics Prime95B sCMOS or Hamamatsu Fusion BT sCMOS camera, and 488, 561 and 647 nm excitation LASERS as described previously (Gaeta et al., 2021; Meenderink et al., 2019). Samples are loaded into a heated stage top incubator system maintained at 5% CO2 (Tokai Hit) and then imaged using a 100× oil 1.49 NA TIRF objective (Nikon instruments).
Before loading samples into the microincubation chamber, confirm with transmitted light microscopy that cells are present in sub-confluent islands or recently reached confluency (Figure 3). Cells that have been confluent for > 1 day will no longer be suitable for observation of nascent microvilli growth events.
-
2.
Load a glass bottom dish into the microincubation chamber maintained at 37°C with 5% CO2 and humidity.
Optional: We typically use CL4 medium with phenol red (DMEM + 10% FBS, + 1% L-glutamine) for imaging. However, Fluorobrite (Gibco) supplemented with 10% FBS and 1% L-glutamine may also be used to decrease background fluorescence.
-
3.Using a 100×/1.49 NA oil immersion objective, find a cell expressing your protein(s) of interest.
-
a.Determine whether the cell is suitable for observing microvillus growth events. Suitable cells typically satisfy the following conditions (Figure 4):
-
i.Protein(s) of interest are expressed at moderate levels. This is defined as high enough expression to produce a SNR adequate for imaging, but low enough to avoid aberrant localization (relative to that observed with endogenous staining) or phenotypes. A microvilli signal ∼1.2–2× the cytoplasmic signal is a good starting point.
-
ii.Microvilli are sparse enough that growth of new protrusions will not be obscured by existing protrusions.
-
iii.A majority of the apical surface volume can be imaged in a 3–5 μm z-depth imaging volume (Figure 5A).
-
i.
-
b.Design acquisition routine in Nikon Elements “ND Acquisition” window (Figure 5).
-
i.Configure lambda (channel series) settings (Figure 5B arrow 1). Select the excitation/filter set of interest. In our case we will choose FITC X1 (488) and TRITC X1 (561).
-
ii.Configure time interval and duration (Figure 5B arrow 2). Standard time interval is 30 s for 30 min duration. This can be adjusted based on experimental needs.
-
iii.Set laser power. For our system, we typically set the laser power to no more than 4% and exposure time to no more than 100 ms to minimize photobleaching. However, the laser power setting may vary depending on the system and total initial power of the LASERs used for excitation. The goal is to achieve a microvilli signal at least 1.2× the cytoplasmic signal using the lowest possible laser power and exposure time settings. We recommend keeping laser power and exposure time settings consistent between cells within an experiment if quantitative intensity measurements will be collected.
-
iv.Set the z-step size (Figure 5C arrow 3). Nikon Elements will suggest a step size to achieve Nyquist sampling (Figure 5C, magenta box). However, to maximize the performance of deconvolution in postprocessing, we typically over-sample in Z; using a 100×/1.49NA objective the step size is set to 0.18 μm or less (Figure 5A).
- v.
-
vi.Set order of experiment to Lambda(Z series) (Figure 5B, arrow 5). This allows for acquisition of the total imaging volume per channel before switching channels.
-
vii.Press “1 time loop” (Figure 5B arrow 6)
-
viii.Scroll through imaging volume to see if a majority of the apical surface volume is captured.
-
ix.Proceed with imaging if a majority of the apical surface volume is captured. If not, adjust the middle focal plane and press “1 time loop”. If a majority of the apical surface volume is still not captured, repeat step 3.
-
i.
-
a.
- 4.
Note: Finding “optimal” imaging conditions requires patience and balancing many factors. For example, a cell may present with sparse microvilli and protein(s) of interest expressed at reasonable levels, however a 5 μm z-depth imaging volume may be insufficient to capture the bulk of the apical surface.
Note: For tips on minimizing cell death during the acquisition, see troubleshooting 4.
Figure 4.

Example of a CL4 cell expressing EGFP-EPS8 (green) and mCherry-ESPN (magenta)
This cell satisfies conditions outlined in Step-by-step details 3a. Most notably, microvilli are sparse enough to observe nascent microvillus growth events and the mCherry-ESPN signal is sufficiently high over background. Scale bar = 10 μm. Zoom inset scale bar = 2 μm.
Figure 5.

Optical sectioning of CL4 cells over time to capture apical surface dynamics
(A) Representative schematic of oversampling in the z dimension to enable deconvolution.
(B) Stepwise acquisition settings.
Expected outcomes
Creation of stable cell lines containing an actin marker such as mCherry-ESPN or mCherry-LifeAct allows for the visualization of individualized protrusions (Figure 1). In our case, co-expression of the actin binding protein EGFP-EPS8 with mCherry-ESPN should result in a punctate localization pattern of EPS8, with EPS8 marking the distal tip of each microvillus (Figure 4).
Quantification and statistical analysis
Acquisition of multi-channel time-lapse datasets in the manner outlined above enables the direct visualization of microvillus growth dynamics (Gaeta et al., 2021; Meenderink et al., 2019). These data offer access to a range of quantitative parameters that provide information not only on the relative concentration of a protein of interest, but also the timing of its accumulation/loss relative to other factors during protrusion growth. Below we outline steps for: performing intensity measurements of EPS8 puncta, analyzing the timing of EPS8 recruitment during microvillus growth, and measuring microvillus growth rate. For other potential measurements including quantification of microvillus length, velocity, and mean squared displacement, see (Meenderink et al., 2019).
Image deconvolution
Before performing quantitative analysis, we typically deconvolve image volumes (Figure 6). Deconvolution is performed to computationally increase image resolution, which in turn improves SNR.
-
1.
Set up the deconvolution in Nikon Elements, Deconvolution→ 3D deconvolution (Figure 6). Parameters and deconvolution type will be empirically determined. For our data, we typically apply “Richardson-Lucy” deconvolution on the “medium” noise setting with 20 iterations in Nikon Elements Software (Figure 6 arrows 1–3). After deconvolution, automated segmentation and binary tracking is performed using Nikon Elements software, or manual analysis of microvillus growth is performed in FIJI, outlined below.
Figure 6.

Stepwise 3D deconvolution setup
Quantification of the distribution of EPS8 puncta intensities
-
2.
First, using Nikon Elements create a maximum intensity projection from the deconvolved image through Image → ND processing → Create maximum intensity projection in Z. Save the file. (Figure 7A). The image does not have to be background subtracted prior to segmentation.
-
3.
Next, segment puncta using “Spot Segmentation” in Nikon Elements. Navigate to Binaries → Spot Detection → Bright Spots (Figure 7B). Select the relevant channel and adjust the “Typical Diameter” and “Contrast” levels (Figure 7B arrows 1–2). If spots are of uniform diameter, set the diameter equal for all cells. Contrast levels, which impact the threshold of spot detection, may be adjusted between cells to account for differences in expression levels. If adjusting contrast levels between cells, it is advised to set levels so that the threshold of detection is approximately the same. For example, roughly the same number of “dim” spots are detected between cells, so that results are not biased towards dimmer or brighter puncta. For EGFP-EPS8, we used 0.45 μm diameter size and ∼3.64 contrast. Click “OK”.
Note: these settings will change depending on the experiment and region tracked.
Note: background subtraction is not necessary.
-
4.
Navigate to Binaries→Track Binaries (Figure 7C). Change settings as desired to optimize tracks. Click “Track”.
Note: Tracking of binaries is sensitive to SNR, object density, and other parameters and therefore may need to be adjusted on a case by case basis.
-
5.
At this point a spreadsheet with several output parameters will be generated. Copy mean intensity data from the first frame of the tracks into Prism 10 (GraphPad) using the “column” format to create a histogram.
-
6.
Navigate to Insert → New Analysis → Frequency distribution. Under “create” select “Frequency distribution” (Figure 7D arrow 1). Under “tabulate” select “Relative frequency (fractions)” (Figure 7D arrow 2).
-
7.
Adjust bin range and bin width as appropriate for data. Graph data (Figure 7E).
Figure 7.

Performing spot detection and tracking binaries
(A) Progression of a deconvolved z stack to a maximum intensity projection. The maximum intensity projected image is then used for spot detection.
(B) Spot detection setup.
(C) Track Binaries setup.
(D) Frequency distribution setup in Prism 10.
(E) Example of EGFP-EPS8 puncta intensity histogram. n = number of puncta from a single representative cell (reprinted with permission from Gaeta et al., 2021).
Quantification of the timing of EPS8 recruitment during microvillus growth
Note: This protocol outlines 2D analysis, performed on maximum intensity projected images.
-
8.
In FIJI, load images as hyperstacks.
-
9.Scroll through time-lapse time to find de novo microvillus growth events. De novo events are defined as microvillus growth events which occur independently of existing protrusions (Gaeta et al., 2021).
-
a.If analyzing cells expressing two or more fluorescent proteins, decide which channel serves as the “fiduciary point”. To understand the temporal sequence of events, it is recommended to begin analysis ∼3 frames before the appearance of the fiduciary signal, however this may be adjusted depending on the experimental goal. In our case, we used EGFP-EPS8 as a fiduciary point due to its punctate localization.
-
a.
-
10.
Use the oval tool to draw an ROI around the tip of the microvillus to track the EGFP-EPS8 signal (Figure 8A arrow 1, 8B). (Alternatively, Linear ROIs or other shapes can be used depending on experimental needs).
-
11.
Save the ROI by opening the ROI manager: Analyze→ Tools→ ROI manager. Record an ROI for each relevant frame of the movie using Add [t] (Figure 8C, arrow 1).
-
12.
Set measurements to choose parameters to measure: Analyze → Set Measurements. To quantify microvillus growth events, we produce intensity vs time plots. In this case select “mean gray value” in the Set Measurements window.
-
13.
Highlight all relevant ROIs in the manager (shift + click) and ensure you are measuring the correct channel (EGFP-EPS8) by noting the position of the channel slider. Press measure (m), or Analyze → Measure. (Figure 8C, arrow 2). A results window will appear (Figure 8D).
-
14.
Repeat ROIs for the second channel if applicable (in our case mCherry-ESPN), using the same circular ROI area. Track the distal end of the microvillus.
-
15.
Save measurements: Results→ File → Save.
-
16.
Copy intensity data into Prism10 using the XY data table. Input time on the “X” column and intensity data in the “Group X” columns.
-
17.
Normalize data. Insert → new analysis → normalize. Define 0% as “smallest mean in each data set” and 100% as “largest mean in data set”. Present results as “fractions”.
-
18.
Perform row stats on normalized data. New analysis→ Row means with SD or SEM. Select Row means with SD.
-
19.
Plot graph (Figure 8E).
Figure 8.

Region tracking of microvillus growth events in FIJI
(A) FIJI user interface with an arrow pointing to oval selection.
(B) Example of manual region tracking of EGFP-EPS8 puncta. Scale bar = 1 μm.
(C) ROI manager interface with arrows pointing to the add and measure selections.
(D) Example of a results table displaying the ROI area, mean, standard deviation, channel and frame.
(E) Normalized intensity vs time plot of EGFP-EPS8 and mCherry-ESPN. N= 14 events from 7 cells (reprinted with permission from Gaeta et al., 2021).
(F) Example montage of a microvillus growth event in a cell expressing mCherry-ESPN. Box width = 4μm. Linitial and Lfinal measurements are overlaid (reprinted with permission from Gaeta et al., 2021).
(G) Quantification of microvillus growth rate. Using ANOVA with Kruskal-Wallis, ∗∗∗p=0.0004 compared to ESPN only control. Bracketed asterisks use ANOVA with Kruskal-Wallis compared to EGFP-EPS8 ∗∗∗p=0.0045. ESPN n = 19 growth events, IRTKS n = 19 growth events, EPS8 n = 23 growth events, LNK:AAA n = 19 growth events, V690D L694D n = 16 growth events. Error bars indicate mean ± SD. Each data point represents a single de novo microvillus growth event. (E–G reprinted with permission from Gaeta et al., 2021).
Quantification of microvillus growth rate
Note: This protocol outlines 2D analysis, performed on maximum intensity projected images.
-
20.
Scroll through time lapse to find de novo microvillus growth events.
-
21.
Using the line tool in FIJI, measure the length of the first frame of the detectable coalesced mCherry-ESPN signal. This is the initial length (Linitial) measurement. Measure the timepoint, this will be the initial time (Tinitial).
-
22.
Next, measure the length of the microvillus at the final frame of the time lapse. This will be the final length (Lfinal) measurement. If the microvillus is perpendicular to the cell surface, measure the last frame with the microvillus mostly parallel to the cell surface. Exclude events where microvilli are only perpendicular to the cell surface. Measure the timepoint, this will be the final time (Tfinal).
-
23.
Calculate the growth rate using the equation (Lfinal − Linitial)/ (Tfinal − Tinitial). Plot the growth rates, with each point representing the growth rate from a single event.
-
24.
Repeat measurements with different protein of interest constructs to allow for quantitative comparisons. For example, in our case, we compared CL4 cells expressing EPSN only to ESPN + WT EPS8, ESPN+EPS8 LNK758-760AAA and ESPN + EPS8 V690D L694D using ANOVA with Kruskal Wallis (Figure 8G, (Gaeta et al., 2021)).
Note: As this analysis is performed on projected 3D data, quantifying events where the microvillus in Lfinal is mostly parallel to the cell surface will ensure the most accurate length measurements.
Limitations
Because this protocol outlines an overexpression approach, we advise confirming that overexpressed proteins of interest exhibit localization similar to endogenous factors, as assessed by standard immunofluorescence staining. We also advise confirming that overexpression does not lead to abnormal cellular morphology. Endogenously tagged cell lines may be substituted in place of overexpression lines.
Troubleshooting
Problem 1
Stable cell lines are slow to recover during antibiotic selection (before you begin: steps 2b v and 5d v).
Potential solutions
If cells are recovering slowly in a large volume flask (T-175), consider splitting total cell volume down to a smaller flask size (T-25 or T-75). Concentrating the cells in this manner should allow them to recover more quickly. Additionally, transfection efficiency may be increased by adjusting the ratio of DNA:FuGENE 6 or performing transfection using Lipofectamine2000.
Problem 2
Finding cells co-expressing constructs of interest is difficult, even after double stable selection (before you begin: step 5d v).
Potential solution
After selection of the first expression construct, sort cells using FACS. This will ensure that all cells transfected or transduced with the second expression construct will co-express the first construct.
Problem 3
293FT cell contamination in cells transduced with lentivirus (before you begin: step 4h).
Potential solution
It is unlikely but possible that 293FT cells are carried over after pelleting cells during viral harvest if the supernatant is not handled carefully. To avoid this, we recommended filtering the virus using a 0.45 μm PES filter after the initial cell pelleting.
Problem 4
Significant phototoxicity and/or cell death during acquisition (Methods video S1; Step by step method details: step 4).
Scale Bar = 10 μm. See troubleshooting 4
Potential solution
It is not uncommon for cells to exhibit phototoxicity or die during time lapse volume acquisition. To minimize this potential, decrease the laser power and/or exposure time while trying to maintain sufficient SNR. Additionally, 35 mm glass bottom dishes may be pretreated with 50 μg/mL laminin for 45 min prior to plating, to decrease the chance of cell lifting. Minimizing the acquisition volume will also help limit phototoxicity. Limit the number of z-slices to the absolute minimum required to capture apical features of interest; in practice we try not exceed 5 μm in z-depth. Note however, that under sampling in z will decrease the benefits of image deconvolution.
Problem 5
Microvilli are too dense to capture individual growth events (Step by step methods details step 3a).
Potential solution
Make sure cells are in subconfluent islands (Figure 3) at the time of imaging. Try plating cells over a range of different densities; this will help ensure that a subset of samples will be within an acceptable range on the day of imaging.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Matthew J. Tyska (matthew.tyska@vanderbilt.edu).
Materials availability
Plasmids and cell lines generated during these studies will be made available upon request directed to the Lead Contact.
Acknowledgments
This work was supported by the Vanderbilt Cellular, Biochemical and Molecular Sciences Training Grant 5T32GM008554-25 (I.M.G.); the NIH NIDDK National Research Service Award F31DK122692 (I.M.G.); NIH grant T32-A1007474 (L.M.M.); Department of Veterans Affairs Career Development Award 1IK2-BX004885 (L.M.M); and NIH grants R01-DK125546, R01-DK111949, and R01-DK095811 (M.J.T.). Figures were partially created using BioRender.com
Author contributions
I.M.G. wrote the manuscript, prepared the figures, and devised methodology. L.M.M. prepared figures and devised methodology. M.J.T. supervised the study. All authors contributed to editing the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2021.100998.
Contributor Information
Isabella M. Gaeta, Email: isabella.m.gaeta@vanderbilt.edu.
Matthew J. Tyska, Email: matthew.tyska@vanderbilt.edu.
Data and code availability
No large-scale datasets or new codes were generated in this study.
References
- Baas A.F., Kuipers J., van der Wel N.N., Batlle E., Koerten H.K., Peters P.J., Clevers H.C. Complete polarization of single intestinal epithelial cells upon activation of LKB1 by STRAD. Cell. 2004;116:457–466. doi: 10.1016/s0092-8674(04)00114-x. [DOI] [PubMed] [Google Scholar]
- Elia N., Lippincott-Schwartz J. Culturing MDCK cells in three dimensions for analyzing intracellular dynamics. Curr. Protoc. Cell Biol. 2009;Chapter 4:4–22. doi: 10.1002/0471143030.cb0422s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaeta I.M., Meenderink L.M., Postema M.M., Cencer C.S., Tyska M.J. Direct visualization of epithelial microvilli biogenesis. Curr. Biol. 2021;31:2561–2575.e2566. doi: 10.1016/j.cub.2021.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meenderink L.M., Gaeta I.M., Postema M.M., Cencer C.S., Chinowsky C.R., Krystofiak E.S., Millis B.A., Tyska M.J. Actin dynamics drive microvillar motility and clustering during brush border assembly. Dev. Cell. 2019;50:545–556.e544. doi: 10.1016/j.devcel.2019.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melak M., Plessner M., Grosse R. Actin visualization at a glance. J. Cell Sci. 2017;130:525–530. doi: 10.1242/jcs.189068. [DOI] [PubMed] [Google Scholar]
- Peterson M.D., Mooseker M.S. Characterization of the enterocyte-like brush border cytoskeleton of the C2BBe clones of the human intestinal cell line, Caco-2. J. Cell Sci. 1992;102:581–600. doi: 10.1242/jcs.102.3.581. [DOI] [PubMed] [Google Scholar]
- Postema M.M., Grega-Larson N.E., Neininger A.C., Tyska M.J. IRTKS (BAIAP2L1) elongates epithelial microvilli using EPS8-dependent and independent mechanisms. Curr. Biol. 2018;28:2876–2888.e2874. doi: 10.1016/j.cub.2018.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyska M.J., Mooseker M.S. MYO1A (brush border myosin I) dynamics in the brush border of LLC-PK1-CL4 Cells. Biophysical J. 2002;82:1869–1883. doi: 10.1016/S0006-3495(02)75537-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Scale Bar = 10 μm. See troubleshooting 4
Data Availability Statement
No large-scale datasets or new codes were generated in this study.