

Summary
Patient-derived organoids (PDOs) have shown the potential to reflect patient sensitivity to chemotherapeutic or targeted drugs. Recently, we showed that organoid models can also serve as a platform to screen for selectivity and potency of oncolytic adenoviruses (OAds). In this protocol, we describe the steps for tumor organoid adenoviral infection and functional assessment of patient-specific responses to OAds. We provide methods to determine OAd relative efficacy by evaluation of PDO viability after infection and adenoviral replication within cancer cells.
For complete details on the use and execution of this protocol, please refer to Raimondi et al. (2020).
Subject areas: Biotechnology and bioengineering, Cancer, Health Sciences, Organoids
Graphical abstract
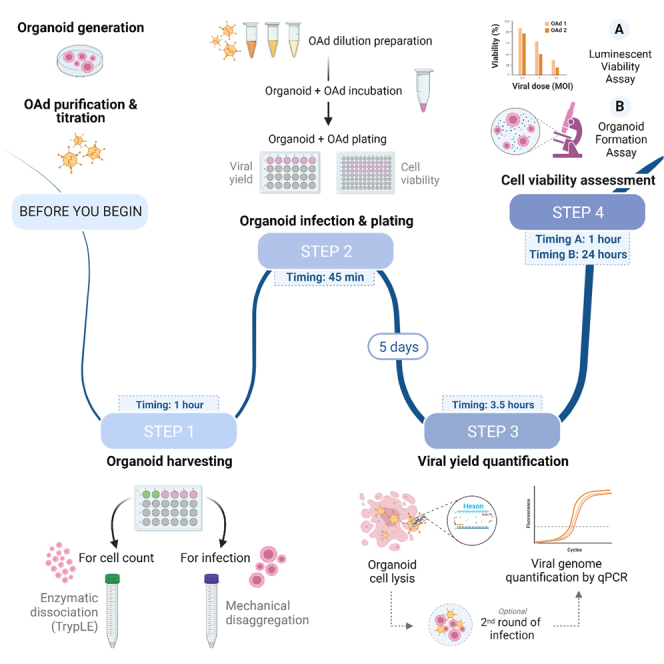
Highlights
-
•
Protocol for infection of organoids with oncolytic adenoviruses
-
•
Steps to quantify adenoviral replication within organoids
-
•
Evaluation of viability after infection to determine relative oncolytic efficacy
Patient-derived organoids (PDOs) have shown the potential to reflect patient sensitivity to chemotherapeutic or targeted drugs. Recently, we showed that organoid models can also serve as a platform to screen for selectivity and potency of oncolytic adenoviruses (OAds). In this protocol, we describe the steps for tumor organoid adenoviral infection and functional assessment of patient-specific responses to OAds. We provide methods to determine OAd relative efficacy by evaluation of PDO viability after infection and adenoviral replication within cancer cells.
Before you begin
For the completion of this protocol, availability of both organoids (PDOs) and oncolytic adenoviruses (OAd) is assumed.
Organoid generation
Timing: 2–6 weeks
-
1.
Generate organoids from tumor tissue following standard protocols. Detailed instructions for the establishment of PDAC organoids can be found in Driehuis et al. (2020).
-
2.
For culture maintenance, grow organoids in 80% Matrigel (diluted in F12+++ medium) and replace organoid growth medium every 3–4 days.
Note: Matrigel lots with a protein concentration > 9 mg/mL are preferred to ensure organoid 3D morphology. Therefore, the final protein concentration in drops containing organoids is 7.0–7.5 mg/mL, both in culture maintenance and in organoid seeding after infection.
-
3.
Split organoids 6–7 days before organoid infection to ensure > 70% confluency at the day of the experiment, saving 2 of the seeded wells for cell count. Change organoid growth medium every 2–3 days.
CRITICAL: To maximize reproducibility of results, organoids should be passaged at a constant split ratio. Each organoid line may require different split ratios depending on its growth rate (for PDAC organoids, split ratios usually range between 1:3–1:6 every 7–10 days). PDOs should ideally have a similar size and confluence at the time of seeding and infection to compare outcomes of different experiments.
Note: Once established, we usually maintain organoid cultures by seeding a drop of 50 μL of Matrigel 80% (10 μL F12+++ + 40 μL Matrigel) containing organoids in each well of a 24-well plate and supplementing with 450 μL of organoid growth medium. Therefore, approximate numbers given in this protocol refer to cultures grown in this format, even though other organoid handling techniques may also be valid.
Generation and titration of oncolytic adenoviruses
-
4.
Oncolytic adenoviruses (OAds) should be available before starting this protocol. The production and titration of OAds used here were performed in-house as described in Rovira-Rigau et al. (2019).
Note: It is advisable to titer OAds again before use if they have been stored for more than 12 months.
Optional: Reporter genes (e.g., EGFP) can be additionally cloned in the OAd genome to easily follow infection and viral spreading within organoids (Figure 1). Alternatively, for non-tagged OAds, viral genomes can be quantified by qPCR (Figure 2). For detailed instructions on viral genome quantification from infected PDOs, see section ‘step-by-step method details’.
Figure 1.

Time-course of PDO infection with EGFP-OAd
Organoids infected with an EGFP-containing OAd (2 MOI) were imaged every 24 h after infection in an inverted fluorescence microscope. Viral replication and spreading within organoids can be confirmed visually by an increase in EGFP expression with time. Scale bar, 100 μm.
Figure 2.

Quantification of viral genomes by qPCR to confirm OAd replication in PDOs
PDOs were infected with 2 different OAds (OAd1 and OAd2) at 2 MOI, subjected to freeze-thaw cycles and to DNA extraction (passage 1, P1). Fifteen μL of lysate-derived supernatants were used to infect fresh PDOs, and the same process was repeated to quantify viral genomes in organoids (passage 2, P2). Adenoviral genomes were quantified by qPCR amplification of hexon genes using a standard curve consisting of DNA dilutions of known copy numbers (102–107). High viral yields were detected in P1 and P2 with both viruses, proving that the OAd effectively replicates within PDOs. Data are represented as mean ± SEM (n = 3).
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Human pancreatic ductal adenocarcinoma tissue (surgical resection specimens, fine-needle biopsies) | Hospital Clínic (Barcelona) | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| A 83-01 | Tocris Bioscience (Bio-Techne) | Cat#2939 |
| Advanced DMEM/F-12 | Gibco | Cat#12634010 |
| B-27 supplement (50x), serum-free | Gibco (Thermo Fisher Scientific) | Cat#17504044 |
| Bovine Serum Albumin | Sigma-Aldrich | Cat#A3294; CAS: 9048-46-8 |
| Gastrin I, human (hGastrin I) | Tocris Bioscience (Bio-Techne) | Cat#3006 |
| GlutaMAX™ Supplement | Gibco | Cat#35050038 |
| HEPES solution (1M, pH 7.0–7.6) | Sigma-Aldrich | Cat#H0887; CAS: 7365-45-9 |
| Human EGF Recombinant Protein (hEGF) | Gibco | Cat#PHG0311 |
| Matrigel Basement Membrane Matrix, Phenol Red-free | Corning | Cat#356231 |
| N-Acetylcysteine | Sigma-Aldrich | Cat#A9615; CAS: 616-91-1 |
| Nicotinamide | Sigma-Aldrich | Cat#N0636; CAS: 98-92-0 |
| Penicillin/Streptomycin (P/S) | Life Technologies | Cat#15140122 |
| Phosphate Buffer Saline (PBS) (10X), pH 7.2 | Gibco | Cat#70013065 |
| Primocin | InvivoGen | Cat#ANT-PM-1 |
| Critical commercial assays | ||
| Blood DNA Isolation Mini Kit | Norgen Biotek | Cat#46380 |
| CellTiter-Glo® 3D Cell Viability Assay | Promega | Cat#G9681 |
| LightCycler® 480 SYBR Green I Master (for qPCR) | Roche | Cat#04707516001 |
| Oligonucleotides | ||
| Hexon Forward (Fw): GCCGCAGTGGTCTTACATGCACATC | (Villanueva et al., 2017) | N/A |
| Hexon Reverse (Rv): CAGCACGCCGCGGATGTCAAAG | (Villanueva et al., 2017) | N/A |
| Software and algorithms | ||
| Excel | Microsoft Office | RRID:SCR_016137 |
| Prism 8 | GraphPad | RRID:SCR_002798 |
| Other | ||
| BackSeal-96/384, White Adhesive Bottom Seal for 96-well and 384-well Microplate | PerkinElmer | Cat#6005199 |
| CulturPlate-96, White Opaque 96-well Microplate, Sterile and Tissue Culture Treated | PerkinElmer | Cat#6005680 |
| Glass Pasteur pipettes (pre-narrowed in the flame) | Deltalab | Cat#702 |
| MicroAmpTM Optical 384-Well Reaction Plate with Barcode | Applied Biosystems | Cat#4309849 |
| MicroampTM Optical Adhesive Film | Applied Biosystems | Cat#4311971 |
| Nanodrop 1000 | Thermo Scientific | RRID:SCR_016517 |
| Olympus IX51 Inverted Microscope | Olympus | N/A |
| SynergyTM HT Multi-mode Microplate Reader | BioTek | RRID:SCR_020536 |
| ViewPlate-96, White 96-well Microplate with Clear Bottom, Sterile and Tissue Culture Treated, Lid Included | PerkinElmer | Cat#6005181 |
| ViiA 7 Real-Time PCR System | Applied Biosystems | N/A |
Materials and equipment
Alternatives: To perform CellTiter-Glo® 3D cell viability assay, white-walled, clear-bottom plates can also be used to facilitate visual inspection of infected PDOs. However, we recommend the use of white-bottom plates to minimize signal crosstalk between wells.
Basal medium, with (F12+++B) or without (F12+++) BSA
| Reagent | Final concentration | Amount |
|---|---|---|
| Advanced DMEM/F-12 | N/A | 500 mL |
| HEPES (1 M) | 10 mM | 5 mL |
| Pen/Strep | 1% | 5 mL |
| GlutaMAX | 2 mM (1%) | 5 mL |
| BSA 30% (for F12+++B) | 0.1% | 1.72 mL |
| Total | n/a | 516.72 mL |
Once prepared, store at 4°C
30% BSA solution
| Reagent | Final concentration | Amount |
|---|---|---|
| BSA | 30% | 6 g |
| PBS 1X (sterile) | N/A | Up to 20 mL |
Once prepared, filter the solution through a 0.22 μm filter and store in 1.8 mL aliquots at −20°C
Pancreatic tumor organoid growth medium
| Reagent | Final concentration | Amount |
|---|---|---|
| F12+++ | N/A | 14 mL |
| Wnt3a-conditioned medium∗ | 50% | 25 mL |
| R-spondin-1-conditioned medium∗ | 10% | 5 mL |
| mNoggin-conditioned medium∗ | 10% | 5 mL |
| B27 (50×) | 1× | 1 mL |
| Nicotinamide (1 M) | 10 mM | 500 μL |
| N-Acetylcysteine (0.5 M) | 1.25 mM | 125 μL |
| hEGF (200 μg/mL) | 50 ng/mL | 12.5 μL |
| hFGF-10 (100 μg/mL) | 100 ng/mL | 50 μL |
| hGastrin I (100 μM) | 10 nM | 5 μL |
| A83-01 (500 μM) | 500 nM | 50 μL |
| Primocin (50 mg/mL) | 100 μg/mL | 100 μL |
| Total | N/A | 50 mL |
Once prepared, store 10 mL aliquots at −20°C for up to 3 months.
∗For production and activity testing of conditioned media, see Farin et al. (2012) and Boj et al. (2017).
Step-by-step method details
In this protocol, we first detail quantification of OAd genomes as a readout of viral replication inside PDOs 5 days after infection. This is a step that can be performed to prove replication capabilities of designed OAds when fluorescent tags are not incorporated in the viral genome. To compare OAd infectivity and cytotoxicity among different PDOs, we explain two complementary approaches to assess PDO cell viability after viral infection: the CellTiter-Glo® 3D Cell Viability Assay and an organoid formation assay. To perform the organoid formation assay, a sublethal dose of OAd is needed to allow for formation of enough organoids to be counted, so previous cell viability experiments may be needed to establish optimal experimental conditions.
Organoid harvesting for infection
Either to evaluate viral replication or effects on cell viability, organoids are infected as whole entities, since in our experience, when infected as single cells, they have significant problems to recover. Even though variability between biological replicates can be an issue, this can be minimized by performing a cell count using 2 additional wells (see ‘before you begin’) to estimate the number of cells to seed. Prior to harvesting, it is very important to check organoids under the microscope and confirm similar confluency between wells used for infection and seeding. At this point, organoid diameter should not exceed 200 μm, as bigger sizes can lead to higher variability (troubleshooting 1). The density to be expected in organoid cultures harvested is usually of about 50,000–100,000 cells/well.
Note: Organoid harvesting and infection times have been calculated for 1 PDO line infected with 2 doses of 2 different OAds to be compared.
-
1.
Thaw Matrigel aliquots needed on ice.
Note: For a 500-μL aliquot to be thawed, 30–45 min are needed, so by the end of the infection protocol Matrigel should already be a pipettable liquid. Calculate the volume of Matrigel needed to seed the experiment to minimize freeze-thaw cycles. See Table 1 for orientation on how much Matrigel will be needed.
Table 1.
Proportion of PDOs, OAds, and Matrigel in 96- and 24-well plates
| Format | Component | Amount (1 well) | Amount per OAd dose (4 wells) | ||
|---|---|---|---|---|---|
| 96-well plate | PDOs | 5000 cells | 2 μL | 20000 cells | 8 μL |
| OAd | MOI ⋅ DF/titer | (MOI ⋅ DF/titer) ⋅ 4 | |||
| Matrigel | 8 μL | 32 μL | |||
| 24-well plate | PDOs | 25,000 cells | 10 μL | 100,000 cells | 40 μL |
| OAd | MOI ⋅ DF/titer | (MOI DF/titer) ⋅ 4 | |||
| Matrigel | 40 μL | 200 μL | |||
DF = dilution factor. Numbers are given for 1 dose of 1 OAd to infect 1 PDO line.
-
2.
Pre-cool a centrifuge to 4°C.
-
3.
Prepare 15 mL-tubes with 10 mL of cold basal medium and keep them on ice.
Note: Due to Matrigel density, a maximum of 3 wells of a 24-well plate (containing 150,000–300,000 cells) can be harvested in a single tube. If more organoids need to be collected, split wells into different tubes to perform the first centrifugation step.
-
4.
Take an aliquot of basal medium to prepare OAd dilutions. This can be kept at room temperature (20°C–25°C) inside the cell culture hood during organoid processing until use.
-
5.
Harvest organoids.
Tube 1: 2 wells for cell count
Tubes 2-X: 2–3 wells per tube for organoid infection-
a.Remove organoid growth medium from the wells without touching the Matrigel dome.
-
b.Add 500 μL of basal medium from the corresponding tube to each well to disaggregate the Matrigel dome with the help of a P1000 pipette, and collect the content into the tube.Note: Either F12+++ or F12+++B basal media can be used for this step. BSA present in F12+++B helps prevent attachment of organoids to the walls of the tube, which can lead to loss of cells during the process.
-
c.Repeat step 5b 2–3 times until removing most of Matrigel remainders inside the wells.
-
d.Centrifuge at 100g 5 min at 4°C.
-
e.Remove supernatant carefully, trying to remove as much Matrigel as possible but without touching the pellet.Note: If organoids are not cleanly pelleted under a Matrigel layer, remove medium leaving the Matrigel and add 10 mL of fresh basal medium to repeat centrifugation in step 5d at higher speed (400g, 5 min, 4°C).
-
a.
-
6.Disaggregate organoids.Note: Organoids that will be used for cell count (Tube 1) are dissociated to single cells using TrypLE, while those used for infection with OAd (Tube 2) are mechanically disaggregated. See Figure 4 in Driehuis et al. (2020) for examples of how organoids look like before and after disaggregation.
-
a.Resuspend the pellet of organoids in Tube 1 (cell count) in 1 mL TrypLE Select by pipetting up and down 10–15 times with a P1000 pipette to facilitate disaggregation.
-
b.Incubate Tube 1 for 5 min at 37°C.
-
c.During the incubation step, disaggregate organoids in Tube 2 (infection) mechanically. For that, resuspend the pellet in 2 mL of basal medium and use a pre-narrowed, pre-wet (with F12+++B) sterile glass Pasteur pipette to pipette the volume up and down 8–10 times and keep the tube on ice. Try not to create bubbles when pipetting.Note: For preparation of pre-narrowed glass Pasteur pipettes, after lighting a Bunsen burner, hold the pipette horizontally with both hands and roll it 3–4 times while keeping the tip in the flame. Make sure that the tip is equally narrowed by all sides, and that it has been reshaped to a quarter of its original diameter. Narrowed tips should be sterilized by autoclaving before being used. For pictures on how pre-narrowed pipettes look like, refer to Driehuis et al. (2020).
-
d.Check the suspension in Tube 1 under a microscope. If big organoids are still visible, pipette up and down a few times and incubate 5 min more at 37°C.
-
e.Add cold basal medium up to 13 mL to Tube 1, and up to 10 mL to Tube 2.
-
f.Centrifuge at 100g 5 min at 4°C. Like before, repeat the centrifugation step at a higher speed if organoids are not nicely pelleted.Note: During this time, thaw ready-to-use virus aliquots (stored at −80°C) on ice, and label one 1.5-mL tube for each experimental condition. E.g., for infection of 1 PDO with 2 doses of 2 different OAds (+ non-infected control), prepare 5 tubes.
-
g.Remove the supernatant of both tubes carefully and resuspend each pellet in 1 mL of basal medium.Note: If more than 3 wells were harvested for infection (more than 1 tube for infection), this is the moment to pool all organoids together in a single tube, maintaining a proportion of 2 wells/mL. It is very important to maintain the same concentration of organoids in Tube 1 and Tube 2 to have a comparable cell number that can be used for calculations of the volume needed for infection and seeding.
-
a.
-
7.
Count cells and calculate the volume of organoid suspension needed for OAd infection (VPDO).
Note: The number of cells to seed in each well and the type of multiwell plate used varies depending on the assay to be performed after infection. For viral genome quantification, we seed 25,000 cells/well in a 24-well plate. For cell viability assessment using CellTiter-Glo 3D or organoid formation assay, we seed 5000 cells/well in a white-walled (with either white or clear bottom) or transparent 96-well plate, respectively.
Note: Doses used for infection vary depending on the experiment to be performed. Generally, doses used to infect PDOs lie in a range between 0.2 MOI and 20 MOI. However, for organoid formation assay a lower dose may be needed to allow for enough structures to grow after infection and re-seeding. Try to add similar volumes of viral dilution to each tube. For this, preparation of several serial dilutions is recommended.
Organoid infection and plating
Since adenoviruses are not able to cross Matrigel, infection of PDOs is performed in the absence of extracellular matrix, which is added right before seeding.
To infect organoids, both cell homogenate-derived supernatants or purified OAds can be used. However, for experiments that will be compared, the virus source used should be the same. Calculate in advance the volume of virus dilution needed for PDO infection (VOAd), considering that it should not represent more than 20% of the total volume to seed. For this reason, purified, high-titer (∼1·1010–1.1011 PFU/mL) OAds are preferred.
-
8.
Add the volume calculated in step 7 (VPDO) to each of the prepared 1.5-mL tubes.
-
9.
Bring the volume up to 700–800 μL with basal medium.
-
10.Centrifuge at 400g 5 min at 4°C.
-
a.During this time, prepare virus dilutions in basal medium according to calculated viral titers and doses to be used for infection.
-
a.
-
11.
Remove the supernatant carefully, leaving organoids in the volume of basal medium needed to infect. We generally perform infections in a final volume of 10 μL per well to seed.
Note: Remember that basal medium will represent 20% of the total volume to be seeded (the remaining 80% is Matrigel, which is added after infection), that is, 2 μL per well. When calculating how much volume to leave in the tube, also subtract VOAd to the total volume to avoid excessive dilution of Matrigel and virus. This will ensure that all PDO infections are performed in the same volume, guaranteeing that the contact between organoids and viral particles will be equivalent, and thus comparable, in all the tested conditions.
-
12.
Add VOAd to each 1.5-mL tube.
-
13.
Mix well by tapping with the finger or by carefully pipetting up and down.
Note: Use the same mixing method (tapping or pipetting) for all the Eppendorf tubes in the experiment to further ensure comparable PDO-OAd contacts.
-
14.
Incubate 30 min at 37°C.
-
15.
Add Matrigel to each tube and mix well by pipetting, avoiding bubble creation.
Note: Once Matrigel has been added, put the tubes containing PDOs and OAds on ice to avoid premature polymerization of Matrigel.
-
16.
Seed organoid-containing Matrigel drops (10 μL in 96-well plates, 50 μL in 24-well plates) (troubleshooting 2).
-
17.
Seed drops of 80% Matrigel (diluted in basal medium) in triplicate to read background signals.
-
18.
Let Matrigel solidify for > 15 min at 37°C (in the incubator).
-
19.
Add 90 μL (96-well plate) or 450 μL (24-well plate) of organoid growth medium to each well.
Optional: If white-bottom plates are used, a representative well per condition can be seeded in a transparent 96-well plate for visual inspection of infected organoids before cell viability assessment (Figure 3).
Figure 3.

Bright-field images of PDAC PDOs infected with OAd
Representative images of PDAC organoids infected with increasing doses of an oncolytic adenovirus (5 days after infection). Arrowheads indicate dead organoids. Scale bar, 200 μm.
Viral yield quantification
OAd replication inside organoids can be assessed through the quantification of viral genomes by qPCR amplification of the adenoviral hexon gene in infected organoids. To this end, organoids infected with known doses from OAd stock (passage 1, P1) are grown for 5 days and lysed by freeze and thaw to release viral particles. After centrifugation of the lysate, a fraction of the resulting virus-containing supernatant is used to infect new organoids (passage 2, P2) and proceed as with P1.
For viral yield quantification, supernatants from both P1 and P2 are analyzed by qPCR for the presence of viral genomes. The detection of viral genomes in the P2 supernatant is a further proof of viral replication in organoids. Before starting, and for absolute quantification of OAd genomes, a standard curve of known viral genome copies must be generated. See ‘quantification and statistical analysis’ for calculations and instructions on how to build the standard curve.
For this procedure, organoids should be infected in a 24-well format to guarantee obtention of enough DNA for qPCR amplification.
-
20.
Prepare a cooling bath by adding dry ice and ethanol to an appropriate container.
Note: Alternatively, organoids can be frozen by storing them in a −80°C freezer (even though the freezing process will be slower).
-
21.
Harvest each well of infected organoids (passage 1, P1) in a 2-mL tube, and 1 well of non-infected organoids as explained in the section ‘organoid harvesting for infection’.
Note: In the case of P1 organoids, for the washing steps, bring the volume up to 1.9 mL with cold basal medium (instead of 10 mL used when harvesting organoids in 15-mL tubes). In addition, after the 1st centrifugation step, remove all the supernatant carefully to resuspend the pellet in the basal medium.
-
22.
After the 2nd centrifugation step, remove the supernatant carefully and resuspend the pellet in 150 μL of cold basal medium.
-
23.Perform 3 freeze-thaw cycles to release viral particles from cells.
-
a.To freeze, put the tubes in the cooling bath.
-
b.To thaw, submerge the tubes partially in a 37°C water bath with gentle agitation.
-
a.
Pause point: Pelleted organoids can be left frozen at −80°C (before 1st freeze-thaw cycle) to continue the protocol in the following weeks.
-
24.
Centrifuge at 1000g 5 min to remove cell debris.
-
25.Transfer 120 μL of P1 supernatant to a new 1.5-mL tube. Not taking the whole volume of supernatant will prevent the transfer of any cell debris. From this volume of supernatant:
-
a.Use 10 μL to infect harvested, non-infected PDOs (P2) as explained in steps 8–19, treating this supernatant as VOAd. Add 10 μL of basal medium and 80 μL of Matrigel to perform a 1:2 split, seeding organoids in 2 wells of a 24-well plate.
-
b.Save 25 μL of P1 supernatant to perform DNA extraction (step 27).
-
a.
-
26.
After 5 days, repeat the process described in steps 20–25 to obtain 120 μL of supernatant containing viral particles produced by P2 PDOs.
Optional: The supernatant derived from P2 organoids can also be used to infect new PDOs (10 μL per well of PDOs growing in a 24-well plate) and obtain viral particles released in subsequent passages.
-
27.
Isolate DNA from P1 and P2 supernatants using the Blood DNA Isolation Mini Kit following the manufacturer's protocol. Since the starting volume required for this kit is 200 μL, bring the supernatant sample up to this volume by adding PBS.
-
28.Quantification of viral genomes by qPCR amplification of hexon gene.
-
a.Prepare a qPCR master mix and add 9 μL to each well of a 384-well microplate. For each sample:
qPCR master mix: 5 μL SYBR Green 2X + 0,3 μL Hexon Primer Mix 10 μM (Fw + Rv) + 3,7 μL Nuclease-Free H2O Note: Extra master mix volume should be prepared, considering that each sample needs to be run in triplicate. As a rule of thumb, make a master mix for 1 extra well for every 9 wells to be prepared.CRITICAL: Protect the master mix from light with aluminum foil and keep it on ice while working. -
b.Add 1 μL of isolated DNA into corresponding wells, trying to minimize bubble formation.
-
c.Carefully seal the microplate with an optical adhesive film.Optional: Centrifuge the microplate briefly to ensure that the volume is correctly placed at the bottom of the wells.
-
d.Insert the microplate in a qPCR system (e.g., ViiA 7 Real-Time PCR System).
-
e.Assign samples and targets to corresponding wells and start the qPCR program:
qPCR cycling conditions
Step Temperature Time Cycles Initial Denaturation 95°C 10 min 1 Denaturation 95°C 10 sec 40 Annealing 58°C 30 sec Extension 72°C 20 sec Final Extension 72°C 10 min 1 (Melt curve) 95°C 5 sec - 65°C 1 min 97°C 15 sec
-
a.
-
29.
Quantify OAd genomes as explained in ‘quantification and statistical analysis’. Detection of OAd genomes in the different passages is indicative of OAd replication within cancer cells (Figure 2).
Cell viability assessment
Adenoviruses complete their lytic infection cycle in 36 h. To observe effects on cell viability derived from the infection of organoids by newly formed viral particles, organoids are incubated with OAds for 5 days (120 h), allowing for the completion of 3 viral cycles (Figure 3).
-
30.Cell viability (CellTiter-Glo® 3D Cell Viability Assay)Note: The protocol is based on the manufacturer's instructions, with minor modifications.CRITICAL: Since this is an ATP-based assay, microbial contamination may alter results. For this reason, steps 30b–e should be performed in a cell culture hood and CellTiterGlo-3D should be handled with care. White-walled, 96-well plates must be used to reduce luminescence crosstalk.
-
a.Thaw CellTiter-Glo® 3D reagent at 4°C (for 2–48 h).
-
b.Equilibrate the plate/s containing infected organoids, CellTiter-Glo® 3D reagent and basal medium (F12+++) for 30 min at room temperature (20°C–25°C).Optional: During this time, take brightfield pictures of organoids seeded in the transparent plate using an inverted microscope.
-
c.Prepare a mix of basal medium and CellTiter-Glo® 3D at a 1:1 ratio.
-
d.Remove medium from the wells of the plate containing infected organoids using a glass Pasteur pipette equipped with a 10 μL-tip, taking care not to disturb the Matrigel domes. Change the tip between different experimental conditions.Optional: If using white-walled, clear-bottom plates, this is the moment to carefully stick the sealing film to the bottom of the plate.
-
e.Transfer the mix to a sterile plastic container and add 100 μL of it to each well using a multichannel pipette.
-
f.Mix the plate/s vigorously (450 rpm) in a microplate shaker for 5 min at room temperature (20°C–25°C) to lyse organoids.
-
g.Incubate the plate for 25 min at room temperature (20°C–25°C).
-
h.Quantify luminescence in a microplate reader (troubleshooting 3).Note: We use a Biotek Synergy HT for quantification, programmed with the following features: Integration Time 0.1 s, Plate Costar 96w white opaque, Sensitivity 100.
-
i.Subtract background (Matrigel) luminescence values and normalize values of infected organoids to non-infected organoids, which correspond to 100% viability (Figure 4, troubleshooting 4).Note: See Table 2 for approximate luminescence values expected under described conditions.
-
a.
-
31.Organoid formation assay
-
a.Harvest organoids as described in the ‘organoid harvesting for infection’ section, performing gentle mechanical disaggregation after the first centrifugation step.Note: Pool 2 wells of every experimental condition into one tube. In this case, since they are seeded in a 96-well plate, smaller tubes (1.5 mL) can be used to harvest organoids. Wash wells carefully with basal medium to recover all organoids.
-
b.After the second centrifugation step, carefully remove the supernatant to leave organoids in a final volume of 8 μL.
-
c.Add 32 μL of Matrigel, pipetting up and down to mix.
-
d.Seed 10 μL drops in a new 96-well plate, performing a 1:2 split (if 2 wells were collected, seed 4).
-
e.Let Matrigel drops solidify for > 15 min at 37°C.
-
f.Add 90 μL of organoid growth medium in each well.
-
g.Incubate at 37°C for 24 h.
-
h.After 24 h, take brightfield pictures of the wells seeded using an inverted microscope.Note: Take several pictures that include different fields so that the whole well can be observed for organoid count.
-
i.Count organoids in the wells by visually inspecting the pictures (troubleshooting 5).Note: Only organoids that have not been infected with OAd, or with a low viral progeny inside, will be able to reassemble and form a new, round-shaped organoid. For manual organoid count, we consider as viable organoids those structures that are rounded and have a diameter bigger than 20 μm (Figure 5). Therefore, organoid count can be interpreted as a readout of cell viability, which is inversely correlated with sensitivity to the OAd.
-
a.
Figure 4.

Comparison of sensitivity to oncolytic adenoviruses between PDOs
Two different PDOs were infected with 2 or 20 MOI of two different oncolytic adenoviruses (OAds) and assessed for viability using CellTiter-Glo 3D 5 days after infection. Data are represented as mean ± SEM (n ≥ 3). Luminescence units were normalized to values of non-infected (mock) organoids.
Table 2.
Example of raw luminescence data of a CellTiter-Glo® 3D cell viability assay
| Technical replicate | Negative control (mock) | OAd 1 |
OAd 2 |
Blank (Matrigel) | ||||
|---|---|---|---|---|---|---|---|---|
| 0.2 MOI | 2 MOI | 20 MOI | 0.2 MOI | 2 MOI | 20 MOI | |||
| #1 | 1065780 | 1140390 | 800990 | 210150 | 954530 | 726390 | 94080 | 40 |
| #2 | 1205090 | 1102890 | 788530 | 212610 | 1120560 | 638680 | 102600 | 10 |
| #3 | 1296840 | 1343930 | 781060 | 222520 | 1302120 | 639860 | 91450 | 50 |
Figure 5.

Organoid formation assay for survival assessment after PDO infection
(A) Representative bright-field images of non-infected (mock) or OAd-infected PDOs (0.02 MOI for 5 days) and re-seeded at a 1:2 split ratio. Examples of organoids (round-shaped, > 20 μm diameter) formed 24 h after reseeding are indicated with arrowheads.
(B) Quantification of organoids formed 24 h after re-seeding. Data are represented as mean ± SEM (n ≥ 5).
Expected outcomes
This protocol describes the steps to infect pancreatic tumor PDOs with OAds, as well as methods to analyze viral replication and assess PDO sensitivity to therapeutic viruses. If the OAd genome encodes a fluorescent protein (e.g., EGFP), spreading of the virus to adjacent cells across organoid structures should be observed within 1–4 days after infection (Figure 1). Alternatively, viral replication can be confirmed by quantification of viral genomes in infected organoids. After a 5-day incubation period with OAds, which allows for the completion of 2 viral lytic cycles, we generally obtain viral genome concentrations that range between 105-106 copies/μL for organoids initially infected at a dose of 2 MOI. If the supernatant of infected organoids (P1) is used to newly infect organoids, similar results should be obtained (P2), confirming that viral particles can be efficiently produced within PDOs (Figure 2).
We suggest 2 different approaches to assess PDO sensitivity to OAds. Five days after infection, PDOs can be either analyzed for cell viability using CellTiter-Glo3D (Figure 4), or harvested and re-seeded to evaluate organoid formation after 24 h (Figure 5).
Since oncolytic viruses should selectively replicate in and kill tumor cells, infection of normal organoids should not lead to viral replication or diminished cell viability. Comparison of viral replication and OAd-induced cell death between tumor and normal organoids can be found in Raimondi et al. (2020).
Quantification and statistical analysis
Quantification of OAd genomes from qPCR data
To quantify OAd genomes present in supernatants derived from infected organoids, it is necessary to first build a standard curve of known genomic DNA copies. In our case, this curve covers a range from 102 to 107 copies of viral genomes.
-
1.
Quantify a sample of viral DNA in a spectrophotometer.
-
2.
Calculate the weight of 1 copy of OAd genome with the following equation:
-
3.
Dilute the sample until obtaining a concentration of 107 copies/μL and prepare serial 1:10 dilutions containing from 102 to 107 copies.
-
4.
Amplify the hexon gene by qPCR as explained in step 28, loading 1 μL of gDNA to each reaction (4 technical replicates).
-
5.
Represent CT (cycle threshold) values versus DNA copies in Excel or GraphPad Prism.
-
6.
Transform DNA concentration to logarithmic values and fit the curve using linear regression (Figure 6).
-
7.In Excel, use the equation obtained to interpolate qPCR results derived from PDO infection in Excel and calculate viral genomes/μL (Table 3; Figure 2):Note: Since CT values represent the number of PCR cycles required for the fluorescent signal to exceed a threshold (i.e., background), the slope of the equation obtained should be negative.
-
a.Calculate the mean and standard deviation (SD) of the technical replicates’ CT values.Note: SD of technical replicates should be < 0.3 for results to be considered.
-
b.Calculate the mean and standard deviation (SD) of the technical replicates’ CT values.
-
c.Using the equation obtained, calculate the DNA copies (log10DNA) corresponding to the mean CT value (y).
-
d.Transform log10DNA to DNA copies (from logarithmic to linear scale).
-
e.Plot results in Excel or GraphPad Prism.
-
a.
Figure 6.

Example of a standard curve for viral genome quantification by qPCR
The viral hexon gene was amplified by qPCR in samples with known DNA copies (from 102 to 107). Results expressed as CT (cycle threshold) for each sample were represented and fitted to a linear regression model. The resulting equation is shown in a green square. Data are represented as mean ± SEM (n = 4).
Table 3.
Example of viral genome quantification by qPCR
| Replicate | CT value | Mean CT | SD | LogDNA | OAd genomes/μL |
|---|---|---|---|---|---|
| #1 | 20.807 | 20.949 | 0.247 | 4.582 | 38205.46 |
| #2 | 20.806 | ||||
| #3 | 21.234 |
Limitations
Even though seeding of PDOs as whole entities is necessary for infection with OAds, this leads to higher variability compared to seeding single cells. If inconsistent seeding densities are observed after plating, a relatively high number of replicates may be required (n ≥ 4). As described in this protocol, we generally keep 2 wells (of a 24-well plate) for cell count and infect an approximate number of organoids by extrapolation of this cell count, even though other strategies to consistently seed whole organoids have been reported elsewhere (Ooft et al., 2019). Importantly, cell numbers described here could vary depending on growth rates of individual PDO lines, so they may need to be adjusted before starting screening of OAds by seeding increasing cell densities. Proper maintenance of organoids and splitting them at a constant ratio and frequency are also key points for consistent results.
Due to time-consuming steps, like organoid harvesting and manual seeding of Matrigel drops, this method is limited to low-throughput screenings of OAds. Comparing more than 4 viruses at a time may become too laborious, especially if testing more than one dose. If a higher throughput is needed, automated seeding in a semi-solid format (5%–10% of Matrigel) might be used.
This protocol only describes methods to compare relative sensitivities to OAds among different PDOs in a preclinical setting. While being a very important aspect for virotherapy, OAd selectivity is not assessed here. Nevertheless, it can be easily evaluated by any of the methods described above by infecting normal and tumor-tissue derived PDOs in parallel. Since OAds are designed to complete their lytic cycle in a tumor background, they should therefore not be able to replicate in normal organoids and kill them. Experiments showing OAd selectivity can be found in Raimondi et al. (2020). To ensure specific replication of OAds inside cancer cells, it is also recommended to check that pathways on which OAd design is based are active (e.g., by qPCR analysis of downstream effectors), especially if a dose-response decrease in cell viability is not observed in tumor PDOs.
CellTiter-Glo 3D evaluates cell viability based on ATP levels. Other viability assays based on the measurement of metabolic activity, such as the MTT assay, can also be used (Grabinger et al., 2014; Raimondi et al., 2020). Moreover, complementary techniques to inspect cell death induced by viral infection could be applied.
This protocol has only been applied to pancreatic ductal adenocarcinoma (PDAC) organoids. However, we anticipate that it could be applied to patient-derived organoids derived from many solid organ malignancies.
Troubleshooting
Problem 1
Organoids are too big at the day of harvesting and infection (organoid harvesting for infection).
Potential solution
Harvest organoids as explained in steps 1–5 and disaggregate them enzymatically by incubating with TrypLE Select for 5–10 min at 37°C (1 mL for every 3 wells of a 24-well plate). Vigorously pipette up and down (about 10 times) using a narrowed Pasteur pipette every 2–3 min and check organoids under the microscope to ensure that they do not become a single-cell suspension, since this may lead to poor recovery from disaggregation.
Problem 2
Matrigel/organoid suspension hardens before seeding all PDO wells (step 16).
Potential solution
Always keep tubes on ice when working with Matrigel and during seeding. Chill pipette tips inside the refrigerator, too.
Problem 3
Too low, or too high, luminescence levels are obtained after CellTiter-Glo® 3D Cell Viability Assay (step 30).
Potential solution
If luminescence levels are too low to compare between experimental conditions, incubation time with CellTiter-Glo® 3D reagent can be extended to maximize organoid lysis. Alternatively, they may indicate that a higher number of cells is needed to detect differences in cell viability (likewise, viral dose should be reduced). In case luminescence levels are too high, the integration time can be reduced. However, if biological replicates show substantial differences in luminescence signals, the culture should be examined for potential microbial contaminations. It is recommended to set up a protocol for luminescence reading in the multiplate reader’s software before starting screenings.
Problem 4
Variability among technical replicates is very high (step 30).
Potential solution
To ensure homogeneous seeding, it is very important to thoroughly pipette the organoid suspension before dividing the volume into different tubes to infect, especially when seeding is performed in a 96-well format. Pipette well the OAd-PDO-Matrigel mixture right before seeding to ensure that the percentage of Matrigel is similar in all wells. Try to seed Matrigel drops in such a way that they do not come in contact with the walls of the wells, since this may lead to irregular growth of organoids (Figure 7).
Figure 7.

Uneven seeding of Matrigel drops
Red crosses indicate Matrigel drops in contact with the well’s walls.
Problem 5
There are almost no organoids in the wells, or no big organoids (with a diameter > 20 μm) are observed (step 31).
Potential solution
Some organoids are always lost during harvesting and seeding. Since the organoid formation assay is performed in a 96-well format, too many organoids could be lost during the process of reseeding. To compensate for that, harvest more wells for re-seeding or perform mechanical disaggregation with a p200 pipette pre-wet in F12+++B medium. The OAd dose may also need to be reduced to ensure that enough organoids are alive for count after re-seeding.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Cristina Fillat (cfillat@clinic.cat).
Materials availability
This study did not generate new unique reagents.
Acknowledgments
This work was supported by grants from the Spanish Ministerio de Ciencia e Innovación BIO2017-89754-C2-2R and PID2020-119692RB-C22, co-financed by Fondo Europeo de Desarrollo Regional (FEDER); from the Fundació La Marató de TV3 (301/C/2019); and from the Fundación Mutua Madrileña (AP172132019), with partial support from the Generalitat de Catalunya SGR17/861. CIBER de Enfermedades Raras is an initiative of the ISCIII. We also acknowledge the support of the CERCA Programme/Generalitat de Catalunya. This work was developed at the Centro Esther Koplowitz, Barcelona, Spain. S.P.-S. is recipient of an FPU predoctoral contract (FPU19/05036) from the Spanish Ministerio de Universidades. G.R. has been recipient of an FPI predoctoral contract (BES-2015-071612). A.M.-B. was the recipient of an FPU predoctoral contract (AP2010-5297). Graphical abstract was created using Biorender.com.
Author contributions
Conceptualization, S.P.-S, G.R, A.M.-B. and C.F.; writing – original draft, S.P.-S.; writing – review & editing, G.R. and C.F.; investigation, S.P.-S, G.R, A.M.-B.; methodology, G.R., S.P.-S., and A.M.-B.; resources, F.A.; funding acquisition, C.F. and E.C.V.; supervision, C.F.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Silvia Pascual-Sabater, Email: sipascual@clinic.cat.
Cristina Fillat, Email: cfillat@clinic.cat.
Data and code availability
This study did not generate or analyze any datasets or code.
References
- Boj S.F., Vonk A.M., Statia M., Su J., Dekkers J.F., Vries R.R.G., Beekman J.M., Clevers H. Forskolin-induced swelling in intestinal organoids: an in vitro assay for assessing drug response in cystic fibrosis patients. J. Vis. Exp. 2017;120:55159. doi: 10.3791/55159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driehuis E., Gracanin A., Vries R.G.J., Clevers H., Boj S.F. Establishment of pancreatic organoids from normal tissue and tumors. STAR Protoc. 2020;1:100192. doi: 10.1016/j.xpro.2020.100192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farin H.F., Van Es J.H., Clevers H. Redundant sources of Wnt regulate intestinal stem cells and promote formation of paneth cells. Gastroenterology. 2012;143:1518–1529. doi: 10.1053/j.gastro.2012.08.031. [DOI] [PubMed] [Google Scholar]
- Grabinger T., Luks L., Kostadinova F., Zimberlin C., Medema J.P., Leist M., Brunner T. Ex vivo culture of intestinal crypt organoids as a model system for assessing cell death induction in intestinal epithelial cells and enteropathy. Cell Death Dis. 2014;5:e1228. doi: 10.1038/cddis.2014.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooft S.N., Weeber F., Dijkstra K.K., McLean C.M., Kaing S., van Werkhoven E., Schipper L., Hoes L., Vis D.J., van de Haar J., et al. Patient-derived organoids can predict response to chemotherapy in metastatic colorectal cancer patients. Sci. Transl. Med. 2019;11:1–10. doi: 10.1126/scitranslmed.aay2574. [DOI] [PubMed] [Google Scholar]
- Raimondi G., Mato-Berciano A., Pascual-Sabater S., Rovira-Rigau M., Cuatrecasas M., Fondevila C., Sánchez-Cabús S., Begthel H., Boj S.F., Clevers H., et al. Patient-derived pancreatic tumour organoids identify therapeutic responses to oncolytic adenoviruses. EBioMedicine. 2020;56:102786. doi: 10.1016/j.ebiom.2020.102786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovira-Rigau M., Raimondi G., Marín M.Á., Gironella M., Alemany R., Fillat C. Bioselection reveals miR-99b and miR-485 as enhancers of adenoviral oncolysis in pancreatic cancer. Mol. Ther. 2019;27:230–243. doi: 10.1016/j.ymthe.2018.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva E., Navarro P., Rovira-Rigau M., Sibilio A., Méndez R., Fillat C. Translational reprogramming in tumour cells can generate oncoselectivity in viral therapies. Nat. Commun. 2017;8:14833. doi: 10.1038/ncomms14833. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate or analyze any datasets or code.