

Abstract
Proteomic investigations of Alzheimer’s and Parkinson’s disease have provided valuable insights into neurodegenerative disorders. Thus far, these investigations have largely been restricted to bottom-up approaches, hindering the degree to which one can characterize a protein’s “intact” state. Top-down proteomics (TDP) overcomes this limitation; however, it is typically limited to observing only the most abundant proteoforms and of a relatively small size. Therefore, fractionation techniques are commonly used to reduce sample complexity. Here, we investigate gas-phase fractionation through high-field asymmetric waveform ion mobility spectrometry (FAIMS) within TDP. Utilizing a high complexity sample derived from Alzheimer’s disease (AD) brain tissue, we describe how the addition of FAIMS to TDP can robustly improve the depth of proteome coverage. For example, implementation of FAIMS with external compensation voltage (CV) stepping at −50, −40, and −30 CV could more than double the mean number of non-redundant proteoforms, genes, and proteome sequence coverage compared to without FAIMS. We also found that FAIMS can influence the transmission of proteoforms and their charge envelopes based on their size. Importantly, FAIMS enabled the identification of intact amyloid beta (Aβ) proteoforms, including the aggregation-prone Aβ1–42 variant which is strongly linked to AD. Raw data and associated files have been deposited to the ProteomeXchange Consortium via the MassIVE data repository with data set identifier PXD023607.
Keywords: FAIMS, differential mobility spectrometry, top-down proteomics, Alzheimer’s, brain tissue, ion mobility
Graphical Abstract
INTRODUCTION
Over the last 2 decades, the incidence of neurodegenerative diseases has more than doubled worldwide, with Alzheimer’s disease (AD) as the most prevalent form.1 Two protein species, amyloid beta (Aβ) peptides and phosphorylated microtubule-associated protein tau (tau),2–5 have been found to be strongly associated with AD. Therefore, detailed proteome characterization of AD has been of particular importance.6,7 Mass spectrometry has played a central role in these investigations,8 predominantly by bottom-up approaches.9–16 However, the protease digestion required for bottom-up analyses impedes the capturing of a protein’s complete state, which can vary due to genetic alleles, alternative splicing, proteolytic processing, and post-translational modifications (referred to as “proteoforms”).17–20 Since top-down proteomic (TDP) approaches analyze proteins in an intact state, the likelihood of capturing proteoforms associated with certain pathologies is greater and allows for a stronger, more direct connection between genotype and phenotype.20–22 For example, TDP is particularly well suited for capturing endogenous proteolytic fragments derived from proteins such as tau, which have been linked to Alzheimer’s pathology.23–26 Several previous TDP applications have shown great promise in revealing regional brain proteome heterogeneity and neuronal changes in response to various stimuli.27–30 However, TDP of complex samples typically requires offline fractionation techniques to reduce sample complexity as the human proteome spans several orders of magnitude in size and abundance, and proteins are generally not well-resolved with reverse-phase high-performance liquid chromatography (LC).31 This offline fractionation can be accomplished with gel-eluted liquid fraction entrapment electrophoresis, size-exclusion chromatography, ion-exchange chromatography, or affinity purification, to name a few.22,32–35 Unfortunately, offline fractionation typically reduces yield and throughput by requiring extended sample handling.36
An attractive fractionation alternative that can be introduced between the LC and MS dimensions without requiring additional sample handling steps is gas-phase ion mobility separation. High-field asymmetric waveform ion mobility spectrometry (FAIMS) is particularly well suited to this task with the recent introduction of the FAIMS Pro device, whose modular design allows facile incorporation of ion mobility onto several current Orbitrap instruments.37 FAIMS, also referred to as differential mobility spectrometry, separates ions in a carrier gas based on combinations of factors such as size, charge, or shape through the introduction of an asymmetric waveform with high and low electric fields.38 To prevent collision of the ions with the electrode, a deviation in the ion’s path is introduced through the application of a DC compensation voltage (CV),39 allowing selective transmission of that ion.38 The application of FAIMS to intact protein analysis has largely been restricted to the separation of conformers of individual proteins or small combinations of proteins;40–45 however, FAIMS has recently been applied to intact and native protein analyses using liquid extraction surface analysis.46–50
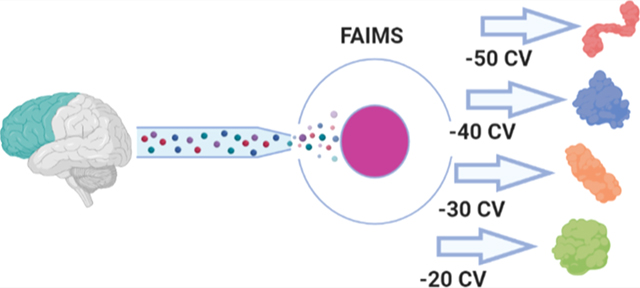
Here, we apply FAIMS-TDP analysis to a whole-tissue sample from the medial frontal cortex (MFC) of an Alzheimer’s patient. Scanning across a CV range of −50 to −20 CV with external stepping allowed us to determine how modulation of FAIMS CV influences the characteristics of the ions transmitted through the cylindrical FAIMS unit, and how this relationship can be exploited to target proteoforms based on size. FAIMS-TDP more than doubled proteoform identifications at a single CV compared to without FAIMS and enabled deeper interrogation of proteoforms relevant to neurodegenerative diseases, including α-, β-, and γ-synucleins, PARK7, tau splice isoforms, and several intact Aβ proteoforms. Taken together, this work describes how greater identifications of proteoforms and proteome coverage can be achieved reproducibly via gas-phase fractionation with FAIMS in TDP.
METHODS
Sample Preparation
The MFC sample was received from Rush University Alzheimer’s Disease Center. Participants were clinic-based older persons who enrolled with memory complaints and/or dementia from 1992 to 2005. They were evaluated at the Rush Memory Clinic for possible dementia and agreed to brain donation as part of the clinical core of the Rush Alzheimer’s Disease Core Center. The study was approved by the Institutional Review Board of the Rush University Medical Center. Following death, next of kin provided consent for autopsy. The patient’s overall cognitive diagnostic category was Alzheimer’s dementia with no other causes of cognitive impairment,51 while the post-mortem interval was 230 min. All sample handling steps were performed with BSL-2+ precautions. 33 mg of MFC tissue stored at −80 °C was transferred to 1.5 mL LoBind Eppendorf tubes (Eppendorf, Cat# 022431081) with the addition of 0.5 mL of homogenization buffer consisting of 8 M urea, 10 mM ammonium bicarbonate (ABC), and 10 mM tris(2-carboxyethyl)phosphine (TCEP) at pH 7.5. Homogenization was achieved through physical disruption by applying a handheld motor-driven pellet pestle for 30 s (BioVortexer with SpiralPestle; Biospec 1083 and 1017), before the addition of 0.5 mL of homogenization buffer at room temperature. The sample was then incubated at 37 °C for 60 min with 1200 rpm using a ThermoMixer to allow for extraction and denaturation of cellular proteins. Pelleting of the urea-insoluble material was accomplished with centrifugation at 18,000g for 20 min at 22 °C, and the resulting supernatant was then added to 3 mL of wash buffer (WB; 8 M urea, 10 mM ABC, pH 7.5) within a 4 mL 100 kDa molecular-weight cutoff (MWCO) filter to remove large MW species. After 1 h of centrifugation at 22 °C and 5000g, the retentate volume was 200 μL and filtrate volume was 3.8 mL. 3.8 mL of the filtrate was transferred to a 3 kDa MWCO filter to remove small-molecule contaminants and low MW peptides. The 3 kDa MWCO filter was centrifuged at 22 °C and 7300g for 1 h, giving a retentate volume of 200 μL. 200 μL of retentate from the 100 kDa MWCO filter was washed once more by diluting to 4 mL with WB and filtering it again through the same filter. This flow through was added to the same 3 kDa MWCO filter as mentioned above and concentrated one more time. The final 200 μL of retentate from the 3 kDa MWCO filter wash was transferred to a 1.5 mL LoBind Eppendorf tube. To acidify the sample, 10 μL of 10% formic acid (FA) was added, resulting in 0.5% FA concentration in the final sample. The sample was centrifuged at 18,000g for 15 min to remove the precipitated material before measuring the protein concentration using the bicinchoninic acid (BCA) assay. To account for any potential buffer contributions to the BCA assay, bovine serum albumin standards were prepared in 8 M urea, 10 mM ABC, and 0.5% FA. The protein concentration by the BCA assay was determined to be 1.1 mg/mL, corresponding to the final yield of 0.22 mg of total recovered protein in the ∼200 μL sample. The sample was diluted to 0.5 mg/mL with WB containing 0.5% FA and stored at −80 °C until LC–MS analysis.
We should note that initial experiments (data unpublished) using a traditional protease/phosphatase inhibitor cocktail within our homogenization buffer led to misleading BCA assay and LC–MS results due to retention of several cocktail components by the 3K MWCO filter. Furthermore, many of the phosphatase and protease inhibitors commonly utilized are chemically incompatible with reducing agents such as TCEP. Considering these incompatibilities, and that the strongly denaturing conditions of 8 M urea with 10 mM TCEP would be expected to inactivate most proteases, we believed that proteolytic degradation would still be minimized under these conditions.
LC–MS/MS Analyses and FAIMS Settings
Samples were analyzed using a Waters NanoACQUITY UPLC system with mobile phases consisting of 0.2% FA in H2O (mobile phase A) and 0.2% FA in ACN (mobile phase B). Both trapping precolumn (100 μm i.d., 5 cm length) and analytical column (75 μm i.d., 50 cm length) were slurrypacked with the C2 packing material (5 and 3 μm for trap/analytical, respectively, 300 Å, Separation Methods Technology). Samples were loaded into a 5 μL loop, corresponding to 2.5 μg of the loaded material amount, and injected onto the trapping column with an isocratic flow of 5% b at 3 μL/min over 20 min. Separation was performed with a 5–50% b gradient over 180 min at 300 nL/min. For MS/MS analysis of proteins, the NanoACQUITY system was coupled to a Thermo Scientific Orbitrap Fusion Lumos Tribrid mass spectrometer equipped with the FAIMS Pro interface. Source parameters included an electrospray voltage of 2.2 kV, a transfer capillary temperature of 275 °C, and an ion funnel radio frequency amplitude of 60%. FAIMS was set to standard resolution without supplementary user-controlled carrier gas flow and a dispersion voltage of −5 kV (equivalent to a dispersion field of −33.3 kV/cm),50 while the CV varied depending on the experiment (referred to as “external stepping”). The Fusion Lumos was set to the “intact protein” application mode, which lowers the higher-energy C-trap dissociation (HCD) cell N2 pressure to 2 mTorr, and data were collected as full profile. MS1 and MS2 data were acquired at a resolution of 120k and 60k, microscans of 3 and 2, across a 500–2,000 m/z and 400–2,000 m/z range, and with AGC targets of 5 × 106 and 5 × 105, respectively. MS1 and MS2 were acquired with a maximum inject time of 400 ms as well. Datadependent settings included selection of top 6 most intense ions, exclusion of ions lower than charge state 5+, inclusion of undetermined charge states, and dynamic exclusion after one observation for 30 s. Ions selected for MS2 were isolated over a ±1.5 m/z window and fragmented through collision-induced dissociation (CID) with a collision energy of 35%. We utilized CID instead of HCD due to the lower dependency of normalized collision energy for achieving interpretable protein fragmentation spectra, thereby prioritizing the breadth of our proteoform observations over sequence coverage.52 Data sets for each FAIMS CV were collected in triplicate. All raw files have been deposited into the MassIVE data repository and can be accessed via accession MSV000086696, or alternatively with PXD023607 on ProteomeXchange.
Data Analysis
Proteoform identification was performed with TopPIC version 1.3.53 Settings for TopPIC included a precursor window of 3 m/z (to account for isotopic envelope), a mass error tolerance of 15 ppm, a max/min unknown mass shift of 500 Da, and a maximum number of allowed unknown modifications of 1. MS2 spectra were searched against a database concatenated with entries from Homo sapiens Swiss-Prot (20,352), Swiss-Prot splice variants (22,000), and TrEMBL (54,436), as well as common contaminants. Identified proteoforms were filtered to an false discovery rate (FDR) of 1% through TopPIC. Downstream data analysis was performed in the R environment for statistical computing and figure generation.54 In downstream analysis, proteoforms from the same gene with the same starting and ending amino acids that were found to be within ±5 Da were combined as a single proteoform in order to increase the stringency of our assignments. Although the 5 Da threshold is arbitrary, this approach compensated for incorrectly assigned monoisotopic peaks, ambiguity in unknown mass shifts, and other artifactual deviations. In order to determine the relative standard deviation (RSD) of proteoforms within replicates, we utilized the “feature intensity” output from TopPIC. Fragmentation sequence coverage maps and associated spectra were generated using the LCMsSpectator software.55 Specific settings for LCMsSpectator included a precursor ion tolerance of 10 ppm, a product ion tolerance of 10 ppm, a minimum S/N threshold of 1.5, a Pearson correlation threshold of 0.7, a default smoothing of 9 points, and a precursor isotope relative intensity threshold of 0.1.
RESULTS AND DISCUSSION
Addition of FAIMS Robustly Increases Proteome Coverage
The cortex tissue sample from a patient diagnosed with Alzheimer’s was processed following the workflow, as shown in Figure 1.
Figure 1.

Workflow demonstrating sample preparation, LC–MS, FAIMS-TDP, and data analysis with TopPIC/R.
We first sought to analyze the performance of FAIMS at various CVs in comparison to without FAIMS (referred to as “No FAIMS”). It is worth remarking that the FAIMS interface produces a modest loss of ion transmission, presumably due to the longer ion path through the source.56 Therefore, to better represent typical conditions and ensure equitable comparisons, the “No FAIMS” data were collected using the same sample without the FAIMS unit installed. These “No FAIMS” data were collected in triplicate and analyzed according to the workflow, as shown in Figure 1 with a top 6 DDA MS2 analysis.
The “No FAIMS” data sets on average identified 754 ± 35 proteoforms (Figure 2A and Table S1) and collectively identified 1073 unique (non-redundant) proteoforms (Figure 2B) derived from 293 unique genes (Figure 2C), covering 29,359 amino acids across the proteome (Figure 2D). The metric “proteome coverage” (Figure 2D) is defined as non-redundant amino acids covered by each proteoform’s sequence. Essentially, this metric accounts for both the length and diversity of the identified proteoforms. We consider this to be a more balanced metric compared to the raw number of proteoforms and genes as it avoids biases toward smaller proteoforms. Although simple counts of proteoforms or genes are intuitive, these metrics appear to be strongly influenced by short proteolytic fragments.
Figure 2.

Bar charts demonstrating several metrics used in the comparison of FAIMS CVs in steps of 5 V with “No FAIMS” data sets. (A) Mean number of proteoform identifications per replicate (n = 3). Error bars represent standard deviation from the mean. (B) Total unique proteoforms found across all three replicates for each FAIMS CV or “No FAIMS”. (C) Total unique genes found across all three replicates for each FAIMS CV or “No FAIMS”. (D) Total proteome coverage or non-redundant amino acids covered by each proteoform’s sequence, found across all three replicates for each FAIMS CV or “No FAIMS”.
The FAIMS data sets were collected from −50 to −20 CV scanned in steps of 5 V. Each of the resulting 7 CVs was collected in triplicate. With respect to proteoforms and genes, FAIMS outperformed “No FAIMS” for all the CVs tested within the −50 to −30 CV range (Figure 2A–C). For example, the three data sets at −50 CV identified 1833 ± 17 proteoforms, an average increase of ∼140% per run compared to “No FAIMS”. Collectively, these three data sets at −50 CV identified 2564 unique proteoforms from 530 unique genes covering a total of 43,437 amino acids, an increase of 95, 69, and 69% over the three “No FAIMS” data sets, respectively (Figure 2B–D and Table S1). Also, although FAIMS at −25 V observes fewer unique proteoforms and genes compared to “No FAIMS”, the proteoforms themselves are much longer on average. Thus, when considering proteoform length, (Figure 2D), the −25 V data sets covers nearly as many amino acids from the proteome (28,398) relative to “No FAIMS” (29,359), doing so with only half as many proteoforms (Figure 2B).
We also investigated whether FAIMS could provide as robust and reproducible an analysis compared to without FAIMS. We assessed quantification reproducibility by calculating the RSD (equivalent to coefficient of variation) of each proteoform’s feature intensity, provided the proteoform met the criterion of being observed across all three replicate data sets for each CV setting. Because the FAIMS and “No FAIMS” data sets were all collected from a single biological sample, variance among the replicates should be exclusively attributed to instrumental factors. The boxplot in Figure 3 (as well as individual histograms in Figure S1) demonstrates how the distributions of RSDs compare between the FAIMS and “No FAIMS” data sets.
Figure 3.

Boxplot displaying the distributions of the RSDs determined from the feature intensities of proteoforms found in FAIMS and “No FAIMS” replicates. The median RSD for each condition is written within the upper box, above the line indicating its position. The upper and lower hinges correspond to the first and third quartiles (25th and 75th percentiles), while the upper and lower whiskers represent 1.5 times the interquartile range (IQR). Outliers beyond the whiskers are not plotted.
Based on median RSD, addition of FAIMS resulted in similar or slightly improved quantification quality relative to “No FAIMS”. The improvement in RSDs among lower voltages (−20 to −30 CV) may be related to the observation that fewer proteoforms are transmitted within that range, providing higher quality MS1 spectra and therefore better estimations of feature intensity. Furthermore, it is worth noting that the RSDs determined from these replicates are similar to previous top-down55 and bottom-up57,58 experiments performed with similar instrumentation. Taken together, these data demonstrate that the addition of FAIMS to TDP robustly increases proteoform identifications, without any sacrifice of quantification quality.
We next investigated the overlap in identifications between the CVs, which could provide the largest non-redundant set of observations. Using overlap coefficients, defined as the intersection between two sets divided by the smaller of the two sets, we determined the similarity between each data set. The overall mean overlap coefficient for both proteoform and gene identifications within each FAIMS CV was 0.76. This is comparable to the overlap coefficient from “No FAIMS” data sets (0.77), suggesting that the technical replicates show similar reproducibility. A heatmap displaying the FAIMS overlap coefficients of proteoforms and genes is shown in Figures 4 and S2, respectively.
Figure 4.

Heatmap generated in R comparing the overlap coefficients of each CV replicate based on proteoforms identified with TopPIC. White lines separate replicates, while black lines separate different CVs. Mean overlap coefficients of each CV’s replicates are shown in the middle block of the CVs being compared. White text color is used on overlap coefficients ≥0.5 and black text color <0.5 to improve visibility.
Notably, the overlap of gene identifications across the CV space is much greater relative to proteoforms, although it is particularly noticeable when comparing the longest CV distances. For example, the overlap between −50 and −20 CV is 0.64 at the gene level and 0.06 at the proteoform level. This was expected as each gene can potentially be represented by multiple proteoforms. Figure 4 demonstrates that while a 5−10 V difference in CV produces overlap coefficients mostly above 0.5, distances greater than 15 V produce larger degrees of dissimilarity and are therefore better spaced for capturing sufficiently different sets of proteoforms. With these data sets, we next pursued determining which combinations of CVs are optimal for achieving maximal gene identifications, proteoform identifications, and sequence coverage with constraints on the number of CVs for each combination. First, we established the optimal combinations for any number of CVs from 1 to 7 using the metrics unique proteoforms, genes, and proteome sequence coverage (Figure 5).
Figure 5.

Combinatorial analysis of FAIMS CVs with regard to (A) number of proteoforms, (B) number of genes, and (C) sequence coverage. For each combination, the highest number plotted is highlighted in red and labeled with the CVs for that specific combination.
Using combinations limited to three CVs as an example, Figure 5A,B details how −35, −40, and −50 V are ideal if one desires to maximize the number of proteoforms or genes. The proteoforms and genes identified in these three CVs (nine data sets) cover 84% of the total unique proteoforms and 92% of the total genes over the entire set of 21 data sets. When weighing a proteoform’s length with sequence coverage as a metric, the ideal combination is at −30, −40, and −50 CV, covering 88% of the unique amino acids from all 21 FAIMS data sets with only a modest loss of proteoform and gene identifications (4221 proteoforms and 723 genes, Figure 5C). However, these combinations include all replicates and represent the maximum identifications that could be achieved for a given combination of CVs. Therefore, to demonstrate the advantage of external CV stepping, we decided to investigate what numbers could reasonably be achieved on average with just three runs, where each run is a different CV.
Using the −50, −40, and −30 V data sets as an example, we determined the average unique proteoforms, unique genes, and proteome sequence coverages based on the 27 unique combinations from the three different replicates within each CV. Based on this analysis, we could achieve 2986 (±46) unique proteoforms and 618 (±12) unique genes covering 64,534 (±1089) amino acids on average. Compared to the “No FAIMS” triplicate data sets, which identified 1073 unique proteoforms and 293 unique genes covering 29,359 amino acids, external CV stepping at −50, −40, and −30 V could more than double each metric. Nevertheless, the optimal combination of CVs will also likely depend on the organism and tissue the sample is derived from, the sample preparation steps applied, as well as any additional offline fractionation implemented before LC–MS analysis. Overall, when taken together with the overlap analysis (Figure 4), suggest that 15 V or greater distances between CVs provide the least amount of overlap between proteoforms, while 5−10 V separation within the −50 to −30 CV maximizes genes, proteoforms, and proteome sequence coverage.
To further determine the impact of FAIMS on the depth of proteome coverage, we compared the genes identified from our top-down data sets to a typical bottom-up analysis of the human brain. Comprehensive bottom-up data sets of human brain tissue allowed us to estimate abundances for 8528 proteins using weighted spectral counting.59–61 Proteins were binned into 10 different abundance percentiles based on spectral counts compiled from several bottom-up data sets of the human brain. By cross-referencing with our top-down data sets, we were able to determine where the genes, and the proteoforms they are derived from, rank in terms of estimated protein abundance (using data set-normalized spectral counts) in the human brain (Figure 6). Genes that were found within our top-down analysis but not the bottom-up reference set were binned as “NA”.
Figure 6.

Bottom-up data sets from human brain tissue were used to estimate protein abundance by weighted spectral counting. These proteins were then binned into 10 different abundance percentiles, as well as a top-down only bin indicated as “NA”. (A) Comparison between the genes found in FAIMS and “No FAIMS” data sets across the 10 different abundance percentiles, as well as genes only found from our top-down analysis. (B) The number of genes for each FAIMS CV was normalized to the number of genes found in the “No FAIMS” data sets for each abundance percentile bin.
One immediate and apparent observation is that the higher the abundance percentile, the higher the probability of identification with TDP. In other words, TDP mostly identifies highly abundant proteins. About half of the genes identified from the top-down data sets (with or without FAIMS) were contained within the top 20% of abundance bins for the bottom-up analysis (Figure 6A). Conversely, 71 genes could only be found in the top-down data sets (“NA” bin) (Figure 6A). These genes appeared to represent proteins that were relatively short (<150 AA) and contained numerous basic Lys/Arg residues which, presumably, precluded their ability to be detected by bottom-up analysis. For example, included within the top-down only bin were histone proteins such as H3C1, H4C1, H1–4, and H2BC12.
As anticipated, we found that FAIMS could improve identifications in lower-abundant percentiles over “No FAIMS”, increasing the depth of the observable proteome. By determining the ratio of FAIMS to “No FAIMS” across all percentiles, an appreciable increase in identifications below the 40th percentile is apparent within CVs in the −40 to −50 range (Figure 6B). However, when considering the absolute number of genes, the major contribution of FAIMS’ advantage is through broadening the spectrum of identifications across all abundance percentiles.
Relationship between FAIMS CV and Transmission of Proteoforms
With FAIMS, we also noted a trend between FAIMS CV and proteoform molecular weight (Figure 7).
Figure 7.

Boxplot displaying distributions of proteoform masses identified with TopPIC against tested FAIMS CVs. Each distribution includes only non-redundant proteoforms found within all three replicates. The median proteoform mass for each condition is written within the upper box, above the line indicating its position. The upper and lower hinges correspond to the first and third quartiles (25th and 75th percentiles), while the upper and lower whiskers represent 1.5 times the IQR. Outliers beyond the whiskers are not plotted.
At −50 CV, the median proteoform mass is ∼5 kDa and increases to ∼15 kDa at −20 CV. Based on these mass distributions (Figure 7), CVs less than −50 V appear well suited for top-down or middle-down proteomic experiments, while CVs greater than −50 V may best benefit peptidomic or bottom-up experiments. The observed molecular-weight trend presumably extends beyond −20 CV; however, we restricted our search at −20 CV based on the drop-off in proteoform and gene identifications which led to diminishing returns in proteome sequence coverage per run. We should note that a previous work using FAIMS found +40 CV to be the ideal voltage for transmitting a NIST mAb heavy chain (∼51 kDa) and −20 CV was best for the corresponding light chain (∼23 kDa), supporting the idea that the trend we observe extends into positive FAIMS CVs.45 Interestingly, although many spectra identifying a single proteoform were limited to being found within a 10 V range (3599 out of 5165), there were seven proteoforms observed across the entire CV range from −50 to −20. Presumably, this can be attributed to their high abundance in the sample, particularly in the case of ubiquitin (UBB), myelin basic protein, and acyl-CoA-binding protein. However, it is also likely that the different charge states of these proteoforms adopt several gas-phase conformations depending on their proton isomerization, impacting their mobilities.42 These seven proteoforms allowed us to investigate how the charge state envelope is differentially transmitted through the modulation of CV. As a proxy for the charge state distribution at each CV, we used the median charge state based on the proteoform-spectrum-matches (PrSMs) identified at each CV and tracked how this value changed as a function of CV starting at −50 CV. The MS1 scans in Figure S3 demonstrate how the median PrSM charge state tracks the charge state distribution of UBB. Of these seven proteoforms, four followed an inverse relationship, with median observed charge state increasing as CV was decreased (Figure 8).
Figure 8.

Heatmap of the median PrSM charge states from seven proteoforms identified across the −50 to −20 CV range, filtered by ≥4 PrSMs in order to increase the confidence in identifications and provide a reasonable estimate of the median charge state. The median PrSM charge state is shown within each block, while the deviation of the PrSM charge state is indicated in red (increasing) or blue (decreasing) relative to −50 CV. Proteoforms on the x-axis are written as the gene name, starting amino acid, and ending amino acid.
It is worth noting that this trend has generally been observed with peptides and small proteins.56,62–64 Surprisingly, three proteoforms showed the opposite relationship where decreasing CV decreased the median charge states observed (Figure 8). A cursory examination of the mean precursor mass between the two groups suggests that larger precursors are more likely to favor higher charge states as CV is decreased. To validate these relationships further, we expanded this analysis to include proteoforms that were identified within a more modest, but still wide, 20–30 CV range (n = 256 proteoforms). Here, proteoforms whose median charge shifted greater than one charge across the entire CV range were binned into the two different groups depending on the direction of that shift. Those that shifted less than one charge were considered “neutral” with respect to changing CV. Similar to the previous results, larger precursors were significantly more likely to be associated with an inverse relationship between observed charge states and CV (Table S2). With decreasing CV, the majority of proteoforms appeared to transmit at lower charge states (n = 177) compared to those that favored higher charge states (n = 39). The remainder (n = 40) were considered “neutral” with respect to changes in CV. Proteoforms binned within the neutral group demonstrated an average precursor mass between the inverse and direct groups, once again suggesting that the mass of the proteoform is intrinsically linked to this behavior. Other primary sequence-based parameters such as basic/acidic amino acid composition and aliphatic index were not significantly correlated however (Table S2). Although the proteins being introduced into the gas-phase are presumably denatured, factors typically associated with native proteins such as dipole moment or collisional-cross section may have better predictive value toward this phenomenon.64–66 Our results demonstrate how CV can be used to filter proteins of different masses, and how a protein’s charge state envelope can be differentially transmitted through FAIMS as well.
Utility of Protein Fragments in TDP Experiments
Surprisingly, a significant number of proteoforms we identified were fragments of larger proteins. We found that only 25% of the unique proteoforms identified covered greater than half of the protein’s sequence from which they were derived. This 25% had an average mass of 11.4 kDa compared to the remaining 75% which had an average mass of 5.7 kDa. Several factors may contribute to this observation, some of which are independent of FAIMS. For example, these fragments themselves may be proteolytic cleavage products produced under normal homeostatic conditions as part of the cellular “degradome”,67 or despite the various precautions taken, introduced during the post-mortem interval and sample handling. Central nervous system tissue is a rich source of signaling peptides known as “neuropeptides” that are commonly derived from much larger precursor proteins as well.68 By cross-referencing our proteoform identifications with an established neuropeptide database (NeuroPedia),69 we were able to identify several such neuropeptides including vasostatin-1, secretoneurin, cholecystokinin-58 desnonopeptide, and neuropeptide y and many non-canonical sequence variants derived from the known neuropeptide-producing genes. Beyond biological factors, certain instrument parameters can influence the observation of protein fragments as well. For example, low in-source “fragmentation” voltages (between 10 and 20 V) can typically be used to remove adducts and desolvate protein ions, while higher voltages can produce source-induced dissociation. However, the susceptibility of amide bonds to dissociation can vary widely, and as the size of a protein increases so does the likelihood of it containing labile amide bonds such as Xaa-Pro.70–73 As b- and y-ions may be produced inadvertently through this mechanism, even at low source voltages, we decided to eschew applying source voltage to reduce the chance of introducing fragment ions into the instrument. It is also possible that proteins are susceptible to fragmentation events as they pass through the electric fields created in FAIMS. However, these fragments would not be expected to have the same mobility as the parent ion, with the caveat that this is likely dependent on the size of the fragment relative to the precursor ion as well.
Finally, it is worth pointing out a few factors that can bias toward the observation of smaller proteoforms (<∼15 kDa). First and foremost is the signal spreading that occurs as the size of a proteoform increases.74 This signal spreading can largely be attributed to the charge state envelope and isotopic distributions of each charge state. Specific to our experimental setup was the use of a size 3K MWCO filter as the final filtration step in our sample prep, which in this case was chosen to ensure that smaller amyloid beta proteoforms could be efficiently captured if they were present. General instrumental factors can create bias toward small proteoforms as well, including the tuning of the quadrupole, electrodynamic capture, and collisions with residual gas molecules in the Orbitrap cell that lead to quicker decay of the transient for larger molecules.75 Irrespective of these biases or the exact origin of the fragments, they can still provide insights into the proteome. These fragments are particularly useful in the context of neurodegenerative disease where disruptions in proteostasis are commonly linked to pathology.76,77 Indeed, fragments of tau are often found to be neurotoxic and play a role in the progression of tauopathies such as AD.23–26 Generally, the fragments we observe are much larger than tryptic peptides and in several cases where there are many fragments for a particular protein, we observe considerable sequence coverage.
This is exemplified by examining the fragments providing coverage for the ∼50 kDa tubulin alpha-1B chain (TUBA1B) and the ∼70 kDa synapsin-1 (SYN1), as shown in Figure 9A,B, both large proteins that would otherwise be difficult to observe in their full-length form in TDP of a complex sample.
Figure 9.

Sequence maps of (A) tubulin alpha-1B chain (TUBA1B) and (B) synapsin-1 (SYN1). Each colored segment represents a proteoform with a unique amino acid start and end site. Color scale represents the number of observed PrSMs that can be mapped to the corresponding segment. Unknown mass shifts were not utilized in this analysis.
Identification of Proteoforms with Relevance to Neurodegenerative Disease
The utilization of FAIMS with our top-down analysis enabled identification of a number of unique Swiss-Prot splice variants and TrEMBL entries, with 267 unique splice variants and 96 TrEMBL entries (Table S3). Notably, we observe multiple PrSMs unambiguously identifying an alternative ORF isoform (A0A0D9SF30) for neural cell adhesion molecule 1 (NCAM1). We also observed proteoforms derived from genes that have known roles in several neurodegenerative diseases, in particular α-synuclein and PARK7. In both of these cases, the predominant proteoforms contain the full-length sequence. Interestingly, the majority of α-synuclein, βsynuclein, and (to a lesser extent) γ-synuclein PrSMs were found to contain a ∼177 Da unknown mass shift near the C-terminus (full-length α-synuclein spectrum with unknown modification, as shown in Figure S4A). This mass shift has been previously observed in open database searches and is generally found at Asp or Glu residues.78 A potential explanation that closely matches the average and monoisotopic mass of the modification consists of one oxygen with three iron atoms as well as the loss of seven hydrogen atoms (based on Unimod accession #1971). Comparison of the isotopic peaks of a y242+ fragment ion from α-synuclein (Figure S4B) with a simulated spectrum containing the aforementioned elements that are believed to belong to the unknown modification (Figure S4C) demonstrates the similarity between the two isotopic distributions, with many of the isotopic peaks aligning within 2 ppm of each other. It should also be pointed out that the left most isotopic peaks are unique to naturally occurring iron isotopes, and the absence of these peaks is readily apparent when the spectrum is simulated without containing the three iron atoms (Figure S4D), strongly suggesting that this unknown modification is very likely composed of the proposed elements. Prior studies have also described the high affinity of α-synuclein for various metal ions, and the region we observe to contain this modification overlaps with residues known to be involved in binding―specifically the 119DPDNEA124 motif (Figure S5).79–81 Our data also suggest that the C-terminal regions of β-synuclein and γ-synuclein have similar roles in metal binding as multiple spectra with the same unknown mass shift were matched to similar sequences within these proteins. With regard to the mitochondrial protein PARK7, we noted a ∼116.0 Da mass shift on its single active site Cys residue, C106 (Figure S6). One potential explanation with a similar delta mass is succinylation, a modification that is attributed to mitochondrial stress and forms due to Michael addition of fumarate onto a Cys thiol group.82
We also identified numerous fragments derived from non-canonical splice isoforms of tau protein known to be expressed in the human brain.83 Briefly, tau isoforms are defined by the number of N-terminal inserts due to alternative splicing of exon 2 and/or 3 (referred to as 0N, 1N, and 2N) and the number of microtubule-binding repeats due to alternative splicing of exon 10 (referred to as 3R or 4R).84 It is also worth noting that endogenous fragments of tau are a common finding in human brain tissue and cerebrospinal fluid, and differential fragmentation of tau may play a role in AD progression.85,86 We observed several fragments that could unambiguously distinguish 0N and 1N tau (Figures 10A, S7, and S8). The fact that we observe high spectral counts for 0N (86 PrSMs) and 1N (91 PrSMs), but do not observe any PrSMs for 2N, is aligned with previous quantitative immunoblotting that found 0N and 1N to be the dominant forms and make up ∼91% of total tau while 2N comprised only ∼9%.87 With regard to unambiguous assignment of the microtubule-binding repeats, we observed spectra that could be assigned to both 3R (Figures 10A and S9) and 4R tau (Figures 10A and S10) as well. Parsimonious inference allowed us to conclude that brain tau is represented mostly by the mixture of 1N3R (Tau-B) followed by 0N4R (Tau-D) splice variants, which is in agreement with previous observations.87,88 While other splice isoforms also had unique fragments, the low number of spectral matches (<4) did not allow us to confidently conclude about their existence.
Figure 10.

(A) Schematic representation of selected tau fragments with the number of PrSMs that unambiguously identify 0/1N or 3/4R tau. 0N and 1N fragments are being shown as sharing the same KKVAV residues at their C-terminus, while 3R and 4R proteoforms share VRTPP at their N-terminus. Residues underlined are the KVAVVR hexapeptide sequence in the second proline-rich region of tau which is cleaved at proteolytic cleavage site #1. The 3R and 4R hexapeptide motifs are not shown but are both cleaved at proteolytic cleavage site #2. (B) Sequence coverage map of MAPT 1N3R (Tau-B) showing all proteoforms that could be mapped to this isoform with a unique amino acid start and end site. Red lines at the bottom represent the positions of proteolytic cleavage sites #1 and #2. Color scale represents the number of observed PrSMs that can be mapped to the corresponding segment. Unknown mass shifts were not utilized in this analysis.
Surprisingly, N-termini of the predominant R-domain containing fragments are a continuation from the C-termini of the predominant N-domain containing fragments, or in other words, they appear to be linked fragments produced from a possible proteolytic cleavage event (referred to as site #1 in Figure 10A). Furthermore, the C-termini of the 3R and 4R domain-containing proteoforms share the same cleavage site (referred to as site #2 in Figure 10A), even though the sequences themselves are different due to splice variation of exon 10. Even more surprising, all of these cleavage sites lie within three well-described hexapeptide motifs.89–95 Figure 10B demonstrates all of the unique fragments we identified from our data sets that could be mapped to the 1N3R tau isoform, as well as the location of the cleavage sites. Cleavage site #1 lies within the second proline-rich region of tau and divides the KVAVVR hexapeptide sequence which is known to provide one of the strongest binding sites for microtubules (Figure 10B).89–91 Cleavage site #2 lies within the 3R and 4R hexapeptide motifs VQIVYK and VQIINK, which have been demonstrated to drive aggregation of tau (Figure 10B).92–95 Interestingly, many of the fragments, as shown in Figure 10B, begin or end within proximity of these two cleavage sites. Each of these hexapeptide motifs is known to form a β-structure, often in the form of β-hairpins which underlie the aggregation involved in various neurodegenerative diseases.89,93,96 Intriguingly, these observations highlight the possibility of a shared proteolytic degradation pathway among tau isoforms that is potentially capable of disrupting their aggregation-seeding regions.25,85,86
Lastly, we found numerous spectral matches for Aβ that could only be observed exclusively by using FAIMS. These Aβ proteoforms, which typically require special fractionation or handling techniques in order to improve recovery due to their hydrophobic and aggregation prone-properties,9,16,97,98 were observed intact using FAIMS within the −40 to −50 CV range. This includes the canonical Aβ1–42 and Aβ1–40 (Figure 11A,B, respectively), as well as several N-terminally truncated forms (specifically Aβ2–42 and Aβ4–42, Figure 11C,D).
Figure 11.

Representative MS2 spectra and MS2 fragment ion coverage maps with matched b and y ions of the 5+ charge state of four Aβ proteoforms: (A) Aβ1–42 from 903.663 m/z precursor with 68.3% sequence coverage (43 PrSMs). (B) Aβ1–40 from 866.4374 m/z precursor with 48.7% sequence coverage (6 PrSMs). (C) Aβ2–42 from 880.6570 m/z precursor with 40.0% sequence coverage (3 PrSMs). (D) Aβ4–42 from 840.6410 m/z precursor with 44.7% sequence coverage (4 PrSMs).
It is worth noting that Aβ proteoforms with various N- and C-terminal cleavages are notoriously difficult to identify in bottom-up analyses due to the extreme hydrophobicity of the tryptic products, which highlights an advantage of intact protein analysis. We believe that the FAIMS-TDP methodology offers a robust way for achieving intact identification of these Aβ proteoforms, allowing for facile determination of the different combinations of N- and C-terminal cleavage products that exist.
CONCLUSIONS
We have described how nano-LC reverse-phase separation of a highly complex sample containing proteins, over a wide mass range, can benefit from the implementation of gas-phase fractionation through FAIMS in the context of top-down mass spectrometry. FAIMS was demonstrated to impact the transmission of proteoforms by size and/or charge, reducing MS1 complexity and allowing greater depth of coverage of the proteome. FAIMS at a single CV (−50) enabled identification of 1833 ± 17 unique proteoforms on average, more than double compared to without FAIMS (754 ± 35). Addition of FAIMS did not result in deterioration of quantification reproducibility and remained near 20% RSD, which is comparable with label-free bottom-up approaches. Decreasing CV of FAIMS was noted to increase the molecular mass of ion species being transmitted to the instrument (median MW of ∼5 kDa at −50 CV vs ∼15 kDa at −20 CV), and modulation of CV was also observed to differentially influence the transmission a proteoform’s charge state envelope. We also defined optimal combinations of CVs that could produce the largest theoretical maximum of proteoforms/genes or proteome sequence coverage. Compared to the three “No FAIMS” data sets, external CV stepping at −50, −40, and −30 V could more than double the number of unique proteoforms, unique genes, and proteome sequence coverage. It is also worth pointing out that our work only explored a relatively small CV range which we felt best suited our TDP approach using a complex sample. Since FAIMS can easily be adapted to other methods, low-resolution MS1 analyses with Orbitrap analyzers may be able to improve on the identification of larger proteoforms within the CV range we tested, as well as beyond −20 V where larger proteoforms would likely be observed. It can also be envisioned that future mass analyzer instrumentation that improves on the acquisition of larger proteoforms and is compatible with FAIMS may benefit from exploring CVs below −20, extending into the positive voltage range.
Our TDP workflow allowed us to identify and characterize unique proteoforms derived from genes with known roles in neurodegenerative diseases. This included determination of the composition of an unknown mass shift present near the iron-binding domain of α-synuclein, which was used to pinpoint the locations of potential iron-binding domains in β- and γsynuclein as well. PARK7 was also found to be modified with a succinyl group on its active site Cys residue. We were able to unambiguously distinguish tau fragments corresponding to 0N, 1N, 3R, and 4R splice variant isoforms individually and describe new proteolytic cleavage sites located within or near several aggregation-seeding hexapeptide repeats. Finally, FAIMS enabled identification of several intact Aβ proteoforms, including the aggregation-prone Aβ1–42, without the need for complex fractionation or purification techniques. In summary, we believe that addition of FAIMS to discover TDP will provide a robust and reproducible method for increasing the proteome available for detection and characterization.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank Tao Liu and Richard Smith for advice and helpful discussions. This work was supported by U01 AG061356 (P.L.D.J.) and R01 AG015819 (D.A.B.). A portion of the research was performed using EMSL (grid.436923.9), a DOE Office of Science User Facility sponsored by the Biological and Environmental Research program. Graphical abstract and image assets within Figure 1 were created using Biorender.com.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.1c00049.
(Table S1) Mean proteoforms observed per condition, total unique proteoforms, unique genes, and proteome sequence coverage; (Figure S1) reproducibility of FAIMS and no FAIMS TDP data; (Figure S2) overlap coefficient heatmap of genes; (Figure S3) consecutive MS1 scans with median charge states of UBB; (Table S2) proteoform characteristics; (Table S3) Uunique Swiss-Prot splice variants and TrEMBL entries; (Figure S4) characterization of unknown modification on α-synuclein; (Figure S5) MS2 fragment coverage map of α-synuclein; (Figure S6-10) and MS2 spectrum and coverage map of succinylated PARK7, Tau(0N-R) fragment, Tau(1N-R) fragment, Tau(−N3R) fragment, and Tau(−N4R) fragment (PDF)
AUTHOR INFORMATION
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jproteome.1c00049
The authors declare no competing financial interest.
Contributor Information
James M. Fulcher, Biological Sciences Division, Pacific Northwest National Laboratory, Richland, Washington 99352, United States.
Aman Makaju, Life Sciences Mass Spectrometry Unit, Thermo Fisher Scientific, San Jose, California 95134, United States.
Ronald J. Moore, Biological Sciences Division, Pacific Northwest National Laboratory, Richland, Washington 99352, United States.
Mowei Zhou, Environmental Molecular Sciences Laboratory, Pacific Northwest National Laboratory, Richland, Washington 99354, United States.
David A. Bennett, Rush Alzheimer’s Disease Center, Rush University Medical Center, Chicago, Illinois 60612, United States
Philip L. De Jager, Department of Neurology, Center for Translational & Computational Neuroimmunology, Columbia University Medical Center, New York 10032, United States
Wei-Jun Qian, Biological Sciences Division, Pacific Northwest National Laboratory, Richland, Washington 99352, United States.
Ljiljana Paša-Tolić, Environmental Molecular Sciences Laboratory, Pacific Northwest National Laboratory, Richland, Washington 99354, United States.
Vladislav A. Petyuk, Biological Sciences Division, Pacific Northwest National Laboratory, Richland, Washington 99352, United States.
REFERENCES
- (1).Nichols E; Szoeke CEI; Vollset SE; Abbasi N; Abd-Allah F; Abdela J; Aichour MTE; Akinyemi RO; Alahdab F; Asgedom SW; Awasthi A; Barker-Collo SL; Baune BT; Béjot Y; Belachew AB; Bennett DA; Biadgo B; Bijani A; Bin Sayeed MS; Brayne C; Carpenter DO; Carvalho F; Catalá-López F; Cerin E; Choi J-YJ; Dang AK; Degefa MG; Djalalinia S; Dubey M; Duken EE; Edvardsson D; Endres M; Eskandarieh S; Faro A; Farzadfar F; Fereshtehnejad S-M; Fernandes E; Filip I; Fischer F; Gebre AK; Geremew D; Ghasemi-Kasman M; Gnedovskaya EV; Gupta R; Hachinski V; Hagos TB; Hamidi S; Hankey GJ; Haro JM; Hay SI; Irvani SSN; Jha RP; Jonas JB; Kalani R; Karch A; Kasaeian A; Khader YS; Khalil IA; Khan EA; Khanna T; Khoja TAM; Khubchandani J; Kisa A; Kissimova-Skarbek K; Kivimäki M; Koyanagi A; Krohn KJ; Logroscino G; Lorkowski S; Majdan M; Malekzadeh R; März W; Massano J; Mengistu G; Meretoja A; Mohammadi M; Mohammadi-Khanaposhtani M; Mokdad AH; Mondello S; Moradi G; Nagel G; Naghavi M; Naik G; Nguyen LH; Nguyen TH; Nirayo YL; Nixon MR; Ofori-Asenso R; Ogbo FA; Olagunju AT; Owolabi MO; Panda-Jonas S; Passos V. M. d. A.; Pereira DM; Pinilla-Monsalve GD; Piradov MA; Pond CD; Poustchi H; Qorbani M; Radfar A; Reiner RC; Robinson SR; Roshandel G; Rostami A; Russ TC; Sachdev PS; Safari H; Safiri S; Sahathevan R; Salimi Y; Satpathy M; Sawhney M; Saylan M; Sepanlou SG; Shafieesabet A; Shaikh MA; Sahraian MA; Shigematsu M; Shiri R; Shiue I; Silva JP; Smith M; Sobhani S; Stein DJ; Tabarés-Seisdedos R; Tovani-Palone MR; Tran BX; Tran TT; Tsegay AT; Ullah I; Venketasubramanian N; Vlassov V; Wang Y-P; Weiss J; Westerman R; Wijeratne T; Wyper GMA; Yano Y; Yimer EM; Yonemoto N; Yousefifard M; Zaidi Z; Zare Z; Vos T; Feigin VL; Murray CJL Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 88–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Beyreuther K; Masters CL Amyloid precursor protein (APP) and beta A4 amyloid in the etiology of Alzheimer’s disease: precursor-product relationships in the derangement of neuronal function. Brain Pathol. 1991, 1, 241–251. [DOI] [PubMed] [Google Scholar]
- (3).Hardy J; Higgins G. Alzheimer’s disease: the amyloid cascade hypothesis. Science 1992, 256, 184–185. [DOI] [PubMed] [Google Scholar]
- (4).Goedert M. Tau protein and the neurofibrillary pathology of Alzheimer’s disease. Trends Neurosci. 1993, 16, 460–465. [DOI] [PubMed] [Google Scholar]
- (5).Iqbal K; Liu F; Gong C-X Tau and neurodegenerative disease: the story so far. Nat. Rev. Neurol. 2016, 12, 15–27. [DOI] [PubMed] [Google Scholar]
- (6).Musiek ES; Holtzman DM Three dimensions of the amyloid hypothesis: time, space and ‘wingmen’. Nat. Neurosci. 2015, 18, 800–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Takeda S. Tau Propagation as a Diagnostic and Therapeutic Target for Dementia: Potentials and Unanswered Questions. Front. Neurosci. 2019, 13, 1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Gregorich ZR; Ge Y. Top-down proteomics in health and disease: challenges and opportunities. Proteomics 2014, 14, 1195–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Zakharova NV; Bugrova AE; Kononikhin AS; Indeykina, The authors declare no competing financial interest. M. I.; Popov IA; Nikolaev EN Mass spectrometry analysis of the diversity of Abeta peptides: difficulties and future perspectives for AD biomarker discovery. Expert Rev. Proteomics 2018, 15, 773–775. [DOI] [PubMed] [Google Scholar]
- (10).Grasso G. Mass spectrometry is a multifaceted weapon to be used in the battle against Alzheimer’s disease: Amyloid beta peptides and beyond. Mass Spectrom. Rev. 2019, 38, 34–48. [DOI] [PubMed] [Google Scholar]
- (11).Andreev VP; Petyuk VA; Brewer HM; Karpievitch YV; Xie F; Clarke J; Camp D; Smith RD; Lieberman AP; Albin RL; Nawaz Z; El Hokayem J; Myers AJ Label-free quantitative LC-MS proteomics of Alzheimer’s disease and normally aged human brains. J. Proteome Res. 2012, 11, 3053–3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Zhang Q; Ma C; Gearing M; Wang PG; Chin LS; Li L. Integrated proteomics and network analysis identifies protein hubs and network alterations in Alzheimer’s disease. Acta Neuropathol. Commun. 2018, 6, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Ping L; Duong DM; Yin L; Gearing M; Lah JJ; Levey AI; Seyfried NT Global quantitative analysis of the human brain proteome in Alzheimer’s and Parkinson’s Disease. Sci Data 2018, 5, 180036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Johnson ECB; Dammer EB; Duong DM; Yin L; Thambisetty M; Troncoso JC; Lah JJ; Levey AI; Seyfried NT Deep proteomic network analysis of Alzheimer’s disease brain reveals alterations in RNA binding proteins and RNA splicing associated with disease. Mol. Neurodegener. 2018, 13, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Ovod V; Ramsey KN; Mawuenyega KG; Bollinger JG; Hicks T; Schneider T; Sullivan M; Paumier K; Holtzman DM; Morris JC; Benzinger T; Fagan AM; Patterson BW; Bateman RJ Amyloid beta concentrations and stable isotope labeling kinetics of human plasma specific to central nervous system amyloidosis. Alzheimers Dement. 2017, 13, 841–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Schindler SE; Bollinger JG; Ovod V; Mawuenyega KG; Li Y; Gordon BA; Holtzman DM; Morris JC; Benzinger TLS; Xiong C; Fagan AM; Bateman RJ High-precision plasma beta-amyloid 42/40 predicts current and future brain amyloidosis. Neurology 2019, 93, e1647–e1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Smith LM; Kelleher NL; Kelleher NL Proteoform: a single term describing protein complexity. Nat. Methods 2013, 10, 186–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Aebersold R; Agar JN; Amster IJ; Baker MS; Bertozzi CR; Boja ES; Costello CE; Cravatt BF; Fenselau C; Garcia BA; Ge Y; Gunawardena J; Hendrickson RC; Hergenrother PJ; Huber CG; Ivanov AR; Jensen ON; Jewett MC; Kelleher NL; Kiessling LL; Krogan NJ; Larsen MR; Loo JA; Ogorzalek Loo RR; Lundberg E; MacCoss MJ; Mallick P; Mootha VK; Mrksich M; Muir TW; Patrie SM; Pesavento JJ; Pitteri SJ; Rodriguez H; Saghatelian A; Sandoval W; Schlüter H; Sechi S; Slavoff SA; Smith LM; Snyder MP; Thomas PM; Uhlén M; Van Eyk JE; Vidal M; Walt DR; White FM; Williams ER; Wohlschlager T; Wysocki VH; Yates NA; Young NL; Zhang B. How many human proteoforms are there? Nat. Chem. Biol. 2018, 14, 206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Smith LM; Kelleher NL Proteoforms as the next proteomics currency. Science 2018, 359, 1106–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Savaryn JP; Catherman AD; Thomas PM; Abecassis MM; Kelleher NL The emergence of top-down proteomics in clinical research. Genome Med. 2013, 5, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Toby TK; Fornelli L; Kelleher NL Progress in Top-Down Proteomics and the Analysis of Proteoforms. Annu. Rev. Anal. Chem. 2016, 9, 499–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Catherman AD; Skinner OS; Kelleher NL Top Down proteomics: facts and perspectives. Biochem. Biophys. Res. Commun. 2014, 445, 683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Chesser AS; Pritchard SM; Johnson GV Tau clearance mechanisms and their possible role in the pathogenesis of Alzheimer disease. Front. Neurol. 2013, 4, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Derisbourg M; Leghay C; Chiappetta G; Fernandez-Gomez FJ; Laurent C; Demeyer D; Carrier S; Buee-Scherrer V; Blum D; Vinh J; Sergeant N; Verdier Y; Buee L; Hamdane M. Role of the Tau N-terminal region in microtubule stabilization revealed by new endogenous truncated forms. Sci. Rep. 2015, 5, 9659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Hanger DP; Wray S. Tau cleavage and tau aggregation in neurodegenerative disease. Biochem. Soc. Trans. 2010, 38, 1016–1020. [DOI] [PubMed] [Google Scholar]
- (26).Quinn JP; Corbett NJ; Kellett KAB; Hooper NM Tau Proteolysis in the Pathogenesis of Tauopathies: Neurotoxic Fragments and Novel Biomarkers. J. Alzheimer’s Dis. 2018, 63, 13–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Delcourt V; Franck J; Quanico J; Gimeno J-P; Wisztorski M; Raffo-Romero A; Kobeissy F; Roucou X; Salzet M; Fournier I. Spatially-Resolved Top-down Proteomics Bridged to MALDI MS Imaging Reveals the Molecular Physiome of Brain Regions. Mol. Cell. Proteomics 2018, 17, 357–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Davis RG; Park H-M; Kim K; Greer JB; Fellers RT; LeDuc RD; Romanova EV; Rubakhin SS; Zombeck JA; Wu C; Yau PM; Gao P; van Nispen AJ; Patrie SM; Thomas PM; Sweedler JV; Rhodes JS; Kelleher NL Top-Down Proteomics Enables Comparative Analysis of Brain Proteoforms Between Mouse Strains. Anal. Chem. 2018, 90, 3802–3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Lubeckyj RA; Basharat AR; Shen X; Liu X; Sun L. Large-Scale Qualitative and Quantitative Top-Down Proteomics Using Capillary Zone Electrophoresis-Electrospray Ionization-Tandem Mass Spectrometry with Nanograms of Proteome Samples. J. Am. Soc. Mass Spectrom. 2019, 30, 1435–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Park H-M; Satta R; Davis RG; Goo YA; LeDuc RD; Fellers RT; Greer JB; Romanova EV; Rubakhin SS; Tai R; Thomas PM; Sweedler JV; Kelleher NL; Patrie SM; Lasek AW Multidimensional Top-Down Proteomics of Brain-RegionSpecific Mouse Brain Proteoforms Responsive to Cocaine and Estradiol. J. Proteome Res. 2019, 18, 3999–4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Shen Y; Tolic N; Piehowski PD; Shukla AK; Kim S; Zhao R; Qu Y; Robinson E; Smith RD; Paša-Tolić L. High-resolution ultrahigh-pressure long column reversed-phase liquid chromatography for top-down proteomics. J. Chromatogr. A 2017, 1498, 99–110. [DOI] [PubMed] [Google Scholar]
- (32).Doucette AA; Tran JC; Wall MJ; Fitzsimmons S. Intact proteome fractionation strategies compatible with mass spectrometry. Expert Rev. Proteomics 2011, 8, 787–800. [DOI] [PubMed] [Google Scholar]
- (33).Ayaz-Guner S; Zhang J; Li L; Walker JW; Ge Y. In vivo phosphorylation site mapping in mouse cardiac troponin I by high resolution top-down electron capture dissociation mass spectrometry: Ser22/23 are the only sites basally phosphorylated. Biochemistry 2009, 48, 8161–8170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Parks BA; Jiang L; Thomas PM; Wenger CD; Roth MJ; Boyne MT 2nd; Burke PV; Kwast KE; Kelleher NL Top-down proteomics on a chromatographic time scale using linear ion trap fourier transform hybrid mass spectrometers. Anal. Chem. 2007, 79, 7984–7991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Sharma S; Simpson DC; Tolic N; Jaitly N; Mayampurath AM; Smith RD; Paša-Tolić L. Proteomic profiling of intact proteins using WAX-RPLC 2-D separations and FTICR mass spectrometry. J. Proteome Res. 2007, 6, 602–610. [DOI] [PubMed] [Google Scholar]
- (36).Smoluch M; Mielczarek P; Drabik A; Silberring J. Online and Offline Sample Fractionation. In Proteomic Profiling and Analytical Chemistry; Ciborowski P, Silberring J, Eds.; Elsevier: Boston, 2016; pp 63–99. [Google Scholar]
- (37).Eiceman GA; Karpas Z; Jr H. Ion Mobility Spectrometry, 3rd ed.; CRC Press, 2013; pp 1–400. [Google Scholar]
- (38).Guevremont R. High-field asymmetric waveform ion mobility spectrometry: a new tool for mass spectrometry. J. Chromatogr. A 2004, 1058, 3–19. [PubMed] [Google Scholar]
- (39).Shvartsburg AA; Noskov SY; Purves RW; Smith RD Pendular proteins in gases and new avenues for characterization of macromolecules by ion mobility spectrometry. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 6495–6500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Purves RW; Guevremont R. Electrospray ionization high-field asymmetric waveform ion mobility spectrometry-mass spectrometry. Anal. Chem. 1999, 71, 2346–2357. [DOI] [PubMed] [Google Scholar]
- (41).Robinson EW; Leib RD; Williams ER The role of conformation on electron capture dissociation of ubiquitin. J. Am. Soc. Mass Spectrom. 2006, 17, 1470–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Shvartsburg AA; Smith RD High-resolution differential ion mobility spectrometry of a protein. Anal. Chem. 2013, 85, 10–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Shvartsburg AA; Li F; Tang K; Smith RD Characterizing the structures and folding of free proteins using 2-D gas-phase separations: observation of multiple unfolded conformers. Anal. Chem. 2006, 78, 3304–3315. [DOI] [PubMed] [Google Scholar]
- (44).Borysik AJH; Read P; Little DR; Bateman RH; Radford SE; Ashcroft AE Separation of beta2-microglobulin conformers by high-field asymmetric waveform ion mobility spectrometry (FAIMS) coupled to electrospray ionisation mass spectrometry. Rapid Commun. Mass Spectrom. 2004, 18, 2229–2234. [DOI] [PubMed] [Google Scholar]
- (45).Melani RD; Srzentic K; Gerbasi VR; McGee JP; Huguet R; Fornelli L; Kelleher NL Direct measurement of light and heavy antibody chains using ion mobility and middle-down mass spectrometry. mAbs 2019, 11, 1351–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Griffiths RL; Hughes JW; Abbatiello SE; Belford MW; Styles IB; Cooper HJ Comprehensive LESA Mass Spectrometry Imaging of Intact Proteins by Integration of Cylindrical FAIMS. Anal. Chem. 2020, 92, 2885–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Griffiths RL; Dexter A; Creese AJ; Cooper HJ Liquid extraction surface analysis field asymmetric waveform ion mobility spectrometry mass spectrometry for the analysis of dried blood spots. Analyst 2015, 140, 6879–6885. [DOI] [PubMed] [Google Scholar]
- (48).Sarsby J; Griffiths RL; Race AM; Bunch J; Randall EC; Creese AJ; Cooper HJ Liquid Extraction Surface Analysis Mass Spectrometry Coupled with Field Asymmetric Waveform Ion Mobility Spectrometry for Analysis of Intact Proteins from Biological Substrates. Anal. Chem. 2015, 87, 6794–6800. [DOI] [PubMed] [Google Scholar]
- (49).Griffiths RL; Creese AJ; Race AM; Bunch J; Cooper HJ LESA FAIMS Mass Spectrometry for the Spatial Profiling of Proteins from Tissue. Anal. Chem. 2016, 88, 6758–6766. [DOI] [PubMed] [Google Scholar]
- (50).Hale OJ; Illes-Toth E; Mize TH; Cooper HJ High-Field Asymmetric Waveform Ion Mobility Spectrometry and Native Mass Spectrometry: Analysis of Intact Protein Assemblies and Protein Complexes. Anal. Chem. 2020, 92, 6811–6816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Schneider JA; Arvanitakis Z; Bang W; Bennett DA Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 2007, 69, 2197–2204. [DOI] [PubMed] [Google Scholar]
- (52).Shliaha PV; Gibb S; Gorshkov V; Jespersen MS; Andersen GR; Bailey D; Schwartz J; Eliuk S; Schwämmle V; Jensen ON Maximizing Sequence Coverage in Top-Down Proteomics By Automated Multimodal Gas-Phase Protein Fragmentation. Anal. Chem. 2018, 90, 12519–12526. [DOI] [PubMed] [Google Scholar]
- (53).Kou Q; Xun L; Liu X. TopPIC: a software tool for top-down mass spectrometry-based proteoform identification and characterization. Bioinformatics 2016, 32, 3495–3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Team RC R: A Language and Environment for Statistical Computing, 2019.
- (55).Park J; Piehowski PD; Wilkins C; Zhou M; Mendoza J; Fujimoto GM; Gibbons BC; Shaw JB; Shen Y; Shukla AK; Moore RJ; Liu T; Petyuk VA; Tolic N; Paša-Tolić L; Smith RD; Payne SH; Kim S. Informed-Proteomics: open-source software package for top-down proteomics. Nat. Methods 2017, 14, 909–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Pfammatter S; Bonneil E; McManus FP; Prasad S; Bailey DJ; Belford M; Dunyach J-J; Thibault P. A Novel Differential Ion Mobility Device Expands the Depth of Proteome Coverage and the Sensitivity of Multiplex Proteomic Measurements. Mol. Cell. Proteomics 2018, 17, 2051–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Piehowski PD; Petyuk VA; Orton DJ; Xie F; Moore RJ; Ramirez-Restrepo M; Engel A; Lieberman AP; Albin RL; Camp DG; Smith RD; Myers AJ Sources of technical variability in quantitative LC-MS proteomics: human brain tissue sample analysis. J. Proteome Res. 2013, 12, 2128–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Bekker-Jensen DB; Martínez-Val A; Steigerwald S; Rüther P; Fort KL; Arrey TN; Harder A; Makarov A; Olsen JV A Compact Quadrupole-Orbitrap Mass Spectrometer with FAIMS Interface Improves Proteome Coverage in Short LC Gradients. Mol. Cell. Proteomics 2020, 19, 716–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Petyuk VA; Chang R; Ramirez-Restrepo M; Beckmann ND; Henrion MYR; Piehowski PD; Zhu K; Wang S; Clarke J; Huentelman MJ; Xie F; Andreev V; Engel A; Guettoche T; Navarro L; De Jager P; Schneider JA; Morris CM; McKeith IG; Perry RH; Lovestone S; Woltjer RL; Beach TG; Sue LI; Serrano GE; Lieberman AP; Albin RL; Ferrer I; Mash DC; Hulette CM; Ervin JF; Reiman EM; Hardy JA; Bennett DA; Schadt E; Smith RD; Myers AJ The human brainome: network analysis identifies HSPA2 as a novel Alzheimer’s disease target. Brain 2018, 141, 2721–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Johnson ECB; Dammer EB; Duong DM; Ping L; Zhou M; Yin L; Higginbotham LA; Guajardo A; White B; Troncoso JC; Thambisetty M; Montine TJ; Lee EB; Trojanowski JQ; Beach TG; Reiman EM; Haroutunian V; Wang M; Schadt E; Zhang B; Dickson DW; Ertekin-Taner N; Golde TE; Petyuk VA; De Jager PL; Bennett DA; Wingo TS; Rangaraju S; Hajjar I; Shulman JM; Lah JJ; Levey AI; Seyfried NT Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat. Med. 2020, 26, 769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Kim M-S; Pinto SM; Getnet D; Nirujogi RS; Manda SS; Chaerkady R; Madugundu AK; Kelkar DS; Isserlin R; Jain S; Thomas JK; Muthusamy B; Leal-Rojas P; Kumar P; Sahasrabuddhe NA; Balakrishnan L; Advani J; George B; Renuse S; Selvan LDN; Patil AH; Nanjappa V; Radhakrishnan A; Prasad S; Subbannayya T; Raju R; Kumar M; Sreenivasamurthy SK; Marimuthu A; Sathe GJ; Chavan S; Datta KK; Subbannayya Y; Sahu A; Yelamanchi SD; Jayaram S; Rajagopalan P; Sharma J; Murthy KR; Syed N; Goel R; Khan AA; Ahmad S; Dey G; Mudgal K; Chatterjee A; Huang T-C; Zhong J; Wu X; Shaw PG; Freed D; Zahari MS; Mukherjee KK; Shankar S; Mahadevan A; Lam H; Mitchell CJ; Shankar SK; Satishchandra P; Schroeder JT; Sirdeshmukh R; Maitra A; Leach SD; Drake CG; Halushka MK; Prasad TSK; Hruban RH; Kerr CL; Bader GD; Iacobuzio-Donahue CA; Gowda H; Pandey A. A draft map of the human proteome. Nature 2014, 509, 575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Schweppe DK; Prasad S; Belford MW; Navarrete-Perea J; Bailey DJ; Huguet R; Jedrychowski MP; Rad R; McAlister G; Abbatiello SE; Woulters ER; Zabrouskov V; Dunyach J-J; Paulo JA; Gygi SP Characterization and Optimization of Multiplexed Quantitative Analyses Using High-Field Asymmetric-Waveform Ion Mobility Mass Spectrometry. Anal. Chem. 2019, 91, 4010–4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Pfammatter S; Bonneil E; McManus FP; Thibault P. Gas-Phase Enrichment of Multiply Charged Peptide Ions by Differential Ion Mobility Extend the Comprehensiveness of SUMO Proteome Analyses. J. Am. Soc. Mass Spectrom. 2018, 29, 1111–1124. [DOI] [PubMed] [Google Scholar]
- (64).Shvartsburg AA Ultrahigh-resolution differential ion mobility separations of conformers for proteins above 10 kDa: onset of dipole alignment? Anal. Chem. 2014, 86, 10608–10615. [DOI] [PubMed] [Google Scholar]
- (65).Shvartsburg AA; Bryskiewicz T; Purves RW; Tang K; Guevremont R; Smith RD Field asymmetric waveform ion mobility spectrometry studies of proteins: Dipole alignment in ion mobility spectrometry? J. Phys. Chem. b 2006, 110, 21966–21980. [DOI] [PubMed] [Google Scholar]
- (66).Shvartsburg AA; Andrzejewski R; Entwistle A; Giles R. Ion Mobility Spectrometry of Macromolecules with Dipole Alignment Switchable by Varying the Gas Pressure. Anal. Chem. 2019, 91, 8176–8183. [DOI] [PubMed] [Google Scholar]
- (67).Pérez-Silva JG; Español Y; Velasco G; Quesada V. The Degradome database: expanding roles of mammalian proteases in life and disease. Nucleic Acids Res. 2016, 44, D351–D355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Hökfelt T; Broberger C; Xu Z-QD; Sergeyev V; Ubink R; Diez M. Neuropeptides–an overview. Neuropharmacology 2000, 39, 1337–1356. [DOI] [PubMed] [Google Scholar]
- (69).Kim Y; Bark S; Hook V; Bandeira N. NeuroPedia: neuropeptide database and spectral library. Bioinformatics 2011, 27, 2772–2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Grewal RN; El Aribi H; Harrison AG; Siu KWM; Hopkinson AC Fragmentation of protonated tripeptides: The proline effect revisited. J. Phys. Chem. b 2004, 108, 4899–4908. [Google Scholar]
- (71).Bleiholder C; Suhai S; Harrison AG; Paizs B. Towards understanding the tandem mass spectra of protonated oligopeptides. 2: The proline effect in collision-induced dissociation of protonated Ala-Ala-Xxx-Pro-Ala (Xxx = Ala, Ser, Leu, Val, Phe, and Trp). J. Am. Soc. Mass Spectrom. 2011, 22, 1032–1039. [DOI] [PubMed] [Google Scholar]
- (72).Breci LA; Tabb DL; Yates JR 3rd; Wysocki VH Cleavage N-terminal to proline: analysis of a database of peptide tandem mass spectra. Anal. Chem. 2003, 75, 1963–1971. [DOI] [PubMed] [Google Scholar]
- (73).Haverland NA; Skinner OS; Fellers RT; Tariq AA; Early BP; LeDuc RD; Fornelli L; Compton PD; Kelleher NL Defining Gas-Phase Fragmentation Propensities of Intact Proteins During Native Top-Down Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2017, 28, 1203–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Donnelly DP; Rawlins CM; DeHart CJ; Fornelli L; Schachner LF; Lin Z; Lippens JL; Aluri KC; Sarin R; Chen B; Lantz C; Jung W; Johnson KR; Koller A; Wolff JJ; Campuzano IDG; Auclair JR; Ivanov AR; Whitelegge JP; Paša-Tolić L; Chamot-Rooke J; Danis PO; Smith LM; Tsybin YO; Loo JA; Ge Y; Kelleher NL; Agar JN Best practices and benchmarks for intact protein analysis for top-down mass spectrometry. Nat. Methods 2019, 16, 587–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Makarov A; Denisov E. Dynamics of ions of intact proteins in the Orbitrap mass analyzer. J. Am. Soc. Mass Spectrom. 2009, 20, 1486–1495. [DOI] [PubMed] [Google Scholar]
- (76).Thibaudeau TA; Anderson RT; Smith DM A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers. Nat. Commun. 2018, 9, 1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Kurtishi A; Rosen B; Patil KS; Alves GW; Møller SG Cellular Proteostasis in Neurodegeneration. Mol. Neurobiol. 2019, 56, 3676–3689. [DOI] [PubMed] [Google Scholar]
- (78).Kong AT; Leprevost FV; Avtonomov DM; Mellacheruvu D; Nesvizhskii AI MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nat. Methods 2017, 14, 513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Binolfi A; Rasia RM; Bertoncini CW; Ceolin M; Zweckstetter M; Griesinger C; Jovin TM; Fernández CO Interaction of alpha-synuclein with divalent metal ions reveals key differences: a link between structure, binding specificity and fibrillation enhancement. J. Am. Chem. Soc. 2006, 128, 9893–9901. [DOI] [PubMed] [Google Scholar]
- (80).Lu Y; Prudent M; Fauvet B; Lashuel HA; Girault HH Phosphorylation of alpha-Synuclein at Y125 and S129 alters its metal binding properties: implications for understanding the role of alpha-Synuclein in the pathogenesis of Parkinson’s Disease and related disorders. ACS Chem. Neurosci. 2011, 2, 667–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Duce JA; Wong BX; Durham H; Devedjian JC; Smith DP; Devos D. Post translational changes to alpha-synuclein control iron and dopamine trafficking; a concept for neuron vulnerability in Parkinson’s disease. Mol. Neurodegener. 2017, 12, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Blatnik M; Thorpe SR; Baynes JW Succination of proteins by fumarate: mechanism of inactivation of glyceraldehyde-3phosphate dehydrogenase in diabetes. Ann. N. Y. Acad. Sci. 2008, 1126, 272–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Goedert M; Spillantini MG; Jakes R; Rutherford D; Crowther RA Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 1989, 3, 519–526. [DOI] [PubMed] [Google Scholar]
- (84).Liu F; Gong C-X Tau exon 10 alternative splicing and tauopathies. Mol. Neurodegener. 2008, 3, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Chen H-H; Liu P; Auger P; Lee SH; Adolfsson O; ReyBellet L; Lafrance-Vanasse J; Friedman BA; Pihlgren M; Muhs A; Pfeifer A; Ernst J; Ayalon G; Wildsmith KR; Beach TG; van der Brug MP Calpain-mediated tau fragmentation is altered in Alzheimer’s disease progression. Sci. Rep. 2018, 8, 16725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Cicognola C; Brinkmalm G; Wahlgren J; Portelius E; Gobom J; Cullen NC; Hansson O; Parnetti L; Constantinescu R; Wildsmith K; Chen H-H; Beach TG; Lashley T; Zetterberg H; Blennow K; Höglund K. Novel tau fragments in cerebrospinal fluid: relation to tangle pathology and cognitive decline in Alzheimer’s disease. Acta Neuropathol. 2019, 137, 279–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Hong M; Zhukareva V; Vogelsberg-Ragaglia V; Wszolek Z; Reed L; Miller BI; Geschwind DH; Bird TD; McKeel D; Goate A; Morris JC; Wilhelmsen KC; Schellenberg GD; Trojanowski JQ; Lee VM Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science 1998, 282, 1914–1917. [DOI] [PubMed] [Google Scholar]
- (88).Goedert M; Jakes R. Expression of separate isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 1990, 9, 4225–4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Kadavath H; Jaremko M; Jaremko Ł; Biernat J; Mandelkow E; Zweckstetter M. Folding of the Tau Protein on Microtubules. Angew Chem. Int. Ed. 2015, 54, 10347–10351. [DOI] [PubMed] [Google Scholar]
- (90).Kadavath H; Hofele RV; Biernat J; Kumar S; Tepper K; Urlaub H; Mandelkow E; Zweckstetter M. Tau stabilizes microtubules by binding at the interface between tubulin heterodimers. Proc. Natl. Acad. Sci. U.S.A. 2015, 112, 7501–7506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Schwalbe M; Kadavath H; Biernat J; Ozenne V; Blackledge M; Mandelkow E; Zweckstetter M. Structural Impact of Tau Phosphorylation at Threonine 231. Structure 2015, 23, 1448–1458. [DOI] [PubMed] [Google Scholar]
- (92).Seidler PM; Boyer DR; Rodriguez JA; Sawaya MR; Cascio D; Murray K; Gonen T; Eisenberg DS Structure-based inhibitors of tau aggregation. Nat. Chem. 2018, 10, 170–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Zhang W; Tarutani A; Newell KL; Murzin AG; Matsubara T; Falcon B; Vidal R; Garringer HJ; Shi Y; Ikeuchi T; Murayama S; Ghetti B; Hasegawa M; Goedert M; Scheres SHW Novel tau filament fold in corticobasal degeneration. Nature 2020, 580, 283–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Ait-Bouziad N; Lv G; Mahul-Mellier AL; Xiao S; Zorludemir G; Eliezer D; Walz T; Lashuel HA Discovery and characterization of stable and toxic Tau/phospholipid oligomeric complexes. Nat. Commun. 2017, 8, 1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).von Bergen M; Friedhoff P; Biernat J; Heberle J; Mandelkow E-M; Mandelkow E. Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif (306VQIVYK311) forming beta structure. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 5129–5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Jeganathan S; von Bergen M; Brutlach H; Steinhoff H-J; Mandelkow E. Global hairpin folding of tau in solution. Biochemistry 2006, 45, 2283–2293. [DOI] [PubMed] [Google Scholar]
- (97).Wildburger NC; Esparza TJ; LeDuc RD; Fellers RT; Thomas PM; Cairns NJ; Kelleher NL; Bateman RJ; Brody DL Diversity of Amyloid-beta Proteoforms in the Alzheimer’s Disease Brain. Sci. Rep. 2017, 7, 9520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Schauer SP; Mylott WR Jr.; Yuan M; Jenkins RG; Rodney Mathews W; Honigberg LA; Wildsmith KR Preanalytical approaches to improve recovery of amyloid-beta peptides from CSF as measured by immunological or mass spectrometry-based assays. Alzheimer’s Res. Ther. 2018, 10, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.