

Abstract
The integration of DNA methylation and transcriptional state within single cells is of broad interest. Several single-cell dual- and multi-omics approaches have been reported that enable further investigation into cellular heterogeneity, including the discovery and in-depth study of rare cell populations. Such analyses will continue to provide important mechanistic insights into the regulatory consequences of epigenetic modifications. We recently reported a new method for profiling the DNA methylome and transcriptome from the same single cells in a cancer research study. Here, we present details of the protocol and provide guidance on its utility. Our Smart-RRBS (reduced representation bisulfite sequencing) protocol combines Smart-seq2 and RRBS and entails physically separating mRNA from the genomic DNA. It generates paired epigenetic promoter and RNA-expression measurements for ~24% of protein-coding genes in a typical single cell. It also works for micro-dissected tissue samples comprising hundreds of cells. The protocol, excluding flow sorting of cells and sequencing, takes ~3 d to process up to 192 samples manually. It requires basic molecular biology expertise and laboratory equipment, including a PCR workstation with UV sterilization, a DNA fluorometer and a microfluidic electrophoresis system.
Introduction
Since the initial report of single-cell transcriptomics1, similar technologies have evolved as central tools in most areas of biology2,3. Many groups independently developed technologies for profiling single-cell genomes3-5, transcriptomes6-8 and various types of epigenomic modifications, including DNA methylation9-12, histone modifications13, chromatin organization and accessibility14-17. Over the past decade, a few examples have focused, for instance, on dissecting cellular heterogeneity, characterizing cell types, discovering rare cells in a heterogeneous population, tracking cell evolution and investigating the diversity of microbial populations2,10,18,19. Applications of these technologies have notably expanded our understanding of fundamental questions in many areas of biology, which cannot be easily answered by sequencing bulk samples18,19. For instance, single-cell genome sequencing of primary tumors has helped refine our view of intra-tumor heterogeneity, clonal evolution and the function of rare cells in tumor development3,20.
To further investigate the connections between multiple layers of genomics information within the same single cell, several groups, including ours, have developed sequencing methods to measure multiple modalities of the cells simultaneously. Some of these methods include joint profiling of genome and transcriptome21,22, methylome and transcriptome10,23,24, methylome and chromatin organization25,26 and genome, methylome and transcriptome27. Dual- and multimodal single-cell ‘omics’ has improved our ability to interrogate and understand the biological networks in heterogeneous tissues and during development. It is foreseeable that these technologies will be of broad interest in many research areas such as cancer, immunology, neurobiology and microbiology in years to come.
Our new method was applied in a recent cancer research study10. It integrates our multiplexed single-cell reduced representation bisulfite sequencing (Msc-RRBS)9 with Smart-seq228 and generates both DNA methylation and transcriptome data for hundreds of flow-sorted single cells in batches of 96 or 192 cells when processed manually. Notably, the exact same protocol can also be applied to low-input multi-cell bulk samples and works well in ranges of 50 to 1,000 cells per sample (e.g., for micro-dissected tissues from mouse embryos)29, without explicit nucleic acid extraction and purification. Using Smart-RRBS, we have generated thousands of joint RRBS and RNA-seq profiles from single cells and low-input tissue samples with highly reproducible results10,29.
Development of the protocol
RRBS was developed more than a decade ago as a cost-effective method to examine a biologically relevant ‘reduced representation’ of mammalian methylomes30 and has been widely used by many groups ever since31-35. The latest improvements optimize and streamline the work flow, thereby allowing hundreds of Msc-RRBS libraries to be generated within only 2 d9,10. Msc-RRBS generates fewer unwanted adapter-only side products and therefore has a higher rate of mapped reads than other single-cell bisulfite-sequencing methods that use random primers with index tails to copy bisulfite-converted single-strand DNA such as single-cell bisulfite sequencing (scBS-Seq)12,36 and single-nucleus methylcytosine sequencing (snmC-seq2)37. Our aim was to examine both methylome and transcriptome and maximize the yield of protein-coding genes with paired epigenetic promoter and RNA-expression measurements at the single-cell level. As such, we set out to combine Smart-seq238, one of the most sensitive single-cell RNA-seq protocols6,8 and Msc-RRBS, which enriches CpG-rich sequences such as CpG islands (CGIs), features that co-localize with ~70% of annotated promoters and whose methylated state is strongly correlated with transcriptional silencing39.
Following the paradigm of single-cell genome and transcriptome (scG&T)22,40 and single-cell methylome and transcriptome (scM&T) sequencing, a method that combines scBS-Seq and Smart-seq223, Smart-RRBS relies on physically separating mRNA and genomic DNA with minor modifications to the original protocol for pulling down poly(A)+ mRNAs hybridized to biotinylated oligo (dT) reverse transcription (RT) primer on streptavidin beads. To optimize this crucial step, we tested the RT-priming activity of biotinylated oligo(dT) primer conjugated to four different types of streptavidin beads (Dynabeads M-280 and M-270 as well as MyOne T1 and C1). To do so, we mixed equal volumes of each bead type with oligo(dT) primer and performed RT reactions in aliquots of a cell lysate. M-280 beads performed best in terms of quantity and size distribution of PCR-amplified cDNA, even though they are larger in size and have a lower biotin-binding capacity than the MyOne streptavidin C1 beads used in scG&T and scM&T protocols. After testing different amounts of RT primer and beads in single-cell Smart-RRBS experiments, we reduced both, thereby lowering cost and improving handling (less bead volume during fiddly washing steps) without affecting library quality. Second, we added Tween-20 at a low concentration to all bead-pretreat buffers and capture reagents to decrease bead clumping, unspecific binding and loss of material. Lastly, unlike earlier RRBS-based dual-omics methods24,27, we sort single cells into 96-well PCR plates and carry out almost all downstream procedures by using multichannel pipettes, thus increasing throughput without expensive automation equipment. Most molecular biology laboratories will thus be able to generate Smart-RRBS libraries following this protocol (Fig. 1).
Fig. 1 ∣. Schematic overview of the Smart-RRBS protocol.

After deposition by flow cytometry of single cells into a 96-well plate containing lysis buffer and optional flash freezing and storage at −80 °C, poly(A)+ mRNAs are pulled down by using M-280 streptavidin beads (shown as black dots) coated with a 5′ biotinylated oligo(dT) RT primer. Genomic DNA (gDNA) in the supernatant is cleaned up on AMPure beads (shown as a red dot). After separation of mRNA from genomic DNA, single-cell RRBS and Smart-seq2 libraries are generated separately. Not counting the time it takes to sort cells into single-cell plates at the beginning (Steps 1-5) and to generate Illumina sequencing data at the end (Step 96), it takes ~3 d to carry out both branches of the Smart-RRBS protocol when Smart-seq2 and RRBS library construction steps are staggered as described in the step-by-step procedure.
Overview of the procedure
Briefly, after physically separating poly(A)+ mRNAs and genomic DNA (Steps 15–21), mRNAs captured from each single cell are reverse transcribed in the presence of a template-switching oligonucleotide (TSO) to enable subsequent PCR amplification using a single primer (Steps 22–27). The PCR products are purified, quantified and normalized (Steps 66–71) and converted into a pool of Illumina single-cell RNA-seq libraries by using the Nextera XT DNA tagmentation kit (Steps 72–87). Separately, the mRNA-depleted nucleic acid fraction is cleaned up on AMPure beads (Steps 28–30), and the genomic DNA is digested by MspI or by MspI and HaeIII (double-digest). Fragment ends are blunted and dA-tailed by Klenow exo- DNA polymerase and ligated to indexed methylated adapters (Steps 31–45). All these reactions are carried out within the same well and without intermediate clean-up steps, to minimize DNA loss. Next, 24 indexed ligation reactions are pooled together (Steps 46–55) and treated with sodium bisulfite to convert unmethylated cytosine to uracil while leaving methylated cytosine unaltered (Steps 56–58). Bisulfite-converted restriction fragments with adapters on both ends are amplified with PCR primers carrying pool-specific indexes (Steps 59–65). Thus, multiple library pools from up to 24 single cells can be combined and sequenced on the same lane of an Illumina sequencer, and the sequencing reads can be unambiguously assigned to individual wells on each original 96-well plate. Size selection on an agarose gel (Steps 88–95) removes adapter dimers and, optionally, allows preparing and sequencing two separate size fractions to minimize size bias on the sequencer. Of note, because the 6-bp well-level indexes in our methylated adapters are in the double-stranded stem and thus attached to both ends of a restriction fragment, each legitimate RRBS read pair has the same inline barcode at the beginning of the read, whereas illegitimate pairs with discordant inline barcodes caused by cross-over events during pooled PCR or barcode ‘hopping’ on patterned sequencing flowcells can be eliminated from the data.
After testing robustness and reproducibility, we used Smart-RRBS in two large biological studies where we analyzed single B lymphocytes from leukemia patient cohorts10,41 and micro-dissected paired murine extraembryonic ectoderm and epiblast tissue samples29. When applied to samples containing multiple cells, cell numbers from different samples should be within a narrow range (ideally within twofold), so that samples can be pooled after adapter ligation. Otherwise, we recommend grouping samples by cell number or processing individual samples all the way through PCR amplification and pooling the final libraries before sequencing.
Applications
DNA methylation within certain promoter contexts is clearly correlated with transcriptional repression42. However, beyond those examples, the relationship between the methylation of different genomic features and genome regulation is more complicated43. Our approach provides a powerful tool to help dissect the direct correlation and interdependencies of DNA methylation and transcription in the same cell. Application of the method in development and disease should provide important new insights that will improve our understanding of the underlying mechanisms. Specifically, simultaneous profiling of methylome and transcriptome from single cells has been applied in our recent study of heterogeneity and clonal evolution in chronic lymphocytic leukemia, where the RNA-seq data also provided a readout of the genetic mutation status of key genes10. In addition, we envision applications in other areas of cancer research, such as identifying rare tumor cells, studying the mechanism of drug resistance and investigating tumor cell subpopulations. Without additional sequencing, another layer of genomic information, copy number variation, has been reported in a similar approach, indicating that this method can supply more genomic information if needed27.
Comparison with other methods
Three alternative methods to sequence both the methylome and transcriptome of the same single cells have been published in the scientific literature (Table 1), all of which worked well in their respective research projects23,24,27. Like Smart-RRBS, scM&T-seq23 involves physically separating mRNA and genomic DNA by using streptavidin-coated magnetic beads and a biotinylated oligo(dT) RT primer and generating RNA-seq libraries by Smart-seq2, while genomic DNA is bisulfite-converted first and then copied by two cycles of random priming and extension followed by PCR. The challenge of scBS-seq includes lower mapping rates (means of 16% for the dual scM&T-seq protocol23 and 8–50% for standalone scBS-Seq12,36) than Smart-RRBS (>80%; see Anticipated results). Of note, unlike RRBS, the intentionally unbiased sampling of the methylome by scBS-seq does not enrich for CpG-containing reads and CpG-rich gene-regulatory regions. Therefore, scM&T-seq covers about three times fewer unique CpGs than Smart-RRBS per 1 million reads (212,000 versus 639,000), but can cover a much larger fraction of CpGs in the genome at the single-cell level than a reduced-representation approach (e.g., 26% with 31 million reads versus up to 9% in our benchmark data set). The other two methods, scTrio-seq27 and single-cell methylome and transcriptome sequencing (scMT-seq)24, require the manual separation of nuclei and cytoplasm from individual cells, which is low throughput and demands special training to carry out the assay. In addition, both methods incorporate the original sc-RRBS protocol, which requires two rounds of PCR amplification and a total of 50 PCR cycles to generate a sequencing library from each single cell11,44.
Table 1 ∣.
Comparison of methods for profiling the methylome and transcriptome of single cells
| Smart-RRBS | scM&T-seq23 | scTrio-seq27 | scMT-seq24 | |
|---|---|---|---|---|
| Cell-isolation technique | FACS or manual isolation | FACS or manual isolation | Manual isolation | Manual isolation |
| Separation of mRNA and genomic DNA | Dynabeads M-280 streptavidin | Dynabeads MyOne streptavidin C1 | Manual isolation of nucleus | Manual isolation of nucleus |
| Batch size (single cells) | 96–192 | 96 | Dozens | Dozens |
| Protocol for methylome | Msc-RRBS9 | scBS-seq12 | sc-RRBS44 | sc-RRBS44 |
| Protocol for transcriptome | Smart-seq228 | Smart-seq228 | mRNA-seq1 | Smart-seq228 |
| CpGs covered per cell (million) | 1.4a | 2.5b | 1.5c | 0.5d |
| Fraction of CpGs covered (%) | 5a | 9b | 5c | 2d |
Our method combines the higher yield and greater throughput of our Msc-RRBS protocol with the widely used Smart-seq2 protocol and features inline barcodes on purposely truncated Illumina adapters, such that 24 samples can be pooled early in the protocol and processed together after adding an inert carrier DNA, thereby simplifying the workflow and minimizing inevitable losses during the procedure. A key benefit of our truncated adapters is the ease of removal of the relatively short adapter dimers by bead clean-up before PCR amplification. Moreover, base-balanced inline indexes at the beginning of each sequencing read obviate non-standard sequencing modes such as the dark cycles required for first generation RRBS libraries45. Most steps in the Smart-RRBS protocol are carried out in 96-well plates and are amenable to automation to increase the throughput further. For instance, we note that clean separation and recovery of mRNA and genomic DNA fractions for scG&T-seq can be performed on conventional liquid-handling robots22, and both Smart-seq2 and Smart-seq346 can be easily implemented on a relatively low-cost automated Formulatrix Mantis liquid dispenser platform that also accommodates reagent additions to 96-well plates during RRBS library construction.
Limitations
To date, we have generated Smart-RRBS data from thousands of single cells and hundreds of samples containing variable cell numbers (50–1,000 cells)10,29,41. Nonetheless, the throughput and scale of Smart-RRBS is limited, and the cost per single cell is high compared to standalone RNA-seq and other omics protocols that use emulsion droplets rather than wells on a plate to physically partition single cells before molecular barcoding47,48, or split-pool combinatorial indexing approaches that allow computational partitioning of thousands of single-cell omics data sets49,50. To our knowledge, neither high-throughput approach has been developed for joint single-cell methylome and transcriptome sequencing. Our sc-RRBS has no unique molecular indexing (UMI) capability. However, UMI-based removal or binning of RRBS PCR duplicates is far less critical than for counting assays such as RNA-seq. Cells have few copies of the genome to begin with, and, given the sparse genome coverage, RRBS reads aligning to the same locus are unlikely to come from different chromosomes. We note that Smart-seq2 also lacks UMI capability for proper removal of PCR duplicates. However, this feature has been implemented in the recent sequel protocol Smart-seq3, which has even better detection sensitivity46. Furthermore, as with other single-cell RNA-seq protocols, the number of distinct genes detected is highly dependent on cell type, with larger cells generally yielding higher numbers of cytoplasmic mRNAs, as well as sample collection, preservation and flow-sorting procedures.
Intrinsic limitations that our protocol shares with others is the low copy number of each genomic locus in a single diploid cell (two copies of double-strand DNA during G1/G0 phases of the cell cycle), the chemically harsh sodium bisulfite treatment that causes DNA degradation and the inability to PCR-amplify before bisulfite conversion because this would erase the methylation marks. Moreover, the mRNA and genomic DNA separation step inevitably leads to some loss of material. The main causes of losses during RRBS library construction are incomplete restriction digestion, end-repair and, particularly, incomplete adapter ligation at extremely low concentrations. In contrast to the post-bisulfite-conversion library construction approach used in scM&T, restriction fragments decorated with adapters on both ends are still vulnerable to fragmentation during the bisulfite conversion and thus exclusion from subsequent PCR amplification. As a consequence, coverage of the methylome is sparse, although not necessarily sparser than for other single-cell methods that suffer from other inefficiencies (e.g., undesired side products and lower mapping efficiencies). The typical CpG coverage at the single-cell level by Smart-RRBS is slightly lower than by standalone Msc-RRBS. It typically reaches 10–20% of the coverage delivered by bulk RRBS or pseudo-bulk data sets aggregated from sequencing reads obtained from multiple single cells. For example, in our benchmark data set from human embryonic stem cells, 1.54 million (median) and 1.38 million (mean) unique CpGs were covered per passing single cell, whereas pseudo-bulk data aggregated from all 80 passing single cells covered 10.5 million CpGs. Because some of these losses appear to be random rather than systematic dropout events, the methylation status of the same CpG site, or of the same locus if it contains few restriction fragments in the RRBS size range, cannot be easily compared across many single cells. In our benchmark data, 51%, 14%, 0.3%, 0.02% and 0.005% of the pseudo-bulk 10.5 million CpGs in the aggregate data set are informative across 4, 8, 16, 32 and 64 cells, respectively. However, one can compare the methylation state of the larger functional unit such as a CGI without comparing the exact same CpG. Hence, at a minimum coverage threshold of one CpG per CGI, 62%, 33%, 15%, 4%, and 0.2% of the 26,687 CGIs in the aggregate sample data set can be compared across 4, 8, 16, 32 or 64 cells, respectively.
Another limitation that our protocol shares with others is low throughput and high cost per cell compared to droplet- or combinatorial indexing–based single-omics assays. Although the costs of both branches of Smart-RRBS are about the same, the main respective cost drivers are not. Amortizing the initial investment cost in methylated adapters over 48 plates (4,608 single cells), ~2/3 of the cost of RRBS is for Illumina sequencing. In contrast, ~80% of the cost for Smart RNA-seq is for other reagents, particularly for Illumina Nextera kits to convert PCR-amplified cDNA from each cell into a sequencing library. Our protocol reduces the cost by scaling the Nextera tagmentation reaction volume down to half. Substantial further cost savings can be achieved by using home-brew reagents including generic Tn5 transposase, which can be produced in the laboratory51,52 or purchased commercially, and standard desalted oligonucleotides as a generic substitute for indexed Nextera PCR primers from Illumina—albeit at the expense of additional development efforts.
Experimental design
Cell preservation before cell sorting
Some biological studies (e.g., examining the downstream effects of perturbations by gene knock-down or drug treatment) encompass a Smart-RRBS time course to follow DNA methylation and transcriptome over time. For such experiments, we recommend preserving the bulk cell samples collected at different time points in Qiagen RNAprotect cell reagent following the manufacturer’s instructions and recommendations for storage. For most cell types, this treatment preserves the RNA and DNA without cell lysis and leakage. The samples can then be stored for ~1 week at room temperature and for ≤4 weeks at 2–8 °C until cell sorting, thereby obviating the need for sorting the cells immediately after each collection time point or freezing and thawing the bulk cell samples before cell sorting. In our limited experience, we did not observe any adverse effects on data quality from stabilization and storage. Our benchmark Smart-RRBS data in Anticipated data came from cells that had been preserved in this manner for ≥9 d before cell sorting.
Processing single cells sorted into alternative lysis buffers
We note that it is not always possible—and not always necessary—to perform a custom cell sort specifically for Smart-RRBS. For example, in our experience, cells that have already been sorted into Qiagen TCL buffer (cat. no. 1031576) and stored at −80 °C for standalone single-cell RNA-seq have worked well for Smart-RRBS.
Restriction digest
The Smart-RRBS protocol and our truncated adapters with 3′-T overhang are compatible with all cut sites that are blunt or can be filled in and appended by a 3′-dA overhang. Traditionally, most RRBS libraries for Illumina sequencing have been prepared from DNA samples digested with MspI (cut site C∣CGG) to enrich the CpG-rich fraction of mammalian genomes and ensure the presence of at least one CpG (within the MspI site) at the beginning of each sequencing read30. An MspI+TaqI (T∣CGA) double digest increases the representation of CpG-dense regions, whereas combining MspI with restriction enzymes that do not cut at CpG sites such as ApeK1 (G∣CWGC) or HaeIII (GG∣CC) adds genomic features with lower CpG frequencies to the reduced representation, including many promoters and enhancers53. We prefer the MspI+HaeIII double-digest option for extended coverage because it covers ≤30% of all CpGs in the human genome by RRBS in silico54 and because the CutSmart buffer recommended for both MspI and HaeIII is compatible with all subsequent enzymatic reactions, including adapter ligation. Of course, the downside of extending the reduced representation fraction is that more sequencing reads are required to approach saturation of the sequencing libraries. For example, ~1.7 million reads are sufficient to reach 80% saturation of the unique CpG content of MspI-only sc-RRBS libraries10, whereas ~5 million reads are required to reach 80% saturation for MspI+HaeIII double digests. Another advantage of the MspI single digest is that it facilitates direct comparison of restriction fragments covered by both single-cell and pre-existing bulk MspI RRBS data.
Control samples
When performing Smart-RRBS for the first time, or the first time with a different cell type, we recommend preparing a test 96-well cell plate with empty control wells and wells that contain more than one single cell (e.g., 3, 10, 30 and 100 cells), generating Smart-RRBS data and comparing the yield of covered CpGs and transcripts detected in these wells with corresponding pseudo-bulk measurements obtained by aggregating data from an equivalent number of cells processed individually. After this initial test, it is generally sufficient to include two empty wells on the first two production plates for a project, one well used as a blank control and the other as a positive control for both RRBS and RNA-seq by adding 1 μl containing 6 pg of purified genomic DNA and 1 μl of a 10−5 dilution of standard RNA mixtures from the External RNA Controls Consortium (ERCC). To use the ERCC RNA as a true internal control for normalizing single-cell RNA-seq data, a 20-fold lower amount (1 μl of a 5 x 10−7 dilution per cell) than when used as a positive control sample may be spiked into the lysis buffer before cell sorting. We assess the efficiency of the bisulfite conversion by determining the C to T conversion of presumably unmethylated Cs in non-CpG (i.e., CpH) context in genomic RRBS reads from the cell itself rather than from spiked-in internal controls such as synthetic oligonucleotides or bacteriophage lambda DNA, which do not display conversion issues due to residual DNA-bound nuclear proteins. CpH dinucleotides include CpA and CpT, which can be naturally methylated to some degree in certain cell types, including neurons and embryonic stem cells. C-to-T conversion analysis on CpC sites is less confounded by true non-canonical methylation.
Quality control (QC) assays
We recommend QC assays at multiple stages of the workflow, particularly when performing Smart-RRBS for the first time. After Step 27, we determine the DNA concentration of all PCR-amplified cDNA samples and perform at least a spot test of the size distribution on eight randomly selected wells per 96-well plate. We recommend a similar QC routine for the PCR-amplified Nextera XT Smart-seq2 libraries in Step 86. Bisulfite conversion and recovery of small amounts of DNA can be challenging without prior experience. We recommend performing the optional semi-quantitative PCR assay (Steps 59–63) on at least one sample of bisulfite-converted material from 24 cells from each batch to determine the minimal PCR cycle number required to generate a sufficient amount of library DNA for downstream sequencing. Real-time qPCR assays typically used for quantification of sequencing libraries after PCR amplification are another option to determine the appropriate cycle number, as long as they do not include uracil-DNA glycosylase to degrade uracil-containing DNA and proofreading DNA polymerases that stall at uracil. However, in our experience, real-time qPCR data are often confounded by adapter dimers. Finally, all PCR-amplified RRBS libraries must be quantified, and their size distribution must be examined by BioAnalyzer or TapeStation before and after the optional final size selection on a gel (Fig. 2). The prominent bands in RRBS libraries represent satellite repeat sequences in the genome. The pattern is highly reproducible and characteristic for each genome and combination of restriction enzymes and allows a simple visual QC of the restriction digest before sequencing.
Fig. 2 ∣. Electrophoretic quality control.

a, Agilent bioanalyzer gel-like images of PCR-amplified cDNA derived from 11 single cells. Typical sizes range from 300 bp to 7 kb with modes between 1 and 2 kb. b, Agilent TapeStation gel-like images of 12 single-cell RNA-seq libraries. Typical Smart-seq2 libraries contain products ranging from 300 bp to 1 kb with a mode around 500 bp, including sequencing adapters. c, Bioanalyzer run of four sets of RRBS libraries after PCR amplification (Step 65). The pronounced bands represent repetitive restriction fragments from satellite sequences in the human genome. d, Final sequencing libraries QCed and quantitated on a Bioanalyzer. Lanes 1 and 2 are small and large size cuts of RRBS library pools; lane 3 is a pooled set of Smart-seq2 libraries. e, Rare examples of Smart-RRBS failures. Shown are gel-like Bioanalyzer images of RRBS libraries (lanes 1 and 2) that contained essentially nothing but PCR-amplified adapter dimers (that run as two pronounced bands), possibly because of improper cell sorting (empty wells or damaged DNA-less debris) or incomplete cell lysis preventing the release of nuclear DNA, and failed cDNA amplifications (lanes 3 and 4) caused by either improper cell sorting (empty wells, cell damage and leakage of cytoplasmic mRNA) or any other conceivable reason as described in the trouble-shooting section.
Size fractionation of RRBS libraries
Running the RRBS libraries on an agarose gel (Steps 88–95) removes adapter dimers before sequencing. Preparing small and large size fractions is optional. We recommend size fractionation when optimal coverage at the single-cell level is crucial for answering the biological research question. Sequencing the two size fractions in separate lanes minimizes under-representation of large restriction fragments because of competitive bias against large fragments during cluster formation on the sequencing flow cell and concomitant reduction of CpG and CGI coverage in the RRBS data set.
Sequencing guidelines
To maximize the number of CpGs per library fragment, we sequence the RRBS libraries with paired-end 75–100-base reads on most Illumina instruments, except for large projects where paired-end 150-base reads on HiSeq X or NovaSeq S4 are more cost effective. The forward read corresponds to the C-poor bisulfite-converted genomic DNA; the reverse read is the G-poor complementary strand. To improve the sequencing quality on HiSeq 2500 instruments, we spike 10% of a PhiX, or of another base-balanced complex library, into the RRBS library pool and run the flow cell at a lower cluster density (~800,000/mm2). For patterned flow cells, we spike in 12% (HiSeq 4000 or HiSeq X) or 20% (NovaSeq) of a balanced library. To minimize adverse effects of sequencing bias against larger fragments, we run the smaller and the larger size-range libraries in separate lanes in a ratio of 3:1 and merge the reads before further analysis. For example, 3 + 1 lanes of HiSeq 2500 for 96 cells, or 3 + 1 lanes of HiSeq 4000 for 192 cells, generate ~2.5 million passing purity-filtered fully demultiplexed RRBS read pairs per cell on average. For RNA-seq, one lane of HiSeq 2500 or HiSeq 4000 with 50–75-base paired-end reads is adequate for 96 or 192 cells, respectively. Increasing the RNA-seq read lengths has negligible effects on detection sensitivity and expression values.
Processing and analysis of sequencing data
Processing and analysis of sequencing data for quality control purposes, as well as for the biological analysis, is an integral part of any Smart-RRBS experiment. In this section, we refer to packages of analysis tools that we use routinely. Briefly, FastQC, a widely used tool available on the Babraham Bioinformatics website (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/), is used for initial assessment of the technical quality of both the RNA-seq and RRBS-seq datasets. We align RNA-seq reads to the GRCh38.p2 genome build (Ensembl v79) file by using STAR55 and process the resulting read counts per gene by the scater package56 to compute additional QC metrics and visualizations of the RNA-seq dataset. RRBS data are processed from raw reads to CpG-level methylation values by using published cloud-based DNA-methylation analysis pipelines that we developed for large bisulfite sequencing data sets, including sc-RRBS data57 (https://github.com/aryeelab/dna-methylation-tools). The pipelines run in the Google cloud and can be accessed for a fee through the Broad Institute’s Terra platform (https://app.terra.bio/#workspaces/aryee-lab/bisulfite-seq-tools-grch38). A schematic of tools and their roles in the workflow and which tools are available in the pre-processing/QC package is provided as Extended Data Fig. 1.
Briefly, after removing low-quality bases and reads, the 6-base inline barcodes at the beginning of each read are identified, extracted and clipped off along with the universal T at position 7. The Trim galore wrapper tool identifies and removes Illumina adapters along with the preceding seven bases (A followed by the reverse complement of the inline barcode) at the 3′ ends of reads. Fully trimmed sequences are aligned to the bisulfite converted GRCh38.p2 genome in Bismark58 (version 0.18.2), with bowtie259 as the aligner module. Bismark’s methylation extractor (-bedgraph --buffer_size 50%) is used to determine the methylation state of individual CpGs across both the strands. The scmeth R package57 in the cloud pipeline provides various HTML (HyperText Markup Language) and plain-text QC data and visualizations to look for potential technical biases and batch effects in large data sets. It flags low-quality samples or batches on the basis of alignment, average methylation per base across the reads, CpG coverage, distribution across various genomic features and mean methylation across samples. Finally, the annotatr R package60 is used to annotate features such as CpG islands, shelves, shores, and promoters in the reference genome. For enhancer annotations, we use the FANTOM5 database61.
Expertise and equipment needed to implement the protocol
This protocol requires basic molecular biology expertise (prior experience in construction of sequencing libraries would be particularly helpful) and laboratory equipment, including a PCR workstation with UV sterilization and thermocyclers for 96-well plates (with temperature-adjustable heated lids) and 384-well plates. The protocol also requires access to instruments commonly used in genomics laboratories to determine DNA concentrations fluorometrically in single tubes and ideally also in 96-well format on a plate reader, and the size distributions on a microfluidic electrophoresis system such as an Agilent BioAnalyzer (for ≤11 samples) or TapeStation (for ≤96 samples). In addition, the protocol requires a FACS sorting facility for sorting single cells into 96-well plates, as well as access to high-output Illumina sequencing instruments in the laboratory or in a dedicated sequencing facility. A high-performance computing environment is required for processing and analysis of sequencing data. Finally, knowledge of a programming language such as Python or R is necessary for further downstream analyses and data interpretation.
Materials
Reagents
24 indexed 5mC-methylated adapters; oligonucleotide sequences in Supplementary Data 1; see Reagent setup
RRBS library amplification PCR primers; sequences in Supplementary Data 2; 100-nmol-scale synthesis; standard desalted (Integrated DNA Technologies (IDT)). At least four single indexes or four unique dual-index (UDI) pairs are required to process 96 single cells ▲ CRITICAL For sequencing on Illumina instruments with patterned flow cells, we recommend using the UDI set of pre-mixed forward and reverse RRBS PCR primers (see Reagent setup) to minimize index hopping and mis-assignments of indexes to library pools in the RRBS data set.
Oligo(dT) RT primer 5′-biotinTEG-AAGCAGTGGTATCAACGCAGAGTACT30VN 100-nmol-scale synthesis; RNAse-free HPLC purification (IDT)
TSO 5′-AAGCAGTGGTATCAACGCAGAGTACrGrG+G containing two ribonucleotide (r) and one locked nucleic acid (+) linkage at the 3′ end; 250-nmol-scale synthesis; RNAse-free HPLC purification (Exiqon)
cDNA amplification PCR primer 5′-AAGCAGTGGTATCAACGCAGAGT; 25-nmol-scale synthesis; standard desalted (IDT)
RNaseZap RNase decontamination solution (Thermo Fisher Scientific, cat. no. AM9780)
RNAprotect cell reagent (Qiagen, cat. no. 76526)
RLT Plus (Qiagen, cat. no. 1053393) ! CAUTION This reagent contains guanidine thiocyanate, which is harmful and should be handled with care (e.g., wear protective gloves, clothes and goggles).
SUPERase In RNase inhibitor, 20 U/μl (Thermo Fisher Scientific, cat. no. AM2694)
Dynabeads M-280 streptavidin (Thermo Fisher Scientific, cat. no. 11205D)
ERCC RNA Spike-In mix (Thermo Fisher Scientific, cat. no. 4456740)
Sonicated salmon sperm DNA (Thermo Fisher Scientific, cat. no. 15632011)
Heat-labile shrimp alkaline phosphatase (New England Biolabs, cat. no. M0371S)
QiaQuick PCR purification kit (Qiagen, cat. no. 28104)
50% Tween 20 (Thermo Fisher Scientific, cat. no. 003005)
NaCl, 5 M (Sigma-Aldrich, cat. no. S5150)
EDTA, 0.5 M, pH 8.0 (VWR, cat. no. 101384-758)
NaOH, 1 N (Sigma-Aldrich, cat. no. S2770) ! CAUTION This reagent is highly corrosive. Drops of sodium hydroxide can burn skin easily. Wear protective gloves, clothing and goggles.
Tris-HCl, 1 M, pH 8.0 (Sigma-Aldrich, cat. no. T-3038)
Tris-HCl, 1 M, pH 7.5 (Thermo Fisher Scientific, cat. no. 15567027)
UltraPure DNase/RNase-free distilled water (Thermo Fisher Scientific, cat. no. 10977015)
SuperScript II RT, 200 U/μl (Thermo Fisher Scientific, cat. no. 18064-071)
DTT, 100 mM (Thermo Fisher Scientific, cat. no. 18064-071)
Superscript II first-strand buffer, 5× (Thermo Fisher Scientific, cat. no. 18064-071)
Betaine, 5 M (Sigma, cat. no. 107-43-7)
MgCl2, 1 M (Thermo Fisher Scientific, cat. no. AM9530G)
dNTP mix, 10 mM (New England Biolabs, cat. no. N0447L)
Proteinase K (New England Biolabs, cat. no. P8107S)
CutSmart buffer, 10× (New England Biolabs, cat. no. P7204S)
MspI (New England Biolabs, cat. no. R0106L)
HaeIII (New England Biolabs, cat. no. R0108S)
Klenow fragment (3′→5′ exo-; New England Biolabs, cat. no. M0212L)
Deoxynucleotide solution set, 25 μM each (New England Biolabs, cat. no. N0447L)
T4 DNA ligase (New England Biolabs, cat. no. M0202M)
ATP solution, 100 mM (Thermo Fisher Scientific, cat. no. R0441)
2× KAPA HiFi HotStart ReadyMix (Roche, cat. no. KK2602)
2× KAPA HiFi HotStart Uracil+ ReadyMix (Roche, cat. no. KK2802)
EpiTect fast DNA bisulfite kit (Qiagen, cat. no. 59824)
Zymo-Spin IC columns (Zymo Research, cat. no. C1004-50)
Quant-iT DNA assay kit, high sensitivity (Invitrogen, cat. no. Q-33120)
Criterion precast 10% polyacrylamide TBE gels (Bio-Rad, cat. no. 3450053)
AMPure XP SPRI beads, 60 ml (Beckman Coulter, cat. no. A63881) ▲ CRITICAL The clean-up and size-selection steps performed by using this protocol have been optimized for AMPure brand magnetic SPRI beads. Before substituting these beads with other brands, determine the size selectivity at different ratios of bead suspension to sample volume by using a DNA size marker for gel electrophoresis.
NuSieve 3:1 agarose (Lonza, cat. no. 50090)
MinElute gel extraction kit (Qiagen, cat. no. 28604)
Low-molecular-weight DNA ladder (New England Biolabs, cat. no. N3233L)
DNA ladder (20-bp extended range; Lonza, cat. no. 50320)
SYBR green I nucleic acid gel stain (Invitrogen, cat. no. S7567) ! CAUTION SYBR Green I is a potential mutagen. Wear protective gloves, clothing and goggles.
PEG 8000 (Millipore Sigma, cat. no. 25322-68-3)
Gel loading dye, blue (6×) (New England Biolabs, cat. no. B7021S)
Nextera XT DNA sample preparation kit, 96 samples (Illumina, cat. no. FC-131-1096)
IDT for Illumina Nextera DNA UDI Set A, B, C or D (Illumina, cat. no. 20027213, 20027214, 20027215 or 20027216) or Sets A–D (Illumina, cat no. 20027217)
Nextera XT 96-Index kit v2, 384 samples, Set A, B, C or D combinatorial dual-index sets (Illumina, cat. no. FC-131-2001, FC-131-2002, FC-131-2003 or FC-131-2004) ▲ CRITICAL For sequencing on Illumina instruments with patterned flow cells, we recommend using the UDI sets, which minimize index hopping and mis-assignments of indexes to samples in the RNA-Seq data set.
BioAnalyzer high-sensitivity DNA kit (Agilent Technologies, cat. no. 5067-4626)
Qubit double-stranded (ds) DNA HS assay kit (Thermo Fisher Scientific, cat. no. Q32854)
TapeStation D1000 and D5000 screen tape (Agilent Technologies, cat. nos. 5067-5584 and 5067-5592)
TapeStation high sensitivity D1000 and D5000 reagents (Agilent Technologies, cat. nos. 5067-5585 and 5067-5593)
PhiX Control v3 spike-in sequencing library (Illumina, cat. no. FC-110-3001)
Equipment
Microseal ‘F’ aluminized foil seals (Bio-Rad, cat. no. MSF1001)
DynaMag-2 magnet (Invitrogen, cat. no. 123-21D)
Tube Rotator HulaMixer sample mixer (Thermo Fisher Scientific, cat. no. 15920D)
Eppendorf MixMate (Thermo Fisher Scientific, cat. no. 2137900)
DynaMag-2 (Thermo Fisher Scientific, cat. no. 12321D)
DynaMag-96 side magnet (Thermo Fisher Scientific, cat. no. 12331D)
Eppendorf twin.tec PCR plate 96 LoBind (Eppendorf, cat. no. 0030129504)
Adhesive tape sheets (Qiagen, cat. no. 19570)
Reagent reservoirs, 50 ml (VWR, cat. no. 53504-035)
Reagent reservoirs, 100 ml (VWR, cat. no. 29442-476)
Zymo-Spin IC columns (Zymo Research, cat. no. C1004-50)
Bioanalyzer 2100 (Agilent, cat. no G2939BA)
Agilent 2200 TapeStation system (Agilent, cat. no. G2964AA)
TapeStation loading tips (Agilent Technologies, cat. no. 5067-5153)
Criterion vertical electrophoresis system for polyacrylamide gels (Bio-Rad, cat. no. 1656001)
96-well deep-well plates (VWR, cat. no. 10755-258)
Qubit assay tubes (Thermo Fisher Scientific, cat. no. Q32856)
Qubit fluorometer (Thermo Fisher Scientific, cat. no. Q33238)
Illumina next-generation sequencing platforms (HiSeq 2500, HiSeq 4000, Hiseq X or NovaSeq 6000)
Mini PCR plate spinner (VWR, cat. no. 89184-608)
Reagent setup
24 indexed 5mC methylated adapters for RRBS
Order 24 top and 24 bottom oligonucleotides (sequences in Supplementary Data 1) with all cytosines substituted by methylated 5mC at 100-nmol scale with standard desalting. Note that these methylated oligos are the single most expensive reagent required for this protocol, but the minimum order (100-nmol scale) is sufficient for processing tens of thousands of ligation reactions, each containing 0.23 pmol of adapter. Combine each pair of top and bottom oligonucleotides at a concentration of 100 μM each in 0.5 ml of annealing buffer (10 mM Tris-HCl (pH 8.0) and 100 mM NaCl). Heat for 5 min at 95 °C in a heat block to denature the oligonucleotides and then switch off the heat block for a slow temperature ramp down to room temperature. Prepare a 0.15-μM dilution in low EDTA TE (Tris EDTA) buffer and prepare working aliquots of 25 μl of diluted adapters in 96-well format (one set of 24 adapters per plate in two rows). Keep annealed adapters that have been diluted in low-salt buffer on ice or below 25 °C to prevent melting of the 19-bp stem sequences. Diluted adapter plates and remaining unannealed and annealed oligonucleotides and intermediate dilutions can be stored at −80 °C for ≥3 years.
UDI RRBS PCR primer pairs.
Prepare a minimum of four UDI primer pairs (25 μM for each primer) by combining equal volumes of 50 μM i5 indexed forward and 50 μM i7 indexed reverse RRBS PCR primers in low-EDTA TE. The 16 UDI RRBS primer pairs in Supplementary Data 2 are sufficient to process 384 single cells in four 96-well plates without barcode collision at the pool level. Primer stocks and aliquots of primer pairs can be stored at −20 °C for ≤1 year if evaporation is prevented, or at −80 °C for ≥3 years.
Dephosphorylated carrier DNA.
To prevent potential traces of residual ligase activity after Step 46 from ligating adapter to the carrier DNA added in Step 50, dephosphorylate the carrier DNA as follows: mix 268 μl of nuclease-free water, 2 μl of sonicated salmon sperm DNA (20 μg), 30 μl of NEB 10× CutSmart buffer and 30 μl of heat-labile shrimp alkaline phosphates and incubate the reaction overnight at 37 °C. Heat-inactivate the phosphatase for 20 min at 65 °C and purify the DNA by using two columns from a QIAquick PCR purification kit as per the manufacturer’s instructions. Elute DNA in a total of 100 μl, measure the DNA concentration by a fluorometric assay (e.g., Qubit), adjust the concentration to 50 ng/μl and store in aliquots at −20 °C for ≤1 year if evaporation is prevented, or at −80 °C for ≥3 years.
Low-EDTA TE buffer
Low-EDTA TE buffer is 10 mM Tris-HCl (pH 8.0) and 0.1 mM EDTA. This buffer can be stored at room temperature for 6 months.
Solution A
Solution A contains 0.1 M NaOH, 0.05 M NaCl and 0.05% (vol/vol) Tween-20. To prepare 10 ml of solution A, add 1 ml of 1 M NaOH, 100 μl of 5 M NaCl and 50 μl of 10% (vol/vol) Tween 20 to 8.85 ml of nuclease-free water. This solution should be made up fresh each time.
Solution B
Solution B is composed of 0.1 M NaCl and 0.05% (vol/vol) Tween-20. To prepare 10 ml of solution B, add 200 μl of 5 M NaCl and 50 μl of 10% (vol/vol) Tween 20 to 9.75 ml of nuclease-free water. Solution B can be stored at room temperature for one month.
2× binding and washing (B&W) buffer
This buffer contains 10 mM Tris-HCl (pH 7.5), 1 mM EDTA, 2 M NaCl and 0.05% (vol/vol) Tween-20. Prepare a 50-ml stock by using nuclease-free water. This buffer can be stored at room temperature for 3 months.
mRNA binding buffer
This buffer consists of 10 mM Tris-HCl (pH 8.0), 50 mM NaCl, 0.1 mM EDTA and 1 U/μl RNase inhibitor. To prepare 30 ml of stock, add 300 μl of 1 M Tris-HCl (pH 8.0), 300 μl of 5 M NaCl and 60 μl of 500 mM of EDTA (pH 8.0) to 29.34 ml of nuclease-free water. Dispense 1.5 ml to each of 20 Eppendorf tubes and store at −20 °C for ≤1 year. Add RNase inhibitor to a final concentration of 1 U/μl before use.
AMPure bead dilution buffer
The bead dilution buffer is 20% (wt/vol) PEG 8000, 2.5 M NaCl and 0.05% (vol/vol) Tween-20. The buffer can be stored at 4 °C for 3 months.
1:4 diluted AMPure beads
1:4 diluted AMPure beads are one volume of AMPure beads and three volumes of the above AMPure bead dilution buffer. To prepare 40 ml of diluted beads, add 30 ml of bead dilution buffer to 10 ml of AMPure beads.
Preparation of 2.5% (wt/vol) NuSieve 3:1 TBE agarose gel
Dissolve 7.5 g of NuSieve 3:1 agarose powder in 300 ml of 0.5× TBE buffer for 5 min by shaking at 120 rpm on an orbital shaker. Microwave this solution for 3 min at full power. Place the bottle containing the melted gel on the orbital shaker and rotate at 120 rpm for ~30 min. After the solution cools down to ~40 °C, pour the gel in a gel-casting tray. ! CAUTION Direct microwaving of a 3% (wt/vol) NuSieve 3:1 agarose solution will lead to excessive foaming.
Procedure
Preparing single-cell plates ● Timing 1 h
▲ CRITICAL STEP RNase is ubiquitous in laboratories. We advise conducting all RNA-related steps in a clean area such as a PCR workstation with UV sterilization. Clean pipettes and the working area with RNAZap before each experiment and turn on a UV light afterwards for 30 min to destroy surface microorganisms or nucleic acids.
Day 0. In the PCR workstation, add 600 μl of RLT Plus buffer to a fresh 1.5-ml Eppendorf tube. Optionally, if an internal spike-in standard in each single cell data set is desired, add 6 μl of a 10−5 dilution of ERCC RNA Spike-In mix.
Place a 48-μl aliquot of RLT Plus in each of the 12 wells in one row of a 96-well plate. Seal the plate with an adhesive tape sheet and then spin it briefly.
Transfer eight aliquots of 5 μl from each of the 12 wells in the above intermediate plate to fill all 96 wells of a fresh plate by using a 20-μl 12-channel pipette (Box 1; Extended Data Fig. 2).
Seal and then centrifuge the plate briefly to bring down the cell lysis buffer.
-
Sort one cell to each well by FACS; seal the plate with one Microseal ‘F’ aluminized foil seal and immediately spin the plate briefly.
▲ CRITICAL STEP Seal the sample plate thoroughly for long-term storage. The Microseal ‘F’ aluminized foil seal is stable at −80 °C.
■ PAUSE POINT Single-cell plates can be stored at −80 °C for ≥1 year.
Box 1 ∣. Process for manually dispensing a master mix across a sample plate ● Timing 10 min.
Procedure
Prepare sufficient master mix for one 96-well sample plate in a microcentrifuge tube, adding an excess of ~20% to compensate for losses during pipetting (e.g., 120 reaction volumes for 96 samples).
Distribute aliquots of nine reaction volumes to one row of 12 wells in a 96-well plate.
Use a 12-channel pipette to dispense the master mix to all wells of the sample plate (Extended Data Fig. 2).
Preparing beads for mRNA capture and RT ● Timing 1 h
-
6
Day 1. Transfer 600 μl of M-280 beads to a 1.5-ml Eppendorf tube, place the tube on a DynaMag-2 magnet for 1 min and then remove and discard the supernatant without disturbing the beads.
▲ CRITICAL STEP To isolate mRNAs from each single-cell, 5 μl of M-280 beads is sufficient. When preparing M-280 beads for 96 single cells, make 20% extra to compensate for bead loss during pipetting.
-
7
Resuspend the M-280 beads in 600 μl of freshly prepared solution A and wash the beads for 2 min in a tube rotator/ sample mixer. Put the tube back to the magnet for 1 min, aspirate and discard the supernatant.
-
8
Repeat Step 7 once.
-
9
Wash the M-280 beads with 600 μl of solution B in the tube rotator for 1 min, spin quickly, collect the beads on the magnet for 2 min and discard the supernatant. The M-280 beads are ready to be coated with the biotinylated RT-primer.
-
10
Add 600 μl of 2 μM biotinylated RT-primer oligonucleotide to the M-280 beads and mix by vortexing gently; then, add 600 μl of 2× B&W buffer.
-
11
Mix the beads and biotinylated RT-primer mixture for 15 min on the sample mixer at room temperature with gentle rotation.
-
12
After conjugating biotinylated RT-primer with M-280 beads, quickly spin and place the tube on the magnet for 2 min to collect the beads.
-
13
Remove and discard the supernatant; then add 1 ml of 1× B&W buffer, vortex gently, spin briefly and collect the beads on a magnet for 2 min. Perform two additional washes in the same manner.
-
14
Remove and discard the supernatant and resuspend the biotinylated RT-primer–coated beads in 1.2 ml of mRNA binding buffer.
Separation of genomic DNA and mRNA ● Timing 1-1.5 h
-
15
Take the cell plate from the −80 °C freezer; thaw the content on ice and spin the plate at 2,500g for 1 min.
-
16
Distribute 90 μl of the biotinylated RT-oligo–coated beads from Step 14 to each of 12 wells in a row on a fresh PCR plate, and transfer 10 μl to each well of the sample plate by using a 12-channel pipette (Box 1). Seal the plate with an adhesive tape sheet.
-
17
Place the sample plate in an Eppendorf MixMate and rotate at 600 rpm for 20 min at room temperature to prevent the beads from settling.
-
18
While the sample plate is incubating, prepare the mRNA-bead washing buffer and the RT master mix on ice according to the following recipes:
mRNA-bead washing buffer
| Stock | Volume (μl) | Final concentration |
|---|---|---|
| 5× SuperScript II first-strand buffer | 720 | 1× |
| 100 mM DTT | 360 | 10 mM |
| 10% (vol/vol) Tween-20 | 180 | 0.5% (vol/vol) |
| DNase/RNase-free H2O | 2,160 | — |
| RNase inhibitor (20 U/μl) | 180 | 1 U/μl |
| Total | 3,600 | — |
RT master mix
| Stock | Per sample (μl) | Per 120 samples (μl) | Final concentration |
|---|---|---|---|
| 5× Superscript II first-strand buffer | 2 | 240 | 1× |
| SuperScript II RT (200 U/μl) | 0.5 | 60 | 10 U/μl |
| RNase inhibitor (20 U/μl) | 0.5 | 60 | 1 U/μl |
| 100 mM DTT | 0.25 | 30 | 2.5 mM |
| 5 M betaine | 2 | 240 | 1 M |
| 100 mM MgCl2 | 0.9 | 108 | 9 mM |
| TSO (10 μM) | 1 | 120 | 1 μM |
| dNTP mix (10 mM of each) | 1 | 120 | 1 mM |
| DNase/RNase-free H2O | 1.85 | 222 | — |
| Total | 10 | 1,200 | — |
-
19
Centrifuge the sample plate briefly, collect the beads containing captured mRNA to the side of the well by using a DynaMag-96 side magnet and transfer the supernatant containing genomic DNA (gDNA) to a 96-well LoBind plate.
▲ CRITICAL STEP When transferring the supernatant, do not let the tips touch the beads. The gDNA plate should be sealed immediately each time to avoid evaporation and sample contamination.
-
20
Resuspend the beads in 15 μl of mRNA-bead washing buffer, briefly spin down and then collect the beads by using the DynaMag-96 side magnet. Transfer the supernatant to the corresponding wells of the above gDNA plate.
▲ CRITICAL STEP The beads accumulated on one side of the well are barely covered by the washing buffer. Sealing and vortexing helps resuspend the beads if necessary. Spin the plate quickly to bring beads to the well bottom before placing it back on the magnet.
-
21
Repeat Step 20. The combined gDNA from each single-cell sample, typically only ~36 μl due to incomplete liquid transfers, will be used for making single-cell RRBS libraries in Steps 28–67.
RT and cDNA amplification ● Timing 5–5.5 h (hands-on time: 0.5–1 h)
-
22
Distribute 95 μl of the pre-prepared RT master mix from Step 18 to each of 12 wells in a row on a PCR plate, transfer 10 μl to each sample in the plate containing bead-bound mRNAs with a 12-channel pipette (Box 1) and then seal the plate.
-
23
Gently vortex the plate to resuspend the M-280 beads and place the plate on a thermocycler with the lid temperature preset to 50 °C. Thermocycle the RT reaction as follows:
| Cycle number | Temperature (°C) | Duration (min) |
|---|---|---|
| 1 | 42 | 90 |
| 10 | 50 | 2 |
| 42 | 2 | |
| 1 | 70 | 15 |
▲ CRITICAL STEP To increase mRNA representation, pause the thermocycler at 10, 20 and 30 min after the initiation of the RT reaction, take the plate out, vortex gently to resuspend the M-280 beads and then place the plate back to the thermocycler and resume the reaction.
-
24
Once the RT reaction is done, take the plate from the thermocycler and spin it briefly to bring any liquid accumulated under the bottom of the sealing membrane top and on the sides of each sample well to the bottom.
-
25
Prepare PCR master mix on ice according to the recipe below. Distribute 140 μl of the master mix to each of the 12 wells in one row of a 96-well plate and transfer 15 μl to each well of the plate above by using a 12-channel pipette (Box 1).
| Component | Per sample (μl) |
Per 120 samples (μl) |
Final concentration in 25-μl reaction |
|---|---|---|---|
| 2× KAPA HiFi HotStart ReadyMix | 12.5 | 1,500 | 1× |
| 10 μM PCR primer | 0.25 | 30 | 100 nM |
| H2O | 2.25 | 270 | — |
| Total | 15 | 1,800 | — |
▲ CRITICAL STEP The M-280 beads are still in sample wells but will not adversely affect the downstream PCR reaction.
-
26
Seal and centrifuge the plate briefly, vortex gently to mix the reagents and centrifuge the plate again.
-
27
Place the plate in the thermocycler and run the following program:
| Cycle number | Denature | Anneal | Extend |
|---|---|---|---|
| 1 | 98 °C, 3 min | — | — |
| 18–22 | 98 °C, 20 s | 67 °C, 20 s | 72 °C, 6 min |
| 1 | — | — | 72 °C, 6 min |
▲ CRITICAL STEP 18 cycles of PCR are generally sufficient to generate sufficient PCR product (~2 ng); however, some cell types, such as human B or T lymphocytes, may require more PCR cycles (e.g., 22) to generate a sufficient amount for downstream reactions.
■ PAUSE POINT Once the PCR is complete, the amplified cDNA can be stored at 4 °C for ≤1 week. For long-term storage (several months), we recommend purifying the PCR product by using AMPure beads as described in Box 2, eluting in 15 μl of 10 mM Tris-HCl (pH 8.0) and storing at −20 °C.
Box 2 ∣. DNA purification by using Agencourt AMPure beads ● Timing 1 h.
Procedure
Take AMPure XP beads (undiluted or 1:4 diluted in AMPure bead dilution buffer if prescribed in the procedure) from the refrigerator and leave at room temperatures for 20 min.
Transfer 10 ml of AMPure XP beads to a 50-ml reagent reservoir and then use a 12-channel pipette to dispense an appropriate amount of the beads to each sample well.
-
Seal the sample plate properly and place the plate in the Eppendorf MixMate for 20-30 min at room temperature with a rotation speed of 700 rpm.
▲ CRITICAL STEP Incubating the DNA sample with beads for ≥20 min will maximize the DNA recovery rate.
Spin the sample plate down briefly and leave it on the DynaMag-96 side magnet for 5 min, allowing beads to accumulate on one side of the well.
Keep the sample plate on the DynaMag-96 side magnet; carefully take the supernatant and discard it by using a 200-μl 12-channel pipette.
Dispense 100 μl of freshly prepared 80% (vol/vol) ethanol to each sample well without disturbing the beads and then incubate for 1 min.
Aspirate the ethanol and discard it. Repeat the ethanol wash once.
Take the remaining ethanol from the sample wells by using a 20-μl 12-channel pipette and discard it.
Let the sample plate air-dry until there is no apparent ethanol residual on the beads’ surface.
Add the amount of elution buffer specified in the procedure for this step to the bead side and let the buffer run down through the beads.
Seal and vortex the sample plate and centrifuge briefly. Leave the beads in the elution buffer unless specified otherwise and perform the downstream reaction as described in the procedure.
Clean-up of gDNA fraction ● Timing 1 h
-
28
While the RT reaction (Step 23) is running, add 1 μl of 1:2 diluted (in water) proteinase K to each well of the gDNA plate (Step 21), dispense an equal volume (~36 μl) of diluted AMPure beads (1:4 in bead dilution buffer) to each well and then seal the plate properly.
▲ CRITICAL STEP Although adding proteinase K at this step may seem redundant given that the cells have been collected in denaturing RLT Plus lysis buffer, we saw a measurable improvement (fewer PCR cycles required for library amplification), most likely because of digestion of residual DNA-bound proteins that inhibit the subsequent reactions.
-
29
Clean up the gDNA fraction according to the instructions in Box 2.
-
30
Resuspend the AMPure beads in 8.5 μl of water for single digest with MspI (or in 8 μl for MspI +HaeIII double digest) to elute the gDNA. With beads still in each well to minimize sample loss, set up the restriction digest as described in Steps 31 and 32. Volumes in parentheses are for the double-digest option.
Restriction digestion of gDNA ● Timing 2 h (hands-on time: 0.5 h)
-
31
Prepare the master mix for the restriction digestion reaction on ice as follows:
| Component | Per reaction (μl) |
Per 120 reactions (μl) |
Final concentration in 10-μl reaction |
|---|---|---|---|
| 10× CutSmart buffer | 1 | 120 | 1× |
| MspI (20 U/μl) | 0.5 | 60 | 1 U/μl |
| (Double digest only) HaeIII (50 U/μl) | (0.5) | (60) | (2.5 U/μl) |
| Total | 1.5 (2) | 180 (240) | — |
▲ CRITICAL STEP Prepare 25% more master mix (e.g., enough for 120 aliquots for 96 samples) to compensate for loss during pipetting.
-
32
Split 14 μl (or 19 μl for double digest) of the master mix to each of 12 wells in a 96-well plate, and then transfer 1.5 μl (or 2 μl for double digest) of the master mix to each of the sample wells by using the 12-channel pipette (Box 1) for a total liquid volume of 10 μl. Seal, vortex and spin the plate briefly at room temperature.
-
33
Put the sample plate in the thermocycler with the lid temperature pre-set to 70 °C. Incubate the reaction according to the program below:
| Order | Temperature (°C) | Duration (min) | Purpose |
|---|---|---|---|
| 1 | 37 | 80 | gDNA digestion |
| 2 | 70 | 10 | Inactivation of restriction enzymes |
| 3 | 4 | Hold | — |
▲ CRITICAL STEP Complete inactivation of HaeIII requires 80 °C for 20 min, whereas MspI cannot easily be heat inactivated. However, any residual enzyme activity after 10 min at 70 °C does not cut and interfere in downstream steps, because neither an MspI nor an HaeIII site is regenerated upon ligation to the adapter.
-
34
Remove the plate from the thermocycler and centrifuge at 1,000g for 30 s.
End repair and A-tailing ● Timing 1.5 h (hands-on time: 0.5 h)
-
35
Prepare the master mix as described in the following recipe:
| Component | Per reaction (μl) |
Per 120 reactions (μl) |
Final concentration in 12-μl reaction |
|---|---|---|---|
| 10× CutSmart buffer | 0.2 | 24 | 1× |
| Klenow exo- (5 U/μl) | 0.5 | 60 | ~0.2 U/μl |
| dNTP mixture (10 mM dATP, 1 mM dCTP and 1 mM dGTP) | 0.4 | 48 | 333 μM dATP, 33 μM dCTP and 33 μM dGTP |
| H2O | 0.9 | 108 | — |
| Total | 2 | 240 | — |
-
36
Transfer 19 μl of the above master mix to each of the 12 wells in one row of a 96-well plate and transfer 2 μl of the master mix to each sample well by using the 20-μl 12-channel pipette (Box 1) for a total liquid volume of 12 μl.
-
37
Seal and vortex the sample plate and spin at 1,000g for 30 s.
-
38
Carry out the reaction in the thermocycler according to the following program:
| Order | Temperature (°C) | Duration (min) | Purpose |
|---|---|---|---|
| 1 | 30 | 25 | Filling in cut MspI sites |
| 2 | 37 | 25 | A-tailing |
| 3 | 70 | 10 | Inactivation of Klenow exo- |
| 4 | 4 | Hold | — |
-
39
Centrifuge the plate at 1,000g for 30 s.
Adapter ligation ● Timing 16–20 h (hands-on time: 0.5 h)
-
40
Take the adapter plate containing 25 μl each of one set of 24 adapters in two rows from the −80 °C freezer, place it on ice or below 25 °C at room temperature to thaw the adapter solution and then centrifuge the adapter plate at 3,000g for 2 min to bring all liquid to the bottom before use.
▲ CRITICAL STEP Keep the adapter plate on ice or below 25 °C and put it back to −80 °C immediately after use. Avoid more than three thaw-freeze cycles.
-
41
Prepare the master mix for adapter ligation as follows:
| Component | Per reaction (μl) | Per 120 reactions (μl) |
Final concentration in 13.5-μl reaction |
|---|---|---|---|
| 10× CutSmart buffer | 0.3 | 36 | 1× |
| T4 ligase (2,000 U/μl) | 0.5 | 60 | ~66.7 U/μl |
| 100 mM ATP | 0.1 | 12 | ~0.67 mM |
| H2O | 0.6 | 72 | — |
| Total | 1.5 | 180 | — |
-
42
Distribute 14 μl of the ligation master mix to each of the 12 wells in one row of a 96-well plate and dispense 1.5 μl to each well of the sample well (Step 39) by using a 12-channel pipette (Box 1) for a total liquid volume of 13.5 μl.
-
43
Carefully add 1.5 μl of the 24 diluted adapters (0.15 μM) to each sample well in rows 1 and 2 of the sample plate with a 12-channel pipette. Add 1.5 μl of the same set of adapters to rows 3 and 4, 5 and 6 as well as 7 and 8 of the sample plate.
-
44
Seal the sample plate and centrifuge briefly, followed by vortexing and spinning down again.
-
45
Incubate the ligation reaction at 16 °C for 16–20 h in the thermocycler with lid temperature preset lowered to 25 °C.
Pooling and clean-up of ligation reactions ● Timing ~2 h
-
46
Day 2. Heat-inactivate the T4 ligase at 70 °C for 15 min, then take the sample plate out from the thermocycler, spin it down at 2,000g for 1 min to collect all liquid at the bottom of the wells and then vortex the plate gently to resuspend the beads.
-
47
Pool each set of 24 samples indexed with different adapters including the AMPure beads in each well to a fresh, labeled low-binding Eppendorf tube for a total of four pools per 96-well plate.
-
48
Wash each set of 24-sample wells (two rows on the 96-well reaction plate) successively with a total of 20 μl of TE buffer and add the 20 μl to the Eppendorf tube containing the respective sample pool.
-
49
Repeat Step 48 once. The total volume for each pool of 24 samples is ~350–380 μl.
-
50
Add 2 μl of dephosphorylated carrier DNA to each pool.
-
51
Add 1.8 volumes of AMPure bead dilution buffer to each library pool to re-precipitate DNA onto the beads; rotate the tube at room temperature for 30 min to prevent the beads from settling and promote DNA binding.
-
52
Spin down library pools briefly and collect the bead-bound DNA on a DynaMag-2 magnet for 10 min; aspirate and discard the supernatant carefully without disturbing the beads.
-
53
Wash the beads twice with 1 ml of 80% (vol/vol) freshly prepared ethanol.
-
54
Leave the sample tubes open and place them in a fume hood with flowing air until the leftover ethanol completely evaporates.
▲ CRITICAL STEP Drying the beads takes ~20–30 min because of the large volume of beads. Watch carefully to make sure that beads are not over-dried as indicated by cracks.
-
55
Elute DNA in 40 μl of 10 mM Tris-HCl (pH 8.0).
Bisulfite conversion ● Timing ~1.5 h
-
56
To each eluate, add 85 μl of Qiagen EpiTect Fast bisulfite solution and 15 μl of Qiagen DNA protect buffer to a total of 140 μl. Split each reaction into 2 × 70 μl and thermocycle the bisulfite reactions as follows:
| Order | Temperature (°C) | Duration (min) | Purpose |
|---|---|---|---|
| 1 | 98 | 5 | Denature dsDNA |
| 2 | 60 | 20 | Convert unmethylated cytosine to uracil |
| 3 | 98 | 5 | Denature dsDNA |
| 4 | 60 | 20 | Convert unmethylated cytosine to uracil |
| 5 | 20 | Hold | — |
-
57
Transfer the bisulfite reactions to a 1.5-ml Eppendorf tube, add 310 μl of Qiagen EpiTect Fast buffer BL (containing 10 μg/μl carrier RNA). Perform the on-column desulfonation and clean-up steps by using the Qiagen EpiTect Fast buffers BW and BD following the kit’s instructions except substituting Zymo-Spin IC fast-spin columns for the columns from the Qiagen kit.
-
58
Elute bisulfate-converted DNA in 30 μl of low-EDTA TE buffer.
▲ CRITICAL STEP Bisulfite-converted DNA can quickly degrade or get lost by sticking to plastic ware. We advise proceeding immediately to optional Steps 59–63 (semiquantitative test PCR) or directly to Step 64 (library amplification) or store the bisulfite-converted DNA frozen (≤1 week at −20 °C; longer term (several months) at −80 °C).
RRBS library amplification ● Timing 2.5 h
Steps 59–63 are optional but strongly recommended when performing the procedure for the first time or from an unfamiliar source of flow-sorted single cells.
-
59
Set up pilot PCR reactions as follows:
| Component | Per reaction (μl) | Final concentration |
|---|---|---|
| Bisulfite-converted DNA | 1.5 | — |
| 2× KAPA HiFi HotStart Uracil+ ReadyMix | 25 | 1× |
| Primer mix (25 μM for each) | 1 | 500 nM |
| H2O | 22.5 | — |
| Total | 50 | — |
-
60
Split the reactions into three 10-μl aliquots in three 384-well plates and vary the total number of PCR cycles between 16 and 22 as shown below.
| Cycle number | Denature | Anneal | Extend |
|---|---|---|---|
| 1 | 98 °C, 45 s | — | — |
| 6 | 98 °C, 20 s | 58 °C, 30 s | 72 °C, 1 min |
| 10, 13 and 16 | 98 °C, 20 s | 65 °C, 30 s | 72 °C, 1 min |
| 1 | — | — | 72 °C, 5 min |
-
61
Run 5 μl of each PCR product on a precast 10% (wt/vol) PAGE gel for 1 h at 130 V. Alternatively, run 1 μl on a high-sensitivity BioAnalyzer chip.
-
62
Stain the PAGE gel by using 1:10,000 diluted SYBR Green I for 15 min.
-
63
Examine the gel image and choose a number close to the minimal PCR cycle number that produced a clearly visible smear of RRBS PCR products between 200 and 500 bp.
? TROUBLESHOOTING
-
64
Prepare the PCR master mix for RRBS library amplification as follows:
| Component | Per reaction (μl) |
Per 4.5 reactions (μl) |
Final concentration in 120-μl reaction |
|---|---|---|---|
| 2× KAPA HiFi HotStart Uracil+ ReadyMix | 60 | 270 | 1× |
| UDI RRBS PCR primer pair (25 μM for each) | 2.5 | 11.25 | ~0.5 μM |
| H2O | 29.5 | 132.75 | — |
| Total | 92 | 414 | — |
▲ CRITICAL STEP For sequencing on Illumina instruments with patterned flow cells, we recommend using unique dual-index combinations of i5 indexed forward primers and i7 indexed reverse primers to minimize index hopping and mis-assignments of indexes to sample pools in the RRBS data set.
-
65
Add 92 μl of master mix to each bisulfite-converted sample (28 μl; Step 58), mix, then split 40 μl to three wells in a 96-well PCR plate and perform the preparative RRBS library amplification. Typically, 10–13 cycles in step 3 of the thermoprofile (16–19 PCR cycles in total) are appropriate.
| Cycle number | Denature | Anneal | Extend |
|---|---|---|---|
| 1 | 98 °C, 45 s | — | — |
| 6 | 98 °C, 20 s | 58 °C, 30 s | 72 °C, 1 min |
| 11 | 98 °C, 20 s | 65 °C, 30 s | 72 °C, 1 min |
| 1 | — | — | 72 °C, 5 min |
■ PAUSE POINT The PCR products can be stored at 4 °C for 1 week. For longer-term storage (several months), keep at −80 °C.
cDNA purification and quantification ● Timing 3-4 h
-
66
Take the plate of amplified cDNA generated in Step 27 from the 4 °C refrigerator (liquid volume: ~25 μl), spin it briefly and then place it on the DynaMag-96 side magnet for 2 min.
-
67
Transfer the supernatant to a clean 96-well LoBind plate and discard the plate with M-280 beads.
-
68
Add 0.8× volume of AMPure beads (~20 ul) to each well of the cDNA plate above and purify the cDNA as described in Box 2.
-
69
Elute cDNA with 15 μl of 10 mM Tris-HCl (pH 8.0) for each sample and transfer the supernatant to a fresh 96-well PCR plate.
-
70
Determine the DNA concentrations and size distribution of each sample on a TapeStation by using D5000 tapes and reagents. Alternatively, determine the concentrations of all samples by a dsDNA high-sensitivity kit on a Qubit fluorometer or a plate reader and run eight samples on a high-sensitivity Bioanalyzer chip as a random spot test of the size distribution (Fig. 2a).
? TROUBLESHOOTING
-
71
Normalize the cDNA concentrations to 0.1 ng/μl by adding low-EDTA TE buffer.
▲ CRITICAL STEP It is important to normalize the cDNA to the correct concentration because the ratio of transposon to DNA substrate will determine the size distribution of Nextera tagmentation in the next step. In our experience, using the DNA default concentration of the Nextera XT protocol (0.5 ng in a down-sized 10-μl tagmentation) generates many fragments that are too large for sequencing.
Generation of Nextera XT RNA-Seq libraries ● Timing 3-4 h
▲ CRITICAL STEP To minimize the reagent cost, we use half the amount of each reagent in the Nextera XT DNA sample preparation kit, including PCR primers.
-
72
Take a LoBind PCR plate and put it on ice and then transfer 2.5 μl of normalized cDNA from each sample to the plate.
-
73
Set up a master mix in a 1.5-ml Eppendorf tube for the tagmentation reaction on ice following the recipe below.
| Component | Per reaction (μl) |
Per 120 reactions (μl) |
Final concentration in 10-μl reaction |
|---|---|---|---|
| 2× tagment DNA buffer | 5 | 600 | 1× |
| Amplicon tagment mix | 2.5 | 300 | — |
| Total volume | 7.5 | 900 | — |
-
74
Distribute 74 μl to each of the 12 wells in one row of a 96-well plate and then transfer 7.5 μl to the plate containing 0.25 ng of cDNA in 2.5 μl in each well (Box 1) for a final volume of 10 μl.
-
75
Seal and vortex the plate gently and spin it down at 1,000g for 30 s.
-
76
Carry out the tagmentation reaction as described below by using a thermocycler with the lid temperature set to 60°C.
| Order | Temperature (°C) | Duration (min) | Purpose |
|---|---|---|---|
| 1 | 55 | 5 | Tagmentation |
| 2 | 10 | Hold | — |
-
77
Distribute a 24-μl aliquot of NT buffer from the Nextera XT DNA sample preparation kit to a different row of 12 wells in the 96-well intermediate aliquot plate from Step 74 above. Then, transfer 2.5 μl of NT buffer to each well with tagmented DNA by using a 12-channel pipette (Box 1) for a total of 12.5 μl, mix the reaction by vortexing and spin the plate.
-
78
Incubate the reaction at room temperature for 5 min to inactivate the transposase.
-
79
Dispense 75 μl of Nextera PCR master mix to one distinct row of 12 wells in the 96-well PCR plate and then transfer 7.5 μl to each well containing the tagmented DNA (Box 1) for a total of 20 μl.
-
80
Centrifuge the Illumina Nextera UDI plate for 1 min at room temperature, place a clean 96-well PCR plate on top of it and press down slowly to puncture the foil seal on all 96 wells.
-
81
Transfer 5 μl from each well of the index plate to the sample plate for a total of 25 μl. Reseal the index plate with an aluminum-backed adhesive plate seal for storage.
-
82
Seal the PCR plate with a tape sheet, briefly vortex and spin the plate at 2,000g for 1 min.
-
83
Perform PCR following the program below.
| Cycle number | Denature | Anneal | Extend |
|---|---|---|---|
| 1 | — | — | 72 °C, 3 min |
| 1 | 95 °C, 30 s | — | — |
| 11 | 95 °C, 30 s | 55 °C, 30 s | 72 °C, 30 s |
| 1 | — | — | 72 °C, 5 min |
■ PAUSE POINT The PCR products can be stored at 4 °C for 1 week or at −80 °C for 1 year.
Cleaning-up and pooling of RNA-seq libraries ● Timing 3 h
-
84
Day 3. Purify the above PCR products by adding 17.5 μl (0.7× volumes) of undiluted AMPure beads following the instructions in Box 2.
-
85
Elute DNA from AMPure beads in 20 μl of low-EDTA TE buffer and transfer the supernatant to a new 96-well PCR plate.
-
86
Quantify and determine the size distribution of the libraries by using the high-sensitivity D1000 (Fig. 2b) or D5000 ScreenTape assay. Alternatively, perform a spot test of the size distribution by running eight randomly selected samples on a high-sensitivity Bioanalyzer chip and measure the library DNA concentration of all libraries by using a dsDNA high-sensitivity kit on a Qubit fluorometer or a plate reader.
-
87
Pool equal amounts (picomoles as determined on the TapeStation or nanograms as determined by the fluorometric assay) of each library for sequencing and run on a high-sensitivity BioAnalyzer chip to determine the size distribution and concentration for sequencing (Fig. 2d).
? TROUBLESHOOTING
Size selection of RRBS library ● Timing ~5 h
-
88
Transfer the PCR products of the same sample from Step 65 to a fresh Eppendorf tube.
-
89
Clean the PCR products by using 1.3 volumes of diluted (1:4 in bead dilution buffer) AMPure beads, incubate at room temperature for 10 min and then wash the beads with 800 μl of 80% (vol/vol) ethanol twice.
-
90
Elute DNA from the beads in 20 μl of low-EDTA TE buffer and run 1 μl on a high-sensitivity Bioanalyzer chip (Fig. 2c).
? TROUBLESHOOTING
-
91
Add 4 μl of 6× gel loading dye to the RRBS libraries and run them in four adjacent wells flanked by an empty well and low-molecular-weight DNA ladder on a 3% (wt/vol) NuSieve 3:1 agarose gel in 0.5× TBE at 5 V/cm in 0.5× TBE buffer until the bromophenol blue marker has migrated ~5 cm.
-
92
Stain the gel with SYBR green I, visualize the DNA by using a DR195 M blue light transilluminator and excise the 170–800-bp-size fraction across all four lanes, avoiding the adapter dimer product at ~135 bp. Optionally (to minimize size bias and maximize CpG coverage by sequencing), excise two size fractions (170–400 and 400–800 bp) and place the two gel slices in separate 5-ml tubes.
▲ CRITICAL STEP Do not use a UV transilluminator to visualize the stained DNA on the gel, because this may damage the DNA.
-
93
Purify the library DNA by using a Qiagen gel purification kit as per the manufacturer’s recommendations.
-
94
Elute the library DNA in 20 μl of low-EDTA TE buffer.
-
95
Examine the RRBS library size fractions on a high-sensitivity Agilent Bioanalyzer chip (Fig. 2d).
Sequencing the Smart-RRBS libraries ● Timing variable (depending on instrument)
-
96
Days 4 and 5. Quantify the RRBS library (or its size fractions) and the RNA-seq library pool by Qubit or high-sensitivity Agilent BioAnalyzer data and sequence in separate lanes on Illumina instruments.
Troubleshooting
Troubleshooting advice can be found in Table 2.
Table 2 ∣.
Troubleshooting table
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 63 | No PCR products (Fig. 2e) | Initial cell sorting was not carried out properly | Ensure that cell sorting is handled properly |
| >25 PCR cycles required and very pronounced satellite DNA bands visible | Bisulfite-converted DNA was degraded | Clean up the bisulfite-converted DNA immediately and perform the PCR amplification as quickly as possible. Keep bisulfite-converted DNA on ice when setting up the PCR reaction or put it in a −20 °C freezer directly if not PCR-amplifying right away | |
| 70 | No PCR products (Fig. 2e) | Initial cell sorting was not carried out properly | Ensure that cell sorting is handled properly |
| <2 ng of amplified cDNA | PCR cycle numbers were not optimized for the cell type | Increase the PCR cycle number up to 22 without jeopardizing mRNA representation | |
| The biotinylated RT-oligo–coated M-280 beads did not adequately bind mRNAs | Confirm that the biotinylated RT-oligo–coated beads are resuspended in the right buffer in Step 13 | ||
| Loss of M-280 beads, thereby bead-bound mRNAs, during washing | Make sure that no M-280 beads are improperly transferred to the plate containing gDNA from Steps 19–21 | ||
| 87 | The sizes of RNA-seq library DNA are too large (>1,000 bp) | Too much cDNA added to tagmentation reaction | Reduce the cDNA concentration to 0.1 ng |
| 90 | No or very little (barely visible) RRBS library DNA (Fig. 2e) | Bisulfite-converted DNA was degraded between Steps 57 (purification of bisulfite-converted DNA) and 65 (RRBS library amplification) | Perform RRBS library amplification sooner rather than later and keep bisulfite-converted DNA frozen while performing Steps 59–63 (test PCR amplifications) |
Timing
Day 0
Steps 1–5, preparing single-cell plates: 1 h hands-on time excluding FACS sorting
Day 1
Steps 6–14, preparing beads for mRNA capture and RT: 1 h
Steps 15–21, separation of gDNA and mRNA: 1–1.5 h
Steps 22–27, RT and cDNA amplification: 5–5.5 h (hands-on time: 0.5–1 h)
Steps 28–30, clean-up of gDNA fraction: 1 h
Steps 31–34, restriction digestion of gDNA: 2 h (hands-on time: 0.5 h)
Steps 35–39, end repair and A-tailing: 1.5 h (hands-on time: 0.5 h)
Steps 40–45, adapter ligation: 16–20 h (hands-on time: 0.5 h)
Day 2
Steps 46–55, pooling and clean-up of ligation reactions: ~2 h
Steps 56–58, bisulfite conversion: ~1.5 h
Steps 59–65, RRBS library amplification: 2.5 h
Steps 66–71, cDNA purification and quantification: 3–4 h
Steps 72–83, generation of RNA-seq libraries by using a Nextera XT DNA sample preparation kit: 3–4 h
Day 3
Steps 84–87, cleaning-up and pooling of RNA-seq libraries: 3 h
Steps 88–95, size selection of RRBS library: ~5 h
Days 4 and 5
Step 96, sequencing the Smart-RRBS libraries: variable (30 h on HiSeq-2500; sequencing on other Illumina instruments may take up to 5 d)
Anticipated results
With training, one person can produce a set of Smart-RRBS libraries from one to two 96-well plates of flow-sorted single cells within 3 d. In this section, we show representative data from 96 single human embryonic stem (ES) cells that were engineered to allow doxycycline-inducible down-regulation of DNA methyl transferase 1 (non-induced day 0 HUES64 cells62) and fixed and stored in RNAprotect cell reagent before FACS sorting. The RRBS libraries were from MspI+HaeIII double-digested gDNA. The underlying sequencing data have been deposited to the NCBI GEO Archive under accession GSE157115 (GSE157113 for RNA-seq and GSE157114 for RRBS data).
Of 96 input cells, 84 cells produced >2 × 105 passing demultiplexed RRBS read pairs and had >105 distinct CpGs covered by at least one read, while 91 cells passed our quality filters for RNA-seq (>7 × 104 reads aligned to exons, >5,000 genes detected and <20% mitochondrial reads). Overall, 80 cells (83%) passed both RRBS and RNA-seq QC (Fig. 3). These 80 cells received a median of 2.3 million passing RRBS read pairs, of which 2.0 million (86%) aligned to the genome; 1.8 million of them (79%) mapped uniquely. C-to-T conversion rates at presumably unmethylated non-CpG (i.e., CpH) dinucleotides were 99.4%, whereas mean CpG methylation levels were 78% in a typical (median) cell (Extended Data Fig. 3a-c). Passing and aligned RNA-seq counts (medians of 4.1 million aligned and 2.0 million uniquely aligned), final library sizes of exonic reads (0.65 million) and the proportion of mitochondrial genes (9.9%) for passing cells are shown in Extended Data Fig. 4.
Fig. 3 ∣. Quality filters for Smart-RRBS data.

Scatter plots of the two primary filters for RRBS (distinct CpGs covered over passing read pairs) and Smart-seq2 (genes detected at TPM >0 over aligned reads) for 96 single cells are shown at the top, with the quality thresholds on both axes indicated by dashed lines. The numbers of passing cells in the upper right quadrant were 84 (RRBS) and 91 (RNA-seq). The Venn diagram at the bottom illustrates the intersection of 80 cells that passed all quality filters for both assays. One cell failed both RRBS and RNA-seq QCs and is not represented in the Venn diagram.
The number of CpGs covered by RRBS ranged from 0.2 to 2.5 million per cell (mean: 1.4 million; median: 1.5 million; Fig. 4a). About half of these CpGs were covered only by reads from the large size fraction, ~40% only by the small and ~10% by both size fractions (Extended Data Fig. 3d). Down-sampling of the sequencing data indicates that we did not saturate the complexity of the RRBS libraries. Extrapolation of the saturation curve suggests that increasing the sequencing effort by fivefold (e.g., by sequencing on a NovaSeq instead of a HiSeq2500 instrument) may increase the number of covered CpGs by ~1.5-fold to 2.0 million per cell on average without an increase in sequencing cost (Extended Data Fig. 5). We note that many more CpGs (10.5 million) are covered in pseudo-bulk data aggregated from all passing cells, indicating largely random losses at the single-cell level during the process.
Fig. 4 ∣. Performance metrics for 80 passing cells.

a, Notched box plot showing the overall and inner-quartile range, median and 95% confidence interval of the number of unique CpG sites covered by at least one RRBS read. b, Box plots showing the fraction of CpGs covered by RRBS reads that map to the eight genome compartments indicated on the x axis. Note that a given CpG can be assigned to more than one genome compartment. Annotations of features were accessed through the annotatr package60. The total number of CpG islands assigned to chromosomes in hg38 is 27,949. CpG shores are defined as 2 kb upstream/downstream from the ends of the CpG islands, and CpG shelves are defined as another 2 kb upstream/downstream of the farthest upstream/downstream limits of the CpG shores. Exons, introns and intergenic regions refer to 25,130 protein-coding genes in hg38. Promoters are 1-kb segments including and upstream of 25,130 annotated transcription start sites of the same protein-coding genes. Enhancers are the permissive enhancer annotations from the FANTOM5 dataset. c, Violin box plots showing the distribution of the fractions of all annotated CGIs, promoters of protein-coding genes and FANTOM5 enhancers in the human genome with RRBS coverage at one or more CpGs at the single-cell level. Bars indicate the fraction of these genomic features covered by RRBS reads aggregated from all 80 passing single cells. d, Notched box plots showing the number of protein-coding genes covered by RRBS at a minimum of one or three CpGs in their promoter region (left panel), detected by Smart-seq2 at TPMs >0, >1 or >3 (middle) and with both promoter methylation and gene expression data at the minimal respective thresholds indicated (right panel).
As expected for RRBS, a large fraction (median: 77%) of covered CpGs mapped to CpG islands. More than half and about one-third of CpGs lie in intergenic regions (55%) or in protein-coding exons (33%), respectively. About 10% of CpGs were found in introns, CGI shelves, promoters and enhancers, and 5% were in CGI shores (Fig. 4b). Almost 40% of all annotated CGIs in the genome have at least one CpG covered in a typical single cell (median: 37%; red violin plot in Fig. 4c), whereas 95% of all CGIs received minimal coverage by pseudo-bulk RRBS data aggregated from the same 80 single cells (red bar in Fig. 4c). This is consistent with dropouts at the single-cell level because of losses during library construction starting from merely two copies of ds gDNA in post-mitotic diploid single cells. A similar fraction of promoter annotations—many of them containing CGIs—is covered in single cells (median: 41%), compared to 70% in aggregated single-cell data. As expected, only a minority of CpG-poorer features, such as permissive Fantom5 enhancers (median: 9.1%), are covered at the single-cell level, and the dropout rate compared to aggregation of single-cell data (49%) is higher, presumably because these elements contain fewer restriction fragments in the RRBS size range than CGIs that may still receive minimal coverage at some fragments despite dropout of other fragments. The percentage of CGIs, promoters and enhancers with methylation information for three or more CpGs is shown in Extended Data Fig. 6.
Dropouts at the single-cell level limit the ability to compare the methylation status of individual CpGs and, to a lesser degree, of larger multi-CpG functional units such as CGIs. Not surprisingly, given that the mean number of CpGs covered at the single-cell level is only 13% of the CpGs covered by the aggregated single-cell data, the number of CpGs covered across multiple cells drops rapidly as the number of intersected single cells goes up. In aggregate, ~5.4 million or 1.5 million CpGs can be compared across four or eight cells, respectively, and 31,132, 2,427 or 509 CpGs are informative across 16, 32 or 64 cells in the data set (Extended Data Fig. 7). Importantly, the methylation status of CGIs can be compared across cells despite lack of shared coverage at the single-CpG level. At a minimum coverage threshold of one CpG per CGI, an aggregate of 16,599; 8,740; 3,883; 1,169 or 64 CGIs are informative across 4, 8, 16, 32 or 64 cells, respectively. Ten percent of the aggregate 26,887 CGIs but only 0.1% of all CpGs in the data set can be compared across 20 cells, and the percentage of CGIs informative across n cells is >50-fold higher for 16 to 64 cells than for common CpGs, which drop to <10% at 9 cells and <1% at 14 cells (Extended Data Fig. 7).
Forty-one percent (10,388) and 33% (8,221) of all promoters had at least one CpG and at least three CpGs covered by RRBS, respectively, in a typical (median) cell (Fig. 4d, left). Similarly, 42% (10,458) of protein coding genes were detected by RNA-seq at a level greater than zero transcripts per million (TPM), whereas fewer genes (medians: 6,509 and 4,544) had TPMs >1 and >3, respectively (Fig. 4d, center). Importantly, and highlighting the strength of our Smart-RRBS protocol, 24% (5,906) of all protein-coding genes had detectable (TPM >0) gene expression and RRBS coverage of at least one CpG in their promoter region in the same single cell (Fig. 4d, right). Raising the RNA-seq threshold to TPM >1 reduced the median number of genes by ~40%, whereas raising the minimum promoter CpG coverage from one to three affected only ~20% of the genes. For example, 4,671 genes had a TPM >0 and a methylation measurement for three or more promoter CpGs. A summary table of these and other key performance metrics is provided in Table 3. We also include a diagnostic plot of single-cell TPM values for the 30 highest-expressed genes (Extended Data Fig. 8) and the distribution of TPM values across 80 single cells for 18 genes expected to be expressed in human ES cells, including genes for DNA methyltransferases, NANOG (a key factor in maintaining pluripotency) and the four original Yamanaka iPS re-programming transcription factors Oct4 (Pou5f1), Sox2, cMyc and Klf4 (Extended Data Fig. 9). Complete numerical data underlying each data figure are available as Source Data files.
Table 3 ∣.
Summary of key performance metrics
| Measurement and metric | Median per cell | Mean per cell |
|---|---|---|
| RRBS | ||
| CpGs covered | 1,543,918 | 1,376,086 |
| Bisulfite conversion at CpH sites (%) | 99.4 | 99.4 |
| Gene promoters covered at ≥1 CpG | 10,388 | 9,631 |
| Gene promoters covered at ≥3 CpGs | 8,221 | 7,542 |
| Smart RNA-seq | ||
| Genes with TPM >0 | 10,458 | 10,068 |
| Genes with TPM >1 | 6,509 | 6,072 |
| Genes with TPM >3 | 4,544 | 4,298 |
| Joint Smart-RRBS | ||
| Genes covered at ≥1 promoter CpG and TPM >0 | 5,906 | 5,683 |
| Genes covered at ≥1 promoter CpG and TPM >1 | 3,584 | 3,316 |
| Genes covered at ≥1 promoter CpG and TPM >3 | 2,464 | 2,298 |
| Genes covered at ≥3 promoter CpGs and TPM >0 | 4,671 | 4,620 |
| Genes covered at ≥3 promoter CpGs and TPM >1 | 2,809 | 2,704 |
| Genes covered at ≥3 promoter CpGs and TPM >3 | 1,961 | 1,872 |
TPM, transcripts per million.
Extended Data
Extended Data Fig. 1 ∣. Schematic of steps involved in the workflow of the RRBS data analysis.
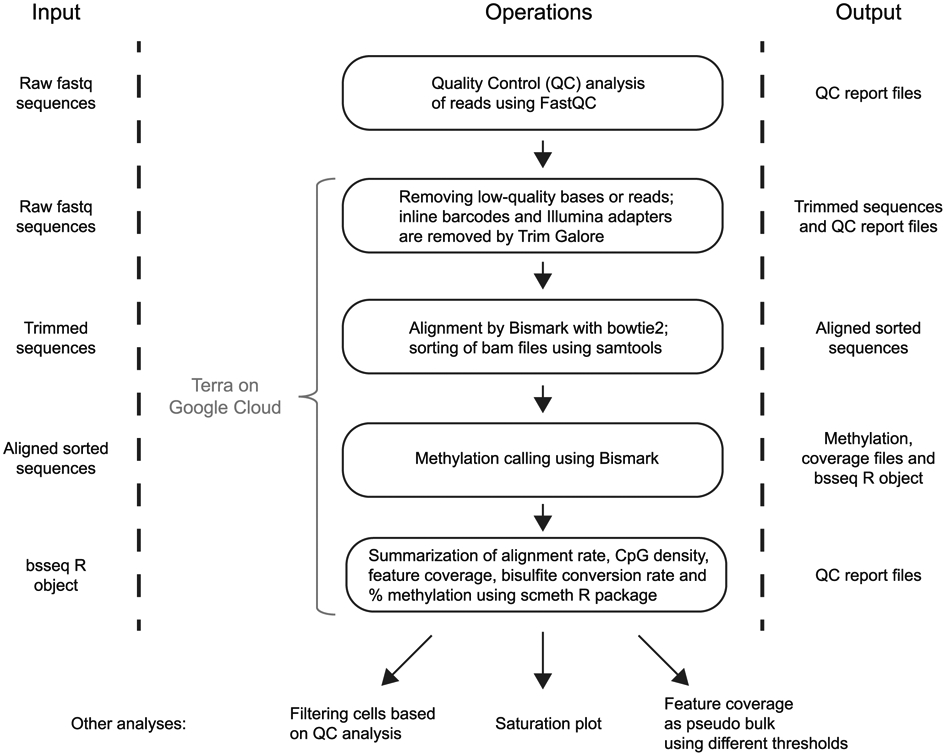
The figure is divided into three sections. In the operations section, each box contains a description of the data analysis step, and arrows indicate the progression through the analysis workflow. First, the user can easily run the FastQC analysis, which provides basic sequencing quality metrics, base composition and Illumina-adapter content. The rest of the workflow is developed in the Workflow Description Language format and is available on Terra, a cloud-native platform that runs in the Google cloud. Each row shows the input files that are required for each operation and the format of result reports or files that will be obtained. The last analysis steps (quality-filtering of cells, saturation plot and pseudo bulk analysis) have been described in the main text.
Extended Data Fig. 2 ∣. Process for manually dispensing a master mix to a sample plate.

This two-step technique is recommended at several steps of the protocol.
Extended Data Fig. 3 ∣. RRBS performance metrics and global CpG methylation across 80 passing cells.

Notched box plots to show the overall and inner-quartile range, median and 95% confidence interval of the number of purity filtered and fully demultiplexed RRBS reads before and after genome alignment (total and unique alignments) (a), the C-to-U bisulfite conversion rate of presumably unmethylated cytosines in non-CpG (CpH) context covered by RRBS reads (b) and the mean methylation level at all CpG sites covered in a given cell (c). d, Venn diagrams showing median and mean numbers of CpG sites covered per passing cell exclusively by reads from the large or small size fraction or covered by both. The approximate total numbers of passing read pairs in 80 passing cells from large and small size fractions were 83 million and 90 million, respectively. Aligned and uniquely aligned read numbers for each size fraction are available in the source data for this figure.
Extended Data Fig. 4 ∣. RNA-seq performance metrics across 80 passing cells.

Notched box plots to show the overall and inner-quartile range, median and 95% confidence interval of purity-filtered demultiplexed RNA-seq reads before and after alignment to the genome (total and unique alignments) by using STAR55 v.2.7.3a with the arguments --quantMode GeneCounts --sjdbGTFtagExonParentGene gene_name --outFilterScoreMinOverLread 0.1 --outFilterMatchNminOverLread 0.1 (a), the library size of unique reads aligning to exons (b) and the fraction of exonic reads that map to the mitochondrial genome (c).
Extended Data Fig. 5 ∣. Down-sampling and extrapolation of RRBS effort and CpG coverage.

a, The aggregate set of passing RRBS read pairs from 80 single cells was randomly down-sampled in steps from the full data (1 on the x-axis; 1.72 billion total read pairs; mean: 2.15 million per cell) to 0.01. The number of CpGs covered in each cell at each step was determined, and the corresponding saturation curves were normalized by the CpGs covered at the original full sequencing effort, interpolated and plotted as a function of the relative sequencing effort (fraction of reads). The blue dots represent the mean normalized fraction of CpGs at each step. b, The blue dots in the saturation curve are the same down-sampled data points as in a. The insert is a linear regression (R2 = 0.99999) of the actual mean normalized CpGs (blue dots) versus calculated mean normalized CpGs, assuming the saturation curve follows a Michaelis-Menten equation: (Fraction of CpGs)calc = (Fraction of reads/0.6004)/(Fraction of reads + 0.4023/0.6004), whereby 0.6004 and 0.4023 were the slope and y-intercept, respectively, of a Hanes-Woolf linearization of the Michaelis-Menten equation (R2 = 0.9999). The red dots are extrapolated mean normalized fractions of RRBS-covered CpGs. The saturation curve approaches a limit of ~1.67 on the y-axis, corresponding to a mean of 2.3 million CpGs per cell. Extrapolation to 5 on the x-axis results in 2.0 million CpGs per cell on average.
Extended Data Fig. 6 ∣. Fraction of all annotated CpG islands, promoters and enhancers covered at three or more CpGs.

Violin box plots show the distribution of the fraction of all annotated CGIs, promoters of protein-coding genes and permissive FANTOM5 enhancers in the human genome with RRBS coverage at three or more CpGs at the single-cell level. Bars indicate the fraction of these genomic features covered by RRBS reads aggregated from all 80 passing single cells.
Extended Data Fig. 7 ∣. Comparability of distinct CpG sites and CpG islands across single cells.

Shown on the left y-axes are the absolute numbers and fractions of their respective pseudo-bulk aggregate numbers of common CpGs (red) or CGIs (blue) that can be compared anywhere in the data set across the number of cells indicated on the x-axis. The absolute numbers were the CpGs or CGIs with n or more hits in a matrix of 80 cells versus all CpGs (10,532,278) or all CGIs (26,887) that were hit at least once in data aggregated from all 80 passing cells (i.e., the pseudo-bulk aggregate, which is by definition the value for comparability across n = 1 cell). The minimum coverage threshold for CGIs is one CpG. The y-axis on the right is the ratio of the fractions of comparable CGIs and CpGs.
Extended Data Fig. 8 ∣. Genes with the highest RNA expression levels in 80 passing cells.

Tick marks on the horizontal lines denote TPM calculated for each single cell. Genes are ordered top to bottom by the Mean TPM value (blue circles) across 80 single cells. The eleven most highly expressed genes are mitochondrial (MT) genes.
Extended Data Fig. 9 ∣. Expression levels of 18 genes associated with pluripotent cells.

Shown are the distribution (violin plots) and single cell values (dots) of expression levels (TPM) on a log2 scale.
Supplementary Material
Acknowledgements
This work was supported by a Broad Institute SPARC award (to A.G.), NIGMS grant no. P01GM099117 (to A.M. and A.G. and a pilot grant to M.J.A.), National Cancer Institute grant no. R01-CA229902 (to D.A.L.) and the Merkin Institute (to M.J.A.). A.M. is supported by the Max Planck Society. All human embryonic stem cell work was approved by Harvard University’s Embryonic Stem Cell Research Oversight (ESCRO #E00021).
Footnotes
Competing interests
The authors declare no competing financial interests.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41596-021-00571-9.
Reporting Summary Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Code availability Our DNA-methylation analysis pipeline for large bisulfite sequencing data sets, including single-cell RRBS data (https://github.com/aryeelab/dna-methylation-tools), runs in the Google cloud and can be accessed for a fee through the Broad Institute’s Terra platform (https://app.terra.bio/#workspaces/aryee-lab/bisulfite-seq-tools-grch38).
Data availability
Sequencing Data underlying Figs. 3 and 4 and Extended Data Figs. 3-9 have been deposited to the NCBI GEO Archive under GSE157115 (GSE157113 for RNA-seq and GSE157114 for RRBS data). Source data are provided with this paper.
References
- 1.Tang F et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 6, 377–382 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Gawad C, Koh W & Quake SR Single-cell genome sequencing: current state of the science. Nat. Rev. Genet 17, 175–188 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Navin N et al. Tumour evolution inferred by single-cell sequencing. Nature 472, 90–94 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu S et al. Probing meiotic recombination and aneuploidy of single sperm cells by whole-genome sequencing. Science 338, 1627–1630 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zong C, Lu S, Chapman AR & Xie XS Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science 338, 1622–1626 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ding J et al. Systematic comparison of single-cell and single-nucleus RNA-sequencing methods. Nat. Biotechnol 38, 737–746 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haque A, Engel J, Teichmann SA & Lönnberg T A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med. 9, 75 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ziegenhain C et al. Comparative analysis of single-cell RNA sequencing methods. Mol. Cell 65, 631–643.e4 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Charlton J et al. Global delay in nascent strand DNA methylation. Nat. Struct. Mol. Biol 25, 327–332 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gaiti F et al. Epigenetic evolution and lineage histories of chronic lymphocytic leukaemia. Nature 569, 576–580 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guo H et al. Single-cell methylome landscapes of mouse embryonic stem cells and early embryos analyzed using reduced representation bisulfite sequencing. Genome Res. 23, 2126–2135 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smallwood SA et al. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat. Methods 11, 817–820 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rotem A et al. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat. Biotechnol 33, 1165–1172 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buenrostro JD et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cusanovich DA et al. Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 348, 910–914 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jin W et al. Genome-wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples. Nature 528, 142–146 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stevens TJ et al. 3D structures of individual mammalian genomes studied by single-cell Hi-C. Nature 544, 59–64 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anaparthy N, Ho YJ, Martelotto L, Hammell M & Hicks J Single-cell applications of next-generation sequencing. Cold Spring Harb. Perspect. Med 9, a026898 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y & Navin NE Advances and applications of single-cell sequencing technologies. Mol. Cell 58, 598–609 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Navin NE The first five years of single-cell cancer genomics and beyond. Genome Res. 25, 1499–1507 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dey SS, Kester L, Spanjaard B, Bienko M & van Oudenaarden A Integrated genome and transcriptome sequencing of the same cell. Nat. Biotechnol 33, 285–289 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Macaulay IC et al. G&T-seq: parallel sequencing of single-cell genomes and transcriptomes. Nat. Methods 12, 519–522 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Angermueller C et al. Parallel single-cell sequencing links transcriptional and epigenetic heterogeneity. Nat. Methods 13, 229–232 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu Y et al. Simultaneous profiling of transcriptome and DNA methylome from a single cell. Genome Biol. 17, 88 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee DS et al. Simultaneous profiling of 3D genome structure and DNA methylation in single human cells. Nat. Methods 16, 999–1006 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li G et al. Joint profiling of DNA methylation and chromatin architecture in single cells. Nat. Methods 16, 991–993 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hou Y et al. Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res. 26, 304–319 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Picelli S et al. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods 10, 1096–1098 (2013). [DOI] [PubMed] [Google Scholar]
- 29.Smith ZD et al. Epigenetic restriction of extraembryonic lineages mirrors the somatic transition to cancer. Nature 549, 543–547 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meissner A et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454, 766–770 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guo H et al. The DNA methylation landscape of human early embryos. Nature 511, 606–610 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Klughammer J et al. The DNA methylation landscape of glioblastoma disease progression shows extensive heterogeneity in time and space. Nat. Med 24, 1611–1624 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schrott R et al. Cannabis use is associated with potentially heritable widespread changes in autism candidate gene DLGAP2 DNA methylation in sperm. Epigenetics 15, 161–173 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stryjewska A et al. Zeb2 regulates cell fate at the exit from epiblast state in mouse embryonic stem cells. Stem Cells 35, 611–625 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Szymczak S et al. DNA methylation QTL analysis identifies new regulators of human longevity. Hum. Mol. Genet 29, 1154–1167 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clark SJ et al. Genome-wide base-resolution mapping of DNA methylation in single cells using single-cell bisulfite sequencing (scBS-seq). Nat. Protoc 12, 534–547 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Luo C et al. Robust single-cell DNA methylome profiling with snmC-seq2. Nat. Commun 9, 3824 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Picelli S et al. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc 9, 171–181 (2014). [DOI] [PubMed] [Google Scholar]
- 39.Deaton AM & Bird A CpG islands and the regulation of transcription. Genes Dev. 25, 1010–1022 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Macaulay IC et al. Separation and parallel sequencing of the genomes and transcriptomes of single cells using G&T-seq. Nat. Protoc 11, 2081–2103 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Pastore A et al. Corrupted coordination of epigenetic modifications leads to diverging chromatin states and transcriptional heterogeneity in CLL. Nat. Commun 10, 1874 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schubeler D Function and information content of DNA methylation. Nature 517, 321–326 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Neri F et al. Intragenic DNA methylation prevents spurious transcription initiation. Nature 543, 72–77 (2017). [DOI] [PubMed] [Google Scholar]
- 44.Guo H et al. Profiling DNA methylome landscapes of mammalian cells with single-cell reduced-representation bisulfite sequencing. Nat. Protoc 10, 645–659 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Boyle P et al. Gel-free multiplexed reduced representation bisulfite sequencing for large-scale DNA methylation profiling. Genome Biol. 13, R92 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hagemann-Jensen M et al. Single-cell RNA counting at allele and isoform resolution using Smart-seq3. Nat. Biotechnol 38, 708–714 (2020). [DOI] [PubMed] [Google Scholar]
- 47.Klein AM et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161, 1187–1201 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Macosko EZ et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cao J et al. Comprehensive single-cell transcriptional profiling of a multicellular organism. Science 357, 661–667 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mulqueen RM et al. Highly scalable generation of DNA methylation profiles in single cells. Nat. Biotechnol 36, 428–431 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hennig BP et al. Large-scale low-cost NGS library preparation using a robust Tn5 purification and tagmentation protocol. G3 (Bethesda) 8, 79–89 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Picelli S et al. Tn5 transposase and tagmentation procedures for massively scaled sequencing projects. Genome Res. 24, 2033–2040 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang J et al. Double restriction-enzyme digestion improves the coverage and accuracy of genome-wide CpG methylation profiling by reduced representation bisulfite sequencing. BMC Genomics 14, 11 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Martinez-Arguelles DB, Lee S & Papadopoulos V In silico analysis identifies novel restriction enzyme combinations that expand reduced representation bisulfite sequencing CpG coverage. BMC Res. Notes 7, 534 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McCarthy DJ, Campbell KR, Lun AT & Wills QF Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics 33, 1179–1186 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kangeyan D et al. A (fire)cloud-based DNA methylation data preprocessing and quality control platform. BMC Bioinforma. 20, 160 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Krueger F & Andrews SR Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27, 1571–1572 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cavalcante RG & Sartor MA annotatr: genomic regions in context. Bioinformatics 33, 2381–2383 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Andersson R et al. An atlas of active enhancers across human cell types and tissues. Nature 507, 455–461 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tsankov AM et al. Loss of DNA methyltransferase activity in primed human ES cells triggers increased cell-cell variability and transcriptional repression. Development 146, dev174722 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Related links
Key references using this protocol
Gaiti, F. et al. Nature 569, 576-580 (2019): https://doi.org/10.1038/s41586-019-1198-z
Pastore, A. et al. Nat. Commun. 10, 1874 (2019): https://doi.org/10.1038/s41467-019-09645-5
Smith, Z. D. et al. Nature 549, 543-547 (2017): https://doi.org/10.1038/nature23891
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing Data underlying Figs. 3 and 4 and Extended Data Figs. 3-9 have been deposited to the NCBI GEO Archive under GSE157115 (GSE157113 for RNA-seq and GSE157114 for RRBS data). Source data are provided with this paper.