

Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the causative agent of the current coronavirus disease 2019 (COVID-19) pandemic. It is known that the receptor-binding domain (RBD) of the spike protein of SARS-CoV-2 interacts with the human angiotensin-converting enzyme 2 (ACE2) receptor, initiating the entry of SARS-CoV-2. Since its emergence, a number of SARS-CoV-2 variants have been reported, and the variants that show high infectivity are classified as the variants of concern according to the US CDC. In this study, we performed both all-atom steered molecular dynamics (SMD) simulations and microscale thermophoresis (MST) experiments to characterize the binding interactions between ACE2 and RBD of all current variants of concern (Alpha, Beta, Gamma, and Delta) and two variants of interest (Epsilon and Kappa). We report that the RBD of the Alpha (N501Y) variant requires the highest amount of force initially to be detached from ACE2 due to the N501Y mutation in addition to the role of N90-glycan, followed by Beta/Gamma (K417N/T, E484K, and N501Y) or Delta (L452R and T478K) variant. Among all variants investigated in this work, the RBD of the Epsilon (L452R) variant is relatively easily detached from ACE2. Our results combined SMD simulations and MST experiments indicate what makes each variant more contagious in terms of RBD and ACE2 interactions. This study could shed light on developing new drugs to inhibit SARS-CoV-2 entry effectively.
Graphical Abstract
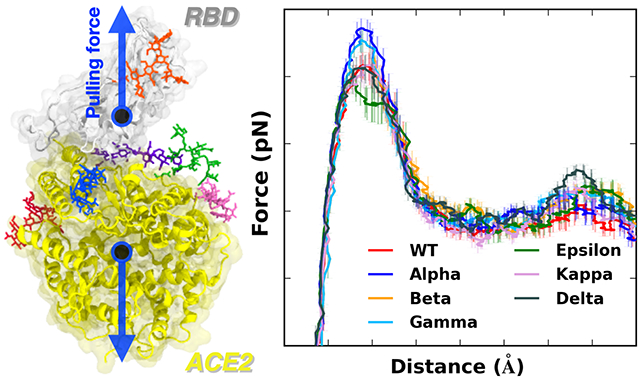
INTRODUCTION
Reported in late 2019, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) emerged and has rapidly infected people worldwide. As of mid-September 2021, 230 million cases and 4.71 million deaths have been reported globally.1 Despite worldwide efforts to overcome the current coronavirus disease 2019 (COVID-19) pandemic, the rise of various SARS-CoV-2 variants may become futile in the efficacy of vaccination and other countermeasures.
SARS-CoV-2 virus utilizes receptor-binding domain (RBD) of S1 protein, a part of trimeric spike (S) glycoprotein,2–3 for viral entry through the RBD interaction with the human receptor angiotensin-converting enzyme 2 (ACE2). Since ACE2 can interact with the RBD of both SARS-CoV-2 and SARS-CoV (or SARS-CoV-1, the virus that caused the 2002-2004 SARS outbreak), there have been many studies not only to understand binding interactions between RBD and ACE2, but also to characterize the difference between SARS-CoV-1 and SARS-CoV-2.4–6
In September 2020, the Alpha variant, lineage B.1.1.7, was first detected in southeast England and quickly became a populated lineage in the United Kingdom. The variant was subsequently detected in the United States in December 2020.7–8 Beta variant, lineage B.1.351, was first detected in South Africa in May 2020 and found in the United States at the end of January 2021.9 At that time, there was another identified Gamma variant, which is known for lineage P.1,10–11 in the United States that was initially found in Japan from a traveler from Brazil. In November 2020, Epsilon variant, lineage B.1.427, was detected in California in the United States.12 Recently, two additional variants, Kappa (lineage B.1.617.1) and Delta (lineage B.1.617.2), first identified in India at the end of 2020, were detected in the United States.13 Since the emergence of diverse SARS-CoV-2 variants, Alpha, Beta, Gamma, and Delta variants are classified as the variants of concern by the US CDC due to their high infectivity.
Several studies have been performed experimentally and computationally to better understand the highly contagious characteristics of these variants.14–16 For example, Tian et al. conducted an experimental and computational study to capture the role of N501Y mutation in Alpha, Beta, and Gamma variants.14 They suggested that the π-π interactions and π-cation interactions are responsible for the enhanced interactions between RBD and ACE2. However, only the N501Y mutation was examined in their study, although other potentially important mutations have emerged. More recently, Socher et al. performed energy decomposition analysis from molecular dynamics simulations to compare the interaction energies between ACE2 and RBD of Alpha, Beta, and Gamma variants.15 They investigated each specific mutation, N501Y, K417N/T, and E484K, and reported that F486, Q498, T500, and Y505 in RBD are important residues across viral variants in the RBD-ACE2 interface.
In this study, using all-atom steered molecular dynamics (SMD) simulations and microscale thermophoresis (MST) experiments (see Supporting Information Methods), we report the differential interactions between human ACE2 and RBD of SARS-CoV-2 of all variants of concern (Alpha, Beta, Gamma, and Delta) as well as two variants of interest (Epsilon and Kappa). The study also provides a better understanding of such differences at the molecular level.
METHODS
Computational Methods
A fully-glycosylated SARS-CoV-2 RBD and ACE2 complex was obtained from COVID-19 Protein Library in CHARMM-GUI Archive (6vsb_1_1_1_6vw1.pdb).17 The complex includes 6 N-linked glycans: five glycans in ACE2 (Asn53, Asn90, Asn103, Asn322, and Asn546) and one glycan in RBD (Asn343). For system generation, parameter setup, and corresponding mutations, we utilized CHARMM-GUI Solution Builder.18–19 From the WT RBD structure, each variant was modeled with the following mutations: Alpha (N501Y), Beta (K417N, E484K, N501Y), Gamma (K417T, E484K, N501Y), Epsilon (L452R), Kappa (L452R, E484Q), and Delta (L452R, T478K). The CHARMM36(m) force field20–21 for protein and carbohydrates with TIP3P water model22 was used with 0.15 M of K+ and Cl− ions for mimicking physiological conditions. The system size was determined to be large enough (about 190 Å × 190 Å × 190 Å) to have the proteins solvated enough when they are fully unattached. The total number of atoms is approximately 550,000.
The overall simulation details are nearly identical to our previous work.4 NAMD simulation software23 was used for the pulling simulations with the COLVARS method. As an initial condition, the SARS-CoV-2 RBD and ACE2 complex structures were aligned along the X-axis, and the center of mass (COM) of each protein was calculated to apply the external force on the proteins. The effective force acting on the COMs of both proteins can be calculated through the following equation:
where k is the spring constant, v is the moving speed of the spring potentials (also called dummy atoms), R(t) is the current position of the selected protein COM, and n is the COM-COM unit vector. This force enables the spring-connected proteins to move in the opposite directions to pull away two proteins. The moving speed of proteins was set to 0.5 Å/ns along the X-axis, and a spring constant of 5 kcal/mol/Å2 was applied to the COM of each protein to have both proteins move along the X direction and restrict moving along the Y and Z directions. For better statistics, 20 independent simulations for each system were performed (140 systems total, 20 replicas of 7 variants) with at least 40 ns of each simulation run. The pulling simulations stopped when the RBD and ACE2 were completely detached from each other.
The van der Waals interactions were switched off smoothly over 10-12 Å using a force-based switching function.24 The electrostatic interactions were calculated by the particle-mesh Ewald method with a mesh size of 1 Å.25 To constrain bond lengths involving hydrogen atoms, the SHAKE algorithm was used.26 The simulation time-step was set to 4 fs with the hydrogen mass repartitioning method.27–28 Equilibration simulations were performed with the NVT (constant particle number, volume, and temperature) ensemble where positional and dihedral restraints were employed. The restraint was gradually decreased during the equilibration simulations. The NPT (constant particle number, pressure, and temperature) ensemble was then applied for the production runs, where the Langevin piston method29 was used for the pressure control. The entire simulation temperature was set to 303.15 K with the Langevin damping control method.
Experimental Methods
Recombinant human ACE2 protein (GenBank accession: AF291820.1, Sino Biological 10108-H08H; Wayne, PA) was labeled with RED-NHS (2nd Generation) dye using the Monolith Protein Labeling Kit (NanoTemper Technologies, MO-L011, München, Germany). Labeled ACE2 (5 nM, final concentration) was mixed with the RBD proteins (WT or variants, 2-fold diluted in a 15-step starting from 1.5-4 μM) in PBS buffer supplanted with 0.1 % Pluronic® F-127. The RBD proteins include: WT (ACRObiosystems, SPD-C52H3, Newark, DE, GenBank accession: QHD43416.1), Alpha (N501Y, ACRObiosystems, SPD-C52Hn), Beta (K417N, E484K, N501Y, ACRObiosystems, SPD-C52Hp), Epsilon (L452R, Sino Biological, 40592-V08H28), Kappa (L452R, E484Q, Sino Biological, 40592-V08H88), and Delta (L452R, T478K, Sino Biological, 40592-V08H90). All the recombinant proteins used in this study were produced in HEK293 cells and presumably fully glycosylated. The mixed RBD+ACE2 samples were separately loaded into 16 premium glass capillaries (NanoTemper Technologies, MO-K025). The 16 capillaries were then placed in the reaction chamber in the order of concentration. Microscale thermophoresis (MST) measurements were conducted on a Monolith NT.115 instrument (NanoTemper Technologies) at 20% excitation power at 24 °C. The measurement was repeated at least three times. Kd calculations were performed using the MO Affinity Analysis software (NanoTemper Technologies).
RESULTS AND DISCUSSION
Almost all variants show increased interactions with ACE2
To gain molecular insight into the difference of all variants that are classified as the variants of concern (Alpha (originated in the United Kingdom, B.1.1.7: N501Y), Beta (originated in South Africa, B.1.351: K417N, E484K, N501Y), Gamma (originated in Japan/Brazil, P.1: K417T, E484K, N501Y), and Delta (originated in India, B.1.617.2: L452R, T478K)) and two additional variants of interest (Epsilon (originated in US-California, B.1.427: L452R) and Kappa (originated in India, B.1.617.1: L452R, E484Q)), pulling force analysis was performed on each RBD-ACE2 complex (Figure 1A) as a function of distance (D) between the centers of mass of RBD and ACE2 proteins. Our fully-glycosylated S RBD-ACE2 complex model (Figure 1B,C) was employed for the pulling simulation.17 As shown in Figure 1A, most variants have increased force profiles than WT except for the Epsilon variant, indicating that the variants have strengthened interactions with ACE2. It should be noted that the amount of initial force at D = 53 Å shows a good match with our previous WT study,4 where we utilized only the N-linked glycan (N-glycan) core structure for all N-glycans. In this study, we used the most probable N-glycan structures (Figure 1C) that are larger than the core structure.
Figure 1.
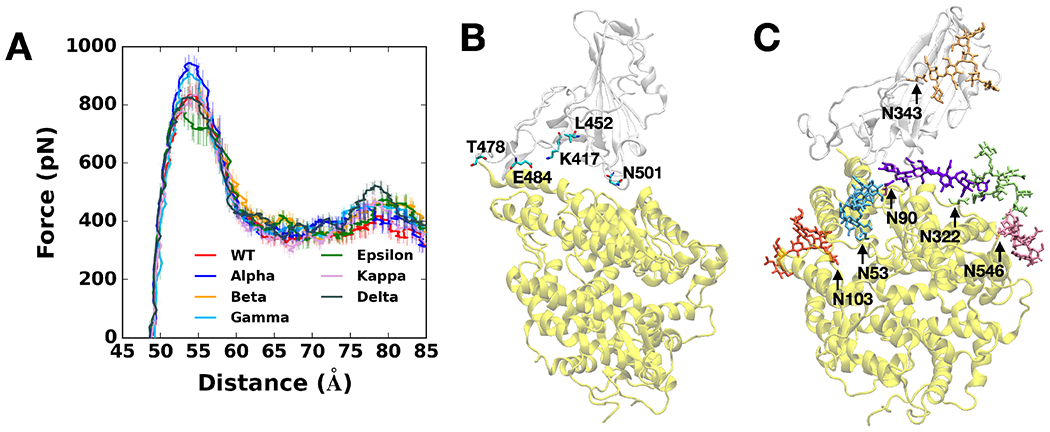
(A) Average force profiles of WT (red), Alpha (blue), Beta (orange), Gamma (sky blue), Epsilon (green), Kappa (pink), and Delta (gray) variants as a function of the distance between the centers of mass of RBD and ACE2. (B) Initial snapshot of WT. Residues subjected to each mutation are shown as solid sticks (N501, K417, E484, L452, and T478). RBD and ACE2 are respectively colored in light gray and yellow. All N-glycans, water, and ions are hidden for clarity. (C) Initial snapshot of WT with clockwise 90° rotation along the normal from (B). All N-glycans are depicted in different colors. Any other residues, water, and ions are not shown for clarity.
The alpha variant could have the highest chance of RBD-ACE2 interaction
Figure 1A shows that at D = 53 Å, the Alpha variant clearly requires the highest initial force to pull the RBD-ACE2 complex in the opposite direction. The difference can be explained in Figure 2B, a two-dimensional contact map between RBDAlpha and ACE2 at D = 53 Å, where RBD Y501 presents increased interactions with ACE2 Q42, Y41, and D38. Such contacts are decreased or even lost in the case of RBDWT or RBDEpsilon lacking the N501Y mutation (Figure 2A,D). To quantify the contact frequency between RBD residue 501 (N501 for WT, Epsilon, Kappa, and Delta; Y501 for Alpha, Beta, and Gamma) and ACE2, the number of heavy atom contacts was calculated (Figure 3A). The contact was counted if RBD residue 501 is positioned within 4.5 Å of heavy atoms of key interacting residues of ACE2 protein. Notably, Y501 of Alpha, Beta, and Gamma variants retain more contacts (about 40%) than N501 of WT, Epsilon, Kappa, and Delta variants. As shown in Figure 3B,C, Alpha Y501 is located closer to ACE2 Y41 and K353 than WT N501 at D = 53 Å and thus, it has the π-π and π-cation interactions with neighboring Y41 and K353, which is in accordance with the recent cyro-EM study.30 On top of the Y501-ACE2 interactions, RBDAlpha also contains the highest amounts of contacts with ACE2 N90-glycan (Figure S3), which could be the reason why it has been reported as the most common lineages by June 19, 2021, among the estimated proportions of SARS-CoV2 lineages according to the US CDC,31 although this study considers only single RBD out of trimeric SARS-CoV2 S protein.
Figure 2.
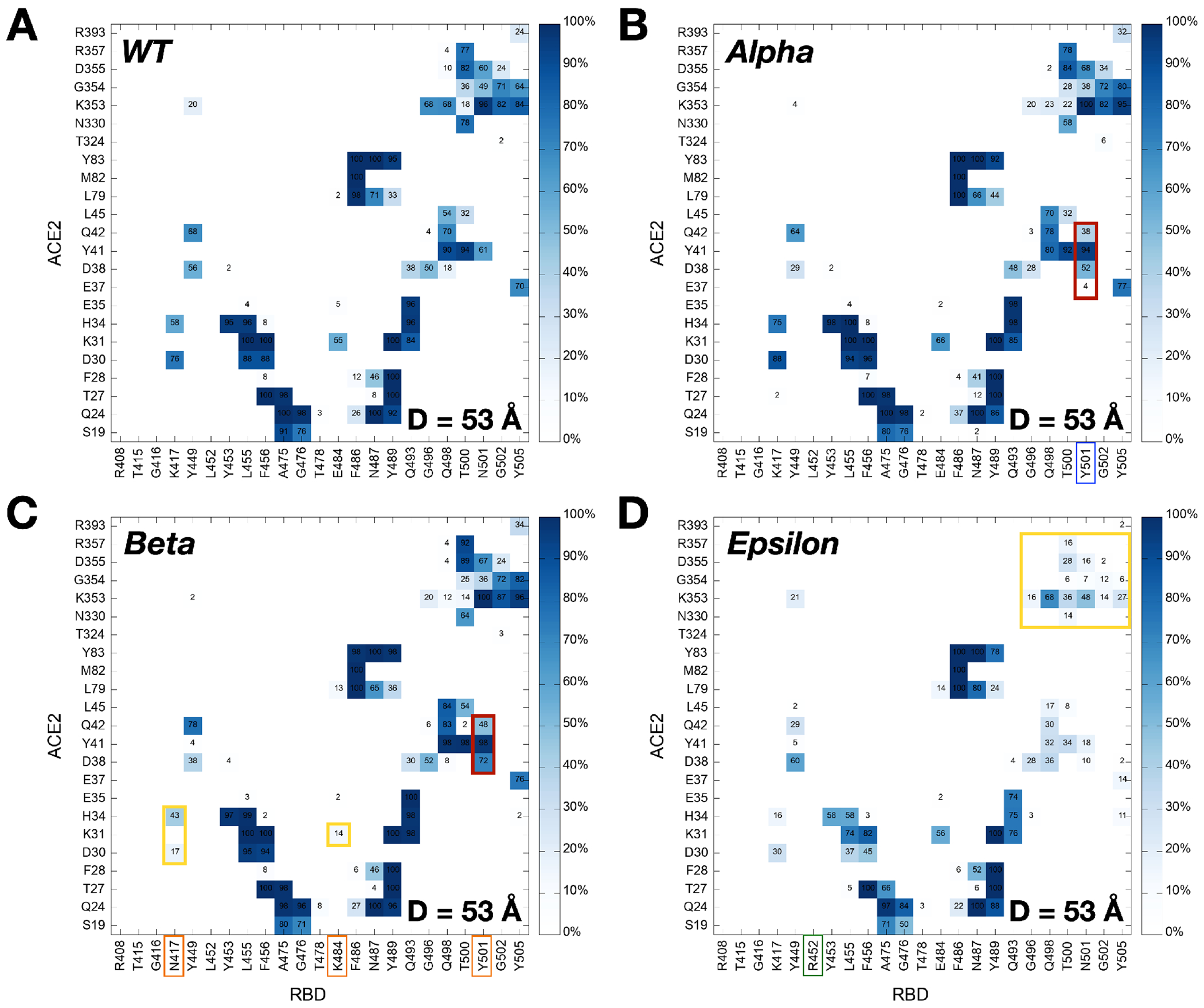
Two-dimensional contact maps at D = 53 Å. (A) Interacting residue pairs between RBDWT and ACE2. RBD residues subjected to mutation are shown in colored boxes at the bottom: (B) blue for Alpha, (C) orange for Beta, and (D) green for Epsilon. The contact frequency is numbered with colors from light blue to dark blue. Dark red and yellow colors on the map respectively represent increased and decreased interactions between RBD and ACE2 upon mutations.
Figure 3.
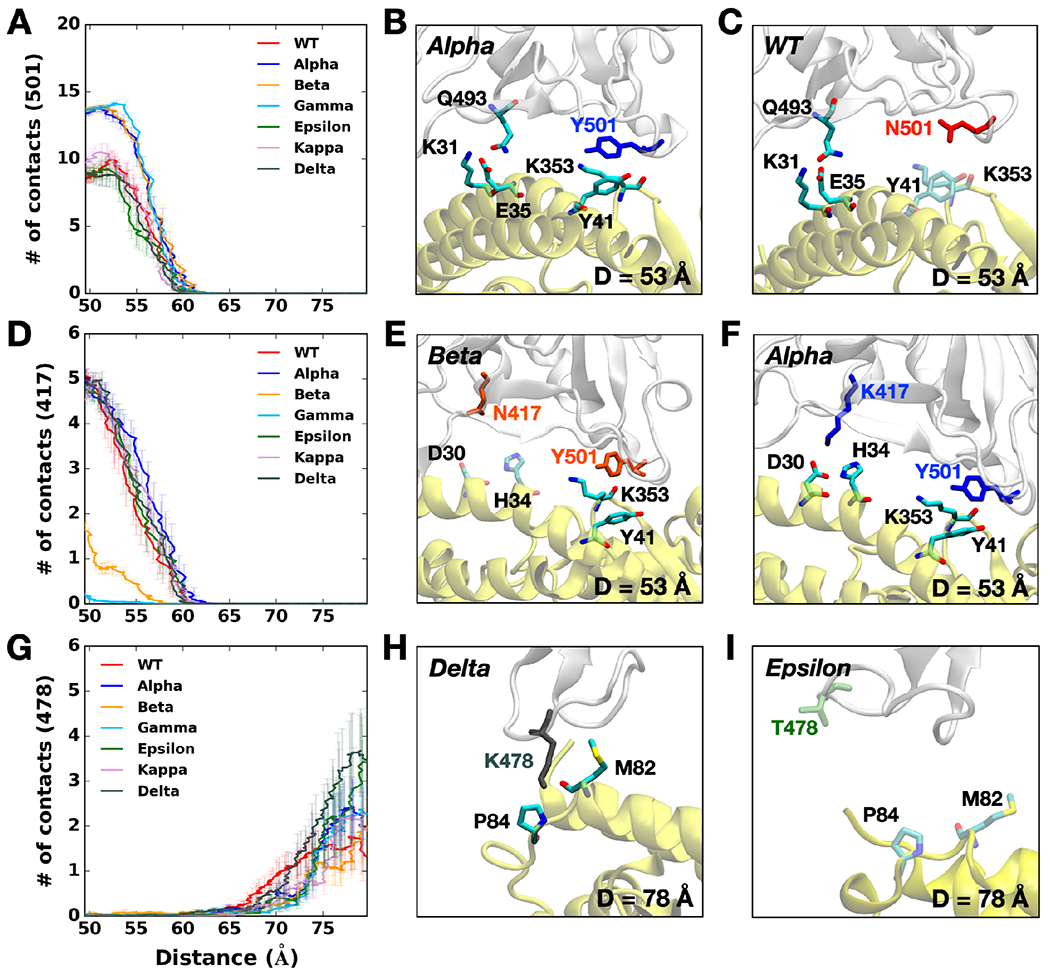
(A) The average number of contacts between RBD residue 501 and ACE2. (B and C) Representative snapshots at D = 53 Å of (B) Alpha variant and (C) WT. (D) The average number of contacts between RBD residue 417 and ACE2 and (E and F) their interacting residue pairs at D = 53 Å of (E) Beta and (F) Alpha variants. (G) The average number of contacts between RBD residue 478 and ACE2 and (H and I) key interaction pairs at D = 78 Å of (H) Delta and (I) Epsilon variants. The overall color scheme is the same as in Figure 1, and each mutated residue in each variant is shown using the same colors (i.e., red for WT, blue for Alpha, orange for Beta, green for Epsilon, and gray for Delta). Interacting residues are depicted as the solid sticks, and residues losing their interactions are shown as the transparent sticks. RBD and ACE2 are presented in light gray and yellow, respectively.
Beta and Gamma variants display weaker RBD-ACE2 interaction than the Alpha variant, yet they are still stronger than WT
The force profiles of Beta and Gamma variants at D = 53 Å present weaker maximum forces than the Alpha variant, albeit they show higher forces than WT at the same distance (Figure 1A). As shown in Figure 2B,C, Alpha and Beta variants include N501Y mutation, while Beta variant involves two additional mutations, K417N and E484K. Clearly, compared to WT or Epsilon, Y501 of Beta variant has increased interactions (colored in dark red box) with ACE2 D38, Y41, and Q42, similar to Alpha variant. However, it entails decreased contact frequency (shown as the yellow box) between RBDBeta N417 and ACE2 D30/H34, as well as RBDBeta K484 and ACE2 K31, which could explain why Beta has relatively weaker interactions than Alpha. Gamma variant also shows decreased contact numbers similar to Beta due to its K417T mutation (Figure S2A). The only difference between Gamma and Beta is the K417 mutation, i.e., K417T vs. K417N. Figure 3D compares the number of contacts of residue 417 of all variants that are in contact with heavy atoms of key interacting residues of ACE2. While all other variants containing K417 (i.e., WT, Alpha, Epsilon, Kappa, and Delta) display some RBD-ACE2 contacts from 50 to 60 Å few interactions were found for the Beta variant. The sidechain-shortening mutation from lysine to asparagine could have an impact on the RBD-ACE2 interface, resulting in fewer interactions at the same distance (Figure 3E,F). Interestingly, T417 of Gamma shows almost no interaction because threonine is even shorter than N417 of Beta. The weakened interactions of RBDBeta N417 and RBDGamma T417 could make them less contagious than the Alpha variant, while the N501Y mutation still allows them to have a strong enough potential to interact with ACE2. Recently, Bhattarai et al. and Gobeil et al. observed similar results from Alpha and Beta variants, and Barton et al. reported corresponding results from Alpha, Beta, and Gamma through different approaches, indicating that our SMD results are reliable enough to investigate the RBD-ACE2 interface of the variants.32–34 The weakened interactions of both RBD residue 417 and 484 with ACE2 possibly provide an ability for the virus to escape from neutralizing antibodies targeting the RBD-ACE2 interface, allowing them to transmit more. This could explain why/how the Gamma variant took the second-highest portion by June 5, 2021, among the estimated proportions of SARS-CoV2 lineages, provided by the US CDC.31
L452 mutation of Epsilon variant destabilizes RBD itself, causing weakened interactions with ACE2
Although most variants show similar maximum forces around D = 53 Å, the Epsilon variant shows decreased forces with more fluctuations than other variants (Figure 1A). The two-dimensional contact map in Figure 2D confirms its distinct interactions at D = 53 Å, as it shows the least number of contacts between RBDEpsilon and ACE2 (n.b., the yellow box represents deceased interactions). For example, K353 residue of all other variants actively interacts with ACE2 Q493, Q496, Q498, T500, N/Y501, G502, and Y505 (Figure 2A,B,C and Figure S2A,B,C). K353 of Epsilon, however, lost its contact with corresponding residues at least by 50%. To investigate the mechanism behind such a big difference, the contact analysis in between RBD residues was performed, where the influence of the L452R mutation was examined by checking its contacts with surrounding residues, L450 and L492 (Figure S4). Interestingly, mutated R452 interacts more with L450 (Figure S4C) and less with L492 (Figure S4A) simultaneously. Note that L450 and L492 are positioned in different β-strands (Figure S4B,D colored in green and orange, respectively), and the L452R mutation makes the RBD-ACE2 interface unstable by shortening each β-strand (i.e., the length of interacting β-strands of Epsilon variant is decreased by almost half). Because of such an unstable RBD structure, the Epsilon variant appears to be detached from ACE2 easier than WT. Indeed, K353 of Epsilon variant lost contacts with ACE2 Q498 and Y505 at D = 55 Å (Figure S4D), but WT holds their interactions at the same distance (Figure S4B). The Epsilon variant has been shown to reduce the neutralization potency of several antibodies in a way that it reduces sensitivity to the antibodies.35 This result indicates that the Epsilon variant has impacted the world by decreasing the antibody sensitivity, not increasing direct RBD-ACE2 interactions. According to the US CDC, as of June 29, 2021, the Epsilon variant deescalated from the variants of concern and became the variants of interest since its considerable decrease in lineage proportion in the United States.
Position of T478 mutation in Delta variant could be responsible for its high infectivity
Newly reported Kappa and Delta variants involve the same L452R mutation as Epsilon, but each variant contains an additional mutation, E484Q (Kappa) or T478K (Delta). Two-dimensional contact maps (Figure S2B,C) display that Kappa and Delta variants have almost identical interaction patterns to WT between ACE2 K353 and RBD residues (i.e., Q493, Q496, Q498, T500, N/Y501, G502, and Y505). The Delta variant, interestingly, shows distinct features that are not found in other variants. Upon the T478K mutation, it requires the highest force for the RBD-ACE2 complex to be completely dissociated at D = 78 Å (Figure 1A). To see what makes the difference, the number of contacts between RBD and ACE2 was calculated (i.e., residue 478 and heavy atoms of selected key interacting residues of ACE2). As shown in Figure 3G, RBDDelta exclusively makes more contacts with ACE2 than other variants. Figure 3H shows that Delta K478 retains contacts with ACE2 P84 and M82 at D = 78 Å, but Epsilon T478 already lost such interactions. The contacts of residue 478 are observed from the pulling simulations, but in terms of virus entry, it is possible that residue 478 located in the flexible loop first has a chance to contact ACE2. The stronger interactions of Delta K478 with ACE2 could explain why the proportion of Delta variant has dramatically increased with high infectivity. Recently, Baral et al. reported that a subtle reorientation of G496 in the Delta induces stronger β-strand interactions and that it could be due to the L452R mutation.36 It should be noted that both Kappa/Delta and Epsilon share L452R mutation. Although our results are accordant with recently published studies,30, 32–33, 35–36 the reason why Kappa/Delta and Epsilon behavior distinctively is remained to be further studied, and it might stem from the limitation in our model, as we only employed L452R mutation in RBD for Epsilon variant without D614G mutation. The Delta variant became the current variant of concern, and it took the highest portion among the estimated variant proportions as of July 3, 2021, according to the US CDC.31
Microscale Thermophoresis study confirms findings from the MD simulation
To validate the SMD simulation results, we conducted an experimental protein binding assay using microscale thermophoresis (MST). MST detects molecular binding kinetics based on the thermophoretic movement of molecules induced by a microscopic temperature gradient inside a glass capillary generated by an infrared laser.37 MST has been used for detecting viral protein-receptor interactions,38 including SARS-CoV-2 S proteins.39 In our assay, human recombinant ACE2 was fluorescently labeled, and various RBD variants were titrated in a two-fold fashion and mixed with the ACE2. The MST signal was first converted to saturated fraction data and subsequently fitted to a first-order 1:1 binding kinetics model using the manufacturer’s software (Figure S5). The binding affinities of ACE2 and RBDWT were detected to be 27.5 ± 4.8 nM (Figure 4). This value is in agreement with a reported Kd range of 5-40 nM measured by surface plasmon resonance.40 Importantly, our MST data indicate that Alpha variant binds ACE2 with a 2.3-fold higher affinity (11.8 ± 0.8 nM) than WT. The rest of the variants show slightly different affinity from WT. Beta and Delta variants display approximately 20-30% higher affinities than WT, and the Epsilon variant shows a 15% lower affinity than WT. In Figure 4, Kd values from MST experiments were directly compared with the FWT/F ratio from the SMD simulations, where FWT and F are the maximum pulling forces of WT and each variant around D = 53 Å (Figure 1A). Our MST affinity data are consistent with the SMD simulation data, indicating Alpha and Epsilon variants possess the strongest and weakest binding to ACE2, respectively.
Figure 4.
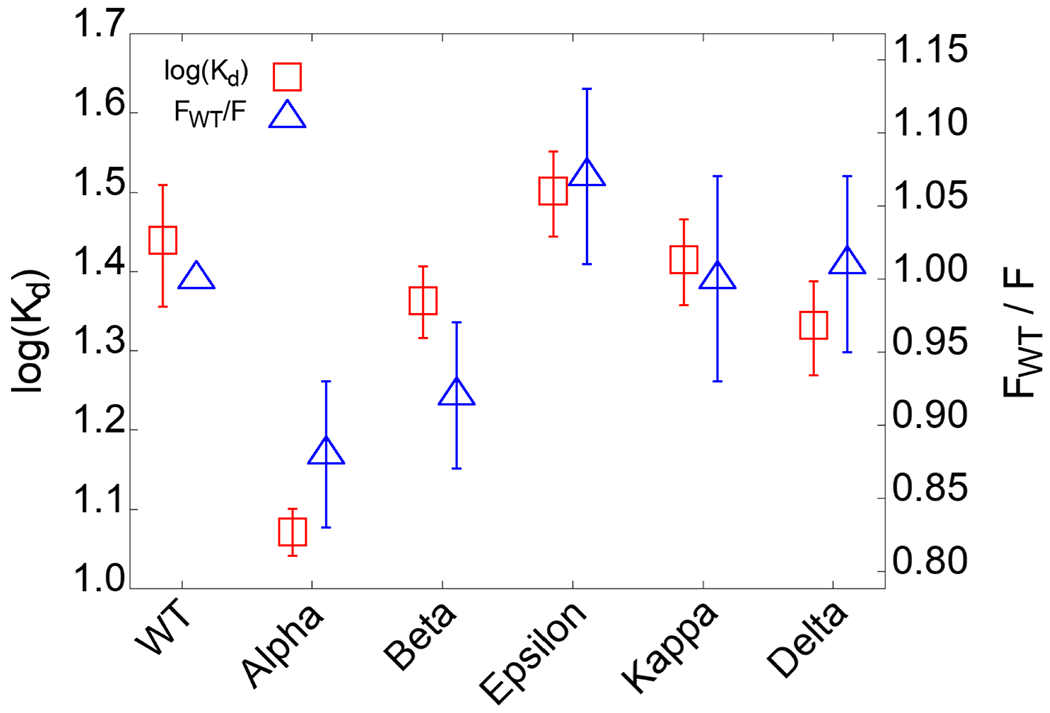
Binding affinities between RBD variants and ACE2 and its comparison with the simulation results. Kd is obtained from microscale thermophoresis experiments. FWT/F is a ratio, where FWT and F are the respective maximum pulling force of WT and of each variant obtained from the SMD simulations.
CONCLUSIONS
This study characterizes interactions between ACE2 and RBD of all variants that the US CDC classifies as the variants of concern and variants of interest. The results indicate that the Alpha variant requires the highest force for initial separation from ACE2, followed by Beta and Gamma variants or Delta variant. K417N/T mutations of Beta and Gamma appear to make the RBD-ACE2 interactions less strong compared to the Alpha variant. In addition, the Epsilon variant is relatively easily dissociated from ACE2 than others due to its destabilized RBD structure upon the L452R mutation. Delta variant specifically shows stronger interactions with ACE2 than other variants at a relatively far distance between RBD and ACE2. The MST experiments show consistent results with the simulation results, where Alpha and Epsilon variants display the strongest and weakest binding to ACE2, respectively.
SARS-CoV-2 variants have been evolving by changing their structures so that they can either strengthen the interactions with the human receptor, i.e., ACE2, or escape from neutralizing antibodies by altering their structures targeting the RBD-ACE2 interface highlighting their complex behaviors. We hope this study could provide valuable information that distinguishes important features of all variants and their interactions with ACE2 and shed light on developing new drugs to inhibit SARS-CoV-2 entry effectively.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported in part by NIH AI133634 and NSF 1804117 (to X.F.Z.), NIH R21 AI163708 (to X.F.Z. and W.I.), NIH GM138472, and MCB-1810695 (to W.I.), and an internal grant from Lehigh University (to X.F.Z, and W.I.).
Footnotes
The supporting information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jctc.1c00965.
Force profiles of all replicas of all variants; two-dimensional contact map of Gamma, Kappa, and Delta; N90-glycan contact; β-strand interactions of WT and Epsilon; MST analysis of variants and affinities.
REFERENCES
- 1.World Health Organization. WHO Coronavirus (COVID-19) Dashboard (accessed on September 23, 2021). https://covid19.who.int.
- 2.Shang J; Ye G; Shi K; Wan Y; Luo C; Aihara H; Geng Q; Auerbach A; Li F, Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wrapp D; Wang N; Corbett KS; Goldsmith JA; Hsieh CL; Abiona O; Graham BS; McLellan JS, Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cao W; Dong C; Kim S; Hou D; Tai W; Du L; Im W; Zhang XF, Biomechanical characterization of SARS-CoV-2 spike RBD and human ACE2 protein-protein interaction. Biophys. J 2021, 120, 1011–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoffmann M, et al. , SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Q, et al. , Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tang JW; Tambyah PA; Hui DSC, Emergence of a new SARS-CoV-2 variant in the UK. J. Infect 2021, 82, e27–e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kirby T, New variant of SARS-CoV-2 in UK causes surge of COVID-19. Lancet Respir. Med 2021, 9, e20–e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tegally H, et al. , Detection of a SARS-CoV-2 variant of concern in South Africa. Nature 2021, 592, 438–443. [DOI] [PubMed] [Google Scholar]
- 10.Faria NR; Claro IM; Candido D; Franco LM; Andrade PS; Coletti TM; Silva CA; Sales FC; Manuli ER; Aguiar RS, Genomic characterisation of an emergent SARS-CoV-2 lineage in Manaus. Science 2021, 372, 815–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Voloch CM, et al. , Genomic characterization of a novel SARS-CoV-2 lineage from Rio de Janeiro, Brazil. J. Virol 2021, 95, e00119–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang W; Davis BD; Chen SS; Sincuir Martinez JM; Plummer JT; Vail E, Emergence of a Novel SARS-CoV-2 Variant in Southern California. JAMA. 2021, 325, 1324–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Singh J; Rahman SA; Ehtesham NZ; Hira S; Hasnain SE, SARS-CoV-2 variants of concern are emerging in India. Nat. Med 2021, 27, 1131–1133. [DOI] [PubMed] [Google Scholar]
- 14.Tian F; Tong B; Sun L; Shi S; Zheng B; Wang Z; Dong X; Zheng P, Mutation N501Y in RBD of Spike Protein Strengthens the Interaction between COVID-19 and its Receptor ACE2. In bioRxiv, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Socher E; Conrad M; Heger L; Paulsen F; Sticht H; Zunke F; Arnold P, Decomposition of the SARS-CoV-2-ACE2 interface reveals a common trend among emerging viral variants. In bioRxiv, 2021. [DOI] [PubMed] [Google Scholar]
- 16.Harvey WT, et al. , SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol 2021, 19, 409–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Woo H, et al. , Developing a Fully Glycosylated Full-Length SARS-CoV-2 Spike Protein Model in a Viral Membrane. J. Phys. Chem. B 2020, 124, 7128–7137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jo S; Kim T; Iyer VG; Im W, CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem 2008, 29, 1859–1865. [DOI] [PubMed] [Google Scholar]
- 19.Lee J, et al. , CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput 2016, 12, 405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hatcher E; Guvench O; MacKerell AD, CHARMM Additive All-Atom Force Field for Aldopentofuranoses, Methyl-aldopentofuranosides, and Fructofuranose. J. Phys. Chem. B 2009, 113, 12466–12476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang J; Rauscher S; Nawrocki G; Ran T; Feig M; de Groot BL; Grubmüller H; MacKerell AD, CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML, Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 1983, 79, 926–935. [Google Scholar]
- 23.Phillips JC; Braun R; Wang W; Gumbart J; Tajkhorshid E; Villa E; Chipot C; Skeel RD; Kale L; Schulten K, Scalable molecular dynamics with NAMD. J. Comput. Chem 2005, 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dion M; Rydberg H; Schröder E; Langreth DC; Lundqvist BI, Van der Waals density functional for general geometries. Phys. Rev. Lett 2004, 92, 246401. [DOI] [PubMed] [Google Scholar]
- 25.Essmann U; Perera L; Berkowitz ML; Darden T; Lee H; Pedersen LG, A smooth particle mesh Ewald method. J. Chem. Phys 1995, 103, 8577–8593. [Google Scholar]
- 26.Ryckaert J-P; Ciccotti G; Berendsen HJ, Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys 1977, 23, 327–341. [Google Scholar]
- 27.Hopkins CW; Le Grand S; Walker RC; Roitberg AE, Long-time-step molecular dynamics through hydrogen mass repartitioning. J. Chem. Theory Comput 2015, 11, 1864–1874. [DOI] [PubMed] [Google Scholar]
- 28.Gao Y; Lee J; Smith IPS; Lee H; Kim S; Qi Y; Klauda JB; Widmalm G. r.; Khalid S; Im W, CHARMM-GUI Supports Hydrogen Mass Repartitioning and Different Protonation States of Phosphates in Lipopolysaccharides. J. Chem. Inf. Model 2021, 61, 831–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feller SE; Zhang Y; Pastor RW; Brooks BR, Constant pressure molecular dynamics simulation: the Langevin piston method. J. Chem. Phys 1995, 103, 4613–4621. [Google Scholar]
- 30.Cai Y, et al. , Structural basis for enhanced infectivity and immune evasion of SARS-CoV-2 variants. Science 2021, 373, 642–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Centers for Disease Control and Prevention. Estimated Proportions of SARS-CoV-2 Lineages (accessed on July 3, 2021). https://covid.cdc.gov/covid-data-tracker/#variant-proportions.
- 32.Gobeil SM-C, et al. , Effect of natural mutations of SARS-CoV-2 on spike structure, conformation, and antigenicity. Science 2021, 373, eabi6226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bhattarai N; Baral P; Gerstman BS; Chapagain PP, Structural and Dynamical Differences in the Spike Protein RBD in the SARS-CoV-2 Variants B.1.1.7 and B.1.351. J. Phys. Chem. B 2021, 125, 7101–7107. [DOI] [PubMed] [Google Scholar]
- 34.Barton MI; MacGowan SA; Kutuzov MA; Dushek O; Barton GJ; van der Merwe PA, Effects of common mutations in the SARS-CoV-2 Spike RBD and its ligand, the human ACE2 receptor on binding affinity and kinetics. Elife 2021, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McCallum M, et al. , SARS-CoV-2 immune evasion by the B.1.427/B.1.429 variant of concern. Science 2021, 373, 648–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baral P; Bhattarai N; Hossen ML; Stebliankin V; Gerstman BS; Narasimhan G; Chapagain PP, Mutation-induced changes in the receptor-binding interface of the SARS-CoV-2 Delta variant B.1.617.2 and implications for immune evasion. Biochem. Biophys. Res. Commun 2021, 574, 14–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Plach MG; Grasser K; Schubert T, MicroScale Thermophoresis as a tool to study protein-peptide interactions in the context of large eukaryotic protein complexes. Bio-Protoc. 2017, 7, e2632–e2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Walls A; Tortorici MA; Bosch BJ; Frenz B; Rottier PJ; DiMaio F; Rey FA; Veesler D, Crucial steps in the structure determination of a coronavirus spike glycoprotein using cryo-electron microscopy. Protein Sci. 2017, 26, 113–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Petruk G, et al. , SARS-CoV-2 Spike protein binds to bacterial lipopolysaccharide and boosts proinflammatory activity. J. Mol. Cell. Biol 2020, 12, 916–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu H; Zhang Q; Wei P; Chen Z; Aviszus K; Yang J; Downing W; Jiang C; Liang B; Reynoso L, The basis of a more contagious 501Y. V1 variant of SARS-COV-2. Cell Res. 2021, 31, 720–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.