

Abstract
Background and Objectives
To review the current evidence on the options available for initiating dopaminergic treatment of motor symptoms in early-stage Parkinson disease and provide recommendations to clinicians.
Methods
A multidisciplinary panel developed practice recommendations, integrating findings from a systematic review and following an Institute of Medicine–compliant process to ensure transparency and patient engagement. Recommendations were supported by structured rationales, integrating evidence from the systematic review, related evidence, principles of care, and inferences from evidence.
Results
Initial treatment with levodopa provides superior motor benefit compared to treatment with dopamine agonists, whereas levodopa is more likely than dopamine agonists to cause dyskinesia. The comparison of different formulations of dopamine agonists yielded little evidence that any one formulation or method of administration is superior. Long-acting forms of levodopa and levodopa with entacapone do not appear to differ in efficacy from immediate-release levodopa for motor symptoms in early disease. There is a higher risk of impulse control disorders associated with the use of dopamine agonists than levodopa. Recommendations on initial therapy for motor symptoms are provided to assist the clinician and patient in choosing between treatment options and to guide counseling, prescribing, and monitoring of efficacy and safety.
Parkinson disease (PD) is a neurodegenerative disorder that causes both motor and nonmotor symptoms and increases in prevalence with age. Motor symptoms in the early stages of PD include tremor, rigidity, and bradykinesia, with gait and balance impairment becoming more prominent with disease progression. The treatment options for the alleviation of motor symptoms in the early stages of PD are based on the enhancement of dopaminergic tone with levodopa, monoamine oxidase inhibitors, dopamine agonists (DAs), or a combination thereof. The choice of initial treatment is influenced by the potential for neuropsychiatric adverse effects associated with DAs and dyskinesia and motor fluctuations associated with levodopa. In 2002, the American Academy of Neurology (AAN) published the “Initiation of Treatment for Parkinson Disease” practice guideline,1 which contains recommendations regarding the use of dopaminergic medications for patients with PD. Since 2002, many new medications and new formulations of older medications have become available for PD treatment. The goal of this guideline is to review the current evidence on initial dopaminergic treatment of motor symptoms in early-stage PD and provide guidance to clinicians. This article is a summary of the key findings of the practice guideline update. The complete practice guideline update, including the full systematic review, is available at aan.com/Guidelines/home/GetGuidelineContent/1048.
Description of the Analytic Process
In August 2017, the AAN Guideline Subcommittee (GS) recruited a multidisciplinary panel of authors to develop this guideline. The panel included content and methodology experts, patient representatives, and a staff representative from the Michael J. Fox Foundation for Parkinson's Research. As required by the AAN, a majority of the members (T.P., R.M.A.d.B., D.A.H., G.S.D., N.L., K.S., L.B., E.R., M.S.F., L.H., M.J.A., J.A.G., M.R., N.C., A.R.-G., T.H.) of the panel and the lead author (T.P.) are free of conflicts of interest (COIs) relevant to this practice guideline. Five of the guideline developers were determined to have COI, but the COI were judged to be not significant enough to preclude them from authorship (A.J.E., J.M.M., A.E.L., R.A.H., J.P.M.). Whereas the development of this guideline primarily followed the 2017 edition of the AAN's Clinical Practice Guideline Process Manual,2 this edition of the manual was not published by the time of the guideline initiation. Therefore, disclosures were reviewed following the previous process found in the 2011 Clinical Practice Guideline Process Manual.3 The full author panel was solely responsible for the final decisions about the design, analysis, and reporting of the systematic review and practice guideline, which was submitted for approval to the AAN GS.
Study Screening and Selection Criteria
Types of Participants
We included studies of participants with PD in the early stages (i.e., Hoehn & Yahr stages 1 or 2, or within 2 years of disease onset).
Types of Interventions
We included studies of DAs, levodopa, monoamine oxidase type B (MAO-B) inhibitors, and catechol-O-methyltransferase (COMT) inhibitors to treat motor symptoms of PD in the early stages of the disease.
Comparison Group
We included studies using active comparators only.
Types of Studies
For clinical questions 1 through 6, we included only randomized controlled trials. For clinical questions 7 and 8, we included randomized controlled trials, population-based epidemiologic studies, and prospective cohort studies.
Types of Outcome Measures
The preferred outcome measure was the Unified Parkinson's Disease Rating Scale (UPDRS) part III, which measures motor symptoms. To determine the change in motor symptoms, the authors calculated the raw mean difference (RMD) between scores on the UPDRS part III at baseline and at follow-up. To determine the change in dyskinesia, hallucinations, adverse event (AE)–related discontinuation, and impulse control disorders (ICDs), the risk differences (RDs) were calculated.
The author panel searched the Medline, Cochrane Central Register of Controlled Trials (CENTRAL), and ClinicalTrials.gov databases from database inception through June 2020 for relevant peer-reviewed articles that met the inclusion criteria. After review of abstracts, 255 articles were identified as potentially relevant and each article was reviewed by 2 independent panel members. The panelists selected 59 articles for inclusion in the analysis.
Each of the 59 selected articles was rated by 2 panel members using the AAN criteria for classification of therapeutic articles.2 A modified form of the Grading of Recommendations Assessment, Development, and Evaluation (GRADE) process was used to develop conclusions. The confidence in the evidence (high, moderate, low, or very low) was anchored to the error domain—class of evidence, indirectness of evidence, and precision of effect estimate—with the highest risk of error.2
Analysis of Evidence
Data Synthesis and Confidence in Evidence Statements for Levodopa vs DAs
1. In people with early PD, what is the comparative efficacy of levodopa vs DAs vs MAO-B inhibitors for motor symptoms?
2. In people with early PD, what is the comparative risk of adverse effects (specifically dyskinesia, hallucinations, and AE-related discontinuation) of levodopa vs DAs vs MAO-B inhibitors?
UPDRS Part III Score
The change in the UPDRS part III score from baseline to endpoint was extracted from studies comparing levodopa to DAs (with or without levodopa) and the RMD between treatments was calculated (Figure 1). Negative values favored levodopa. Where possible, estimates were combined using meta-analysis at specific time points. The minimal clinically important difference (MCID) in the UPDRS part III score was determined by consensus to be 3 points; changes of 1 point or less were considered unimportant.
Figure 1. Levodopa vs Dopamine Agonist: Change in Unified Parkinson's Disease Rating Scale Motor Score.
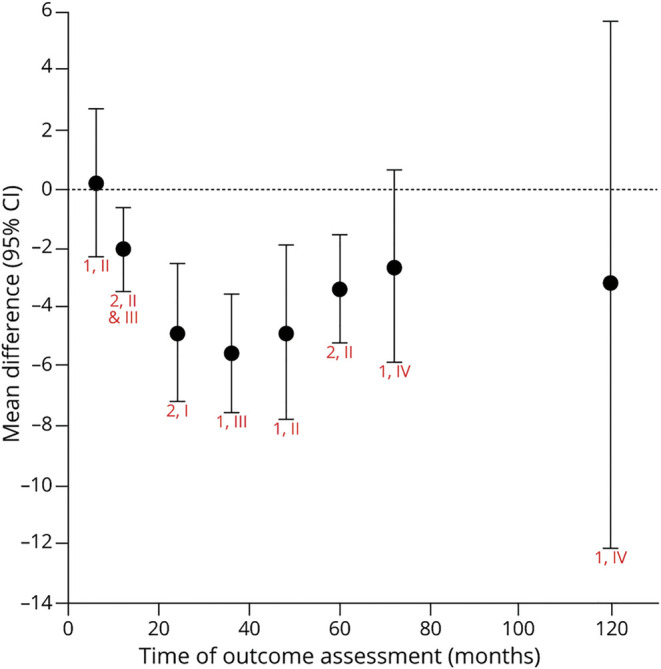
Chart shows random effects meta-analysis for each time (red text = number of articles, class). eTable 1 (links.lww.com/WNL/B569) shows raw mean difference (levodopa – dopamine agonist) for each study and time. For change in motor function, levodopa as compared to dopamine agonists is possibly no more effective at 6 months (raw mean difference 0.2 [95% confidence interval (CI) −2.3 to 2.7], low confidence); possibly more effective at 1 year (−2.1 [−3.6 to −0.7], low confidence); likely more effective at 2 years (−5.0 [−7.2 to −2.5], moderate confidence); possibly more effective at 4 years (−4.9 [−7.8 to −1.9], low confidence); and likely more effective at 5 years (−3.4 [−5.2 to −1.6], moderate confidence). There is insufficient evidence to determine whether levodopa is better or worse than dopamine agonists at 3 years (−5.6 [−7.6 to −3.6]), 6 years (−2.7 [−5.9 to 0.6]), and 10 years (−3.2 [−12.1 to 5.6]). The confidence in the evidence is algorithmically determined, as outlined in the Clinical Practice Guideline Process Manual.2
The trend over time demonstrates that levodopa provides greater benefit for motor symptoms than DAs, with the majority of studies demonstrating significantly greater improvement in the participants' UPDRS part III score for up to 5 years of follow-up. Data beyond 5 years are scarce and of low quality. With longer periods of follow-up, an increasing proportion of participants (90% of patients at 6 years and 100% at 10 years) originally randomized to DAs were taking supplemental levodopa, therefore minimizing the difference between groups for this outcome.
Dyskinesia
The proportion of participants who developed dyskinesia in each treatment group was extracted from studies comparing levodopa with DAs (with or without levodopa) and the RD was calculated (Figure 2). A positive value indicates that the risk of dyskinesia was higher with levodopa. Where possible, estimates were combined using meta-analysis at specific time points. The MCID in the risk of dyskinesia was determined by consensus to be 15%; RDs ≤5% were considered clinically unimportant.
Figure 2. Levodopa vs Dopamine Agonist: Risk Difference for Dyskinesia.
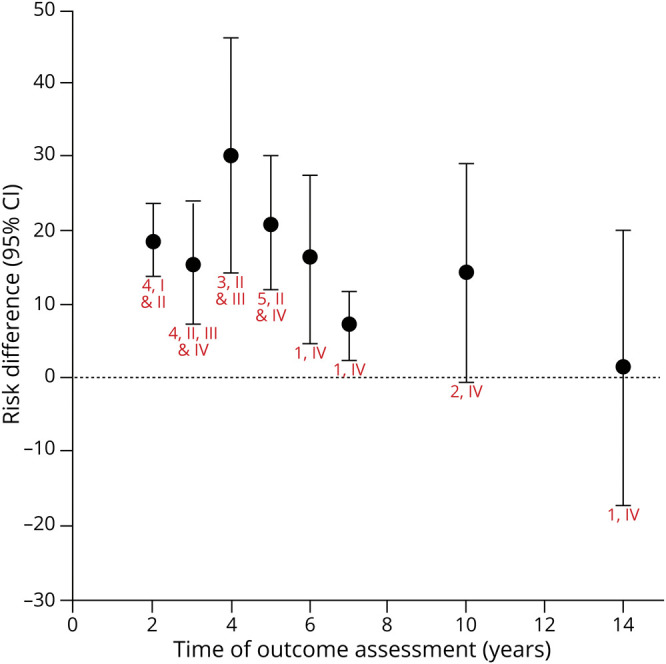
Chart shows random effects meta-analysis for each time (red text = number of articles, class). eTable 2 (links.lww.com/WNL/B569) shows risk difference (levodopa – dopamine agonist) for each study and time. The induction of dyskinesia, with levodopa as compared to dopamine agonists, is probably more likely at 2 years (risk difference 18.7% [95% confidence interval (CI) 13.7%–23.8%], moderate confidence); possibly more likely at 3 years (12.5% [2.8%–22.1%], low confidence); probably more likely at 4 years (29.2% [19.6%–38.8%], moderate confidence); and possibly more likely at 5 years (17.5% [4.5%–30.5%], low confidence). There is insufficient evidence to determine whether levodopa is more or less likely than dopamine agonists to induce dyskinesia at 6 years (16.5% [4.6%–27.7%]), 7 years (7.1% [2.4%–11.8%]), 10 years (14.3% [−0.4% to 29.1%]), and 14 years (1.6% [−17.3% to 20%]), all with very low confidence. The confidence in the evidence is algorithmically determined, as outlined in the Clinical Practice Guideline Process Manual.2
The trend over time demonstrates that levodopa is more likely to induce dyskinesia than DAs. Data beyond 5 years of follow-up (when patients are generally taking a combination of treatments) is of low quality, leading to insufficient evidence to make a conclusion. Clinical question 8 addresses long-term disabling dyskinesia.
Hallucinations
The proportion of participants who developed hallucinations in each treatment group was extracted from studies comparing levodopa with DAs (with or without levodopa) and the RD was calculated (Figure 3). A negative value indicates that the risk of hallucinations was higher with DAs. Where possible, estimates were combined using meta-analysis at specific time points. The MCID in the risk of hallucinations was determined by consensus to be 10%; RDs ≤3% were considered clinically unimportant.
Figure 3. Levodopa vs Dopamine Agonist: Risk Differences for Development of Hallucinations.
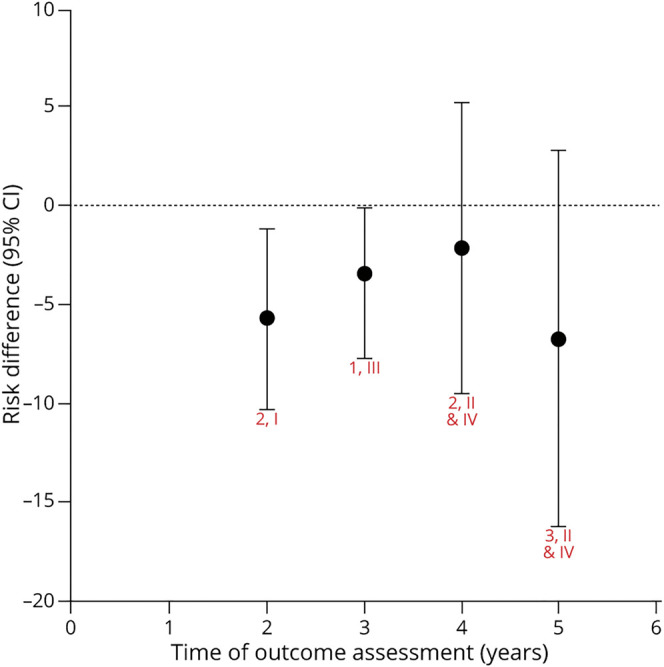
Chart shows random effects meta-analysis for each time (red text = number of articles, class). eTable 3 (links.lww.com/WNL/B569) shows risk difference (levodopa – dopamine agonist) for each study and time. Hallucinations with dopamine agonists as compared to levodopa are possibly more likely at 2 years (risk difference −5.7% [95% confidence interval (CI) −10.3% to −1.2%], low confidence); possibly no more likely at 4 years (6.6% [−13.9% to 0.7%], low confidence); and possibly no more likely at 5 years (−5.6% [−16.6% to 5.5%], low confidence). There is insufficient evidence to determine whether dopamine agonists are more or less likely than levodopa to induce hallucinations at 3 years (−3.4% [−7.7% to −0.2%], very low confidence). The confidence in the evidence is algorithmically determined, as outlined in the Clinical Practice Guideline Process Manual.2
The trend demonstrates that although DAs are more likely than levodopa to cause hallucinations at some time points, the difference between treatments for this outcome is small in early PD for the first 5 years of treatment. This may be related to the inclusion of younger patients without cognitive impairment in early PD trials.
AE-Related Discontinuation of Treatment
The proportion of participants who discontinued treatment due to adverse effects in each group was extracted from studies comparing levodopa with DAs (with or without levodopa) and the RD was calculated (Figure 4). A negative value indicates that the risk of AE-related discontinuation was higher with DAs. Where possible, estimates were combined using meta-analysis at specific time points. The MCID was determined by consensus to be 15%; RDs ≤5% were considered clinically unimportant.
Figure 4. Levodopa vs Dopamine Agonist: Risk Difference for Discontinuation Due to Adverse Events.
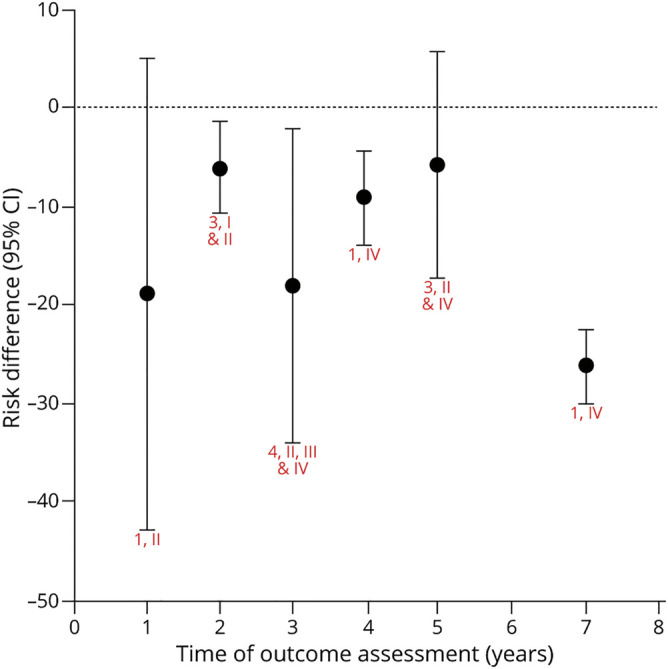
Chart shows random effects meta-analysis for each time (red text = number of articles, class). eTable 4 (links.lww.com/WNL/B569) shows risk difference (levodopa – dopamine agonist) for each study and time. The discontinuation of medication due to adverse effects, with dopamine agonists as compared to levodopa, is possibly more likely at 2 years (risk difference −6.1% [95% confidence interval (CI) −10.7% to −1.4%], low confidence); possibly no more likely at 3 years (−4.3% [−9.6% to 0.9%], low confidence); and probably no more likely at 5 years (−1% [−12.3% to 10.4%], moderate confidence). There is insufficient evidence to determine whether dopamine agonists are more or less likely than levodopa to cause medication discontinuation due to adverse effects at 1 year (−18.8% [−43% to 5%]), 4 years (−9.1% [−14% to −4.4%]), and 10 years (−26.2% [−30% to −22.5%]), all with very low confidence. The confidence in the evidence is algorithmically determined, as outlined in the Clinical Practice Guideline Process Manual.2
Although the trend suggests that AE-related discontinuation of treatment is higher with DAs than with levodopa at all time points, confidence in the evidence is low or very low due to study quality or poor precision of estimates.
Data Synthesis and Confidence in Evidence Statements for Levodopa vs MAO-B Inhibitors
There were inadequate data on the effect of levodopa vs MAO-B inhibitors on motor symptoms, preventing effect size calculations (Table 1).
Table 1.
Levodopa vs Monoamine Oxidase Type B Inhibitors: Dyskinesia and Adverse Event–Related Discontinuation

Whereas the trend over time suggests that the risk of dyskinesia is higher with levodopa than with MAO-B inhibitors, confidence in the evidence is very low due to study quality and, over time, most patients treated initially with MAO-B inhibitors were also receiving levodopa.
Over time, AE-related discontinuation of treatment is higher with MAO-B inhibitors than with levodopa, but confidence in the evidence is very low due to the quality of studies.
Data Synthesis and Confidence in Evidence Statements for DA Comparisons
3. In people with early PD, what is the comparative efficacy of different formulations of DAs for motor symptoms?
4. In people with early PD, what is the comparative risk of adverse effects (specifically dyskinesia, hallucinations, and AE–related discontinuation) of different formulations of DAs?
The confidence in evidence statements for comparisons of different formulations of DAs are summarized in Tables 2 and 3. It is important to note that bromocriptine is not routinely used in clinical practice and piribedil is not widely used.
Table 2.
Dopamine Agonist Comparisons: Change in Unified Parkinson's Disease Rating Scale Part III Score From Baseline to Endpoint

Table 3.
Dopamine Agonist Comparisons: Risk of Dyskinesia, Hallucinations, and AE-Related Discontinuation

The available evidence suggests minimal differences in efficacy between formulations of DAs for improving motor function, with the exception of ropinirole being possibly more effective than rotigotine. The available evidence suggests that there are minimal differences between formulations of DAs in the risk of dyskinesia, hallucinations, and AE-related discontinuation.
Data Synthesis and Confidence in Evidence Statements for Long-Acting vs Immediate-Release (IR) Levodopa
5. In people with early PD, what is the comparative efficacy of long-acting formulations of levodopa (including sustained-release or controlled-release (CR) formulations of levodopa and levodopa plus entacapone) vs IR levodopa for motor symptoms?
6. In people with early PD, what is the comparative risk of adverse effects (specifically dyskinesia, wearing-off, hallucinations, and AE-related discontinuation) of long-acting formulations of levodopa vs IR levodopa?
The confidence in evidence statements for comparisons of different formulations of levodopa is summarized in Table 4.
Table 4.
Long-Acting vs Immediate-Release Levodopa: Change in Unified Parkinson's Disease Rating Scale Part III Score From Baseline to Endpoint, Risk of Dyskinesia, Hallucinations, and Adverse Event–Related Discontinuation

The available evidence is insufficient to make conclusions regarding the relative efficacy of long-acting vs IR levodopa for improvement in motor function or the risk of hallucinations. There do not appear to be major differences between long-acting and IR levodopa in the risk of dyskinesia or AE-related discontinuation.
Data Synthesis and Confidence in Evidence Statements for Risk of ICDs
7. In people with early PD, what is the risk of ICDs with medications used for the treatment of motor symptoms and does the risk differ between drug formulations?
The MCID in the risk of ICDs was determined by consensus to be 2%; RDs ≤1% were considered clinically unimportant.
Confidence in Evidence Statements
Conclusion (moderate confidence): In early PD, DAs are probably more likely than levodopa to cause ICDs at 2 years (3 Class III studies, RD 23.2%, 95% confidence interval [CI] 14.3–32.1).
Conclusion (low confidence): In early PD, combination treatment with levodopa and DAs is possibly more likely than levodopa alone to cause ICDs at 2 years (1 Class III study, RD 11.2%, 95% CI 4.2–17.3).
Conclusion (low confidence): In early PD, DAs are possibly more likely than levodopa to cause ICDs at 5 years (2 Class III studies, RD 23.7%, 95% CI 1.1–46.3).
Conclusion (low confidence): In early PD, combination treatment with levodopa and DAs is possibly more likely than levodopa alone to cause ICDs at 5 years (1 Class III study, RD 24.4%, 95% CI 7.0–39.0).
Data Synthesis and Confidence in Evidence Statements for Long-term Risk of Disabling Dyskinesia
8. In people with early PD initially treated with DAs vs levodopa, what is the long-term risk of disabling dyskinesia?
The MCID in the risk of disabling dyskinesias was determined by consensus to be 15%; RDs ≤5% were considered clinically unimportant.
Confidence in Evidence Statements
Conclusion (low confidence): In early PD, levodopa is possibly more likely than ropinirole to cause disabling dyskinesia at 5 years (1 Class II study, RD 14.7%, 95% CI 5.8–24.8).
Conclusion (very low confidence): In early PD, there is insufficient evidence to determine whether levodopa is more or less likely than pramipexole to cause disabling dyskinesia at 6 years (1 Class IV study, RD 0.7%, 95% CI −4.8% to 6.2%).
Conclusion (very low confidence): In early PD, there is insufficient evidence to determine whether levodopa is more or less likely than ropinirole to cause disabling dyskinesia at 10 years (1 Class IV study, RD 11.6%, 95% CI −7.0 to 31.9).
Practice Recommendations
The following recommendations pertain to the initiation of pharmacologic treatment for motor symptoms in early PD, presuming that patients have received a correct diagnosis. There are no current disease-modifying pharmacologic treatments for PD14-16; current PD pharmacologic therapy is symptomatic only. When symptoms are not causing disability, most individuals with PD and clinicians are comfortable with a “wait and see” approach, although this requires careful monitoring and advising patients not to tolerate disability or reduction in quality of life unnecessarily. In the Parkinson's Progression Markers Initiative dataset of individuals with new PD diagnoses who were expected to be able to remain untreated for at least 6 months, 283 of 423 (67%) individuals with PD started treatment within 2 years of study onset, an average of 0.78 (SD 0.5) years after study entry.17 This provides an opportunity for interested individuals with de novo PD to participate in clinical trials targeting this population.
The panel developed rationale statements that precede each recommendation. Four types of premises can be used to support recommendations: (1) evidence-based conclusions from the systematic review, (2) generally accepted principles of care, (3) strong evidence from related conditions, and (4) deductive inferences from other premises. Recommendations must always be supported by at least one premise.
In addition to the evidence-based conclusions from the systematic review, the guideline developers consider the following non–evidence-based factors when formulating recommendations: (1) the relative value of the benefit compared with the risk, (2) the feasibility of complying with the intervention, (3) the cost of the intervention, and (4) the expected variation in patient preferences relative to the risks, burdens, and benefits of the intervention. After drafting recommendations, the panel assigns levels of obligation (A, B, or C) to each recommendation using a modified Delphi process that synthesizes all previously listed factors. Each designation corresponds to a helping verb that denotes the recommendation's strength level. Level A is the strongest recommendation level and is denoted by the verb must. These recommendations are based on high confidence in the evidence and require both a high magnitude of benefit and low risk. Level B corresponds to the verb should. The requirements for these recommendations are less stringent but still based on the evidence and benefit–risk profile. Level C corresponds to the verb may. This is the lowest allowable recommendation level that the AAN considers useful within the scope of clinical practice and accommodates the highest degree of practice variation.
Levodopa vs DAs vs MAO-B Inhibitors
Recommendation 1 Rationale
Clinical trials have failed to provide evidence of disease modification when the initial therapy prescribed is levodopa,23 a DA,24 or an MAO-B inhibitor.25 Studies comparing treatment with levodopa to treatment with MAO-B inhibitors early in the disease course provide Class IV evidence. These studies demonstrate greater improvement in mobility with levodopa than with MAO-B inhibitors, a higher risk of AE-related discontinuation with MAO-B inhibitors, and that >60% of individuals randomized to MAO-B inhibitors will require additional therapy within 2 to 3 years.
Initial treatment of early PD with levodopa provides greater benefit for motor symptoms than initial treatment with DAs, as shown in the majority of studies that demonstrate greater improvement in the UPDRS part III score for the first 5 years of follow-up. Initial treatment with levodopa is more likely to induce dyskinesia than initial treatment with DAs for up to 5 years of follow-up, but the prevalence of severe or disabling dyskinesia during this 5-year period is low. Although initial treatment with DAs is possibly more likely to cause hallucinations than treatment with levodopa, the difference between treatments for this outcome is small for the first 5 years of treatment. Treatment with DAs in early PD is associated with a higher risk of ICDs.
Patient and disease characteristics influence the risk of adverse effects related to the use of levodopa and DAs and may affect initial treatment choices. Younger age at disease onset,26 lower body weight,27,28 female sex,26 and increased disease severity29-31 are all predisposing factors for the development of levodopa-induced dyskinesia. Predisposing patient characteristics for ICDs are male sex, younger age, history of ICDs, history of mood disorders (particularly depression), apathy, and a family history of ICDs and addiction.32-35 Older patients are at greater risk for cognitive and behavioral adverse effects of DAs.36 DAs are associated with a greater risk of excessive daytime somnolence and sleep attacks; therefore, patients whose employment requires driving or operating heavy machinery may face greater impairment from these adverse effects.37
Recommendation 1 Statements
1a. Clinicians should counsel patients with early PD on the benefits and risks of initial therapy with levodopa, DAs, and MAO-B inhibitors based on the individual patient's disease characteristics to inform treatment decisions (Level B).
1b. In patients with early PD who seek treatment for motor symptoms, clinicians should recommend levodopa as the initial preferential dopaminergic therapy (Level B).
1c. Clinicians may prescribe DAs as the initial dopaminergic therapy to improve motor symptoms in select early PD patients <60 years who are at higher risk for the development of dyskinesia (Level C).
1d. Clinicians should not prescribe DAs to patients with early-stage PD at higher risk of medication-related adverse effects, including individuals >70 years, patients with a history of ICDs, and patients with preexisting cognitive impairment, excessive daytime sleepiness (EDS), or hallucinations (Level B).
Prescribing Levodopa
Recommendation 2 Rationale
The evidence comparing IR levodopa to CR levodopa or levodopa/carbidopa/entacapone is either of very low confidence or did not detect differences between formulations for improvement in motor symptoms, dyskinesia, hallucinations, or AE-related discontinuation in early PD. There are no studies comparing IR levodopa to extended-release (ER) carbidopa/levodopa in early PD.
Although there is no evidence to support superiority of one formulation of levodopa over another, there are other reasons to favor initiating treatment with IR levodopa. CR levodopa has lower bioavailability and less predictable symptom relief compared to IR levodopa,38,39 which may necessitate treatment discontinuation in later stages of the disease due to dose failures. Whereas levodopa/carbidopa/entacapone can be helpful for patients who experience end-of-dose wearing-off,40 this is not a usual clinical feature in early PD. IR levodopa is less costly than other levodopa formulations. Clinical trials in early PD demonstrate symptomatic benefit with levodopa/carbidopa at dosages of 150–300 mg/day and a lower risk of dyskinesia with dosages <400 mg/day. Although the risk is higher with DAs, levodopa may cause ICDs, hallucinations, and EDS.37 Levodopa may exacerbate postural hypotension.
Nausea is a common early and dose-dependent adverse effect of levodopa.41 Taking levodopa with meals affects the absorption of levodopa in the gut by slowing gastric emptying; dietary protein intake and resulting concentrations of large neutral amino acids may decrease entry of levodopa into the brain.42 In early PD, taking levodopa with meals may decrease nausea and improve compliance with therapy. In later disease stages, taking levodopa with meals may decrease therapeutic efficacy.
Recommendation 2 Statements
2a. Clinicians should initially prescribe IR levodopa rather than CR levodopa or levodopa/carbidopa/entacapone in patients with early PD (Level B).
2b. In patients with early PD, clinicians should prescribe the lowest effective dose of levodopa (i.e., the lowest dose that provides adequate symptomatic benefit) to minimize the risk of dyskinesia and other adverse effects (Level B).
2c. Clinicians should routinely monitor patients taking levodopa for their motor response to treatment and for the presence of dyskinesia, motor fluctuations, ICDs, EDS, postural hypotension, nausea, and hallucinations, to guide dosage titration over time (Level B).
2d. Clinicians should counsel patients taking levodopa that higher dosages are more likely to cause dyskinesia (Level B).
2e. Clinicians should counsel patients that in later disease stages, taking levodopa with meals may affect levodopa absorption and efficacy, but this is usually not problematic at the time of levodopa initiation in early PD (Level B).
Prescribing DAs
Recommendation 3 Rationale
Before prescribing a medication, it is important to inform patients and caregivers of medication-associated adverse effects and to screen for preexisting conditions, personality traits, concurrent medication use, and other relevant exposures that are associated with increased risk of medication-related adverse effects. DAs (vs levodopa) are associated with an increased risk of ICDs, EDS, sudden-onset sleep, nausea, and hallucinations in patients with early PD.37 DAs may exacerbate postural hypotension.
Patients may not always report certain nonmotor symptoms associated with PD or its treatment due to lack of awareness, embarrassment, or other concerns.43 Systematic and specific interrogation by practitioners concerning impulsive behaviors, sleep-related behaviors, and perceptual disturbances may set expectations and normalize reporting of embarrassing behaviors, leading to improved recognition of problematic adverse effects associated with DA use.
Recommendation 3 Statements
3a. Clinicians should inform the patient and caregiver (when present) of important side effects of DAs before prescribing; this discussion should specifically include ICDs, EDS, sudden-onset sleep, nausea, postural hypotension, and hallucinations (Level B).
3b. Clinicians should screen patients for cognitive impairment, EDS, sudden-onset sleep, hallucinations, orthostatic hypotension, and the presence of risk factors for ICDs before prescribing a DA (Level B).
3c. Clinicians should screen patients for the presence of adverse effects related to DAs, including ICDs, EDS, sudden-onset sleep, orthostatic hypotension, cognitive impairment, and hallucinations repeatedly in follow-up of patients prescribed DAs (Level B).
3d. Clinicians should involve caregivers in assessments for ICDs, EDS, sudden-onset sleep, orthostatic hypotension, cognitive impairment, and hallucinations in patients with PD (Level B).
Recommendation 3e Rationale
Standardized measures may be used to systematically screen patients for risk factors for adverse effects associated with medication use or disease progression; questionnaires can be especially useful when screening for or grading the severity of complex adverse effects that exist along a spectrum, such as ICDs and EDS. “Positive” scores on standard questionnaires should trigger the clinician to further explore the symptom through a focused clinical interview to determine the range and severity of symptoms, as well as need for clinical management. Effective management may necessitate tapering or discontinuation of DAs to mitigate morbidity associated with medication-related adverse effects.
The Questionnaire for Impulsive-Compulsive Disorders in Parkinson's Disease (QUIP) is a validated self-assessment screening instrument for a range of ICDs and other compulsive behaviors that occur in patients with PD, including gambling, sexual behaviors, buying, eating behaviors, punding, hobbyism, walkabout, and compulsive medication use. Patients with higher QUIP scores are at higher risk of impulsive-compulsive behaviors.44
The Epworth Sleepiness Scale (ESS) is a self-report questionnaire consisting of 8 questions and responses on a 4-point Likert scale. Patients rate their usual chances of dozing off or falling asleep as they engage in different activities. The ESS score is the sum of the 8-item scores ranging from 0 to 24, where a higher score represents greater sleepiness. ESS scores above 10 are considered to represent EDS.45
The QUIP and ESS are patient-completed scales with an administration time of <10 minutes and are publicly available for clinical use.
Recommendation 3e Statement
3e. Clinicians may screen patients for the presence of adverse effects associated with DAs using questionnaires validated for this purpose, including the QUIP for ICDs and the ESS for the assessment of impaired wakefulness (Level C).
Recommendation 4 Rationale
Multiple DA medications and formulations (e.g., short-acting, long-acting, oral, and transdermal) are approved for the treatment of patients with early PD. This systematic review did not uncover strong evidence supporting the use of ropinirole vs pramipexole for the treatment of early PD. Furthermore, there was no compelling evidence that pramipexole ER vs pramipexole IR was associated with a more favorable UPDRS score or a different rate of AE-related treatment discontinuation at 18 weeks. There are preliminary observational data that long-acting and transdermal formulations of DAs have lower rates of ICDs than short-acting formulations.46 In the absence of compelling evidence concerning safety or efficacy, the selection of a medication and formulation should take into account patient preferences with the goal of optimizing compliance with treatment recommendations. Specific to DAs, relevant patient preferences may include the cost and the frequency (once daily, twice daily, or 3 times daily) and mode (oral vs transdermal) of administration.
Regardless of the formulation, the practice of prescribing a DA has been to start at the lowest possible dosage and increase slowly until the desired effect or adverse effect occurs. Clinicians may opt to increase dosages gradually, stopping at the lowest dosage that is recognized to have clinical efficacy (6–9 mg/day of ropinirole, 1.5 mg/day of pramipexole, or 4 mg/24 hours of rotigotine).47
Recommendation 4 Statements
4a. Clinicians should integrate patient preferences concerning formulation, mode of administration, and cost when prescribing a DA (Level B).
4b. Clinicians should prescribe the lowest dose of DA required to provide therapeutic benefit (Level B).
Tapering and Discontinuing DAs
Recommendation 5 Rationale
Adverse effects associated with DAs can lead to substantial impairments in psychosocial functioning, interpersonal relationships, and quality of life for the patient and caregivers. The consequences of medication-related adverse effects may be mitigated through adjustments to prescribed medications, including DAs, or through additional behavioral or pharmacologic interventions, if appropriate.
Patients may experience undesirable side effects when attempting to decrease dopaminergic medications, especially DAs, including dopamine agonist withdrawal syndrome (DAWS) or low mood and apathy.48 These side effects can make it difficult to taper or discontinue DAs. Staged reduction in dosing may reduce the severity of withdrawal symptoms and improve compliance with medication recommendations.
Recommendation 5 Statements
5a. Clinicians should recommend tapering or discontinuation of DAs if patients experience disabling medication-related adverse effects, including ICDs, EDS, sudden-onset sleep, cognitive impairment, or hallucinations (Level B).
5b. When DAs must be discontinued due to adverse effects, clinicians should monitor patients for symptoms of DAWS and, when possible, gradually decrease the dosage to minimize symptoms (Level B).
Prescribing MAO-B Inhibitors
Recommendation 6 Rationale
Initial treatment of early PD with levodopa provides greater benefit for mobility than initial treatment with MAO-B inhibitors. Initial treatment with levodopa may be more likely to induce dyskinesia than initial treatment with MAO-B inhibitors. Most patients on monotherapy with a MAO-B inhibitor will require additional therapy within 2 to 3 years compared to those being treated with levodopa or DAs. Treatment of early PD with MAO-B inhibitors is associated with a higher risk of AE-related discontinuation compared with treatment with levodopa.
There are no studies comparing the efficacy of selegiline and rasagiline in the treatment of early PD. Studies of monotherapy with selegiline and rasagiline have demonstrated superiority to placebo for treatment of motor symptoms in people with early PD.49,50 Prescribing information for selegiline and rasagiline caution against their use with selective serotonin reuptake inhibitors (SSRIs); however, serotonin syndrome is rarely reported in patients with PD on concomitant therapy with an MAO-B inhibitor and an SSRI.51-53
Recommendation 6 Statements
6a. Clinicians should counsel patients with early PD on the greater motor benefits of initial therapy with levodopa compared with MAO-B inhibitors to inform treatment decisions (Level B).
6b. Clinicians may prescribe MAO-B inhibitors as the initial dopaminergic therapy for mild motor symptoms in patients with early PD (Level C).
Suggestions for Future Research
Future research will hopefully establish effective disease-modifying therapy that would be initiated when the diagnosis is made, and possibly in patients with probable prodromal PD before motor features are evident. The role of nonpharmacologic therapy, such as exercise and physiotherapy, in patients not receiving pharmacotherapy needs to be established using carefully controlled research designs. Further studies are required to address whether quality of life is significantly improved with the earlier initiation of symptomatic treatment vs following a “wait and watch” strategy. Research is needed to determine whether genetic status should influence decisions on how to initiate therapy. Personalized medicine approaches must be considered in future research, with the goal of moving away from a one-size-fits-all therapeutic approach to initiating treatment for motor symptoms in early PD. For example, further work is required to advance initial pharmacogenomic studies that have suggested patient-specific differences in response to some anti-Parkinson drugs, such as rasagiline and entacapone. Similarly, further research is required to establish definitive genetic predispositions to important treatment complications such as the risk of developing ICDs with DAs or a greater risk of earlier severe dyskinesia with levodopa. This would then guide the use of these agents in early treatment. This might also permit more definitive research studies on the relative risk of levodopa vs DAs in inducing the pathogenetic mechanisms that underlie dyskinesia. Finally, a high priority of future research should be to determine whether newer, more effective methods of providing stable levodopa plasma levels initiated soon after diagnosis will delay the onset of dyskinesia. These could include the use of newer ER levodopa formulations, alternative modes of levodopa administration (e.g., transdermal), or longer-acting COMT inhibitors.
Disclaimer
Clinical practice guidelines, practice advisories, systematic reviews, and other guidance published by the AAN and its affiliates are assessments of current scientific and clinical information provided as an educational service. The information (1) should not be considered inclusive of all proper treatments or methods of care, or as a statement of the standard of care; (2) is not continually updated and may not reflect the most recent evidence (new evidence may emerge between the time information is developed and when it is published or read); (3) addresses only the questions specifically identified; (4) does not mandate any particular course of medical care; and (5) is not intended to substitute for the independent professional judgment of the treating provider, as the information does not account for individual variation among patients. In all cases, the selected course of action should be considered by the treating provider in the context of treating the individual patient. Use of the information is voluntary. The AAN provides this information on an “as is” basis and makes no warranty, expressed or implied, regarding the information. The AAN specifically disclaims any warranties of merchantability or fitness for a particular use or purpose. AAN assumes no responsibility for any injury or damage to persons or property arising out of or related to any use of this information or for any errors or omissions.
Conflict of Interest
The AAN is committed to producing independent, critical, and trustworthy clinical practice guidelines and evidence-based documents. Significant efforts are made to minimize the potential for COIs to influence the recommendations of this evidence-based document. Management and disclosure of document developer relationships is conducted in compliance with the 2017 AAN process manual section titled “Implementing the AAN Conflict of Interest Policy for Guidelines and Case Definitions,” which can be viewed at aan.com.
Glossary
- AAN
American Academy of Neurology
- AE
adverse event
- CI
confidence interval
- COI
conflict of interest
- COMT
catechol-O-methyltransferase
- CR
controlled-release
- DA
dopamine agonist
- DAWS
dopamine agonist withdrawal syndrome
- EDS
excessive daytime sleepiness
- ER
extended-release
- ESS
Epworth Sleepiness Scale
- GS
Guideline Subcommittee
- ICD
impulse control disorder
- IR
immediate-release
- MAO-B
monoamine oxidase type B
- MCID
minimal clinically important difference
- PD
Parkinson disease
- QUIP
Questionnaire for Impulsive-Compulsive Disorders in Parkinson's Disease
- RD
risk difference
- RMD
raw mean difference
- SSRI
selective serotonin reuptake inhibitor
- UPDRS
Unified Parkinson's Disease Rating Scale
Appendix. Authors
Footnotes
Podcast: NPub.org/Podcast9720
Study Funding
This guideline was developed with financial support from the AAN. Authors who have served as AAN subcommittee members or methodologists (T.P., G.S.D., D.B.S., A.R.-G., N.L., M.J.A., G.G., L.B., K.S.), or who are AAN staff members (M.D.O., H.S.), were reimbursed by the AAN for expenses related to travel to subcommittee meetings where drafts of manuscripts were reviewed.
Disclosure
T. Pringsheim has received travel funding from the AAN to attend Guideline Subcommittee meetings; is a paid evidence-based medicine consultant for the AAN; has received research funding from the Maternal Newborn Child and Youth Strategic Clinical Network, the Owerko Center of Alberta Children's Hospital Research Institute, and the Canadian Institutes of Health Research; and serves as an editor for Neurology Clinical Practice. G. Day serves as clinical director of the Anti-NMDA Receptor Encephalitis Foundation; has received travel funding from the AAN and the Texas Neurologic Society; serves as the dementia subsection topic editor for DynaMed; has received honoraria from the North Carolina Neurologic Society, the AAN, and the Texas Neurologic Society for grand rounds presentations and academic grant reviews; has received research support from Avid Radiopharmaceuticals, the American Brain Foundation, and the Weston Brain Institute; owns stock in ANI Pharmaceuticals and stock options for Parabon NanoLabs; and has served as an expert consultant pertaining to Wernicke encephalopathy and thiamine deficiency. D. Smith is a paid evidence-based medicine consultant for the AAN. A. Rae-Grant has received publishing royalties from 2 publications relating to health care and has received travel funding from the AAN to attend Guideline Subcommittee meetings. N. Licking has received travel funding from the AAN to attend Guideline Subcommittee meetings. M. Armstrong is supported by grants from the National Institute on Aging (P30AG047266, R01AG068128) and the Florida Department of Health (grant 20A08) and was previously supported by a grant from the Agency for Healthcare Research and Quality (K08HS24159). She serves as a consultant for the Parkinson's Foundation on a grant from the Patient-Centered Outcomes Research Institute (PCORI). She serves as an investigator for a Lewy Body Dementia Association Research Center of Excellence. She on the level of evidence editorial board for Neurology and related publications (uncompensated) and previously received compensation from the AAN for work as an evidence-based medicine methodology consultant. She has received honoraria for teaching at conferences for the AAN and the International Parkinson and Movement Disorder Society, participating in creating educational materials for the Parkinson's Foundation, and serving on drug safety monitoring boards for the Alzheimer's Clinical Trials Consortium and Alzheimer's Therapeutic Research Institute as well as the Alzheimer's Disease Cooperative Study. R.M.A. de Bie has received funding for travel from the International Parkinson and Movement Disorder Society and Rush University and has received research support from GE Healthcare, Medtronic, Lysosomal Therapeutics, NeuroDerm, and the Netherlands Organization for Health Research and Development. E. Roze has received research support from Merz Pharma, ORKYN, Aguettant, Elivie, Ipsen, EVER Pharma, Fondation Desmarest, AMADYS, Fonds de Dotation Patrick de Brou de Laurière, Agence Nationale de la Recherche, and the Société Française de Médecine Esthétique; has served on scientific advisory boards for ORKYN, Aguettant, and Merz Pharma; has received honoraria for speeches from ORKYN, Aguettant, Merz Pharma, EVER Pharma, and the International Parkinson and Movement Disorder Society; and has received travel grants from VitalAire, PEPS Developpement, Aguettant, Merz Pharma, Ipsen, Merck & Co., ORKYN, Elivie, Adelia Medical, the Dystonia Medical Research Foundation, the International Parkinson and Movement Disorders Society, the European Academy of Neurology, and the International Association of Parkinsonism and Related Disorders. J. Miyasaki receives grant funding from the National Institute of Neurologic Disorders and Stroke, the Patient-Centered Outcomes Research Institute, Allergan, and the University Hospital Foundation; receives honoraria from the Davis Phinney Foundation and Sunovion; receives publishing royalties from UpToDate; and has done paid consulting work for GE Consulting and Cynapsus Therapeutics and unpaid consulting for Sunovion Pharmaceuticals. R. Hauser has served on a scientific advisory board for Inhibikase Therapeutics, Impel NeuroPharma, CereSpir Incorporated, and Vivifi Biotech; has received funding for travel from AbbVie, Acadia Pharmaceuticals, Acorda Therapeutics, Adamas Pharmaceuticals, Affiris, Amneal Pharmaceuticals, ApoPharma, BioMarin International, Bracket, Britannia Pharmaceuticals Ltd., Cadent Therapeutics, Cleveland Clinic, CNS Ratings, Cowen and Company, Cynapsus Therapeutics, Denali Therapeutics, Eli Lilly and Company, F. Hoffmann-La Roche, GE Healthcare, Impax Laboratories, Impel NeuroPharma, Intrance Medical Systems, Jazz Pharmaceuticals, Kyowa Kirin, Lundbeck, Medtronic, Michael J. Fox Foundation for Parkinson's Research, Neurocrine Biosciences, NeuroDerm, NeuroPore Therapies, Orion, Parkinson Study Group, Pfizer, Prexton Therapeutics, Sarepta Therapeutics, Sunovion Pharmaceuticals, Supernus Pharmaceuticals, Takeda Pharmaceuticals, Teva Pharmaceutical Industries, the Tremor Research Group, UCB Biosciences, and US WorldMeds; serves on the editorial board for the Journal of Clinical Trials and Clinical Parkinsonism & Related Disorders; has received financial compensation for work as a consultant from AbbVie, Academy for Continued Healthcare Learning, Acadia Pharmaceuticals, Acorda Therapeutics, Adamas Pharmaceuticals, Affiris, AlphaSights, Amneal Pharmaceuticals, ApoPharma, Aptinyx, Aranca, AstraZeneca, Axovant Gene Therapies, Back Bay Life Sciences, Biomarin International, Biotie Therapies, Bracket, Britannia Pharmaceuticals Ltd., Cadent Therapeutics, CAVR, Cerevance, Cerevel Therapeutics, ClearView Healthcare Partners, ClinicalMind Medical and Therapeutic Communications, Clinical SCORE, CNS Ratings LLC, Compass Consulting Group, Cowen and Company, Curium, Cynapsus Therapeutics, DDB Health, Decision Resources Group, The Dedham Group, Defined Health, Denali Therapeutics, Eli Lilly and Company, Enterin, eResearch Technology, Expert Connect, Extera Partners, F. Hoffmann-La Roche Ltd., FirstWord Pharma, GE Healthcare, Gerson Lehrman Group (GLG), Global Kinetics Business Consultants, Global Life Sciences Solutions, GuidePoint Global, Health Advances, HealthLogix, Huron Consulting Group, Impax Laboratories, Impel NeuroPharma, Inhibikase Therapeutics, InSearch Consulting, Insignia Strategies, Intec, Intrance Medical Systems, International Stem Cell Corporation (ISCO), IQVIA, Jazz Pharmaceuticals, Kaiser Permanente, Kashiv BioSciences, KeifeRx, KeyQuest Health, Kx Advisors, Kyowa Kirin, LCN Consulting, L.E.K. Consulting, LifeMax, LifeSciences Consultants (LSC), Lundbeck, Medscape, Merz Pharma, Michael J. Fox Foundation for Parkinson's Research, Mitsubishi Tanabe Pharma America, National Parkinson Foundation, Neuro Challenge Foundation for Parkinson's, Neurocrine Biosciences, NeuroDerm, Neuropore Therapies, Novus Biologicals, Outcomes Insights, Orion, Parkinson Study Group, Peerview Press, Pennside Partners, Perception OpCo (Cerevel Therapeutics), Pfizer, Pharma Two B, PharmaTher, Putnam Associates, Phase Five Communications, Projects in Knowledge, Prexton Therapeutics, P\S\L Group, Quintiles, Regenera Pharma, Research Catalyst, Revance Therapeutics, RMEI Medical Education, Sanofi-Aventis, Sarepta Therapeutics, Schlesinger Group, Scion NeuroStim, LLC, Seagrove Partners, Seelos Therapeutics, Sio Gene Therapies, Slingshot Insights, Sunovion Pharmaceuticals, Supernus Pharmaceuticals, Syneos Health, Takeda Pharmaceuticals, Teva Pharmaceutical Industries, Tolmar Pharmaceuticals, UCB Biosciences, US WorldMeds, Vista Research, Vivifi Biotech, and Windrose Consulting Group; serves on a speakers bureau for AbbVie, Acorda Therapeutics, Adamas Pharmaceuticals, Amneal Pharmaceuticals, Kyowa Kirin, Lundbeck, Neurocrine Biosciences, Sunovion Pharmaceuticals, Teva Pharmaceuticals, and US WorldMeds; has received research support from AbbVie, Adamas Pharmaceuticals, AstraZeneca, Atlantic Research/Chelsea Therapeutics, Axovant Sciences Ltd., Biogen, Biotie Therapies, Cavion, Centogene, Cerevance, Cerevel Therapeutics, Civitas Therapeutics, Cynapsus Therapeutics, Dart NeuroScience, Enterin, F. Hoffman-La Roche, Global Kinetics Corporation, Impax Laboratories, Intec Pharma, Integrative Research Laboratories Sweden AB, Jazz Pharmaceuticals, Kyowa Kirin, Lundbeck, Michael J. Fox Foundation for Parkinson's Research, NIH, National Institute of Neurologic Disorders and Stroke, Neuraly, NeuroDerm, Northwestern University, Parkinson Study Group, Pfizer, Pharma Two B, Prexton Therapeutics, Revance Therapeutics, Sanofi US Services, Sun Pharma Advanced Research, Sunovion Pharmaceuticals, and UCB Biopharma; holds stock in Axial Biotherapeutics and Inhibikase Therapeutics; and has received license for fee payments from USF for Parkinson's Disease Diary. A. Espay has received grant support from the NIH and the Michael J. Fox Foundation for Parkinson's Research; has received financial compensation for work as a consultant or scientific advisory board member for AbbVie, NeuroDerm, Neurocrine Biosciences, Amneal Pharmaceuticals, Adamas Pharmaceuticals, Acadia Pharmaceuticals, Acorda Therapeutics, Intrance Medical Systems, Kyowa Kirin, Sunovion Pharmaceuticals, Lundbeck, and US WorldMeds; has received publishing royalties from Lippincott Williams & Wilkins, Cambridge University Press, and Springer Publishing; and has received honoraria from US WorldMeds, Acadia Pharmaceuticals, Amneal Pharmaceuticals, and Sunovion Pharmaceuticals. J. Martello has received honoraria from AbbVie, Acorda Therapeutics, Lundbeck, Neurocrine Biosciences, and Sunovion Pharmaceuticals for being on their speakers bureau. J. Gurwell has received travel funding from Boston Scientific and the AAN; serves on the editorial advisory board for Neurology Today; has received honoraria from Boston Scientific; and has received research support from AbbVie, Cynapsus Therapeutics, Sunovion Pharmaceuticals, and NeuroDerm. L. Billinghurst has received travel funding from the AAN to attend Guideline Subcommittee meetings. K. Sullivan has received intellectual property interests from a discovery or technology relating to health care and has received travel funding from the AAN to attend Guideline Subcommittee meetings. M. Fitts reports no disclosures. N. Cothros has received research support from Parkinson Association of Alberta and the Canadian League Against Epilepsy. D. Hall has received research support from the NIH, the Parkinson's Foundation, the Huntington's Disease Society of America, the CHDI Foundation, AbbVie, Biogen, Biohaven Pharmaceuticals, Pfizer, Neurocrine Biosciences, and uniQure; and has received editorial board support from the AAN. M. Rafferty has received research support from the National Institute on Disability, Independent Living, and Rehabilitation Research, the U.S. Department of Defense, the National Institute on Aging, and the Parkinson's Foundation. L. Hagerbrant reports no disclosures. T. Hastings serves on a patient advisory board for Kyowa Kirin. M. Dolan O'Brien is an employee of the AAN. H. Silsbee is an employee of the AAN. G. Gronseth has received travel funding from the AAN to attend Guideline Subcommittee meetings, serves as an associate editor for Neurology, has served as chief evidence-based medicine consultant for the AAN, and serves as an editorial advisory board member of Brain & Life. A. Lang has received financial compensation for work as a consultant from AbbVie, AFFiRiS, Biogen, Janssen, Lilly, Lundbeck, Merck & Co., Paladin Labs, Roche, Sun Pharma, Theravance Biopharma, and CBD Solutions; has received advisory board support from Jazz Pharmaceuticals, PhotoPharmics, Sunovion Pharmaceuticals; has received other honoraria from Sun Pharma, AbbVie, Sunovion Pharmaceuticals, the AAN, and the International Parkinson and Movement Disorder Society; has received grants from Brain Canada, the Canadian Institutes of Health Research, CBD Solutions, the Edmond J. Safra Philanthropic Foundation, the Michael J. Fox Foundation for Parkinson's Research, the Ontario Brain Institute, the Parkinson's Foundation, Parkinson Canada, and the W. Garfield Weston Foundation; and has received royalties from Elsevier, Saunders, Wiley-Blackwell, Johns Hopkins University Press, and Cambridge University Press. Go to Neurology.org/N for full disclosures.
References
- 1.Miyasaki JM, Martin W, Suchowersky O, Weiner WJ, Lang AE. Practice parameter: initiation of treatment for Parkinson's disease: an evidence-based review: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2002;58(1):11-17. [DOI] [PubMed] [Google Scholar]
- 2.Gronseth GS, Cox J, Gloss D, et al. Clinical Practice Guideline Process Manual, 2nd Edition. American Academy of Neurology; 2017. Accessed June 9, 2021. aan.com/siteassets/home-page/policy-and-guidelines/guidelines/about-guidelines/17guidelineprocman_pg.pdf [Google Scholar]
- 3.Gronseth GS, Woodroffe LM, Getchius TSD. Clinical Practice Guideline Process Manual, 2011 Edition. American Academy of Neurology; 2011. Accessed June 9, 2021. aan.com/siteassets/home-page/policy-and-guidelines/guidelines/about-guidelines/11guidelinedevmanual_v408_web.pdf [Google Scholar]
- 4.Caraceni T, Musicco M. Levodopa or dopamine agonists, or deprenyl as initial treatment for Parkinson's disease. A randomized multicenter study. Parkinsonism Relat Disord. 2001;7(2):107-114. [DOI] [PubMed] [Google Scholar]
- 5.PD Med Collaborative Group, Gray R, Ives N, Rick C, et al. Long-term effectiveness of dopamine agonists and monoamine oxidase B inhibitors compared with levodopa as initial treatment for Parkinson's disease (PD MED): a large, open-label, pragmatic randomised trial. Lancet. 2014;384(9949):1196-1205. [DOI] [PubMed] [Google Scholar]
- 6.Thomas A, Bonanni L, Di Iorio A, et al. End-of-dose deterioration in non ergolinic dopamine agonist monotherapy of Parkinson's disease. J Neurol. 2006;253(12):1633-1639. [DOI] [PubMed] [Google Scholar]
- 7.Hauser RA, Schapira AH, Rascol O, et al. Randomized, double-blind, multicenter evaluation of pramipexole extended release once daily in early Parkinson's disease. Mov Disord. 2010;25(15):2542-2549. [DOI] [PubMed] [Google Scholar]
- 8.Poewe W, Rascol O, Barone P, et al. Extended-release pramipexole in early Parkinson disease: a 33-week randomized controlled trial. Neurology. 2011;77(8):759-766. [DOI] [PubMed] [Google Scholar]
- 9.Kieburtz K, Parkinson Study Group Prami BIDI. Twice-daily, low-dose pramipexole in early Parkinson's disease: a randomized, placebo-controlled trial. Mov Disord. 2011;26(1):37-44. [DOI] [PubMed] [Google Scholar]
- 10.Stocchi F, Hersh BP, Scott BL, Nausieda PA, Giorgi L; Ease-PD Monotherapy Study Investigators. Ropinirole 24-hour prolonged release and ropinirole immediate release in early Parkinson's disease: a randomized, double-blind, non-inferiority crossover study. Curr Med Res Opin. 2008;24(10):2883-2895. [DOI] [PubMed] [Google Scholar]
- 11.Korczyn AD, Brunt ER, Larsen JP, Nagy Z, Poewe WH, Ruggieri S. A 3-year randomized trial of ropinirole and bromocriptine in early Parkinson's disease. The 053 Study Group. Neurology. 1999;53(2):364-370. [DOI] [PubMed] [Google Scholar]
- 12.Giladi N, Boroojerdi B, Korczyn AD, et al. Rotigotine transdermal patch in early Parkinson's disease: a randomized, double-blind, controlled study versus placebo and ropinirole. Mov Disord. 2007;22(16):2398-2404. [DOI] [PubMed] [Google Scholar]
- 13.Castro-Caldas A, Delwaide P, Jost W, et al. The Parkinson-Control study: a 1-year randomized, double-blind trial comparing piribedil (150 mg/day) with bromocriptine (25 mg/day) in early combination with levodopa in Parkinson's disease. Mov Disord. 2006;21(4):500-509. [DOI] [PubMed] [Google Scholar]
- 14.Markovic V, Stankovic I, Petrovic I, et al. Dynamics of impulsive-compulsive behaviors in early Parkinson's disease: a prospective study. J Neurol 2020;267(4):1127-1136. [DOI] [PubMed] [Google Scholar]
- 15.Corvol JC, Artaud F, Cormier-Dequaire F, et al. Longitudinal analysis of impulse control disorders in Parkinson disease. Neurology 2018;91(3):e189-e201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haaxma CA, Horstink MW, Zijlmans JC, Lemmens WA, Bloem BR, Borm GF. Risk of disabling response fluctuations and dyskinesias for dopamine agonists versus levodopa in Parkinson's disease. J Parkinsons Dis 2015;5(4):847-853. [DOI] [PubMed] [Google Scholar]
- 17.Bjornestad A, Forsaa EB, Pedersen KF, Tysnes OB, Larsen JP, Alves G. Risk and course of motor complications in a population-based incident Parkinson's disease cohort. Parkinsonism Relat Disord 2016;22:48-53. [DOI] [PubMed] [Google Scholar]
- 18.Dupont E, Andersen A, Boas J, et al. Sustained-release Madopar HBS compared with standard Madopar in the long-term treatment of de novo parkinsonian patients. Acta Neurol Scand 1996;93(1):14-20. [DOI] [PubMed] [Google Scholar]
- 19.Hauser RA, Panisset M, Abbruzzese G, et al. Double-blind trial of levodopa/carbidopa/entacapone versus levodopa/carbidopa in early Parkinson's disease. Mov Disord 2009;24(4):541-550. [DOI] [PubMed] [Google Scholar]
- 20.Koller WC, Hutton JT, Tolosa E, Capilldeo R. Immediate-release and controlled-release carbidopa/levodopa in PD: a 5-year randomized multicenter study. Carbidopa/Levodopa Study Group. Neurology 1999;53(5):1012-1019. [DOI] [PubMed] [Google Scholar]
- 21.Fung VS, Herawati L, Wan Y. Movement Disorder Society of Australia Clinical R, Trials G, Group Q-AS. Quality of life in early Parkinson's disease treated with levodopa/carbidopa/entacapone. Mov Disord 2009;24(1):25-31. [DOI] [PubMed] [Google Scholar]
- 22.Stocchi F, Rascol O, Kieburtz K, et al. Initiating levodopa/carbidopa therapy with and without entacapone in early Parkinson disease: the STRIDE-PD study. Ann Neurol 2010;68(1):18-27. [DOI] [PubMed] [Google Scholar]
- 23.Verschuur CVM, Suwijn SR, Boel JA, et al. Randomized delayed-start trial of levodopa in Parkinson's disease. N Engl J Med 2019;380(4):315-324. [DOI] [PubMed] [Google Scholar]
- 24.Schapira AH, McDermott MP, Barone P, et al. Pramipexole in patients with early Parkinson's disease (PROUD): a randomised delayed-start trial. Lancet Neurol 2013;12(8):747-755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Olanow CW, Rascol O, Hauser R, et al. A double-blind, delayed-start trial of rasagiline in Parkinson's disease [erratum in 2011;364(19):1882]. N Engl J Med 2009;361(13):1268-1278. [DOI] [PubMed] [Google Scholar]
- 26.Warren Olanow C, Kieburtz K, Rascol O, et al. Factors predictive of the development of Levodopa-induced dyskinesia and wearing-off in Parkinson's disease. Mov Disord 2013;28(8):1064-1071. [DOI] [PubMed] [Google Scholar]
- 27.Sharma JC, Ross IN, Rascol O, Brooks D. Relationship between weight, levodopa and dyskinesia: the significance of levodopa dose per kilogram body weight. Eur J Neurol 2008;15(5):493-496. [DOI] [PubMed] [Google Scholar]
- 28.Rascol O, Brooks DJ, Korczyn AD, De Deyn PP, Clarke CE, Lang AE. A five-year study of the incidence of dyskinesia in patients with early Parkinson's disease who were treated with ropinirole or levodopa. N Engl J Med 2000;342(10):1484-1491. [DOI] [PubMed] [Google Scholar]
- 29.Cilia R, Akpalu A, Sarfo FS, et al. The modern pre-levodopa era of Parkinson's disease: insights into motor complications from sub-Saharan Africa. Brain 2014;137(pt 10):2731-2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nadjar A, Gerfen CR, Bezard E. Priming for l-dopa-induced dyskinesia in Parkinson's disease: a feature inherent to the treatment or the disease? Prog Neurobiol 2009;87(1):1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hauser RA, McDermott MP, Messing S. Factors associated with the development of motor fluctuations and dyskinesias in Parkinson disease. Arch Neurol 2006;63(12):1756-1760. [DOI] [PubMed] [Google Scholar]
- 32.Marin-Lahoz J, Sampedro F, Martinez-Horta S, Pagonabarraga J, Kulisevsky J. Depression as a risk factor for impulse control disorders in Parkinson disease. Ann Neurol 2019;86(5):762-769. [DOI] [PubMed] [Google Scholar]
- 33.Smith KM, Xie SX, Weintraub D. Incident impulse control disorder symptoms and dopamine transporter imaging in Parkinson disease. J Neurol Neurosurg Psychiatry 2016;87(8):864-870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gatto EM, Aldinio V. Impulse control disorders in Parkinson's disease: a brief and comprehensive review. Front Neurol 2019;10:351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Antonini A, Barone P, Bonuccelli U, Annoni K, Asgharnejad M, Stanzione P. ICARUS study: prevalence and clinical features of impulse control disorders in Parkinson's disease. J Neurol Neurosurg Psychiatry 2017;88(4):317-324. [DOI] [PubMed] [Google Scholar]
- 36.Latt MD, Lewis S, Zekry O, Fung VSC. Factors to consider in the selection of dopamine agonists for older persons with Parkinson's disease. Drugs Aging 2019;36(3):189-202. [DOI] [PubMed] [Google Scholar]
- 37.Yeung EYH, Cavanna AE. Sleep attacks in patients with Parkinson's disease on dopaminergic medications: a systematic review. Mov Disord Clin Pract 2014;1(4):307-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hsu A, Yao HM, Gupta S, Modi NB. Comparison of the pharmacokinetics of an oral extended-release capsule formulation of carbidopa-levodopa (IPX066) with immediate-release carbidopa-levodopa (Sinemet®), sustained-release carbidopa-levodopa (Sinemet® CR), and carbidopa-levodopa-entacapo. J Clin Pharmacol 2015;55(9):995-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dhall R, Kreitzman DL. Advances in levodopa therapy for Parkinson disease: review of RYTARY (carbidopa and levodopa) clinical efficacy and safety. Neurology 2016;86(14 suppl 1):S13-S24. [DOI] [PubMed] [Google Scholar]
- 40.Ruottinen HM, Rinne UK. Entacapone prolongs levodopa response in a one month double blind study in parkinsonian patients with levodopa related fluctuations. J Neurol Neurosurg Psychiatry 1996;60(1):36-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fahn S, Oakes D, Shoulson I, et al. Levodopa and the progression of Parkinson's disease. N Engl J Med 2004;351(24):2498-2508. [DOI] [PubMed] [Google Scholar]
- 42.Wang L, Xiong N, Huang J, et al. Protein-restricted diets for ameliorating motor fluctuations in Parkinson's disease. Front Aging Neurosci 2017;9:206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hurt CS, Rixon L, Chaudhuri KR, Moss-Morris R, Samuel M, Brown RG. Barriers to reporting non-motor symptoms to health-care providers in people with Parkinson's. Parkinsonism Relat Disord 2019;64:220-225. [DOI] [PubMed] [Google Scholar]
- 44.Weintraub D, Mamikonyan E, Papay K, Shea JA, Xie SX, Siderowf A. Questionnaire for impulsive-compulsive disorders in Parkinson's disease rating scale. Mov Disord 2012;27(2):242-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johns MW. A new method for measuring daytime sleepiness: the Epworth Sleepiness Scale. Sleep 1991;14(6):540-545. [DOI] [PubMed] [Google Scholar]
- 46.Rizos A, Sauerbier A, Antonini A, et al. A European multicentre survey of impulse control behaviours in Parkinson's disease patients treated with short- and long-acting dopamine agonists. Eur J Neurol 2016;23(8):1255-1261. [DOI] [PubMed] [Google Scholar]
- 47.Choi J, Horner KA. Dopamine Agonists. StatPearls Publishing; 2020. [PubMed] [Google Scholar]
- 48.Okai D, Samuel M, Askey-Jones S, David AS, Brown RG. Impulse control disorders and dopamine dysregulation in Parkinson's disease: a broader conceptual framework. Eur J Neurol 2011;18(12):1379-1383. [DOI] [PubMed] [Google Scholar]
- 49.Parkinson Study Group. A controlled trial of rasagiline in early Parkinson disease: the TEMPO Study. Arch Neurol 2002;59(12):1937-1943. [DOI] [PubMed] [Google Scholar]
- 50.Parkinson Study Group. Effects of tocopherol and deprenyl on the progression of disability in early Parkinson's disease. N Engl J Med 1993;328(3):176-183. [DOI] [PubMed] [Google Scholar]
- 51.Hilli J, Korhonen T, Laine K. Lack of clinically significant interactions between concomitantly administered rasagiline and escitalopram. Prog Neuropsychopharmacol Biol Psychiatry 2009;33(8):1526-1532. [DOI] [PubMed] [Google Scholar]
- 52.Panisset M, Chen JJ, Rhyee SH, Conner J, Mathena J; STACCATO study investigators. Serotonin Toxicity Association with Concomitant Antidepressants and Rasagiline Treatment: retrospective study (STACCATO). Pharmacotherapy 2014;34(12):1250-1258. [DOI] [PubMed] [Google Scholar]
- 53.Richard IH, Kurlan R, Tanner C, et al. Serotonin syndrome and the combined use of deprenyl and an antidepressant in Parkinson's disease” Parkinson Study Group. Neurology 1997;48(4):1070-1077. [DOI] [PubMed] [Google Scholar]