

Abstract
Background
Characterizing the longevity and quality of cellular immune responses to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) enhances understanding of coronavirus disease 2019 (COVID-19) immunity that influences clinical outcomes. Prior studies suggest SARS-CoV-2–specific T cells are present in peripheral blood 10 months after infection. Analysis of the function, durability, and diversity of cellular response long after natural infection, over a range of ages and disease phenotypes, is needed to identify preventative and therapeutic interventions.
Methods
We identified participants in our multisite longitudinal, prospective cohort study 12 months after SARS-CoV-2 infection representing a range of disease severity. We investigated function, phenotypes, and frequency of T cells specific for SARS-CoV-2 using intracellular cytokine staining and spectral flow cytometry, and compared magnitude of SARS-CoV-2–specific antibodies.
Results
SARS-CoV-2–specific antibodies and T cells were detected 12 months postinfection. Severe acute illness was associated with higher frequencies of SARS-CoV-2–specific CD4 T cells and antibodies at 12 months. In contrast, polyfunctional and cytotoxic T cells responsive to SARS-CoV-2 were identified in participants over a wide spectrum of disease severity.
Conclusions
SARS-CoV-2 infection induces polyfunctional memory T cells detectable at 12 months postinfection, with higher frequency noted in those who experienced severe disease.
Keywords: COVID-19, SARS-CoV-2, 12 months, T cell, antibody, memory, cytotoxicity, polyfunctionality, durability
Circulating SARS-CoV-2–specific T cells could be measured 12 months postinfection. Severe acute COVID-19 was associated with a higher magnitude of SARS-CoV-2–specific CD4 T cells and a polyfunctional, central memory phenotype predominated across the disease spectrum.
Understanding the development and durability of protective immune responses against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) remains critical as we seek global reduction in disease burden. Antibody responses induced during a primary SARS-CoV-2 infection have been shown to wane, but may be present in the circulation up to 12 months post symptoms onset (PSO) [1, 2]. Thus far, SARS-CoV-2–specific T cells have been detected up to 10 months PSO [3, 4]. Intriguingly, SARS-CoV-1–specific T cells have been identified 17 years postinfection, suggesting the potential for very long-lived T-cell memory [5]. To utilize the specificity and potent viral clearance of T cells, further knowledge is required regarding SARS-CoV-2 antigen specificity, memory differentiation, and longevity [6].
Studies on SARS-CoV-2–specific T cells within 2 months PSO suggest that potent antigen-specific T cells are associated with mild disease, whereas a lack of these antiviral cells or a delay in development is associated with severe disease [7, 8]. Peng et al [9] showed that individuals who had mild disease had a higher ratio of polyfunctional CD8:CD4 T cells around 42 days PSO, suggesting that the quality of the T-cell response played a role in clinical disease. Interestingly, SARS-CoV-2 convalescent T-cell transfer, performed in a recent clinical trial, indicated improved clinical outcomes and no serious side effects [10]. Analysis of functional T-cell responses at memory time points is needed to provide insight into their durability and cytolytic potential that prior studies relying on activation-induced markers [6] could not.
The T-cell response to SARS-CoV-2 has been shown to recognize epitopes across multiple viral proteins, including the spike glycoprotein (S), nucleocapsid (N), membrane (M), and small envelope (E) proteins, as well as other nonstructural proteins (nsp) [11, 12]. Due to the ability of T cells to recognize structural and nonstructural proteins that are less susceptible to antibody-dependent mutational pressure, T cells may provide cross-reactive immunity against other coronaviruses, as well as SARS-CoV-2 variants [13, 14].
In this study, we analyzed the durability and functional characteristics of the SARS-CoV-2–specific memory T-cell response at 12 months PSO derived from a prospective, longitudinal cohort of United States Military Health System (MHS) beneficiaries, including active-duty military and dependents with varying severity of disease, to help determine how the humoral and cellular components of antiviral immunity work synergistically to prevent future infection, inform testing platforms [15], and vaccine strategies.
METHODS
Study Participants
Individuals were enrolled into the Epidemiology, Immunology, and Clinical Characteristics of Emerging Infectious Diseases with Pandemic Potential (EPICC) study, a prospective, longitudinal cohort study, if they were exposed to, had symptoms consistent with, or had documented SARS-CoV-2 infection, beginning in March of 2020. Samples from 4 of 10 military treatment facilities, that is Walter Reed National Military Medical Center (Bethesda, MD), Brooke Army Medical Center (San Antonio, TX), Naval Medical Center San Diego (San Diego, CA), and Madigan Army Medical Center (Tacoma, WA), were included based on peripheral blood mononuclear cells (PBMCs) obtained 12 months PSO and absence of evidence for reinfection. Infection was confirmed by reverse-transcriptase polymerase chain reaction (RT-PCR) and serologic response. After primary infection, biological samples and clinical questionnaire data were collected at 1, 6, and 12 months PSO. Receipt of a SARS-CoV-2 vaccine was identified by report and confirmed by medical data repository review. Those who received a SARS-CoV-2 vaccine prior to the 12-month peripheral blood collection were excluded from S-specific T-cell and antibody analysis. Study participants were evaluated for reinfection by documented PCR positive for SARS-CoV-2 or a >2-fold rise in both S- and N-specific antibody responses after primary infection. Participants who were reinfected were excluded for comparative T-cell and antibody analysis in the current study. The protocol was approved by the Uniformed Services University Institutional Review Board (IDCRP-085), and all subjects or their legally authorized representative provided informed consent to participate.
PBMC and Serum Preparation
PBMCs were isolated from peripheral whole blood collected in acid citrate dextrose tubes at 12 months PSO. PBMCs were purified using a Ficoll-Histopaque (Fisher Scientific) gradient. Cells were preserved in 90% fetal bovine serum (Sigma-Aldrich) and 10% dimethyl sulfoxide (Sigma-Aldrich) and stored in liquid nitrogen. Serum was isolated from blood collection in EDTA tubes.
Measurement of Antigen-Presenting Cells
One million thawed PBMCs were stained in phosphate-buffered saline (PBS) at 4°C for 20 minutes for antigen-presenting cells (APCs) using the antibodies listed in Supplementary Table 1 and were acquired on a CYTEK Aurora 5-laser spectral flow cytometer (CYTEK Biosciences).
Antigen-Specific T-Cell Analysis
For identification of antigen-specific T cells, isolated PBMCs were cultured in 96-well plates and stimulated overnight with peptide pools derived from selected viral proteins. Peptide pools were comprised of 15-mer peptides overlapping by 11 amino acid residues covering the S, M, N, and E proteins of SARS-CoV-2 reference strain Wuhan-Hu-1 (JPT); the S protein of endemic human coronavirus (hCoV) strains HKU1, 229E, NL63, and OC43; and a cytomegalovirus (CMV) peptide pool including pp50, pp65, IE1, IE2, and envelope glycoprotein B (Mabtech) at a final concentration of 1mg/mL for each individual peptide (Supplementary Table 2). Monensin (BD Biosciences, BioLegend) was added 1 hour after peptide addition to prevent cytokine secretion as recommended by the manufacturer. Ten minutes after peptide administration, CD107a antibody (BioLegend) was added. Antibodies for cell surface marker analysis are listed in Supplementary Table 3. Surface staining was followed by cell permeabilization using FoxP3/transcription factor staining buffer set (Thermo Fisher Scientific) at 4°C for at least 30 minutes. Antibodies used for intracellular cytokine staining (Supplementary Table 3) were then added. PBMCs cultured in medium without peptide stimulation or with CytoStim (Miltenyi Biotec) served as the negative and positive control for the assay, respectively. At least 105 T cells were acquired on a CYTEK Aurora. Cytometric data were analyzed using FlowJo software (BD Biosciences). Samples were considered positive if the frequency of interferon-γ (IFN-γ)-positive T cells was 2-fold higher than the medium control and greater than 0.01% of CD4 or CD8 T cells after subtracting the medium control value [16].
Multiplex Microsphere-Based Immunoassay Screening Procedures
Detailed experimental procedures of SARS-CoV-2 S and N protein-based multiplex microsphere immunoassays have been previously described [2, 17]. Briefly, diluted serum and capillary blood samples were added to 96-well microtiter plates containing antigen-coupled microspheres and tested in technical duplicates. After 45 minutes of agitation, wells were washed, and biotin-conjugated goat anti-human IgG (Thermo Fisher Scientific) diluted in PBS plus 0.05% Tween20 (PBST) was added to each well. Wells were subsequently washed again, then streptavidin-phycoerythrin (1:1000 in PBST) (Bio-Rad) was added to each well. A final wash was performed after 45 minutes of incubation, and antigen-antibody complexes were analyzed on BioPlex 200 multiplexing systems (Bio-Rad) for immunoglobulin G (IgG) binding, and median fluorescence intensity values were reported for specificity to SARS-CoV-2 S and N protein. Participants were excluded from 12 months PSO evaluation based on observed increases in SARS-CoV-2 S and N protein binding from 6–12 months collections across longitudinal serum samples, suggesting possible reinfections.
Statistical Analysis
Data were analyzed using GraphPad Prism 9 (GraphPad Software). Two-group test significance levels were calculated using Mann-Whitney analysis. Correlation coefficients and significance levels were calculated using Spearman rank correlation. A P value < .05 was considered statistically significant.
RESULTS
Study Cohort
From our cohort, we identified 29 patients who had COVID-19 approximately 1 year prior, and did not have evidence of reinfection prior to a peripheral blood collection at 12 months PSO. Disease severity was classified by inpatient or outpatient status during the acute phase of illness. The age range of the study participants was 20–72 years and the overall distribution of age did not vary significantly (P = .1) between inpatients (median age 50.6 years [minimum-maximum, 21.4–72.4 years]) and outpatients (median age 44.6 years [minimum-maximum, 20.1–60.1 years]), nor did sex (Table 1 and Supplementary Table 4). Inpatients, however, were more likely to have a comorbidity with respiratory and pulmonary illnesses being the most common. Six of 14 study inpatients received intermittent intranasal or inhaled corticosteroids for underlying conditions, in contrast to 1 of 15 outpatient. Two study participants received systemic steroids during acute illness (Table 1 and Supplementary Table 4). Data on S-specific humoral and cellular responses were not included for individuals vaccinated prior to the 12-month blood draw. Inpatients were further dichotomized into those who required supplemental oxygen during hospitalization and those who did not. Further demographic and clinical information are provided in Supplementary Table 4.
Table 1.
Demographic Characteristics of Participants
| Characteristics | Hospitalized (n = 14) | Outpatient (n = 15) | Total (n = 29) |
|---|---|---|---|
| Sex, male, No. (%) | 8 (57.1) | 8 (53.3) | 16 (55.2) |
| Race, No. (%) | |||
| Asian | 2 (14.3) | 2 (13.3) | 4 (13.8) |
| Black | 8 (57.1) | 1 (6.7) | 9 (31.0) |
| Hispanic | 1 (7.1) | 5 (33.3) | 6 (20.7) |
| Other | 0 (0.0) | 1 (6.7) | 1 (3.4) |
| White | 3 (21.4) | 6 (40.0) | 9 (31.0) |
| Age, y | |||
| Median (Q1, Q3) | 50.6 (45.3, 55.8) | 44.6 (39.7, 50.9) | 46.4 (40.1, 53.5) |
| Min-max | 21.4–72.4 | 20.1–60.2 | 20.1–72.4 |
| Age group, y, No. (%) | |||
| <18 | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| 18–44 | 3 (21.4) | 8 (53.3) | 11 (37.9) |
| 45–64 | 10 (71.4) | 7 (46.7) | 17 (58.6) |
| 65+ | 1 (7.1) | 0 (0.0) | 1 (3.4) |
| Military affiliation, No. (%) | |||
| Active duty | 2 (14.3) | 5 (33.3) | 7 (24.1) |
| Dependent | 6 (42.9) | 6 (40.0) | 12 (41.4) |
| Retired military | 6 (42.9) | 4 (26.7) | 10 (34.5) |
| Any comorbidities, No. (%) | 13 (92.9) | 7 (46.7) | 20 (69.0) |
| Multiple comorbidities, No. (%) | 4 (28.6) | 2 (13.3) | 6 (20.7) |
| Charlson comorbidity index | |||
| Median (Q1, Q3) | 1.0 (0.0, 1.5) | 0.0 (0.0, 1.5) | 1.0 (0.0, 1.75) |
| Min-max | 0–6 | 0– 3 | 0–6 |
Convalescent T-Cell and Antibody Responses Are Detectable 12 Months PSO
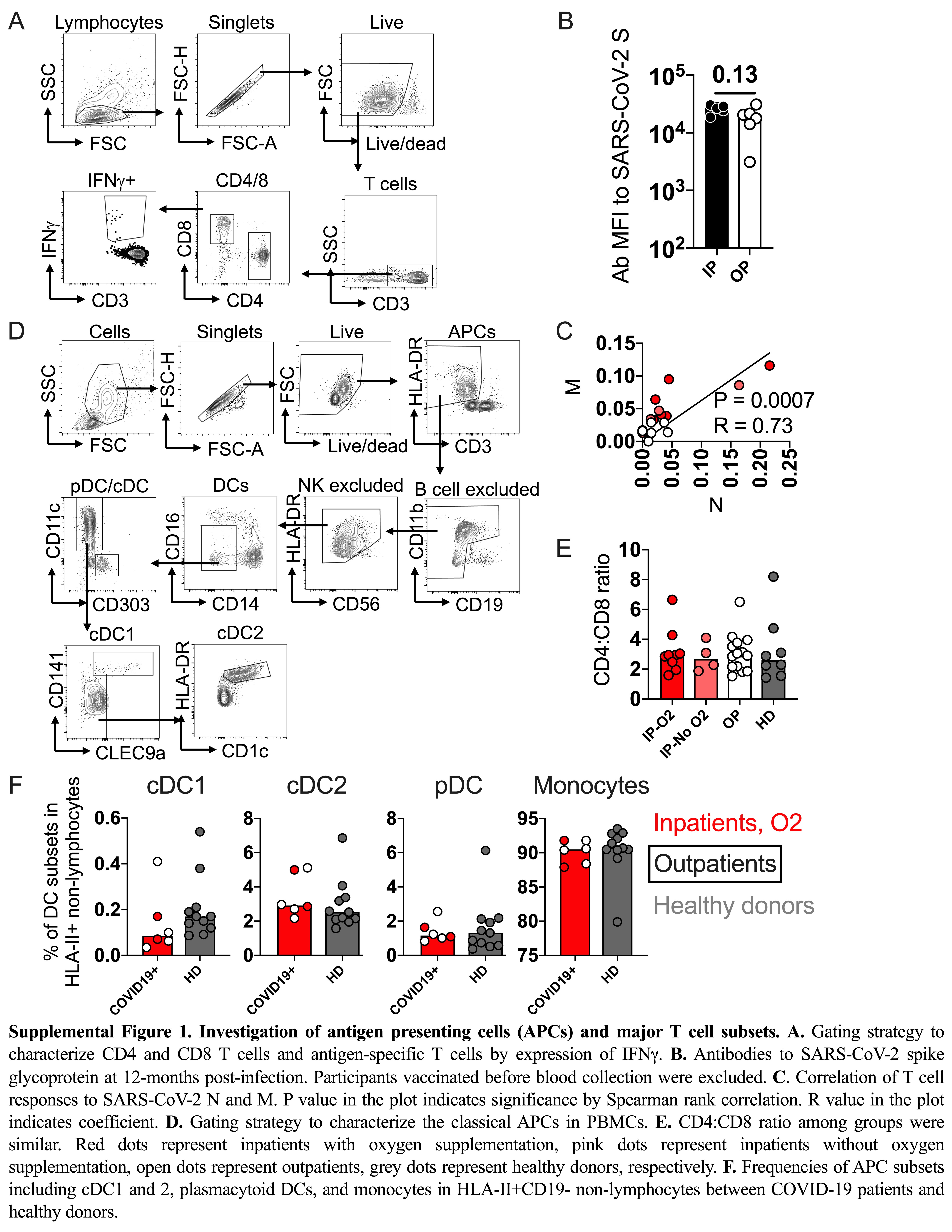
Through stimulation of PBMCs with peptide pools from S, N, M, and E, we demonstrated the presence of SARS-CoV-2–specific CD4 and CD8 T cells at 12 months PSO by intracellular cytokine staining in 22/29 (75.9%) of study participants (Figure 1A and Supplementary Figure 1A). Only 3 of 29 participants had a measurable response to E (data not shown). Stimulation with the CMV peptide pool demonstrated CMV-specific CD4 and CD8 T-cell responses in 73.9% of study participants. Overall, SARS-CoV-2–specific CD4 T cells were more frequently identified in the peripheral blood compared to SARS-CoV-2–specific CD8 T cells. In addition, T-cell responses to endemic hCoV strains HKU1, 229E, OC43, and NL63 were sporadic, with no differential distribution or magnitude differences between inpatients and outpatients identified (Table 2).
Figure 1.

Adaptive immune responses to SARS-CoV-2 viral components at 12 months PSO. A, SARS-CoV-2–specific CD4 and CD8 T-cell responses were detected at 12 months PSO by expression of IFN-γ upon peptide stimulation and intracellular cytokine staining. B, Antibodies to SARS-CoV-2 nucleocapsid protein at 12 months postinfection. Threshold shows the global MFI of antibodies to endemic hCoVs using the same sera in the same assay. C, Frequency of CD4 T-cell responses to the tested viral components N, M, and S in all patient groups. D, Frequency of CD8 T-cell responses to the tested viral components N, M, and S in all patient groups. Bars represent the median value in each group. P values in the plots indicate the significance by Mann-Whitney test. Abbreviations: CMV, cytomegalovirus; hCoV, human coronavirus; IFN-γ, interferon-γ; IP, inpatient; M, membrane; MFI, mean fluorescence intensity; N, nucleocapsid; O2, oxygen supplementation; OP, outpatient; PSO, post symptoms onset; S, spike; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
Table 2.
Frequencies of T-Cell Responses to Endemic CoV and SARS-CoV-2 Viral Components
| HKU-S | NL63-S | OC43-S | 229E-S | SARS-CoV-2 N | SARS-CoV-2 M | SARS-CoV-2 E | |
|---|---|---|---|---|---|---|---|
| Inpatient, O2 | 3 of 7 | 4 of 6 | 3 of 6 | 2 of 7 | 7 of 11 | 9 of 11 | 0 of 11 |
| Inpatient, no O2 | 3 of 6 | 3 of 6 | 3 of 6 | 2 of 6 | 3 of 3 | 3 of 3 | 2 of 3 |
| Outpatient | 4 of 14 | 4 of 14 | 5 of 14 | 4 of 14 | 6 of 15 | 7 of 15 | 1 of 15 |
Data are number of participants positive of total number of participants tested in each group.
Abbreviations: CoV, coronavirus; E, envelope; M, membrane; N, nucleocapsid; O2, oxygen supplementation; S, spike; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
SARS-CoV-2–specific antibodies against S and N proteins were also present across disease severity groups at 12 months PSO (Figure 1B and Supplementary Figure 1B). The antibody response against N in inpatients was higher than in outpatients, in whom it waned below the threshold level of endemic hCoV N responses (P = .01). These findings demonstrate that the humoral and cellular responses to SARS-CoV-2 exhibit durability at 12 months PSO, but vary by severity of disease and antigen specificity.
Stratification of study participants by inpatient and outpatient status at peak disease severity demonstrated that individuals with more severe disease exhibited the highest frequency of CD4 T cells responsive to N (P = .02), M (P = .002), and S (P = .064) proteins, regardless of oxygen requirement (Figure 1C). Of the inpatients, 11 of 13 (84.6%) exhibited T-cell responses specific for either N or M (patient 30 and patient 41 did not exhibit T-cell responses). In contrast, 7 of 14 (50.0%) outpatients exhibited CD4 T-cell responses to N or M. Individuals who exhibited T-cell specificity for either N or M at 12 months were also more likely (P = .0007) to simultaneously recognize other SARS-CoV-2 epitopes in N or M (Supplementary Figure 1C). Compared with the CD4 T-cell response, the CD8 T-cell response, measured by IFN-γ production, was lower in magnitude and did not correlate with severity of disease (Figure 1D).
Quantification of the APCs (Supplementary Figure 1D) and frequency of total T cells at 12 months PSO were measured by flow cytometry to determine whether differences exhibited between study groups were influenced by APC or T-cell availability. Our data show that the ratio of total CD4 and CD8 T cells (Supplementary Figure 1E), and the frequency of classical APCs, including conventional dendritic cells 1 and 2, plasmacytoid dendritic cells, and monocytes (Supplementary Figure 1F), were similar among patient groups at 12 months PSO and healthy controls.
SARS-CoV-2–Specific T Cells at 12 Months PSO Predominantly Exhibit Central Memory Characteristics
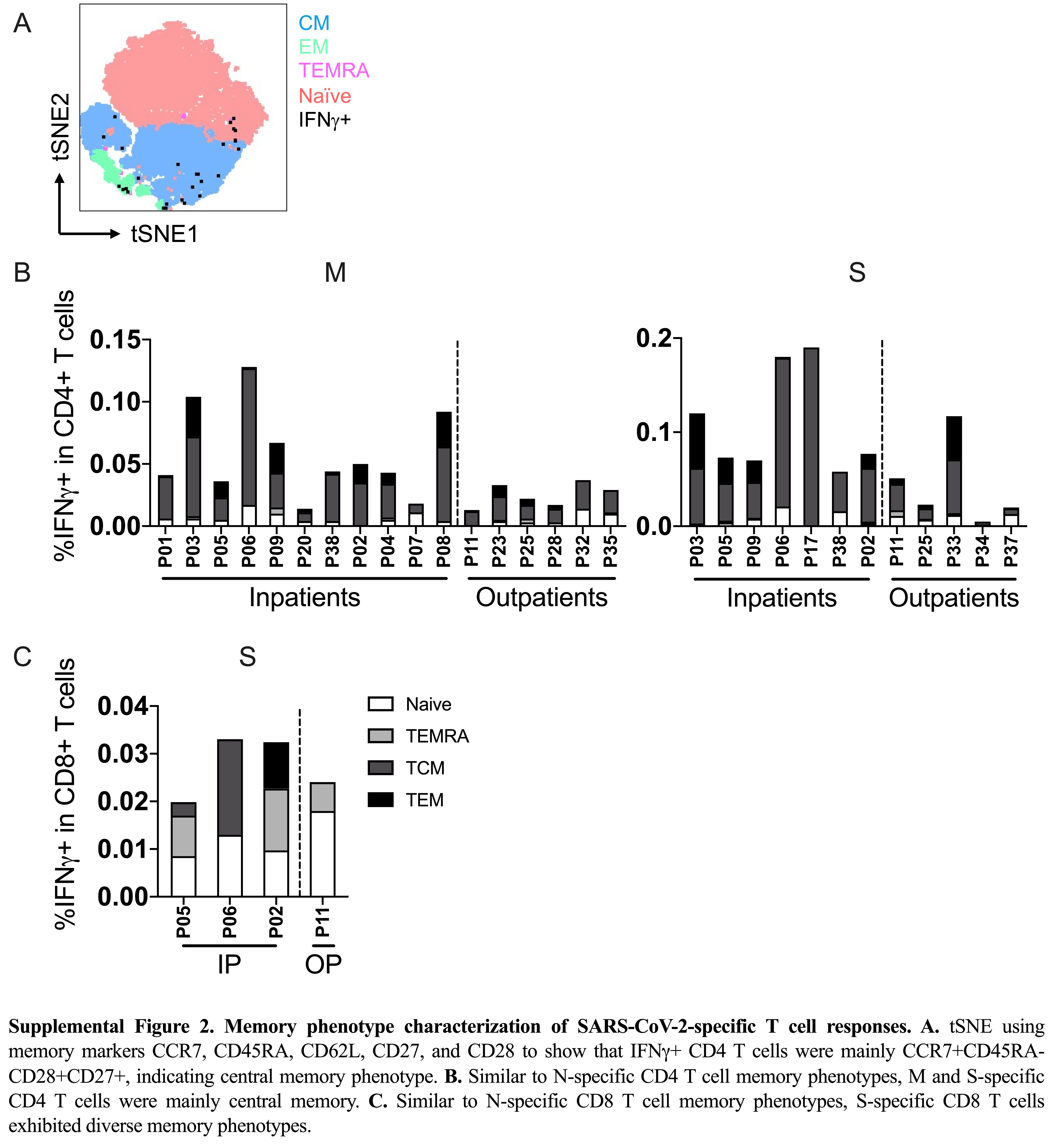
SARS-CoV-2–specific T cells identified by IFN-γ production in response to SARS-CoV-2 peptides were characterized with established phenotypic markers of differentiation as previously described [6]. Expression of the lymph node homing receptor, CCR7, coupled with the absence of CD45RA expression is characteristic of central memory T cells [18] (Figure 2A), which are also CD27+CD28+ [19]. This was the predominant phenotype exhibited by CD4 T cells responding to N (Figure 2B and Supplementary Figure 2A) and was distinct from the predominance of naive T cells in the total CD4 T-cell population. The phenotypic distribution of CD4 T cells responding to M and S was similarly skewed towards a central memory phenotype (Supplementary Figure 2B). In contrast, CD8 T cells specific for N and S derived from multiple compartments, including naive (CCR7+CD45RA+), effector memory T cells reexpressing CD45RA (TEMRA), central, and effector memory phenotypes (Figure 2C and Supplementary Figure 2C).
Figure 2.

Memory phenotype characterization of SARS-CoV-2–specific T-cell responses. A, A representative flow plot of the essential markers used for memory phenotyping. B, Memory phenotypes of IFN-γ+ CD4 T cells to SARS-CoV-2N protein as an example in each patient separated by patient group. C, Memory phenotypes of IFN-γ+ CD8 T cells to SARS-CoV-2N protein in each patient separated by patient group. Abbreviations: IFN-γ, interferon-γ; IP, inpatient; N, nucleocapsid; O2, oxygen supplementation; OP, outpatient; P, patient; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; TCM, central memory T cells; TEM, effector memory T cells; TEMRA, effector memory T cells reexpressing CD45RA.
Together these findings demonstrate that the SARS-CoV-2–specific T-cell responses exhibit a longevity of at least 12 months, with a predominance of central memory CD4 T cells specific for S, N, and M proteins. SARS-CoV-2–specific CD8 T cells were less frequent in the peripheral blood and exhibited more diverse memory phenotypes. For CD4 and CD8 T cells, the severity of disease during acute illness was not reflected in distinct memory phenotype differentiation at 12 months PSO.
Cytokine Profiles Indicate SARS-CoV-2–Specific Memory T Cells Are Polyfunctional
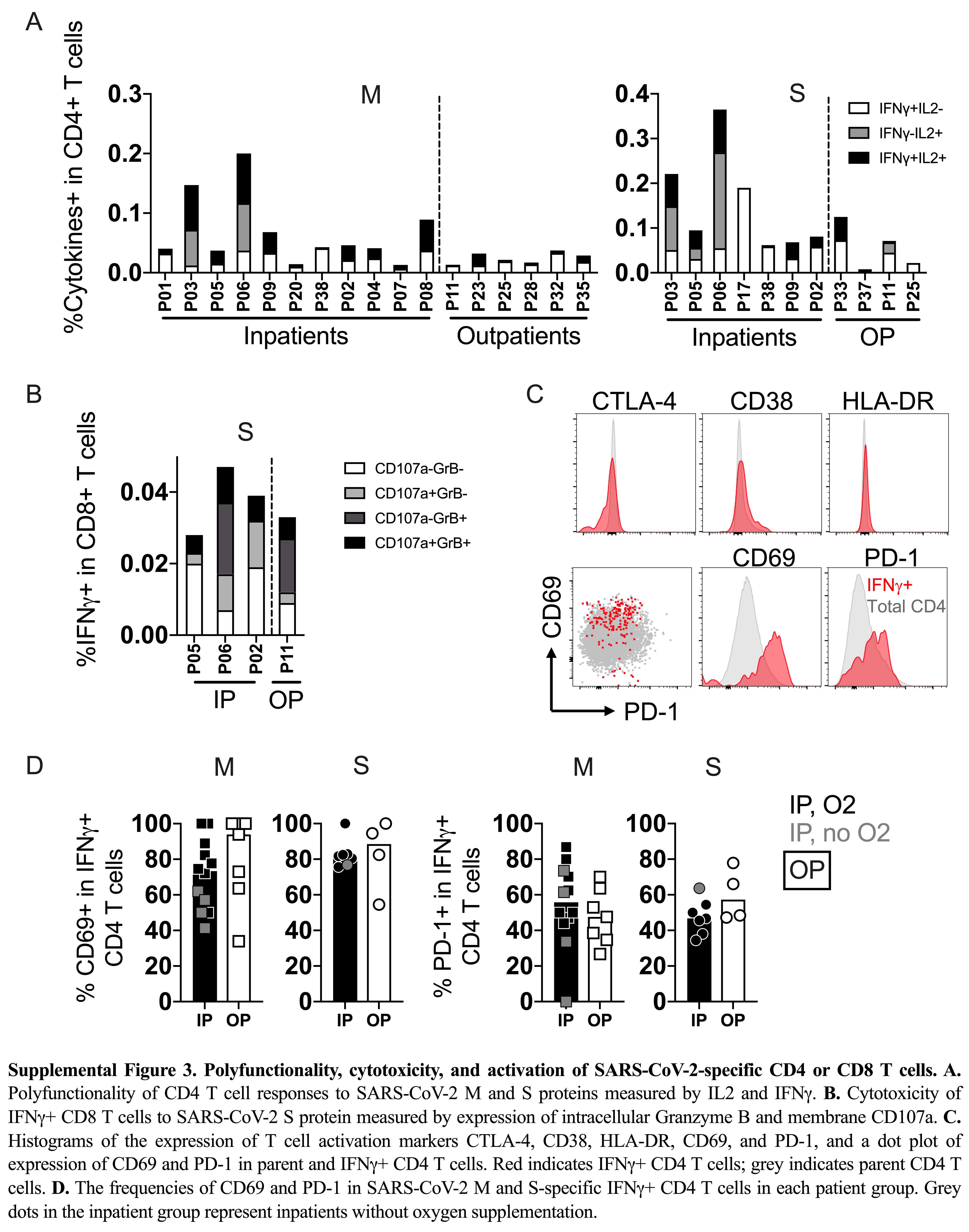
T cells were evaluated for the expression of multiple cytokines in response to the distinct SARS-CoV-2 peptide pools to determine their functional potential. CD4 T cells responsive to N exhibited expression of IFN-γ and interleukin 2 (IL-2), indicating a polyfunctional response in all study participants in both inpatient and outpatient severity groups at 12 months PSO (Figure 3A). Overall, inpatients had a higher frequency of cytokine-expressing CD4 T cells, but the frequency of polyfunctional CD4 T cells did not differ significantly between severity groups (Figure 3A). CD4 T-cell responses against M and S exhibited similar frequencies of polyfunctional T cells, suggesting that epitope specificity did not predict polyfunctionality (Supplementary Figure 3A and 3B). At 12 months, we did not detect expression of IL-17A, IL-21, IL-4, or IL-13 by CD4 T cells in response to the evaluated peptide pools (data not shown) based on the assay cutoff, suggesting that type 1 helper (Th1) CD4 T cells were dominant over Th2, Th17, or circulating T follicular helper cells.
Figure 3.

Polyfunctionality, cytotoxicity, and activation of SARS-CoV-2–specific CD4 or CD8 T cells. A, Polyfunctionality of CD4 T-cell responses to SARS-CoV-2N protein measured by IL-2 and IFN-γ. B, Cytotoxicity of IFN-γ+ CD8 T cells to SARS-CoV-2N protein measured by expression of intracellular granzyme B and membrane CD107a. C, Frequencies of CD69 and PD-1 in SARS-CoV-2 N-specific IFN-γ+ CD4 T cells in each patient group. Gray symbols in the inpatient group represent inpatients without oxygen supplementation. D, A representative flow plot of cytotoxic CD8 T cells by the expression of granzyme B and CD107a. Abbreviations: GrB, granzyme B; IFN-γ, interferon-γ; IL, interleukin; IP, inpatient; O2, oxygen supplementation; OP, outpatient; P, patient; PD-1, programmed cell death protein 1; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
CD8 T cells expressing IFN-γ were further evaluated for cytolytic potential by the expression of granzyme B and surface exposure of the lysosome-associated membrane protein CD107a, a marker of degranulation (Figure 3D). The CD8 T-cell response to N and S displayed heterogeneity in cytolytic potential, with cells generally expressing either marker in response to their cognate antigens (Figure 3B and Supplementary Figure 3B). Again, we saw no difference in frequency and function of the SARS-CoV-2–specific CD8 T cells based on disease severity.
We further looked into the activation features of the IFN-γ+ CD4 T cells. Upon peptide stimulation, SARS-CoV-2–specific IFN-γ+ CD4 T cells upregulated CD69 and PD-1 compared to the total CD4 T-cell population, and was similar in inpatients and outpatients (Figure 3C and Supplementary Figure 3C and 3D). The other activation markers, including CD38, HLA-DR, or CTLA-4, in the IFN-γ+ T cells were comparable to the total CD4 T-cell population (Supplementary Figure 3C).
Together these data suggest that the memory CD4 T-cell response against SARS-CoV-2 predominantly exhibits a Th1-type profile, and that SARS-CoV-2–specific memory CD8 T cells display cytolytic potential at 12 months.
DISCUSSION
Here we show that SARS-CoV-2–specific T-cell responses are detected at 12 months PSO, extended our prior understanding of the durability of the T-cell response against SARS-CoV-2 [4]. The frequency of SARS-CoV-2–specific CD4, but not CD8, T cells was higher at 12 months PSO in individuals who experienced severe disease compared to those who had mild disease at the acute phase. Importantly, memory SARS-CoV-2–specific CD4 T and CD8 T cells exhibited polyfunctionality and cytotoxicity, respectively, suggesting strong recall responses. Our findings and those of others also show that antibodies to SARS-CoV-2 persist 12 months PSO, but wane rapidly [1, 2, 20] and in the case of N-specific antibodies may be undetectable.
Overarchingly, we identified SARS-CoV-2–specific T-cell responses in 75.9% of study participants at 12 months PSO using peptide pools derived from SARS-CoV-2 structural proteins N, M, E, and S. Other studies have shown that epitopes in SARS-CoV-2N, M, and S, together with nsp3, nsp4, ORF3a are recognized by CD4 and CD8 T cells representing the majority of T-cell responses at convalescence, while SARS-CoV-2 E was less frequently recognized [12, 21, 22]. Although we did not map T-cell epitopes, overall we observed the same trend of T-cell responses to corresponding peptide pools, but at a much later time point. As the magnitude of CD8 T-cell responses contracts within a month of disease onset in humans vaccinated with live yellow fever virus and smallpox vaccines [23, 24], similarly in COVID-19 patients [25], SARS-CoV-2–specific CD8 T cells in the peripheral blood are of low frequency at 1 year PSO. Importantly, T -cell recognition of multiple epitopes was common in study participants.
Study participants who were hospitalized during acute infection demonstrated the highest frequency of SARS-CoV-2–specific CD4 T-cell responses and antibodies to SARS-CoV-2N, while in outpatients with mild or minimal symptoms these were statistically lower. Why severe COVID-19 is associated with a higher memory response for CD4 T cells remains unclear. Because severe disease is associated with higher viral burden [26–28], one hypothesis is that higher viral load at acute infection increases stimulation and antigen availability for T cells, expanding more durable responses. However, due to differences in timing of nasal swab acquisition and the size of this subcohort, we were unable to correlate T-cell longevity and viral load at acute infection. Published data have yet to show that initial SARS-CoV-2 viral load correlates with the longevity of cellular immunity. Indeed, the longevity of memory T cells is affected by multiple factors including the cytokines, such as IL-7 and IL-15 (reviewed in [29]), as well as the density of antigens presented by APCs, including B cells [30].
Importantly, our data show that SARS-CoV-2–specific CD4 T cells present 12 months postinfection exhibit differentiation toward central memory phenotypes, activation by expression of CD69 and PD-1, and polyfunctionality by expression of IFN-γ and IL-2. In other viral infections such as influenza, polyfunctional CD4 T cells expressing IFN-γ, IL-2, and tumor necrosis factor-α (TNF-α) are more frequent in convalescent patients who better controlled infection [31]. COVID-19 patients with milder symptoms also had more polyfunctional T cells expressing IFN-γ, IL-2, and TNF-α [32] while patients with severe and critical disease tended to have restricted functional T cells 1–2 months PSO [33]. CD4 T cells 1–2 months after SARS-CoV-2 infection also upregulated CD69, CD38, HLA-II, CTLA-4, and PD-1 as an indication of activation [33, 34]. While the polyfunctionality and activation of long-term memory CD4 T cells is less well characterized in other viral infections and could be impacted by factors such as age and antigen concentration [35–37], in our study, the SARS-CoV-2–specific CD4 T cells between severity study groups were similarly polyfunctional and equally expressed CD69 and PD-1 at 12 months PSO. Although CD8 T cells were less frequent compared to CD4 T cells at 12 months PSO, SARS-CoV-2–specific CD8 T cells exhibited cytotoxicity while CD4 T cells did not (data not shown). Memory CD4 T cells are, however, important in promoting CD8 T-cell cytotoxicity and viral clearance (reviewed in [38]).
SARS-CoV-2 shares epitopes with endemic hCoVs including HKU1, 229E, NL63, and OC43 [34, 39]. Our analysis was restricted to hCoV peptide pools composed of S from these viruses and we detected low frequencies of T-cell recognition of endemic hCoV that did not differ between patient severity groups. Analysis of additional structural and nonstructural protein-derived peptides in larger study cohorts may yield more information regarding the correlation of cross-reactive T-cell responses. Moreover, T cells can ostensibly provide broad coverage over SARS-CoV-2 variants of concern, whereas antibodies primarily target S [14]. Data have shown that associated mutations can develop and emerge in the receptor binding domain and other sites of the S glycoprotein that limit prior antibody binding [40–42]. However, due to the unique requirements of peptide presentation in the context of an HLA molecule, distinct epitope-HLA interactions between individuals presenting a diversity of unique viral mutational sites constraining the success of host evasion.
Limitations to our study include the challenge of obtaining biological samples at 12 months PSO prior to vaccination in our MHS cohort, which had timely and reliable access to vaccines. Fifty percent of individuals in our cohort were vaccinated prior to a 12 months draw (Table 1). For these individuals we did not include their S-specific T-cell or antibody responses, but we included their N, M, and E humoral or cellular responses because these are not components of the mRNA-based vaccines (the vaccines primarily available to our study population). A strength of our cohort is that study participants are followed within the MHS and their epidemiologic, clinical and COVID-19 testing records, and vaccination status are maintained primarily from electronic health record. In addition, sera were analyzed longitudinally to avoid reinfected participants for 12-month T-cell analysis.
In summary, our data show that SARS-CoV-2 humoral and cellular responses are measurable 12 months PSO in individuals across a spectrum of disease phenotypes. The magnitude of the CD4 T-cell and antibody responses more closely correlated with acute disease severity. Importantly, the memory phenotype and polyfunctional response of the SARS-CoV-2 specific T cells did not differ by disease severity. These findings indicate that SARS-CoV-2–specific T cells exhibit longevity and polyfunctionality at least 12 months postinfection and their use in therapeutics, immune testing, and vaccine design warrants further evaluation.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online. Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
{kind=link}
{kind=link}
{kind=link}
Notes
Acknowledgments. We thank Camille Estupigan for her editorial assistance in the preparation of this manuscript. We thank the members of the EPICC COVID-19 Cohort Study Group for their many contributions in conducting the study and ensuring effective protocol operations. The following members were all closely involved with the design, implementation, and oversight of the study. Brooke Army Medical Center, Fort Sam Houston, TX: Col J. Cowden; S. Deleon, LTC A. Markelz, K. Mende, T. Merritt, S. Merritt, R. Walter, CPT T. Wellington; Carl R. Darnall Army Medical Center, Fort Hood, TX: MAJ S. Bazan, P. Kay Love; Fort Belvoir Community Hospital, Fort Belvoir, VA: L. Brandon, N. Dimascio-Johnson, MAJ E. Ewers, LCDR K. Gallagher, LCDR D. Larson, MAJ M. Odom, A. Rutt; Henry M. Jackson Foundation, Inc., Bethesda, MD: D. Clark; Madigan Army Medical Center, Joint Base Lewis McChord, WA: S. Chambers, CPT C. Conlon, CPT K. Everson, COL P. Faestel, COL T. Ferguson, MAJ L. Gordon, LTC S. Grogan, CPT S. Lis, COL C. Mount, LTC D. Musfeldt, W. Robb-McGrath, MAJ R. Sainato, C. Schofield, COL C. Skinner, M. Stein, MAJ M. Switzer, MAJ M. Timlin, MAJ S. Wood; Naval Medical Center Portsmouth, Portsmouth, VA: G. Atwood, S. Banks, R. Carpenter, LCDR C. Eickhoff, CAPT K. Kronmann, T. Lalani, LCDR T. Lee, LCDR A. Smith, R. Tant, CDR T. Warkentien; Naval Medical Center San Diego, San Diego, CA: CAPT J. Arnold, CDR C. Berjohn, S. Cammarata, LCDR S. Husain, N. Kirkland, LCDR A. Lane, J. Parrish, CAPT (ret.) G. Utz; Tripler Army Medical Center, Honolulu, HI: S. Chi, MAJ E. Filan, K. Fong, CPT T. Horseman, MAJ M. Jones, COL A. Kanis, LTC A. Kayatani, MAJ W. Londeree, LTC C. Madar, MAJ J. Masel, MAJ M. McMahon, K. Miyasato, G. Murphy, COL V. Ngauy, MAJ E. Schoenman, C. Uyehara, LTC R. Villacorta Lyew; Uniformed Services University of the Health Sciences, Bethesda, MD: C. Byrne, COL (ret.) K. Chung, C. Coles, C. Fox, M. Grother, D. Gunasekera, COL P. Hickey, LTC J. Livezey, C. Morales, COL (ret.) T. Oliver, E. Parmelee, J. Rusiecki, M. Sanchez-Edwards, A. Scher; United States Air Force School of Aerospace Medicine, Dayton, OH: A. Fries; Walter Reed National Military Medical Center, Bethesda, MD: I. Barahona, D. Gunasekera, M. Oyeneyin; William Beaumont Army Medical Center, El Paso, TX: CPT M. Banda, CPT B. Davis, MAJ T. Hunter, CPT O. Ikpekpe-Magege, CPT S. Kemp, R. Mody, R. Resendez, P. Sandoval, COL M. Wiggins.
Disclaimer. E. L., M. S., C. D., A. S., R. M., D. L., C. C., C. L., D. T., T. B., and A. M. are service members or employees of the US Government. This work was prepared as part of their official duties. Title 17 U.S.C. §105 provides that “Copyright protection under this title is not available for any work of the United States Government.” Title 17 U.S.C. §101 defines a US Government work as a work prepared by a military service member or employee of the US Government as part of that person’s official duties. The contents of this publication are the sole responsibility of the authors and do not necessarily reflect the views, opinions, or policies of Uniformed Services University of the Health Sciences; National Institutes of Health or the Department of Health and Human Services; the Henry M. Jackson Foundation for the Advancement of Military Medicine, Inc; the Department of Defense; the US Army Medical Department; the US Army Office of the Surgeon General; the Departments of the Army, Navy, or Air Force; Brooke Army Medical Center; Walter Reed National Military Medical Center; Naval Medical Center San Diego; and Madigan Army Medical Center. Mention of trade names, commercial products, or organizations does not imply endorsement by the US Government. The investigators have adhered to the policies for protection of human subjects as prescribed in 45 CFR 46.
Financial support. This work was supported by the Defense Health Program (grant number HU00012020067) and the National Institute of Allergy and Infectious Diseases (grant number HU00011920111). The protocol was executed by the Infectious Disease Clinical Research Program, a Department of Defense program executed by the Uniformed Services University of the Health Sciences through a cooperative agreement by the Henry M. Jackson Foundation for the Advancement of Military Medicine, Inc. This project has been funded in part by the National Institute of Allergy and Infectious Diseases at the National Institutes of Health, under an interagency agreement (Y1-AI-5072).
Potential conflicts of interest. S. D. P., T. H. B., D. T., J. S. R., and M. P. S. report that the Uniformed Services University of the Health Sciences (USU) Infectious Diseases Clinical Research Program (IDCRP), a US Department of Defense institution, and the Henry M. Jackson Foundation for the Advancement of Military Medicine, Inc. (HJF) were funded under a Cooperative Research and Development Agreement to conduct an unrelated phase III COVID-19 monoclonal antibody immunoprophylaxis trial sponsored by AstraZeneca. The HJF, in support of the USU IDCRP, was funded by the Department of Defense Joint Program Executive Office for Chemical, Biological, Radiological, and Nuclear Defense to augment the conduct of an unrelated phase III vaccine trial sponsored by AstraZeneca. Both of these trials were part of the US Government COVID-19 response. Neither is related to the work presented here. All other authors report no potential conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
Contributor Information
EPICC COVID-19 Cohort Study Group:
J Cowden, S Deleon, A Markelz, K Mende, T Merritt, S Merritt, R Walter, T Wellington, S Bazan, P Kay, L Brandon, N Dimascio-Johnson, E Ewers, K Gallagher, D Larson, M Odom, A Rutt, D Clark, S Chambers, C Conlon, K Everson, P Faestel, T Ferguson, L Gordon, S Grogan, S Lis, C Mount, D Musfeldt, W Robb-McGrath, R Sainato, C Schofield, C Skinner, M Stein, M Switzer, M Timlin, S Wood, G Atwood, S Banks, R Carpenter, C Eickhoff, K Kronmann, T Lalani, T Lee, A Smith, R Tant, T Warkentien, J Arnold, C Berjohn, S Cammarata, S Husain, N Kirkland, A Lane, J Parrish, G Utz, S Chi, E Filan, K Fong, T Horseman, M Jones, A Kanis, A Kayatani, W Londeree, C Madar, J Masel, M McMahon, K Miyasato, G Murphy, V Ngauy, E Schoenman, C Uyehara, R Villacorta Lyew, C Byrne, K Chung, C Coles, C Fox, M Grother, D Gunasekera, P Hickey, J Livezey, C Morales, T Oliver, E Parmelee, J Rusiecki, M Sanchez-Edwards, A Scher, A Fries, I Barahona, D Gunasekera, M Oyeneyin, M Banda, B Davis, T Hunter, O Ikpekpe-Magege, S Kemp, R Mody, R Resendez, P Sandoval, and M Wiggins
References
- 1. De Giorgi V, West KA, Henning AN, et al. Naturally acquired SARS-CoV-2 immunity persists for up to 11 months following infection. J Infect Dis 2021; 224:1294–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Laing ED, Sterling SL, Richard SA, et al. Antigen-based multiplex strategies to discriminate SARS-CoV-2 natural and vaccine induced immunity from seasonal human coronavirus humoral responses. medRxiv, doi: 10.1101/2021.02.10.21251518, 12 February 2021, preprint: not peer reviewed. [DOI] [Google Scholar]
- 3. Kang CK, Kim M, Lee S, et al. Longitudinal analysis of human memory T-cell response according to the severity of illness up to 8 months after severe acute respiratory syndrome coronavirus 2 infection. J Infect Dis 2021; 224:39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jung JH, Rha MS, Sa M, et al. SARS-CoV-2-specific T cell memory is sustained in COVID-19 convalescent patients for 10 months with successful development of stem cell-like memory T cells. Nat Commun 2021; 12:4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Le Bert N, Tan AT, Kunasegaran K, et al. SARS-CoV-2-specific T cell immunity in cases of COVID-19 and SARS, and uninfected controls. Nature 2020; 584:457–62. [DOI] [PubMed] [Google Scholar]
- 6. Dan JM, Mateus J, Kato Y, et al. Immunological memory to SARS-CoV-2 assessed for up to 8 months after infection. Science 2021; 371:eabf4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rydyznski Moderbacher C, Ramirez SI, Dan JM, et al. Antigen-specific adaptive immunity to SARS-CoV-2 in acute COVID-19 and associations with age and disease severity. Cell 2020; 183:996–1012.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhou R, To KK, Wong YC, et al. Acute SARS-CoV-2 infection impairs dendritic cell and T cell responses. Immunity 2020; 53:864–77.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Peng Y, Mentzer AJ, Liu G, et al. ; Oxford Immunology Network Covid-19 Response T Cell Consortium; ISARIC4C Investigators. Broad and strong memory CD4+ and CD8+ T cells induced by SARS-CoV-2 in UK convalescent individuals following COVID-19. Nat Immunol 2020; 21:1336–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pérez-Martínez A, Mora-Rillo M, Ferreras C, et al. Phase I dose-escalation single centre clinical trial to evaluate the safety of infusion of memory T cells as adoptive therapy in COVID-19 (RELEASE). EClinicalMedicine 2021; 39:101086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Saini SK, Hersby DS, Tamhane T, et al. SARS-CoV-2 genome-wide T cell epitope mapping reveals immunodominance and substantial CD8+ T cell activation in COVID-19 patients. Sci Immunol 2021; 6:eabf7550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tarke A, Sidney J, Kidd CK, et al. Comprehensive analysis of T cell immunodominance and immunoprevalence of SARS-CoV-2 epitopes in COVID-19 cases. Cell Rep Med 2021; 2:100204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mateus J, Grifoni A, Tarke A, et al. Selective and cross-reactive SARS-CoV-2 T cell epitopes in unexposed humans. Science 2020; 370:89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Geers D, Shamier MC, Bogers S, et al. SARS-CoV-2 variants of concern partially escape humoral but not T-cell responses in COVID-19 convalescent donors and vaccinees. Sci Immunol 2021; 6:eabj1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Elyanow R, Snyder TM, Dalai SC, et al. T-cell receptor sequencing identifies prior SARS-CoV-2 infection and correlates with neutralizing antibody titers and disease severity. medRxiv, doi: 10.1101/2021.03.19.21251426, 22 March 2021, preprint: not peer reviewed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Amara RR, Nigam P, Sharma S, Liu J, Bostik V.. Long-lived poxvirus immunity, robust CD4 help, and better persistence of CD4 than CD8 T cells. J Virol 2004; 78:3811–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lalani T, Lee TK, Laing ED, et al. SARS-CoV-2 infections and serologic responses among military personnel deployed on the USNS COMFORT to New York city during the COVID-19 pandemic. Open Forum Infect Dis 2021; 8:ofaa654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sallusto F, Lenig D, Förster R, Lipp M, Lanzavecchia A.. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 1999; 401:708–12. [DOI] [PubMed] [Google Scholar]
- 19. Tomiyama H, Matsuda T, Takiguchi M.. Differentiation of human CD8+ T cells from a memory to memory/effector phenotype. J Immunol 2002; 168:5538–50. [DOI] [PubMed] [Google Scholar]
- 20. Xiang T, Liang B, Fang Y, et al. Declining levels of neutralizing antibodies against SARS-CoV-2 in convalescent COVID-19 patients one year post symptom onset. Front Immunol 2021; 12:708523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nelde A, Bilich T, Heitmann JS, et al. SARS-CoV-2-derived peptides define heterologous and COVID-19-induced T cell recognition. Nat Immunol 2021; 22:74–85. [DOI] [PubMed] [Google Scholar]
- 22. Grifoni A, Weiskopf D, Ramirez SI, et al. Targets of T cell responses to SARS-CoV-2 coronavirus in humans with COVID-19 disease and unexposed individuals. Cell 2020; 181:1489–501.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Akondy RS, Johnson PL, Nakaya HI, et al. Initial viral load determines the magnitude of the human CD8 T cell response to yellow fever vaccination. Proc Natl Acad Sci U S A 2015; 112:3050–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Miller JD, van der Most RG, Akondy RS, et al. Human effector and memory CD8+ T cell responses to smallpox and yellow fever vaccines. Immunity 2008; 28:710–22. [DOI] [PubMed] [Google Scholar]
- 25. Schulien I, Kemming J, Oberhardt V, et al. Characterization of pre-existing and induced SARS-CoV-2-specific CD8+ T cells. Nat Med 2021; 27:78–85. [DOI] [PubMed] [Google Scholar]
- 26. Zheng S, Fan J, Yu F, et al. Viral load dynamics and disease severity in patients infected with SARS-CoV-2 in Zhejiang province, China, January-March 2020: retrospective cohort study. BMJ 2020; 369:m1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. To KK, Tsang OT, Leung WS, et al. Temporal profiles of viral load in posterior oropharyngeal saliva samples and serum antibody responses during infection by SARS-CoV-2: an observational cohort study. Lancet Infect Dis 2020; 20:565–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pujadas E, Chaudhry F, McBride R, et al. SARS-CoV-2 viral load predicts COVID-19 mortality. Lancet Respir Med 2020; 8:e70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Surh CD, Sprent J.. Homeostasis of naive and memory T cells. Immunity 2008; 29:848–62. [DOI] [PubMed] [Google Scholar]
- 30. Welsh RA, Song N, Sadegh-Nasseri S.. How does B cell antigen presentation affect memory CD4 T cell differentiation and longevity? Front Immunol 2021; 12:677036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pizzolla A, Nguyen TH, Sant S, et al. Influenza-specific lung-resident memory T cells are proliferative and polyfunctional and maintain diverse TCR profiles. J Clin Invest 2018; 128:721–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sekine T, Perez-Potti A, Rivera-Ballesteros O, et al. Robust T cell immunity in convalescent individuals with asymptomatic or mild COVID-19. Cell 2020; 183:158–68.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schub D, Klemis V, Schneitler S, et al. High levels of SARS-CoV-2-specific T cells with restricted functionality in severe courses of COVID-19. JCI Insight 2020; 5:e142167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Braun J, Loyal L, Frentsch M, et al. SARS-CoV-2-reactive T cells in healthy donors and patients with COVID-19. Nature 2020; 587:270–4. [DOI] [PubMed] [Google Scholar]
- 35. Rudolph ME, McArthur MA, Barnes RS, Magder LS, Chen WH, Sztein MB.. Differences between pediatric and adult T cell responses to in vitro staphylococcal enterotoxin B stimulation. Front Immunol 2018; 9:498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chiu YL, Shan L, Huang H, et al. Sprouty-2 regulates HIV-specific T cell polyfunctionality. J Clin Invest 2014; 124:198–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. van Haren SD, Ganapathi L, Bergelson I, et al. In vitro cytokine induction by TLR-activating vaccine adjuvants in human blood varies by age and adjuvant. Cytokine 2016; 83:99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Swain SL, McKinstry KK, Strutt TM.. Expanding roles for CD4+ T cells in immunity to viruses. Nat Rev Immunol 2012; 12:136–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Woldemeskel BA, Kwaa AK, Garliss CC, Laeyendecker O, Ray SC, Blankson JN.. Healthy donor T cell responses to common cold coronaviruses and SARS-CoV-2. J Clin Invest 2020; 130:6631–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ou J, Zhou Z, Dai R, et al. V367F mutation in SARS-CoV-2 spike RBD emerging during the early transmission phase enhances viral infectivity through increased human ACE2 receptor binding affinity. J Virol 2021; 95:e0061721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jangra S, Ye C, Rathnasinghe R, et al. ; Personalized Virology Initiative Study Group. SARS-CoV-2 spike E484K mutation reduces antibody neutralisation. Lancet Microbe 2021; 2:e283–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gobeil SM, Janowska K, McDowell S, et al. Effect of natural mutations of SARS-CoV-2 on spike structure, conformation, and antigenicity. Science 2021; 373:eabi6226. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.