

Abstract
Hypertension frequently coexists with diabetes and the cardiometabolic syndrome. β‐Blockers have been a mainstay for controlling blood pressure for nearly 4 decades. However, β‐blockers are perceived to cause glucose and lipid metabolism dysregulation, including hypoglycemia masking, reduced glycemic control, insulin resistance, and dyslipidemia. It should be noted, however, that β‐blockers are diverse in their effects on glucose and lipid metabolism. Potential mechanisms that contribute to these metabolic effects include hemodynamic differences, anti‐inflammatory and anti‐oxidative pathways, and/or weight changes. Traditional β‐blockers decrease cardiac output while peripheral vascular resistance increases or remains unchanged, which may result in glucose and lipid abnormalities. In contrast, vasodilating β‐blockers reduce peripheral vascular resistance but have little effect on cardiac output. Vasodilating β‐blockers may therefore result in less impact on insulin sensitivity and glycemic control, a reduced new‐onset diabetes risk, and improved dyslipidemia compared with traditional β‐blockers. Because of these effects, vasodilating β‐blockers may represent a favorable option in the treatment of high‐risk patients with hypertension. J Clin Hypertens (Greenwich). 2011;13:52–59. ©2010 Wiley Periodicals, Inc.
Hypertension frequently coexists with diabetes and the cardiometabolic syndrome, conditions that are characterized by metabolic abnormalities including hyperglycemia, insulin resistance (IR), dyslipidemia, and abdominal obesity. 1 Patients with diabetes are 2 to 4 times more likely to develop cardiovascular (CV) disease compared with patients without diabetes. 2 Similarly, patients with the cardiometabolic syndrome have a 5‐ to 9‐fold increased risk for diabetes and a 2‐ to 4‐fold increased risk for CV disease. 3 , 4 , 5 Analysis of the Treating to New Targets (TNT) study revealed that patients with the cardiometabolic syndrome and coronary heart disease had a significantly higher incidence of major CV events compared with patients without the cardiometabolic syndrome. 6
Blood pressure (BP) control is the most effective means of reducing CV risk in patients with diabetes and comorbid hypertension. The United Kingdom Prospective Diabetes Study (UKPDS) demonstrated that aggressive BP control in patients with hypertension and diabetes resulted in clinically meaningful reductions in the risk of diabetes‐related deaths and complications and the progression of diabetic retinopathy. 7 Various agents may be used to control elevated BP, including angiotensin‐converting enzyme inhibitors, angiotensin receptor blockers, α1 inhibitors, β‐blockers, calcium channel blockers, and thiazide diuretics. 1
Individual agents within the β‐blocker class differ in terms of their effects on glucose and lipid metabolism. From a metabolic standpoint, reduction in cardiac output by traditional β‐blockers decreases glucose delivery to peripheral tissues, which may lead to detrimental effects on glucose and lipid metabolism. 8 Vasodilating β‐blockers reduce peripheral vascular resistance and are associated with neutral effects on glucose and lipid profiles. 9 This review will discuss the pathophysiologic relationship between IR, dyslipidemia, and hypertension and the differences between traditional and vasodilating β‐blockers regarding their effects on glucose and lipid metabolism.
Pathophysiologic Relationship Between Hypertension, IR, and Dyslipidemia
Insulin is a pleiotropic hormone that plays a pivotal role in the development of hypertension, diabetes, and the cardiometabolic syndrome. The main metabolic actions of insulin are to stimulate glucose uptake in skeletal muscle and heart and to suppress the production of glucose and very low‐density lipoprotein (VLDL) in the liver. 10 Under fasting conditions, insulin secretion is suppressed, leading to increased glucose synthesis in the liver and kidneys (gluconeogenesis) and increased conversion of glycogen to glucose in the liver (glycogenolysis). After eating, insulin is released from pancreatic β‐cells and inhibits gluconeogenesis and glycogenolysis. Insulin stimulates the sympathetic nervous system (SNS) to increase cardiac output and the delivery and utilization of glucose in the peripheral tissues.
Insulin Resistance
IR denotes an impaired response to insulin in skeletal muscle, liver, adipose, and CV tissue. IR arises as a result of various genetic, acquired, and environmental factors, including the presence of obesity, especially central obesity (Figure 1). 11 Renin‐angiotensin‐aldosterone system increased activation may also contribute to the development of IR via the stimulation of angiotensin II type 1 receptors, which trigger increased production reactive oxygen species (ROS) in adipocytes, skeletal muscle, and CV tissue of obese individuals. 12 , 13 Excess free fatty acids (FFAs) may further exacerbate ROS generation. 12 The resulting increase in oxidative stress induces a shift toward endothelial dysfunction and atherogenesis 12 (Figure 1). 11 Additionally, inflammation has been linked to IR development and diabetes pathogenesis. 14
Figure 1.
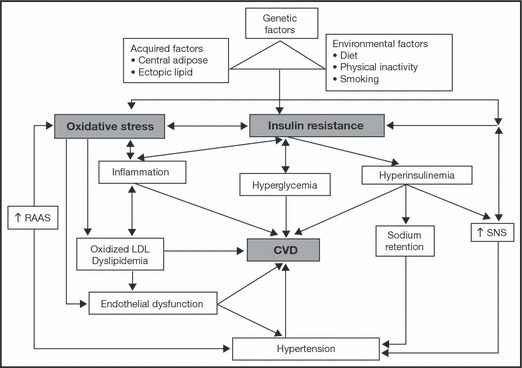
Relationship between insulin resistance, dyslipidemia, and hypertension. CVD indicates cardiovascular disease; LDL, low‐density lipoprotein; RAAS, renin‐angiotensin‐aldosterone system; SNS, sympathetic nervous system. Reproduced with permission from Stump and colleagues. 11
The cellular consequences of IR are complex and not entirely understood. IR is associated with abnormalities in key components of insulin‐signaling pathways, including phosphatase overexpression and downregulation of protein kinase cascades. 15 Impaired insulin signaling may result in abnormalities in the expression and action of various cytokines, growth factors, and peptides. 16 In addition, impaired insulin signaling stimulates VLDL overproduction, which promotes the dyslipidemic profile commonly observed in the insulin‐resistant state. 15 IR may also result in impaired fibrinolysis, which is characterized by elevated levels of fibrinogen and plasminogen activator inhibitor type 1 (an inhibitor of fibrinolysis) and hypercoagulability. 17 , 18 Impaired fibrinolysis may contribute to increased thrombosis and CV event risk. 16
Systemic Consequences of IR
Dyslipidemia. Dyslipidemia is one of the most common IR complications and is characterized by elevated levels of low‐density lipoprotein cholesterol (LDL‐C) and triglycerides and reduced high‐density lipoprotein cholesterol (HDL‐C) levels. 15 These lipid abnormalities arise as a result of excess FFAs, which trigger increased triglyceride production in the liver. 19 Hypertriglyceridemia reduces HDL‐C levels and promotes the formation of small, dense LDL‐C particles from VLDL, which are more atherogenic than their larger, more buoyant counterparts because of an increased ability to cross the endothelium and a susceptibility to oxidation. 19 Dyslipidemia results in a highly atherogenic profile that increases CV disease risk. 15
Hypertension. Various mechanisms have been proposed to explain how IR contributes to hypertension development. Vasoconstriction may be exacerbated by reduced endothelium‐derived nitrous/nitric oxide (NO) production that occurs in the insulin‐resistant state. Activation of the SNS and reabsorption of sodium by the kidney is also increased in insulin‐resistant patients. 19 , 20 Oxidized LDL‐C accumulation in the arterial wall may result in decreased arterial elasticity and increased peripheral vascular resistance. 21 Increased oxidized LDL‐C production may be related to hypertension through sympathetic activation and decreased endothelial‐dependent NO production.
Cardiometabolic Syndrome. Numerous effects related to IR have been observed in patients with the cardiometabolic syndrome, including hyperglycemia, hyperinsulinemia, and dyslipidemia. 19 Oxidative stress, which is increased in the cardiometabolic syndrome, may contribute to alterations in adipose‐derived cytokine secretion such as adiponectin. 22 Low adiponectin levels decrease VLDL catabolism and increase HDL‐C catabolism, resulting in elevated FFAs secretion from adipose tissue. 22 Proinflammatory cytokines such as tumor necrosis factor α may also contribute to the development of the cardiometabolic syndrome by increasing plasminogen activator inhibitor type 1 levels, which initiate and sustain the low inflammatory states that characterize hypertension and atherosclerosis progression. 22
Target Organ Damage. Hyperglycemia and the compensatory hyperinsulinemia required to maintain glucose homeostasis in insulin‐resistant patients cause target organ damage. Hyperglycemia increases ROS production and advanced glycation end products. 23 Increased oxidative stress suppresses endothelium‐derived vasodilation by converting NO into peroxynitrite. In addition, advanced glycation end products promote the development of inflammation and induce vascular cell adhesion molecule production, which enhances the interaction between the vascular endothelium and circulating monocytes. 24 Endothelial dysfunction, in the form of increased ROS production and decreased NO bioavailability, may contribute to the development of atherosclerosis. 25
Effect of β‐Blockers on IR and Dyslipidemia
β‐Blockers are recommended among the first‐line treatments for hypertension in patients with compelling indications, including heart failure, high coronary disease risk, diabetes, and following myocardial infarction. 1 This recommendation is based on numerous clinical studies that demonstrated β‐blockers lower elevated BP and reduce CV morbidity and mortality in patients with heart failure and in patients with a prior myocardial infarction. 26 β‐Blockers reduce BP by inhibiting SNS activity 26 ; however, individual agents within this class differ in terms of their mechanism of action and physiologic effects. Traditional β‐blockers include agents that inhibit both β1‐ and β2‐adrenergic receptors (eg, propranolol) or agents that specifically inhibit β1‐adrenergic receptors (eg, atenolol and metoprolol). Traditional β‐blockers reduce BP via a reduction in cardiac output, whereas peripheral vascular resistance is maintained or increased. 8 Administration of traditional β‐blockers may be associated with unfavorable side effects (eg, depression, fatigue, sexual dysfunction, and cold extremities) 27 and loss of glycemic control and dyslipidemia. In contrast, vasodilating β‐blockers (nebivolol, labetalol, and carvedilol) lower BP with a reduction of peripheral vascular resistance but have little or no effect on cardiac output. 9 Consequently, vasodilating β‐blockers can increase peripheral blood flow, which may result in improved tolerability and metabolic profiles compared with traditional β‐blockers. 28
Traditional β‐Blockers
Effects on Glucose and Lipid Metabolism. Traditional β‐blockers may reduce insulin sensitivity, lessen glycemic control, and increase the risk of new‐onset diabetes. For example, metoprolol decreased insulin sensitivity by 14% from baseline in 72 nondiabetic patients with hypertension after 12 weeks of treatment. 29 Additionally, metoprolol was associated with increased triglyceride levels and decreased HDL‐C levels. In another study that included 42 patients with hypertension, metoprolol increased glycosylated hemoglobin (HbA1c) levels by 5% from baseline after 6 months of treatment (P=.04). 30 This finding is clinically meaningful because the Norfolk Cohort of the European Prospective Investigation Into Cancer and Nutrition (EPIC‐Norfolk; n=10,232, of whom 243 had diabetes) demonstrated that a 1% HbA1c increase was associated with a 26% increase in mortality risk (P<.02) when patients with diabetes or known history of CV disease were excluded. 31
The International Verapamil–Trandolapril Study (INVEST) of 22,576 patients with hypertension and coronary artery disease demonstrated that long‐term therapy with atenolol was associated with a 15% higher risk of new‐onset diabetes compared with verapamil. 32 In the Losartan Intervention For Endpoint (LIFE) study, the risk of developing diabetes was 25% higher among patients with hypertension and left ventricular hypertrophy who received atenolol‐based therapy compared with patients who received losartan‐based therapy (N=9193). 33 A substudy of the Atherosclerosis Risk in Communities (ARIC) study (3804 patients with hypertension; 6‐year follow‐up) demonstrated that, compared with no medication, patients receiving traditional β‐blockers had a 28% higher risk of developing diabetes. 34
Potential Mechanisms Contributing to the Metabolic Effects of Traditional β‐Blockers. Various mechanisms have been proposed to explain the negative effects of traditional β‐blockers on glucose and lipid metabolism. Traditional β‐blockers produce unopposed α1‐adrenergic receptor activity, which may induce vasoconstriction, decrease skeletal blood flow, and reduce insulin‐stimulated peripheral glucose uptake. 35 Additionally, traditional β‐blockers—atenolol in particular—may not improve oxidative stress and NO production, which are involved in the structural integrity and endothelial function of arteries. 36 Traditional β‐blockers may also inhibit insulin secretion from pancreatic β‐cells. 37 Weight gain, which has been reported in patients treated with traditional β‐blockers, is closely linked to reduced insulin sensitivity. 38 , 39
Vasodilating β‐Blockers
Effects on Glucose and Lipid Metabolism. Evidence suggests that nebivolol, a selective β1‐blocker, mediates vasodilation via the stimulation of endothelium‐derived NO release. 40 In patients with hypertension, nebivolol has been associated with improvements in glycemic control and dyslipidemia. Among 30 patients with hypertension and hyperlipidemia, atenolol 50 mg daily raised lipoprotein(a) levels by 30% (P=.028 vs baseline) and triglyceride levels by 19% after 24 weeks (P=.05 vs baseline), whereas nebivolol 5 mg daily produced no significant changes in either parameter. 41 While insulin level reduction by nebivolol was not significant (10%), the Homeostasis Model Assessment (HOMA) index was reduced by 20% (P=.05). 41 In another study among 72 patients with hypertension, nebivolol 5 mg daily significantly reduced baseline insulin levels (P=.001) and the HOMA of IR (HOMA‐IR) (P=.003) compared with metoprolol 100 mg daily after 6 months of treatment. 40 , 42 In 2838 patients with hypertension and diabetes, nebivolol 2.5 mg, 5.0 mg, or 10 mg daily (monotherapy or as add‐on therapy for 3 months) was significantly associated with decreased fasting glucose, HbA1c, total cholesterol, LDL‐C, and triglyceride levels and increased HDL‐C levels compared with baseline (Table I). 43
Table I.
Metabolic Parameters of Patients Treated With Nebivolol (N=2838)
| mean change from baseline (sd) | p value | |
|---|---|---|
| Fasting glucose, mg/dL | −13.1 (27.0) | <.001 |
| HbA1c, % | −0.25 (0.59) | <.001 |
| Serum cholesterol, mg/dL | ||
| Total | −16.3 (31.3) | <.001 |
| LDL | −13.3 (27.5) | <.001 |
| HDL | 2.4 (18.0) | <.001 |
| Triglycerides, mg/dL | −24.1 (75.4) | <.001 |
Abbreviations: HbA1c, glycosylated hemoglobin; HDL, high‐density lipoprotein; LDL, low‐density lipoprotein; SD, standard deviation. Adapted with permission from Schmidt and colleagues. 43
Labetalol, a nonselective β‐blocker with α1‐adrenergic receptor–blocking activity, is effective in the long‐term management of mild, moderate, and severe hypertension and during hypertensive emergencies. 44 Although the effect of labetalol on glucose and lipid metabolism in hypertension has not been extensively studied, two small studies suggest that labetalol treatment is associated with neutral effects on glucose and lipid profiles. 45 , 46
Carvedilol is a nonselective β‐blocker whose vasodilating activity has been attributed to α1‐adrenergic receptor blockade. 28 A number of recent studies have reported that carvedilol exerts neutral effects on glucose and lipid metabolism. The Glycemic Effects in Diabetes Mellitus: Carvedilol–Metoprolol Comparison in Hypertensives (GEMINI) study of 1235 patients with hypertension and diabetes demonstrated that carvedilol 6.25 mg to 25 mg twice daily did not appreciably affect HbA1c levels vs baseline (0.02%; P=.65), whereas metoprolol tartrate 50 mg to 200 mg twice daily significantly increased HbA1c levels vs baseline (0.15%; P<.001) after 35 weeks of treatment. 47 Carvedilol resulted in significant improvement in insulin sensitivity (HOMA‐IR) compared with metoprolol tartrate (−9.1% vs −2.0%; P=.004), as well as larger decreases in total serum cholesterol levels and smaller increases in triglyceride levels (P≤.001; Table II). 47 Fewer patients discontinued treatment because of deteriorating glycemic control with carvedilol (0.6% vs 2.2% with metoprolol tartrate; P=.04). Of note, in the GEMINI study, carvedilol‐treated patients did not experience weight gain from baseline (0.17±0.19 kg; P=.36), whereas significant weight gain from baseline occurred among metoprolol‐treated patients (1.2±0.16 kg; P<.001). 48 Carvedilol‐treated patients were also more likely to experience no weight change (44% vs 35%; P=.005) and less likely to experience a weight gain >7% (1.1% vs 4.5%; P=.006) compared with metoprolol‐treated patients. 48
Table II.
Metabolic Parameters of Patients Treated With Carvedilol or Metoprolol Tartrate in the GEMINI Study
| Parameter | carvedilol (n=454) | metoprolol (n=657) | between treatmentsa | |
|---|---|---|---|---|
| difference from baseline, % | difference from baseline, % | difference, % (95% cl) | P Value | |
| Mean ACRb | −14.0 | 2.5 | −16.2 (−25.3 to −5.9) | .003 |
| Mean HOMA‐IRb | −9.1 | −2.0 | −7.2 (−13.8 to −0.2) | .004 |
| Mean plasma glucosec | 6.6 | 10.6 | −4.0 (−8.7 to 0.8) | .10 |
| Mean serum insulinc | −19.4 | −15.1 | −4.2 (−16.7 to 8.2) | .51 |
| Mean body weightc | 0.17 | 1.2 | −1.0 (−1.4 to −0.6) | <.001 |
| Mean serum cholesterolb | ||||
| Total | −3.3 | −0.4 | −2.9 (−4.6 to −1.2) | .001 |
| LDL | −4.0 | −2.7 | −1.3 (−4.31 to 1.78) | .40 |
| HDL | −5.5 | −5.7 | 0.2 (−1.68 to 2.12) | .83 |
| Mean triglyceridesb | 2.2 | 13.2 | −9.8 (−13.7 to −5.8) | <.001 |
Abbreviations: ACR, urinary albumin/creatinine ratio; CI, confidence interval; GEMINI, Glycemic Effects in Diabetes Mellitus: Carvedilol–Metoprolol Comparison in Hypertensives; HDL, high‐density lipoprotein; HOMA‐IR, Homeostatic Model Assessment of Insulin Resistance ([fasting plasma insulin concentration (μU/mL)×fasting plasma glucose]/22.5); LDL, low‐density lipoprotein. aTreatment difference from metoprolol. bGeometric means based on exponentiation of the least‐squares means adjusted by the analysis model of natural log‐transformed parameter. cLeast‐squares mean adjusted by the terms in the analysis model. Adapted with permission from Bakris and colleagues. 47
Carvedilol was also associated with beneficial effects on lipid parameters in 77 patients with the cardiometabolic syndrome. 49 Atenolol‐ and doxazosin‐treated patients (50 mg and 2 mg daily, respectively) experienced greater reductions in HDL‐C levels (−8.0% and −5.6%, respectively) compared with carvedilol‐treated patients (−0.1%; P<.05). In a recently reported trial involving 568 patients with hypertension and normal lipid profiles or mild dyslipidemia, patients treated with extended‐release metoprolol 50 mg to 200 mg daily had greater increases from baseline in triglyceride levels (10.39%) compared with patients treated with extended‐release carvedilol 20 mg to 80 mg daily (2.65%; P=.014 between‐treatment comparison) after 6 months of treatment. 50 Changes from baseline in HDL‐C levels were similar between the treatment groups (−5.1% vs −4.4% for carvedilol; P=.607). Subgroup analyses among patients with higher CV risks such as low HDL‐C levels, body mass index ≥30 kg/m2, or the metabolic syndrome showed improvements in triglyceride level changes with extended‐release carvedilol compared with extended‐release metoprolol (Figure 2A). 50 Improvements in insulin and C‐peptide level changes were also reported with extended‐release carvedilol compared with extended‐release metoprolol. Comparable results from the overall population were observed for HDL‐C levels in the subgroup analyses (Figure 2B). 50
Figure 2.
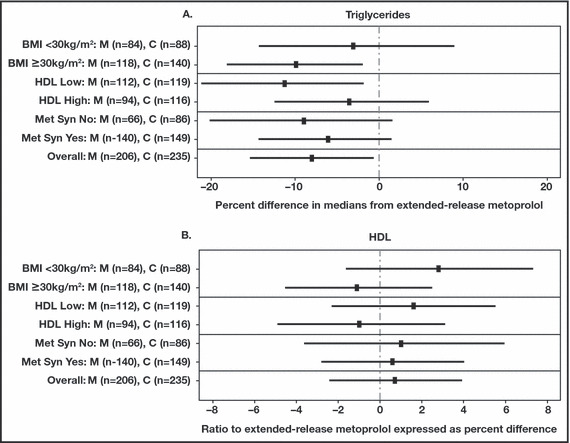
Comparison of extended‐release carvedilol (C) and extended‐release metoprolol (M) median change from baseline to treatment end for (A) triglycerides and (B) high‐density lipoprotein (HDL) in patient subgroups. BMI indicates body mass index; Met Syn, metabolic syndrome. Confidence interval overall is 97.5%. Reprinted from Fonarow and colleagues.50
Potential Mechanisms Contributing to the Metabolic Effects of Vasodilating β‐Blockers. Various mechanisms have been proposed to account for the favorable effects of vasodilating β‐blockers on glucose and lipid metabolism, including α1‐adrenergic receptor blockade and vasodilation, anti‐inflammatory activity, reduced oxidative stress, and lack of weight gain. Carvedilol and labetalol prevent norepinephrine binding to α1‐adrenegric receptors, which decreases peripheral vascular resistance and increases peripheral blood flow and glucose uptake. 26 , 28 Carvedilol and nebivolol also promote endothelial‐dependent vasodilation via enhanced NO synthesis. 40 , 51
Carvedilol possesses antioxidant properties, including the ability to scavenge free oxygen radicals, suppress free radical generation, and prevent ferric ion‐induced oxidation. 52 The antioxidant activity of carvedilol may also be related to stimulation of endothelial NO production or a reduction in NO inactivation. 53 Antioxidant activity is not limited to carvedilol, as nebivolol also decreases oxidative stress via a reduction in ROS generation and NO inactivation. 54
Vasodilating β‐blockers have been shown to reduce pro‐inflammatory mediators. Nebivolol administration to patients with hypertension and dyslipidemia is associated with reduced levels of C‐reactive protein. 41 Carvedilol administration to patients with hypertension and diabetes is associated with reductions in C‐reactive protein and monocyte chemotactic protein‐1. 52
Metabolic Effects of Vasodilating β‐Blockers and the CV Disease Management Guidelines. The American Association of Clinical Endocrinologists (AACE) guidelines recognize that β‐blockers such as carvedilol and nebivolol are suitable for the treatment of diabetes because of their ability to induce vasodilation and increase insulin sensitivity. 55 The European Society of Hypertension/European Society of Cardiology (ESH/ESC) guidelines note that vasodilating β‐blockers have no appreciable effects on insulin sensitivity, lipid profiles, or body weight and are less likely to result in new‐onset diabetes compared with traditional β‐blockers. 56 Although the National Institute for Health and Clinical Excellence (NICE) guidelines no longer recommend β‐blockers as first‐step therapy, it is noted that clinical outcome data are derived primarily from atenolol, and it is unclear whether the conclusions can be generalized to all β‐blockers. 57
Conclusions
Hypertension, diabetes, and the cardiometabolic syndrome frequently coexist, exerting a cumulative effect on CV disease risk. Although the pathophysiologic relationship between these disease entities is not entirely understood, IR and dyslipidemia appear to play a pivotal role in their development. Current guidelines recommend various agents such as β‐blockers to lower elevated BP and reduce the risk of CV morbidity and mortality.
β‐Blockers are among the most diverse class of antihypertensive agents, differing in terms of their mechanism of action and their effects on glucose and lipid metabolism. Traditional β‐blockers reduce BP via a reduction in cardiac output but maintain or increase peripheral vascular resistance. Furthermore, atenolol showed no improvement in endothelial function through anti‐oxidative activities or NO production. These agents are associated with the development of IR and dyslipidemia. In contrast, vasodilating β‐blockers reduce peripheral vascular resistance, have little or no effect on cardiac output, and improve endothelial function through anti‐oxidative properties and/or NO production. Vasodilating β‐blockers do not adversely affect insulin sensitivity, glycemic control, or lipid profiles. Moreover, use of vasodilating β‐blockers may reduce the risk of new‐onset diabetes compared with traditional β‐blockers. Because of the increased risk of CV disease in high‐risk patients with hypertension, the beneficial metabolic effects of vasodilating β‐blockers beyond BP control should be considered when selecting antihypertensive therapy.
Acknowledgments and disclosures: I thank Ann Marie Fitzmaurice, PhD, ProEd Communications, Inc, for her medical editorial assistance, for which funding was provided by GlaxoSmithKline, Research Triangle Park, North Carolina. I am on the speakers’ bureau for Novartis, GlaxoSmithKline, and Forrest Laboratories.
References
- 1. Chobanian AV, Bakris GL, Black HR, et al. Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Hypertension. 2003;42:1206–1252. [DOI] [PubMed] [Google Scholar]
- 2. Fagan TC, Sowers J. Type 2 diabetes mellitus: greater cardiovascular risks and greater benefits of therapy. Arch Intern Med. 1999;159:1033–1034. [DOI] [PubMed] [Google Scholar]
- 3. Lakka HM, Laaksonen DE, Lakka TA, et al. The metabolic syndrome and total and cardiovascular disease mortality in middle‐aged men. JAMA. 2002;288:2709–2716. [DOI] [PubMed] [Google Scholar]
- 4. Laaksonen DE, Lakka H‐M, Niskanen LK, et al. Metabolic syndrome and development of diabetes mellitus: application and validation of recently suggested definitions of the metabolic syndrome in a prospective cohort study. Am J Epidemiol. 2002;156:1070–1077. [DOI] [PubMed] [Google Scholar]
- 5. Wilson PW, D’Agostino RB, Parise H, et al. Metabolic syndrome as a precursor of cardiovascular disease and type 2 diabetes mellitus. Circulation. 2005;112:3066–3072. [DOI] [PubMed] [Google Scholar]
- 6. Deedwania P, Barter P, Carmena R, et al. Reduction of low‐density lipoprotein cholesterol in patients with coronary heart disease and metabolic syndrome: analysis of the Treating to New Targets study. Lancet. 2006;368:919–928. [DOI] [PubMed] [Google Scholar]
- 7. Tight blood pressure control and risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 38. UK Prospective Diabetes Study Group. BMJ. 1998;317:703–713. [PMC free article] [PubMed] [Google Scholar]
- 8. Messerli FH, Grossman E. Beta‐blockers in hypertension: is carvedilol different? Am J Cardiol. 2004;93:7B–12B. [DOI] [PubMed] [Google Scholar]
- 9. Sundberg S, Tiihonen K, Gordin A. Vasodilatory effects of carvedilol and pindolol. J Cardiovasc Pharmacol. 1987;10(suppl 11):S76–S80. [PubMed] [Google Scholar]
- 10. Yki‐Jarvinen H. Nonglycemic effects of insulin. Clin Cornerstone. 2003;5(suppl 4):S6–S12. [DOI] [PubMed] [Google Scholar]
- 11. Stump CS, Clark SE, Sowers JR. Oxidative stress in insulin‐resistant conditions: cardiovascular implications. Treat Endocrinol. 2005;4:343–351. [DOI] [PubMed] [Google Scholar]
- 12. Leiter LA, Lewanczuk RZ. Of the renin‐angiotensin system and reactive oxygen species Type 2 diabetes and angiotensin II inhibition. Am J Hypertens. 2005;18:121–128. [DOI] [PubMed] [Google Scholar]
- 13. Sharma AM, Engeli S. The role of renin‐angiotensin system blockade in the management of hypertension associated with the cardiometabolic syndrome. J Cardiometab Syndr. 2006;1:29–35. [DOI] [PubMed] [Google Scholar]
- 14. Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116:1793–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Avramoglu RK, Basciano H, Adeli K. Lipid and lipoprotein dysregulation in insulin resistant states. Clin Chim Acta. 2006;368:1–19. [DOI] [PubMed] [Google Scholar]
- 16. Fonseca V, Desouza C, Asnani S, et al. Nontraditional risk factors for cardiovascular disease in diabetes. Endocr Rev. 2004;25:153–175. [DOI] [PubMed] [Google Scholar]
- 17. Meigs JB, Mittleman MA, Nathan DM, et al. Hyperinsulinemia, hyperglycemia, and impaired hemostasis: the Framingham Offspring Study. JAMA. 2000;283:221–228. [DOI] [PubMed] [Google Scholar]
- 18. Grundy SM, Brewer HB Jr, Cleeman JI, et al. Definition of metabolic syndrome: report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition. Circulation. 2004;109:433–438. [DOI] [PubMed] [Google Scholar]
- 19. Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet. 2005;365:1415–1428. [DOI] [PubMed] [Google Scholar]
- 20. Anderson EA, Hoffman RP, Balon TW, et al. Hyperinsulinemia produces both sympathetic neural activation and vasodilation in normal humans. J Clin Invest. 1991;87:2246–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Osei K. Insulin resistance and systemic hypertension. Am J Cardiol. 1999;84:33J–36J. [DOI] [PubMed] [Google Scholar]
- 22. Palomo I, Alarcon M, Moore‐Carrasco R, et al. Hemostasis alterations in metabolic syndrome (review). Int J Mol Med. 2006;18:969–974. [PubMed] [Google Scholar]
- 23. Nishikawa T, Edelstein D, Du XL, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–790. [DOI] [PubMed] [Google Scholar]
- 24. Schmidt AM, Yan SD, Wautier JL, et al. Activation of receptor for advanced glycation end products: a mechanism for chronic vascular dysfunction in diabetic vasculopathy and atherosclerosis. Circ Res. 1999;84:489–497. [DOI] [PubMed] [Google Scholar]
- 25. Kawano H, Motoyama T, Hirashima O, et al. Hyperglycemia rapidly suppresses flow‐mediated endothelium‐dependent vasodilation of brachial artery. J Am Coll Cardiol. 1999;34:146–154. [DOI] [PubMed] [Google Scholar]
- 26. Packer M. Beta‐adrenergic blockade in chronic heart failure: principles, progress, and practice. Prog Cardiovasc Dis. 1998;41(suppl 1):39–52. [DOI] [PubMed] [Google Scholar]
- 27. Ko DT, Hebert PR, Coffey CS, et al. Beta‐blocker therapy and symptoms of depression, fatigue, and sexual dysfunction. JAMA. 2002;288:351–357. [DOI] [PubMed] [Google Scholar]
- 28. Pedersen ME, Cockcroft JR. The vasodilatory beta‐blockers. Curr Hypertens Rep. 2007;9:269–277. [DOI] [PubMed] [Google Scholar]
- 29. Jacob S, Rett K, Wicklmayr M, et al. Differential effect of chronic treatment with two beta‐blocking agents on insulin sensitivity: the Carvedilol–Metoprolol study. J Hypertens. 1996;14:489–494. [PubMed] [Google Scholar]
- 30. Haenni A, Lithell H. Treatment with a beta‐blocker with beta 2‐agonism improves glucose and lipid metabolism in essential hypertension. Metabolism. 1994;43:455–461. [DOI] [PubMed] [Google Scholar]
- 31. Khaw KT, Wareham N, Bingham S, et al. Association of hemoglobin A1c with cardiovascular disease and mortality in adults: the European prospective investigation into cancer in Norfolk. Ann Intern Med. 2004;141:413–420. [DOI] [PubMed] [Google Scholar]
- 32. Pepine CJ, Handberg EM, Cooper‐DeHoff RM, et al. A calcium antagonist vs a non‐calcium antagonist hypertension treatment strategy for patients with coronary artery disease. The International Verapamil–Trandolapril Study (INVEST): a randomized controlled trial. JAMA. 2003;290:2805–2816. [DOI] [PubMed] [Google Scholar]
- 33. Dahlof B, Devereux RB, Kjeldsen SE, et al. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet. 2002;359:995–1003. [DOI] [PubMed] [Google Scholar]
- 34. Gress TW, Nieto FJ, Shahar E, et al. Hypertension and antihypertensive therapy as risk factors for type 2 diabetes mellitus. Atherosclerosis Risk in Communities Study. N Engl J Med. 2000;342:905–912. [DOI] [PubMed] [Google Scholar]
- 35. Lund‐Johansen P, Omvik P, Nordrehaug JE. Long‐term hemodynamic effects of antihypertensive treatment. Clin Investig. 1992;70(suppl 1):S58–S64. [DOI] [PubMed] [Google Scholar]
- 36. Agabiti‐Rosei E, Porteri E, Rizzoni D. Arterial stiffness, hypertension, and rational use of nebivolol. Vasc Health Risk Manag. 2009;5:353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. DeFronzo RA, Mandarino L, Ferrannini E. Metabolic and molecular pathogenesis of type 2 diabetes mellitus. In: DeFronzo RA, Ferrannini E, Keen H, Zimmet P, eds. International Textbook of Diabetes Mellitus, Vol 1, 3rd ed. Chichester: John Wiley & Sons Ltd; 2004:389–438. [Google Scholar]
- 38. Rossner S, Taylor CL, Byington RP, et al. Long term propranolol treatment and changes in body weight after myocardial infarction. BMJ. 1990;300:902–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Caro JF. Clinical review 26: insulin resistance in obese and nonobese man. J Clin Endocrinol Metab. 1991;73:691–695. [DOI] [PubMed] [Google Scholar]
- 40. Agabiti Rosei E, Rizzoni D. Metabolic profile of nebivolol, a beta‐adrenoceptor antagonist with unique characteristics. Drugs. 2007;67:1097–1107. [DOI] [PubMed] [Google Scholar]
- 41. Rizos E, Bairaktari E, Kostoula A, et al. The combination of nebivolol plus pravastatin is associated with a more beneficial metabolic profile compared to that of atenolol plus pravastatin in hypertensive patients with dyslipidemia: a pilot study. J Cardiovasc Pharmacol Ther. 2003;8:127–134. [DOI] [PubMed] [Google Scholar]
- 42. Celik T, Iyisoy A, Kursaklioglu H, et al. Comparative effects of nebivolol and metoprolol on oxidative stress, insulin resistance, plasma adiponectin and soluble P‐selectin levels in hypertensive patients. J Hypertens. 2006;24:591–596. [DOI] [PubMed] [Google Scholar]
- 43. Schmidt AC, Graf C, Brixius K, et al. Blood pressure‐lowering effect of nebivolol in hypertensive patients with type 2 diabetes mellitus: the YESTONO study. Clin Drug Investig. 2007;27:841–849. [DOI] [PubMed] [Google Scholar]
- 44. Louis WJ, McNeil JJ, Drummer OH. Pharmacology of combined alpha‐beta‐blockade. I. Drugs. 1984;28(suppl 2):16–34. [DOI] [PubMed] [Google Scholar]
- 45. Siwach SB, Dahiya SS, Seth S, et al. Effect of atenolol and labetalol on serum lipids. J Assoc Physicians India. 1993;41:293–294. [PubMed] [Google Scholar]
- 46. Ohman KP, Weiner L, von Schenck H, et al. Antihypertensive and metabolic effects of nifedipine and labetalol alone and in combination in primary hypertension. Eur J Clin Pharmacol. 1985;29:149–154. [DOI] [PubMed] [Google Scholar]
- 47. Bakris GL, Fonseca V, Katholi RE, et al. Metabolic effects of carvedilol vs metoprolol in patients with type 2 diabetes mellitus and hypertension: a randomized controlled trial. JAMA. 2004;292:2227–2236. [DOI] [PubMed] [Google Scholar]
- 48. American Diabetes Association . Standards of medical care for patients with diabetes mellitus. Diabetes Care. 2003;26(suppl 1):S33–S50. [DOI] [PubMed] [Google Scholar]
- 49. Uzunlulu M, Oguz A, Yorulmaz E. The effect of carvedilol on metabolic parameters in patients with metabolic syndrome. Int Heart J. 2006;47:421–430. [DOI] [PubMed] [Google Scholar]
- 50. Fonarow GC, Deedwania P, Fonseca V, et al. Differential effects of extended‐release carvedilol and extended‐release metoprolol on lipid profiles in patients with hypertension: results of the Extended‐Release Carvedilol Lipid Trial. J Am Soc Hypertens. 2009;3:210–220. [DOI] [PubMed] [Google Scholar]
- 51. Kozlovski VI, Lomnicka M, Chlopicki S. Nebivovol and carvedilol induce NO‐dependent coronary vasodilatation that is unlikely to be mediated by extracellular ATP in the isolated guinea pig heart. Pharmacol Rep. 2006;58(suppl):103–110. [PubMed] [Google Scholar]
- 52. Dandona P, Ghanim H, Brooks DP. Antioxidant activity of carvedilol in cardiovascular disease. J Hypertens. 2007;25:731–741. [DOI] [PubMed] [Google Scholar]
- 53. Toda N. Vasodilating beta‐adrenoceptor blockers as cardiovascular therapeutics. Pharmacol Ther. 2003;100:215–234. [DOI] [PubMed] [Google Scholar]
- 54. Fratta Pasini A, Garbin U, Nava MC, et al. Nebivolol decreases oxidative stress in essential hypertensive patients and increases nitric oxide by reducing its oxidative inactivation. J Hypertens. 2005;23:589–596. [DOI] [PubMed] [Google Scholar]
- 55. American Association of Clinical Endocrinologists Medical Guidelines for Clinical Practice for the diagnosis and treatment of hypertension. Endocr Pract. 2006;12:193–222. [PubMed] [Google Scholar]
- 56. Mancia G, De Backer G, Dominiczak A, et al. 2007 Guidelines for the management of arterial hypertension: the Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). Eur Heart J. 2007;28:1462–1536. [DOI] [PubMed] [Google Scholar]
- 57. Sever P. New hypertension guidelines from the National Institute for Health and Clinical Excellence and the British Hypertension Society. J Renin Angiotensin Aldosterone Syst. 2006;7:61–63. [DOI] [PubMed] [Google Scholar]