

Abstract
The metabolic syndrome (MS) has been associated with hyperactivity of the renin‐angiotensin‐aldosterone system (RAAS). To assess the hypothesis that diuretic therapy in MS patients through further stimulation of RAAS would elicit greater potassium (K) depletion, two groups of hypertensive patients with (MS group [MSG]; n=20) and without (control group [CG]; n=19) MS were studied. Plasma renin activity (PRA), aldosterone (PA), and K levels were determined and an oral glucose tolerance test with plasma insulin determinations for calculation of homeostasis model assessment of insulin resistance (HOMA‐IR), sensitivity (ISI), and secretion (HOMA‐β) was performed, both before and 12 weeks after hydrochlorothiazide (HCT; 25 mg/d) therapy. At baseline, higher HOMA IR and HOMA‐β and lower ISI and plasma K were found in the MSG than in the CG, with no differences in PA and PRA between groups. With therapy, PRA increased similarly in both groups while PA increased only in the MSG. However, greater reduction in plasma K occurred in the CG, and the 2 groups reached similar final K values. Impairment in glucose tolerance occurred in both groups, with no change in HOMA‐β in the CG and reduction in the MSG, suggesting that diuretic therapy increases insulin resistance and impairs insulin secretion independent of abdominal obesity. These alterations could not be attributed to hyperactivity of RAAS.
With the increase of obesity prevalence, several studies have identified the connection between abdominal visceral fat and insulin resistance. 1 , 2 , 3 From this relationship, a new clinical identity, currently known as the metabolic syndrome (MS), 3 has been shown to increase the risk of coronary artery disease and cardiovascular mortality. 4
Arterial hypertension as part of MS, presents a strong connection with obesity and is associated with a greater risk of diabetes and glucose intolerance. 5 , 6 Therefore, when choosing the appropriate antihypertensive treatment for obese or MS patients, the metabolic effects of these drugs should be considered. Diuretic therapy, even in low doses, may induce the development of unwanted adverse effects such as glucose intolerance with a subsequent increased risk of diabetes. 7 , 8 , 9 , 10
Potassium depletion, induced by thiazide diuretics, is described as the main cause of alterations in glucose homeostasis. 10 , 11 Hypokalemia affects glucose metabolism through 2 different mechanisms: impairment in insulin secretion 11 , 12 and decrease in peripheral insulin sensitivity. 13 Thiazide diuretics lead to potassium depletion through the activation of the renin‐angiotensin‐aldosterone system (RAAS) in response to reductions in circulating blood volume, which increases urinary potassium excretion. In addition, the penetration of potassium in the cells, due to diuretic‐induced alkalosis, contributes to decreased plasma potassium levels. 14
Activation of the RAAS is one of the mechanisms responsible for arterial hypertension associated with obesity. 15 , 16 The increase of several circulating components of the RAAS, such as angiotensinogen, renin, angiotensin‐converting enzyme, and aldosterone, have been observed in obese patients. 16 In addition, the presence of several RAAS components have been demonstrated in the adipose tissue of animals and humans. 17 , 18 However, the contribution of the adipose tissue RAAS in the increases of circulating RAAS components is still a matter of debate.
Assuming that patients with abdominal obesity/MS, compared with nonobese patients, present greater insulin resistance and activation of RAAS, diuretic therapy in these patients that promotes even greater activation of RAAS could potentially induce a higher degree of potassium depletion, thus increasing the risk of type 2 diabetes. The objective of the present study was to assess this hypothesis.
Methods
This prospective clinical study (case control study) was conducted in 39 hypertensive patients assigned to 2 different groups: the case group (the MS group [MSG]), which was composed of 20 patients with MS; and the control group (CG), which included 19 patients without MS. The diagnostic criteria used to define MS were those defined by the International Diabetes Federation (IDF). 19 Participants were recruited from the hypertension and cardiovascular disease outpatient clinic at the Hospital do Rim e Hipertensão in Sao Paulo, Brazil.
The study protocol was approved by the ethics committee of the institution where the study was conducted. According to the research ethics criteria “in anima nobili,” patients were informed about the purposes of the study, all doubts were cleared in advance, and a written informed consent was obtained from each patient.
Male and female hypertensive patients, aged 30 to 60 years, were eligible. In both groups, sitting systolic and diastolic arterial blood pressure (BP) values, in the absence of antihypertensive treatment and after a 5‐minute rest, were to be ≥140 mm Hg and 90 mm Hg but not >160 mm Hg and 100 mm Hg, respectively. Hypertensive patients taking antihypertensive therapy could have values <140/90 mm Hg. Thus, only patients with mild and moderate arterial hypertension were considered eligible.
In the MSG, waist circumference in men and women were ≥94 cm and 80 cm, respectively. In addition to this criterion (already added to the presence of arterial hypertension), at least 1 other condition needed to be present to characterize MS, such as triglyceride levels ≥150 mg/dL and/or high‐density lipoprotein (HDL) cholesterol <40 mg/dL and 50 mg/dL in men and women respectively, and/or fasting glycemia ≥100 mg/dL. In the CG, waist circumference in men and women were <94 cm and 80 cm, respectively. In addition, except for arterial hypertension, patients could present only 1 more criterion of MS.
The exclusion criteria for this study were hypokalemia (plasma potassium <3.5 mEq/L), body mass index (BMI) >40 kg/m2, fasting glycemia ≥110 mg/dL, glycemia at 120 minutes post‐glucose load ≥200 mg/dL or diabetes while on treatment, triglycerides >400 mg/dL, congestive heart failure, chronic renal failure, hepatopathy, or severe psychiatric disease. Also, patients who presented with cardiovascular events (myocardial infarction or stroke) within the previous 6 months and women using oral contraceptives or who were pregnant did not take part in this study. In addition, patients who presented any evidence of secondary hypertension, malignant hypertension, or sitting systolic and diastolic BP levels >160 mm Hg and 100 mm Hg, respectively, were excluded.
The study protocol had a baseline period of 4 weeks for washout of antihypertensive drugs. After the first 2 weeks, patients were reevaluated and if their systolic and/or diastolic BPs were >160 mm Hg and 100 mm Hg respectively, alpha‐methyldopa (maximum dosage of 500 mg twice a day) was introduced for the next 2 weeks to reduce BP, according to the ethics committee recommendation.
At the end of the baseline period, alpha‐methyldopa was withdrawn and all patients received hydrochlorothiazide (HCT) 25 mg/d, maintaining their usual diet for a 12‐week term. During HCT therapy, patients were evaluated every 4 weeks for BP, heart rate, and anthropometric determinations.
BP was measured 3 times after 5 minutes of rest in the sitting position and after 5 minutes in the upright position using a sphygmomanometer with an appropriate cuff size. The values presented correspond to the arithmetic average of each 3 determinations.
The BMI was calculated as weight in kilograms divided by height in squared meters. The waist circumference was determined in centimeters at the middle point between the costal margin and the iliac crest.
Fasting plasma potassium (K), uric acid, total cholesterol, high‐density lipoprotein (HDL) cholesterol, low‐density lipoprotein (LDL) cholesterol, triglycerides (TGs), creatinine, plasma aldosterone (PA), and plasma renin activity (PRA) after at least 2 hours of deambulation were determined and an oral glucose tolerance test (OGTT) was performed both after the washout period (baseline) and after a 12‐week period of HCT therapy. During OGTT, glycemia and insulinemia were determined both after fasting and 120 minutes after an oral 75‐g glucose load. Based on these parameters, homeostasis model assessment of insulin resistance (HOMA‐IR), 20 insulin secretion (HOMA‐β), 20 and insulin sensitivity (ISI) 21 were calculated. Urinary potassium excretion (mEq/24 h) was determined both before and after diuretic therapy.
PA was determined through Active DSL‐8600 kit assay (Webster, TX), with analytical sensitivity of 7.64 pg/mL, intra‐assay variability of 3.3% to 4.5%, and interassay variability of 5.9% to 9.9%. The kit assay used for PRA determination was GammaCoat, (Stillwater, Minnesota, MN), with analytical sensitivity of 0.018 ng per tube, intra‐assay variability of 4.6% and 10.0%, and interassay variability of 5.6% and 7.6%. Plasma and urinary potassium values were determined by ion selective electrode method and expressed as mEq/L and mEq/24 h, respectively. Plasma glucose, uric acid, total cholesterol, and TGs were determined by enzymatic colorimetric method. HDL cholesterol was measured by homogeneous colorimetric enzymatic method and LDL cholesterol was calculated by the Friedwald formula: LDL cholesterol = total cholesterol − (HDL cholesterol + TG/5). The analyzer used was HITACHI 912 (Roche Diagnostics, Basel, Switzerland). Plasma creatinine was measured through alkaline picrate assay. Plasma insulin was determined by microparticle enzyme immunoassay.
Statistical Analysis
Statistical analyses were performed using SPSS version 13.0 software for Windows (SPSS, Inc, Chicago, IL). Paired t test for dependent measures and a t test for independent measures were used for comparison of variables within and between groups, respectively. Pearson coefficient was calculated to determine correlations between different variables. Data were expressed as mean ± SD and statistical significance was defined as P<.05.
Results
Thirty‐nine hypertensive patients of a miscigenated Brazilian population were included in this study. The Table shows the clinical and laboratory characteristics of all patients divided into the MSG (n=20) and the CG (n=19). There was no significant statistical difference in sex, age, smoking habit, or use of hormonal replacement therapy between the 2 study groups. Also, the number of participants who required alpha‐methyldopa for BP control before HCT therapy (baseline) did not differ between the MSG (7 of 20) and the CG (7 of 19). Higher BMI (34.0±3.4 vs 24.6±2.6 kg/m2; P<.05) and waist circumference (110.7±9.8 vs 82.9±6.6 cm; P<.05) were observed in the MSG than in the CG.
Table.
Clinical and Laboratory Findings in Patients With and Without the Metabolic Syndrome
| Groups | Control Group | Metabolic Syndrome Group | ||
|---|---|---|---|---|
| No. | 19 | 20 | ||
| Age, y | 49.32±8.69 | 47.10±9.81 | ||
| Women, % | 47.4 | 50 | ||
| ΔK, mEq/L | −0.69±−0.35 | −0.29±−0.27a | ||
| Status | Baseline | Post‐HCT | Baseline | Post‐HCT |
| Body mass index, kg/m2 | 24.58±2.58 | 24.54±2.60 | 34.01±3.39a | 33.88±3.53a |
| Waist circumference, cm | 82.89±6.64 | 82.42±6.84 | 110.65±9.78a | 110.50±9.81a |
| Heart rate, bpm | 73.84±8.45 | 71.05±8.91 | 76.65±8.72 | 77.65±5.62a |
| Systolic BP, mm Hg | 141.89±10.70 | 132.16±12.66b | 139.45±7.41 | 130.50±11.41b |
| Diastolic BP, mm Hg | 93,42±3.02 | 88.42±7.43b | 91.85±5.60 | 88.05±4.78b |
| Plasma K, mEq/L | 4,50±0.28 | 3.80±0.31b | 4.24±0.31a | 3.95±0.38b |
| PA, ng/dL | 15.44±7.53 | 20.90±12.12 | 16.25±6.87 | 28.67±15.00b |
| PRA, ng/mL/h | 1.14±1.00 | 3.30±4.12b | 0.90±0.85 | 2.35±1.72b |
| Glucose 0 min, mg/dL | 91.26±13.25 | 100.53±24.04 | 94.70±11.22 | 101.20±10.77b |
| Glucose 120 min, mg/dL | 95.21±29.90 | 107.74±34.43b | 117.05±16.23a | 130.40±27.11a,b |
| Insulin 0 min, μU/mL | 5.78±3.17 | 6.74±4.31 | 9.31±3.30a | 10.38±3.30a |
| Insulin 120 min, μU/mL | 35.12±30.10 | 46.92±35.16 | 67.61±44.82a | 69.54±43.21 |
| Plasma creatinine, mg/dL | 1.02±0.12 | 1.06±0.17 | 1.00±0.12 | 1.00±0.16 |
| Total cholesterol, mg/dL | 195.16±32.74 | 200.90±44.51 | 201.30±39.87 | 201.30±41.49 |
| HDL cholesterol, mg/dL | 58.42±12.35 | 58.32±17.07 | 49.90±15.58a | 47.55±12.47a |
| LDL cholesterol, mg/dL | 114.53±28.39 | 118.53±39.30 | 118.90±29.33 | 119.35±30.01 |
| Triglycerides, mg/dL | 100.32±38.93 | 120.42±48.89 | 156.55±74.01a | 172.25±78.54a |
| Triglycerides/HDL ratio | 1.84±1.00 | 2.30±1.39b | 3.35±1.69a | 3.80±2.00a,b |
| Uric acid, mg/dL | 4.87±1.03 | 5.62±1.38b | 5.48±1.35 | 6.13±1.57b |
| Urine volume, mL | 1478.68±531.96 | 1401.58±668.04 | 1678.90±785.57 | 1337.25±498.75 |
| Urinary K, mEq/24 h | 51.07±28.89 | 40.62±26.58 | 52.23±25.10 | 39.51±14.80b |
Abbreviations: 0 min, fasting on the oral glucose tolerance test; 120 min, 120 minutes after glucose load on the oral glucose tolerance test; BP, blood pressure; bpm, beats per minute; HCT, hydrochlorothiazide; HDL, high‐density lipoprotein; K, potassium; LDL, low‐density lipoprotein; PA, plasma aldosterone; PRA, plasma renin activity. Values are expressed as mean ± standard deviation. a P<.05 vs control group. b P<.05 vs baseline.
In the baseline period, compared with the CG, the MSG presented greater fasting insulinemia (9.3±3.3 vs 5.8±3.2 μU/mL; P<.05) and, at 120 minutes after glucose load, higher plasma glucose (117.0±16.2 vs 95.2±29.9 mg/dL; P<.05) and insulin levels (67.6±44.8 vs 35.1±30.1 μU/mL; P<.05). Thus, the MSG showed greater HOMA‐IR (2.18±0.84 vs 1.29±0.74; P<.05) (Figure, A) and lower ISI (0.69±0.30 vs 1.05±0.29; P<.05) (Figure, B) than the CG.
Figure.
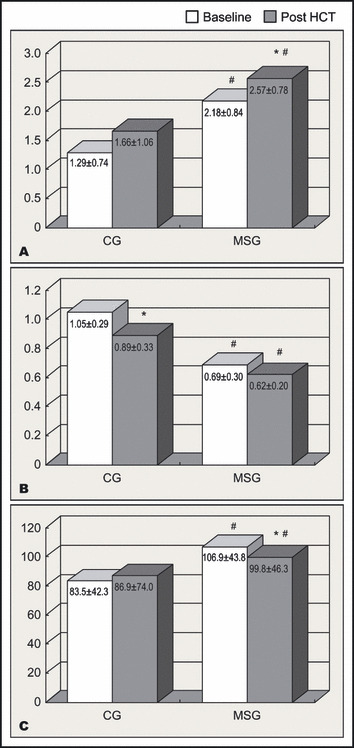
(A) Homeostasis model assessment of insulin resistance, (B) insulin sensitivity index, (C) and insulin secretion index in the control group (CG) and metabolic syndrome group (MSG). HCT indicates hydrochlorothiazide. * P<.05 vs baseline #P<.05 vs CG.
Before diuretic therapy, no differences were observed between the MSG and CG in PRA (0.9±0.8 vs 1.1±1.0 ng/mL/h; P=not significant [NS]), PA (16.2±6.9 vs 15.4±7.5 ng/dL; NS), and 24‐hour potassium urinary excretion (52.2±25.1 vs 51.1±28.9 mEq/24 h; NS), respectively. Baseline plasma K level, was lower in the MSG compared with the CG (4.24±0.31 vs 4.50±0.28 mEq/L; P<.05) and when the 2 groups were analyzed together, no correlation was found between baseline plasma K and PA (r=−0.17; P=.29). However, in the whole group, plasma K showed a negative and significant correlation with fasting plasma insulin (r=−0.46; P=.003) and HOMA‐IR (r=−0.44; P=.005).
After 12 weeks of HCT therapy, a smaller fall in plasma K levels was observed in the MSG compared with the CG (−0.29±−0.27 vs −0.69±−0.35 mEq/L, respectively; P<.05), resulting in similar levels of final plasma K in MSG and CG (3.95±0.38 vs 3.80±0.31 mEq/L, respectively; NS). As shown in the Table, this was associated with similar increases in PRA in both groups, while a significant increase in PA was observed only in the MSG.
Following HCT therapy, increases in blood glucose occurred at fasting and 120 minutes after glucose load in both groups, reaching higher levels in the MSG. These increases in glycemia, however, were not followed by elevations in plasma insulin levels. Consequently, a significant increase in HOMA‐IR was noted in the MSG (2.18±0.84 to 2.57±0.78; P<.05) (Figure, A) while ISI index decreased in the CG (1.05±0.29 to 0.89±0.33; P<.05) (Figure, B). Also, no significant change was observed in HOMA‐β in the CG (83.5±42.3 to 86.9±74.0; NS) (Figure, C), while in the MSG, HOMA‐β index decreased significantly (106.9±43.8 to 99.8±46.3; P<.05) (Figure, C). These were associated with increases in TG/HDL ratio and uricemia in both groups, also indicating increases in insulin resistance.
Discussion
In contrast to other studies, our results did not show evidence of hyperactivity of RAAS in patients with abdominal obesity and MS. Some published data suggest a direct association between higher levels of PA and MS, although this association has been demonstrated predominantly in black individuals. 22 , 23 Apart from differences in ethnicity, the reason for our different results is not known. Although we have studied a miscigenated population, our data are consistent with studies involving predominantly Caucasian individuals. 24 , 25 Kathiresan and colleagues 24 did not find any correlation between levels of PA and BMI values, while Egan and colleagues 25 did not find differences in mean PA levels between patients with and without MS.
In a previous study, our group also observed that plasma potassium levels in abdominal obese patients, even without diuretic therapy, were lower than in nonobese hypertensive patients. 26 This was attributed to a potential hyperactivity of the RAAS, not confirmed in the present study. Thus, the reasons for the lower levels of plasma K in our abdominal obese patients are not clear. One could argue that aldosterone levels were relatively high for the levels of plasma potassium found in the MSG and that these lower plasma potassium levels would be masking a hyperactive RAAS by suppressing aldosterone levels. However, this possible mechanism has yet to be proven. The significant negative correlation found between baseline plasma insulin and plasma K in the present study could also suggest a role of hyperinsulinemia in the reduction of plasma potassium levels in abdominal obesity. This has been shown acutely during euglycemic glucose clamp 27 and OGTT, 28 but there is no report describing long‐term hyperinsulinemia‐inducing hypokalemia.
After HCT therapy, the mean plasma K level in the MSG, which was lower than in the CG at baseline, showed a drop that was smaller than in the CG, despite a significant increase in PA. This resulted in similar plasma K levels at the end of the study in the 2 study groups, suggesting that these lower final plasma K levels may have similarly limited the elevation of PA levels induced by HCT therapy, thereby hindering greater K depletion.
The increased risk of diabetes associated with the use of thiazides has been described in several studies. 7 , 29 , 30 In our study, HCT induced glucose intolerance not only in patients with MS but also in nonobese patients. Those patients with MS who were more insulin‐resistant prior to HCT therapy showed more evident alterations, particularly in plasma glucose levels post‐glucose overload. Our results suggest a worsening of insulin resistance associated with impairment in insulin secretion. In fact, in other studies, the changes that occurred in glucose metabolism after thiazide diuretic administration have been partially attributed to K depletion, 11 decrease in insulin secretion, 11 , 12 and reduction in peripheral insulin sensitivity. 13 Diuretic‐induced increases in angiotensin II associated with hypokalemia may account for reductions in β‐cell function and increases in insulin resistance. 11 , 12 , 31 Experimental studies have shown that angiotensin II interferes with both insulin action 32 and secretion. 33 Accordingly, it has been observed in clinical trials that angiotensin II blockade in hypertensive patients reduces the incidence of type 2 diabetes. 34
The TG/HDL ratio showed significant increases in both groups, after HCT therapy, which, according to other studies, reflects a worsening in insulin resistance. 35 , 36 The increase in this ratio seems to be strongly associated with increase in the number of small LDL cholesterol particles, which are more atherogenic than normal‐sized LDL particles, thereby predicting a greater risk of coronary arterial disease. 37
Conclusions
Our results indicate that patients with MS do not present systemic hyperactivity of the RAAS, which cannot explain the lower levels of plasma K found in abdominal obese patients. Although more pronounced in obese patients, the disturbances in glucose metabolism induced by thiazide diuretic therapy seem independent of the presence of MS and can be attributed to worsening in both insulin resistance and secretion.
References
- 1. Reaven GM. Role of insulin resistance in human disease. Diabetes. 1999;37:1595–1607. [DOI] [PubMed] [Google Scholar]
- 2. DeFronzo RA, Ferraninni E. Insulin resistance – a multifaceted syndrome responsible for NIDDM, obesity, hypertension, dyslipidemia and atherosclerotic cardiovascular disease. Diabetes Care. 1991;14:173–194. [DOI] [PubMed] [Google Scholar]
- 3. Timar O, Sestier F, Levy E. Metabolic syndrome X: a review. Can J Cardiol. 2000;16:779–789. [PubMed] [Google Scholar]
- 4. Lakka HM, Laaksonen DE, Lakka TA, et al. The metabolic syndrome and total and cardiovascular disease mortality in middle‐aged men. JAMA. 2002;288:2709–2716. [DOI] [PubMed] [Google Scholar]
- 5. Wilsgaard T, Shirmer H, Arnesen E. Impact of body weight on blood pressure with a focus on sex differences. Arch Intern Med. 2000;160:2847–2853. [DOI] [PubMed] [Google Scholar]
- 6. Sironi AM, Gastaldelli A, Mari A, et al. Visceral fat in hypertension. influence on insulin resistance and β‐cell function. Hypertension. 2004;44:127–133. [DOI] [PubMed] [Google Scholar]
- 7. Elliot WJ, Meyer PM. Incident diabetes in clinical trials of antihypertensive drugs: a network meta‐analyses. Lancet. 2007;369:201–207. [DOI] [PubMed] [Google Scholar]
- 8. Padwal R, Laupacis A. Antihypertensive therapy and incidence of type 2 diabetes. a systematic review. Diabetes Care. 2004;27:247–255. [DOI] [PubMed] [Google Scholar]
- 9. Zillich AJ, Garg J, Basu S, et al. Thiazide Diuretics, Potassium, and the development of diabetes: a quantitative review. Hypertension. 2006;48:219–224. [DOI] [PubMed] [Google Scholar]
- 10. Plavinik FL, Rodrigues CI, Zanella MT, et al. Hypokalemia, glucose intolerance, and hyperinsulinemia during diuretic therapy. Hypertension. 1992;19 (2 suppl):II 26–II 29. [DOI] [PubMed] [Google Scholar]
- 11. Helderman JH, Elahi D, Andersen DK, et al. Prevention of the glucose intolerance of thiazide diuretics by maintenance of body potassium. Diabetes. 1983;32:106–111. [DOI] [PubMed] [Google Scholar]
- 12. Rowe JW, Tobin JD, Rosa RM, et al. Effects of experimental potassium deficiency on glucose and insulin metabolism. Metabolism. 1980;29:498–502. [DOI] [PubMed] [Google Scholar]
- 13. Pollare T, Lithel H, Berne C. A comparison of the effects of hydroclortiazide and captopril on glucose and lipids metabolism in patients with hypertension. N Engl J Med. 1989;321:868–873. [DOI] [PubMed] [Google Scholar]
- 14. Prichard BNC, Owen CWI, Woolf AS. Adverse reactions to diuretics. Eur Heart J. 1992;13 (suppl G):96–103. [DOI] [PubMed] [Google Scholar]
- 15. Boustany CM, Bharadwaj K, Daugherty A, et al. Activation of the systemic and adipose renin‐angiotensin system in rats with diet‐induced obesity and hypertension. Am J Physiol Regul Integr Comp Physiol. 2004;287:R943–R949. [DOI] [PubMed] [Google Scholar]
- 16. Engeli S, Bohnke J, Gorzelniak K, et al. Weight loss and the renin‐angiotensin‐aldosterone system. Hypertension. 2005;45:356–362. [DOI] [PubMed] [Google Scholar]
- 17. Goodfriend TL, Kelley DE, Goodpaster BH, et al. Visceral obesity and insulin resistance are associated with plasma aldosterone levels in women. Obesity Res. 1999;7:355–362. [DOI] [PubMed] [Google Scholar]
- 18. Giacchetti G, Faloia E, Mariniello B, et al. Overexpression of the Renin‐Angiotensin system in human visceral adipose tissue in normal and overwight subjects. Am J Hypertens. 2002;15:381–388. [DOI] [PubMed] [Google Scholar]
- 19. International Diabetes Federation . The IDF Consensus Worldwide Definition of the Metabolic Syndrome. http://www.idf.org/webdata/ docs/IDF_Metasyndrome_definition.pdf. Accessed October 4, 2005. [Google Scholar]
- 20. Matthews D, Hosker JP, Rudenski AS, et al. Homeostasis model assessment: insulin resistance and β‐cell function from fasting plasma glucose and insulin concentration in man. Diabetologia. 1985;28:412–419. [DOI] [PubMed] [Google Scholar]
- 21. Belfiore F, Iannello S, Volpicelli G. Insulin sensitivity indices calculated from basal and OGTT‐induced insulin, glucose and FFA levels. Mol Genet Metab. 1998;63:134–141. [DOI] [PubMed] [Google Scholar]
- 22. Kidambi S, Kotchen JM, Grim CE, et al. Association of adrenal steroids with hypertension and the metabolic syndrome in blacks. Hypertension. 2007;49:704–711. [DOI] [PubMed] [Google Scholar]
- 23. Bochud M, Nussberger J, Bovet P, et al. Plasma aldosterone is independently associated with the metabolic syndrome. Hypertension. 2006;48:239–245. [DOI] [PubMed] [Google Scholar]
- 24. Kathiresan S, Larson MG, Benjamin EJ, et al. Clinical and genetic correlates of serum aldosterone in the community: the Framingham Heart Study. Am J Hypertens. 2005;18:657–665. [DOI] [PubMed] [Google Scholar]
- 25. Egan BM, Papademetriou V, Wofford M, et al. Metabolic syndrome and insulin resistance in the TROPHY sub‐study: contrasting views in patients with high‐normal blood pressure. Am J Hypertens. 2005;18:3–12. [DOI] [PubMed] [Google Scholar]
- 26. Mariosa LSS, Ribeiro Filho FF, Batista M, et al. Abdominal Obesity is Associated with potassium depletion and changes in glucose homeostasis during diuretic therapy. J Clin Hypertens (Greenwich). 2008;10:443–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Minaker KL, Rowe JW. Potassium homeostasis during hyperinsulinemia: effect of insulin level, beta‐blockade, and age. Am J Physiol. 1982;242(6):E373–E377. [DOI] [PubMed] [Google Scholar]
- 28. Natali A, Quiñones Galvan A, Santoro D, et al. Relationship between insulin release, antinatriureuresis and hypokalemia after glucose ingestion in normal and hypertensive man. Clin Sci. 1993;85(3):327–335. [DOI] [PubMed] [Google Scholar]
- 29. ALLHAT Officers coordinators for the ALLHAT collaborative research group . The Antihypertensive and Lipid‐Lowering Treatment to prevent Heart Attack trial. Major outcomes in high‐risk hypertensive patients randomized to angiotensin‐converting enzyme inhibitor or calcium channel blocker vs diuretic: The Antihypertensive and Lipid‐Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). JAMA. 2002;288:2981–2987. [DOI] [PubMed] [Google Scholar]
- 30. Verdecchia P, Reboldi G, Angeli F, et al. Adverse prognostic significance of new diabetes in treated hypertensive subjects. Hypertension. 2004;43:963–969. [DOI] [PubMed] [Google Scholar]
- 31. Andersson OK, Gudbrandsson T, Jamerson K. Metabolic adverse effects of thiazide diuretics. The importance of normokalemia. J Intern Med. 1991;229 (suppl 2):89–96. [PubMed] [Google Scholar]
- 32. Folli F, Saad MJ, Velloso L, et al. Crosstalk between insulin and angiotensin II signaling systems. Exp Clin Endocrinol Diabetes. 1999;107:133–139. [DOI] [PubMed] [Google Scholar]
- 33. Chu KY, Lau T, Carlsson PO, et al. AT1 receptor blockade improves β‐cell function and glucose tolerance in mouse model of type 2 diabetes. Diabetes. 2006;55:367–374. [DOI] [PubMed] [Google Scholar]
- 34. Scheen AJ. Renin‐angiotensin system inhibition prevents type 2 diabetes mellistus. Part 1. A meta‐analysis of randomized clinical trials. Diabetes Metab. 2004;30:487–496. [DOI] [PubMed] [Google Scholar]
- 35. McLaughlin T, Abbasi F, Cheal K, et al. Use of metabolic markers to identify overweight individuals who are insulin resistant. Ann Intern Med. 2003;139:802–809. [DOI] [PubMed] [Google Scholar]
- 36. Boizel R, Benhamou PY, Lardy B, et al. Ratio of triglycerides to HDL cholesterol an indicator of LDL particle size in patients with type 2 diabetes and normal HDL cholesterol levels. Diabetes Care. 2000;23:1679–1685. [DOI] [PubMed] [Google Scholar]
- 37. Lamarche B, Tchernof A, Moorjani S, et al. Small, dense low‐density lipoprotein particle as a predictor of the risk of ischemic heart disease in men. Prospective results from the Quebec cardiovascular study. Circulation. 1997;95:69–75. [DOI] [PubMed] [Google Scholar]