

Abstract
Activation of the renin‐angiotensin‐aldosterone system (RAAS) is the primary etiologic event in the development of hypertension in people with diabetes mellitus. Modulation of the RAAS has been shown to slow the progression and even cause regression of the microvascular and macrovascular complications associated with diabetes mellitus. Early pharmacotherapy with agents that decrease RAAS activation in the adipose tissue have had a dramatic impact on the prevalence of diabetes related complications. Recent data show that preventing the development of “angry fat” can prevent not just hypertension but also type 2 diabetes mellitus and its associated complications. This review updates what is known about angry fat and the role of RAAS inhibition in preventing the metabolic sequelae of local RAAS activation. J Clin Hypertens (Greenwich). 2011;13:224–237. © 2011 Wiley Periodicals, Inc.
The growing worldwide epidemic of obesity and diabetes mellitus continues to expand, with a rapid decrease in the age at which diabetes is being diagnosed. Early initiation of comprehensive diabetes management is needed to prevent not only the macrovascular complications, including coronary artery disease, stroke, and peripheral arterial disease, but also the microvascular complications, encompassing diabetic neuropathy, nephropathy, and retinopathy. 1 , 2 , 3 , 4 The macrovascular and microvascular complications are more common in persons who have type 2 diabetes (T2DM) with pre‐existing hypertension (HTN), thus a common pathophysiology has been proposed, with the renin‐angiotensin‐aldosterone system (RAAS) emerging as a likely unifying mechanism. The objective of this review is to discuss the role of the RAAS in the pathophysiology of diabetes and hypertension and the associated complications.
As the body mass index (BMI) increases in a population, average blood pressure (BP) also rises. 5 Obesity is an independent predictor of the development of hypertension. It has been estimated that up to 40% of patients with hypertension have a BMI in the obese range and about half of individuals with essential hypertension are considered insulin‐resistant. 5 Likewise, insulin resistance and hyperinsulinemia increase the risk of hypertension. 5 Among patients being treated for hypertension, the risk of new‐onset diabetes is doubled in those with uncontrolled BP. 6 With the common thread of obesity, attention has turned to the role of adipose tissue in the development of T2DM and HTN. Adipose tissue, which is now recognized to be an endocrinologically active tissue, 5 , 7 , 8 , 9 that expresses all components of the RAAS and has its own local RAAS that is involved in regulation of BP. 10
RAAS as an Endocrine System
Renin is secreted by the kidney in response to decreases in blood volume and renal perfusion. Renin cleaves angiotensinogen to form the inactive decapeptide angiotensin I (Ang I). Ang I is converted to the active octapeptide angiotensin II (Ang II) by angiotensin‐converting enzyme (ACE) and non‐ACE pathways. Ang II is a powerful vasoconstrictor and leads to the release of catecholamines from the adrenal medulla and prejunctional nerve endings. It also promotes aldosterone secretion and sodium reabsorption, thereby increasing blood pressure. Ang II also inhibits renin release, thus providing a negative feedback to the system. This cycle, from renin through angiotensin to aldosterone and its associated negative feedback loop, is known as the RAAS. Systemically, an increase in pressure occurring after vascular injury or during stretch, stress, turbulence, and mechanical injury can result in mechanical stress and physical injury to the vessel wall. The resulting localized upregulation of Ang II synthetic pathways ultimately leads to alterations in the structure and function of blood vessels. Initially thought to be a general systemic process, this cycle is also now recognized to occur on a tissue level, utilizing Ang II produced locally to regulate the vascular structure and tone in the microenvironment of each tissue.
In addition to its pressor, proinflammatory, pro‐oxidative and salt‐retaining effects, Ang II is capable of inhibiting the action of insulin in vascular and skeletal muscle tissue via the angiotensin type 1 receptor (AT1R), interfering with insulin signaling through phosphatidylinositol 3‐kinase (PI3K) and its downstream protein kinase B (Akt) signaling pathway. This inhibition can result in decreased NO production in endothelial cells, increased vasoconstriction, and decreased glucose transport in skeletal muscle. 11 Evidence is emerging that Ang II has detrimental islet cell effects, including structural and functional damage induced by Ang II–mediated oxidative stress. 12 Ang II–mediated transforming growth factor β1 (TGF‐β1) production may also contribute to the progression of hepatic fibrosis, such as in the patient with underlying fatty liver or nonalcoholic fatty liver disease (NAFLD). 13 In the rat bile duct ligation model, RAAS blockade with captopril attenuated the progression of hepatic fibrosis and reduced hepatic hydroxyproline levels and TGF‐β1 and procollagen α1(I) messenger RNA. 13
Overactivation of the RAAS may lead to metabolic alterations that not only impact BP but also insulin resistance through increasing vasoconstriction, increasing renal sodium reabsorption, and stimulating aldosterone hormone secretion. 14 Aldosterone has been implicated in alterations in the insulin regulatory pathway metabolic diseases through its ability to impair insulin signaling by downregulating insulin receptor substrate‐1 (IRS‐1) in vascular smooth muscle cells. 15 Local aldosterone has also been implicated in worsening vascular disease. 15 Additionally, there is evidence that aldosterone may worsen pre‐existing alterations in glucose homeostasis, as in those with metabolic syndrome or prediabetes. 16
Implications of the RAAS in Obesity‐Associated Hypertension
Angiotensinogen Production by the Adipocyte
Hypertension is more common in prediabetic individuals who subsequently convert to diabetes than in those who do not convert to diabetes. 17 The intersections of the insulin signaling, the RAAS and the nitric oxide (NO) signaling pathways, predict that dysfunction at multiple locations is likely to occur in persons with T2DM and HTN. 18 , 19 , 20 The activity of the RAAS appears to be regulated by food intake, and overfeeding has been reported to increase formation of Ang II in adipocytes in studies in rodents. 21 Plasma aldosterone levels, likely driven by elevated Ang II, are increased in obese hypertensive patients, especially in patients with excess visceral fat. A large multicenter prospective study of newly diagnosed hypertensive patients, the Primary Aldosteronism Prevalence in Hypertension (PAPY) study, addressed the relationship between BMI, aldosterone, plasma renin activity, and aldosterone renin ratio. 22 In the 1125 hypertensive participants there was a positive correlation between BMI and plasma aldosterone, independent of age, sex, BP, or sodium intake in essential hypertensive patients. The association was stronger in patients with overweight and obesity, thus suggesting a pathophysiologic link between aldosterone production and fat deposition. This association was not found in patients with primary aldosteronism (PA), although such populations are enriched in the metabolic syndrome, suggesting that local modulation of the RAAS plays an etiologic role as opposed to pharmacologic aldosterone levels, as seen in PA. These data suggest that overactivation of the local RAAS likely combines with modest overproduction of aldosterone to interfere with insulin signaling, leading to insulin resistance and inflammation.
Fat is both a dynamic endocrine organ and a highly active metabolic tissue. 23 Adipose tissue produces and secretes an increasingly recognized number of inflammatory factors (adipose‐derived cytokines or adipokines), which play an important role not only in the process of atherosclerosis but also in the development of insulin resistance and diabetes. 7 , 22 , 24 , 25 There are at least 3 dozen substances identified as coming from fat. These include angiotensinogen, and a variety of other inflammatory factors, including plasminogen activating factor‐1; leptin; interleukin (IL) 6, which stimulates C‐reactive protein production in the liver; and IL‐1. In obesity, altered adipokine secretion with increased production of inflammatory factors, such as tumor necrosis factor α (TNF‐α), IL‐1, IL‐6, plasminogen activator inhibitor‐1 (PAI‐1), angiotensinogen and others, and decreased adiponectin, create a proinflammatory milieu.
Adipocytes enlarge as more food is ingested. With excessive eating, adipocyte storage capacity of fatty acids becomes overwhelmed and adipocytes undergo apoptosis and “burst.” Monocytes are then recruited into the adipose tissue, most commonly in visceral fat, to engulf dead adipocytes. These adipose tissue macrophages surround dead adipocytes to form crown‐like structures and control cytokine and hormone production by neighboring fat cells. In healthy individuals, 5% to 10% of visceral adipocytes are macrophages. Additionally, after recruitment into the adipose tissue, the monocytes begin to store the free fatty acids (FFAs) and, as they increase in size, they increase production of monocyte chemoattractant protein‐1 (MCP‐1), thus attracting more monocytes to enter the adipose tissue, become activated, and further exacerbate inflammation and promote lipolysis. Overeating also increases fat cell production of MCP‐1 and other factors, which recruit more monocytes into the adipose tissue. In obese individuals, up to 50% of visceral adipocytes can be macrophages. 26
A beneficial hormone produced from fat is adiponectin. Adiponectin promotes insulin sensitivity by enhancing glucose intake into skeletal muscle and decreasing hepatic glucose production through activation of the APM kinase pathway. 27 Adiponectin also has potent inflammatory effects to inhibit TNF‐α and to maintain endothelial function. 28 Low adiponectin levels have been clinically shown to predict cardiovascular (CV) events. In a 10‐year study of a Hong Kong population, Chow and colleagues 29 showed that low adiponectin levels predict hypertension in a Chinese population. In the United States, Pischon and colleagues 30 showed that low levels of adiponectin predicted myocardial infarction in men. Our group found that adiponectin predicts coronary flow abnormalities that are endothelial‐dependent in Mexican Americans (W. Hsueh, C. Lyon, Unpublished data). Adipose tissue macrophages promote increased fat secretion of inflammatory factors and decreased adiponectin. 31
High levels of angiotensinogen and low levels of adiponectin cause adipocytes to become a pressor organ. The hyperinsulinemia of insulin resistance has also been implicated in obesity‐associated HTN. Endothelial dysfunction can also contribute to vascular damage and HTN. Leptin increases central sympathetic nerve activity, leading to a rise in BP, but does not suppress appetite, since most obese people become leptin‐resistant. Obese humans have a 3‐fold increased prevalence of HTN, which is also a component of the diagnosis of the metabolic syndrome. Hypertension and the RAAS markedly increase vascular oxidative stress. 32
Angry Fat in Obesity
Angry fat is a term for adipocytes that not only produce increased amounts of adipokines, but undergo excess unregulated lipolysis leading to an increase in circulating FFAs. When insulin is unable to prevent this breakdown of fat, increased circulating fatty acid levels develop. This leads to the ectopic deposition of fat, which, in liver, skeletal muscle, and heart causes insulin resistance, mitochondrial dysfunction, and tissue injury, and in pancreatic β‐cells, tissue injury leading to apoptosis. Taken together, the result of the ectopic fat deposition is tissue inquiry, insulin resistance, and T2DM (Figure 1).
Figure 1.
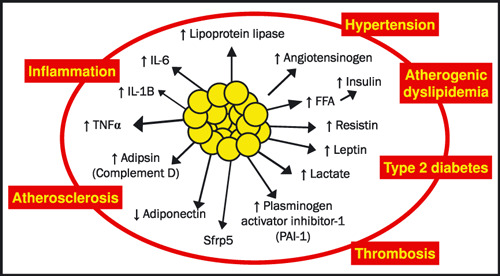
Adipose is an endocrine organ. Adipose tissue produces adipokines that send signals to many organs to maintain basic metabolic functions. IL‐6 = interleukin‐6; CRP = C‐reactive protein; TNFα = tumor necrosis factor‐alpha.
Attenuation of inflammatory capacity of adipose tissue macrophages decreases insulin resistance and liver steatosis, whereas enhanced inflammation, such as that created in mouse models of macrophage knockout of nuclear receptors such as peroxisome proliferator–activated receptors (PPARs), estrogen receptor α, and others, increases insulin resistance. 24 , 33 , 34 , 35 , 36 Excess tissue fat coupled with inflammation inhibits insulin signaling and stresses mitochondrial β oxidation capacity, leading to mitochondrial dysfunction and more oxidative stress followed by more inflammation, 26 , 37 creating a vicious cycle leading to further buildup of ectopic fat. Genetic factors additionally appear to be important determinants of mitochondrial function 38 and decreased fatty acid oxidation. The pathophysiologic processes potentially link: (1) the development of obesity and adipose tissue macrophages to the initiation and propagation of inflammation, and (2) inflammation to the induction of tissue and systemic insulin resistance are illustrated in Figure 2. 39
Figure 2.
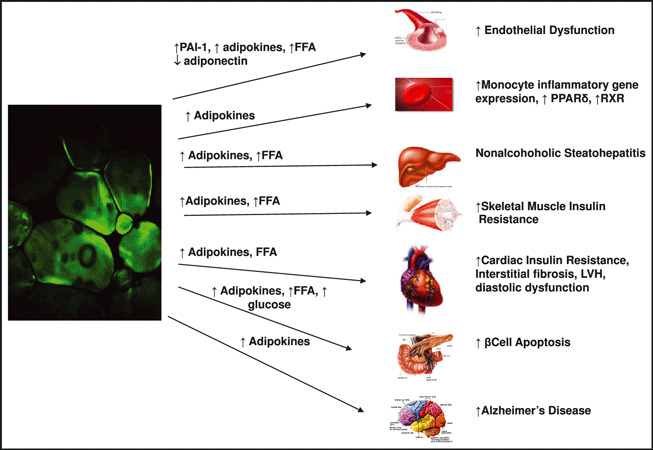
Targets of angry fat. Activation of the RAAS in adipose tissue is associated with an increase in production of cytokines that communicate the health of the adipose tissue to the nonadipose organs throughout the body. PAI‐1 = plasminogen activator inhibitor 1; PPARs = peroxisome proliferators‐activated receptors; RXR = retinoid × receptor; LVH = left ventricular hypertrophy.
One of the major targets of angry fat is the liver. As the liver becomes full of fat, it becomes nonresponsive to insulin and causes enhanced hepatic glucose production. Sixty million people in the United States are estimated to have fatty liver with inflammation and fibrosis, referred to as nonalcoholic steatohepatitis (NASH). 40 These sequelae of obesity are the second most common cause of liver cirrhosis and the third most common reason for liver transplants in the United States. It has been suggested that elevated liver enzymes and NASH are predictors of T2DM, HTN, and atherosclerosis. 41
Another organ that takes up fat is skeletal muscle. Skeletal muscle that accumulates fat becomes insulin‐resistant due to an inability of insulin to stimulate glucose uptake in skeletal muscle. But with exercise, skeletal muscle fat is minimized and skeletal muscles can again metabolize glucose. Whenever skeletal muscle is insulin‐resistant, cardiac muscle is also insulin‐resistant, since the heart also depends on insulin for glucose utilization. 42 The result is obesity‐associated cardiomyopathy, manifested as left ventricular hypertrophy, diastolic dysfunction, and a diffuse interstitial fibrosis leading to heart failure.
The pancreas is also an important target of angry fat. β‐Cells undergo apoptosis in the presence of high FFAs and glucose, resulting in a loss of insulin and hyperglycemia. In the presence of insulin resistance, even if increased insulin is produced, it may not be enough to overcome the insulin resistance so that type 2 diabetes ultimately develops. Fat storage is a yin yang between PPAR‐γ and inflammatory cytokines, like TNF‐α. Activation of PPAR‐γ promotes fat storage by transcriptional control of genes that mediate this process. Thus, one of the side effects of thiazolidinediones (TZDs) is weight gain. However, these drugs decrease visceral adiposity and redistribute adipose tissue to subcutaneous areas. TNF‐α inhibits the PPAR‐γ effect. Rather than storing FFAs, there is a greater release of FFAs, leading to accumulation of ectopic fat. 26 TNF‐α also inhibits insulin signaling, which both attenuates glucose uptake and promotes lipolysis. Overall, inflammation has multiple effects in adipose tissue to inhibit fat cell differentiation, lipid storage formation, PPAR‐γ activity, and insulin action. 26
Clinical Impact of the RAAS in Angry Fat
All components of the RAAS are expressed on the adipocyte. Adipose angiotensinogen overexpression increases plasma levels of Ang and raises BP, underscoring the endocrine function of the adipocyte in hypertension. However, there is confusion about the role of Ang II in adipocyte metabolism, as Ang II inhibits adipocyte differentiation but promotes triglyceride storage. 21 Ang II has also been shown to stimulate leptin release from adipose tissue. 43 The Ang II type 2 receptor (AT2R) is expressed in fat and appears to increase fat mass in mice. Knocking out the AT2R decreased adipocyte diameter. 44 Ang‐II inhibition decreases weight gain in the angiotensinogen transgenic mouse. In the angiotensinogen knockout mouse, adding back angiotensinogen to the fat cell resulted in larger fat cells. Combining the angiotensinogen fat overexpressing mouse with the AT2R knockout attenuates fat mass but increases adipocyte number. Absence of the AT2R increases BP; however, the impact of AT2R on fat appears independent of the BP effects.
RAAS and the Metabolic Syndrome
The metabolic syndrome is identified by the presence of and/or being on treatment for at least 3 of the 5 components (Table). The local adipose RAAS is a common pathophysiologic factor to all of the 5 components. As the waist circumference begins to expand, it is a marker for the accumulation of visceral fat, which is the most metabolically active adipose depot. Up to 50% of the cells in visceral adipose tissue can be macrophages, thus this is likely the site where the process of insulin resistance and unregulated lipolysis initiate the activation of the angry fat, which then spreads to other adipose depots and directly contributes to the cascade of inflammation, dyslipidemia, hypertension, and hyperglycemia. The overproduction of Ang II and activation of the AT1R, leads to increased production of aldosterone, which impairs insulin signaling, thus worsening insulin resistance and fueling this vicious cycle. RAAS blockade has the potential to break this cycle and, if started early enough, possibly stop the progression of the metabolic syndrome.
Table.
Criteria for Metabolic Syndrome
| Component Factors | IDF Definition Central Obesity Plus (Presence of or Treatment for any 2 of 4 Factors Below) | NHLBI/AHA Definition (Presence of or Treatment for any 3 of 5 Total Factors) |
|---|---|---|
| Central obesity (waist circumference) | ≥94 cm (in) in men or ≥80 cm (in) in womena | ≥102 cm (40 in) in men or ≥88 cm (35 in) in women |
| Elevated triglycerides | ≥150 mg/dL (1.7 mmol/L) | ≥150 mg/dL (1.7 mmol/L) |
| Reduced HDL cholesterol | <40 mg/dL (1.03 mmol/L) in men or <50 mg/dL (1.29 mmol/L) in women | <40 mg/dL (1.03 mmol/L) in men or <50 mg/dL (1.3 mmol/L) in women |
| Hypertension | Systolic BP ≥130 or diastolic BP ≥85 mm Hg | Systolic BP ≥130 or diastolic BP ≥85 mm Hg |
| Elevated FPG | ≥100 mg/dL (5.6 mmol/L) | ≥100 mg/dL (5.6 mmol/L) |
Abbreviations: AHA, American Heart Association; FPG, fasting plasma glucose; HDL, high‐density lipoprotein; IDF, International Diabetes Federation; NHLBI, National Heart, Lung and Blood Institute. aWaist circumference criteria are for individuals of Caucasian descent; for individuals of Asian descent, values are ≥90 cm (in) in men or ≥80 cm (in) in women. Adapted from references 111, 112, 113.
RAAS activation is important in the development of obesity‐related hypertension, dyslipidemia, and impaired glucose regulation, suggesting that agents that inhibit the RAAS are excellent first‐line therapy. 45 Treatment with RAAS blockers improves parameters of glucose metabolism 46 and can delay or prevent the occurrence of insulin resistance and subsequent type 2 diabetes, and has been shown to decrease the incidence of new‐onset diabetes in patients with or without hypertension who are at high risk for diabetes development. 47 Antihypertensive treatment with agents that modulate the RAAS can reduce the incidence of new‐onset diabetes as well as decrease BP, 11 , 48 , 49 as shown in multiple trials including HOPE, 50 LIFE, 51 ALLHAT, 52 ALPINE, 53 SOLVD, 54 , CHARM, 55 and VALUE. 56
RAAS blockade through ACE inhibitors and Ang II receptor blockers (ARBs) has been studied extensively in hypertension and coronary artery disease and has been demonstrated to prevent CV disease and diabetic nephropathy, while the recently introduced direct renin inhibitor (DRI), aliskiren, is still in the process of being studied for potential benefits beyond BP lowering. 57 , 58 The initial studies with RAAS inhibition in people with diabetic nephropathy clearly demonstrated that there was an effect beyond BP lowering. When compared with conventional antihypertensive therapy, those who received RAAS blockade consistently had greater improvement in their diabetic nephropathy despite attaining similar BP control. 59 , 60 , 61 , 62 , 63 The benefits of ACE inhibitors and ARBs likely extend beyond the direct vascular effects of RAAS blockade through clinically relevant effects of RAAS blockade on insulin resistance and glucose homeostasis. 58 , 64 For example, a recent study in hypertensive, obese individuals with the metabolic syndrome showed equal efficiency of valsartan (320 mg/d) and high‐dose hydrochlorothiazide (25 mg/d) in BP lowering, but showed lower fasting glucose and high‐sensitivity C‐reactive protein (hsCRP) with the ARB. 65 This, and other similar studies clinically support the possibility that not only a reduced incidence of diabetes but also a reduction of progression of the metabolic syndrome can occur though the decrease in the inflammatory response combined with substantial increases in adiponectin levels, enhanced endothelial function, and increased NO activation; improvement in insulin‐mediated glucose uptake; as well as structural and functional improvements in pancreatic islets and enhancement of the recruitment and differentiation of adipocytes, with consequent improvement in the FFA storage capacity of adipose tissue. 66 , 67 , 68 , 69 These then translate into clinically relevant outcomes, such as that seen in a subanalysis of diabetic patients from the HOPE trial, wherein treatment with ramipril was associated with lower CV mortality, even after adjustment for BP reduction. 70
RAAS and Insulin Resistance and Diabetes Prevention
When hypertension trials using RAAS blockade began reporting a decrease in the incidence of T2DM during the trials, a great deal of interest became focused on the possibility that an easy method of diabetes prevention had finally emerged. This was first formally examined with a meta‐analysis that confirmed the observation of a decrease in T2DM in nondiabetic individuals treated with an ACE inhibitor or ARB. 49 , 71 This hypothesis was then tested in the Diabetes Reduction Approaches With Ramipril and Rosiglitazone Medications (DREAM) trial, where 3 years of treatment with ramipril did not significantly decrease the incidence of diabetes, but it did significantly enhance regression to normoglycemia. 64 This was also tested in the Nateglinide and Valsartan in Impaired Glucose Tolerance Outcomes Research (NAVIGATOR) trial, which was a prospective, double‐blind, international, randomized clinical trial that assigned 9306 patients with impaired glucose tolerance in 22 randomization to either valsartan or matching placebo and the short‐acting oral hypoglycemic nateglinide or matching placebo that was performed in 806 centers in 40 countries, with a primary end point of reducing the risk of developing T2DM and CV events. 72 In NAVIGATOR, use of the ARB valsartan led to a modest but significant 14% reduction in new‐onset diabetes. Differences in the trial populations may explain the positive result in NAVIGATOR as opposed to in DREAM, where the patients had less metabolic syndrome and fewer CV risk factors. These trials have raised a number of questions, as the hypothesis tested was well supported by clinical observational data. The pathophysiology supports a preventative mechanism, thus it is possible that the ideal population has not been identified or perhaps that RAAS blockade with just an ACE inhibitor or ARB alone is not sufficient. Based on the physiology reviewed above, it is possible that an ACE inhibitor or ARB plus mineralocorticoid receptor (MR) antagonism, to decrease aldosterone, could be effective. Clinical trial data from the Randomized Aldactone Evaluation Study (RALES) and Eplerenone Post‐Acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS) trials has shown that MR antagonism improves CV outcomes. 73 , 74 However, clinical trials using spironolactone in people with T2DM have not shown an improvement in glucose control. These data suggest that prevention likely needs to begin long before elevated glucose levels emerge. The possibility that the combination of the beneficial effects of RAAS blockade on local RAAS plus MR antagonism, which can improve insulin sensitivity and oxidative stress, will be sufficient to slow the disease process in prediabetic or perhaps in metabolic syndrome patients who are insulin resistant, is an exciting hypothesis that now needs formal testing. A combination therapy approach plus modest weight loss could potentially have long‐lasting beneficial effects on decreasing insulin resistance and endothelial dysfunction thus slowing the progression to T2DM and HTN. 21 , 75 , 76
Implications of the RAAS in Diabetes Complications
Hyperglycemia has been demonstrated to play a role in the complications associated with diabetes. Intervention trials have shown that lowering glucose levels significantly improves the microvascular complications associated with diabetes. Glucose lowering has a modest impact on macrovascular complications, which only becomes apparent after many years. 77 , 78 Conversely, initiation of BP control in patients with newly diagnosed T2DM with pre‐existing HTN showed significant decreases in both microvascular and macrovascular complications. While BP control has been demonstrated to prevent macrovascular complications in diabetic and nondiabetic populations, RAAS blockade has an additional significant impact on slowing or stopping diabetic nephropathy and a more modest but still clinically relevant impact on diabetic retinopathy. Angry fat plays a role in the development of these complications through the adipokines it releases into the circulation. Although the retina and the kidney have not been studied in as great a detail as has adipose, the recent development of techniques to isolate and culture podocytes is providing insight into the communication between the angry fat and other tissues involved in diabetic complications.
Recent studies have demonstrated that loss of podocytes is an early feature of diabetic nephropathy. Podocytes are highly differentiated, pericyte‐like cells that are essential for normal function of the kidney filter. Damage to podocytes is a key initiating factor in the pathogenesis of glomerulosclerosis and proteinuria. Culturing podocytes in high glucose leads to increases in Ang II, AT1R expression, and prorenin receptors. These effects can be blocked by pretreatment with an ACE inhibitor or a DRI. 79 , 80 Podocytes are a direct target for Ang II–mediated injury, as evidenced by altered expression and distribution of podocyte proteins. Podocyte apoptosis followed the onset of hyperglycemia in db/db and Akita mice and coincided with the onset of urinary albumin excretion (UAE). Increased extracellular glucose (30 mmol/L) rapidly stimulated generation of intracellular reactive oxygen species (ROS) through nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and mitochondrial pathways and led to activation of proapoptotic p38 mitogen‐activated protein kinase and caspase 3 and to apoptosis of conditionally immortalized podocytes in vitro. Chronic inhibition of NADPH oxidase prevented podocyte apoptosis and ameliorated podocyte depletion, urinary albumin excretion, and mesangial matrix expansion in db/db mice. In addition to activation of the local RAAS, the podocyte is susceptible to the distant effects of the angry fat through the detrimental effects of low adiponectin and elevated FFAs. As with many other tissues, elevated FFAs block glucose uptake by podocytes. 81 In contrast, adiponectin has a direct protective effect on podocytes that is lost when levels are low, such as in obesity, DM, and CVD. This direct effect was discovered when studies of adiponectin knockout mice found effacement and fusion of podocyte foot processes as well as albuminuria and high oxidative stress in the kidney. 82 Treatment of cultured podocytes from adiponectin knockout mice with adiponectin resulted in a decrease in albumin permeability and a reduction in NADPH oxidase 4 (Nox4), which is the dominant NADPH oxidase in the kidney. These effects were then verified with in vivo studies showing that exogenous adiponectin administration to the adiponectin knockout mice reduces albuminuria and oxidative stress and attenuates podocyte damage. Thus, podocytes are a physically distant but quite relevant target of angry fat. RAAS blockade in both angry fat and the podocyte can help to prevent the diabetic nephropathy. Similar studies are needed in the retina to confirm the clinical observations that RAAS blockade with ACE inhibitors or ARBs can prevent or slow retinopathy.
Inhibition of the RAAS in Diabetes
Role of RAAS Blockade in Prevention of Diabetes Complications
The etiology of diabetic complications was hotly debated until 1993 when the Diabetes Control and Complications Trial (DCCT) demonstrated that intensive glucose control in T1DM decreased microvascular complications and the ACE inhibitor captopril was shown to inhibit the progression of diabetic nephropathy independent of BP lowering. 60 , 83 The UKPDS reaffirmed the role of glucose control in 1998 when it demonstrated that intensive glucose control in T2DM also decreased microvascular complications. 77 However, glucose control did not prevent all complications, thus further studies were needed to determine whether RAAS inhibition could also prevent the microvascular complications in T2DM.
RAAS Blockade and Nephropathy
At the time of publication of the study by Lewis et al in 1993, there were multiple other ongoing trials treating people with either T1DM or T2DM and microalbuminuria and one with normotensive, nonalbuminuric T2DM that have confirmed the finding of a decrease in nephropathy and expanded the observation to other ACE inhibitors, suggesting that this is a class effect. 59 , 84 , 85 , 86 , 87 The introduction of the ARB class provided an opportunity for large trials to assess renal and CV benefits.
Three trials, the Irbesartan in Patients With Type 2 Diabetes and Microalbuminuria (IRMA‐2) study, the Reduction of End Points in Non‐Insulin Dependent Diabetes Mellitus With the Angiotensin II Antagonist Losartan (RENAAL) study, and the Irbesartan Type 2 Diabetic Nephropathy Trial (IDNT) demonstrated that ARBs can impact the progression of diabetic nephropathy in T2DM at all stages of renal disease, ranging from microproteinuria to overt nephropathy and ESRD. 61 , 62 , 63 These studies clearly demonstrated that this effect was in addition to that attributable to BP control alone and was associated with a delay in the need for dialysis or kidney transplant by at least 2 years. This benefit was confirmed in the HOPE study, which included 1808 people with diabetes treated with ramipril, and in the 1059 ACE‐intolerant patients treated with telmisartan in the Telmisartan Randomized Assessment Study in ACE Intolerant Subjects With Cardiovascular Disease (TRANSCEND). 88 , 89 Differences between the specific agents for RAAS blockade have really not been addressed in clinical trials. The Efficacy of Telmisartan Compared With Losartan in Reducing Proteinuria in Hypertensive Type 2 Diabetic Patients With Overt Nephropathy (AMADEO) study showed that telmisartan was more effective than losartan in reducing proteinuria in T2DM patients with nephropathy, but such comparisons have not been further explored. 90 These clinical trials have led to the adoption of ACE inhibitors or ARBs as first‐line therapy both for hypertension and for treatment of albuminuria in patients with diabetes.
Not all diabetic nephropathy has been prevented by treatment with ACE inhibitors or ARBs, thus studies have been designed to test whether combination ACE/ARB therapy may be more beneficial and to ascertain a threshold below which BP lowering provides no further benefit. The HOT study suggested that for high‐risk patients (ie, those with diabetes), it may be beneficial to lower BP even if it is already within the “normal” range. 91 This was formally tested in the recent Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial, which did not show such a benefit for the composite of CV events but did demonstrate a significant decrease in the incidence of macroalbuminuria in the intensive therapy group (4733 patients followed for 4.7 years), which attained an average systolic BP of 119.3 mm Hg (95% confidence interval [CI], 118.9–119.7). However, there was no difference between the groups in the frequency of end‐stage renal disease or the need for dialysis. 92 In contrast, the Action in Diabetes and Vascular Disease: Preterax and Diamicron MR Controlled Evaluation (ADVANCE) study, which randomized 5569 people to the addition of a fixed combination of perindopril and indapamide demonstrated a reduction in the risks of death and major macrovascular or microvascular complications. 93 Comparison of these two trial populations has not revealed an explanation for this discrepancy in effect. Combination ACE/ARB therapy was tested in the Ongoing Telmisartan Alone and in Combination With Ramipril Global Endpoint Trial (ONTARGET), with the combination of ramipril and telmisartan showing superior control of albuminuria as compared with the individual therapies for the general study group. However, the data on the diabetes subgroup of 9612 people (3220 treated with combination therapy) have not yet been released. 94
RAAS Blockade and Retinopathy
The role of the RAAS in diabetic retinopathy has not been as well studied as that in the kidney, thus evidence for RAAS inhibition in retinopathy is still much weaker than for nephropathy. Several recently published studies, the Daily‐Dose Consensus Interferon and Ribavirin: Efficacy of Combined Therapy (DIRECT) trial, the Renin‐Angiotensin System Study (RASS), and the ADVANCE Retinal Measurements (AdRem), have provided evidence that RAAS inhibition may also be beneficial in diabetic retinopathy. 95 , 96 , 97
The AdRem study is a substudy of ADVANCE, involving the 1602 patients with T2DM from ADVANCE centers with access to retinal cameras between 2001 and 2008. At baseline and the final visit, 7‐field stereoscopic retinal photographs were taken and graded by blinded readers (gradeable baseline and final photographs from 1241 patients). Progression of ≥2 steps in the Early Treatment of Diabetic Retinopathy Study (ETDRS) classification (using the eye with worst grading) was the primary outcome. Fewer patients on BP‐lowering treatment (n=623) experienced incidence or progression of retinopathy compared with patients taking placebo (n=618), but the difference was not significant (odds ratio [OR], 0.78; 95% CI, 0.57–1.06; P=.12). 95 BP‐lowering treatment did specifically reduce the occurrence of macular edema (OR, 0.50; 95% CI, 0.29–0.88; P=.016) and arteriovenous nicking compared with placebo (OR, 0.60; 95% CI, 0.38–0.94; P=.025). These results must be interpreted cautiously as there was no reduction (P=.27) in the incidence and progression of retinopathy in the intensive glucose control (n=630) as compared with standard glucose control group. In contrast, the ACCORD Eye Study Group found that 4 years of intensive glycemic control or combination therapy for dyslipidemia but not intensive BP control slowed the progression of diabetic retinopathy in the subgroup of 2856 of 10,251 participants who underwent eye evaluations. 98
Data from studies with type 1 diabetes mellitus (T1DM) appear to shed some light on these discrepancies as they suggest that RAAS blockade is most effective when initiated early in the development of retinopathy. The Diabetic Retinopathy Candesartan Trials Programme (DIRECT) was designed to assess whether the ARB, candesartan, could reduce the incidence and progression of retinopathy in T1DM. 96 Participants with normotensive, normoalbuminuric T1DM without retinopathy were recruited to the DIRECT‐Prevent 1 trial and those with existing retinopathy were recruited to DIRECT‐Protect 1. The primary end points were incidence and progression of retinopathy and were defined as at least a two‐step and at least a three‐step increase on the Early Treatment Diabetic Retinopathy Study (ETDRS) scale, respectively. In these two studies, candesartan was able to reduce the incidence of retinopathy but did not provide a beneficial effect on retinopathy progression. 99 Progression of retinopathy occurred in 127 (13%) participants in the candesartan group vs 124 (13%) in the placebo group. The hazard ratio for candesartan vs placebo was 0.82 (95% CI, 0.67–1.00; P=.0508) for incidence of retinopathy and 1.02 (95% CI, 0.80–1.31; P=.85) for progression of retinopathy.
However, these results were modest, however, raising the question as to how early in the development of retinopathy does the initiation of the RAAS blockade need to occur. The Renin‐Angiotensin System Study (RASS), a multicenter diabetic nephropathy primary prevention trial, evaluated the severity and response to therapy of retinopathy in 285 normoalbuminuric patients with T1DM, who would not normally have been treated with RAAS blockade. 97 Retinopathy was present in 64% of participants and was not related to albumin excretion rate, BP, serum creatinine, or glomerular filtration rate. However, all renal anatomical end points were associated with increasing severity of diabetic retinopathy, while controlling for other risk factors. These data demonstrate a significant association between diabetic retinopathy and preclinical morphologic changes of diabetic nephropathy in T1DM patients. The participants were then randomized to receive losartan (100 mg daily), enalapril (20 mg daily), or placebo and followed for 5 years. A total of 90% and 82% of patients had complete renal biopsy and retinopathy data, respectively. 100 As compared with placebo, the odds of retinopathy progression by two steps or more was reduced by 65% with enalapril (OR, 0.35; 95% CI, 0.14–0.85) and by 70% with losartan (OR, 0.30; 95% CI, 0.12–0.73), independent of changes in BP, thus demonstrating a specific benefit of early RAAS blockade in the progression of retinopathy.
There are beginning to emerge data to support the role of the RAAS in these observed clinical effects, using the model of oxygen‐induced retinopathy (OIR) in rats and cultured bovine retinal endothelial cells (BRECs) and bovine retinal pericytes (BRPs). Aldosterone stimulated proliferation and tubulogenesis in BRECs and exacerbated angiogenesis in OIR, which was attenuated with spironolactone. The MR and aldosterone‐modulated retinal inflammation, with leukostasis and MCP‐1 mRNA and protein in OIR being reduced by spironolactone and increased by aldosterone. A functional retinal MR‐aldosterone system was evident with MR expression, translocation of nuclear MR, and aldosterone synthase expression, which was modulated by RAAS blockade. These data demonstrate that there is a functional RAAS in the retina and that further studies are needed to determine the optimal timing of therapy to help prevent both onset and progression of diabetic retinopathy. 101
Angry fat also plays a role in retinal disease through modulation of inflammatory cytokines and adiponectin levels. The adiponectin knockout mice have been valuable for studying mechanisms of not just nephropathy but also retinopathy. When neonatal mice were subjected to ischemia‐induced retinopathy, pathological retinal neovascularization during ischemia was more common in the adiponectin knockout mice than controls. This effect could be attenuated by provision of adiponectin knockout mice through adenovirus‐mediated overexpression of adiponectin or treatment of wild‐type (WT) mice with TZDs, which are known to increase adiponectin levels, while the TZDs had no effect on ischemia‐induced pathological retinal vessel formation in the adiponectin knockout mice. Pioglitazone reduced TNF‐α expression in ischemic retina in WT mice but not in APN‐knock out mice. 102 , 103 These data support the clinical trial data, that RAAS blockade is effective in combating diabetic retinopathy when started early in the disease process.
RAAS Blockade and CV Disease
RAAS blockade, in the form of ACE inhibitors, were introduced in the United States in 1981 for hypertension management. The recognition that RAAS blockade could prevent or slow diabetic nephropathy led to the design of trials with progression of renal disease as an end point that were not adequately powered to detect CV events. Large trials are either ongoing or recently completed that may provide CV outcomes data in patients with diabetes treated with RAAS blockade as first‐line therapy.
The UKPDS‐BP control substudy demonstrated that lowering BP decreased macrovascular complications, including mortality; however, it was not powered to ascertain a difference between treatment with captoprilor atenolol. Subsequently, the diabetic subgroup of the Captopril Prevention Project (CAPPP), in a prespecified analysis, demonstrated that first‐line therapy with captopril resulted in a decrease in the primary end point, fatal and nonfatal myocardial infarction and stroke as well as other CV deaths, as compared with the conventional therapy group (relative risk, 0.59; P=.018). 91 Studies such as ACCORD, ADVANCE, ONTARGET, and TRANSCEND were then designed to investigate the CV benefits of RAAS blockade with ACE inhibitors, ARBs, or the combination of both.
The ADVANCE trial is an international multicenter study of 11,140 people with type 2 diabetes who were randomized to treatment with a fixed combination of perindopril and indapamide or matching placebo, in addition to current therapy. 104 The primary end points were composites of major macrovascular and microvascular events, defined as death from CV disease, nonfatal stroke or nonfatal myocardial infarction, and new or worsening renal or diabetic eye disease, and analysis was by intention to treat. After a mean of 4.3 years of follow‐up, the relative risk of a major macrovascular or microvascular event was reduced by 9% (P=.04). The separate reductions in macrovascular and microvascular events were similar but were not independently significant. The relative risk of death from CV disease was reduced by 18% (P=.03) and death from any cause was reduced by 14% (P=.03). There was no evidence that the effects of the study treatment differed by initial BP level or concomitant use of other treatments at baseline. While ADVANCE showed a benefit on mortality and macrovascular events, ACCORD did not. 92 As with the nephropathy data, the reasons for this discrepancy are not yet apparent. It is not yet known whether the ACE/ARB combination will provide any greater CV benefit as the data from the diabetic subgroup are not yet available from ONTARGET. TRANSCEND, which studied candidates eligible for ONTARGET who were intolerant to ACE inhibitors, also has a large diabetic subgroup from whom the data are not yet available.
A potential challenge in demonstrating a CV benefit of RAAS blockade in diabetes is that it may need to be initiated very early, to block the effects of the angry fat, and be of long duration to prevent the end‐organ damage in the heart. Nonetheless, once heart failure is present, RAAS inhibition clearly has an impact on multiple CV outcomes, whether the person has diabetes mellitus or not. 105 The argument for early RAAS blockade to prevent the development of heart failure is well grounded in animal data. The aging mouse model has been used to demonstrate that chronic RAAS inhibition reduces cardiac fibrosis, which would then help to prevent the diastolic dysfunction that progresses to heart failure. 106 This effect is likely mediated both through inhibition of the local and circulating RAAS. An overactive cardiac RAAS promotes heart remodeling through the development of left ventricular hypertrophy (LVH) and myocardial fibrosis and leads to an increase in perivascular inflammation that is associated with worsening of the myocardial fibrosis. 107 Increased activity of the circulating RAAS, such as that promoted by the angry fat, contributes to the development of volume overload and vasoconstriction, with consequent increases in left ventricular diastolic filling pressures and increased stress on the stiff heart. 108 , 109 Despite the clearly beneficial effects of RAAS blockade on the underlying pathophysiology, the clinical trial data do not yet show the expected benefits on CV outcomes. Thus, many questions remain as we work to determine a clinical strategy for optimal RAAS blockade. The fact that none of our current therapies provide complete RAAS inhibition raises the possibility that the addition of a DRI to the currently utilized combinations may have further benefit. 110 Studies are ongoing to test this strategy.
Conclusions
Overactivity of the RAAS, both on a systemic and local level, has been shown to play a role in the complications associated with diabetes. The RAAS activity level is increased prior to the onset of hyperglycemia, thus consideration must be given to initiating RAAS inhibition either in prediabetes or even earlier in patients with the metabolic syndrome. Such early treatment has the potential to prevent the progression of the metabolic disorders of obesity, hypertension, and diabetes, which are becoming a global pandemic.
References
- 1. McGuire D, Granger C. Diabetes and ischemic heart disease. Am Heart J. 1999;138:S366–S375. [DOI] [PubMed] [Google Scholar]
- 2. Vinik A, Ullal J, Parson H, et al. Diabetic neuropathies: clinical manifestations and current treatment options. Nat Clin Pract Endocrinol Metab. 2006;2:269–281. [DOI] [PubMed] [Google Scholar]
- 3. Gall M, Hougaard P, Borch‐Johnsen K, et al. Risk factors for development of incipient and overt diabetic nephropathy in patients with non‐insulin dependent diabetes mellitus: prospective, observational study. BMJ. 1997;314:783–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Association AD . Standards of Medical Care in Diabetes‐2011. Diabetes Care. 2011;34:S11–S61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Blaj S, Stanciu S, Jurcut C, et al. Hypertension in obese patients: a dysmetabolic hypertension with a possible adipocyte dysfunction mechanism. Rom J Intern Med. 2003;41:103–111. [PubMed] [Google Scholar]
- 6. Izzo R, de Simone G, Chinali M, et al. Insufficient control of blood pressure and incident diabetes. Diabetes Care. 2009;32(5):845–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bakhai A. Adipokines – targeting a root cause of cardiometabolic risk. QJM. 2008;101:767–776. [DOI] [PubMed] [Google Scholar]
- 8. Kershaw E, Flier J. Adipose tissue as an endocrine organ. J Clin Endocrinol Metab. 2004;89:2548–2556. [DOI] [PubMed] [Google Scholar]
- 9. Mathieu P, Poirier P, Pibarot P, et al. Visceral obesity: the link among inflammation, hypertension, and cardiovascular disease. Hypertension. 2009;53:577–584. [DOI] [PubMed] [Google Scholar]
- 10. Achard V, Boullu‐Ciocca S, Desbriere R, et al. Renin receptor expression in human adipose tissue. Am J Physiol Regul Integr Comp Physiol. 2007;292:R274–R282. [DOI] [PubMed] [Google Scholar]
- 11. Sowers J. Insulin resistance and hypertension. Am J Physiol Heart Circ Physiol. 2004;286:H1597–H1602. [DOI] [PubMed] [Google Scholar]
- 12. Leung KK, Leung PS. Effects of hyperglycemia on angiotensin II receptor type 1 expression and insulin secretion in an INS‐1E pancreatic beta‐cell line. JOP. 2008;9(3):290–299. [PubMed] [Google Scholar]
- 13. Jonsson J, Clouston A, Ando Y, et al. Angiotensin‐converting enzyme inhibition attenuates the progression of rat hepatic fibrosis. Gastroenterology. 2001;121:148–155. [DOI] [PubMed] [Google Scholar]
- 14. Weir M, Dzau V. The renin‐angiotensin‐aldosterone system: a specific target for hypertension management. Am J Hypertens. 1999;12:205S–213S. [DOI] [PubMed] [Google Scholar]
- 15. Hitomi H, Kiyomoto H, Nishiyama A, et al. Aldosterone suppresses insulin signaling via the downregulation of insulin receptor substrate‐1 in vascular smooth muscle cells. Hypertension. 2007;50:750–755. [DOI] [PubMed] [Google Scholar]
- 16. Krug A, Ehrhart‐Bornstein M. Aldosterone and metabolic syndrome: is increased aldosterone in metabolic syndrome patients an additional risk factor? Hypertens Pregnancy. 2008;51:1252–1258. [DOI] [PubMed] [Google Scholar]
- 17. Haffner SM, Mykkanen L, Festa A, et al. Insulin‐resistant prediabetic subjects have more atherogenic risk factors than insulin‐sensitive prediabetic subjects: implications for preventing coronary heart disease during the prediabetic state. Circulation. 2000;101(9):975–980. [DOI] [PubMed] [Google Scholar]
- 18. McGuire D, Winterfield J, Rytlewski J, et al. Blocking the renin‐angiotensin‐aldosterone system to prevent diabetes mellitus. Diab Vasc Dis Res. 2008;5:59–66. [DOI] [PubMed] [Google Scholar]
- 19. Yanai H, Tomono Y, Ito K, et al. The underlying mechanisms for development of hypertension in the metabolic syndrome. Nutr J. 2008;7:7–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Duvnjak L, Bulum T, Metelko Ž. Hypertension and the metabolic syndrome. Diabetol Croat. 2008;37:83–89. [Google Scholar]
- 21. Engeli S, Schling P, Gorzelniak K, et al. The adipose‐tissue renin‐angiotensin‐aldosterone system: role in the metabolic syndrome? Int J Biochem Cell Biol. 2003;35:807–825. [DOI] [PubMed] [Google Scholar]
- 22. Rossi G, Belfiore A, Bernini G, et al. Body mass index predicts plasma aldosterone concentrations in overweight‐obese primary hypertensive patients. J Clin Endocrinol Metab. 2008;93:2566–2571. [DOI] [PubMed] [Google Scholar]
- 23. Lyon CJ, Law RE, Hsueh WA. Minireview: adiposity, inflammation, and atherogenesis. Endocrinology. 2003;144(6):2195–2200. [DOI] [PubMed] [Google Scholar]
- 24. Waki H, Tontonoz P. Endocrine functions of adipose tissue. Annu Rev Pathol. 2007;2:31–56. [DOI] [PubMed] [Google Scholar]
- 25. Cassis L, Police S, Yiannikouris F, et al. Local adipose tissue renin‐angiotensin system. Curr Hypertens Rep. 2008;10:93–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Guilherme A, Virbasius J, Puri V, et al. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol. 2008;9:367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hsueh W, Tangirala R. Adiponectin. In: Izzo J, HR B, eds. Hypertension Primer, 4th edn. Philadelphia, PA: Lippincott Williams and Wilkins; 2008:113–115. [Google Scholar]
- 28. Yamauchi T, Kamon J, Waki H, et al. Globular adiponectin protected ob/ob mice from diabetes and ApoE‐deficient mice from atherosclerosis. J Biol Chem. 2003;278:2461–2468. [DOI] [PubMed] [Google Scholar]
- 29. Chow W, Cheung B, Tso A, et al. Hypoadiponectinemia as a predictor for the development of hypertension: a 5‐year prospective study. Hypertens Pregnancy. 2007;49:1455–1461. [DOI] [PubMed] [Google Scholar]
- 30. Pischon T, Girman C, Rifai N, et al. Association between dietary factors and plasma adiponectin concentrations in men. Am J Clin Nutr. 2005;81:780–786. [DOI] [PubMed] [Google Scholar]
- 31. Suganami T, Nishida J, Ogawa Y. A paracrine loop between adipocytes and macrophages aggravates inflammatory changes: role of free fatty acids and tumor necrosis factor alpha. Arterioscler Thromb Vasc Biol. 2005;25:2062–2068. [DOI] [PubMed] [Google Scholar]
- 32. Doughan A, Harrison D, Dikalov S. Molecular mechanisms of angiotensin II‐mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res. 2008;102:488–496. [DOI] [PubMed] [Google Scholar]
- 33. Solinas G, Vilcu C, Neels J, et al. JNK1 in hematopoietically derived cells contributes to diet‐induced inflammation and insulin resistance without affecting obesity. Cell Metab. 2007;6:386–397. [DOI] [PubMed] [Google Scholar]
- 34. Hevener A, Olefsky J, Reichart D, et al. Macrophage PPAR gamma is required for normal skeletal muscle and hepatic insulin sensitivity and full antidiabetic effects of thiazolidinediones. J Clin Invest. 2007;117:1658–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schenk S, Saberi M, Olefsky J. Insulin sensitivity: modulation by nutrients and inflammation. J Clin Invest. 2008;118:2992–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Patsouris D, Li P, Thapar D, et al. Ablation of CD11c‐positive cells normalizes insulin sensitivity in obese insulin resistant animals. Cell Metab. 2008;8:301–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Muoio D, Newgard C. Mechanisms of disease: molecular and metabolic mechanisms of insulin resistance and beta‐cell failure in type 2 diabetes. Nat Rev Mol Cell Biol. 2008;9:193–205. [DOI] [PubMed] [Google Scholar]
- 38. Petersen K, Dufour S, Shulman G. Decreased insulin‐stimulated ATP synthesis and phosphate transport in muscle of insulin‐resistant offspring of type 2 diabetic parents. PLoS Med. 2005;2:e233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hsueh WA, Orloski L, Wyne K. Prediabetes: the importance of early identification and intervention. Postgrad Med. 2010;122(4):129–143. [DOI] [PubMed] [Google Scholar]
- 40. Turkish A. Nonalcoholic fatty liver disease: emerging mechanisms and consequences. Curr Opin Clin Nutr Metab Care. 2008;11:128–133. [DOI] [PubMed] [Google Scholar]
- 41. Schwimmer J, Deutsch R, Kahen T, et al. Prevalence of fatty liver in children and adolescents. Pediatrics. 2006;118:1388–1393. [DOI] [PubMed] [Google Scholar]
- 42. McGavock J, Victor R, Unger R, et al. Adiposity of the heart, revisited. Ann Intern Med. 2006;144:517–524. [DOI] [PubMed] [Google Scholar]
- 43. Schorr U, Blaschke K, Turan S, et al. Relationship between angiotensinogen, leptin and blood pressure levels in young normotensive men. J Hypertens. 1998;16:1475–1480. [DOI] [PubMed] [Google Scholar]
- 44. Yvan‐Charvet L, Massiéra F, Lamande N, et al. Deficiency of angiotensin type 2 receptor rescues obesity but not hypertension induced by overexpression of angiotensinogen in adipose tissue. Endocrinol Metab Clin North Am. 2009;150:1421–1428. [DOI] [PubMed] [Google Scholar]
- 45. Kurukulasuriya LR, Stas S, Lastra G, et al. Hypertension in obesity. Endocrinol Metab Clin North Am. 2008;37(3):647–662, ix. [DOI] [PubMed] [Google Scholar]
- 46. Nagel JM, Tietz AB, Goke B, et al. The effect of telmisartan on glucose and lipid metabolism in nondiabetic, insulin‐resistant subjects. Metabolism. 2006;55(9):1149–1154. [DOI] [PubMed] [Google Scholar]
- 47. Jandeleit‐Dahm KA, Tikellis C, Reid CM, et al. Why blockade of the renin‐angiotensin system reduces the incidence of new‐onset diabetes. J Hypertens. 2005;23(3):463–473. [DOI] [PubMed] [Google Scholar]
- 48. Engeli S, Boschmann M, Frings P, et al. Influence of salt intake on renin‐angiotensin and natriuretic peptide system genes in human adipose tissue. Hypertension. 2006;48(6):1103–1108. [DOI] [PubMed] [Google Scholar]
- 49. Scheen A. Prevention of type 2 diabetes mellitus through inhibition of the Renin‐Angiotensin system. Drugs. 2004;64:2537–2565. [DOI] [PubMed] [Google Scholar]
- 50. Yusuf S, Sleight P, Pogue J, et al. Effects of an angiotensin‐converting‐enzyme inhibitor, ramipril, on cardiovascular events in high‐risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med. 2000;342(3):145–153. [DOI] [PubMed] [Google Scholar]
- 51. Lindholm LH, Ibsen H, Borch‐Johnsen K, et al. Risk of new‐onset diabetes in the Losartan Intervention For Endpoint reduction in hypertension study. J Hypertens. 2002;20(9):1879–1886. [DOI] [PubMed] [Google Scholar]
- 52. Major outcomes in high‐risk hypertensive patients randomized to angiotensin‐converting enzyme inhibitor or calcium channel blocker vs diuretic. JAMA. 2002;288(23):2981–2997. [DOI] [PubMed] [Google Scholar]
- 53. Lindholm LH, Persson M, Alaupovic P, et al. Metabolic outcome during 1 year in newly detected hypertensives: results of the Antihypertensive Treatment and Lipid Profile in a North of Sweden Efficacy Evaluation (ALPINE study). J Hypertens. 2003;21(8):1563–1574. [DOI] [PubMed] [Google Scholar]
- 54. Vermes E, Ducharme A, Bourassa MG, et al. Enalapril reduces the incidence of diabetes in patients with chronic heart failure: insight from the Studies Of Left Ventricular Dysfunction (SOLVD). Circulation. 2003;107(9):1291–1296. [DOI] [PubMed] [Google Scholar]
- 55. Pfeffer MA, Swedberg K, Granger CB, et al. Effects of candesartan on mortality and morbidity in patients with chronic heart failure: the CHARM‐Overall programme. Lancet. 2003;362(9386):759–766. [DOI] [PubMed] [Google Scholar]
- 56. Julius S, Kjeldsen SE, Weber M, et al. Outcomes in hypertensive patients at high cardiovascular risk treated with regimens based on valsartan or amlodipine: the VALUE randomised trial. Lancet. 2004;363(9426):2022–2031. [DOI] [PubMed] [Google Scholar]
- 57. Scheen AJ. Renin‐angiotensin system inhibition prevents type 2 diabetes mellitus. Part 2. Overview of physiological and biochemical mechanisms. Diabetes Metab. 2004;30(6):498–505. [DOI] [PubMed] [Google Scholar]
- 58. Gillespie EL, White CM, Kardas M, et al. The impact of ACE inhibitors or angiotensin II type 1 receptor blockers on the development of new‐onset type 2 diabetes. Diabetes Care. 2005;28(9):2261–2266. [DOI] [PubMed] [Google Scholar]
- 59. Ravid M, Brosh D, Levi Z, et al. Use of enalapril to attenuate decline in renal function in normotensive, normoalbuminuric patients with type 2 diabetes mellitus. A randomized, controlled trial. Ann Intern Med. 1998;128(12 Pt 1):982–988. [DOI] [PubMed] [Google Scholar]
- 60. Lewis EJ, Hunsicker LG, Bain RP, et al. The effect of angiotensin‐converting‐enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med. 1993;329(20):1456–1462. [DOI] [PubMed] [Google Scholar]
- 61. Lewis EJ, Hunsicker LG, Clarke WR, et al. Renoprotective effect of the angiotensin‐receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med. 2001;345(12):851–860. [DOI] [PubMed] [Google Scholar]
- 62. Brenner BM, Cooper ME, de Zeeuw D, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001;345(12):861–869. [DOI] [PubMed] [Google Scholar]
- 63. Parving HH, Lehnert H, Brochner‐Mortensen J, et al. The effect of irbesartan on the development of diabetic nephropathy in patients with type 2 diabetes. N Engl J Med. 2001;345(12):870–878. [DOI] [PubMed] [Google Scholar]
- 64. Bosch J, Yusuf S, Gerstein HC, et al. Effect of ramipril on the incidence of diabetes. N Engl J Med. 2006;355(15):1551–1562. [DOI] [PubMed] [Google Scholar]
- 65. Zappe DH, Sowers JR, Hsueh WA, et al. Metabolic and antihypertensive effects of combined angiotensin receptor blocker and diuretic therapy in prediabetic hypertensive patients with the cardiometabolic syndrome. J Clin Hypertens (Greenwich). 2008;10(12):894–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Cooper ME. The role of the renin‐angiotensin‐aldosterone system in diabetes and its vascular complications. Am J Hypertens. 2004;17(11 Pt 2):16S–20S; quiz A2–4. [DOI] [PubMed] [Google Scholar]
- 67. Vijayaraghavan K, Deedwania PC. The renin angiotensin system as a therapeutic target to prevent diabetes and its complications. Cardiol Clin. 2005;23(2):165–183. [DOI] [PubMed] [Google Scholar]
- 68. Segura J, Ruilope LM. Obesity, essential hypertension and renin‐angiotensin system. Public Health Nutr. 2007;10(10A):1151–1155. [DOI] [PubMed] [Google Scholar]
- 69. Sharma AM, Engeli S. The role of renin‐angiotensin system blockade in the management of hypertension associated with the cardiometabolic syndrome. J Cardiometab Syndr. 2006;1(1):29–35. [DOI] [PubMed] [Google Scholar]
- 70. Sowers JR, Stump CS. Insights into the biology of diabetic vascular disease: what’s new? Am J Hypertens. 2004;17(11 Pt 2):2S–6S; quiz A2–4. [DOI] [PubMed] [Google Scholar]
- 71. Scheen AJ. Renin‐angiotensin system inhibition prevents type 2 diabetes mellitus. Part 1. A meta‐analysis of randomised clinical trials. Diabetes Metab. 2004;30(6):487–496. [DOI] [PubMed] [Google Scholar]
- 72. Holman RR, Haffner SM, McMurray JJ, et al. Effect of nateglinide on the incidence of diabetes and cardiovascular events. N Engl J Med. 2010;362(16):1463–1476. [DOI] [PubMed] [Google Scholar]
- 73. Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med. 1999;341(10):709–717. [DOI] [PubMed] [Google Scholar]
- 74. Pitt B. Effect of aldosterone blockade in patients with systolic left ventricular dysfunction: implications of the RALES and EPHESUS studies. Mol Cell Endocrinol. 2004;217(1–2):53–58. [DOI] [PubMed] [Google Scholar]
- 75. Engeli S. Role of the renin‐angiotensin‐aldosterone system in the metabolic syndrome. Contrib Nephrol. 2006;151:122–134. [DOI] [PubMed] [Google Scholar]
- 76. Engeli S, Bohnke J, Gorzelniak K, et al. Weight loss and the renin‐angiotensin‐aldosterone system. Hypertension. 2005;45(3):356–362. [DOI] [PubMed] [Google Scholar]
- 77. Intensive blood‐glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998;352(9131):837–853. [PubMed] [Google Scholar]
- 78. Holman RR, Paul SK, Bethel MA, et al. 10‐year follow‐up of intensive glucose control in type 2 diabetes. N Engl J Med. 2008;359(15):1577–1589. [DOI] [PubMed] [Google Scholar]
- 79. Susztak K, Raff AC, Schiffer M, et al. Glucose‐induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes. 2006;55(1):225–233. [PubMed] [Google Scholar]
- 80. Durvasula RV, Shankland SJ. Activation of a local renin angiotensin system in podocytes by glucose. Am J Physiol Renal Physiol. 2008;294(4):F830–F839. [DOI] [PubMed] [Google Scholar]
- 81. Lennon R, Pons D, Sabin MA, et al. Saturated fatty acids induce insulin resistance in human podocytes: implications for diabetic nephropathy. Nephrol Dial Transplant. 2009;24(11):3288–3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Sharma K, Ramachandrarao S, Qiu G, et al. Adiponectin regulates albuminuria and podocyte function in mice. J Clin Invest. 2008;118(5):1645–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. The effect of intensive treatment of diabetes on the development and progression of long‐term complications in insulin‐dependent diabetes mellitus. N Engl J Med. 1993;329(14):977–986. [DOI] [PubMed] [Google Scholar]
- 84. Viberti G, Mogensen CE, Groop LC, et al. Effect of captopril on progression to clinical proteinuria in patients with insulin‐dependent diabetes mellitus and microalbuminuria. European Microalbuminuria Captopril Study Group. JAMA. 1994;271(4):275–279. [PubMed] [Google Scholar]
- 85. Laffel LM, McGill JB, Gans DJ. The beneficial effect of angiotensin‐converting enzyme inhibition with captopril on diabetic nephropathy in normotensive IDDM patients with microalbuminuria. North American Microalbuminuria Study Group. Am J Med. 1995;99(5):497–504. [DOI] [PubMed] [Google Scholar]
- 86. Brichard SM, Santoni JP, Thomas JR, et al. Long term reduction of microalbuminuria after 1 year of angiotensin converting enzyme inhibition by perindopril in hypertensive insulin‐treated diabetic patients. Diabetes Metab. 1990;16(1):30–36. [PubMed] [Google Scholar]
- 87. Chaturvedi N. Randomised placebo‐controlled trial of lisinopril in normotensive patients with insulin‐dependent diabetes and normoalbuminuria or microalbuminuria. Lancet. 1997;349(9068):1787–1792. [PubMed] [Google Scholar]
- 88. Mann JF, Gerstein HC, Yi QL, et al. Progression of renal insufficiency in type 2 diabetes with and without microalbuminuria: results of the Heart Outcomes and Prevention Evaluation (HOPE) randomized study. Am J Kidney Dis. 2003;42(5):936–942. [DOI] [PubMed] [Google Scholar]
- 89. Mann JF, Schmieder RE, Dyal L, et al. Effect of telmisartan on renal outcomes: a randomized trial. Ann Intern Med. 2009;151(1):1–10, W1–2. [DOI] [PubMed] [Google Scholar]
- 90. Bakris G, Burgess E, Weir M, et al. Telmisartan is more effective than losartan in reducing proteinuria in patients with diabetic nephropathy. Kidney Int. 2008;74(3):364–369. [DOI] [PubMed] [Google Scholar]
- 91. Hansson L, Zanchetti A, Carruthers SG, et al. Effects of intensive blood‐pressure lowering and low‐dose aspirin in patients with hypertension: principal results of the Hypertension Optimal Treatment (HOT) randomised trial. HOT Study Group. Lancet. 1998;351(9118):1755–1762. [DOI] [PubMed] [Google Scholar]
- 92. Cushman WC, Evans GW, Byington RP, et al. Effects of intensive blood‐pressure control in type 2 diabetes mellitus. N Engl J Med. 2010;362(17):1575–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Patel A, MacMahon S, Chalmers J, et al. Effects of a fixed combination of perindopril and indapamide on macrovascular and microvascular outcomes in patients with type 2 diabetes mellitus (the ADVANCE trial): a randomised controlled trial. Lancet. 2007;370(9590):829–840. [DOI] [PubMed] [Google Scholar]
- 94. Mann JF, Schmieder RE, McQueen M, et al. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomised, double‐blind, controlled trial. Lancet. 2008;372(9638):547–553. [DOI] [PubMed] [Google Scholar]
- 95. Beulens JW, Patel A, Vingerling JR, et al. Effects of blood pressure lowering and intensive glucose control on the incidence and progression of retinopathy in patients with type 2 diabetes mellitus: a randomised controlled trial. Diabetologia. 2009;52(10):2027–2036. [DOI] [PubMed] [Google Scholar]
- 96. Wright AD, Dodson PM. Diabetic retinopathy and blockade of the renin‐angiotensin system: new data from the DIRECT study programme. Eye. 2009;24(1):1–6. [DOI] [PubMed] [Google Scholar]
- 97. Klein R, Zinman B, Gardiner R, et al. The relationship of diabetic retinopathy to preclinical diabetic glomerulopathy lesions in type 1 diabetic patients. Diabetes. 2005;54(2):527–533. [DOI] [PubMed] [Google Scholar]
- 98. Chew EY, Ambrosius WT, Davis MD, et al. Effects of medical therapies on retinopathy progression in type 2 diabetes. N Engl J Med. 2010;363(3):233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Chaturvedi N, Porta M, Klein R, et al. Effect of candesartan on prevention (DIRECT‐Prevent 1) and progression (DIRECT‐Protect 1) of retinopathy in type 1 diabetes: randomised, placebo‐controlled trials. Lancet. 2008;372(9647):1394–1402. [DOI] [PubMed] [Google Scholar]
- 100. Mauer M, Zinman B, Gardiner R, et al. Renal and retinal effects of enalapril and losartan in type 1 diabetes. N Engl J Med. 2009;361(1):40–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Wilkinson‐Berka JL, Tan G, Jaworski K, et al. Identification of a retinal aldosterone system and the protective effects of mineralocorticoid receptor antagonism on retinal vascular pathology. Circ Res. 2009;104(1):124–133. [DOI] [PubMed] [Google Scholar]
- 102. Higuchi A, Ohashi K, Kihara S, et al. Adiponectin suppresses pathological microvessel formation in retina through modulation of tumor necrosis factor‐alpha expression. Circ Res. 2009;104(9):1058–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Higuchi A, Ohashi K, Shibata R, et al. Thiazolidinediones reduce pathological neovascularization in ischemic retina via an adiponectin‐dependent mechanism. Arterioscler Thromb Vasc Biol. 2010;30(1):46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Study rationale and design of ADVANCE: action in diabetes and vascular disease‐‐preterax and diamicron MR controlled evaluation. Diabetologia. 2001;44(9):1118–1120. [DOI] [PubMed] [Google Scholar]
- 105. Baruch L, Anand I, Cohen IS, et al. Augmented short‐ and long‐term hemodynamic and hormonal effects of an angiotensin receptor blocker added to angiotensin converting enzyme inhibitor therapy in patients with heart failure. Vasodilator Heart Failure Trial (V‐HeFT) Study Group. Circulation. 1999;99(20):2658–2664. [DOI] [PubMed] [Google Scholar]
- 106. Stein M, Boulaksil M, Jansen JA, et al. Reduction of fibrosis‐related arrhythmias by chronic renin‐angiotensin‐aldosterone system inhibitors in an aged mouse model. Am J Physiol. 2010;299(2):H310–H321. [DOI] [PubMed] [Google Scholar]
- 107. Kai H, Kuwahara F, Tokuda K, et al. Diastolic dysfunction in hypertensive hearts: roles of perivascular inflammation and reactive myocardial fibrosis. Hypertens Res. 2005;28(6):483–490. [DOI] [PubMed] [Google Scholar]
- 108. Dzau VJ. Tissue renin‐angiotensin system in myocardial hypertrophy and failure. Arch Intern Med. 1993;153(8):937–942. [PubMed] [Google Scholar]
- 109. Chinnaiyan KM, Alexander D, McCullough PA. Role of angiotensin II in the evolution of diastolic heart failure. J Clin Hypertens (Greenwich). 2005;7(12):740–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Stanton A, Jensen C, Nussberger J, et al. Blood pressure lowering in essential hypertension with an oral renin inhibitor, aliskiren. Hypertension. 2003;42(6):1137–1143. [DOI] [PubMed] [Google Scholar]
- 111. Alberti G, Zimmet P, Shaw J, et al. The IDF Consensus Worldwide Definition of the Metabolic Syndrome. Brussels, Belgium: International Diabetes Federation; 2006. [Google Scholar]
- 112. Alberti KG, Zimmet P, Shaw J. Metabolic syndrome – a new world‐wide definition. A Consensus Statement from the International Diabetes Federation. Diabet Med. 2006;23(5):469–480. [DOI] [PubMed] [Google Scholar]
- 113. Grundy SM, Cleeman JI, Daniels SR, et al. Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation. 2005;112(17):2735–2752. [DOI] [PubMed] [Google Scholar]