

Case History
A 63‐year‐old woman with a long history of hypertension presented in 2006 with hypertensive urgency associated with palpitations and headache. A 24‐hour urine collection had markedly elevated total urine metanephrines (Table). A computed tomographic (CT) scan with contrast revealed an irregular, 3.6 cm, high attenuation, enhancing left adrenal mass with delayed washout (Figure 1). Laparoscopic left adrenalectomy in 2006 and pathological analysis confirmed a pheochromocytoma (PCC) with a PCC of the adrenal gland scaled score (PASS) 1 of 3 based on adipose tissue and capsular invasion. Follow‐up 24‐hour urine collections showed normal urine catecholamines.
Table.
Changes of Hormone Levels and Treatment
| 04/30/2006 | 06/08/2007 | 12/13/2007 | 12/16/2008 | 03/25/2009 | 08/08/2009 | |
|---|---|---|---|---|---|---|
| Urine free catecholamines | ||||||
| Epinephrine, μg/d Nl 0–20 | H 54 | H 26 | 11 | 15 | H 32 | <3 |
| Norepinephrine, μg/d Nl 15–80 | H 175 | 74 | 65 | 76 | 72 | 24 |
| Dopamine, μg/d Nl 65–400 | 347 | 350 | 274 | 339 | H 485 | 300 |
| Urine fractionated metanephrines | ||||||
| Total metanephrine, μg/d Nl <1300 | H 4580 | 804 | 727 | 1089 | 956 | 331 |
| Metanephrines, μg/d Nl <400 | H 2258 | 359 | 263 | 367 | H 459 | 34 |
| Normetanephrines, μg/d Nl <900 | H 2322 | 445 | 464 | 722 | 497 | 297 |
| Vanillylmandelic acid, mg/d | 4.6 | 4.8 | 5 | |||
| Free plasma metanephrines | ||||||
| Metanephrines, nmol/L Nl <0.5 | H 1.94 | H 0.52 | ||||
| Normetanephrines, nmol/L Nl <0.9 | H 2.65 | H 1.22 | ||||
| Dopamine, pg/mL Nl 0–20 | H 27 | |||||
Clinical course: After initial left adrenalectomy in June 2006 there was normalization of urinary catecholamines.
In March 2009 there was elevation of metanephrines and dopamine leading to the diagnosis of right adrenal pheochromocytoma and right adrenalectomy in July 2009.
In August 2009 there was evidence of normalization of urinary catecholamines and metanephrines.
Figure 1.
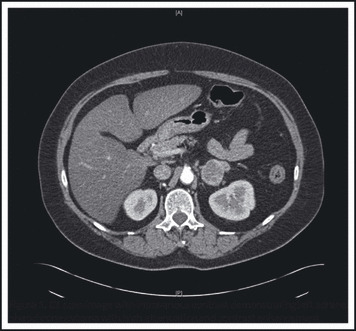
Computed tomography scan image with intravenous contrast demonstrating left adrenal pheochromocytoma with high attenuation and contrast enhancement.
In 2009 the patient was evaluated for impaired glucose tolerance and persistent hypertension despite triple drug therapy. She noted episodes of lightheadedness upon standing associated with palpitations and sweating, as well as weight loss. She then revealed a family history of thyroid cancer in her father and grandmother (Figure 2). Due to suspicion of recurrent or asynchronous second primary PCC and multiple endocrine neoplasia type 2 (MEN 2) syndrome, a 24‐hour urine was collected and showed mild elevation of catecholamines, including dopamine, in both plasma and urine. Genetic testing revealed a RET proto‐oncogene mutation at the Cys634Arg site. A calcitonin level was measured and found to be elevated.
Figure 2.

Family pedigree: patient’s father and grandmother had died and had known diagnosis of thyroid cancer (uncertain if medullary thyroid carcinoma [MTC]). Her 2 sisters and 2 daughters tested negative for RET mutation. Her brother died of heart failure complications and never developed thyroid cancer; however he had 4 sons who tested positive for RET mutation. Two of them had prophylactic thyroidectomy, one died of MTC, and the other one was found to have metastatic MTC.
A CT scan of the abdomen showed postoperative changes on the left side, and an enhancing mild nodularity in the right adrenal gland. An I‐131 iodine meta‐iodobenzylguanidine (MIBG) scan revealed mild uptake, superior and medial to the right kidney. Ultrasound and CT scan of the neck revealed a heterogeneous and multinodular thyroid with calcification (Figure 3). Following α‐blockade with phenoxybenzamine, she underwent a laparoscopic right adrenalectomy. Pathology confirmed a 2 cm right adrenal PCC (Figure 4) with a PASS of 6 due to a pattern of over 10% of the tumor volume, extension into periadrenal fat, profound nuclear pleomorphism, and nuclear hyperchromasia (Figure 5).
Figure 3.
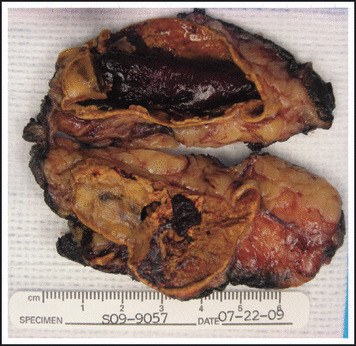
Right adrenal, adrenalectomy: evidence of 2.0×1.5×0.7 cm pheochromocytoma.
Figure 4.

Neck computed tomography scan image: evidence of enlarged, multinodular thyroid with calcification. No demonstrable cervical lymphadenopathy.
Figure 5.

Immunohistochemical staining of medullary thyroid cancer demonstrating positivity for calcitonin.
A total thyroidectomy specimen yielded a calcitonin positive medullary carcinoma without lymph node involvement. Currently, the patient is no longer taking blood pressure medications and a follow‐up 24‐hour catecholamine collection and serum calcitonin were normal. Family screening revealed a RET proto oncogene mutation in her brother and two of her nephews (Figure 2).
Discussion
MEN 2 is an autosomal dominant familial syndrome with age related penetrance. 2 The 3 recognized subtypes include MEN 2A, MEN 2B, and familial medullary thyroid carcinoma (MTC). MEN 2A (accounts for 60%–90% of MEN 2 cases), characterized by MTC, PCC, and hyperparathyroidism. MEN 2B is characterized by MTC, PCC, and a marfanoid habitus, mucosal neuromas, and ganglioneuromatosis of the gastrointestinal tract. Familial MTC has MTC as its only phenotypic expression. 3
Germ‐line mutations of the RET proto‐oncogene have been identified as the underlying cause of the syndrome. 4 This oncogene is expressed in normal and neoplastic tissue of neural crest origin which includes MTC and PCC. 5 It is located on chromosome 10q11.2 6 and it encodes for membrane receptor tyrosine kinase. 7 It consists of 21 exons and has an N‐terminal extracellular region with 4 cadherin‐like repeats and a cysteine rich region; a transmembrane domain; a cytoplasmic tyrosine kinase domain; and a C‐terminal tail. 8 The majority of families with MEN 2A have RET mutations in one of the cysteine domains of codons 609, 611, 618, 620 of exon 10, or codon 634 of exon 11. 4 This RET proto‐oncogene mutation results in a gain of function by permitting dimerization, cross phosphorylation and hence constitutive activation of the tyrosine kinase receptor in the absence of its known ligands. 9
Different mutations result in different phenotypical expressions. In a correlative survey study of 477 MEN 2 families, a mutation at codon 634 and the presence of MTC, PCC, and hyperparathyroidism was established. 10 In a series of 63 patients, a significant correlation was established between the specific RET proto‐oncogene mutation and the patient’s age at diagnosis of MTC. 11 Based on the youngest age at diagnosis, a classification into 3 groups was established: a high risk group (codons 634 and 618 with youngest age at diagnosis of 3 and 7 years, respectively), an intermediate risk group (codons 790, 620, 611 with corresponding youngest age at diagnosis of 12, 34, and 42 years), and a low risk group (codons 768 and 804 with youngest age at diagnosis of 47 and 60 years). No correlations were found between the RET proto‐oncogene mutation and the tumors, nodes, and metastases category or basal serum calcitonin levels before surgery.
The most common mutation in MEN 2A occurs in codon 634 according to the results of the International RET mutation consortium analysis. 10 Specific mutations in codon 634 also affect the disease course. This was demonstrated in a study conducted in Brazil in which 69 individuals with codon 634 mutation were analyzed. 12 Of the 8 families classified as MEN 2A, all but one had a mutation at codon 634. Individuals were classified according to the particular codon/amino acid substitution: C634Y, C634R, or C634W. Individuals with the specific C634R mutation had more significant metastases at diagnosis and presented at a younger age.
For the above reasons, molecular genetic testing has not only replaced screening of MTC with intravenous pentagastrin or calcium stimulation test but is also now being considered as an important tool that will optimize timing of prophylactic thyroidectomy at an earlier age in the high risk groups, particularly those with the C634R amino acid substitution. 11 , 12
MTC is generally the first manifestation, usually presenting as a thyroid nodule or cervical lymphadenopathy. Usual treatment consists of total thyroidectomy with central lymph node dissection due to the multicentric and bilateral nature of MTC in all the MEN 2 syndromes. 3 Screening for PCC should always be done prior to thyroidectomy as adrenalectomy should be performed first.
The majority of catecholamine secreting tumors are sporadic, however they are present in approximately 40% of patients with MEN 2 syndrome. 3 PCCs associated with MEN 2A are usually bilateral, with a lesser propensity toward malignancy, and only rarely are the first manifestation of this familiar syndrome. Diagnosis involves urine collection of total metanephrines (the most specific test) and fractionated plasma free metanephrines (the most sensitive test) with subsequent radiologic tests including CT and magnetic resonance imaging being both very sensitive (98%–100%) but only 70% specific. Finally MIBG has a higher specificity but is associated with a 10% false negative rate. 13
There is ongoing controversy as to whether patients with apparent sporadic PCC without family history and unilateral disease should undergo RET mutation testing. A review of 271 patients with apparent sporadic PCCs from population‐based registries in Germany and Poland revealed that 13 patients (4.8%) had a RET mutation. 14 On follow‐up it was found that 6 had a family history. Similarly, in our patient, family history was elicited for the first time when she was seen in our clinic. In addition, mutation carriers in sporadic PCC may be different according to the population studied. For that reason, genetic testing in the United States might not be necessary in those with unilateral disease, absence of syndrome signs or symptoms, and absence of family history.
Treatment of PCC consists of adrenalectomy. Some advocate unilateral adrenalectomy if the contralateral gland is normal in appearance, however others suggest bilateral adrenalectomy for apparent unilateral PCCs in MEN 2 syndromes. 15 Our patient is an example of someone who would have benefited from the latter approach.
The accepted definition for malignant PCC is the presence of metastatic disease, however a PASS grading system based on histological features has been created to distinguish benign from malignant PCCs. 1 Patients with clinically aggressive neoplasm had a PASS of ≥4 and those with a combined score of ≤3 didn’t develop metastatic disease in one study. 1 Thus, this score can potentially separate aggressive from more benign tumors.
Our patient had elevated levels of dopamine and urinary catecholamines on repeat studies in 2009. Pure dopamine secreting tumors are rare and only 10 cases have been described in the literature. 16 They are characterized by absence of dopamine β‐hydroxylase and a vesicular monoamine transporter required for uptake of norepinephrine into synaptic vesicles. They are usually not associated with hypertension and may present, as did our patient, with symptoms of orthostasis. Since the vesicular monoamine transporter is also used for uptake of MIBG, this could have explained the low uptake in our patient’s scan. Additionally, dopamine secreting tumors have a greater malignant potential which correlates with the PASS of 6 seen in our patient.
The current case underscores the importance of interpreting results in the setting of a high pretest probability of having MEN. After the patient underwent left adrenalectomy in 2006, repeat laboratory assessment showed only mild elevations in urine and plasma catecholamines and metanephrines. In addition, the CT scan showed no changes in the right adrenal gland when compared to previous studies, and an MIBG showed minimal uptake. In our patient’s case it was the persistence of symptoms, the genetic diagnosis of MEN 2A syndrome, and the accompanying higher likelihood of bilateral PCCs in this setting, in conjunction with the above laboratory and imaging studies that prompted a right adrenalectomy that evidenced a PCC.
Even though our patient presented with a high risk mutation and the C634R amino acid substitution, the diagnosis of MTC was made after diagnosis of PCC and there was no evidence of lymph node metastasis. However this certainly places her at a higher recurrence risk and follow‐up with calcitonin is required at least annually. Similarly, since she had an elevated PASS, screening of PCC with urine catecholamines is mandated at least yearly or with recurrence of symptoms.
Finally, the importance of genetic screening was evidenced in our patient’s family. Her 2 daughters and 2 sisters were negative for a RET mutation. Her brother died of congestive heart failure without known history of MTC or PCC. However, his 4 sons were positive for RET mutation and are currently being evaluated for MTC and PCC.
In summary, this case underscores the importance of ascertaining a family history of this familial syndrome in conjunction with molecular genetic testing. The diagnosis of this index case impacted positively on the patient and her family. Additionally, though virtually all PCCs associated with MEN 2 are benign, it is possible to develop recurrence or metastatic disease many years after resection. 17 In our patient, vigilant monitoring will be required given the C634R mutation of the RET proto‐oncogene, the elevated PASS, and the secretion of dopamine by the patient’s PCC.
References
- 1. Thompson L. Pheochromocytoma of the Adrenal gland Scaled Score (PASS) to separate benign from malignant neoplasms: a clinicopathologic and immunophenotypic study of 100 cases. Am J Surg Pathol. 2002;26:551–566. [DOI] [PubMed] [Google Scholar]
- 2. Ponder BAJ, Ponder MA, Coffer R, et al. Risk estimation and screening in families of patients with medullary thyroid carcinoma. Lancet. 1988;1:397–401. [DOI] [PubMed] [Google Scholar]
- 3. Raue F, Frank‐Raue K, Grauer A. Multiple endocrine neoplasia type 2: clinical features and screening. Endocrinol Metab Clin North Am. 1994;23:137–156. [PubMed] [Google Scholar]
- 4. Mulligan LM, Eng C, Healey CS, et al. Specific mutations of the RET proto‐oncogene are related to disease phenotype in MEN‐2A and FMTC. Nat Genet. 1994;6:70. [DOI] [PubMed] [Google Scholar]
- 5. Nakamura T, Ishizaka Y, Nagao M, et al. Expression of the ret proto‐oncogene product in human normal and neoplastic tissues of neural crest origin. J Pathol. 1994;172:255–260. [DOI] [PubMed] [Google Scholar]
- 6. Mole SE, Mulligan LM, Healey CS, et al. Localization of the gene for multiple endocrine neoplasia type 2A to a 480 kb region in chromosome band 10q11.2. Hum Mol Genet. 1993;2:247–252. [DOI] [PubMed] [Google Scholar]
- 7. Marsh DJ, Mulligan LM, Eng C. RET proto‐oncogene mutations in multiple endocrine neoplasia type 2 and medullary thyroid carcinoma. Horm Res. 1997;47:168–178. [DOI] [PubMed] [Google Scholar]
- 8. Iwamoto T, Taniguchi M, Asai N, et al. cDNA cloning of mouse ret proto‐oncogene and its sequence similarity to the cadherin superfamily. Oncogene. 1993;8:1087–1091. [PubMed] [Google Scholar]
- 9. Santoro M, Carlomagno F, Romano A, et al. Activation of RET as a dominant transforming gene by germline mutation of MEN2A and MEN2B. Science. 1995;267:381–383. [DOI] [PubMed] [Google Scholar]
- 10. Eng C, Clayton D, Schuffenecker L, et al. The relationship between specific RET proto‐oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. International RET mutation consortium analysis. JAMA. 1996;276:1575–1579. [PubMed] [Google Scholar]
- 11. Machens A, Gimm O, Hinze R, et al. Genotype‐phenotype correlations in hereditary medullary thyroid carcinoma: oncological features and biochemical properties. J Clin Endocrinol Metab. 2001;86:1104–1109. [DOI] [PubMed] [Google Scholar]
- 12. Punales MK, Granf H, Gross JL, et al. RET codon 634 mutations in multiple endocrine neoplasia type 2: variable clinical features and clinical outcome. J Clin Endocrinol Metab. 2003;88:2644–2649. [DOI] [PubMed] [Google Scholar]
- 13. Reisch N, Peczkowska M, Januszewicz A, et al. Pheochromocytoma: presentation, diagnosis and treatment. J Hypertens. 2006;24:2331–2339. [DOI] [PubMed] [Google Scholar]
- 14. Neumann HP, Bausch B, McWhinney SR, et al. Germ‐line mutations in nonsyndromic pheochromocytoma. N Engl J Med. 2002;346:1459–1466. [DOI] [PubMed] [Google Scholar]
- 15. Evans DB, Lee JE, Merrel RC. Adrenal medullary disease in multiple endocrine neoplasia type 2: appropriate management. Endocrinol Metab Clin North Am. 1994;23:167–176. [PubMed] [Google Scholar]
- 16. Dubois LA, Gray DK. Dopamine‐secreting pheochromocytomas: in search of a syndrome. World J Surg. 2005;29:909–913. [DOI] [PubMed] [Google Scholar]
- 17. Van Heerden JA, Roland CF, Carney JA, et al. Long‐term evaluation following resection of apparently benign pheochromocytoma(s)/paraganglioma(s). World J Surg. 1990;14:325–329. [DOI] [PubMed] [Google Scholar]