

Abstract
Arterial hypertension is associated with increased plasma levels of complement C3, C4, and C‐reactive protein (CRP). The aim of the study was to compare these laboratory markers in patients with resistant arterial hypertension (RAH) and controlled arterial hypertension (CAH). Patients with RAH (n=34), those with CAH (n=34), and 26 normotensive controls were included. White blood cell count, erythrocyte sedimentation rates, and blood levels of complement components C3, C4, and high‐sensitivity C‐reactive protein (hs‐CRP) were compared among the study groups. In the RAH group, serum C3 (183.9±47.5 mg/dL) and hs‐CRP (6.9±5.8 mg/L) were higher than in the CAH group (C3, 123.1±42.3 mg/dL; P<.001, hs‐CRP, 4.2±4.8; P=.021, respectively). Significant positive correlations between systolic blood pressure and C3 (r= 0.6481; P<.001) and hs‐CRP (r=0.3968; P=.02) were observed in the RAH group. RAH is associated with higher blood levels of C3 and CRP.
Refractory or resistant arterial hypertension (RAH) is conventionally defined as a systolic or diastolic blood pressure (BP) that remains uncontrolled (ie, >140/90 mmHg in the general population or >130/80 mm Hg in patients with diabetes or renal disease [serum creatinine >1.5 mg/dL in men or >1.3 mg/dL in women, or urinary protein excretion >300 mg over a 24‐hour period]) despite sustained therapy with at least 3 different classes of antihypertensive agents, one of which is a diuretic given in appropriate doses. 1 Although the prevalence of RAH is unknown, data from recent hypertension outcome trials indicate that it is greater than previously estimated. 1 , 2 Moreover, its prevalence increases with increasing severity of initial BP. A possible link between chronic vascular inflammation and arterial hypertension is now a subject of intensive study. Several inflammatory markers have been associated with arterial hypertension, including C‐reactive protein (CRP), 3 , 4 , 5 , 6 total white blood cells (WBCs), 7 , 8 and interleukin (IL)‐6. 9 Hypertension has been also associated with raised plasma levels of complement factors 3 and 4 (C3 and C4). 10 , 11 We hypothesized that RAH might be characterized as a condition with higher levels of “low‐grade systemic inflammation” than controlled arterial hypertension (CAH). Therefore, we compared blood levels of clinically relevant markers of inflammation such as CRP, WBC count, erythrocyte sedimentation rate (ESR), C3, and C4 between patients with RAH and CAH.
Materials and Methods
Study Population
The local ethics committee approved the protocol and the participants gave written informed consent according to Helsinki guidelines. Data were collected in primary clinics in the Ashdod region and medicine and geriatric departments of the Barzilai Medical Center (Ashkelon, Israel) between April 2005 and January 2007.
A total number of 700 individuals with a history of arterial hypertension of ≤3 years’ duration were screened. Patients had RAH diagnosed if they had persistent uncontrolled arterial hypertension, based on conventional office BP measurements of systolic BP (SBP) >140 mm Hg or/and diastolic BP (DBP) >90 mm Hg and had been receiving treatment for >3 months with a stable scheme consisting of ≥3 antihypertensive drugs of appropriate combination (including a diuretic) and dose (RAH group). In patients with type 2 diabetes (13 individuals), the definition of RAH was a BP level >130/80 mm Hg.
Patients receiving treatment for >3 months with ≤3 antihypertensive drugs with a stable regimen and with office SBP <140 mm Hg and DBP <90 mm Hg were considered to have CAH and were randomly recruited into the CAH group.
Normotensive controls were recruited from patients referred to the outpatient primary care clinics with several unrelated medical conditions, without known cardiovascular disorders and not receiving antihypertensive medications, whose office BP was <140/85 mm Hg.
Exclusion criteria were (1) fasting hyperglycemia (blood glucose >250 mg/dL); (2) liver or kidney disease; (3) cancer; (4) acute myocardial infarction or unstable angina within 6 months; (5) heart failure; (6) use of corticosteroids or other immunosuppressive therapy; (7) abnormal levels of plasma aldosterone, supine and standing plasma renin activity, or 24‐hour urinary catecholamines; and (8) anomalies on renal ultrasonography or renal arterial duplex scanning, which were done to exclude renal artery stenosis.
Blood Pressure
Measurements of BP were carried out at study entry, in the sitting position and after 10 minutes of rest, on the dominant arm with an automatic electronic sphygmomanometer (OMRON M4; OMRON Matsuzaka Co. Ltd., Matsuzaka City, Japan). Three consecutive BP measurements were performed and a mean of the measurements was used to define SBP and DBP. The clinic BP value was calculated as the average of the 3 readings taken at 3 different visits on days 0, 7, and 14.
To exclude the occurrence of “false” RAH due to a persistent white‐coat effect, all the patients in the RAH group also underwent home BP monitoring to assess both office and out‐of‐office BP. All patients were taught to measure BP correctly with their own sphygmomanometer at home. The home BP value was calculated as the average of 14 readings over 14 days. The white‐coat phenomenon was considered to be present when the difference between clinic BP and home BP was >20/10 mm Hg.
Body height and weight measurements were obtained with the patients wearing light clothing but no shoes. Body mass index was calculated.
Laboratory Analyses
Blood Hematology and Chemistry Analyses
Analyses were performed at the clinical laboratory of Barzilai Medical Center, Ashkelon, Israel.
Complement C3 and C4 Components
Venous blood was collected from all patients in heparin tubes. Tubes were centrifuged at 4°C for 15 minutes at 2000 g, and the plasma was collected and stored at −70°C within an hour of collection and subsequently thawed as required. The C3 and C4 protein components were stable at −70°C up to 3 months from the date of collection. Measurement of the complement C3 and C4 was performed by the ABICAP assay (Biognosis, Jülich, Germany) in serum obtained from venous blood of patients with RAH, patients with CAH, and controls. The normal standardized values for each component were 80 to 180 mg/dL for C3 and 10 to 40 mg/dL for C4. The assay was reproducible and accurate.
Blood CRP
The blood levels of high‐sensitivity CRP (hs‐CRP) were measured by immunonephelometry using BN systems (Dade Behring, Marburg, Germany) and the reference interval for hs‐CRP was 1 to 3 mg/L.
Data Analysis
Results are presented as mean±SD. Paired Student’s t‐tests were used to compare differences between means as appropriate. The Pearson test for correlations was used to analyze correlations between the variables. A P value of <.05 was considered significant. The statistical analysis was performed using the software Statistica 6 for Windows (StatSoft, Tulsa, OK).
Results
The demographic, clinical, and laboratory data of the study are shown in the Table. The group of the patients with RAH (n=34) and the group of patients with CAH (n=34) were similar with respect to their baseline demographic and laboratory characteristics (Table). Twenty‐six healthy individuals without arterial hypertension served as a control group.
Table.
Demographics of the Study Population
| Characteristics | RAH Group (n=34) | CAH Group (n=34) | P Value a | Control Group (n=26) | P Value b | P Value c |
|---|---|---|---|---|---|---|
| Age, y | 54.7±13.2 | 52.9±14.6 | .73 | 51.4±18.1 | ||
| Male/female | 19/15 | 16/18 | 13/13 | |||
| Arterial hypertension, No. (%) | 34 (100) | 34 (100) | 0 (0%) | |||
| Diabetes mellitus, No. (%) | 13 (38.2) | 11 (32.3) | .21 | 6 (23.1) | ||
| Smokers, No. (%) | 17 (50) | 15 (44.1) | .57 | 9 (34.6) | ||
| Body mass index, kg/m2 | 27.5±1.9 | 27.3±2.1 | .63 | 26.8±2.5 | ||
| SBP, mm Hg | 152.1±10.7 | 133.4±5.2 | .009 | 130.8±8.3 | ||
| DBP, mm Hg | 89.2±8.9 | 78.6±3.8 | .002 | 76.7±4.6 | ||
| Heart rate, beats/min | 75±8 | 73±7 | .28 | 73±9 | ||
| Medication, No. (%) | ||||||
| ACE inhibitors/ARBs | 34 (100) | 34 (100) | 0 (0%) | |||
| Hydrochlorothiazide | 34 (100) | 25 (73.5) | .038 | 0 (0%) | ||
| β‐Blockers | 19 (55.9) | 7 (20.6) | .014 | 0 (0%) | ||
| Calcium antagonists | 26 (76.5) | 18 (53) | .027 | 0 (0%) | ||
| α‐Blockers | 17 (50) | 5 (14.7) | .006 | 0 (0%) | ||
| Statins | 13 (38.2) | 9 (26.5) | .062 | 4 (15.4) | ||
| Laboratory values | ||||||
| C3, mg/dL | 183.9±47.5 | 123.1±42.3 | <.001 | 129.2±39.7 | <.001 | .46 |
| C4, mg/dL | 25.7±10.7 | 26.2±8.1 | .49 | 26.3±9.8 | .83 | .91 |
| CRP | 6.9±5.8 | 4.2±4.8 | .021 | 3.9±4.3 | .004 | .18 |
| ESR | 13.9±6.9 | 16.4±6.4 | .79 | 14.4±7.1 | .09 | .57 |
| WBCs, cells3/mL | 7.1±1.3 | 6.9±1.7 | .52 | 6.3±1.5 | .31 | .72 |
| Total cholesterol, mg/dL | 215.6±26.8 | 213.0±21.7 | .46 | 221.1±24.2 | .61 | .84 |
| LDL‐C, mg/dL | 121.8±20.5 | 117.1±18.6 | .61 | 122.5±17.9 | .79 | .81 |
| HDL‐C, mg/dL | 47.4±9.6 | 48.2±8.1 | .83 | 45.8±9.3 | .85 | .76 |
| Glucose, mg/dL | 110.8±22.4 | 112.4±20.9 | .39 | 104.9±18.1 | .52 | .29 |
Abbreviations: ACE, angiotensin‐converting enzyme; ARB, angiotensin receptor blocker; CAH, controlled arterial hypertension; CRP, C‐reactive protein; DBP, diastolic blood pressure; ESR, erythrocyte sedimentation rate; HDL‐C, high‐density lipoprotein cholesterol; LDL‐C, low‐density lipoprotein cholesterol; RAH, resistant arterial hypertension; SBP, systolic blood pressure; WBC, white blood cell. aBetween RAH and CAH groups; bbetween RAH and control groups; cbetween CAH and control groups.
BP Measurements
In the RAH group, mean clinic SBP and mean clinic DBP were 152.1±10.7 and 89.2±8.9 mm Hg, while in the CAH group BPs were 133.4±5.2 and 78.6±3.8 mm Hg (Table). In the RAH group, the mean home SBP was 147.9±8.3 mm Hg, and the mean home DBP was 87.9±9.2 mm Hg. None of the patients in the RAH group had a diagnosis of the white‐coat phenomenon.
Complement C3 and C4 Components
In the RAH group, there was a statistically significant positive correlation between SBP and blood levels of C3 (r=.6481; P=.001) (Figure 1). No correlation was observed between blood levels of C4 and BP in either study group. The mean and range of plasma complement C3 and C4 concentrations in the 3 groups are shown in Figure 2. Serum C3 in the RAH group (183.9±47.5 mg/dL) was significantly increased compared with both patients in the CAH group (C3, 123.1±42.3 mg/dL; P<.001) and normotensive controls (control group, C3, 127.3±50.1 mg/dL; P<.001) (Figure 2). There was no statistically significant difference between the C3 levels in the CAH and control groups. No significant differences in C4 were observed among any of the groups (Table).
Figure 1.
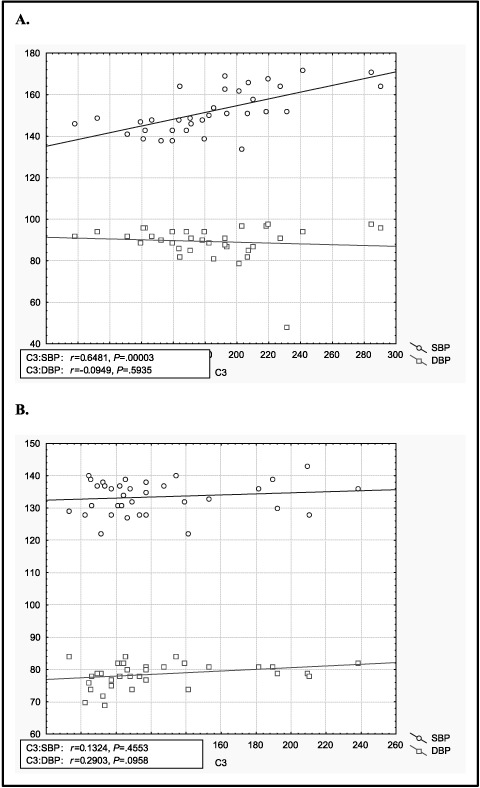
Correlation between C3 and blood pressure. (A) Resistant arterial hypertension group, (B) controlled arterial hypertension group. C3 indicates, blood C3 complement component level (mg/dL); SBP systolic blood pressure (mm Hg); DBP, diastolic blood pressure (mm Hg).
Figure 2.
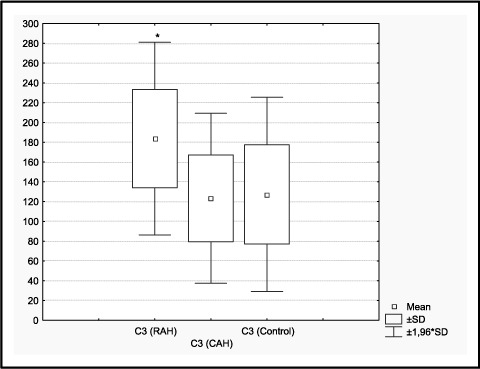
RAH indicates resistant arterial hypertension; CAH, controlled arterial hypertension.
C‐Reactive Protein
An increased level of serum hs‐CRP (6.9±5.8 mg/dL) was observed in the RAH group when compared with the CAH group (4.2±4.8 mg/dL; P=0.021) and control group (3.9±4.3 mg/dL; P=.004). Also, a significant positive correlation between SBP and CRP (r=0.3968; P=.02) was observed in the RAH group, but not in either the CAH or control group.
ESR and WBCs
There were no differences between ESR and WBC count between the 2 groups (Table). No correlation was observed between ESR, WBCs, and BP in all study groups.
Discussion
Inflammatory mechanisms are important participants in the pathophysiology of arterial hypertension. The aim of the study was to compare the laboratory markers of so‐called low‐grade systemic inflammation in RAH and CAH. We hypothesized that RAH may represent a state of increased inflammation and that at least part of antihypertensive treatment resistance is associated with higher levels of innate immune activation/inflammation. To check the hypothesis, we tested readily available and well‐recognized markers of low‐grade systemic inflammation, such WBC count, ESR, complement C3 and C4 components, and hs‐CRP in patients with RAH and CAH.
All patients with RAH underwent an extensive evaluation directed toward confirming true treatment resistance, including secondary causes of hypertension, assessment of treatment adherence, and use of good BP measurement techniques and home BP readings to exclude white‐coat effect. There were no significant differences between the RAH and CAH patients in duration of the hypertension, treatment adherence, or current medication use, including herbal and over‐the‐counter medications. Biochemical evaluation of the RAH and CAH groups included a routine metabolic profile (sodium, potassium, chloride, bicarbonate, glucose, blood urea nitrogen, and creatinine) and urinalysis. Paired morning plasma aldosterone and or plasma renin activity were similar in the RAH and CAH patients.
The results of our study indicate significantly higher levels of complement C3 and hs‐CRP in RAH than in CAH. The mechanisms by which hs‐CRP and more generally low‐grade systemic inflammation may render arterial hypertension resistant to treatment remains to be elucidated. Several mechanisms may be at work: CRP has been reported to decrease production of nitric oxide by endothelial cells 12 and thus might indirectly promote vasoconstriction, leukocyte adherence, platelet activation, oxidation, and thrombosis. 13 CRP also has proatherosclerotic properties by up‐regulating angiotensin type 1 and down‐regulating angiotensin type 2 receptor expression, thereby affecting the renin‐angiotensin system and contributing to the pathogenesis of hypertension. 14 CRP causes structural and functional changes in the endothelium via IL‐6, intercellular adhesion molecule 1, tumor necrosis factor α, and plasminogen activator inhibitor 1, and this may possibly lead to the development of resistant hypertension. 15 CRP, IL‐6, and tumor necrosis factor α further increase arterial stiffness and BP. 16 In addition, patients with lower CRP levels after statin therapy have better clinical outcomes than those with higher CRP levels, regardless of the resultant level of low‐density lipoprotein cholesterol. 17 , 18
The role of complement proteins in the pathophysiology of arterial hypertension is mainly unknown. The synthesis of complement C3 and C4 proteins is increased in response to inflammation and infection but at a slower rate than for traditional acute phase proteins. 19 , 20 , 21 In a recently published population‐based longitudinal study, plasma C3 was found to be associated with future BP increase and the development of hypertension. 22
Although complement factors C3 and C4 have been associated with atherosclerosis and cardiovascular risk factors, the precise pathophysiologic role of C3 in hypertension is largely speculative. Higher plasma C3 levels have been reported to be increased in hypertensive patients compared with normotensive controls. 10 In this study, we found significant differences in C3 levels between patients with RAH and normotensive persons but not between patients with CAH and normotensive persons. Although C4 serum concentration was reported to be increased in hypertensive patients in one previously published study, 23 we found no significant differences when compared with either normotensive controls or patients with CAH. It is to be noted, however, that the relative increase in serum C3 protein and decrease in C4 protein, as expressed in serum complement C3/C4 ratio, is considered to be a novel marker for recurrent cardiovascular events. 24
Among the components that mediate complement activation, C3 is the most abundant protein in the complement system and is also the most essential for the critical steps of the complement cascade. C4, which belongs to the classical pathway, is the second most abundant component in the complement system. Although the major site of C3 and C4 synthesis is in the liver, 25 , 26 other cells such as endothelial cells 27 and vascular smooth muscle cells 28 have also been shown to synthesize C3 and C4. Regulation of C3 and C4 production differed according to smooth muscle cell type. 26 In aortic smooth muscle cells, C3 production was enhanced by interferon γ, tumor necrosis factor α, and phorbol‐12‐myristate‐13‐acetate. 28 On the other hand, C4 production was stimulated only by interferon γ, but was reduced by tumor necrosis factor α and phorbol‐12‐myristate‐13‐acetate. 28 This dichotomy may indeed explain our finding of increased levels of C3, but not C4, associated with increased vascular mediated inflammation.
There are only a few experimental studies that have investigated a possible role of complement components on BP. In spontaneously hypertensive rats, selective expression of the complement C3 in arterial cells and clear involvement of this protein in mediating enhanced arterial smooth muscle growth and BP elevation was found 29 ; this suggests that the complement system is critical for the exaggerated growth and altered phenotype observed in arterial smooth muscle cells in arterial hypertension.
Resistant hypertension is a specific subgroup of essential arterial hypertension that remains understudied and is a major clinical problem despite the development of increasingly effective medications. Approximately 15% of the hypertensive population may be classified as having resistant hypertension. 1 The Antihypertensive and Lipid‐Lowering Treatment to Prevent Heart Attack Trial (ALLHAT) 30 reported that after 5 years of follow‐up, BP in 34% of participants remained uncontrolled (>140/90 mm Hg) on an average of 2 medications, with 27% of participants receiving ≥3 medications. 31 At the end of the study, approximately 8% of ALLHAT patients received prescriptions for ≥4 medications. In the Controlled Onset Verapamil Investigation of Cardiovascular End Points (CONVINCE) trial, 33% of patients had BP that remained uncontrolled (>140/90 mm Hg), with 18% of patients receiving ≥3 medications. 32 In the Valsartan Antihypertensive Long‐Term Use Evaluation (VALUE) study, goal BP had not been achieved in 40% of patients at 30 months of follow‐up. 33
The prognosis of patients with RAH compared with patients with CAH has not been specifically evaluated. Presumably, prognosis with long‐standing poorly controlled hypertension is associated with higher cardiovascular morbidity and mortality. The role of inflammation in the pathophysiology of RAH is unknown, though inflammation potentially could promote increased target organ damage. For example, increased levels of hs‐CRP in hypertensive patients have been associated with left ventricular hypertrophy and albuminuria. 34
This study has several significant limitations. First, the number of participants is too small to exclude chance findings as well as the occurrence of a type 2 error in relation to a number of negative findings (C4, ESR, and WBC count). Overcoming such a challenge will likely require a larger study.
As RAH may represent an extreme phenotype of a multifaceted and complex disease, it seems reasonable that more than one or perhaps a significant number of factors, including inflammatory and noninflammatory ones, may play a role in the pathogenesis of this syndrome. Although based on a relatively small number of patients, our results are provocative and support the importance of additional studies aimed at identifying laboratory markers that may relate to treatment resistance and ultimately to the development of new therapeutic strategies.
References
- 1. Moser M, Setaro J. Resistant or difficult to control hypertension. N Engl J Med. 2006;355:385–392. [DOI] [PubMed] [Google Scholar]
- 2. Cifkova R, Erdine S, Fagard R. Practice guidelines for primary care physicians: 2003 ESH/ESC hypertension guidelines. J Hypertens. 2003;21:1779–1786. [DOI] [PubMed] [Google Scholar]
- 3. Thorand B, Lowel H, Schneider A, et al. C‐reactive protein as a predictor for incident diabetes mellitus among middle‐aged men: results from the MONICA Augsburg cohort study, 1984–1998. Arch Intern Med. 2003;163:93–99. [DOI] [PubMed] [Google Scholar]
- 4. Freeman DJ, Norrie J, Caslake MJ, et al. West of Scotland Coronary Prevention Study: C‐reactive protein is an independent predictor of risk for the development of diabetes in the West of Scotland Coronary Prevention Study. Diabetes. 2002;51:1596–1600. [DOI] [PubMed] [Google Scholar]
- 5. Barzilay JI, Abraham L, Heckbert SR, et al. The relation of markers of inflammation to the development of glucose disorders in the elderly: the Cardiovascular Health Study. Diabetes. 2001;50:2384–2389. [DOI] [PubMed] [Google Scholar]
- 6. Han TS, Sattar N, Williams K, et al. Prospective study of C‐reactive protein in relation to the development of diabetes and metabolic syndrome in the Mexico City Diabetes Study. Diabetes Care. 2002;25:2016–2021. [DOI] [PubMed] [Google Scholar]
- 7. Schmidt MI, Duncan BB, Sharrett AR, et al. Markers of inflammation and prediction of diabetes mellitus in adults (Atherosclerosis Risk in Communities study): a cohort study. Lancet. 1999;353:1649–1652. [DOI] [PubMed] [Google Scholar]
- 8. Ford ES. Leukocyte count, erythrocyte sedimentation rate, and diabetes incidence in a national sample of US adults. Am J Epidemiol. 2002;155:57–64. [DOI] [PubMed] [Google Scholar]
- 9. Pradhan AD, Manson JE, Rifai N, et al. C‐reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA. 2001;286:327–334. [DOI] [PubMed] [Google Scholar]
- 10. Bozzoli C, Muscari A, Puddu GM, et al. Association of serum C3 and essential hypertension. G Ital Cardiol. 1992;22(12):1361–1366. [PubMed] [Google Scholar]
- 11. Schaadt O, Sorensen H, Krogsgaard AR. Association between the C3F‐gene and essential hypertension. Clin Sci (Lond). 1981;61(suppl 7):363s–365s. [DOI] [PubMed] [Google Scholar]
- 12. Verma S, Wang CH, Li SH, et al. A self‐fulfilling prophecy: C‐reactive protein attenuates nitric oxide production and inhibits angiogenesis. Circulation. 2002;106:913–919. [DOI] [PubMed] [Google Scholar]
- 13. Venugopal SK, Devaraj S, Yuhanna I, et al. Demonstration that C‐reactive protein decreases eNOS expression and bioactivity in human aortic endothelial cells. Circulation. 2002;106:1439–1441. [DOI] [PubMed] [Google Scholar]
- 14. Vongpatanasin W, Thomas GD, Schwartz R, et al. C‐reactive protein causes downregulation of vascular angiotensin subtype 2 receptors and systolic hypertension in mice. Circulation. 2007;115(8):1020–1028. [DOI] [PubMed] [Google Scholar]
- 15. Verma S, Li SH, Badiwala MV, et al. Endothelin antagonism and interleukin‐6 inhibition attenuate the proatherogenic effects of C‐reactive protein. Circulation. 2002;105:1890–1896. [DOI] [PubMed] [Google Scholar]
- 16. Mahmud A, Feely J. Arterial stiffness is related to systemic inflammation in essential hypertension. Hypertension. 2005;46(5):1118–1222. [DOI] [PubMed] [Google Scholar]
- 17. Magen E, Viskoper R, Mishal J, et al. Resistant arterial hypertension and hyperlipidemia: atorvastatin, not vitamin C, for blood pressure control. Isr Med Assoc J. 2004;6(12):742–746. [PubMed] [Google Scholar]
- 18. Ridker PM. Rosuvastatin in the primary prevention of cardiovascular disease among patients with low levels of lowdensity lipoprotein cholesterol and elevated high‐sensitivity C‐reactive protein: rationale and design of the JUPITER trial. Circulation. 2003;108:2292–2297. [DOI] [PubMed] [Google Scholar]
- 19. Ritchie RF, Palomaki GE, Neveux LM, et al. Reference distributions for complement proteins C3 and C4: a practical, simple and clinically relevant approach in a large cohort. J Clin Lab Anal. 2004;18:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Moshage H. Cytokines and the hepatic acute phase response. J Pathol. 1997;181:257–266. [DOI] [PubMed] [Google Scholar]
- 21. Gabrielsson BG, Johansson JM, Lönn M, et al. High expression of complement components in omental adipose tissue in obese men. Obes Res. 2003;11:699–708. [DOI] [PubMed] [Google Scholar]
- 22. Engstrom G, Hedblad B, Berglund G, et al. Plasma levels of complement C3 is associated with development of hypertension: a longitudinal cohort study. J Hum Hypertens. 2007;21(4):276–282. [DOI] [PubMed] [Google Scholar]
- 23. Tomaszewski M, Zukowska‐Szczechowska E, Grzeszczak W. Evaluation of complement component C4 concentration and immunoglobulins IgA, IgG, and IgM in serum of patients with primary essential hypertension. Pol Arch Med Wewn. 2000;103(5–6):247–251. [PubMed] [Google Scholar]
- 24. Palikhe A, Sinisalo J, Seppänen M, et al. Serum complement C3/C4 ratio, a novel marker for recurrent cardiovascular events. Am J Cardiol. 2007;99(7):890–895. [DOI] [PubMed] [Google Scholar]
- 25. Alper CA, Johnson AM, Birtch AG, et al. Human C’3: evidence for the liver as the primary site of synthesis. Science. 1969;163(864):286–288. [DOI] [PubMed] [Google Scholar]
- 26. Kulics J, Colten HR, Perlmutter DH. Counterregulatory effects of interferon‐gamma and endotoxin on expression of the human C4 genes. J Clin Invest. 1990;85(3):943–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ueki A, Sai T, Oka H, et al. Biosynthesis and secretion of the third component of complement by human endothelial cells in vitro. Immunology. 1987;61(1):11–14. [PMC free article] [PubMed] [Google Scholar]
- 28. Ueda Y, Nagasawa K, Tsukamoto H, et al. Production of the third and fourth component of complement (C3, C4) by smooth muscle cells. Immunology. 1996;89(2):183–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lin ZH, Fukuda N, Jin XQ, et al. Complement 3 is involved in the synthetic phenotype and exaggerated growth vascular smooth muscle cells from spontaneously hypertensive rats. Hypertension. 2004;44:42–47. [DOI] [PubMed] [Google Scholar]
- 30. Major outcomes in highrisk hypertensive patients randomized to angiotensin‐converting enzyme inhibitor or calcium channel blocker vs diuretic: the Antihypertensive and Lipid‐Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). JAMA. 2002;288:2981–2997. [DOI] [PubMed] [Google Scholar]
- 31. Cushman WC, Ford CE, Cutler JA. Success and predictors of blood pressure control in diverse North American settings: the Antihypertensive and Lipid‐Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). J Clin Hypertens (Greenwich). 2002;4:393–404. [DOI] [PubMed] [Google Scholar]
- 32. Black HR, Elliott WJ, Grandits G. Principal results of the Controlled Onset Verapamil Investigation of Cardiovascular End Points (CONVINCE) trial. JAMA. 2003;289: 2073–2082. [DOI] [PubMed] [Google Scholar]
- 33. Julius S, Kjeldsen SE, Brunner H. VALUE trial: long‐term blood pressure trends in 13,449 patients with hypertension and high cardiovascular risk. Am J Hypertens. 2003;16:544–548. [DOI] [PubMed] [Google Scholar]
- 34. Salles GF, Fiszman R, Cardoso CR, et al. Relation of left ventricular hypertrophy with systemic inflammation and endothelial damage in resistant hypertension. Hypertension. 2007;50(4):723–728. [DOI] [PubMed] [Google Scholar]