

Abstract
Insulin resistance plays a major role in the pathogenesis and clinical course of patients with type 2 diabetes (2DM) and essential hypertension. However, the syndromes differ in prevalence of insulin resistance, and associated insulin secretory response. Essentially all patients with type 2 diabetes are insulin resistant, whereas only approximately 50% of those with essential hypertension are insulin resistant. Furthermore, 2DM develops when the pancreatic β‐cell can no longer maintain the degree of compensatory hyperinsulinemia needed to prevent hyperglycemia. In contrast, the compensatory hyperinsulinemia that prevents most insulin resistant individuals from developing 2DM acts on normally insulin sensitive tissues in a manner that predisposes to the development of essential hypertension. This review will discuss these similarities and differences in some detail, as well as exploring the relationship among insulin resistance and related metabolic abnormalities in the pathogenesis of cardiovascular disease in patients with 2DM and essential hypertension. J Clin Hypertens (Greenwich). 2011;13:238–243. © 2011 Wiley Periodicals, Inc.
Insulin‐mediated glucose uptake by muscle varies more than 6‐fold in apparently healthy individuals, 1 with approximately 50% of the variability in insulin action 2 resulting from differences in degree of adiposity (25%) and physical fitness (25%). The remaining 50% is likely to be of genetic origin, with powerful familial and ethnic influences. 3 , 4 Type 2 diabetes develops when insulin‐resistant individuals cannot secrete the increased amounts of insulin needed to compensate for the insulin resistance. 5 , 6 , 7 However, the majority of insulin‐resistant individuals are able to maintain the degree of hyperinsulinemia required to prevent manifest decompensation of glucose homeostasis. Although compensatory hyperinsulinemia prevents the development of frank hyperglycemia in insulin‐resistant persons, insulin‐resistant/hyperinsulinemic individuals are at greatly increased risk of being somewhat glucose‐intolerant, with dyslipidemia characterized by a high plasma triglyceride (TG) and low high‐density lipoprotein cholesterol (HDL‐C) concentration and an increase in blood pressure (BP). 5 , 8 Glucose intolerance, the dyslipidemia associated with insulin resistance, and essential hypertension represent important risk factors for cardiovascular disease (CVD). The goal of this overview is to summarize the role that insulin resistance plays in the pathogenesis of type 2 diabetes and essential hypertension, as well as demonstrate the link between them and CVD. In the process, the similarities and the differences between these relationships will be emphasized.
Insulin Resistance in the Pathogenesis of Type 2 Diabetes and Essential Hypertension
Type 2 Diabetes
Although the role of insulin resistance in the pathogenesis of diabetes was initially demonstrated prior to World War II by Himsworth and colleagues, 9 , 10 , 11 it was not until approximately 45 years later that it became generally recognized that the majority of patients with impaired glucose tolerance or type 2 diabetes were insulin‐resistant. 12 , 13 In addition, it was shown that insulin resistance existed in nondiabetic first‐degree relatives of patients with type 2 diabetes 14 and that insulin resistance predicted the onset of frank type 2 diabetes. 6 , 7
Essential Hypertension
In 1966, Welborn and colleagues 15 demonstrated that 19 individuals with essential hypertension had significantly higher plasma insulin concentrations than a control population, but approximately another 20 years elapsed before there was confirmation of this finding. 16 During the next few years, it became clear that patients with essential hypertension, as a group, were insulin‐resistant, that insulin resistance predicted the development of essential hypertension, and that normotensive first‐degree relatives of patents with essential hypertension were insulin‐resistant. 17 , 18 , 19
On the other hand, there is a fundamental difference in the relationship between insulin resistance and the development of type 2 diabetes as compared with its role in the pathogenesis of essential hypertension. Namely, the overwhelming majority of patients with type 2 diabetes are insulin‐resistant and this is not the case as regards essential hypertension. There is no absolute definition of insulin resistance, but prospective studies in which the insulin suppression test (IST) was used to quantify insulin‐mediated glucose uptake indicated that one third of apparently healthy individuals who were most insulin‐resistant developed an adverse clinical outcome. 20 Insulin action as quantified by the IST is based on determining the steady‐state plasma glucose (SSPG) concentration in response to a continuous infusion of glucose, insulin, and octreotide—the higher the SSPG, the more insulin‐resistant the individual. Insulin action was quantified with this method in 126 patients with essential hypertension, 21 treated (n=70) or untreated (n=56), using SSPG concentration cut‐points derived from earlier studies to divide patients into tertiles. The two groups were not different in demographic characteristics, and the data in Figure 1 indicate that they also had a similar distribution of SSPG concentrations. Thus, approximately 15% of patients with essential hypertension, treated or untreated, were insulin‐sensitive (SSPG concentration <96 mg/dL), whereas approximately 50% of both groups were insulin‐resistant (SSPG concentration >180 mg/dL). Thus, the prevalence of insulin resistance in patients with essential hypertension is much lower than is the case in patients with type 2 diabetes, in which essentially all patients are insulin‐resistant.
Figure 1.
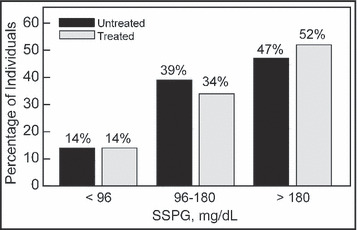
Comparison of the proportion (percent) of untreated and treated patients with essential hypertension in the 3 SSPG categories: <96 mg/dL=most insulin‐sensitive; 96mg/dL–180 mg/dL=intermediate; >180 mg/dL=most insulin‐resistant. Reprinted with permission.21
What is the “Second Hit”?
Type 2 Diabetes
Although the vast majority of patients with type 2 diabetes are insulin‐resistant, insulin resistance, per se, does not explain why fasting hyperglycemia develops. The magnitude of insulin resistance varies dramatically in nondiabetic individuals over a narrow range of fasting plasma glucose concentrations, 1 , 22 and the most insulin‐resistant nondiabetic individuals have values that overlap those of patients with frank type 2 diabetes. Whatever it is that protects the majority of insulin‐resistant individuals from developing type 2 diabetes remains a mystery, but it is clear that failure of the pancreatic β‐cell to maintain the degree of compensatory hyperinsulinemia required to overcome the insulin resistance is what leads to gross decompensation of glucose homeostasis. It is this inability of the insulin secretory mechanism to compensate for insulin resistance that provides the “second hit” that renders insulin‐resistant individuals frankly diabetic. This statement should not be interpreted to mean that absolute insulin deficiency characterizes patients with type 2 diabetes, and plasma insulin concentrations in patients with type 2 diabetes are often as high, or higher, than those of glucose‐tolerant individuals. 23
Essential Hypertension
Identifying the second hit that explains why normotensive, insulin‐resistant individuals develop elevated BP is much more elusive, at least partly because probably no more than half of patients with essential hypertension are insulin‐resistant. 21 However, there is information that may help explain some of the mechanistic links between insulin resistance and an increase in BP. For example, a number of relevant variables were compared in 19 healthy individuals in response to 5‐day periods of high (200 mmol/d) and low (25 mmol/d) sodium diets. 24 Mean changes found with a high‐sodium diet were not surprising and included an increase in weight, sodium excretion, and plasma concentrations of atrial natriuretic peptide (ANP) and plasma renin activity (PRA), and a decrease in plasma aldosterone concentration. The dramatic difference in salt intake did not affect either insulin action or the insulin response to an oral glucose challenge, but there was an increase in urinary nitrate excretion on the high‐salt diet of marginal statistical significance (P=.06).
Regression analysis of the whole group, however, showed that the more insulin‐resistant the person, the less the natriuretic response to the high‐sodium diet (−0.50, P=.04), and the greater the sodium‐induced weight gain (r=0.54, P=.03). Of interest, changes in PRA, ANP, and aldosterone were not significantly related to either sodium excretion or weight gain. Finally, the only two variables significantly associated with a sodium‐induced increase in mean arterial BP were weight gain (r=0.51, P<.05) and a decrease in urinary nitrate excretion (r=−0.77, P<.001). As before, there was no association between increases in BP and changes in PRA, aldosterone, or ANP. These results provide one possible explanation for what the second hit might be, at least in the case of salt‐sensitive hypertension, which increases the chances of an insulin‐resistant person developing essential hypertension. Specifically, the more insulin‐resistant the individual, the more likely they will be to retain salt and water, with subsequent volume expansion, independent of changes in ANP, PRA, or aldosterone. Furthermore, the greater the degree of volume expansion (weight gain), the higher the sodium‐induced increase in BP. Finally, since the increase in BP was inversely related to the change in urinary nitrate excretion, it can be argued that insulin‐resistant individuals will be at risk for volume‐related hypertension, and their ability to compensate will depend on how effective they are at increasing nitric oxide production. Consistent with this possibility is the finding that the plasma concentration of asymmetric dimethylarginine (ADMA), an endogenous inhibitor of nitric oxide synthase, is increased in the subgroup of patients with essential hypertension who are also insulin‐resistant. 25 An impaired nitric oxide response is only one possible second hit that explains why insulin resistant individuals are at increased risk for developing hypertension, and we clearly lack the knowledge to understand why some insulin‐resistant individuals become hypertensive and others do not.
Compensatory Hyperinsulinemia. Good? Bad? It Depends!
Type 2 Diabetes
As discussed above, type 2 diabetes occurs when insulin‐resistant individuals are not able to maintain the degree of compensatory hyperinsulinemia necessary to prevent frank decompensation of glucose tolerance. 5 , 6 , 7 , 12 If the evolutionary function of the pancreatic β‐cell is to secrete enough insulin to prevent the development of type 2 diabetes, it seems reasonable to conclude that compensatory hyperinsulinemia be considered as a beneficial adaptive response to the presence of insulin resistance: “a good guy.”
Essential Hypertension
One way to view compensatory hyperinsulinemia in insulin‐resistant, nondiabetic individuals is as a philanthropic effort on the part of the pancreatic β‐cell to stave‐off the ravages of type 2 diabetes. On the other hand, chronic hyperinsulinemia is not without its price. This paradoxical situation arises because not all tissues are equally insulin‐resistant. More specifically, the presence of defects in insulin‐mediated glucose uptake by muscle and insulin inhibition of adipose tissue lipolysis, 26 subsumed under the rubric of “insulin resistance,” does not mean that insulin‐regulated processes are abnormal in other tissues. For example, the kidney is not resistant to the ability of insulin to enhance sodium reabsorption, explaining why insulin‐resistant/hyperinsulinemic, nondiabetic individuals are at increased risk to retain salt and water (salt–sensitive). 24 Similarly, the sympathetic nervous system retains normal insulin sensitivity, favoring vasoconstriction as well as sodium retention. 27 , 28 Thus, the pancreatic β‐cell response mounted in an effort to maintain normal glucose homeostasis in an individual with adipose tissue and muscle insulin resistance increases the likelihood of that person developing essential hypertension.
Insulin resistance and CVD
Type 2 Diabetes
Although there is general agreement that CVD is the major cause of morbidity and morality in type 2 diabetes, controversy remains as to why this is the case. An obvious contender is hyperglycemia, accounting for the many studies in which efforts have been made to decrease CVD by achieving better glycemic control. Although the results of these studies could best be described as mixed, a pattern seems to emerge when a number of studies are subjected to a meta‐analysis. 29 With this approach, it appears that there is “a modest reduction in major macrovascular events with greater glucose lowering.” The greatest benefit was seen in nonfatal myocardial infarction, with no significant effect on stroke. These findings are not surprising. The CVD risk factors that are present in insulin‐resistant, nondiabetic individuals 5 , 8 do not disappear when fasting hyperglycemia ensues. Consequently, it is also not surprising that the multifactorial approach used in the Steno‐2 Study 30 was shown to decrease both cardiovascular mortality and cardiovascular events in patients with type 2 diabetes. It should also be noted that there was less emphasis on an aggressive improvement of glycemia in this study, and the decrease in hemoglobin A1c concentrations was not as great as in the studies focused only on intensive control of glucose concentrations. Thus, there appear to be a number of CVD risk factors that are operating in increasing risk of CVD in patients with type 2 diabetes.
Essential Hypertension
The link between essential hypertension and vascular disease differs in two important ways from the relationship seen in patients with type 2 diabetes.
Just as the focus in prevention of CVD in patients with type 2 diabetes has been on lowering glucose concentrations, efforts to improve vascular outcomes in patients with hypertension have focused on lowering BP. However, in contrast to the results of studies in type 2 diabetes, it appears that the clinical benefit of lowering BP has been much more dramatic in decreasing risk of stroke as compared with CVD. 31 There is no current consensus for this discordance (CVD vs stroke) in the effects of lowering BP on vascular outcome, and several different explanations have been proposed to account for this finding. For example, it has been argued that stroke is more directly related to BP than is myocardial infarction, and/or that the apparent difference in outcome is a function of the relatively short duration of the intervention trials. Another suggestion has been that thiazide diuretics and/or β‐receptor antagonists, drugs used in many of the controlled clinical trials, are associated with adverse changes in carbohydrate and lipid metabolism that tended to mitigate their beneficial effect on BP. Both of these possibilities may contribute to this apparent paradox, but there is a simpler explanation for the observation that the beneficial effect of lowering BP on CVD risk is less than might have been anticipated, one that is related to the heterogeneity of the essential hypertension phenotype.
Insulin resistance is present in the vast majority of patients with type 2 diabetes, whereas many patients with essential hypertension are insulin‐sensitive. 21 , 32 It is only the approximately 50% of patients with essential hypertension who are insulin‐resistant/hyperinsulinemic who are likely to have some degree of glucose intolerance, an atherogenic lipoprotein phenotype characterized by high triglycerides (TGs) and low high‐density lipoprotein cholesterol (HDL‐C) concentrations, smaller and denser low‐density lipoprotein (LDL) particles, and an exaggerated degree of postprandial lipemia, 21 , 31 , 32 and evidence of endothelial dysfunction. 25
An example of the impact of differences in insulin action on CVD risk factors is given in the Table, 21 in which the 126 patients described in Figure 1 were divided into two groups: IR (the 50% who were most insulin‐resistant; SSPG concentration >180 mg/dL) vs non‐IR patients (the insulin‐sensitive and intermediate tertiles; SSPG concentrations <180 mg/dL). By selection, the IR group had much higher SSPG concentrations, associated with significantly higher fasting plasma glucose, insulin, and TG concentrations, and lower HDL‐C concentrations. As clear as were the differences in CVD risk factors between the two groups, they were actually attenuated by including the intermediate group with the sensitive group.
Table.
Effect of IR on CVD Risk Factors in Patients With Essential Hypertension (Treated and Untreated)
| Variable | Group | P Value | |
|---|---|---|---|
| Non‐IR (n=64) | IR (n=62) | ||
| SSPG, mg/dL | 119±36 | 245±32 | <.001 |
| SBP, mm Hg | 142±17 | 143±18 | .60 |
| DBP, mm Hg | 81±9 | 85±13 | .11 |
| Glucose, mg/dL | 95±11 | 101±16 | .02 |
| >100 mg/dL, % | 28 | 47 | .03 |
| Insulin, μU/mL | 9±4 | 17±6 | <.001 |
| Cholesterol, mg/dL | 197±43 | 197±35 | .95 |
| LDL‐C, mg/dL | 124±36 | 120±31 | .52 |
| HDL‐C, mg/dL | 50±16 | 43±12 | .02 |
| <40 mg/dL, men, % | 39 | 52 | .35 |
| <40 mg/dL, women, % | 31 | 63 | .02 |
| TGs, mg/dL | 115±71 | 186±97 | <.001 |
| ≤150 mg/dL, % | 19 | 65 | <.001 |
Abbreviations: CVD, cardiovascular disease; DBP, diastolic blood pressure; HDL‐C, high‐density lipoprotein cholesterol; IR, insulin resistant (steady‐state plasma glucose concentration [SSPG] concentration ≥180 mg/dL); LDL‐C, low‐density lipoprotein cholesterol; non‐IR, not insulin‐resistant (SSPG concentration <180 mg/dL); SBP, systolic blood pressure; TGs, triglycerides. Values are expressed as mean ± standard deviation unless otherwise indicated.
Furthermore, there is evidence that it is the subset of patients with essential hypertension who are also insulin‐resistant that are more likely to develop CVD. Thus, patients with untreated essential hypertension, without clinical evidence of CVD, but with ischemic heart disease by Minnesota Code criteria, were insulin‐resistant (significantly higher SSPG concentrations during the IST, P<.001) as compared with a matched group of equally hypertensive individuals with normal electrocardiographic (ECG) findings. 33
The results of the oral glucose tolerance tests shown in Figure 2 indicate that the plasma glucose responses to a 75‐mg oral glucose challenge were also somewhat higher in the hypertensive patients with abnormal ECG findings as compared with the equally hypertensive group with normal ECGs, as well as a control group of normal individuals. Reflecting the higher SSPG concentrations, the plasma insulin responses were approximately twice as high in those with high BP and abnormal ECGs as compared with the other two groups. In addition, patients with high BP and abnormal ECG findings had higher TG concentrations (1.86±0.22 mmol/L vs 1.26±0.13 mmol/L; P<.03) and ratios of total cholesterol/HDL‐C (5.09±0.31 vs 3.85±0.27, P<.03) than did those with high BP and normal ECG findings.
Figure 2.
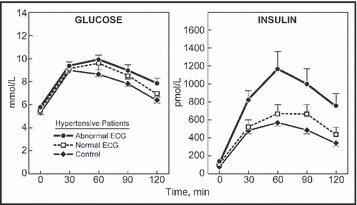
Plasma glucose and insulin concentrations before and after a 75‐g oral glucose challenge in control patients and two groups of patients with essential hypertension with normal or abnormal electrocardiograms (ECGs). Reprinted with permission.33
The link between insulin resistance and CVD in patients with essential hypertension is further supported by studies showing that a high TG value and a low HDL‐C concentration were significant predictors of CVD in patients with high BP, 34 , 35 and that patients with essential hypertension, who had low TG and high HDL‐C concentrations, were no more at risk for CVD than patients with normal BP and low TG and high HDL‐C concentrations. 35
Conclusions
Resistance to insulin‐mediated glucose uptake plays a major role in the pathogenesis and clinical course of patients with type 2 diabetes and essential hypertension. However, there are two fundamental differences in the impact of insulin resistance on the development of the clinical syndromes. First, insulin resistance is a fundamental defect in patients with type 2 diabetes; essentially all patients with type 2 diabetes are insulin‐resistant. In contrast, essential hypertension occurs in insulin‐sensitive individuals, and probably only 50% of persons with essential hypertension are insulin‐resistant. The second major difference is the importance of compensatory hyperinsulinemia in the genesis of type 2 diabetes vs essential hypertension. Type 2 diabetes occurs when the pancreatic β‐cell is no longer able to maintain the degree of hyperinsulinemia needed to overcome resistance to insulin‐mediated glucose disposal by muscle and insulin‐induced inhibition of adipose tissue lipolysis. Insulin‐resistant individuals who are able to secrete enough insulin to maintain normal or near‐normal glucose tolerance do not get type 2 diabetes, but the ongoing state of compensatory hyperinsulinemia acts on normally insulin‐sensitive tissues in a manner that predisposes to the development of essential hypertension. Perhaps the most important similarity is that therapeutic interventions directed only at improving the abnormality that defines the clinical syndrome—plasma glucose concentration or BP—are less effective than might have been anticipated. In that context, it seems important to address all of the CVD risk factors associated with insulin resistance/compensatory hyperinsulinemia that occur in patients with type 2 diabetes and in the subset of patients with hypertension who are also insulin‐resistant.
References
- 1. Yeni‐Komshian H, Carantoni M, Abbasi F, et al. Relationship between several surrogate estimates of insulin resistance and quantification of insulin‐mediated glucose disposal in 490 healthy, nondiabetic volunteers. Diabetes Care. 2000;23:71–175. [DOI] [PubMed] [Google Scholar]
- 2. Bogardus C, Lillioja S, Mott DM, et al. Relationship between degree of obesity and in vivo insulin action in man. Am J Physiol. 1985;248(Endocrinol Metab 11):E286–E291. [DOI] [PubMed] [Google Scholar]
- 3. Lillioja S, Mott DM, Zawadzki J, et al. In vivo insulin action is a familial characteristic in nondiabetic Pima Indians. Diabetes. 1987;36:1329–1335. [DOI] [PubMed] [Google Scholar]
- 4. Zoratti R, Godsland IF, Chaturvedi N, et al. Relation of plasma lipids to insulin resistance, nonesterified fatty acids, and body fat in men from three ethnic groups: relevance to variations in risk of diabetes and coronary artery disease. Metabolism. 2000;49:245–252. [DOI] [PubMed] [Google Scholar]
- 5. Reaven G. Role of insulin resistance in human disease. Diabetes. 1988;37:1595–1607. [DOI] [PubMed] [Google Scholar]
- 6. Warram JH, Martin BC, Krolewski AS, et al. Slow glucose removal rate and hyperinsulinemia precede the development of type II diabetes in the offspring of the diabetic parents. Ann Intern Med. 1990;113:909–912. [DOI] [PubMed] [Google Scholar]
- 7. Lillioja S, Mott DM, Spraul M, et al. Insulin resistance and insulin secretory dysfunction as precursors of non‐insulin‐dependent diabetes mellitus. N Engl J Med. 1993;329:1988–1992. [DOI] [PubMed] [Google Scholar]
- 8. Zavaroni I, Bonora E, Pagliara M, et al. Risk factors for coronary artery disease in healthy persons with hyperinsulinemia and normal glucose tolerance. N Engl J Med. 1989;320:702–706. [DOI] [PubMed] [Google Scholar]
- 9. Himsworth HP. The mechanism of diabetes mellitus. I. Lancet. 1939;2:1–6. [Google Scholar]
- 10. Himsworth HP. The mechanism of diabetes mellitus. II. The control of the blood sugar level. Lancet. 1939;2:65–68. [Google Scholar]
- 11. Himsworth HP. The mechanism of diabetes mellitus. III. Human diabetes mellitus. Lancet. 1939;2:171–175. [Google Scholar]
- 12. Ginsberg H, Olefsky JM, Reaven GM. Further evidence that insulin resistance exists in patients with chemical diabetes. Diabetes. 1974;23:674–678. [DOI] [PubMed] [Google Scholar]
- 13. Ginsberg H, Kimmerling G, Olefsky JM, et al. Demonstration of insulin resistance in untreated adult onset diabetic subjects with fasting hyperglycemia. J Clin Invest. 1975;55:454–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Laws A, Stefanick ML, Reaven GM. Insulin resistance in non‐diabetic relatives of patients with non‐insulin dependent diabetes mellitus. J Clin Endocrinol Metab. 1989;69:343–347. [DOI] [PubMed] [Google Scholar]
- 15. Welborn TA, Breckenridge A, Rubinstein AH, et al. Serum‐insulin in essential hypertension and in peripheral vascular disease. Lancet. 1966;1:1136–1137. [DOI] [PubMed] [Google Scholar]
- 16. Lucas CP, Estigarribia JA. Darga LL, Reaven GM. Insulin and blood pressure in obesity. Hypertension. 1985;7:702–706. [DOI] [PubMed] [Google Scholar]
- 17. Pollare T, Lithell H, Berne C. Insulin resistance is a characteristic feature of primary hypertension independent of obesity. Metabolism. 1990;39:167–174. [DOI] [PubMed] [Google Scholar]
- 18. Raitakari OT, Porkka KVK, Rönnemaa T, et al. The role of insulin in clustering of serum lipids and blood pressure in children and adolescents. Diabetologia. 1995;38:1042–1050. [DOI] [PubMed] [Google Scholar]
- 19. Allemann Y, Horber FF, Colombo M, et al. Insulin sensitivity and body fat distribution in normotensive offspring of hypertensive parents. Lancet. 1993;341:327–331. [DOI] [PubMed] [Google Scholar]
- 20. Facchini FS, Hua N, Abbas F, et al. Insulin resistance as a predictor of age‐related diseases. J Clin Endocrinol Metab. 2001;86:3574–3578. [DOI] [PubMed] [Google Scholar]
- 21. Lima NKC, Abbasi F, Lamendola C, et al. Prevalence of insulin resistance and related risk factors for cardiovascular disease in patients with essential hypertension. Am J Hypertens. 2009;22:106–111. [DOI] [PubMed] [Google Scholar]
- 22. Reaven GM, Hollenbeck CB, Chen Y‐DI. Relationship between glucose tolerance, insulin secretion, and insulin action in non‐obese individuals with varying degrees of glucose tolerance. Diabetologia. 1989;32:52–55. [DOI] [PubMed] [Google Scholar]
- 23. Reaven GM, Chen Y‐DI, Hollenbeck CB, et al. Plasma insulin, C‐peptide, and proinsulin concentrations in obese and nonobese individuals with varying degrees of glucose tolerance. J Clin Endocrinol Metab. 1993;76:44–48. [DOI] [PubMed] [Google Scholar]
- 24. Facchini FS, DoNascimento C, Reaven GM, et al. Blood pressure, nsulin resistance, and urinary nitrate excretion. Hypertension. 1999;33:1008–1012. [DOI] [PubMed] [Google Scholar]
- 25. Stuhlinger MC, Abbasi F, Chu JW, et al. Relationship between insulin resistance and an endogenous nitiric oxide synthase inhibitor. JAMA. 2002;287:1420–1426. [DOI] [PubMed] [Google Scholar]
- 26. Abbasi F, McLaughlin T, Lamendola C, et al. The relationship between glucose disposal in response to physiological hyperinsulinemia and basal glucose and free fatty acid concentrations in healthy volunteers. J Clin Endocrinol Metab. 2000;85:1251–1254. [DOI] [PubMed] [Google Scholar]
- 27. Reaven GM, Lithell H, Landsberg L. Hypertension and associated metabolic abnormalities – the role of insulin resistance and the sympathoadrenal system. N Engl J Med. 1996;334:374–381. [DOI] [PubMed] [Google Scholar]
- 28. Reaven GM. The kidney: an unwilling accomplice in syndrome X. Am J Kidney Dis. 1997;30:928–931. [DOI] [PubMed] [Google Scholar]
- 29. Turnbull FM, Abraira C, Anderson RJ, et al. Intensive glucose control and macrovascular outcomes in type 2 diabetes. Diabetologia. 2009;52:2288–2298. [DOI] [PubMed] [Google Scholar]
- 30. Gaede P, Lund‐Andersen H, Parving H‐H, et al. Effect of a multifactorial intervention on mortality in type 2 diabetes. N Engl J Med. 2008;358:580–591. [DOI] [PubMed] [Google Scholar]
- 31. Reaven GM. Insulin resistance/compensatory hyperinsulinemia, essential hypertension, and cardiovascular disease. J Clin Endocrinol Metab. 2003;88:2399–2403. [DOI] [PubMed] [Google Scholar]
- 32. Zavaroni I, Mazza S, Dall’Aglio E, et al. Prevalence of hyperinsulinaemia in patients with high blood pressure. J Intern Med. 1992;231:235–240. [DOI] [PubMed] [Google Scholar]
- 33. Sheuh WH‐H, Jeng C‐Y, Shieh S‐M, et al. Insulin resistance and abnormal electrocardiograms in patients with high blood pressure. Am J Hypertens. 1992;5:444–448. [DOI] [PubMed] [Google Scholar]
- 34. Gaziano JM, Sesso HD, Breslow JL, et al. Relations between systemic hypertension and blood lipids on risk of myocardial infarction. Am J Cardiol. 1999;84:768–773. [DOI] [PubMed] [Google Scholar]
- 35. Jeppesen J, Hein HO, Suadicani P, et al. Low triglycerides‐high high‐density lipoprotein cholesterol and risk of ischemic heart disease. Arch Intern Med. 2001;161:361–366. [DOI] [PubMed] [Google Scholar]