

Abstract
Background and Objective
Multiple studies highlighting the diagnostic utility of neurofascin-155 (NF155)–immunoglobulin G4 (IgG4) in chronic demyelinating inflammatory polyradiculoneuropathy (CIDP) have been published. However, few studies comprehensively address the long-term outcomes or clinical utility of NF155–immunoglobulin M (IgM) or NF155–immunoglobulin G (IgG) in the absence of NF155-IgG4. We evaluated phenotypic and histopathologic specificity and differences in outcomes between these NF155 antibody isotypes or IgG subclasses. We also compare NF155-IgG4-seropositive cases to other seropositive demyelinating neuropathies.
Methods
Neuropathy patient sera at Mayo Clinic were tested for NF155-IgG4, NF155-IgG, and NF155-IgM autoantibodies. Demographic and clinical data of all seropositive cases were reviewed.
Results
We identified 32 NF155 cases (25 NF155-IgG-positive [20 NF155-IgG4-positive], 7 NF155-IgM-seropositive). NF155-IgG4-seropositive patients clinically presented with distal more than proximal muscle weakness, positive sensory symptoms (prickling, asymmetric paresthesia, neuropathic pain), and gait ataxia. Cranial nerve involvement (11/20 [55%]) and papilledema (4/12 [33%]) occurred in many. Electrodiagnostic testing (EDX) demonstrated demyelinating polyradiculoneuropathy (19/20 [95%]). Autonomic involvement occurred in 45% (n = 9, median composite autonomic scoring scale score 3.5, range 1–7). Nerve biopsies from the NF155-IgG4 patients (n = 11) demonstrated grouped segmental demyelination (50%), myelin reduplication (45%), and paranodal swellings (50%). Most patients needed second- and third-line immunosuppression but had favorable long-term outcomes (n = 18). Among 14 patients with serial EDX over 2 years, all except one demonstrated improvement after treatment. NF155-IgG-positive, NF155-IgG4-negative (NF155-IgG-positive) and NF155-IgM-positive patients were phenotypically different from NF155-IgG4-seropositive patients. Sensory ataxia, neuropathic pain, cerebellar dysfunction, and root/plexus MRI abnormalities were significantly more common in NF155-IgG4-positive compared to myelin-associated glycoprotein (MAG)–IgM neuropathy. Chronic immune sensory polyradiculopathy (CISP)/CISP-plus phenotype was more common among contactin-1 neuropathies compared to NF155-IgG4-positive cases. NF155-IgG4-positive cases responded favorably to immunotherapy compared to MAG-IgM-seropositive cases with distal acquired demyelinating symmetric neuropathy (p < 0.001) and had better long-term clinical outcomes compared to contactin-1 IgG (p = 0.04).
Discussion
We report long-term follow-up and clinical outcome of NF155-IgG4 cases. NF155-IgG4 but not IgM or IgG cases have unique clinical–electrodiagnostic signature. We demonstrate NF155-IgG4-positive patients, unlike classical CIDP with neuropathic pain and dysautonomia common at presentation. Long-term outcomes were favorable.
Classification of Evidence
This study provides Class III evidence that NF155-IgG4-seropositive patients, compared to patients with typical CIDP, present with distal more than proximal muscle weakness, positive sensory symptoms, and gait ataxia.
Neurofascin-155 (NF155) autoantibodies are among the most common nodal and paranodal antibodies, comprising 4%1 to 18% of all chronic demyelinating polyradiculoneuropathy (CIDP) cases.2-9 Despite the growing utilization of these antibodies in clinical practice, studies evaluating long-term outcomes and histopathologic characterization are limited.10
In this study, we determine frequency of NF155 autoantibodies in a large demyelinating neuropathy cohort. We evaluate phenotypic and histopathologic specificity and differences in outcomes between NF155–immunoglobulin G4 (IgG4)–seropositive, NF155-pan–immunoglobulin G (IgG), and NF155–immunoglobulin M (IgM)–seropositive cases. We also compare phenotypic differences and outcomes in NF155-IgG4-positive cases to myelin-associated glycoprotein (MAG)–IgM and contactin-1–IgG–associated demyelinating neuropathies.
Methods
Our primary research question was to evaluate the clinical utility of NF155-IgG4 and NF155-IgM or NF155-IgG (in the absence of NF155-IgG4) autoantibodies and assess phenotypic and histopathologic differences in long-term outcomes among patients with these autoantibodies.
Patient Selection
We retrospectively reviewed the Mayo Clinic Rochester database for diagnostic codes designating demyelinating neuropathies from January 1, 1986, to January 1, 2019. On review of electronic medical records of the screened cases, 237 acute or chronic inflammatory demyelinating polyradiculoneuropathies (AIDP [n = 23], CIDP [n = 214]) were identified (cohort 1). An additional 173 patients with stored sera identified during this initial screening with an alternative neuropathy etiology or clinical presentation not consistent with AIDP/CIDP/chronic immune sensory polyradiculopathy (CISP)/CISP-plus phenotype were used as disease controls (eFigure 1, http://links.lww.com/WNL/B609). Peripheral nerve specialists (S.S., D.D., C.K., M.L.M., S.E.B., P.J.B.D.) prospectively sent samples for NF155 autoantibody evaluation among cases where nodal and paranodal antibody-mediated neuropathy was suspected between January 1, 2019, and March 31, 2021 (cohort 2).
NF155 Testing
Cohort 1
Sera of all patients were tested on in-house flow cytometry–based assay that utilized a stable cell line coexpressing human NF155 and GFP using a previously described approach.11 Patient and control sera were tested at 1:10 and 1:40 dilutions for NF155-IgG4 and NF155-IgG, respectively. The median fluorescence intensity of Alexa Fluor 647–conjugated anti-human IgG4 or IgG was determined for both nontransfected and transfected cells. The ratio of median fluorescence intensity values for green fluorescent protein (GFP)+ and GFP− cells was calculated as IgG or IgG4 binding index. IgG binding index of ≥5 and IgG4 binding index ≥2 were considered positive.
Cohort 2
Prospectively collected patient sera were evaluated by flow cytometry for NF155-IgG4 in-house or for NF155 IgG4, pan-IgG, and IgM antibodies at the Washington University clinical service neuromuscular laboratory by Western blot analysis.
Nerve Histology
Nerve biopsies from NF155 autoantibody–positive cases were performed and processed in our peripheral nerve laboratory using standard histologic stains and immunohistochemistry stains (CD45, CD68) prepared on paraffin and epoxy sections.12,13 Teased fibers were prepared and graded by previously defined pathologic criteria.14 Morphometric analysis was performed on epoxy sections using our Imaging System for Nerve Morphometry.15
Clinical Outcome
Inflammatory Neuropathy Cause and Treatment (INCAT) disability scores and gait assistance (assessed at final neurologic visit) were calculated for all patients. Favorable outcomes were defined by improvement ≥1 in INCAT disability scores calculated before and after immunotherapy.16 We defined disease relapse as >3 units increase in INCAT disability score after at least 3 months of disease stability when follow-up data were available.
Clinical Comparison Group
NF155-IgG4-seropositive (NF155-IgG4-positive) cases were compared to contactin-1-IgG and MAG-IgM-seropositive patients evaluated at Mayo Clinic within the study period. Contactin-1-IgG cases evaluated at Mayo Clinic between January 1, 1993, and March 31, 2021 (4 Mayo patients previously published)17 were included for clinical comparisons. Contactin-1 Euroimmun CBA was utilized for contactin-1 IgG and IgG4 testing.17 We searched the Mayo Clinic electronic database for patients seen in the neuromuscular and peripheral nerve specialty clinics between 2004 and 2018 with a distal acquired demyelinating symmetric neuropathy phenotype in the setting of an IgM monoclonal gammopathy of uncertain significance (n = 86). MAG IgM antibodies were measured using a clinically validated ELISA from Bühlmann Diagnostics. Positivity was defined using a cutoff of 1,500 Bühlmann titer units.
Statistical Analysis
For comparisons between groups, qualitative and continuous variables were analyzed using the Fisher exact test and Mann-Whitney U test, respectively.
Standard Protocol Approvals, Registration, and Patient Consents
The study was approved by the institutional Review Board of the Mayo Clinic, Rochester, Minnesota (institutional review board number 08-006647). Permission to participate was obtained from the patients.
Data Availability
Anonymized data not published within this article will be made available by request from any qualified investigator.
Results
Retrospective and Prospective Cohorts: Classification of NF155 Isotypes
Cohort 1
Among 214 patients with CIDP and 23 patients with AIDP evaluated from the retrospective cohort, 14 patients with CIDP were NF155-IgG4-positive (6.5%); none of the patients with AIDP tested positive. Three patients were NF155-IgG-positive and NF155-IgG4-negative (NF155-IgG-positive). All 173 disease control sera tested for NF155-IgG4 by flow cytometry were negative; 1 patient was NF155-IgG-positive.
Cohort 2
Fourteen NF155 autoantibody–positive patients were evaluated prospectively by peripheral nerve specialists. Six patients were NF155-IgG4-positive. One patient was only NF155-IgG-positive. An additional 7 patients were NF155-IgM-positive on Western blot but NF155-IgG- and NF155-IgG4-negative.
Neuropathic Pain, Distal Weakness, Sensory Ataxia, Tremor, and Cranial Nerve Involvement Were Common Among NF155-IgG4-Positive Patients
Demographic and clinical features for all NF155-IgG4-positive patients are shown in Table 1. Median age at symptom onset was 45.5 years, with 1 patient having symptom onset at 4 years of age. Nadir of symptoms severity occurred up to 12 months from symptom onset. Median time from symptom onset to demyelinating neuropathy diagnosis was 10 months (range 1–208 months). As a group, the most common clinical features were distal more than proximal muscle weakness (14 [70%]). A majority of the patients had distal more than proximal weakness (14 [70%]). Motor deficits involving lower extremities (18 [90%]) were much more common than upper extremity predominant presentations. Positive sensory disturbances (including prickling, asymmetric paresthesia, and neuropathic pain) with sensory as well as cerebellar ataxia and tremor were observed (10 [50%]). The tremor was an action tremor affecting the distal extremities, most often the fingers and hands. Cranial nerve and bulbar symptoms were common in 55% (11/20) and included one or more of the following: dysphagia or dysarthria (n = 7), diplopia or ptosis (n = 2), blurry vision (n = 1), and unilateral facial numbness (n = 2). Papilledema was also present (4/12 [33%]). Neurologic examination findings are shown in Table 1.
Table 1.
Demographic Clinical Characteristics of NF155-IG4-Seropositive Patients
Evaluation of NF155-IgG4-Seropositive Patients Revealed Demyelinating Polyradiculoneuropathy, Frequently With Autonomic Dysfunction
Nerve conduction studies (NCS) and needle EMG examinations were performed in all patients. All NF155-IgG4-seropositive cases had electrodiagnostic features consistent with demyelinating polyradiculopathy with slowed motor conduction velocities or prolonged distal latencies. All 20 patients met the European Federation of Neurologic Societies/Peripheral Nerve Society demyelinating electrophysiologic criteria for CIDP (18 definite, 1 probable, and 1 possible).18
Polyradiculoneuropathy with evidence of root and peripheral nerve involvement was the most common electrodiagnostic phenotype (n = 19 [95%]). More specifically, all patients had reduced or absent motor amplitudes (n = 20 [100%]), reduced or absent sensory amplitudes (n = 19 [95%]), and often prominent slowing of motor conduction velocities (n = 18 [90%]). F-wave latencies were prolonged (n = 16) or absent (n = 2) in 90% of patients (n = 18 [90%]). Other features noted in a minority of cases were temporal dispersion (n = 4 [20%]) and conduction block (n = 4 [20%]). Blink reflex studies were performed in 19 cases and were prolonged in a majority of patients (84% [16/19]). Median R1 latency was 17.3 milliseconds (range 11.6–27.4 milliseconds). In nearly all cases, fibrillation potentials were identified in proximal/paraspinal muscles, supporting radicular localization (n = 19 [95%]).
Autonomic symptoms were present in 45% (9/20) of patients and included orthostatic intolerance (defined by clinical history of syncope or lightheadedness) and blood pressure changes (n = 3), urinary incontinence and constipation (n = 3), and erectile dysfunction temporally associated with the date of symptom onset (n = 2). Six patients demonstrated abnormal autonomic reflex screen testing with median composite autonomic scoring scale (CASS) of 3.5 (range 1–7), indicating a moderate autonomic neuropathy. Testing demonstrated sudomotor dysfunction (n = 5) as well as cardiovascular adrenergic impairment (n = 3).
CSF studies were available in 19 NF155-IgG4-positive patients. CSF protein was elevated in all patients with median CSF protein of 224 g/dL (range 60–834 g/dL, normal <50 mg/dL). Nine patients had mild lymphocytic pleocytosis (median 5 cell/mm3; range 4–100 cells/mm3).
MRI Showed Diffuse Symmetric Enlargement, Nerve Root Thickening, and Nerve Root/Plexus Gadolinium Enhancement Among NF155-IgG4-Seropositive Patients
MRIs were available to review in 17 NF155-IgG4 (spine, n = 13 and plexus, n = 9) patients. Diffuse symmetric enlargement of the lumbosacral plexus or lumbosacral roots with marked T2 hyperintensity were a common finding in 82% (14/17) of patients. The most common abnormality identified on lumbar spine MRI was nerve root thickening (85% [11/13]) followed by nerve root gadolinium enhancement on axial images (46% [6/13]). In one patient, patchy, nonenhancing T2 hyperintensity was seen throughout the cervical spinal cord up to the brainstem. One patient had abnormal signal involving the oculomotor, trigeminal, facial, and vestibulocochlear nerves bilaterally on head MRI. Six patients had abnormal white matter changes that were considered as nonspecific, presumed to be secondary to small vessel disease. Overall, there were no radiologic findings supporting a concurrent central demyelinating disease in the 9 patients who underwent head MRI.
Histopathologic Analysis of NF155-IgG4-Seropositive Patients Showed Increased Rate of Segemental Demyelination, Active Axonal Degeneration, and Paranodal Expansions
Nerve biopsies of 11 NF155-IgG4-positive patients were performed at our institution. Median time from symptom to biopsy was 9 months (range <1–43 months). The biopsies included lumbar dorsal rootlet (n = 1), fascicular sciatic (n = 1), and distal cutaneous nerve biopsies (n = 11). Electron microscopy was performed to better demonstrate ultrastructural changes in one patient who had proximal and distal biopsy.
Teased fiber preparations were available for review in 10 of 11 NF155-IgG4-positive biopsies and showed increased rates of segmental demyelination (n = 5), highest in the fascicular sciatic biopsy (48%; Figure 1D) and lowest in distal cutaneous biopsies (mean 6%; range 0–25%, normal <2%). Increased frequencies of myelin reduplication were common and observed in 50% (n = 5) of biopsies, with areas of paranodal swellings (areas of thickened myelin adjacent to the node of Ranvier) in 50% (n = 5) (Figure 1, A–C), ranging in severity from most severe in the proximal site (Figure 1C) to less severe areas distally (Figure 1, A and B).
Figure 1. Neuropathic Histologic Changes in Neurofascin-155–Immunoglobulin G4–Positive Patients.

Teased fiber preparations from 4 patients taken from the sural (A, B) and sciatic (C, D) nerves. Shown are variable degrees of paranodal separation and myelin swelling (thickening) from least (A) to most involved (C) in the fascicular proximal sciatic biopsy. Also seen in the fascicular sciatic biopsy is frank segmental demyelination (D) where segments are void of osmium staining (light regions). Electron micrographs show myelin separation within the paranodal region (E) and the internodal areas (F). Also observed were Schwann cell cytoplasm around sites of prior unmyelinated fibers now filled with collagen-forming collagen pockets (G) indicative of small fiber involvement.
On the paraffin-embedded sections, the main pathologic abnormality was loss of large, myelinated nerve fibers seen in both distal and proximal biopsies (Figure 1A). Onion-bulb formations were not observed in any of the distal biopsies. Multifocal fiber loss was seen in 4/11 biopsies and subperineurial edema (Figure 2B) was present in 2 biopsies. Individual to small endoneurial and epineurial inflammatory collections were seen in 81% (9/11) of the distal nerve biopsies. In one patient (with severe sensory ataxia, tremor, and pseudoathetosis) who had 2 biopsies, a proximal fascicular sciatic biopsy and a sural nerve biopsy, large collections of epineurial mononuclear cells were seen in the proximal fascicular sciatic biopsy (Figure 2, C and D), with less inflammatory infiltrate in the distal sural site. Fascicular sciatic biopsy from another patient demonstrated multiple small endoneurial and epineurial inflammatory cell collections. The overall density of myelinated fibers ranged from normal to severely reduced.
Figure 2. Interstitial Histologic Alternations in NF155-IgG4-Positive and NF155-IgG-Positive (IgG4-Negative) Patients.
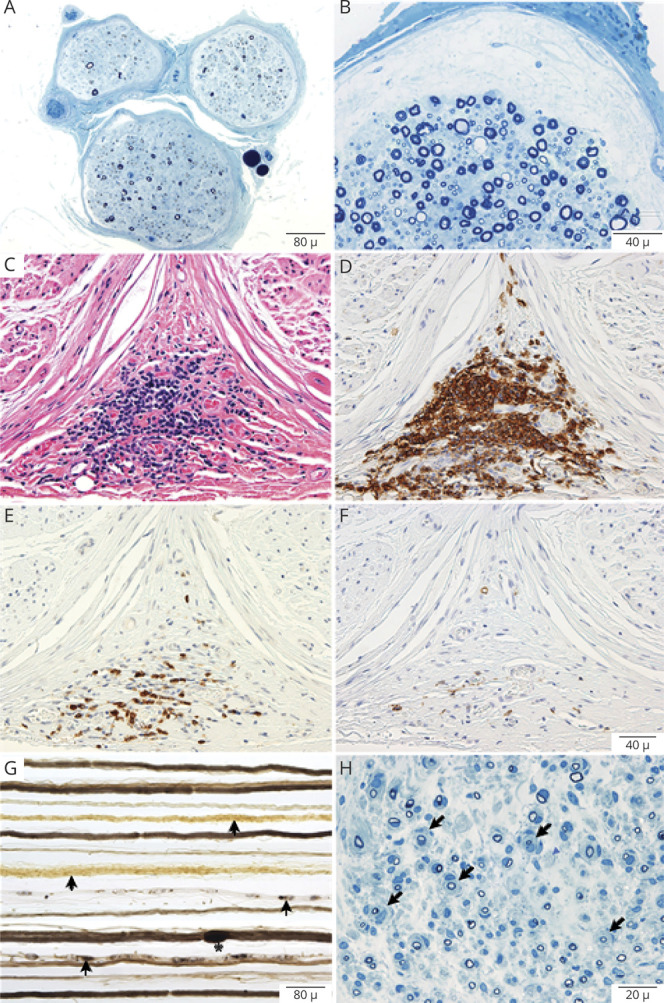
Representative photomicrographs from neurofascin-155 (NF155)–immunoglobulin G4 (IgG4) (A–F) and NF155–immunoglobulin G (IgG) (G–H) patients. Semithin epoxy sections (methylene blue) from a sural nerve biopsy show myelinated nerve fiber loss, active axonal degeneration, and slight multifocality. Sections B–F are taken from a targeted fascicular sciatic nerve biopsy of a 53-year-old man, NF155-IgG4-positive, with rapid onset of numbness, sensory and cerebellar ataxia, cranial nerve involvement, dysarthric speech, tremor, and pseudoathetosis, who was refractory to IV immunoglobulin (IVIg) treatment. However, with prolonged ongoing (several times per week) plasma exchange, he had dramatic recovery and regained independence and ability to ambulate. Semithin epoxy section (methylene blue) (B) shows subperineural edema and reduced density of myelinated nerve fibers with degenerating profiles (mild to moderate degree). Serial paraffin cross-sections (C–F) show a large perineurial inflammatory cell collection on hematoxylin & eosin (C) and on CD-45 (D) that was positive for T cells (CD3) (E) and B cells (CD20) (F). Photomicrographs G and H come from a 35-year-old man with progressive chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) and progressive onset of proximal more than distal weakness with falls who was NF155-IgG-positive (NF155-IgG4-negative). He initially was responsive to IVIg but over 3 years required increasing IVIg doses and was not helped with rituximab, and worsened with plasma exchange. (G) Teased nerve fiber preparations show chronic de- and remyelination with onion bulbs formations (arrows) and tomaculae (myelin reduplication, Asterix) as well as active axonal degeneration (arrowheads). (H) Methylene blue epoxy section shows the mixed pattern of onion bulbs (moderate sized onion bulbs interspersed among myelinated fibers without onion bulb), the typical pattern seen in inflammatory demyelinating neuropathies (CIDP). These findings show the different pathologic patterns seen in NF155-IgG4 (no onion bulbs) compared to non NF155-IgG4 disease (frequent onion bulb formations).
NF155-IgG4-Seropositive Cases Were IVIg Refractory, With Favorable Response to Second-Line Immunosuppressive Agents
All NF155-IgG4-positive patients were treated with immunotherapy. First-line immunotherapies included one or more of the following: IV immunoglobulin (IVIg, n = 18) alone or in combination with IV methylprednisolone (IVMP, n = 7) and plasmapheresis (PLEX, n = 2). Most cases (12/18) had initially favorable responses to IVIg but later became IVIg refractory despite continued treatments. Six patients had worsening of their symptoms even with initial IVIg treatments.
A subset of these patients required secondary long-term immunosuppression (n = 18 [90%]): mycophenolate mofetil (n = 7), rituximab (n = 5), azathioprine (n = 4), or cyclosporine (n = 2). Eight cases needed a third immunotherapy—corticosteroids (n = 4), plasmapheresis (n = 2), or IVIg (n = 2)—while on immunosuppression medications. As a group, most patients improved at the time of last follow-up (n = 18).
Relapses Were Common in NF155-IgG4-Seropositive Patients and Half of the Patients Required Gait Aid by 1 year, Indicating High Morbidity
Median duration of follow-up among NF155-IgG4-positive cases was 7 years (range 5–411 months) measured from symptom onset to last follow-up or death. Most of these cases had >3 years of follow-up with repeated examinations (n = 18 [90%]). Despite refractoriness to IVIg, long-term outcomes in most cases were favorable (n = 18 [90%]). One patient stabilized and one continued to deteriorate despite IVMP and azathioprine treatment. Death was reported in 3 patients (12%) by the end of the study period. One (patient with fascicular sciatic biopsy) died from disease complication due to central port infection after plasmapheresis treatment, 1 from septic shock due to diverticulitis, and cause of death was unclear in 1 patient who was lost to neurology follow-up.
Median number of relapses per patient was 1 (range 1 to 3 times). Median time to relapse was 58 months (range 11–220 months). Nearly 50% of patients experienced relapses in the first 4 years from symptom onset and in the first 2 years from starting treatment (Figure 3A). There was a significant difference in Inflammatory Neuropathy Cause and Treatment (INCAT) disability scores comparing patients who had relapse vs patients who did not experience relapses in their disease course (p = 0.02; Figure 3B).
Figure 3. Relapses Versus Time From Diagnosis in NF155-IgG4-Positive Patients.

Survival analysis comparison showing time to relapse from symptoms onset compared between 2 para/nodal antigens contactin-1 (CNTN1) and neurofascin-155 (NF155)–immunoglobulin G4 (IgG4)–positive patients. We demonstrate 50% of relapses occur in the first year from symptoms onset. The broken line stands for the median for each group (A). Whisker box plot comparison of Inflammatory Neuropathy Cause and Treatment (INCAT) scores at first neuromuscular visit shows higher score (more deficits) in those with relapses (p = 0.02) (B).
Most patients were using gait aids at neurologic presentation (n = 15 [75%]): cane (n = 1), walker (n = 5), or wheelchair (n = 9). At the time of last follow-up, 7 patients who previously used gait aids were independently ambulatory. Of the remaining 8 patients, 1 was using a cane, 3 wheelchairs, and 4 walkers at the time of last follow-up. Neurologic clinical outcomes measured at last neurology visit including needing assistance in daily tasks was markedly better after treatment (p < 0.001, confidence interval 1–5). Time from symptom onset to maximal gait aid dependence was reached in the first year of the disease in half of patients, signifying disease nadir (Figure 4A). Electrophysiologic testing with serial nerve conduction evaluations was available in 14 patients and the summated compound motor action potential (CMAP), a calculation in which CMAP amplitudes of all recurrent motor NCS are summated (a rough measure of axonal integrity), showed improvement after >24 months of treatment in NF155-IgG4-positive cases (Figure 4B).
Figure 4. Electrophysiologic and Clinical Outcomes NF155-IgG4 and Contactin-1–IgG Cohorts.

Time to maximal gait aid needed from symptoms onset comparing neurofascin-155 (NF155) and contactin-1 (CNTN1) positive patients (A), demonstrating that in the first 6 months, 50% of patients will reach disease nadir. (B) Compound motor action potential (CMAP) comparison with 4 intervals of time showing return to baseline more than 2 years after symptoms onset evaluated at our EMG laboratory.
Clinical Comparison of Nodo-Paranodopathies to Other Seropositive Demyelinating Polyradiculoneuropathies
To further characterize the clinical phenotype and to identify distinguishing features, we compared NF155-IgG4-positive patients to those with contactin-1–IgG-positive and MAG-IgM-seropositive demyelinating polyneuropathy (Table 2). NF155-IgG4-positive onset age was significantly younger compared to MAG-IgM (p < 0.001). Sensory ataxia, neuropathic pain, cerebellar dysfunction, and root/plexus MRI abnormalities were significantly more common in NF155-IgG4-positive compared to MAG-IgM neuropathy. CISP/CISP-plus phenotype was more common among contactin-1 neuropathies compared to NF155-IgG4-positive cases. INCAT disability scores at first encounter were significantly worse in the NF155-IgG4-positive compared to MAG-IgM groups (p < 0.001). Improvement in INCAT disability score between first and last INCAT was significantly greater in NF155-IgG4-positive patients vs the MAG-IgM patients (Figure 5). Gait aid at last follow-up improved in the NF155-IgG4 (from 75% to 40%) and worsened in the MAG-IgM group (10%–40%).
Table 2.
Demographic and Clinical Comparison of NF155-IgG4, Contactin-1–IgG, and MAG-IgM Patients

Figure 5. Functional Outcome of NF155-IgG4 Cases in Comparison to Other Seropositive Demyelinating Neuropathies.
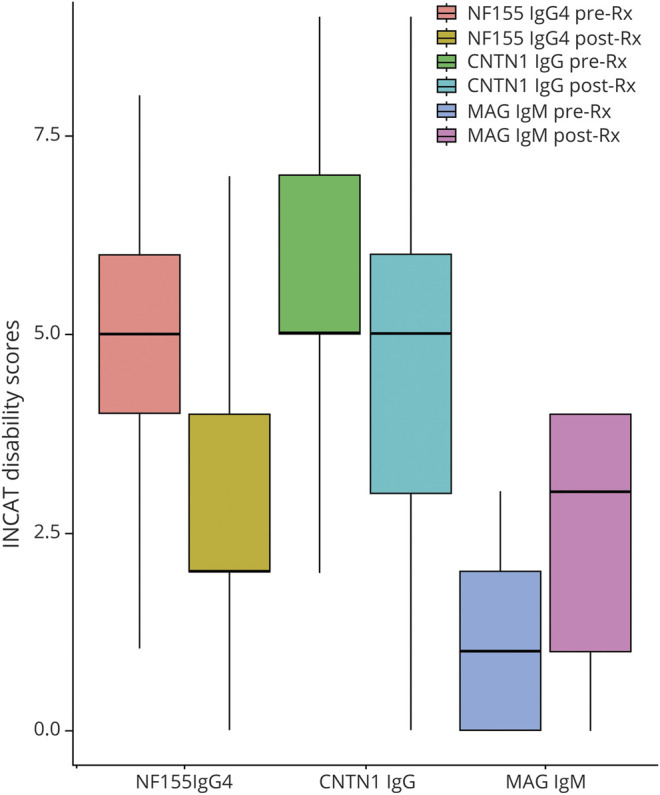
Inflammatory Neuropathy Cause and Treatment (INCAT) disability scores compared between first and last follow-up among 3 groups of patients, all with distal predominant presentation. Neurofascin-155 (NF155)–immunoglobulin G4 (IgG4)–positive and contactin-1–immunoglobulin G (IgG)–positive patients showed improvement in INCAT disability scores compared to myelin-associated glycoprotein (MAG)–immunoglobulin M (IgM) neuropathy patients.
NF155-IgG4-Negative Patient Characteristics
NF155-IgG-Positive or IgM-Positive Patients Were Not Clinically Distinct
A total of 5 patients were NF155-IgG-positive (NF155-IgG4-negative): 4 in retrospective cohort (1 disease control) and 1 in prospective cohort. The median age at diagnosis was 56.5 years (range 30–66 years). Median time from symptom onset to diagnosis was 11 months (range 4–61 months). Clinical presentations were consistent with motor predominant polyradiculoneuropathy (n = 2), subacute motor polyradiculoneuropathy (n = 1), length-dependent sensorimotor peripheral neuropathy (n = 1), and length-dependent sensory neuropathy (n = 1).
We identified 7 cases prospectively who were negative for NF55-IgG but NF55-IgM-positive. The median age at presentation of this group was 54 years (range 21–63 years). Clinically, they had various neurologic presentations, including motor-predominant axonal polyradiculoneuropathy (n = 2), sensory motor predominant axonal polyradiculoneuropathy (n = 1), sensorimotor length-dependent axonal polyneuropathies (n = 1), length-dependent sensory predominant axonal polyneuropathy (n = 1), brachial plexopathy (n = 1), and facial numbness (n = 1).
NF155-IgG-Positive or IgM-Positive Cases had No Distinctive Electrophysiologic, CSF, or Imaging Findings
Electrodiagnostic
Electrodiagnostic findings included prolonged F-wave latency (n = 1), reduced distal sensory and motor amplitudes (n = 2), and reduced motor amplitudes (n = 2). Conduction velocity showed slowing (n = 2) or was normal (n = 3). Blink studies were abnormal in 2 of 3 patients tested.
Among the NF155-IgM-positive patients, electrodiagnostic findings were length-dependent axonal polyneuropathy (n = 3), mixed axonal and demyelinating polyneuropathy (n = 2), upper trunk brachial plexopathy (n = 1), and normal (n = 1). Blink studies were normal in all tested patients (n = 5).
CSF Studies
CSF studies in NF155-IgG-positive patients were performed in 4 patients. CSF protein levels were elevated in 2 of the 4 patients; median CSF protein level was 200 g/dL (range 43–496 g/dL). Among the NF155-IgM-positive patients, CSF protein was only elevated in 1 patient; median CSF protein level was 43 g/dL (range 22–190 g/dL).
Imaging
Two NF155-IgG-positive patients with polyradiculoneuropathy had lumbar plexus MRIs; both showed enlarged proximal nerves and 1 also had increased T2 signal. MRI lumbar spine performed in 3 patients was unremarkable. Head MRI (n = 4) showed nonspecific T2/fluid-attenuated inversion recovery hyperintensity suggestive of microvascular changes (n = 2) or was normal (n = 2).
Among all 5 NF155-IgM-positive cases, MRI lumbar spine was available for review. One patient showed nonenhancing T2 hyperintensity. MRI lumbosacral plexus was abnormal in 2 of the 4 patients showing T2 hyperintensities of the proximal nerves without gadolinium enhancement. Head MRI brain showed nonspecific T2 hyperintensities (n = 2) or was normal (n = 2).
NF155-IgG-Positive or IgM-Positive Nerve Biopsy Findings Were Variable
Two sural nerve biopsies from NF155-IgG-positive cases were performed. Teased fiber preparations did not show evidence of active demyelination; teased fiber preparations and semithin sections showed onion bulbs (Figure 2, G and H) and subperineurial edema (n = 1). Small epineural inflammation was present in one and absent in the other. The density of myelinated fibers was normal in one and borderline normal in the other with normal fiber size distribution.
Two NF155-IgM-positive patients underwent nerve biopsies. One patient had superficial radial nerve biopsy, which showed severe axonal loss with no interstitial abnormalities. The other patient had a sural biopsy showing severely decreased density of myelinated fibers in a diffuse pattern with increased rate of axonal degeneration and multiple moderate perivascular epineurial mononuclear inflammatory cell collections. This was suggestive of inflammatory-immune process, which appeared to involve small vessel walls, raising the possibility of a microvasculitis.
NF155-IgG-Positive or IgM-Positive Clinical Outcomes Were Favorable; Few did Not Require Immune Treatment
Among the NF155-IgG-positive cases, 4 patients were treated with immunotherapy, and one did not require treatment. First-line treatments included IVIg (n = 4) and PLEX (n = 1). Two patients needed additional long-term immunosuppression (azathioprine and mycophenolate mofetil).
Among NF155-IgM-positive cases (n = 7), 6 were treated with immunotherapy. One patient who had facial numbness and borderline normal neurologic examination was not treated. Treatment included IVIg (n = 5) or IVMP (n = 1).
Among the NF155-IgG-positive cases, median follow-up time was 7 years (range 52–253 months). As a group, all had favorable outcome. One patient was only treated for pain (diagnosis length-dependent large fiber sensory peripheral neuropathy). Gait aids were utilized at presentation in 2 patients (walker and wheelchair) and these 2 patients continued to require a walker at last follow-up.
Among the NF155-IgM-positive cases, median follow-up time was 4 years (range 2–128 months). Treatment response evaluation was available in 5 NF155-IgM-positive cases who had neuromuscular clinic follow-ups. Four cases significantly improved with treatment. One patient with brachial plexopathy did not demonstrate muscle strength improvement but his neuropathic pain significantly improved. Gait aids at presentation were needed in 4 patients (cane = 3, wheelchair = 1) and at last follow-up 2 patients still required a cane.
Class of Evidence
This study provides Class III evidence that NF155-IgG4-seropositive patients, compared to typical CIDP, present with distal more than proximal muscle weakness, positive sensory symptoms, and gait ataxia.
Discussion
The occurrence of NF155 IgG4 seropositivity in our US-based tertiary center for patients with immune-mediated demyelinating neuropathies was 6.5%. This is similar to other studies, which reported a frequency of 4%–18%.1,4,8 Serologically, NF155 autoantibodies observed in this study were mainly IgG4 subtype, with a distinct clinical pathologic phenotype. Although a considerable proportion of NF155-IgM-positive or NF155-IgG-positive patients who were NF155-IgG4-negative had electrophysiologic, radiologic, and histopathologic findings suggestive of an immune or inflammatory neuropathy, but no consistent clinical phenotype, histopathologic findings or treatment response was observed (Table 2). More specifically, compared to NF155-IgG4-positive patients, there was significantly lower CSF protein (p = 0.01) and INCAT score at presentation (p < 0.001). This was also shown by other groups, with few patients with chronic indolent idiopathic or genetic neuropathies testing positive for NF155-IgG/NF155-IgM antibodies.19 One hypothesis is that there is a transient IgM or IgG1-3 humoral response to NF155 antigens among neuropathies of varied etiologies that may be secondary to paranodal damage and antigenic release.20
Our study shows high frequency of neuropathic pain and autonomic symptoms (mainly sudomotor dysfunction, 45%) in NF155-IgG4-positive patients, which clinically suggests small fiber involvement in this disease. Median CASS scores were higher than described in classic CIDP.21 Small fiber involvement was also a part of the pathologic abnormalities seen on electron microscopy, showing nonmyelinating Schwann cell cytoplasm infiltrated by bundles of collagen fibers instead of axons, so-called “collagen pockets” (Figure 1G). This indicates that in addition to more distal pattern of muscle weakness and sensory ataxia, autonomic involvement or pain as initial symptom should raise a concern for NF155-IgG4 positivity.21 Presence of papilledema and demyelinating features on electrodiagnostic testing with only a minority of studies demonstrating conduction block suggests that NF155-IgG4 testing should also be considered among patients clinically suspected to have POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, and skin changes).
Blink reflex latency measurements as a reflection of subclinical demyelination were recently reported to be prolonged in a small cohort of NF155-IgG4-positive patients.22 Our findings agree with this observation and we show that the majority of our patients (84%) had prolonged blink responses. This can be a useful tool to detect proximal demyelination in the presence of severe axonal degeneration in distal nerves, which is encountered in many NF155-IgG4-positive patients.
Fascicular sciatic biopsy taken from a severely affected patient with sensory ataxia and tremor demonstrated diffuse loss of myelinated fibers, thickened perineurium, and a large degree of subperineurial edema, similar to what has been previously shown in distal sensory NF155 nerves (Figure 2A).9 What has not been previously shown were the high frequency of segmental demyelination (48%) and associated large epineurial perivascular inflammatory collections supportive of proximal inflammatory pathology (Figure 2C). Pathologically, more severe neurologic involvement in a proximal fascicular sciatic biopsy than in distal cutaneous nerve biopsies of NF55-IgG4-positive patients is similar to what is seen in classical CIDP.23 We also show various degrees of paranodal swellings (myelin thickening) on teased nerve fibers from proximal and distal biopsies among NF55-IgG4-positive patients. The demyelination on NCS and hypertrophy of nerves on MRI made us expect biopsy specimens would show typical hypertrophic features of onion bulb formation, but those were absent in NF155-IgG4-positive cases but present in an NF155-IgG-positive patient (Figure 2H), highlighting that distinct histopathologic changes may be limited to NF155-IgG4-positive cases, and NF155-IgG-positive or NF155-IgM-positive nerve biopsy findings may vary based on associated neuropathy phenotype.
Treatment response in our cohort confirms previous observations that NF155-IgG4-positive patients are IVIg refractory,4 with most patients worsening after initial partial improvement. As a group, they require more extensive immunotherapy and show clinical improvement after second- or third-line immunotherapies are added, comparable to what has been previously described.9 Time to clinical improvement was prolonged compared to classic CIDP24 and in most patients it was longer than 2 years from symptom onset. Similarly, electrophysiologic improvement in the form of summated CMAP was slow and was typically not seen until 12 months after treatment initiation (Figure 4B). The requirement of more aggressive and prolonged immunotherapy course to bring about significant improvement is likely secondary to early axonal loss as shown by histopathologic preparation and electrodiagnostic studies.25
Relapse rates in NF155 nodo/paranodopathies are not well described, with several reports describing clinical declines only when immune treatment was stopped.8 In our study, the number of patients with relapsing course was high (45%), and the relapses commonly occurred in the first 2 years after diagnosis (Figure 3A). Furthermore, we found that patients with longer time from symptom onset to treatment and more neuropathic disability at initial assessment had more relapses, suggesting that early recognition and appropriate treatment is an important factor in outcome.
Both NF155 and contactin-1 proteins play a role in the formation and stability of sodium channel clusters located at nodes of Ranvier.26 Antibodies against these proteins affect the conduction, probably by disturbing axoglial contacts and myelin insulation.27,28 Supported by in vitro studies,27 we speculate the mechanisms of nerve injury are secondary to channel dysfunction, eventually causing axonal damage.29 This is also supported by our pathologic studies demonstrating myelin swelling and thickening in the paranodal areas with multiple focal demyelinating changes (Figure 1C).
We compared the clinical features of other seropositive demyelinating polyradiculoneuropathies to NF155-IgG4-positive patients (Table 2). Younger age at neuropathy onset, sensory ataxia, cerebellar signs, and neuropathic pain at onset were more common among NF155-IgG4-positive patients in comparison to MAG-IgM-positive distal neuropathy. Presenting features sensory ataxia and neuropathic pain were common among NF155-IgG4 and contactin-1–IgG-seropositive patients, making clinical distinction between these 2 conditions during initial presentation difficult. However, 4 of the 9 contactin-1 cases had a CISP or CISP-plus phenotype17,30 and 2 of the 9 cases had membranous glomerulonephropathy versus none of the NF155-IgG4-positive patients. Disease progression among NF155-IgG4 and contactin-1 IgG tends to be more rapid than in MAG-IgM, which has an insidious progression. Furthermore, both NF155-IgG4 and contactin-1 IgG cases were more immunotherapy responsive in comparison to MAG-IgM-associated neuropathy (Figure 5). Comparing neurologic outcomes between NF155-IgG4- and contactin-1–IgG-seropositive patients showed that the latter had a trend towards a more aggressive clinical course.
A limitation of our study is its retrospective nature. The data collected are not population-based; therefore, the frequency of NF155 may be skewed. Evaluation of assay clinical specificity during assay validation is critical; these data are not readily available for antibody evaluations performed at outside laboratories. As has been demonstrated in the past for other cell surface antibodies,31,32 ELISAs may generate a higher number of false-positive results. Therefore, multicenter studies comparing assay metrics of the different testing methodologies utilized for detection of NF155 autoantibodies should be performed.
NF155-IgG4 is a highly specific biomarker of clinically, electrophysiologically, and histopathologically distinct demyelinating polyradiculoneuropathy. However, the clinical utility of NF155-IgM or NF155-IgG without coexisting NF155-IgG4 remains unclear. Therefore, NF155-IgG-positive/NF155-IgM-positive patients should be managed only on the basis of their clinical and electrodiagnostic phenotype. The clinical course and outcome are different from typical CIDP, with most patients requiring long-term immune treatment (<2 years) and the utilization of more aggressive immunotherapy.
Acknowledgment
The authors thank S. Vinje for administrative assistance.
Glossary
- AIDP
acute inflammatory demyelinating polyradiculoneuropathy
- CASS
composite autonomic scoring scale
- CIDP
chronic inflammatory demyelinating polyradiculoneuropathy
- CISP
chronic immune sensory polyradiculopathy
- CMAP
compound motor action potential
- GFP
green fluorescent protein
- IgG
immunoglobulin G
- IgG4
immunoglobulin G4
- IgM
immunoglobulin M
- INCAT
Inflammatory Neuropathy Cause and Treatment
- IVIg
IV immunoglobulin
- IVMP
IV methylprednisolone
- MAG
myelin-associated glycoprotein
- MRS
modified Rankin Scale
- NCS
nerve conduction study
- NF155
neurofascin-155
- PLEX
plasmapheresis
Appendix. Authors
Footnotes
Class of Evidence: NPub.org/coe
CME Course: NPub.org/cmelist
Study Funding
This work was supported by the Mayo Clinic Foundation and the Department of Laboratory Medicine and Pathology, The Center of Individualized Medicine, and the Center for MS and Autoimmune Neurology.
Disclosures
S. Shelly reports no disclosures relevant to the manuscript. C.J. Klein is on the therapeutic CMTA advisory board; has received teaching honorarium from Ackea Pharmaceuticals for lectures on hereditary TTR amyloidosis and Fabry disease; is a paid consultant to Pfizer regarding tafamidis but has received no personal compensation; and has participated in clinical trials for inotersen and patisiran but received no personal compensation for his participation. P.J.B. Dyck receives honorarium from Ionis Pharmaceuticals. P. Paul reports no disclosures relevant to the manuscript. M.L. Mauermann receives research funding from IONIS, Alnylam, and EIDOS Pharmaceuticals; receives royalties from Oxford Publishing; and serves on the editorial board of the Mayo Clinic Proceedings. S.E. Berini, B.M. Howe, J. Fryer, E. Basal, M.B. Hammami, and R.S. Laughlin report no disclosures relevant to the manuscript. A. McKeon reports grant support from Euroimmun, AG, Alexion, and Grifols; has consulted for Roche Pharmaceuticals without personal compensation; and has patents pending for the following IgG biomarkers of autoimmune neurologic disease: septin-5, septin-7, MAP1B, PDE10A, GFAP, and KLCHL11. S.J. Pittock reports grants, personal fees, and nonfinancial support from Alexion Pharmaceuticals, Inc.; grants from Grifols and Autoimmune Encephalitis Alliance; grants, personal fees, nonfinancial support, and other from MedImmune, Inc.; other support from Astellas; personal fees from UCB; and has patent 8,889,102 (application 12-678350): Neuromyelitis Optica Autoantibodies as a Marker for Neoplasia (issued); patent 9,891,219B2 (application 12-573942): Methods for Treating Neuromyelitis Optica (NMO) by Administration of Eculizumab to an Individual That is Aquaporin-4 (AQP4)-IgG Autoantibody Positive (issued); and GFAP-IgG, Septin-5-IgG, MAP1B-IgG, and KLHL11 patents pending. J.R. Mills holds patents on the use of mass spectrometry to measure monoclonal immunoglobulins and has received royalties related to these patents from The Binding Site. D. Dubey has consulted for UCB, Alexion, and Immunovant Pharmaceuticals, with all compensation for consulting activities paid directly to Mayo Clinic; and has patents pending for KLHL11 and LUZP4 as biomarkers of neurologic autoimmunity and germ cell tumor. Go to Neurology.org/N for full disclosures.
References
- 1.Ng JK, Malotka J, Kawakami N, et al. Neurofascin as a target for autoantibodies in peripheral neuropathies. Neurology. 2012;79(23):2241-2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yan W, Nguyen T, Yuki N, et al. Antibodies to neurofascin exacerbate adoptive transfer experimental autoimmune neuritis. J Neuroimmunol. 2014;277(1-2):13-17. [DOI] [PubMed] [Google Scholar]
- 3.Pillai AM, Thaxton C, Pribisko AL, Cheng JG, Dupree JL, Bhat MA. Spatiotemporal ablation of myelinating glia-specific neurofascin (Nfasc NF155) in mice reveals gradual loss of paranodal axoglial junctions and concomitant disorganization of axonal domains. J Neurosci Res. 2009;87(8):1773-1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Querol L, Nogales-Gadea G, Rojas-Garcia R, et al. Neurofascin IgG4 antibodies in CIDP associate with disabling tremor and poor response to IVIg. Neurology. 2014;82(10):879-886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Doppler K, Appeltshauser L, Villmann C, et al. Auto-antibodies to contactin-associated protein 1 (Caspr) in two patients with painful inflammatory neuropathy. Brain. 2016;139(10):2617-2630. [DOI] [PubMed] [Google Scholar]
- 6.Cortese A, Lombardi R, Briani C, et al. Antibodies to neurofascin, contactin-1, and contactin-associated protein 1 in CIDP: clinical relevance of IgG isotype. Neurol Neuroimmunol Neuroinflamm. 2020;7(1):e639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kadoya M, Kaida K, Koike H, et al. IgG4 anti-neurofascin155 antibodies in chronic inflammatory demyelinating polyradiculoneuropathy: clinical significance and diagnostic utility of a conventional assay. J Neuroimmunol. 2016;301:16-22. [DOI] [PubMed] [Google Scholar]
- 8.Devaux JJ, Miura Y, Fukami Y, et al. Neurofascin-155 IgG4 in chronic inflammatory demyelinating polyneuropathy. Neurology. 2016;86(9):800-807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ogata H, Yamasaki R, Hiwatashi A, et al. Characterization of IgG4 anti-neurofascin 155 antibody-positive polyneuropathy. Ann Clin Transl Neurol. 2015;2(10):960-971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu W, Xin Y, He Z, Zhao Y. Association of neurofascin IgG4 and atypical chronic inflammatory demyelinating polyneuropathy: a systematic review and meta-analysis. Brain Behav. 2018;8(10):e01115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fryer JP, Lennon VA, Pittock SJ, et al. AQP4 autoantibody assay performance in clinical laboratory service. Neurol Neuroimmunol Neuroinflamm. 2014;1(1):e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dyck PJB, Klein CJ, Phillip L. Companion to Peripheral Neuropathy E-Book: Illustrated Cases and New Developments: Expert Consult. Saunders; 2010. [Google Scholar]
- 13.Dyck PJTP, Low PA, Griffin JW, Poduslo JF. Pathologic Alterations of Nerves. Elsevier; 1993. [Google Scholar]
- 14.Xu M, Pinto M, Sun C, et al. Expanded teased nerve fibre pathological conditions in disease association. J Neurol Neurosurg Psychiatry. 2019;90(2):138-140. [DOI] [PubMed] [Google Scholar]
- 15.Zimmerman IR, Karnes JL, O'Brien PC, Dyck PJ. Imaging system for nerve and fiber tract morphometry: components, approaches, performance, and results. J Neuropathol Exp Neurol. 1980;39(4):409-419. [DOI] [PubMed] [Google Scholar]
- 16.Hughes R, Bensa S, Willison H, et al. Randomized controlled trial of intravenous immunoglobulin versus oral prednisolone in chronic inflammatory demyelinating polyradiculoneuropathy. Ann Neurol. 2001;50(2):195-201. [DOI] [PubMed] [Google Scholar]
- 17.Dubey D, Honorat JA, Shelly S, et al. Contactin-1 autoimmunity: serologic, neurologic, and pathologic correlates. Neurol Neuroimmunol Neuroinflamm. 2020;7(4):e771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Joint Task Force of the European Federation of Neurological Societies/Peripheral Nerve Society. European Federation of Neurological Societies/Peripheral Nerve Society guideline on management of multifocal motor neuropathy. Report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society: first revision. J Peripher Nerv Syst. 2010;15(3):295-301. [DOI] [PubMed] [Google Scholar]
- 19.Burnor E, Yang L, Zhou H, et al. Neurofascin antibodies in autoimmune, genetic, and idiopathic neuropathies. Neurology. 2018;90(1):e31–e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Collins AM, Jackson KJ. A temporal model of human IgE and IgG antibody function. Front Immunol. 2013;4:235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Figueroa JJ, Dyck PJ, Laughlin RS, et al. Autonomic dysfunction in chronic inflammatory demyelinating polyradiculoneuropathy. Neurology. 2012;78(10):702-708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ogata H, Zhang X, Inamizu S, et al. Optic, trigeminal, and facial neuropathy related to anti-neurofascin 155 antibody. Ann Clin Transl Neurol. 2020;7(11):2297-2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dyck PJ, Lais AC, Ohta M, Bastron JA, Okazaki H, Groover RV. Chronic inflammatory polyradiculoneuropathy. Mayo Clin Proc. 1975;50:621-637. [PubMed] [Google Scholar]
- 24.Dyck PJ, Taylor BV, Davies JL, et al. Office immunotherapy in chronic inflammatory demyelinating polyneuropathy and multifocal motor neuropathy. Muscle Nerve. 2015;52(4):488-497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koike H, Kadoya M, Kaida KI, et al. Paranodal dissection in chronic inflammatory demyelinating polyneuropathy with anti-neurofascin-155 and anti-contactin-1 antibodies. J Neurol Neurosurg Psychiatry. 2017;88(6):465-473. [DOI] [PubMed] [Google Scholar]
- 26.Freeman SA, Desmazieres A, Fricker D, Lubetzki C, Sol-Foulon N. Mechanisms of sodium channel clustering and its influence on axonal impulse conduction. Cell Mol Life Sci. 2016;73(4):723-735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Manso C, Querol L, Lleixa C, et al. Anti-Neurofascin-155 IgG4 antibodies prevent paranodal complex formation in vivo. J Clin Invest. 2019;129(6):2222-2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Manso C, Querol L, Mekaouche M, Illa I, Devaux JJ. Contactin-1 IgG4 antibodies cause paranode dismantling and conduction defects. Brain. 2016;139(6):1700-1712. [DOI] [PubMed] [Google Scholar]
- 29.Querol L, Nogales-Gadea G, Rojas-Garcia R, et al. Antibodies to contactin-1 in chronic inflammatory demyelinating polyneuropathy. Ann Neurol. 2013;73(3):370-380. [DOI] [PubMed] [Google Scholar]
- 30.Shelly S, Shouman K, Paul P, et al. CISP-plus: expanding the spectrum of chronic immune sensory polyradiculopathy (CISP). Neurology. 2021;96(16):e2078–e2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reindl M, Schanda K, Woodhall M, et al. International multicenter examination of MOG antibody assays. Neurol Neuroimmunol Neuroinflamm. 2020;7(2):e674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Waters PJ, Pittock SJ, Bennett JL, Jarius S, Weinshenker BG, Wingerchuk DM. Evaluation of aquaporin-4 antibody assays. Clin Exp Neuroimmunol. 2014;5(3):290-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized data not published within this article will be made available by request from any qualified investigator.