

Abstract
Bedaquiline (BDQ) has shown great value in the treatment of multidrug‐resistant tuberculosis (MDR‐TB) in recent years. However, exposure–safety relationships must be explored to extend the use of BDQ. Two reported safety findings for BDQ are prolongation of the QTc interval and elevation of transaminase levels. In this study, we investigated the potential relationships between BDQ and/or its main metabolite (M2) pharmacokinetic (PK) metrics and QTcF interval or transaminase levels in patients with MDR‐TB using the approved dose regimen. Data from 429 patients with MDR‐TB from two phase IIb studies were analyzed via nonlinear mixed‐effects modeling. Individual model‐predicted concentrations and summary PK metrics were evaluated, respectively, in the QTcF interval and transaminase level exposure–response models. Investigation of further covariate effects was performed in both models. M2 concentrations were found to be responsible for the drug‐related QTcF increase in a model accounting for circadian rhythm patterns, time on study, effect of concomitant medication with QT liability, and patient demographics. Simulations with the final model suggested that doses higher than the approved dose (leading to increased M2 concentrations) are not expected to lead to a critical QTcF interval increase. No exposure–safety relationship could be described with transaminase levels despite previous reports of higher levels in patients treated with BDQ. The developed longitudinal models characterized the role of M2 concentrations in QTc interval prolongation and found no concentration dependency for transaminase level elevation, together suggesting that BDQ exposure at the high end of the observed range may not be associated with a higher risk of safety events.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
The following two main adverse effects have been associated with bedaquiline use: QTc prolongation and elevated transaminase levels. Previous analyses have reported that higher doses of bedaquiline could be beneficial for treatment response, but the safety aspects of increased doses have not been explored.
WHAT QUESTION DID THIS STUDY ADDRESS?
What are the relationships between bedaquiline and/or its main metabolite exposures and QTcF prolongation or transaminase level increases?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
The main metabolite of bedaquiline is the driver of the drug‐induced QTcF prolongation in a multifactorial context influenced by circadian rhythm, time on study, comedication with QT liability, and patient demographics. M2 concentrations up to 1600 ng/mL are not expected to lead to higher risks of safety events. Hepatic levels increase was not found to be correlated with bedaquiline or M2 exposures.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
Now that both exposure–response and exposure–safety relationships have been defined, a dose‐exposure‐efficacy‐safety framework can be established for bedaquiline dose optimization.
INTRODUCTION
With approximately 10 million new cases and 1.4 million related deaths reported in 2019, tuberculosis (TB) was the most common cause of death attributed to an infectious disease globally before the emergence of coronavirus disease 2019. 1 The situation is aggravated by drug resistance to first‐line treatment and limited therapeutic options for patients with multidrug‐resistant TB (MDR‐TB), which represent approximately 3.5% of all new TB cases per year. In 2012, bedaquiline (BDQ) was approved for the treatment of MDR‐TB and is now recommended as one of the first‐line options for MDR‐TB treatment and is widely used at the programmatic level. 2 , 3 , 4 An exposure–response relationship has been established between BDQ exposures and the rate of decline in mycobacterial load in patients, indicating that increased exposures could be beneficial for the treatment response because higher BDQ concentrations generally lead to faster culture conversion. 5 , 6 When considering higher doses, it is of importance to investigate if they can be safely administered.
The main potential adverse effect linked to BDQ administration is cardiac QTc interval prolongation. QTc interval prolongation is the result of the blockade of potassium channels that delays the cardiac repolarization and can lead to arrhythmias, such as torsade de pointes, a risk factor of sudden death. BDQ’s main metabolite (M2) levels were found to significantly correlate with QTcF prolongation, whereas only a positive trend (nonsignificant positive correlation) was observed with BDQ concentrations. 7 , 8 In the Nix‐TB trial (NCT02333799), where BDQ was administered in a combination therapy, the effect of M2 concentrations on QTc interval was described by a linear model. 9 Besides the potential effects of BDQ and M2 concentrations, the QTc interval is subjected to intrinsic variability because of the subject‐related factors such as, for example, sex, age, high weight, circadian rhythm, food intake, or electrolyte disturbances. 10 Furthermore, BDQ is always administered in combination with other TB drugs, and some of those have their own QT liability (e.g., clofazimine, moxifloxacin). 11 Hence, there is a need to characterize the relationship between QTcF interval prolongation and BDQ and/or M2 levels in a multifactorial context.
A second adverse effect that has been reported with BDQ use is an hepatic‐related adverse drug reaction with the increase of transaminase enzyme levels (alanine aminotransferase [ALT] and aspartate aminotransferase [AST]). Indeed, in stage 2 of the placebo‐controlled clinical trial C208, transaminase elevation (≥3 × upper limit of normal) occurred more frequently in the BDQ‐treated arm compared with the placebo arm (4 patients treated with BDQ versus none placebo‐treated patients), but could not be related to BDQ intake on a case‐by‐case basis (using Hy's Law criteria to determine potential BDQ‐induced liver injury). 11
This work aimed to describe the role of BDQ and/or M2 exposure on safety aspects of BDQ administration and to establish exposure–safety relationships with QTc interval prolongation and/or transaminase level increases if relevant.
METHODS
Patients and data
Data from two phase IIb studies (C208, 12 C209 13 ) were obtained through the European PredictTB consortium. The trials were conducted in accordance with Good Clinical Practice standards and received ethical approvals from appropriate local authorities. A total of 335 patients were treated with BDQ and 105 patients were treated with placebo, both on top of a background regimen of anti‐TB drugs. The C208 study was a 2‐stage, randomized, double‐blinded, placebo‐controlled trial that enrolled patients who were newly diagnosed with MDR‐TB. The C209 study was a single‐arm, open‐label trial in patients who were newly diagnosed with MDR‐TB or treatment‐experienced for MDR‐TB. Patients received BDQ (400 mg once daily for 2 weeks, then 200 mg three times a week) or placebo for 24 weeks (or 8 weeks in stage 1 of the C208 study) in combination with a background regimen of 5–7 anti‐TB drugs.
QT interval prolongation was assessed by electrocardiogram (ECG). QT interval is inversely proportional to heart rate; hence it must be corrected to reliably detect abnormalities. In this analysis, QT measurements were corrected by using the Fridericia method (noted QTcF; Equation 1). 14
| (1) |
where the RR interval represents the heart rate. Correction using Bazett's method (QTcB) was also considered, but exploratory plots showed that QTcF was less correlated with heart rate than QTcB (Figure S1).
Hepatic measurements and ECG were recorded in all patients, and the frequency is described in Figure S2. Hepatic measurements were to be taken before drug intake and after a fasting period of 10 h. Single ECGs were performed before the drug intake and triplicate ECGs before the drug intake as well as 5 h postdose.
Pharmacokinetics
Individual BDQ and M2 pharmacokinetic (PK) metrics were derived using a previously published PK model together with the observed PK data, which included rich sampling in the C208 study for most of the patients and single samples at three different timepoints for all patients in the C209 study. 15 BDQ and M2 concentrations were predicted for all timepoints where ECG measurements were conducted. Summary PK metrics such as individual BDQ and M2 average concentrations over 2, 8, and 24 weeks as well as BDQ and M2 weekly average concentrations during the study period were also computed.
Model development
Model development of the time course of QTcF interval and transaminase levels was performed using a nonlinear mixed‐effect approach. For QTcF interval, it comprised structural and stochastic model components (including exposure–safety exploration and structural covariates based on existing knowledge and clinical relevance) followed by an exploratory covariate model development (covariates selected based on statistical significance). For transaminase levels, the analysis consisted of the inclusion of the structural and stochastic components as well as the exploration of exposure–safety relationships and structural covariates, such as effect of TB background regimen or effect of time on treatment.
Modeling methodology, software used, and methods for model qualification are described in Supplementary Document S1.
QTcF interval model
Structural model
Time effect
Relationships where QTcF interval is influenced by time on BDQ treatment were investigated. Linear, power, Emax, and negative exponential models were evaluated (Equations (2), (3), (4), (5)).
| (2) |
| (3) |
| (4) |
| (5) |
where TE is the time effect, α is the slope coefficient of the linear function, TIMW is the time after start of treatment in weeks, γ is the power coefficient of the power function, QTmax is the Emax on QTcF, and T 50 and t 1/2 are the times needed to achieve 50% of the Emax on QTcF.
Circadian rhythm
Within‐day circadian variations of QTcF were evaluated with cosine functions, with one to three oscillations, defined with amplitude and acrophase parameters (Equation (6), (7)).
| (6) |
where CIRC is the circadian variation, CTIME is the clock time, Ai , φ i , and T period, i represent the amplitude, the acrophase, and time period (e.g., 24, 12, 6 h) of the i:th oscillator, respectively.
Drug effect
Both BDQ and its main metabolite M2 were explored for their ability to influence QTcF interval, alone or jointly. Linear Emax models as well as competitive interactions models (Equation (6), (7)) such as full agonist, partial agonist, antagonist, and inverse agonist models were tested. 16 , 17
| (7) |
where DEAB is the combined drug effect of two entities acting on the same receptor (e.g., the Parent Drug A and Metabolite B), E max, A and E max, B are the Emax of each entity, and EC50, A and EC50, B are the concentrations of each entity needed to achieve half of the Emax.
When E max, A = E max, B , a full agonist interaction model is described. If E max, B is less than E max, A , then B has a lower intrinsic activity, and the model describes a partial agonist interaction. If E max, B = 0, B has no intrinsic activity, and it is an antagonist interaction model. If E max, B is less than 0, the intrinsic activity of B is reverse, and an inverse agonist relationship is defined. If E max, B /EC50, B and EC50, B goes toward zero and infinity, respectively, B does not influence the QTc interval. The same logic would apply for entity A.
Concomitant TB medication with QT liability
Intake of concomitant TB drugs with known QT liability such as clofazimine 18 or moxifloxacin 19 was included a priori in the model as prespecified structural covariates on baseline QTcF interval (QTcF0). The presence or absence of clofazimine and/or moxifloxacin at each visit was recorded and handled as binary information and modeled with a step function.
Exploratory covariates
Age at baseline, gender, race, baseline albumin levels, time‐varying and baseline electrolytes levels (potassium and calcium), drug‐resistance severity (drug‐sensitive TB, MDR‐TB, pre‐extensively drug‐resistant tuberculosis (XDR‐TB), and XDR‐TB), and HIV coinfection (negative or positive with CD4+ count ≥250 cells/μL at baseline) were considered as exploratory covariates with linear or power functions.
Transaminase levels model
Background TB treatment regimen
Step functions were considered to describe the potential effect of the background TB treatment regimen. Newly diagnosed patients started BDQ and the background TB regimen concomitantly when entering the trial. For patients treatment‐experienced, the possibilities of different background TB treatments before and after entering the clinical trial and between the C208 and C209 studies were evaluated.
Time effect
Effect of time on BDQ treatment was investigated as linear and Emax relationships (Equations 2 and 4).
Drug effect
Both BDQ and M2 were explored for their ability to influence transaminase levels. As transaminase levels are not expected to directly correlate with drug concentrations, summary metrics (constant or time‐varying within individuals) were preferred. A simple treatment arm function, weekly average concentrations over 2, 8, and 24 weeks, as well as weekly average concentrations during the study period were considered in linear or Emax functions.
RESULTS
Patients and data
Data from 324 patients of the 335 patients receiving BDQ were analyzed (98/102 in the C208 study, 226/233 in the C209 study). All 105 patients in C209 receiving placebo were included in the analysis. The reasons for not including a patient were no PK sample recorded (10 patients) or no ECG measurement after start of treatment (one patient). For the included patients, a total of 18,306 ECGs and 5833 ALT/AST measurements were recorded on the day before the start of treatment and during the treatment period. Patients’ characteristics are summarized in Table 1, and box plots of the raw data of ALT and AST levels at different timepoints are displayed in Figure S3.
TABLE 1.
Summary of patients’ characteristics presented as the median (range) or number of subjects (%)
| Variable | C208–PLC, n = 105 | C208–BDQ, n = 98 | C209–BDQ, n = 226 | Total patients, n = 429 |
|---|---|---|---|---|
| Sex | ||||
| Male | 66 (63) | 67 (68) | 146 (65) | 279 (65) |
| Female | 39 (37) | 31 (32) | 80 (35) | 150 (35) |
| Age, years | 34 (18–61) | 31 (18–63) | 32 (18–68) | 33 (18–68) |
| Weight, kg | 53 (35–83) | 53 (37–81) | 57 (30–113) | 55 (30–113) |
| Race/ethnicity | ||||
| Caucasian or White | 13 (12) | 8 (8) | 56 (25) | 77 (18) |
| Black | 40 (38) | 40 (41) | 73 (32) | 153 (36) |
| Hispanic | 15 (14) | 13 (13) | 0 (0) | 28 (7) |
| Asian | 6 (6) | 9 (9) | 89 (39) | 104 (24) |
| Other | 31 (30) | 28 (29) | 8 (4) | 67 (16) |
| TB type | ||||
| Drug‐sensitive TB | 4 (4) | 3 (3) | 3 (1) | 10 (2) |
| MDR‐TB | 63 (60) | 70 (71) | 89 (39) | 222 (52) |
| pre‐XDR‐TB | 16 (15) | 17 (17) | 43 (19) | 76 (18) |
| XDR‐TB | 5 (5) | 3 (3) | 37 (16) | 45 (10) |
| Missing | 17 (16) | 5 (5) | 54 (24) | 76 (18) |
| HIV | ||||
| Negative | 86 (82) | 85 (87) | 210 (93) | 381 (89) |
| Positive | 19 (18) | 11 (11) | 11 (5) | 41 (9.5) |
| Missing | 0 (0) | 2 (2) | 5 (2) | 7 (1.5) |
| Baseline albumin, g/L | 31 (17–46) | 34 (15–49) | 38 (24–49) | 35 (15–49) |
| Baseline (albumin‐corrected) calcium, IU/L | 2.53 (2.28–2.84) | 2.53 (2.30–2.81) | 2.43 (2.15–2.86) | 2.48 (2.15–2.86) |
| Baseline potassium, IU/L | 4.30 (3.40–5.80) | 4.40 (3.60–5.80) | 4.10 (2.70–5.40) | 4.30 (2.70–5.80) |
| Concomitant medication with QT liability | ||||
| None | 101 (96) | 95 (97) | 200 (88) | 396 (92) |
| Clofazimine | 0 (0) | 0 (0) | 24 (11) | 24 (6) |
| Moxifloxacin | 4 (4) | 3 (3) | 2 (1) | 9 (2) |
Abbreviations: BDQ, bedaquiline; MDR, multidrug resistant; PLC, placebo; TB, tuberculosis; XDR, extensively drug resistant.
Pharmacokinetics
Model‐predicted individual BDQ and M2 concentrations at each ECG timepoint used in the QTcF interval model; model‐predicted individual BDQ and M2 average concentrations over 2, 8, and 24 weeks; and individual BDQ and M2 weekly average concentrations during the study period used in the transaminase level model are presented in Table S1.
QTcF interval model
Final structural and stochastic model
The final structural model comprised the following components: the baseline QTcF interval (QTcF0), a time effect (TE), the circadian rhythm (CIRC), a drug effect (DE), and effects of the presence of comedication with QT liability (clofazimine [CLOFA] and moxifloxacin [MOXI]). It is described by Equation (8) and illustrated in Figure 1, and the final parameter estimates and respective standard errors are reported in Table 2.
| (8) |
FIGURE 1.
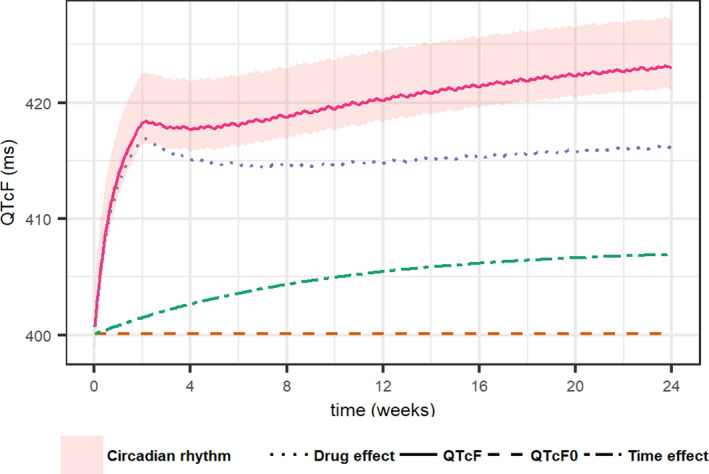
Illustration of the typical profile of QTcF interval over time without comedication (full line) as well as the typical profile of the drug effect (dotted line), the time effect (dot‐dashed line), and the QTcF interval at baseline (QTcF0, dashed line). The circadian rhythm is depicted by the shaded area around the typical profile of QTcF interval
TABLE 2.
Parameter estimates and uncertainty of the final QTcF interval model
| Submodel | Parameters (unit) | Value (RSE%) | IIV %CV (RSE%) |
|---|---|---|---|
| Baseline | QTcF0 (ms) | 400 (0.328) | 3.75 (3.80) |
| Drug effect | Emax,M2 (ms) | 28.6 (13.6) | |
| EC50,M2 (ng/mL) | 855 (24.4) | 148 (11.8) | |
| Time effect | QTmax (ms) | 6.50 (11.8) | 167 a (12.7) |
| T1/2 (weeks) | 6.44 (17.9) | ||
| Circadian rhythm | A24 (ms) | 2.76 (43.9) | |
| φ24 (h) | 4.91 (26.6) | ||
| A12 (ms) | 1.46 (26.7) | ||
| φ12 (h) | 4.50 (23.4) | ||
| Comedication b | Effect of clofazimine (ms) | 11.8 (15.6) | |
| Effect of moxifloxacin (ms) | 2.47 (98.4) | ||
| Covariates | Effect of calcium (ms per IU/L) | −8.74 (28.3) | |
| Effect of potassium (ms per IU/L) | −1.25 (38.5) | ||
| Effect of sex (female) (ms) | 7.75 (19.1) | ||
| Effect of being black (ms) | −6.86 (21.3) | ||
| Effect of age (ms per year) | 0.349 (17.0) | ||
| Residual error model | Additive RUV (ms) | 8.19 (1.81) | 21.2 (11.2) |
| Box–Cox IIV c RUV | 4.11 (24.0) | ||
| Additive RUVrepl (ms) | 6.87 (1.47) | 23.9 (5.57) | |
| Box–Cox IIV c RUVrepl | 0.825 (40.5) |
The %CV is reported as the square root of the variance. RSE of IIV and RUV is reported on the approximate standard deviation scale (standard error/variance estimate)/2.
Abbreviations: A12, amplitude for the 12‐h circadian rhythm cycles; A24, amplitude for the 24‐h circadian rhythm cycles; %CV, percent coefficient of variation; Emax,M2, maximal effect of M2 concentrations; EC50,M2, M2 concentration needed to achieve 50% of Emax,M2; IIV, interindividual variability; QTcF0, baseline QTcF interval; QTmax, maximal effect of time; %RSE, percent relative standard error; RUV, residual unexplained variability; RUVrepl: replicate‐specific residual unexplained variability; T1/2: time needed to achieve 50% of QTmax; φ12: acrophase for the 12‐h circadian rhythm cycles; φ24: acrophase for the 24‐h circadian rhythm cycles.
IIV is coded with a proportional model, whereas the others are coded with an exponential model.
Covariates included a priori given the prior knowledge of these drugs on QTc interval.
Parameter estimate of the Box–Cox transformed distribution of IIV on ε components.
The QTcF0 was estimated to be 400 ms with a variability between patients of 3.75 percent coefficient of variation (%CV), which corresponds to a standard deviation of ca. 15 ms. A variability between triplicates per occasion and patient was observed (Figure S4) and handled with the two components of the residual variability (c.f. residual unexplained variability [RUV] and RUVrepl in Table 2). Interindividual variability (IIV) was added on ε elements, allowing residual variability to be different between patients.
Time effect
The effect of time on BDQ treatment (Figure S5) was described by an asymptotic exponential function (Equation 5) with a maximal increase of QTcF interval of 6.5 ms and an estimated half‐life of 6.44 weeks. A large proportional variability between patients of 166 %CV was estimated, indicating that the time effect may result in a decrease in QTcF interval for some patients or a higher increase than 6.5 ms for others (Figure S6).
Circadian rhythm
Diurnal circadian pattern was characterized by a dual oscillation cosine function (Equation 9), previously described by Piotrovsky 10 and following the same trend as previously reported results in healthy volunteers (Figure S7).
| (9) |
where A 24, A 12, φ24, and φ12 represent the amplitudes (in ms) and acrophases (in h) of the 24‐hand 12‐h circadian rhythm cycles.
Drug effect
The final drug effect model was characterized by an effect of M2 concentrations on QTcF interval via an Emax relationship (Equation 10).
| (10) |
where Emax,M2 represents the maximal QTcF interval prolongation related to M2 concentrations (in ms), EC50,M2 represents the M2 concentration needed to achieve half of the Emax, and ConcM2 represents the individual model‐predicted M2 concentrations at ECG timepoints.
A competitive interaction model involving both BDQ and M2 concentrations was not found to be better than the M2‐only Emax model to describe the data, and a model including only BDQ as driving the QTcF interval prolongation BDQ only was markedly worse, as depicted by the Akaike information criterion (AIC): AICInteraction model (30 degrees of freedom [DF]) =105164, AIC BDQ model (28DF) =105335, AICM2 model (28DF) =105160. In the final model, Emax,M2 was estimated to 28.6 ms, and the EC50,M2 was estimated to 855 ng/mL which was associated with a large between‐subject variability of 147.8 %CV.
Concomitant TB medication with QT liability
Nine and 24 patients of 429 received moxifloxacin or clofazimine, respectively, administered at some point during the study period (on average, during 28% and 57% of the study period, respectively). The number of patients with comedications per study is described in Table 1. These concomitant medications were estimated to increase the QTcF interval by 2.47 ms for moxifloxacin and 11.8 ms for clofazimine. Of the 24 patients receiving clofazimine, 10 experienced QTcF interval measurements higher than 450 ms, but none had to discontinue BDQ treatment. None of the studied patients had coadministrations of both clofazimine and moxifloxacin.
Final covariate model
Covariate relationships with baseline QTcF interval were identified with time‐varying electrolytes levels, sex, race, and age on baseline QTcF interval, as described in Equation (11).
| (11) |
None of the evaluated covariates were found to impact EC50,M2 or QTmax, and no effect of the disease‐related factors (such as degree of TB resistance or being coinfected with HIV) was detected. The direction and size of the covariate effects are illustrated in Figure 2. In short, the QTcF interval increased with higher age, in non‐Black patients and females, and by hypokalemia and hypocalcemia.
FIGURE 2.
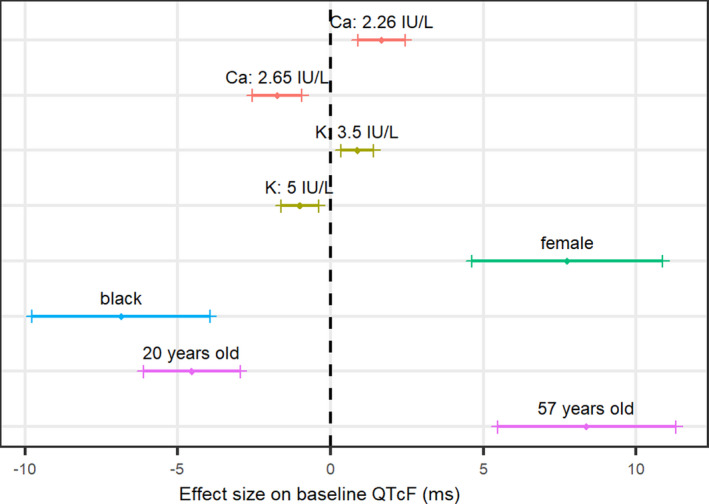
Illustration of the direction and size of each covariate effect on baseline QTcF interval (QTcF0). Continuous covariates are displayed as the 5th and 95th percentiles of the covariate range. Categorical covariates are compared with the other category. The bands show the 90% confidence interval around the point estimate. The vertical dashed line represents the typical individual: a 33‐year‐old non‐Black male subject with a potassium (K) level of 4.2 IU/L and a calcium (Ca) level of 2.45 IU/L
Visual predictive checks of the final model are shown in Figure 3, and the model code is presented in Supplementary Model Code S1.
FIGURE 3.
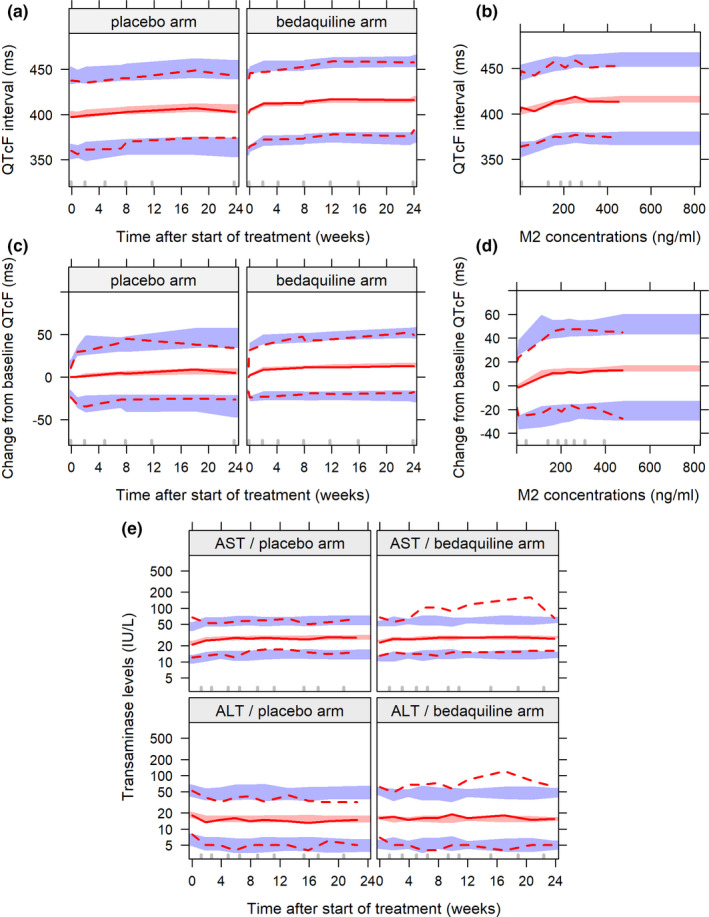
Visual predictive checks of the final models. (a) QTcF interval over time after start of treatment per arm. (b) QTcF interval over individual predicted M2 concentrations in the bedaquiline group. (c) Change from baseline QTcF interval over time after start of treatment per arm. (d) Change from baseline QTcF interval over individual predicted M2 concentrations in the bedaquiline group. (e) Alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels over time after start of treatment per arm. The solid and dashed lines represent the observed data, and the shaded areas represent the simulation‐based 95% confidence intervals for the corresponding percentiles
Transaminase levels model
The final models for both ALT and AST are described by Equations (12) and (13). Both transaminase levels at baseline (TRANS0) are affected by an effect of the background TB treatment regimen (BGtreat) accounting for the fact that patients were either treatment naïve or treatment experienced before the start of BDQ. The possibility of background TB treatment regimens being different between the C208 and C209 studies was not statistically significant for either of the analytes. The time course of AST levels was further described by an effect of time after the start of treatment, characterized by an Emax function (Equation 13).
Interindividual variability components were added on the baseline parameters (TRANS0) and the RUV parameters, of the ALT and AST submodels. Correlation parameters were estimated between the corresponding IIVs of the ALT and AST submodels. The final parameter estimates and respective standard errors of the transaminase levels model can be found in Table 3, and visual predictive checks are presented in Figure 3.
TABLE 3.
Parameters estimates and uncertainty of the final transaminase levels model
| Submodel | Parameters (unit) | Value (RSE%) | IIV %CV (RSE%) | Correlation IIV % (RSE%) |
|---|---|---|---|---|
| Baseline | TRANS0,ALT (IU/L) | 18.4 (3.35) | 44.4 (3.96) | 57.6 (5.82) |
| TRANS0,AST (IU/L) | 22.6 (2.12) | 29.1 (4.17) | ||
| Effect of the background TB treatment | BGtreat,ALT (%) | −16.1 (18.2) | ||
| BGtreat,AST (%) | 12.4 (13.9) | |||
| Time effect | Tmax,AST (%) | 14.8 (11.0) | ||
| T50,AST (weeks) | 3.92 (34.6) | |||
| Residual error model | Proportional RUVALT (%CV) | 32.6 (2.28) | 31.3 (6.28) | 85.4 (6.05) |
| Proportional RUVAST (%CV) | 20.7 (2.65) | 38.1 (5.38) | ||
| Correlation RUVALT − RUVAST (%) | 65.6 (2.69) |
The %CV is reported as the square root of the variance. RSE of IIV and RUV is reported on the approximate standard deviation scale (standard error/variance estimate)/2.
Abbreviations: φ12, φ24, A12, and A24 represent the acrophases (in h) and amplitudes (in ms) of the 12‐h and 24‐h circadian rhythm cycles; %CV, percent coefficient of variation; %RSE, percent relative standard error; ALT, alanine aminotransferase; AST, aspartate aminotransferase; BGtreat, background TB treatment regimen; IIV, interindividual variability; RUV, residual unexplained variability; T50, time needed to achieve 50% of the maximal effect; TB, tuberculosis; Tmax, maximal time effect; TRANS0, transaminase levels at baseline.
No difference between the treatment arms as well as no exposure–safety relationship could be discriminated with either constant or time‐varying PK metrics for both ALT and AST time‐course levels. This is illustrated in Figure S8 (visual predictive check of change from baseline transaminase levels versus BDQ or M2 weekly concentrations).
DISCUSSION
This analysis provides quantitative information on the relationship between BDQ and/or M2 exposure and QTcF interval prolongation after BDQ administration. With a representative population of about 400 patients with MDR‐TB and approximatively 18000 ECG measurements, M2 metabolite concentrations were predicted to be the driver of the treatment‐induced prolongation of the QTcF interval. In vitro experiments indicate that BDQ and M2 act on the same cardiac receptor with a common mechanism, supporting the use of competitive interaction models. 20 However, after inclusion of all the different components in the model (circadian rhythm, secular trend, and other covariates), the more complex interaction model was not significantly better than an Emax model driven by M2 concentrations alone. Model predictions from the final QTcF interval model, using data from thorough QT and drug–drug interaction studies, where BDQ was administered as a single dose, are in line with those findings (see Supplementary Document S2). Indeed, only low M2 concentrations were predicted (as the M2 metabolite accumulates considerably with multiple dosing), and no QTcF increase was detected. The final parameter estimates of the drug effect model were 28.6 ms and 855 ng/mL for Emax,M2 and EC50,M2, respectively (Table 2). Note that the EC50,M2 value of 855 ng/mL is slightly above the highest predicted M2 concentrations of 827 ng/mL under the approved dosing regimen. However, despite the limited number of datapoints near the EC50 value, the Emax model was better than a linear model at describing the data, and the EC50 parameter was estimated with acceptable precision (relative standard error of 24.4%). The model includes IIV on EC50,M2 and not on Emax,M2 (tested but not significant), which suggests that fluctuations are expected more in terms of affinity than amplitude of the maximum effect that the drug can produce. In addition, although the ECG measurements were not drawn at the expected M2 peak concentrations, given the flat PK profile of M2, 15 little differences between trough and peak concentrations were observed. So, as depicted in Figure 4, doubling the M2 concentrations compared with the levels seen with the approved dosing regimen (0–827 ng/mL) is not expected to lead to a QTcF increase larger than 30 ms change from baseline (cutoff point defined in the International Council for Harmonisation E14 guidance).
FIGURE 4.

Simulation of the drug effect profile accounting for variability between patients (median black line surrounded by the 90% confidence interval shaded area) under the currently approved dosing regimen (squared light‐gray area) and after doubling the concentrations. The typical values of Emax,M2 (28.6 ms) and EC50,M2 (855 ng/mL) for the final model are shown by the horizontal and vertical black dashed lines, respectively. Emax,M2, maximal M2 effect; EC50,M2, M2 concentration needed to achieve 50% of Emax,M2
This analysis also characterized other sources of QTcF interval variability. First, a large variability between triplicates was observed per occasion and patient (Figure S4), and handled by two RUV components (a common RUV and a replicate‐specific RUV). Second, the circadian pattern of the QTcF interval displayed an amplitude, which allowed the mean QTcF interval to vary between +4 ms and −2 ms within a day, as shown in Figure 1. The diurnal variations follow the same trend as previously reported results in healthy volunteers (Figure S7) and are important to take into account when interpreting change from baseline QTcF results. 21 The effect of time in study was estimated to be responsible for a maximum increase of 6.5 ms in the QTcF interval with a half‐life of 6.44 weeks, suggesting that at the end of the 24‐week treatment period, 92% of the maximal QT effect would be achieved. This effect of time can be observed in both arms as depicted in Figure S5. A time trend following the same dynamics has been previously reported by Li et al. in patients treated with anti‐TB drugs, 9 but the reason remains unclear. It attributed the “secular trend” effect to possible “accumulation of other metabolites” or “other long‐term physiological/biological changes as a result of treatment” and patients becoming healthier. The first part of the interpretation could hold for patients treated with BDQ as another metabolite, M3, is produced by the subsequent N‐demethylation of M2, bacteriologically inactive but with a suggested similar toxicity profile as M2, 8 but never reported in the literature. The second part of the interpretation could also be the result of an insufficient correction of the QT interval for changes in heart rate. Although the Fridericia correction method is widely used and one of the most accurate correction methods to provide a corrected QT interval independent of the heart rate, 22 , 23 it has been reported to be less reliable at high heart rates where it undercorrects the QT interval 24 , 25 (Figure S1). A patient with tachycardia (>100 beats per min [bpm]) will appear to have a short/low QTcF interval, and when this patient will be becoming healthier (i.e., decreasing heart rate), there will be the appearance of QTcF being prolonged compared with baseline. With this in mind and the fact that 12% of the patients in our analysis have a baseline heart rate >100 bpm, the miss‐correction of QT interval may for some patients explain the effect of time on treatment.
The covariate analysis confirmed the effect of the commonly reported factors affecting QTcF interval (such as age, race, sex, or electrolyte levels). 10 No covariates were found significant on EC50,M2 or QTmax, and no effect of the disease‐related factors (such as degree of TB resistance or being coinfected with HIV) was detected, despite the large IIV on both EC50,M2 or QTmax. Note that around 10% of the study population was HIV positive (Table 1), and none of the patients received antiretroviral drugs during the study period, as per protocol. In the present analysis, concomitant medication such as moxifloxacin and clofazimine were evaluated as categorical covariates and included a priori as structural covariates (not tested for significance). The estimated effects (Table 2) were smaller than previously described in the literature 18 , 26 , 27 , 28 , 29 but considered relevant in this analysis to highlight the impact of TB comedication with known QT liability. 26 , 30
The second focus of this safety analysis was the evaluation of transaminase levels over time. Analysis of the quantitative information of ALT and AST levels during the treatment period did not identify any significant difference between BDQ and placebo arms (also when considering C208 data only). Furthermore, none of the considered constant or time‐varying PK metrics could explain the transaminase level elevation. Previous analysis of those data on a case‐by‐case basis in patients experiencing Grade 3 transaminase increases revealed the presence of confounding factors (such as underlying hepatic disease, alcohol use, concomitant hepatotoxic medication use) in most of the cases. 11 Information about confounding factors was not available for this analysis, but the effects of being treated with several drugs and time after start of treatment (on the AST level time course) implemented in our model might depict a long‐term effect of concomitant hepatotoxic medication use or a slow progressing underlying hepatic disorder.
In conclusion, a safety analysis of both QTcF interval and the transaminase levels time course after BDQ administration has been performed. A relationship between drug exposure levels and the cardiac safety profile of BDQ has been established, where the M2 metabolite concentrations drive the prolongation of the QTcF interval, but no relationship could be identified between different PK metrics and the transaminase levels. Projections beyond the range of observed M2 exposures (up to twice the highest observed value) suggested that the QTcF interval would not reach critical limits (at least 95% of patients with ΔQTcF <30 ms; see Figure 4). This exposure–QTcF model with M2 concentrations can, together with previously developed models for population PK 15 , 31 , 32 , 33 , 34 , 35 and an exposure–efficacy model with BDQ concentrations, 5 , 6 inform an integrated dose‐exposure‐efficacy‐safety framework for BDQ dose evaluation.
CONFLICT OF INTEREST
S.R. is a former employee of Janssen Pharmaceutica NV, Beerse, Belgium. All other authors declared no competing interests for this work.
AUTHOR CONTRIBUTIONS
L.T., E.M.S., S.R., and M.O.K. wrote the manuscript. L.T., E.M.S., and M.O.K. designed the research. L.T., E.M.S., S.R., and M.O.K. performed the research. L.T. analyzed the data.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
We thank the Swedish Foundation for International Cooperation in Research and Higher Education (STINT), jointly with the South African National Research Council, the National Research Foundation (NRF) (STINT Grant SA2015‐6259 and NRF Grant 101575), for the traveling support to Cape Town, South Africa.
Tanneau L, Svensson EM, Rossenu S, Karlsson MO. Exposure–safety analysis of QTc interval and transaminase levels following bedaquiline administration in patients with drug‐resistant tuberculosis. CPT Pharmacometrics Syst Pharmacol. 2021;10:1538–1549. doi: 10.1002/psp4.12722
Funding information
This work was supported by the Swedish Research Council (Grant 521–2011‐3442) in addition to the Innovative Medicines Initiative Joint Undertaking (www.imi.europa.eu) for the PredictTB consortium, the resources of which are composed of financial contributions from the European Union's Seventh Framework Programme (FP7/2007–2013; Grant 115337) and the European Federation of Pharmaceutical Industries and Associations companies’ in‐kind contribution. This work was also supported by Janssen Pharmaceutica NV.
REFERENCES
- 1. World Health Organization . WHO Global tuberculosis report 2020. Published 2020. Accessed October 11, 2021. https://www.who.int/publications/i/item/9789240013131
- 2. World Health Organization . WHO consolidated guidelines on tuberculosis, module 4: treatment ‐ drug‐resistant tuberculosis treatment. Accessed October 12, 2021. https://www.who.int/publications‐detail‐redirect/9789240007048 [PubMed]
- 3. Cox V, Brigden G, Crespo RH, et al. Global programmatic use of bedaquiline and delamanid for the treatment of multidrug‐resistant tuberculosis. Int J Tuberc Lung Dis. 2018;22:407‐412. [DOI] [PubMed] [Google Scholar]
- 4. Hafkin J, Hittel N, Martin A, Gupta R. Compassionate use of delamanid in combination with bedaquiline for the treatment of multidrug‐resistant tuberculosis. Eur Respir J. 2019;53:1801154. [DOI] [PubMed] [Google Scholar]
- 5. Svensson EM, Karlsson MO. Modelling of mycobacterial load reveals bedaquiline’s exposure–response relationship in patients with drug‐resistant TB. J Antimicrob Chemother. 2017;72:3398‐3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tanneau L, Karlsson MO, Svensson EM. Understanding the drug exposure–response relationship of bedaquiline to predict efficacy for novel dosing regimens in the treatment of multidrug‐resistant tuberculosis. Br J Clin Pharmacol. 2020;86:913‐922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Janssen Therapeutics . SIRTURO (bedaquiline) label. Published online December 2012. Accessed October 11, 2021. https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/204384s000lbl.pdf
- 8. Food and Drug Administration . Clinical pharmacology and biopharmaceutics review(s). Application number 204384Orig1s000. Accessed October 12, 2021. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/204384Orig1s000ClinPharmR.pdf
- 9. Li H, Salinger DH, Everitt D, et al. Long‐term effects on QT prolongation of pretomanid alone and in combinations in patients with tuberculosis. Antimicrob Agents Chemother. 2019;63(10):e00445‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Piotrovsky V. Pharmacokinetic‐pharmacodynamic modeling in the data analysis and interpretation of drug‐induced QT/QTc prolongation. AAPS J. 2005;7:E609‐E624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Food and Drug Administration . Medical review(s). Application number 204384Orig1s000. Accessed October 12, 2021. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/204384Orig1s000MedR_.pdf
- 12. Diacon AH, Pym A, Grobusch MP, et al. Multidrug‐resistant tuberculosis and culture conversion with bedaquiline. N Engl J Med. 2014;371:723‐732. [DOI] [PubMed] [Google Scholar]
- 13. Pym AS, Diacon AH, Tang S‐J, et al. Bedaquiline in the treatment of multidrug‐ and extensively drug‐resistant tuberculosis. Eur Respir J. 2016;47:564‐574. [DOI] [PubMed] [Google Scholar]
- 14. Fridericia LS. The duration of systole in the electrocardiogram of normal subjects and of patients with heart disease. Acta Med Scand. 1920;53:469‐486. [Google Scholar]
- 15. Svensson E, Dosne A, Karlsson M. Population pharmacokinetics of bedaquiline and metabolite M2 in patients with drug‐resistant tuberculosis: the effect of time‐varying weight and albumin. CPT Pharmacomet Syst Pharmacol. 2016;5:682‐691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ariëns EJ, van Rossum JM, Simonis AM. Affinity, intrinsic activity and drug interactions. Pharmacol Rev. 1957;9:218‐236. [PubMed] [Google Scholar]
- 17. Holford NHG, Sheiner LB. Kinetics of pharmacologic response. Pharmacol Ther. 1982;16:143‐166. [DOI] [PubMed] [Google Scholar]
- 18. Wallis RS. Cardiac safety of extensively drug‐resistant tuberculosis regimens including bedaquiline, delamanid and clofazimine. Eur Respir J. 2016;48:1526‐1527. [DOI] [PubMed] [Google Scholar]
- 19. Rubinstein E, Camm J. Cardiotoxicity of fluoroquinolones. J Antimicrob Chemother. 2002;49:593‐596. [DOI] [PubMed] [Google Scholar]
- 20. Food and Drug Administration . Pharmacology review(s). Application number 204384Orig1s000. Accessed October 12, 2021. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/204384Orig1s000PharmR.pdf
- 21. Minocha M, Li H, Chiu Y‐L, Carter D, Othman AA. Models of variability and circadian rhythm in heart rate, blood pressure, and QT interval for healthy subjects who received placebo in phase I trials. Clin Transl Sci. 2019;12:470‐480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vandenberk B, Vandael E, Robyns T, et al. Which QT correction formulae to use for QT monitoring? J Am Heart Assoc. 2016;5:e003264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Andršová I, Hnatkova K, Šišáková M, et al. Influence of heart rate correction formulas on QTc interval stability. Sci Rep. 2021;11:14269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Karjalainen J, Viitasalo M, Mänttäri M, Manninen V. Relation between QT intervals and heart rates from 40 to 120 beats/min in rest electrocardiograms of men and a simple method to adjust QT interval values. J Am Coll Cardiol. 1994;23:1547‐1553. [DOI] [PubMed] [Google Scholar]
- 25. Luo S, Michler K, Johnston P, Macfarlane PW. A comparison of commonly used QT correction formulae: the effect of heart rate on the QTc of normal ECGs. J Electrocardiol. 2004;37:81‐90. [DOI] [PubMed] [Google Scholar]
- 26. Bayer HealthCare Pharmaceuticals, Inc . AVELOX (moxifloxacin) label. Published online 2010. Accessed October 12, 2021. https://www.accessdata.fda.gov/drugsatfda_docs/Label/2010/021277s038lbl.pdf
- 27. Dannemann B, Bakare N, De Marez T. QTcF prolongation in a phase II trial of TMC207 plus background regimen as treatment for multidrug‐resistant tuberculosis: effect of co‐administration with clofazimine. In: 52nd Interscience Conference on Antimicrobial Agents and Chemotherapy, San Francisco, CA, USA; 2012:9‐12.
- 28. Zweijpfenning SMH, van Groningen H, van Ingen J, et al. Clofazimine does not lead to significant QT interval prolongation: a multicentre study. Eur Respir J. 2018;52:1801386. [DOI] [PubMed] [Google Scholar]
- 29. Yan LK, Zhang J, Ng MJ, Dang Q. Statistical characteristics of moxifloxacin‐induced QTc effect. J Biopharm Stat. 2010;20:497‐507. [DOI] [PubMed] [Google Scholar]
- 30. Novartis Pharmaceuticals Corporation . LAMPRENE (CLOFAZIMINE) label. Published online 2016. Accessed October 12, 2021. https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/019500s013lbl.pdf
- 31. Maartens G, Brill MJE, Pandie M, Svensson EM. Pharmacokinetic interaction between bedaquiline and clofazimine in patients with drug‐resistant tuberculosis. Int J Tuberc Lung Dis. 2018;22:26‐29. [DOI] [PubMed] [Google Scholar]
- 32. Brill MJE, Svensson EM, Pandie M, Maartens G, Karlsson MO. Confirming model‐predicted pharmacokinetic interactions between bedaquiline and lopinavir/ritonavir or nevirapine in patients with HIV and drug‐resistant tuberculosis. Int J Antimicrob Agents. 2017;49:212‐217. [DOI] [PubMed] [Google Scholar]
- 33. Svensson EM, Aweeka F, Park J‐G, Marzan F, Dooley KE, Karlsson MO. Model‐based estimates of the effects of efavirenz on bedaquiline pharmacokinetics and suggested dose adjustments for patients coinfected with HIV and tuberculosis. Antimicrob Agents Chemother. 2013;57:2780‐2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Svensson EM, Dooley KE, Karlsson MO. Impact of lopinavir‐ritonavir or nevirapine on bedaquiline exposures and potential implications for patients with tuberculosis‐HIV coinfection. Antimicrob Agents Chemother. 2014;58:6406‐6412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Svensson EM, Murray S, Karlsson MO, Dooley KE. Rifampicin and rifapentine significantly reduce concentrations of bedaquiline, a new anti‐TB drug. J Antimicrob Chemother. 2015;70:1106‐1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material