

Abstract
Effector CD4+ T lymphocytes contribute to inflammation and tissue damage in psoriasis, but the underlying molecular mechanisms remain poorly understood. The transcription factor CREMα controls effector T cell function in people with systemic autoimmune diseases. The inhibitory surface co-receptor PD-1 plays a key role in the control of effector T cell function and its therapeutic inhibition in patients with cancer can cause psoriasis. Here we show that CD4+ T cells from patients with psoriasis and psoriatic arthritis exhibit increased production of IL-17 but decreased expression of IL-2 and PD-1. In genetically modified mice and Jurkat T cells CREMα expression was linked to low PD-1 levels. We demonstrate that CREMα is recruited to the proximal promoter of PDCD1 where it trans-represses gene expression and co-recruits DNMT3a mediating DNA methylation. As keratinocytes limit inflammation by PD-1 ligand expression, and here reported reduced expression of PD-1 on CD4+ T cells is linked to low IL-2 and high IL-17A production, our studies reveal a molecular pathway in T cells from people with psoriasis which can deserve clinical exploitation.
Keywords: PD-1, CREM, psoriasis, psoriatic arthritis, molecular, pathophysiology, chromatin, epigenetic, DNMT, inflammation, effector T cell, cytokine, DNA methylation
1. Introduction
Psoriasis is a systemic autoimmune/inflammatory condition that primarily affects the skin, but also other organs (1). Skin disease is characterized by increased proliferation of keratinocytes and immune cell infiltration with neutrophils, dendritic cells, macrophages and T lymphocytes. In psoriasis, effector CD4+ and CD8+ T lymphocytes contribute to skin inflammation and tissue damage. However, the underlying molecular mechanisms remain poorly understood (1–4).
In response to antigen contact and subsequent activation, CD4+ T lymphocytes proliferate and differentiate into effector cells. Surface co-receptors play a key role in the regulation of the immune response and the control of self-reactivity and inflammation through stimulatory and inhibitory signals (5, 6). Effector T lymphocytes are characterized by their surface co-receptor repertoire which contributes to their activation and homing characteristics, and lineage-specific cytokine expression patterns. CD3+TCR+CCR7−CD45RA− T lymphocytes are referred to as effector memory (EM) cells as they respond quickly to T cell receptor ligation. CD3+TCR+CCR7−CD45RA+ T cells are terminally differentiated cells and are referred to as terminally differentiated effector memory cells re-expressing CD45RA (EMRA). Central memory (CM) CD3+TCR+CCR7+CD45RA− T lymphocytes home in secondary lymphoid tissues where they wait for secondary challenges to raise an immune response (5). Undifferentiated naïve CD4+ T cells express low levels of cytokines, but when exposed to antigen they express effector cytokines, including IL-17A, IL-17F and/or interferon (IFN)-γ (5, 6).
The transcription factor cAMP response element modulator (CREM)α belongs to a superfamily of transcription factors comprising >50 homologues, including inducible cAMP early repressor (ICER), cAMP responsive element binding proteins (CREB)-1 and -2, and the CREM/activating transcription factors (ATF)-1, -2, and -3. All CREM family transcription factors share high sequence homology within their DNA binding domains (a leucine zipper domain) and recruit to the palindromic consensus element 5’-TGACGTCA-3’, the cAMP responsive element (CRE), or its 5’-half-site. The CREMα isoform is expressed at increased levels in effector CD4+ T cells (7–9). Expression of CREMα in T cells has been linked with effector phenotypes that contribute to inflammation and damage in T cell mediated autoimmune/inflammatory disease, namely systemic lupus erythematosus (4). CD4+ T cells from patients with SLE express increased amounts of CREMα which has been claimed to be responsible for the increased production of the effector cytokine IL-17A and the limited production of IL-2 (7–12) through epigenetic remodeling of the two loci (13–15). Epigenetic modifications regulate gene expression; the two most well studied epigenetic mechanisms and DNA methylation and histone modifications. Methylation of CpG dinucleotides affects negatively the recruitment of transcription factors and other elements of the transcriptional complex (16). Post-translational modifications to N-terminal amino acid residues affect nucleosome charge and three-dimensional arrangement of chromatin thereby regulating its accessibility to transcription factors and RNA polymerases (13). Through complex interactions involving a range of molecules, DNA methylation and histone modifications are linked, which can augment the “epigenetic code” (17–19). Notably, CREMα physically interacts with and directs the recruitment of epigenetic modifiers to target genes, thereby altering T cell responses in SLE (9). This includes the “epigenetic silencers” DNA methyltransferase (DNMT)3a and histone deacetylase (HDAC)1 (7, 10, 20), and histone methyltransferase G9a as well as the transcriptional co-activator p300 (21, 22), a histone acetyltransferase. These interactions define CREMα as a transcription factor with epigenetic modification functions.
The key involvement of effector CD4+ T cells in the pathophysiology of psoriasis has been established. CD4+ T cells contribute to inflammation and damage through the production of effector cytokines, including IL-17A (23, 24). The generation of effector T helper (Th)17 lymphocytes and IL-17A production is dependent on the transcription factor STAT3 (25). An imbalance between STAT3 and STAT5 contributes to inflammation and damage in autoimmune/inflammatory disease through the promotion of effector T cells (22, 26). The key involvement of IL-17A producing effector CD4+ T cells in psoriasis has recently been further underscored by the observation that blockade of the STAT3 interaction partner Rho-associated kinase (ROCK)2 reduces effector cytokine expression, inflammation and damage by correcting the altered balance between STAT3 and STAT5, thereby inhibiting Th17 effector T cells (27–30).
Effector T cell differentiation, function and survival is regulated by a tightly controlled network of activating and inhibitory signals that include surface co-receptors. The programmed cell death (PD-)1 co-receptor regulates T lymphocyte function down-stream of the CD3/TCR complex (31). PD-1 inhibits T cell activation, cytokine expression and effector T cell differentiation through a multitude of molecular mechanisms, including regulation of protein kinases, metabolic pathways, transcription factor activation, among others (32, 33). Because PD-1 expression is upregulated in response to T cell activation it has been suggested to reflect T cell exhaustion. Although inhibition of PD-1 has been used successfully in the treatment of certain cancers including melanoma, non-small cell lung cancer and renal cell carcinoma, not invariably it results in the appearance of autoimmune/inflammatory diseases including psoriasis in a subset of patients. Reversely, limited expression or function of PD-1 may contribute to the propagation of inflammatory T cells and autoimmune disease including psoriasis (31–38).
In this communication we demonstrate that the transcription factor CREMα, through trans-repression of the PDCD1 promoter and the induction of DNA methylation, represses the expression of PD-1 which associates with increased IL-17A but reduced IL-2 production. The identified pathway offers opportunities for the development of novel therapeutics and biomarkers in people with psoriasis.
2. Materials and Methods
Patients and controls
All psoriasis and psoriatic arthritis patients included in this study were diagnosed and recruited by the Division of Dermatology, Faculty of Medicine Carl Gustav Carus, TU Dresden, Germany. Matched healthy controls (blood donors) were recruited from the clinical research laboratories at Children’s Hospital Dresden. The study was reviewed and approved by the local ethics committee, and all included patients and controls gave written informed consent.
All experiments were carried out with blood samples from either treatment-naïve patients with plaque-type psoriasis or psoriatic arthritis or individuals who did not receive systemic treatment in the 12 weeks prior to sample collection (Supplement Table 1). Patients had moderate to severe skin disease; Psoriasis Area and Severity Index (PASI) scores were evaluated at the time of blood sampling.
Animals
C57BL/6 mice were purchased from The Jackson Laboratory. FVB.CREMα Transgenic (Tg) mice were originally cloned by Klaus Tenbrock, RWTH Aachen University, Germany. Animals were crossed to C57BL/6J mice for over ten generations to transfer the CREMα-CD2 locus to the C57BL/6 background. Animals were sacrificed at the age of 8-12 weeks to perform in vitro culture experiments. All mice were maintained in an SPF animal facility (Beth Israel Deaconess Medical Center). Experiments were approved by the Institutional Animal Care and Use Committee of BIDMC. Spleen and lymph node lymphocytes were isolated as previously described (39).
In vitro T cell stimulation
Naive CD4+ T cells were enriched using the untouched mouse naïve CD4+ T cells isolation kit (Miltenyi Biotec) from freshly isolated mice splenocytes. Naïve CD4+ T cells were stimulated with plate-bound anti-CD3 (0.25 μg/ml, 145–2C11; Biolegend) and anti-CD28 (0.5 μg/ml, 37.51; Biolegend) antibodies for 24 h. For some experiments, genetically modified Jurkat T cells were used, and stimulated with plate-bound CD3 (0.25 μg/ml, 145–2C11; Biolegend) and CD28 (0.5 μg/ml, 37.51; Biolegend) antibodies as indicated in the appropriate sections. Genetically modified CREM-deficient (Cell line 1D4 from (22)) and CREMα overexpressing cells were described and characterized previously, including their respective CREMα mRNA and protein expression levels CREMα (22).
Flow Cytometry of mouse cells
The following antibodies were used for flow cytometry analysis; anti-mouse CD8a (53–6.7), CD90.2 (53–2.1), and PD1 (29F.1A12) were purchased from BioLegend. Anti-CD4 (GK1.5) was purchased from eBioscience. A Zombie Aqua™ Fixable Viability Kit (Biolegend) staining was performed to eliminate dead cells from analysis. Surface staining was performed on ice for 20-30 min. Absolute cell numbers were calculated on the basis of the percentage of each cell population. All flow cytometry data were acquired on a Cytoflex LX (Beckman Coulter) and analyzed with FlowJo (FlowJo, llc). All procedures were performed according to the manufacturer’s instructions.
Human cell collection and culture
Human peripheral blood mononuclear cells (PBMCs) were isolated from peripheral venous blood using Biocoll (Merck) and Leucosep Tubes (Greiner, Bio-One), following standard protocols. Immune cell composition was analysed using flow cytometry and the following antibodies: FITC anti-human CD3, Pacific Blue™ anti-human CD4, APC/Cy7 anti-human CD8, PE anti-human CD197/CCR7, APC anti-human CD45RA antibody, PerCP/Cy5.5 anti-human CD279/PD-1 (all BioLegend), FITC TCR alpha/beta (eBioscience).
Primary human PBMCs were kept in RPMI 1640 medium supplemented with 10% FBS and 2 mM L-glutamine. Cells were incubated in the absence or presence of plate-bound anti-CD3 (OKT3; BioLegend) and anti-CD28 (CD28.2; BioLegend) antibodies for 120 h. All cells where then stimulated with PMA and ionomycin for 4 h and CD4+ T cells were collected using flow cytometry for RNA (gene expression) and DNA (methylation) analyses.
Some experiments were performed in previously reported genetically modified CD4+ Jurkat T cells (22). Jurkat T cells were kept in RPMI 1640 medium supplemented with 10% FBS and 2 mM l-glutamine. As indicated below, cells were incubated in the absence or presence of plate-bound anti-CD3 (OKT3; BioLegend) and anti-CD28 (CD28.2; BioLegend) antibodies for 24 h. Where indicated, cells were re-stimulated with PMA and ionomycin for 4 h.
mRNA expression analysis
Total RNA from primary human T cells or Jurkat T cells was isolated using the QIAGEN RNeasy Mini Kit (QIAGEN) or the Quick-DNA/RNA Miniprep Kit (Zymo Research). For most experiments, Affymetrix QuantiGene mRNA probes were used and analyzed on a Luminex 200 platform according to the manufacturer’s instructions. mRNA copy numbers were normalized to three housekeeping genes (HPRT1, PPIB, and ACTB). For some PD-1 and all CREMα-specific quantification experiments in Jurkat T cells (as indicated), cDNA was generated using a first-strand cDNA synthesis kit (Clontech Laboratories). For gene expression analyses, cDNA was used for quantitative RT-PCR (qRT-PCR) using TaqMan systems. Primer and probe sequences will be shared upon request.
Protein quantification
Cytokine IL-2, IL-17A and IL-17F and PD-1 proteins in cell culture supernatants were quantified using the Meso Scale Discovery Electrochemiluminescence R-PLEX Immunoassay (MSD, Maryland, USA). All assays were performed as per the manufacturer’s instructions using MSD GOLD 96-well Small Spot Streptavidin and assay diluents. Plates were read using the MSD QuickPlex SQ 120 platform.
Luciferase reporter assays in Jurkat T cells
One million CD4+ Jurkat T cells (wild-type, CREM-deficient, or CREMα-overexpressing as indicated) were transfected with 500 ng/106 cells plasmid DNA using the Amaxa transfection system (Lonza). Effector:reporter transfection experiments were performed at a molar ratio of 3:1. Each reporter experiment included 10 ng of Renilla luciferase construct as an internal control. Five hours after transfection, cells were collected and lysed, and luciferase activity was quantified using the Promega Dual Luciferase Assay System following the manufacturer’s instructions.
Chromatin immunoprecipitation assays
Anti-H3K18 acetylation (H3K18ac; Abcam), anti-DNMT3a (Abcam), anti-HA (EMD Millipore), and nonspecific normal rabbit IgG were obtained from EMD Millipore. Chromatin immunoprecipitation (ChIP) assays were carried out according to the manufacturer’s instructions (Invitrogen, Life Technologies). Briefly, cells were cross-linked with 1% formaldehyde, washed with cold PBS, and lysed in buffer containing protease inhibitors (28). Cell lysates were sonicated to shear DNA and sedimented, and diluted supernatants were immunoprecipitated with the indicated Abs. A proportion (10%) of the diluted supernatants was kept as input control. After the recovery of ChIP DNA, real-time quantitative PCR was performed.
Knock-down of DNMT3a with siRNAs
Three million WT or CREM-overexpressing Jurkat CD4+ T cells were transfected with 30 nM scrambled control short inhibiting RNA (siRNA) or DNMT3a-specific siRNA (OriGene) using Lipofectamine 2000 (Invitrogen, Life Technologies). Cells were collected after overnight culture (24 h) and processed for mRNA analysis.
Bisulfite pyrosequencing
Pyrosequencing assays were performed to investigate DNA methylation of CpG sites identified in the PDCD1 promoter (PCR and sequencing primers will be shared upon request), following previously described methodology (40) and manufacturers’ instructions (Pyromark).
Statistical analysis
The paired two-tailed Student t test or Mann Whitney U tests were used for pairwise comparisons as indicated. Nonparametric Kruskal-Wallis tests were used to determine differences between multiple groups, followed by Dunne’s post hoc tests where statistical significance was reached.
3. Results
Normal numbers of total CD4+ T cells in people with psoriasis
CD4+ T cells were isolated from PBMCs collected from patients with psoriasis, psoriatic arthritis and healthy controls. Cells were kept in culture for 5 days under resting conditions or stimulated with plate-bound CD3 and CD28 antibodies to mimic antigen contact. The absolute numbers and proportions of CD4+ T cells were comparable between all groups at the time of sample collection and 5 days after stimulation. Stimulation of CD4+ T cells resulted in decreased numbers that, however, was comparable between the three groups (Supplement Figure 1A, B). Because psoriasis and psoriatic arthritis are characterized by effector T cell responses, we evaluated the subset composition of the peripheral blood CD4+ T cell compartment. While ex vivo isolated and resting CD4+ T cells were largely comparable between patients and controls, stimulation with plate-bound CD3 and CD28 antibodies resulted in reduced numbers of naïve CD4+ T cells in samples from psoriatic arthritis patients alongside increased numbers of effector memory CD4+ T cells that, however, did not reach statistical significance (Supplement Figure 2C, D).
CD4+ T cells from patients with psoriasis exhibit effector T cell gene expression profiles
CD4+ T cells from patients with psoriasis, psoriatic arthritis and healthy controls were cultured for 5 days in the absence or presence of plate-bound CD3 and CD28 antibodies. After stimulation, cells from patients with psoriatic arthritis displayed significantly reduced mRNA and protein expression of IL-2 (Figure 1A), but increased expression of the effector cytokine IL-17A (Figure 1B). Furthermore, unstimulated CD4+ T cells from patients with psoriasis exhibited reduced IL-2 protein expression as compared to cells from control subjects.
Figure 1: CD4+ T cells from patients with psoriasis exhibit effector cytokine expression profile.
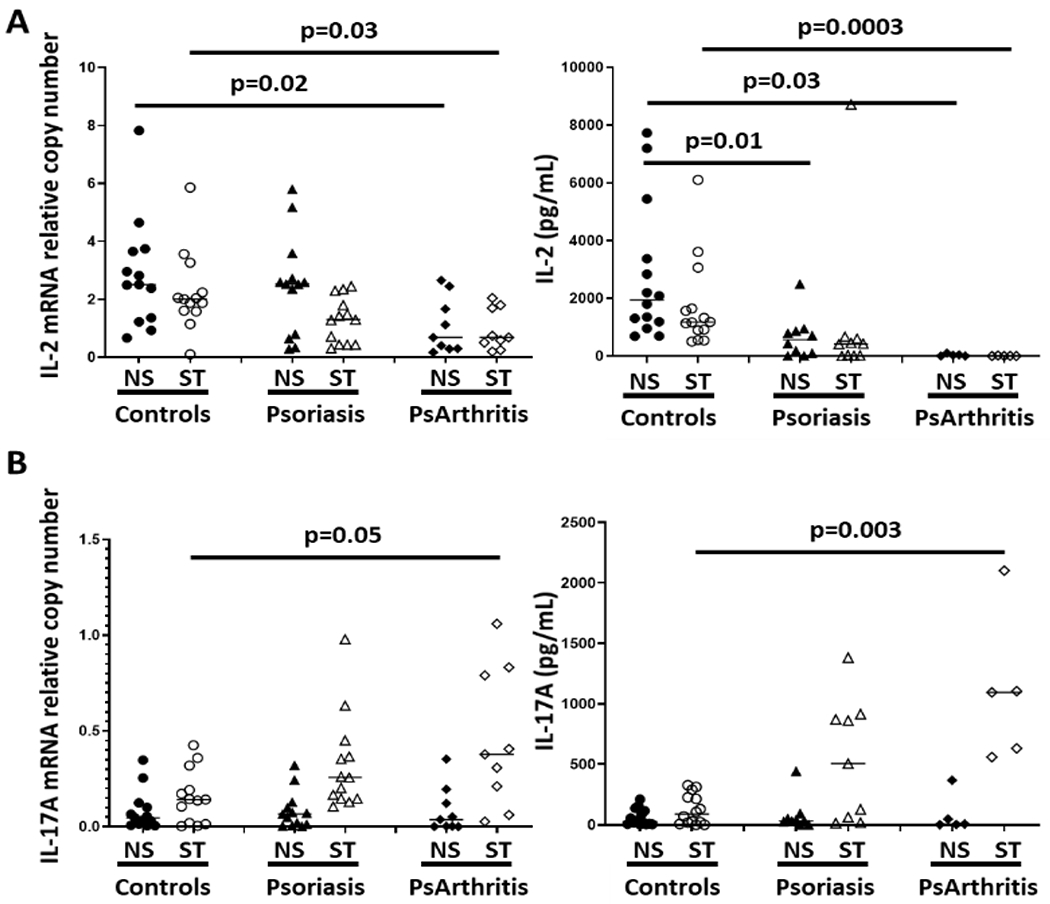
CD4+ T cells from patients with psoriasis (N=13), psoriatic arthritis (N=9) and healthy controls (N=13) were cultured for 5 days in the absence or presence of plate-bound CD3 and CD28 antibodies. A) mRNA and protein IL-2 levels; B) mRNA and protein expression of IL-17A (Kruskal Wallis and Dunne’s tests).
As effector CD4+ T cells express increased levels of the transcription factor CREMα, the expression of total CREM (Figure 2A) and CREMα (Figure 2B) were measured. While total CREM expression (all isoforms) was increased after 5 days of simulation with plate-bound CD3 and CD28 antibodies at comparable levels between the three groups, CREMα expression was significantly increased in CD4+ T cells from patients with psoriasis or psoriatic arthritis.
Figure 2: CD4+ T cells from patients with psoriasis express increased levels of CREMα.
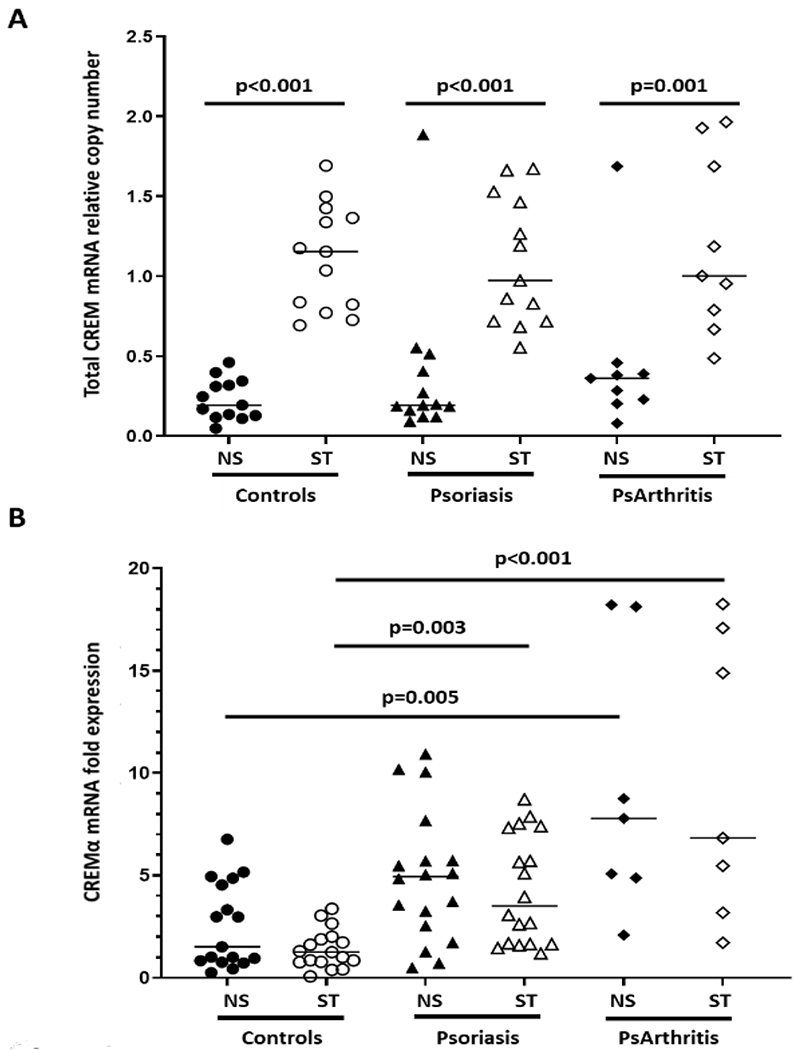
A) CREM expression (all isoforms) in CD4+ T cells from patients with psoriasis (N=13), psoriatic arthritis (N=9) and healthy controls (N=13) after 5 days of simulation with plate-bound CD3 and CD28 antibodies (Mann Whitney U test for unstimulated, NS vs stimulated, ST; Kruskal Wallis between groups). B) Expression of the CREMα isoform in CD4+ T cells from patients with psoriasis or psoriatic arthritis and control subjects (Kruskal Wallis and Dunne’s tests).
CD4+ T cells from patients with psoriasis fail to express the co-inhibitory molecule PD-1
Because the surface co-receptor PD-1 regulates T cell differentiation and limits effector T cell generation and PD-1 inhibition in cancer patients can trigger psoriasis (41), we investigated PD-1 expression on CD4+ T cells from patients with psoriasis. In the absence of CD3/TCR stimulation, PD-1 expression decreased over the 5 d culture period in CD4+ T cells from healthy controls and patients with psoriasis (Figure 3A). Notably, this reduction was less pronounced in CD4+ T cells from patients with psoriasis or psoriatic arthritis. Stimulation with plate-bound CD3 and CD28 antibodies over 5 days resulted in significantly increased numbers of PD-1+ cells in the healthy subjects, which was less pronounced in patients with psoriasis or psoriatic arthritis (Figure 3B). Notably, PD-1 density on the surface membrane of CD4+ T cells, as determined by recording the mean fluorescence intensity (MFI), was significantly reduced in cells from psoriasis and psoriatic arthritis patients as compared to controls (Figure 3C and 3D).
Figure 3: PD-1 surface expression on CD4+ T cells from patients with psoriasis and control subjects.
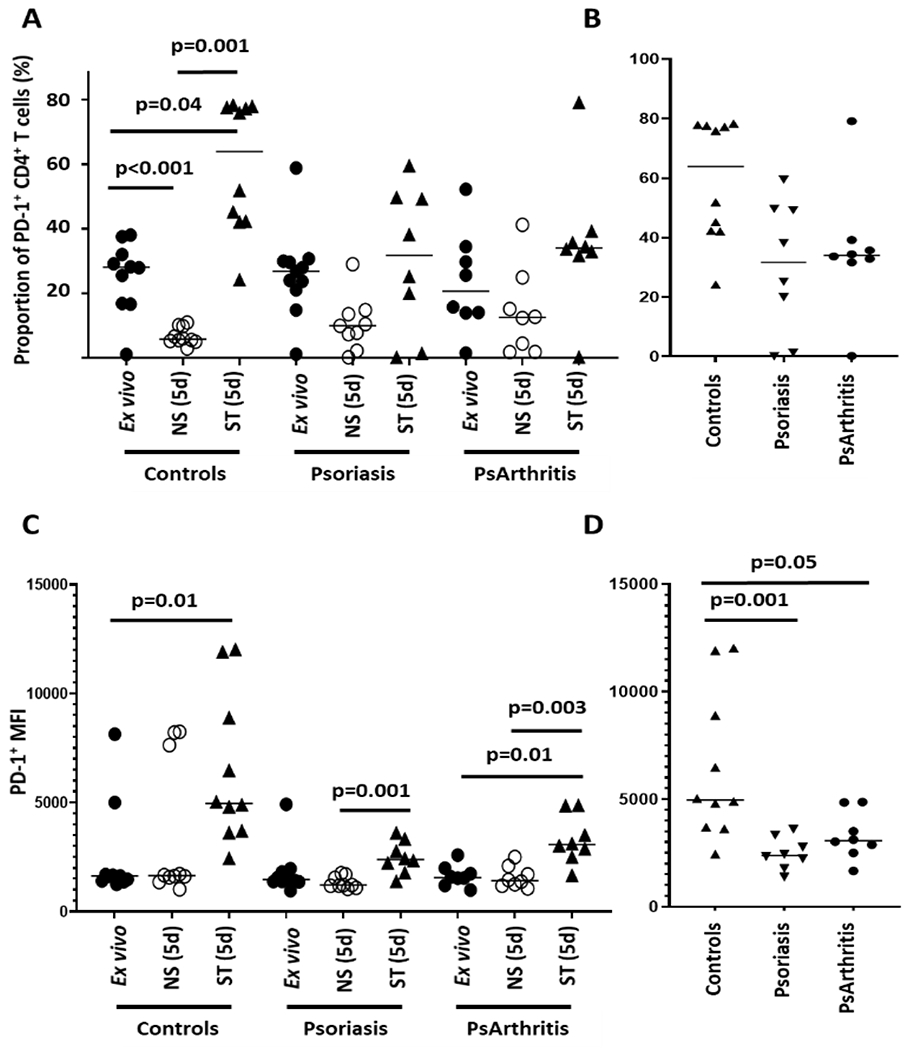
A) Left panel: PD-1 expression in CD4+ T cells from healthy controls (N=10), psoriasis (N=9) and psoriatic arthritis (N=8) patients after ex vivo isolation and cultured for 5 days (resting: NS; stimulated: ST with plate-bound CD3/CD28). Right panel: Data from the same experiments, emphasizing the proportion of PD-1+ CD4+ T cells in patients with psoriasis or psoriatic arthritis and controls after 5d of stimulation. B) Left panel: Mean fluorescence intensity (MFI) of PD-1 surface expression on CD4+ T cells in stimulated cells from control subjects (N=10) and patients with psoriasis (N=9) or psoriatic arthritis (N=8) after ex vivo isolation and cultured for 5 days (resting: NS; stimulated: ST with plate-bound CD3/CD28). Right panel: Data from the same experiments, emphasizing PD-1 surface density in cells from psoriasis and psoriatic arthritis patients and control subjects (Kruskal Wallis and Dunne’s tests).
Next, we tested whether PD-1 surface expression was altered across CD4+ T cell subsets. In the absence of antigen contact under resting conditions after 5 days, naïve, central memory and effector memory CD4+ T cells from patients with psoriasis or psoriatic arthritis exhibited increased PD-1 expression when compared to controls that did, however, not reach statistical significance (Supplement Figure 2A). Stimulation with plate-bound CD3 and CD28 antibodies reversed this and resulted in a trend towards reduced PD-1 expression on all T cell subsets investigated (Supplement Figure 2B). Study of PD-1 receptor density (MFI) revealed reduced PD-1 surface expression on cells from patients which extended across naïve, CM and EM subsets in patients with psoriasis and psoriatic arthritis (Supplement Figure 2C, D).
CREMα regulates PD-1 expression at the transcriptional level
To investigate mechanisms that affect PD-1 expression, we assessed PD-1 mRNA expression on resting and stimulated CD4+ T cells (Figure 4A). In CD4+ T cells from control subjects, patients with psoriasis or psoriatic arthritis that were stimulated for 5 days PD-1 mRNA expression was increased when compared to resting cells (left panel). PD-1 mRNA expression in stimulated cells from psoriasis (trend) and psoriatic arthritis (p=0.002) patients was reduced as compared to cells from healthy subjects (right panel). Notably, PD-1 co-receptors can be shed from the cell surface. The biological relevance of this mechanism is not understood completely (42), but soluble molecules may negatively affect PD-1 surface density on T cells. To test whether reduced PD-1 surface expression was affected by receptor shedding, we measured PD-1 levels in cell culture supernatants from the same experiments. PD-1 levels were not different between controls or patients suggesting that surface receptor shedding does not contribute significantly to reduced numbers of PD-1 positive cells (Figure 4B).
Figure 4: PD-1 mRNA expression and protein shedding in CD4+ T cells from patients with psoriasis and control subjects.
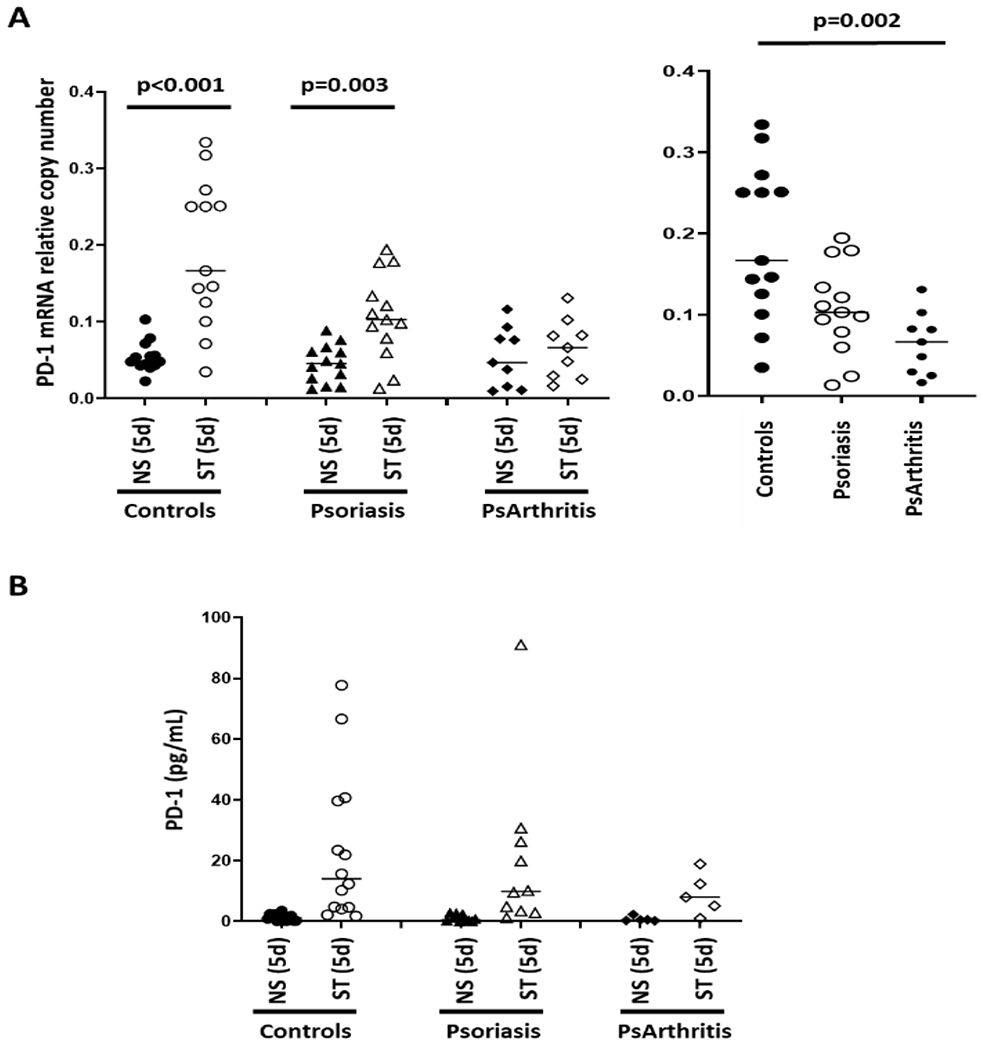
A) PD-1 mRNA expression in CD4+ T cells from controls (N=13), patients with psoriasis (N=13) or psoriatic arthritis (N=9) (Left panel: PD-1 mRNA expression in resting (NS) and stimulated (for 5 days, ST) T cells; right panel: stimulated T cells from patients with psoriasis, psoriatic arthritis and control subjects; Kruskal Wallis and Dunne’s tests). B) PD-1 levels in cell culture supernatants (Mann Whitney U test).
As the transcription factor CREMα is expressed at increased levels in CD4+ T cells from patients with psoriasis and psoriatic arthritis, we tested whether CREMα contributes to reduced PD-1 expression using previously reported genetically modified CD4+ Jurkat T cells (22). Jurkat T cells were cultured for 24 h under resting conditions or in the presence of plate-bound CD3 and CD28 antibodies. Stimulation of Jurkat T cells resulted in significant upregulation of PD-1 mRNA (Figure 5A) and surface protein (Figure 5B) expression. Furthermore, CREMα overexpressing Jurkat T cells resulted in reduced PD-1 mRNA and protein expression compared to wild-type control cells, while CREMα-deficient cells expressed more PD-1 at the mRNA and protein levels. This observation was confirmed also using CD4+ T cells from transgenic CREMα overexpressing C57BL/6 mice which were found to express reduced amounts of PD-1 (Figure 5C).
Figure 5: CREMα inversely correlates with PD-1 expression in genetically modified cells.
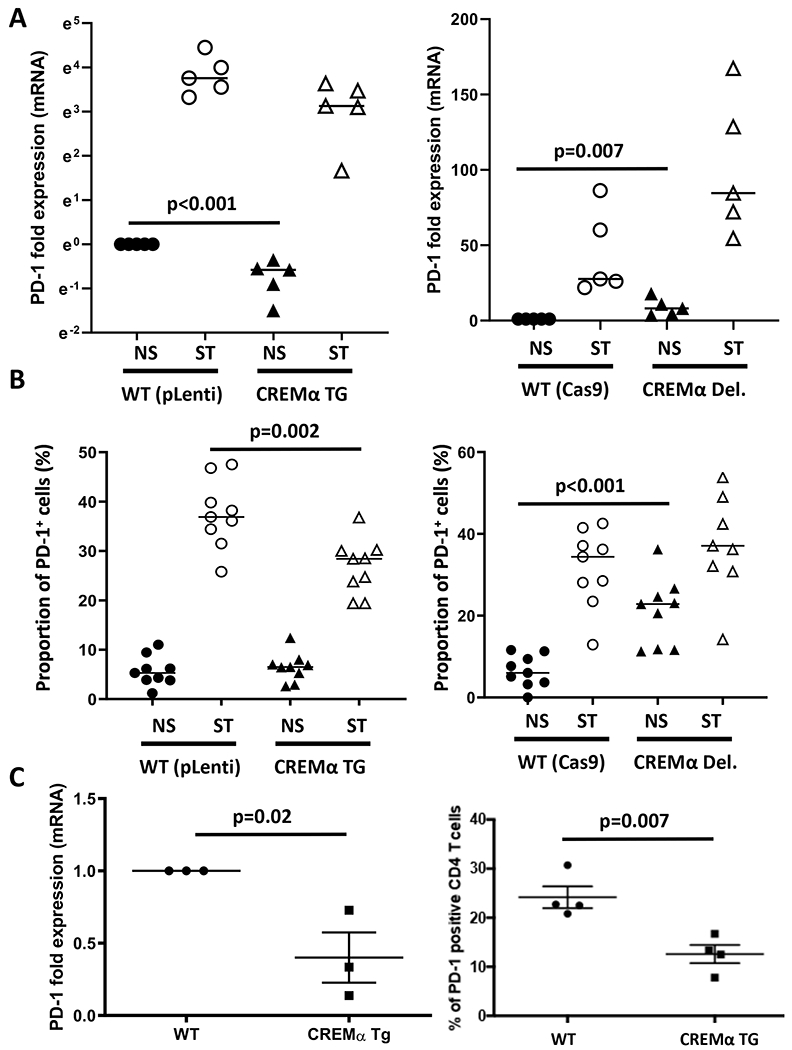
Wild-type, CREMα overexpressing (CREMα TG, left panel), and CREMα-deficient (CREMα Del., right panel) Jurkat T cells were cultured for 24h under resting conditions or in the presence of plate-bound CD3 and CD28 antibodies (N=5 independent experiments). A) PD-1 mRNA levels (logarithmic scale on Y axis). B) Proportion of PD-1 positive cells as assessed by flow cytometry (N=9 independent experiments). C) PD1 mRNA (N=3) and protein levels (N=4) in CD4+ T cells from C57/Bl6 mice overexpressing CREMα in T cells stimulated with plate-bound CD3 and CD28 antibodies for 24h.
PDCD1 is under the control of a 5’ proximal promoter. In silico analyses (Vista Genome Browser) predicted a cluster of three CREM binding elements (CRE) within the previously described core promoter region (43) (Figure 6A). Using Jurkat T cells overexpressing HA-tagged CREMα, ChIP with anti-HA antibodies revealed recruitment of HA-CREMα to the PDCD1 promoter (Figure 6B). Anti-HA antibodies were used to control for cross-reactivity between the approximately 50 isoforms of CREM. Next, using a previously reported luciferase reporter construct of the PDCD1 core promoter regions (43), we tested the effect of CREMα on the PDCD1 promoter activity (Figure 6C). The PDCD1 promoter construct showed spontaneous promoter activity in Jurkat T cells (left). While differential CREMα expression between wild-type, CREMα-deficient and CREM-α overexpressing Jurkat T cells did not alter luciferase activity of “empty” pGL3 plasmids (middle), luciferase activity of the PDCD1 promoter construct was significantly reduced in CREMα overexpressing cells and increased in CREMα-deficient Jurkat T cells (right).
Figure 6: CREMα trans-regulates the PDCD1 proximal promoter.
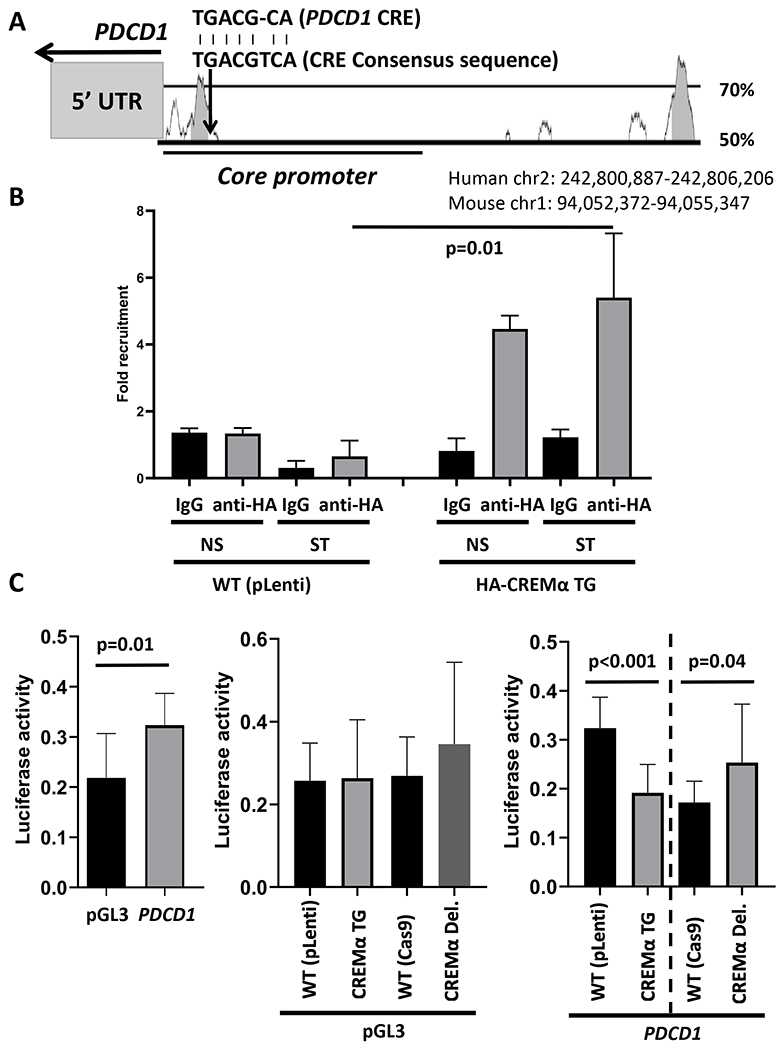
A) PDCD1 is under the control of a 5’ proximal promoter region. The level of inter-species conservation (human vs mouse) is provided as % conservation over 200 base pairs (Vista Genome browser). The core promoter region an almost complete (7/8, 87.5%) cAMP response element (CRE) -942 base pairs upstream the transcriptional initiation sequence, which is indicated above the graph. B) ChIP with antibodies directed against HA in genetically modified Jurkat T cells overexpressing HA-tagged CREMα, CREMα recruitment to the aforementioned -942 CRE is shown (p values from t tests are shown) (N=4). C) PDCD1 promoter region-driven luciferase activity in wild-type (WT) and genetically modified Jurkat T cells (t tests) (N=6).
CREMα regulates PD-1 expression through DNMT3a
Because CREMα physically interacts with DNMT3a at the IL2 and CD8 genes and causes epigenetic silencing by methylating DNA (7, 8, 10, 16, 21), we tested whether this also contributes to the down-regulation of PD-1 expression. Using ChIP assays, we detected DNMT3a recruitment at the same region within the PDCD1 promoter where CREMα is recruited too. This was increased in CREMα overexpressing cells as compared to WT Jurkat T cells (Figure 7A). To test whether CREMα functionally interacts with DNMT3a at the PDCD1 promoter, we silenced DNMT3a expression in WT, CREMα overexpressing and CREMα-deficient cells using siRNAs. When compared to cells transfected with scrambled siRNAs, transfection of DNMT3a specific siRNAs, followed by stimulation with plate-bound CD3 and CD28 antibodies, resulted in increased expression of PD-1 mRNA in WT and CREMα overexpressing cells (Figure 7B). This, however, was not the case in CREM-deficient cells, where DNMT3a silencing did not affect PD-1 mRNA expression.
Figure 7: CREMα regulates PD-1 expression through DNMT3a.
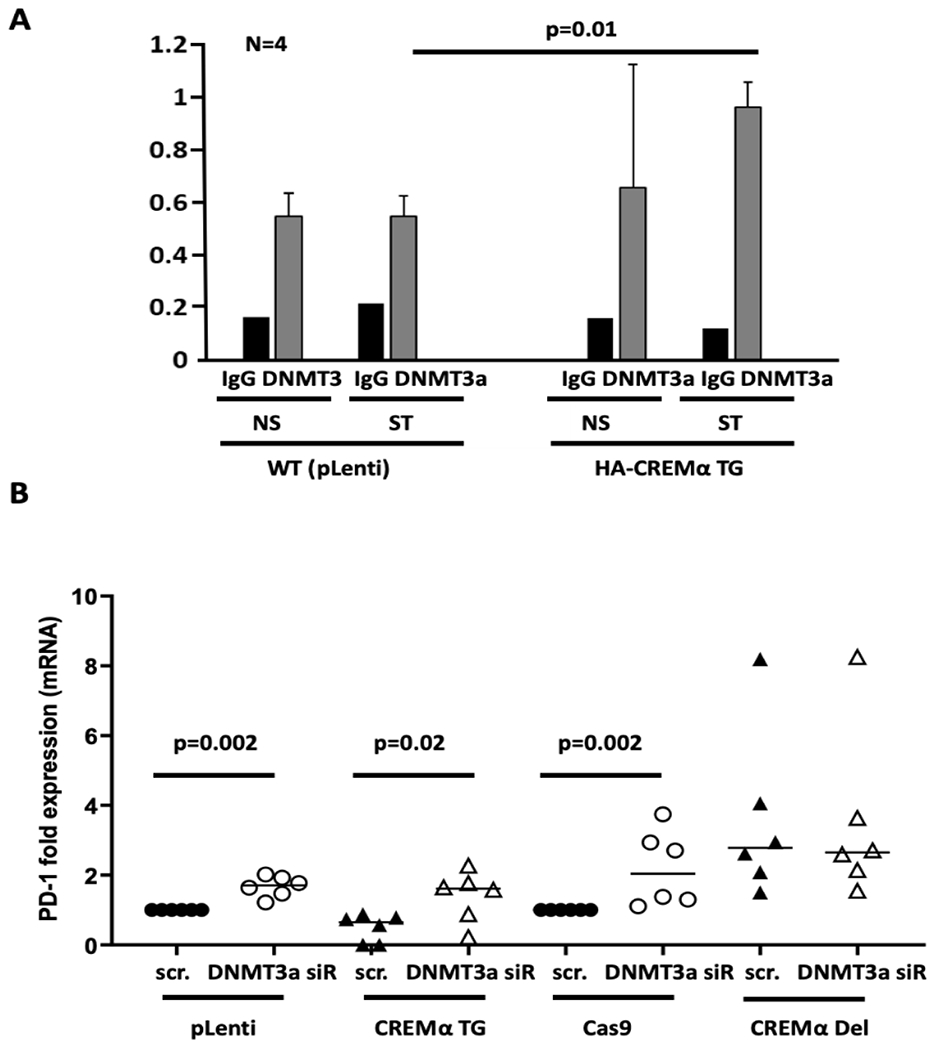
A) ChIP assays demonstrating the co-recruitment of DNMT3a and CREMα to the same region within the PDCD1 promoter in Jurkat T cells transfected with HA-CREMα or wild-type WT pLenti vectors (N=4). B) Jurkat T cells transfected with DNMT3a-specific or scrambled miRNAs and stimulated with plate-bound CD3 and CD28 antibodies (Kruskal Wallis and Dunne’s tests) (N=6).
Next, we assessed CpG DNA methylation at the PDCD1 promoter regions CR-B and CR-C which had previously been associated with differential DNA methylation patterns in PD-1+ vs PD-1− cells (44) (Figure 8A). CREM-deficient Jurkat T cells under resting or stimulated (CD3/CD28 antibodies overnight) conditions exhibited reduced DNA methylation at PDCD1 promoter regions CR-B and CR-C1 when compared to CREMα expressing WT or CREMα overexpressing cells (Figure 8B). No differences were seen between WT and CREM overexpressing cells. This observation was partially reflected by DNA methylation patterns of PDCD1 CR-B at CpG1–3 in stimulated CD4+ T cells from patients with psoriasis or psoriatic arthritis (trend, missing statistical significance) when compared to controls (Figure 8C).
Figure 8: CREMα expression correlates with PDCD1 methylation.
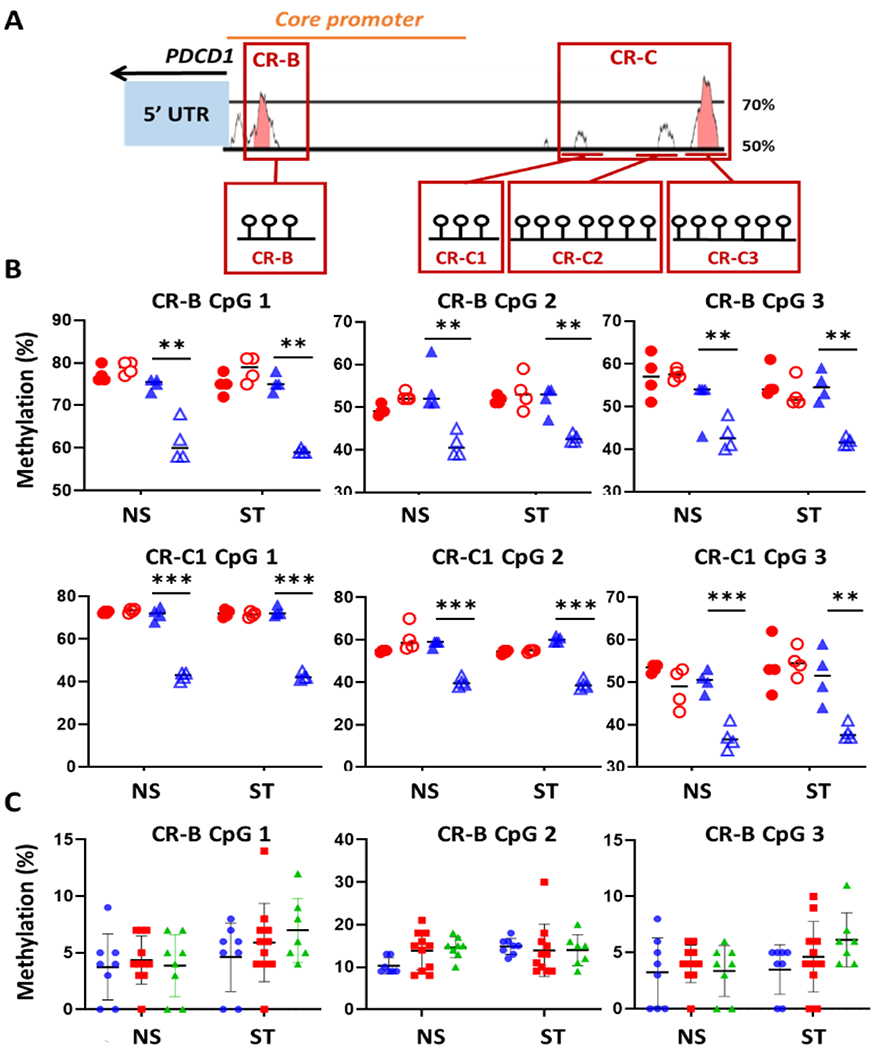
A) PDCD1 promoter region CpG rich regions CR-B and CR-C. DNA methylation of these regions was tested using bisulfite pyrosequencing (CR-B including 7 CpGs, CR-C1 (3 CpGs), CR-C2 (7 CpGs), CR-C3 (6 CpGs). B) DNA methylation of regions CR-B (CpG 1–3) and CR-C1 (CpG 1–3) in CREMα-deficient Jurkat T cells (blue open triangles) wild-type control cells (Cas9; blue filled triangles) under resting conditions (NS) and in response to stimulation with plate-bound CD3 and CD28 antibodies (ST) (N=4). Only CpGs with variable methylation levels are displayed. pLenti: filled red circles, filled red circles: CREMα overexpressing T cells. C) PDCD1 DNA methylation in CD4+ T cells from patients with psoriasis (red, N=11)) and PsA (green, N=8)) and control cells (blue, N=8)). (Mann Whitney U tests; significance levels: **p<0.01; ***p<0.001).
PD-1 negative T cells associate with effector T cell cytokine expression profile
To investigate whether reduced PD-1 expression associates with reduced IL-2 and increased IL-17A expression in the same cells and associates with CREMα expression, we FACS-sorted PD-1+ and PD-1− Jurkat T cells using wild-type, CREMα overexpressing and CREMα-deficient cell lines (Figure 9, Supplement Figure 3). As expected, PD-1− WT and CREMα-deficient Jurkat T cells produced significantly fewer copy numbers of PD-1 mRNA when compared to their PD-1+ equivalents (Figure 9A). Furthermore, WT Jurkat T cells produced fewer PD-1 mRNA copies when compared to CREMα-deficient cells under resting conditions and after stimulation with plate-bound CD3 and CD28 antibodies for 24 h (Supplement Figure 3A). This was mirrored by cytokine expression patterns. PD-1− T cells expressed less IL-2 mRNA (Figure 9B) and higher levels of IL-17A mRNA (Figure 9C) when compared to PD-1+ cells across cell lines (WT vs CREMα-deficient cells). Overall, CREMα-deficient cells exhibited increased IL-2 and reduced IL-17A expression across PD-1+ and PD-1− subsets when compared to WT cells (Supplement Figure 3B, C).
Figure 9: PD-1 negative T cells display effector T cell cytokine expression profile.
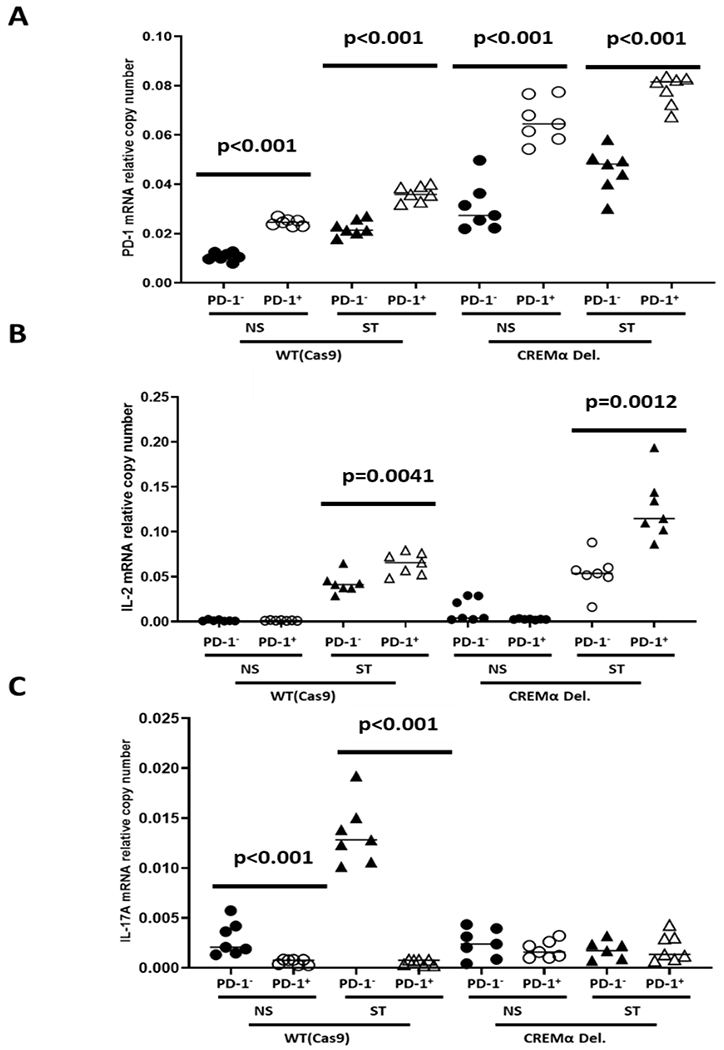
PD-1+ and PD-1− Jurkat T cells using wild-type and CREMα-deficient cell. A) PD-1 mRNA expression in PD-1- WT and CREMα-deficient Jurkat T cells (N=7). B) IL-2 mRNA expression in PD-1− T cells (N=7) and C) mRNA levels of IL-17A (N=7) (Mann Whitney U tests).
4. Discussion
Effector CD4+ T cells contribute to skin inflammation and tissue damage in psoriasis. We and others have demonstrated effector T cell invasion to inflamed skin, and altered effector cytokine expression in psoriasis (2, 45–47). This is in agreement with the response of patients to effector cytokine blocking therapeutic strategies in psoriasis and psoriatic arthritis (46, 48). However, the underlying molecular mechanisms have remained poorly understood (1–4).
In the study presented here, we identified physiological distribution of naïve and effector subsets in the peripheral blood as defined by surface markers CD45 and CCR7 (5). However, CD4+ T cells from patients with psoriasis and psoriatic arthritis exhibited reduced expression of immune regulatory cytokine IL-2 and increased expression of pro-inflammatory cytokine IL-17A. Imbalanced expression of IL-2 and IL-17A is a hallmark of effector T cells in other systemic inflammatory conditions including systemic lupus erythematosus (7, 10, 12). While increased IL-17A expression was known to contribute to the pathophysiology of psoriasis, associated tissue inflammation and damage in the skin, as well as bone remodeling (3, 4, 25, 45–47, 49), reduced IL-2 expression, to our knowledge, has not been previously linked with the molecular pathophysiology of the disease. The transcription factor CREMα is a key factor contributing to immune dysregulation and imbalanced cytokine expression in the T cell-mediated autoimmune disease SLE through trans-regulation of cytokine genes and the induction of epigenetic remodeling (9). Thus, we asked whether it may also be involved in psoriasis and demonstrated that, as in SLE, increased CREMα expression in T cells is associated with effector cytokine expression patterns in psoriasis and psoriatic arthritis patients.
Therapeutic blockade of PD-1 in cancer causes psoriasis in a subset of patients (38), and a recent study reported beneficial effects of PD-1 agonists in the imiquimod-induced murine model of psoriasis (50). The programmed cell death (PD-)1 co-receptor limits T cell differentiation and activation, as well as effector cytokine expression (31). Thus, we tested whether PD-1 expression is reduced in CD4+ T cells from patients with psoriasis, thus contributing to effector cytokine expression. Indeed, PD-1 surface expression/density, particularly in response to CD3/TCR complex activation, was altered in CD4+ T cells from patients with psoriasis and psoriatic arthritis. This is likely of significance to the inflammatory phenotype seen in people with psoriasis, as infiltrating T cells closely interact with keratinocytes and stimulate pro-inflammatory chemokine expression (including CXCL9, CXCL10) that (auto-)amplifies the recruitment of T cells (51–54). Notably, keratinocytes limit pro-inflammatory programs in effector T cells through the up-regulation of PD-1 ligand expression (55, 56). As mentioned above, PD-1 ligand through its interaction with PD-1 inhibits T cell activation, cytokine expression and effector T cell differentiation (32, 33).
Because the transcription factor CREMα, such as PD-1, has been linked with inverse cytokine expression patterns in SLE and psoriasis (9, 35, 57), we tested whether CREMα suppresses PD-1 expression. Using previously characterized genetically modified CD4+ Jurkat T cells (22) and CD4+ T cells from CREMα overexpressing transgenic C57BL/6 mice, we showed that CREMα overexpression leads to the suppression of mRNA and surface protein expression of PD-1. Conversely, biallelic deletion of CREMα in CD4+ Jurkat T cells (22) leads to increased PD-1 expression. This study demonstrates that CREMα is recruited to the PDCD1 proximal promoter where it trans-represses promoter activity and through the co-recruitment of DNMT3a, results in increased DNA methylation and robust transcriptional silencing. Indeed, knock-down of DNMT3a reversed partially CREMα-mediated transcriptional suppression of PDCD1. The absence of significant differences in the PDCD1 DNA methylation status of wild type (pLenti) and CREMα overexpressing Jurkat T cells is most likely caused by the fact that wild type cells already express significant amounts of CREMα, which may not further affect DNA methylation at the promoter region (22). CREMα-deficient Jurkat T cells, however, exhibited significantly reduced PDCD1 DNA methylation stringy supporting this conclusion. Notably, CD4+ T cells collected from patients with psoriasis or psoriatic arthritis, which both display increased CREMα expression compared to healthy subjects, exhibit a trend towards increased DNA methylation at the same region within the PDCD1 promotor (CR-B), suggesting importance of CREMα-mediated epigenetic remodeling and inhibition of PD-1 in psoriasis. Similar CREMα-mediated molecular mechanisms mediating transcriptional silencing through trans-repression and epigenetic remodeling have been reported at the IL2 and CD8 genes in SLE (7, 10, 21, 58).
PD-1 is involved in the regulation of cytokine production in response to CD3/TCR receptor complex engagement (57). Using modified Jurkat T cells and T cells form CREMα transgenic mice we have demonstrated that CREMα represses PD-1 expression through transcriptional and epigenetic mechanisms, which associates with suppressed IL-2 and increased IL-17A production. Our observations along with the report that PD-1 stimulation alleviates Th17 mediated inflammation in murine psoriasis (49), have translational value. In psoriasis, CD4+ T cells are readily found in skin lesions and contribute to tissue inflammation and damage. Inhibition of Rho-associated kinase (ROCK)2 limits CD4+ T cell entry by inhibiting their ability to migrate (59), and corrects the altered balance between pro- and anti-inflammatory cytokine expression thereby suppressing clinical disease (27–29). Here presented observations document additional pathways which account for the development of effector CD4+ T cell subsets that may be targeted therapeutically in people with psoriasis.
Our study offers new insights into the molecular pathophysiology of psoriasis yet, it is limited by relatively small sample size, and certain variation in gender distribution between the three groups. However, immune cell subset distribution did not vary significantly between genders (not shown). Concerns about artefacts caused by slightly variable gender distribution are limited, as women are under-represented in the psoriasis and psoriatic arthritis cohorts. Women exhibit stronger effector phenotypes and increased CREMα expression when compared to men, which suggests that some of the findings reported here, are likely to be even stronger in a cohort perfectly matched for gender distribution (60, 61).
In summary, the present study links increased expression of CREMα with reduced PD-1 expression in CD4+ T cells from patients with psoriasis and psoriatic arthritis. As up-regulation of PD-1 ligand expression in keratinocytes has been recognized as an anti-inflammatory mechanism that dampens effector T cell activation in psoriasis, failure to express PD-1 on CD4+ T cells may be a key contributor to uncontrolled inflammation and damage. CREMα favors effector cytokine production through transcriptional and epigenetic silencing of PDCD1. This promises potential for the previously not appreciated CREMα:PD-1 axis as a future disease biomarker and/or treatment target in psoriasis and other inflammatory conditions characterized by effector CD4+ T cells.
Supplementary Material
Key points.
CD4+ T cells from psoriasis patients exhibit decreased PD-1 expression
The transcription factor CREMα trans-represses PDCD1 and mediates DNA methylation
Reduced expression of PD-1 is linked to low IL-2 and high IL-17A production
Acknowledgments
Funding statement
This study was supported by Novartis (unrestricted research grant) and the University of Liverpool Translational Research Access Programme (TRAP) to CMH, the National Institutes of Health grant R37 AI49954 (to G.C.T.), and Gilead Sciences Research Scholars Program in Rheumatology (to N.Y.).
References
- 1.Khairutdinov VR, Mikhailichenko AF, Belousova IE, Kuligina ES, Samtsov AV, and Imyanitov EN. The role of intradermal proliferation of T-cells in the pathogenesis of psoriasis. An Bras Dermatol. 2017;92(1):41–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brandt D, Sergon M, Abraham S, Mabert K, and Hedrich CM. TCR(+)CD3(+)CD4(−)CD8(−) effector T cells in psoriasis. Clin Immunol. 2017;181:51–59. [DOI] [PubMed] [Google Scholar]
- 3.Hijnen D, Knol EF, Gent YY, Giovannone B, Beijn SJ, Kupper TS, Bruijnzeel-Koomen CA, and Clark RA. CD8(+) T cells in the lesional skin of atopic dermatitis and psoriasis patients are an important source of IFN-gamma, IL-13, IL-17, and IL-22. J Invest Dermatol. 2013;133(4):973–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krueger JG, Fretzin S, Suarez-Farinas M, Haslett PA, Phipps KM, Cameron GS, McColm J, Katcherian A, Cueto I, White T, Banerjee S, and Hoffman RW. IL-17A is essential for cell activation and inflammatory gene circuits in subjects with psoriasis. J Allergy Clin Immunol. 2012;130(1):145–154 e149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sallusto F, Lenig D, Forster R, Lipp M, and Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401(6754):708–712. [DOI] [PubMed] [Google Scholar]
- 6.Wilson CB, Rowell E, and Sekimata M. Epigenetic control of T-helper-cell differentiation. Nat Rev Immunol. 2009;9(2):91–105. [DOI] [PubMed] [Google Scholar]
- 7.Hedrich CM, Crispin JC, Rauen T, Ioannidis C, Apostolidis SA, Lo MS, Kyttaris VC, and Tsokos GC. cAMP response element modulator alpha controls IL2 and IL17A expression during CD4 lineage commitment and subset distribution in lupus. Proc Natl Acad Sci U S A. 2012;109(41):16606–16611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hedrich CM, Crispin JC, and Tsokos GC. Epigenetic regulation of cytokine expression in systemic lupus erythematosus with special focus on T cells. Autoimmunity. 2014;47(4):234–241. [DOI] [PubMed] [Google Scholar]
- 9.Rauen T, Hedrich CM, Tenbrock K, and Tsokos GC. cAMP responsive element modulator: a critical regulator of cytokine production. Trends Mol Med. 2013;19(4):262–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hedrich CM, Rauen T, and Tsokos GC. cAMP-responsive element modulator (CREM)alpha protein signaling mediates epigenetic remodeling of the human interleukin-2 gene: implications in systemic lupus erythematosus. J Biol Chem. 2011;286(50):43429–43436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koga T, Hedrich CM, Mizui M, Yoshida N, Otomo K, Lieberman LA, Rauen T, Crispin JC, and Tsokos GC. CaMK4-dependent activation of AKT/mTOR and CREM-alpha underlies autoimmunity-associated Th17 imbalance. J Clin Invest. 2014;124(5):2234–2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rauen T, Hedrich CM, Juang YT, Tenbrock K, and Tsokos GC. cAMP-responsive element modulator (CREM)alpha protein induces interleukin 17A expression and mediates epigenetic alterations at the interleukin-17A gene locus in patients with systemic lupus erythematosus. J Biol Chem. 2011;286(50):43437–43446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Charras A, and Hedrich CM. The role of epigenetics in paediatric rheumatic disease. Curr Opin Rheumatol. 2019;31(5):450–463. [DOI] [PubMed] [Google Scholar]
- 14.Hedrich CM, and Tsokos GC. Epigenetic mechanisms in systemic lupus erythematosus and other autoimmune diseases. Trends Mol Med. 2011;17(12):714–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Surace AEA, and Hedrich CM. The Role of Epigenetics in Autoimmune/Inflammatory Disease. Front Immunol. 2019;10:1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hedrich CM, Mabert K, Rauen T, and Tsokos GC. DNA methylation in systemic lupus erythematosus. Epigenomics. 2017;9(4):505–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brenner C, and Fuks F. A methylation rendezvous: reader meets writers. Dev Cell. 2007;12(6):843–844. [DOI] [PubMed] [Google Scholar]
- 18.Brenner C, Luciani J, Bizet M, Ndlovu M, Josseaux E, Dedeurwaerder S, Calonne E, Putmans P, Cartron PF, Defrance M, Fuks F, and Deplus R. The interplay between the lysine demethylase KDM1A and DNA methyltransferases in cancer cells is cell cycle dependent. Oncotarget. 2016;7(37):58939–58952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morey L, Brenner C, Fazi F, Villa R, Gutierrez A, Buschbeck M, Nervi C, Minucci S, Fuks F, and Di Croce L. MBD3, a component of the NuRD complex, facilitates chromatin alteration and deposition of epigenetic marks. Mol Cell Biol. 2008;28(19):5912–5923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tenbrock K, Juang YT, Leukert N, Roth J, and Tsokos GC. The transcriptional repressor cAMP response element modulator alpha interacts with histone deacetylase 1 to repress promoter activity. J Immunol. 2006;177(9):6159–6164. [DOI] [PubMed] [Google Scholar]
- 21.Hedrich CM, Crispin JC, Rauen T, Ioannidis C, Koga T, Rodriguez Rodriguez N, Apostolidis SA, Kyttaris VC, and Tsokos GC. cAMP responsive element modulator (CREM) alpha mediates chromatin remodeling of CD8 during the generation of CD3+ CD4-CD8-T cells. J Biol Chem. 2014;289(4):2361–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hofmann SR, Mabert K, Kapplusch F, Russ S, Northey S, Beresford MW, Tsokos GC, and Hedrich CM. cAMP Response Element Modulator alpha Induces Dual Specificity Protein Phosphatase 4 to Promote Effector T Cells in Juvenile-Onset Lupus. J Immunol. 2019;203(11):2807–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lowes MA, Russell CB, Martin DA, Towne JE, and Krueger JG. The IL-23/T17 pathogenic axis in psoriasis is amplified by keratinocyte responses. Trends Immunol. 2013;34(4):174–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lowes MA, Bowcock AM, and Krueger JG. Pathogenesis and therapy of psoriasis. Nature. 2007;445(7130):866–873. [DOI] [PubMed] [Google Scholar]
- 25.Blauvelt A, and Chiricozzi A. The Immunologic Role of IL-17 in Psoriasis and Psoriatic Arthritis Pathogenesis. Clin Rev Allergy Immunol. 2018;55(3):379–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Korn T, Bettelli E, Oukka M, and Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. [DOI] [PubMed] [Google Scholar]
- 27.Flynn R, Paz K, Du J, Reichenbach DK, Taylor PA, Panoskaltsis-Mortari A, Vulic A, Luznik L, MacDonald KK, Hill GR, Nyuydzefe MS, Weiss JM, Chen W, Trzeciak A, Serody JS, Aguilar EG, Murphy WJ, Maillard I, Munn D, Koreth J, Cutler CS, Antin JH, Ritz J, Waksal SD, Zanin-Zhorov A, and Blazar BR. Targeted Rho-associated kinase 2 inhibition suppresses murine and human chronic GVHD through a Stat3-dependent mechanism. Blood. 2016;127(17):2144–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zanin-Zhorov A, Weiss JM, Nyuydzefe MS, Chen W, Scher JU, Mo R, Depoil D, Rao N, Liu B, Wei J, Lucas S, Koslow M, Roche M, Schueller O, Weiss S, Poyurovsky MV, Tonra J, Hippen KL, Dustin ML, Blazar BR, Liu CJ, and Waksal SD. Selective oral ROCK2 inhibitor down-regulates IL-21 and IL-17 secretion in human T cells via STAT3-dependent mechanism. Proc Natl Acad Sci U S A. 2014;111(47):16814–16819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zanin-Zhorov A, Weiss JM, Trzeciak A, Chen W, Zhang J, Nyuydzefe MS, Arencibia C, Polimera S, Schueller O, Fuentes-Duculan J, Bonifacio KM, Kunjravia N, Cueto I, Soung J, Fleischmann RM, Kivitz A, Lebwohl M, Nunez M, Woodson J, Smith SL, West RF, Berger M, Krueger JG, Ryan JL, and Waksal SD. Cutting Edge: Selective Oral ROCK2 Inhibitor Reduces Clinical Scores in Patients with Psoriasis Vulgaris and Normalizes Skin Pathology via Concurrent Regulation of IL-17 and IL-10. J Immunol. 2017;198(10):3809–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen W, Nyuydzefe MS, Weiss JM, Zhang J, Waksal SD, and Zanin-Zhorov A. ROCK2, but not ROCK1 interacts with phosphorylated STAT3 and co-occupies TH17/TFH gene promoters in TH17-activated human T cells. Sci Rep. 2018;8(1):16636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Okazaki T, and Okazaki IM. Stimulatory and Inhibitory Co-signals in Autoimmunity. Adv Exp Med Biol. 2019;1189:213–232. [DOI] [PubMed] [Google Scholar]
- 32.Patsoukis N, Wang Q, Strauss L, and Boussiotis VA. Revisiting the PD-1 pathway. Sci Adv. 2020;6(38). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aksoylar HI, and Boussiotis VA. PD-1(+) Treg cells: a foe in cancer immunotherapy? Nat Immunol. 2020;21(11):1311–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nishimura H, Nose M, Hiai H, Minato N, and Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11(2):141–151. [DOI] [PubMed] [Google Scholar]
- 35.Michot JM, Bigenwald C, Champiat S, Collins M, Carbonnel F, Postel-Vinay S, Berdelou A, Varga A, Bahleda R, Hollebecque A, Massard C, Fuerea A, Ribrag V, Gazzah A, Armand JP, Amellal N, Angevin E, Noel N, Boutros C, Mateus C, Robert C, Soria JC, Marabelle A, and Lambotte O. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer. 2016;54:139–148. [DOI] [PubMed] [Google Scholar]
- 36.Naidoo J, Page DB, Li BT, Connell LC, Schindler K, Lacouture ME, Postow MA, and Wolchok JD. Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann Oncol. 2015;26(12):2375–2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Naidoo J, Page DB, Li BT, Connell LC, Schindler K, Lacouture ME, Postow MA, and Wolchok JD. Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann Oncol. 2016;27(7):1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Naidoo J, Page DB, and Wolchok JD. Immune checkpoint blockade. Hematol Oncol Clin North Am. 2014;28(3):585–600. [DOI] [PubMed] [Google Scholar]
- 39.Yoshida N, Comte D, Mizui M, Otomo K, Rosetti F, Mayadas TN, Crispin JC, Bradley SJ, Koga T, Kono M, Karampetsou MP, Kyttaris VC, Tenbrock K, and Tsokos GC. ICER is requisite for Th17 differentiation. Nat Commun. 2016;7:12993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Najgebauer H, Liloglou T, Jithesh PV, Giger OT, Varro A, and Sanderson CM. Integrated omics profiling reveals novel patterns of epigenetic programming in cancer-associated myofibroblasts. Carcinogenesis. 2019;40(4):500–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sibaud V, Meyer N, Lamant L, Vigarios E, Mazieres J, and Delord JP. Dermatologic complications of anti-PD-1/PD-L1 immune checkpoint antibodies. Curr Opin Oncol. 2016;28(4):254–263. [DOI] [PubMed] [Google Scholar]
- 42.Zhu X, and Lang J. Soluble PD-1 and PD-L1: predictive and prognostic significance in cancer. Oncotarget. 2017;8(57):97671–97682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Park BV, Freeman ZT, Ghasemzadeh A, Chattergoon MA, Rutebemberwa A, Steigner J, Winter ME, Huynh TV, Sebald SM, Lee SJ, Pan F, Pardoll DM, and Cox AL. TGFbeta1-Mediated SMAD3 Enhances PD-1 Expression on Antigen-Specific T Cells in Cancer. Cancer Discov. 2016;6(12):1366–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bally AP, Austin JW, and Boss JM. Genetic and Epigenetic Regulation of PD-1 Expression. J Immunol. 2016;196(6):2431–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eysteinsdottir JH, Sigurgrimsdottir H, Einarsdottir HK, Freysdottir J, Agnarsson BA, Olafsson JH, Sigurgeirsson B, and Luethviksson BR. Effective treatment with balneophototherapy and narrowband UVB monotherapy reduces skin homing Th17/Tc17 and Th22/Tc22 effector cells in peripheral blood in patients with psoriasis. J Dermatol Sci. 2019;96(2):110–112. [DOI] [PubMed] [Google Scholar]
- 46.Ly K, Smith MP, Thibodeaux Q, Reddy V, Liao W, and Bhutani T. Anti IL-17 in psoriasis. Expert Rev Clin Immunol. 2019;15(11):1185–1194. [DOI] [PubMed] [Google Scholar]
- 47.Chen L, and Shen Z. Tissue-resident memory T cells and their biological characteristics in the recurrence of inflammatory skin disorders. Cell Mol Immunol. 2020;17(1):64–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lebwohl M Interleukin-23 blockade: another breakthrough in the treatment of psoriasis. Lancet. 2019;394(10198):544–546. [DOI] [PubMed] [Google Scholar]
- 49.Kim JH, Choi YJ, Lee BH, Song MY, Ban CY, Kim J, Park J, Kim SE, Kim TG, Park SH, Kim HP, Sung YC, Kim SC, and Shin EC. Programmed cell death ligand 1 alleviates psoriatic inflammation by suppressing IL-17A production from programmed cell death 1-high T cells. J Allergy Clin Immunol. 2016;137(5):1466–1476 e1463. [DOI] [PubMed] [Google Scholar]
- 50.Peng S, Cao M, Sun X, Zhou Y, Chen CY, Ma T, Li H, Li B, Zhu B, and Li X. Recombinant programmed cell death 1 inhibits psoriatic inflammation in imiquimodtreated mice. Int J Mol Med. 2020;46(2):869–879. [DOI] [PubMed] [Google Scholar]
- 51.Griffith JW, Sokol CL, and Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol. 2014;32:659–702. [DOI] [PubMed] [Google Scholar]
- 52.Lacotte S, Brun S, Muller S, and Dumortier H. CXCR3, inflammation, and autoimmune diseases. Ann N Y Acad Sci. 2009;1173:310–317. [DOI] [PubMed] [Google Scholar]
- 53.Rauschenberger T, Schmitt V, Azeem M, Klein-Hessling S, Murti K, Gran F, Goebeler M, Kerstan A, Klein M, Bopp T, Serfling E, and Muhammad K. T Cells Control Chemokine Secretion by Keratinocytes. Front Immunol. 2019;10:1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Villarroel VA, Okiyama N, Tsuji G, Linton JT, and Katz SI. CXCR3-mediated skin homing of autoreactive CD8 T cells is a key determinant in murine graft-versus-host disease. J Invest Dermatol. 2014;134(6):1552–1560. [DOI] [PubMed] [Google Scholar]
- 55.Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, Horton HF, Fouser L, Carter L, Ling V, Bowman MR, Carreno BM, Collins M, Wood CR, and Honjo T. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192(7):1027–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Okiyama N, and Katz SI. Programmed cell death 1 (PD-1) regulates the effector function of CD8 T cells via PD-L1 expressed on target keratinocytes. J Autoimmun. 2014;53:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shimizu K, Sugiura D, Okazaki IM, Maruhashi T, Takegami Y, Cheng C, Ozaki S, and Okazaki T. PD-1 Imposes Qualitative Control of Cellular Transcriptomes in Response to T Cell Activation. Mol Cell. 2020;77(5):937–950 e936. [DOI] [PubMed] [Google Scholar]
- 58.Hedrich CM, Rauen T, Crispin JC, Koga T, Ioannidis C, Zajdel M, Kyttaris VC, and Tsokos GC. cAMP-responsive element modulator alpha (CREMalpha) trans-represses the transmembrane glycoprotein CD8 and contributes to the generation of CD3+CD4-CD8-T cells in health and disease. J Biol Chem. 2013;288(44):31880–31887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li Y, Harada T, Juang YT, Kyttaris VC, Wang Y, Zidanic M, Tung K, and Tsokos GC. Phosphorylated ERM is responsible for increased T cell polarization, adhesion, and migration in patients with systemic lupus erythematosus. J Immunol. 2007;178(3):1938–1947. [DOI] [PubMed] [Google Scholar]
- 60.Moulton VR, Holcomb DR, Zajdel MC, and Tsokos GC. Estrogen upregulates cyclic AMP response element modulator alpha expression and downregulates interleukin-2 production by human T lymphocytes. Mol Med. 2012;18:370–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Oertelt-Prigione S The influence of sex and gender on the immune response. Autoimmun Rev. 2012;11(6–7):A479–485. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.