

Abstract
T helper 17 (Th17) cells have emerged as a chief pathogenic cell type in murine models of autoimmunity and human autoimmune diseases. Th17 cells are markedly plastic in their pathogenic potential as they can adopt pro- or anti-inflammatory programming under distinct conditions. The specific mechanism underlying the plasticity of Th17 pathogenesis remains elusive. In this study, we found that Th17-lineage specific transcription factor RORγt directly bound to the promoters of genes engaged in the ubiquitination pathway and thus upregulated their expression in pathogenic Th17 (pTh17) cells. We observed that ubiquitination activity correlated with Th17-related pathology in the context of autoimmunity. Consistent with this finding, the deubiquitinase USP19 was shown to suppress pTh17 differentiation in vitro and Th17-mediated pathogenesis in vivo. Mechanistically, USP19 removed the K63-linked ubiquitin chain from RORγt lysine 313, which is essential for recruiting the coactivator SRC3. Collectively, our findings indicate that USP19 selectively suppresses the pathogenic potential of Th17 cells and offer novel strategies for treating autoimmune diseases.
Introduction
Interleukin (IL)-17-producing T helper (Th17) cells are a subset of CD4+ T cells whose differentiation is determined by the transcription factor RORγt1. The Th17 subset have emerged as a chief pathogenic cell type in murine models of autoimmunity, including experimental autoimmune encephalomyelitis (EAE)2–3 and T-cell-transfer colitis4, as well as in several human inflammatory diseases including Multiple Sclerosis5–6 and Crohn’s disease7. Th17 cells are remarkably plastic in their pathogenic potential as they can adopt a pro- or anti-inflammatory programming under distinct conditions8–11. Stimulation of antigen-activated T cells with transforming growth factor β (TGF-β) and IL-6 in vitro generates non-pathogenic Th17 cells (npTh17)1,12, which produce abundant levels of IL-17A and IL-10 but are not capable of provoking autoimmune disorders following transfer into mice. However, Th17 cells polarized in the absence of TGF-β, with a combination of IL-6, IL-1β, and IL-23, are highly pathogenic (pTh17) and characterized by the production of IFN-γ and IL-17A13–14.
pTh17 cells adopt a transcriptional programming different from that of npTh17 cells, with an elevated expression of pathology-related genes14, some of which are regulated by lineage-specific transcription factor RORγt. RORγt is a member of the nuclear receptor (NR) family, directing the differentiation of Th17 cells through binding DNA as well as coactivators to effectively promote chromatin remodeling and transcriptional activation1,13, leading to the transcription of IL-17A, IL-17F and IL-23R. The steroid receptor coactivator (SRC) family represents well-established NR coactivators, comprised of 3 homologous proteins: SRC1, SRC2, and SRC315-20. An AF-2 domain located in the c-terminus of RORγt binds to SRCs15. Although these coactivators are homologous, they are not functionally redundant. Neither SRC1 nor SRC3 deficient mice are able to generate functional Th17 cells in the context of autoimmunity17–20. SRC1 is indispensable for the initial differentiation of Th17 cells17–18, while the engagement of SRC3 enhances RORγt-mediated pathogenic gene expression19–20.
Dysregulation of RORγt-directed Th17 differentiation has been linked to the development of autoimmunity5–7; thus, several mechanisms are in place to control the activity of RORγt. We recently reported ubiquitination as an important systematic mechanism regulating RORγt activities. Ubiquitination refers to the post-translational modification of lysine residues via covalent attachment of one (monoubiquitination) or more (polyubiquitination) ubiquitin monomers. To form a polymer chain, ubiquitin can be conjugated through one of their lysine residues (e.g. K48 and K63) or the N-terminal methionine residue. Distinct types of linkages are associated with different cellular functions. For example, K48-linked polyubiquitin chains are engaged in proteasomal degradation, whereas K63-linked chains have many well-studied non-degradative roles and are engaged in inflammatory signaling and NF-κB activation through affiliating protein interactions21–22. We have previously shown that ubiquitination of RORγt at lysine 69 is essential for Th17 differentiation23, while ubiquitination of lysine 446 interrupts the recruitment of SRC1 and thus blocks the generation of Th17 cells16. Ubiquitination is a reversible process, and several deubiquitinases precisely cleave ubiquitin from their substrates24. The dynamic balance of these opposing events make it possible for Th17 cells to rapidly respond to complex environments. We previously revealed that USP15 promotes Th17 differentiation through removing a ubiquitin chain conjugated to Lys446 of RORγt which prevents the recruitment of SRC116. These observations provide a proof of concept that ubiquitination can be directly involved in the balance of pro- and anti-inflammatory states of Th17 cells.
Here, we investigated the roles of USP19 in the differentiation and function of Th17 cells. We showed that Th17-lineage specific transcription factor RORγt directly bound to the promoters of genes engaged in the ubiquitination pathway and upregulated their expression in pTh17 cells. The ubiquitination level directly correlates with Th17-related pathology in the context of autoimmunity. Consistent with these findings, the deubiquitinase USP19 suppressed pTh17 differentiation in vitro as well as Th17-mediated pathogenesis in vivo. Mechanistically, USP19 removed K63-linked ubiquitin chain from RORγt lysine 313, which is essential for recruiting coactivator SRC3. Collectively, our findings indicate that the USP19 selectively suppresses the pathogenic potential of Th17 cells, and provides novel strategies for treating autoimmune diseases.
Materials & Methods
Mice
The Rorγt−/− (Rorc2−/−), Rag1−/−, and 2D2 mouse strains were described previously23,25. Mice at 10–12 weeks of age were used for inducing Experimental Autoimmune Encephalomyelitis (EAE). For all other experiments, mice were at 6–8 weeks of age; age-matched littermates were used in all experimental groups. Mice were bred and housed under specific pathogen-free (SPF) conditions in the Animal Resource Center at the Wake Forest School of Medicine under Institutional Animal Care and Use Committee approved protocols.
Antibodies, cytokines and plasmids
Antibodies to RORγt (Q31–378, BD Bioscience), SRC3 (ab2831, Abcam), FLAG (M2, Sigma-Aldrich), and HA (HA-7, Sigma-Aldrich) were used for immunoblot analysis. PE-conjugated anti-RORγt (B2D), Allophycocyanin (APC)-conjugated anti-IL-17A (TC11–18H10.1), APC-conjugated anti-CD3 (145–2C11), FITC-conjugated anti-CD19 (1D3), BV421-conjugated anti-CD11b (M1/70), FITC-conjugated anti-CD4 (GK1.5), APC-conjugated anti-Foxp3 (FJK-16s), PE-conjugated anti-Ly6G (1A8), FITC-conjugated anti-IFN-γ (XMG1.2), PE-conjugated anti-GM-CSF (MP1–22E9), APC-Cy7-conjugated anti-CD45 (104), PE-conjugated anti-CD25 (PC61.5), monoclonal antibodies to mouse CD3 (145–2C11), and CD28 (37.51) were purchased from Biolegend. Monoclonal antibodies to mouse IFN-γ (XMG1.2) was purchased from Bioxcell. Goat anti-hamster antibody (MP0856984) was from MP Biomedicals. Recombinant mouse IL-12, IL-4, IL-6, IL-23 and TGF-β were purchased from Miltenyi Biotech.
Plasmids encoding SRC3, RORγt, ubiquitin and ubiquitin mutants have been described previously19,23. RORγt-K69, RORγt-K313, and RORγt-K446 were constructed by inserting mutated RORγt sequence into the retroviral vector pMSCV. These RORγt mutants contained one single intact lysine residue (K69, 313 or 446) by replacing all other lysine (K) to arginine (R). LMP vector-based retroviral short hairpin RNA (shRNA)-expressing vectors were constructed using the oligonucleotide whose sequences were listed in Table 1.
Table 1.
List of oligonucleotide sequences for constructing LMP vector-based retroviral short hairpin RNA (shRNA)-expressing vectors
| LMP construct | Oligonucleotide sequences (5’ -> 3′) |
|---|---|
| shUSP7–1 | TGCTGTTGACAGTGAGCGCGCAGTGCTGAAGATAATAAATTAGTGAAGCCACAGATGTAATTTATTATCTTCAGCACTGCTTGCCTACTGCCTCGGA |
| shUSP7–2 | TGCTGTTGACAGTGAGCGACGGCCTGCAATGTTAGATAATTAGTGAAGCCACAGATGTAATTATCTAACATTGCAGGCCGCTGCCTACTGCCTCGGA |
| shUSP9X-1 | TGCTGTTGACAGTGAGCGACCTTAAATCCTCATTGCAAATTAGTGAAGCCACAGATGTAATTTGCAATGAGGATTTAAGGCTGCCTACTGCCTCGGA |
| shUSP9X-2 | TGCTGTTGACAGTGAGCGCCCTTGCAAGATCTTGATAATATAGTGAAGCCACAGATGTATATTATCAAGATCTTGCAAGGTTGCCTACTGCCTCGGA |
| shUSP13–1 | TGCTGTTGACAGTGAGCGAGGCCTGTTTCGGATCCTCATTTAGTGAAGCCACAGATGTAAATGAGGATCCGAAACAGGCCCTGCCTACTGCCTCGGA |
| shUSP13–2 | TGCTGTTGACAGTGAGCGCCCTGAGGAAATCGTAGCTATTTAGTGAAGCCACAGATGTAAATAGCTACGATTTCCTCAGGATGCCTACTGCCTCGGA |
| shUSP15–1 | TGCTGTTGACAGTGAGCGACCAGACTGTGGAACAAGTATATAGTGAAGCCACAGATGTATATACTTGTTCCACAGTCTGGCTGCCTACTGCCTCGGA |
| shUSP15–2 | TGCTGTTGACAGTGAGCGCGGACAGGTGTTAGTGATAGAATAGTGAAGCCACAGATGTATTCTATCACTAACACCTGTCCTTGCCTACTGCCTCGGA |
| shUSP19–1 | TGCTGTTGACAGTGAGCGCCGAGCAAACCTGGAAAGTAAATAGTGAAGCCACAGATGTATTTACTTTCCAGGTTTGCTCGATGCCTACTGCCTCGGA |
| shUSP19–2 | TGCTGTTGACAGTGAGCGCCCTGCTTCACGCCTCACTTATTAGTGAAGCCACAGATGTAATAAGTGAGGCGTGAAGCAGGTTGCCTACTGCCTCGGA |
| shUSP22–1 | TGCTGTTGACAGTGAGCGCCCTGATGTACGGAGGTATTTATAGTGAAGCCACAGATGTATAAATACCTCCGTACATCAGGTTGCCTACTGCCTCGGA |
| shUSP22–2 | TGCTGTTGACAGTGAGCGCGCAGCGCAAGGCTTGGAAGATTAGTGAAGCCACAGATGTAATCTTCCAAGCCTTGCGCTGCTTGCCTACTGCCTCGGA |
| shUSP37–1 | TGCTGTTGACAGTGAGCGAGCTTTGCAAACCTGCTTATTATAGTGAAGCCACAGATGTATAATAAGCAGGTTTGCAAAGCGTGCCTACTGCCTCGGA |
| shUSP37–2 | TGCTGTTGACAGTGAGCGACGGCTTTGCAGAAGATGATATTAGTGAAGCCACAGATGTAATATCATCTTCTGCAAAGCCGGTGCCTACTGCCTCGGA |
| shUCHL3–1 | TGCTGTTGACAGTGAGCGACCTGAACTTCTTAGCATGGTATAGTGAAGCCACAGATGTATACCATGCTAAGAAGTTCAGGCTGCCTACTGCCTCGGA |
| shUCHL3–2 | TGCTGTTGACAGTGAGCGAGCTATTCGAGTTACTCATGAATAGTGAAGCCACAGATGTATTCATGAGTAACTCGAATAGCGTGCCTACTGCCTCGGA |
| shVCIP135–1 | TGCTGTTGACAGTGAGCGAGCAGAATGTTTATTGTGAATATAGTGAAGCCACAGATGTATATTCACAATAAACATTCTGCCTGCCTACTGCCTCGGA |
| shVCIP135–2 | TGCTGTTGACAGTGAGCGCCCAGACTATGGATTGAGTAATTAGTGAAGCCACAGATGTAATTACTCAATCCATAGTCTGGTTGCCTACTGCCTCGGA |
Retrovirus Transduction
Retroviral transduction was achieved as previously described24. Platinum-E packaging cells (Cell Biolabs) were plated in a 10 cm dish in 10 ml DMEM medium plus 10% FBS. 24 hours later, cells were transfected with an empty pMSCV vector or the appropriate retroviral expression plasmids with BioT transfection reagent (Bioland). After overnight incubation, the medium was replaced and cultures were maintained for another 24 h. Viral supernatants were collected 48 h and 72 h later, passed through 0.4-μm filters (Millipore), and supplemented with 8 μg/ml of polybrene (Sigma-Aldrich). Naïve CD4+ T cells were first activated with 0.25 μg/ml hamster anti-CD3, 1 μg/ml hamster anti-CD28 in 24-well plates pre-coated with 0.2 mg/ml goat anti-hamster antibody for 24 h, then spin-infected with viral supernatants (1200 g, 30°C for 2 h). After spin infection, viral supernatant was replaced by culture medium with polarizing cytokines for in vitro differentiation.
In vitro differentiation
Naïve CD4+ T cells were purified from C57BL/6, RORγt−/− or 2D2 mice by negative selection (EasySep™ Mouse Naïve CD4+ T Cell Isolation Kit, Stem Cell Technologies). Suspensions of 4×105 cells/ml in Iscove’s modified DMEM (Cellgro) containing 2 mM L-glutamine, 50 mM 2-ME, 100 U/ml penicillin, 100 mg/ml streptomycin, and 10% FBS were cultured in 24-well plates pre-coated with 0.2 mg/ml goat anti-hamster antibody for 3 days. The medium was supplemented with 0.25 μg/ml hamster anti-CD3, 1 μg/ml hamster anti-CD28, and polarizing cytokines: 2 ng/ml TGF-β, 20 ng/ml IL-6, 5 μg/ml anti-IL-4, and 5 μg/ml anti-IFNγ for npTh17 differentiation; 20 μg/ml IL-6, 20 μg/ml IL-1β, 20 μg/ml IL-23, 5 μg/ml anti-IL-4, and 5 μg/ml anti-IFNγ for pTh17 differentiation; or 5 ng/ml TGF-β for Treg differentiation. For the treatment with deubiquitinase inhibitor, cells received 10μM PR-619 (SML0430, Sigma-Aldrich) for 1 hour, followed by extensive washing. Cells were then transferred to Rag1−/− mice or subjected to immunoprecipitation as indicated. For analysis of cytokine production, cells obtained from in vitro cultures were incubated for 4–5 h with 50 ng/ml PMA, 750 ng/ml ionomycin (both from Sigma-Aldrich), and 10 μg/ml brefeldin A (BD Biosciences) in a tissue culture incubator at 37°C followed by intracellular cytokine staining.
Chromatin immunoprecipitation
Chromatin immunoprecipitation was performed according to the manufacturer’s instruction (Active Motif). Briefly, 2×107 cells were incubated with 1% formaldehyde to cross-link proteins with chromatin for 5 min at room temperature. 125 mM glycine was added to stop the cross-linking reaction. To shear genomic DNA into 200–500 bp fragments, cell lysates were sonicated using a water-bath sonicator (Covaris S200). Cell lysates were centrifuged at 12000 g for 10 min and incubated with antigen-specific antibody or IgG control and protein A/G-sepharose beads (Millipore). After extensive washing, DNA was eluted followed by reversion of the protein–DNA cross-linking. DNA was recovered for qRT-PCR to quantify precipitated DNA fragments of interest.
Quantitative real-time PCR (qRT-PCR)
qRT-PCR was performed using SYBR Green PCR Master Mix (Applied Biosystems) in an ABI 7500 Real-Time PCR Detection System (Applied Biosystems) using primers as described previously21. The amplification efficiency of all primers have been tested and optimized conditions were used in all qRT-PCR reactions. Expression was calculated with the ΔΔCt method normalized to β-acti with all measurements performed in triplicates.
Isolation of active deubiquitinases
Pathogenic Th17 cells, differentiated in vitro, were lysed in 0.1% NP-40, 150 mM NaCl, 20 mM CaCl2 and 50 mM Tris pH 7.4 buffer containing 1 mM phenylmethanesulfonyl fluoride (PMSF). Samples containing equal amounts of protein were subjected to the enzymatic reaction with ubiquitin vinyl sulfone HA-tagged probe (Ub-VS-HA; Boston Biochem, USA) at a ratio of ~1:200 for 45 minutes at 37°C. Proteins were separated by SDS-PAGE and immunoblotted with anti-HA antibody to visualize active deubiquitinases. For proteomics analysis, 8 mg of protein per sample was subjected to the enzymatic reaction with the Ub-VS-HA probe as described above and immunoprecipitated as described in the section below.
Induction and assessment of Experimental Autoimmune Encephalomyelitis (EAE)
For Th17-induced passive EAE, donor mice were immunized with 200 μl of myelin oligodendrocyte glycoprotein 35–55 (MOG35–55) peptide emulsion (Hooke Laboratories) subcutaneously. 10 days later, cells were isolated from the spleen and lymph node and cultured with 20 μg/ml MOG35–55 for 3 days under Th17-polarizing conditions (20 ng/ml IL23). CD4+ T cells were then isolated and incubated with PR-619 for 1 hour. Rag1−/− recipient mice were then transferred intraperitoneally with 3.0×107 MOG35–55-specific Th17 cells.
For 2D2-transfer induced EAE, transduced 2D2 T cells were inoculated with IL-2 for 48 hours and CD4+ GFPhi cells were sorted with Canto flow cytometer and intravenously injected into Rag1−/− mice (5.0×104 cells/mouse). Recipient mice were immunized subcutaneously on day 0 with MOG35–55 and received pertussis toxin (200 ng/mouse) intraperitoneally at day 0 and 2.
The severity of EAE was monitored and evaluated on a scale from 0 to 5 according to Hooke Laboratories’ guideline. In brief: 0 = no disease; 1= paralyzed tail; 2= hind limb weakness; 3= hind limb paralysis; 4= hind and fore limb paralysis; 5= moribund and death. Severity of clinical symptoms in a mouse resulting in euthanasia was reported as a score of 5 for the remainder of the experiment. Spinal cords from mice were collected at the 6th day after onset of symptoms and mashed through a 70-μM cell strainer. Cell suspensions were then subjected to 30–70% percoll density gradient centrifugation. Isolated cells were investigated by using flow cytometry with indicated antibodies specific for cell surface markers and cytokines. The absolute number of cellular infiltrates was calculated using the percentage of individual immune population among isolated cells and the total number of isolated cells.
Immunoprecipitation and immunoblot analysis
Cells were lysed in lysis buffer (1% Triton X-100, 20 mM Tris-cl, pH 7.4, 150 mM NaCl, 5 mM EDTA) supplemented with protease inhibitors (cocktail, Sigma-Aldrich and 1 mM PMSF). Cell extracts were incubated overnight with 1μg of described antibodies and proteins were immunoprecipitated for an additional 1 hour at 4°C with protein A/G-Sepharose beads (Milipore). For analyzing ubiquitinated proteins, cell lysates were incubated with TUBE2 agarose beads (UM402, Lifesensor) for 1 hour at 4°C. After incubation, beads were washed four times with lysis buffer. Proteins pulled down by beads were resolved using SDS-PAGE and analyzed using via immunoblotting.
Statistical analysis
Prism software (Graphpad) was used for all statistical analyses. Two-tailed unpaired Student’s t test and one-way analysis of variance (ANOVA) were used to compare experimental groups. A P value of less than 0.05 was reported as statistically significant.
Results
RORγt transactivates ubiquitination-related genes in Th17 cells.
RORγt is a transcription factor essential for Th17 differentiation which initiates a cascade of Th17 signature gene expression, including IL-17A, IL-17F, and IL-221,13. To comprehensively understand the genes directly regulated by RORγt, we analyzed our published sequencing data collected from chromatin immunoprecipitation (ChIP) using antibodies specifically targeting RORγt in Th17 cells (accession code SRP150962, https://www.ncbi.nlm.nih.gov/sra/?term=SRP150962)18. Beyond the promoters of Il17a, RORγt directly bound to a vast amount of loci throughout the genome. An analysis of the genomic sites which ChIP-Seq peaks were observed with significant enrichment (> 4 fold) fell into the promoters, coding regions and intergenic areas of genes bound by RORγt (Fig. 1A). Of noted, a significant portion (8%) of genes whose promoters are bound by RORγt is engaged in the ubiquitination pathway, including Ube2g2 and Ube2j2 with the most significant enrichment (Fig. 1B).
Figure 1. RORγt transactivates ubiquitination-related genes in Th17 cells.

(A) The distribution of RORγt binding sites across the genome in Th17 cells. (B) IGV browser view of ChIP-seq of RORγt occupancy in Il17a, Ube2g2, and Ube2j2 loci from in vitro polarized Th17 cell samples. (C) Independent ChIP-qPCR validation of ChIP-seq results confirming the binding of RORγt to Il17a, Ube2g2, and Ube2j2 loci in polarized npTh17 and pTh17 cells. (D) WT and Rorγt−/− CD4+ T cells polarized under pTh17 conditions were evaluated in amounts of Ube2g2 and Ube2j2 mRNA detected by RT-qPCR. (E-F) RT-qPCR measurement of the expression of E1 (E) and E2 (F) genes in npTh17 and pTh17 cells. Graphs shown mean ± s.d. (G) Naïve T cells from C57BL/6 mice on day 3 post-incubation with indicated cytokine combination (Methods), immunoblotted for ubiquitinated RORγt and total RORγt. The levels of ubiquitinated RORγt relative to that in npTh17 were calculated. IP, immunoprecipitation. IB: Immunoblot. WCL, whole cell lysate. Data are representative of two independent experiments. Each experiment was performed with two technical replicates. Graphs (C, D, E and F) shown mean ± s.d. P values calculated by student’s t-test, ns: not significant, * P<0.05, ** P<0.01, *** P<0.001.
These interactions were confirmed by quantitative real-time PCR (Fig. 1C). The promoters of Il17a and Hbb were also included as control. RORγt showed strong affinity to the promoters of Ube2g2 and Ube2j2, comparable with that to the promoter of Il17a. The affinity of RORγt to these binding sites were significantly increased in pTh17 cells in comparison to npTh17 cells when polarized under in vitro conditions (Fig. 1C). As expected, the expression level of Ube2g2 and Ube2j2 was dramatically lower in RORγt−/− cells primed under pTh17 conditions, indicating that RORγt directly transactivates these genes (Fig. 1D).
Protein ubiquitination is initiated with the priming of a ubiquitin onto a ubiquitin activating enzyme (E1) and the transfer to a ubiquitin conjugating enzyme (E2)26–28. Using quantitative real-time PCR, we found that the transcription levels of several E1 (Fig. 1E) and E2 (Fig. 1F) enzymes were significantly higher in pTh17 than in npTh17 cells, suggesting elevated ubiquitination activity in Th17 cells with greater pathogenic potential. This led us to explore the ubiquitination levels of RORγt in npTh17 and pTh17 cells. Ubiquitinated proteins were immunoprecipitated with TUBE2-beads which bind ubiquitin with high affinity and were detected with an RORγt-specific antibody. We observed a clear laddering of bands, representing the varying levels of ubiquitinated RORγt in Th17 cells (Fig. 1G). Consistent with the expression level of ubiquitination-related genes, RORγt in pTh17 cells was heavily ubiquitinated. These data demonstrate that pTh17 cells possess an overactive ubiquitination pathway.
Blocking deubiquitination enhances Th17-mediated pathology in autoimmunity.
Since pTh17 cells exhibited increased activity in the ubiquitination pathway, we speculated that promoting ubiquitination would enhance Th17-mediated pathology in autoimmunity. To test this hypothesis, murine model of experimental autoimmune encephalomyelitis (EAE) was used, in which Th17 cells play a chief pathogenic role. C57BL/6 mice were immunized subcutaneously with MOG35–55. The cells, collected from the spleens and draining lymph nodes on day 11 after immunization, were stimulated in vitro with MOG35–55 in the presence of IL-23 for 3 days. CD4+ T cells were then purified and treated for 1 hour with PR-619, a cell-permeable and broad-spectrum deubiquitinating enzyme inhibitor. The enrichment of ubiquitinated RORγt was confirmed by immunoblotting, indicating the accumulation of ubiquitinated proteins after PR-619 treatment (Fig. 2A). Notably, PR-619 treatment increased the percentile of highly pathogenic Th17 cells which produce both IL-17A and IFN-γ (Supplementary Fig. 1A) without affecting cell viability (Fig. 2B).
Figure 2. Blocking deubiquitination enhances Th17-mediated pathology in autoimmunity.

(A) Cells isolated from the spleen and lymph node from C57BL/6 mice on day 11 post-immunization with MOG35–55, cultured with 20 ng/ml MOG35–55 for 3 days under Th17-polarizing conditions (20 ng/ml IL23). CD4+ T cells were then isolated and incubated with DMSO or PR-619 for 1 hour and immunoblotted for ubiquitinated RORγt and total RORγt. The levels of ubiquitinated RORγt relative to that in DMSO-treated pTh17 were calculated. Data are representative of two independent experiments. (B) Left: Representative FACS plot showing the viability of cells treated with DMSO or PR-619 as described in A. Right: The percentage of living cells in DMSO or PR-619 treated T cells. (C) EAE clinical scores in Rag1−/− recipients of control or PR619-treated T cells from MOG35–55 immunized donor C57BL/6 mice in a transfer model of encephalomyelitis (n = 6 for each group). Data are combined from two independent experiments and represent the means ± SEMs. (D) The number of lymphocytes, macrophages and microglia in spinal cord of Rag1−/− recipient mice as described in C. (E) CD4+ T cells sorted from cellular infiltrates in spinal cord of Rag1−/− recipient mice as described in C were evaluated for amounts of representative pathogenic gene mRNA by RT-qPCR. Results (B, D and E) are representative of two independent experiments. Each experiment was performed with two technical replicates. Graphs shown mean ± s.d. Statistical analysis was performed by one-way ANOVA followed by the Tukey-Kramer test (C), or by student’s t-test, * P<0.05, ** P<0.01, *** P<0.001.
Treated CD4+ T cells were adoptively transferred to Rag1−/− mice (which have a congenital deficiency in mature B cells and T cells) to elicit EAE. Compared to the vehicle-treated counterpart, PR-619-treated Th17 cells induced more severe encephalomyelitis (Fig. 2C). PR-619-treated Th17-induced EAE showed an increased number of infiltrating inflammatory cells in the Central Nervous System (CNS), including primarily T lymphocytes and macrophages (Fig. 2D). Furthermore, the expression of proinflammatory cytokines and chemokines from these infiltrates were also significantly boosted (Fig. 2E and Supplementary Fig. 1B), consistent with the clinical observations.
Deubiquitinase inhibitor treatment was previously reported to regulate immune responses through the destabilization of Treg cells29. We therefore measured the amount of Treg cells prior to adoptive transfer. Since the Treg population was hardly detected within both vehicle and PR-619-treated donor T cells, it is unlikely that the worsened EAE is due to the reduction of Treg transferred to recipient mice (data not shown). These data collectively indicate that the activity of ubiquitination pathway correlates with Th17 pathogenic potential.
USP19 selectively suppresses pTh17 cell differentiation in vitro.
Enhanced pathogenicity of PR-619-treated Th17 cells indicates the involvement of deubiquitinases in pTh17 differentiation and functions. To determine any specific deubiquitinases modulating pTh17 cells, we first identified active deubiquitinases from pTh17 cells using an HA-tag-labelled probe29, Ub-VS-HA, which forms a covalent bond with active deubiquitinases. The probe-enzyme complex was then immunoprecipitated with an anti-HA antibody. The active deubiquitinases of pTh17 were identified by mass spectrometry and were listed in Fig. 3A. The most abundant active deubiquitinases in pTh17 cells were screened using a small-hairpin RNA (shRNA)-mediated gene silencing assay (Fig. 3B & C) for their potential to regulate pTh17 differentiation.
Figure 3. USP19 selectively suppresses pTh17 cell differentiation in vitro.

(A) List of active deubiquitinases in pTh17 cells. Active deubiquitinases were precipitated by a probe, Ub-VS-HA, from murine pTh17 cells polarized in vitro (Method) and identified using mass spectrometry. (B) Gating strategy for analyzing pTh17 differentiated in vitro as described in C. (C) Percentage of pTh17 (IL-17A+ IFN-γ+) population in shRNA-transduced T cells. Murine T cells transduced to express control or specific shRNAs targeting indicated deubiquitinase were polarized in vitro under pTh17 conditions. On day 3 post transduction, pTh17 differentiation was measured by flow cytometry. Data are representative of two independent experiments. Results are representative of two independent experiments. Each experiment was performed with two technical replicates. Graphs shown mean ± s.d. (D) Efficacy of small hairpin RNA-mediated knock down of USP19 in murine T cells was evaluated with immunoblotting. Murine T cells transduced to express control or specific shRNAs targeting USP19, on day 3 post transduction, were immunoblotted for USP19 and beta-actin loading control. Data are representative of two independent experiments. (E) shRNA-expressing pTh17 cells polarized in vitro as described in B were evaluated for amounts of pathogenic gene mRNA by RT-qPCR. Data were shown by the ratio of mRNA in shUSP19-expressing Th17 versus control shRNA-expressing Th17 cells. Results are representative of two independent experiments. Each experiment was performed with two technical replicates. Graphs shown mean ± s.d. (F) Left, representative FACS plot showing cytokine production of shRNA-transduced T cells polarized in vitro with npTh17-skewing cytokines. Right, percentage of IL-17A+ population in shRNA-transduced T cells. (G) Left, representative FACS plot showing Foxp3 expression of shRNA-expressing T cells polarized in vitro with Treg-priming cytokines. Right, percentage of Foxp3+ population in shRNA-expressing T cells. P values calculated by student’s t-test, * P<0.05, ** P<0.01, *** P<0.001.
Two species of shRNAs were used to knock down each deubiquitinase from murine splenic CD4+ T cells polarized with pTh17 priming cytokine milieu. The retroviral vector for shRNA delivery encoded a Green fluorescent protein (GFP), which served as a label for transduced cells. The generation of highly pathogenic Th17 cells was measured by flow cytometry analysis of the percentile of Th1-like IFN-γ+IL-17A+ population amongst CD4+GFP+ cells. Consistent with our previous findings that USP15 is essential for the initial differentiation of Th17 cells, silencing USP15 effectively blocks the polarization of pTh17 cells. In contrast, reducing USP19 notably fostered pTh17 differentiation (Fig. 3C and Supplementary Fig. 2A), increasing IL-17A+IFN-γ+ population by approximately 2-fold. The shRNA species shUSP19–1 was more potent in promoting pTh17 differentiation, suggesting greater efficacy to knock down target gene expression (Fig. 3D). The transcription of Th17-pathology-related genes in shUSP19–1-expressing pTh17 cells was significantly upregulated (Fig. 3E). Strikingly, silencing USP19 has a marginal impact on npTh17 differentiation, measured by the generation of IL-17A-producing cells (Fig. 3F), indicating USP19 selectively suppresses Th17 proinflammatory potential.
The shared requirement for TGF-β in Treg and Th17 cell differentiation led us to explore the function of USP19 in the Treg cell population. Transduction of T cells with either USP19-targeting shRNA species did not affect Treg differentiation, as shown by the percentile of Foxp3+ cells. This suggests USP19 may not be engaged in Treg skewing (Fig. 3G).
Suppression of USP19 enhances Th17-mediated pathogenesis of autoimmunity.
Since silencing USP19 in in vitro polarized pTh17 cells led to an increase of pathogenic effector molecules, we therefore examined the engagement of USP19 in regulating Th17-mediated pathology in vivo. We used a T-cell adoptive transfer model of encephalomyelitis, in which disease severity is dependent on the proinflammatory potential of Th17 cells. Primary myelin-reactive T cells from 2D2 mice (genetically modified to generate only type of TCR, which is reactive to myelin) were activated and transduced in vitro to express USP19-targeting or control shRNA, and transduced cells (sorted CD4+GFPhi T cells) were adoptively transferred into Rag1−/− mice, which were then immunized with MOG35–55.
Inhibition of USP19 in T cells let to more severe encephalomyelitis (Fig. 4A), characterized by increased number of infiltrating neutrophils (Ly6G+) and immature myeloid cells (Ly6ChiCD11b+Ly6G-) in CNS (Fig. 4B & C). Consistent with the clinical observations, shUSP19-expressing T cells gave rise to an enlarged population coexpressing IL-17A and IFN-γ, an important feature of pathogenic T cells in several inflammatory disease settings (Fig. 4D & E). In line with our in vitro findings, these in vivo observations indicate that USP19 suppresses pathogenic potential of Th17 cells, and therefore ameliorates autoimmunity.
Figure 4. USP19 restrains Th17-mediated pathogenesis in inflammatory encephalomyelitis.

(A) EAE clinical scores in Rag1−/− recipients of control or shUSP19-expressing 2D2-T cells in a transfer model of encephalomyelitis (n = 7 for each group). Naïve myelin-specific T cells (2D2) were activated in vitro and transduced to express control or specific USP19-targeting shRNA. Transduced cells (CD4+GFP+) were adoptively transferred to Rag1−/− recipient mice, which were then immunized with MOG35–55. The clinical symptoms were scored for 30 days. Data are combined from two independent experiments. (B) Representative FACS plot showing gating strategy for analysis of cellular infiltrates in spinal cord from Rag1−/− recipients as described in A. (C) Left: Representative FACS plot showing distinct populations of cellular infiltrates in spinal cord from Rag1−/− recipients as described in A. Right: Cell number of individual populations infiltrated into the spinal cord of Rag1−/− recipient mice as described in A. (D) Representative FACS plot showing IL-17A and IFN-γ expression of CD4+GFP+ cells in spinal cord from Rag1−/− recipients as described in A. (E) Cell number of IL-17A+IFNγ+ pathogenic population in the spinal cord of Rag1−/− recipient mice. Results (C and E) are representative of two independent experiments. Each experiment was performed with two technical replicates. Graphs shown mean ± s.d. Statistical analysis was performed by one-way ANOVA followed by the Tukey-Kramer test (A), or by student’s t-test, ns: not significant, ** P<0.01, *** P<0.001.
USP19 specifically removes ubiquitin chain from RORγt Lys313.
The upregulation of RORγt downstream genes, including Il17f, Ccl20, and Ccr6 (Fig. 3E) in response to USP19 silencing implicated RORγt as a substrate of USP19. To test this possibility, we first examined whether USP19 would bind with RORγt under pTh17 polarizing conditions. The interaction between USP19 and RORγt in pTh17 cells was validated by co-immunoprecipitation (co-IP) (Fig. 5A). An N-terminal 31 amino acid truncation of RORγt disrupted its interaction with USP19, indicating specific domains in the RORγt were required for this interaction (Fig. 5B).
Figure 5. USP19 removes ubiquitin chain from RORγt-K313.

(A) USP19 association with immunoprecipitated RORγt in polarized Th0 or pTh17 cells. IP, immunoprecipitation. IB: Immunoblot. WCL, whole cell lysate. Representative of two independent experiments. (B) Upper, the scheme of RORγt truncations used in immunoprecipitation. Bottom, detection of USP19 associated with immunoprecipitated RORγt truncations in co-transfected HEK293T cells. Representative of two independent experiments. (C) HEK293T cells were transfected to express an indicated RORγt mutant (Methods) and ubiquitin together with USP19, immunoblotted for ubiquitinated RORγt. K69/313/446 is a RORγt mutant with only single lysine 69/313/446 and all other lysine residues replaced with arginine. Data are representative of two independent experiments. (D) HEK293T cells transfected to express RORγt-K313 and indicated deubiquitinase, were immunoblotted for ubiquitinated RORγt. Data are representative of two independent experiments.
We next tested the capability of USP19 to deubiquitinate RORγt. Lysine residues 69, 313, and 446 of RORγt were previously identified as ubiquitin acceptor sites in Th17 cells. To find out which lysine residue was critical for USP19-targeted RORγt protein deubiquitination, a series of RORγt mutants containing one single intact lysine residue was generated (RORγt-K69, RORγt-K313, and RORγt-K446) by replacing all other lysine (K) to arginine (R). HEK293T cells were transfected with ubiquitin, USP19 and RORγt mutants, followed by immunoprecipitation with anti-RORγt, and immunoblot analysis with anti-ubiquitin. USP19 decreases the ubiquitination level on RORγt-K313, but not that on RORγt-K69 or K446 (Fig. 5C). Therefore, lysine residue 313 could be the major acting site of USP19 to prevent RORγt ubiquitination. Interestingly, USP19 docked on the N-terminus of RORγt while acted on the residue in its C-terminus, suggesting that N and C termini of RORγt might reside in close proximity.
Several more deubiquitinases with detected activities in pTh17 were also tested for their capability to deubiquitinate RORγt-K313. Only USP37 could partially reduce the ubiquitination on RORγt-K313, to the extent significantly less than USP19 did (Fig. 5D). None of other tested deubiquitinases were capable of removing ubiquitin from this residue, indicating that USP19 is a specific deubiquitinase preventing ubiquitination of RORγt lysine 313.
USP19 breaks RORγt-SRC3 interaction through the removal K63-linked polyubiquitin moieties from RORγt.
To obtain a better understanding of the roles of RORγt K313 ubiquitination in pTh17 differentiation, we first deciphered the type of ubiquitin chain conjugated to this residue with the aid of ubiquitin mutants. To form a polymer chain, ubiquitin can be conjugated through one of their lysine residues (e.g. K48 and K63) or the N-terminal methionine residue. Ub-K/R is a ubiquitin variant with all lysine residues replaced by arginine and can only form linear ubiquitin chains through the N-terminal methionine residue. Ub-K63R, in which K63 was replaced by arginine, was deficient in forming poly-ubiquitin chains via the K63 (K63-linked ub chain). In contrast, Ub-K63 contains only a single lysine, K63, with all other lysine residues mutated to arginine. This mutation renders ubiquitin able to form exclusively K63-linked polymers. These mutants were cotransfected with RORγt-K313. RORγt-K69 and RORγt-K446 were also included as controls. Ub-K/R and Ub-K63R failed to form polymers on RORγt-K313, whereas Ub-K63 restored ubiquitination of this residue, indicating K63-linked chain type was dominant at residue 313, similar to that on RORγt-K69 as published previously. Interestingly, the ubiquitin polymers on RORγt-K446 was not K63-linked, distinct from those on lysine 69 and 313 (Fig. 6A).
Figure 6. USP19 breaks RORγt-SRC3 interaction through the removal of K63-linked polyubiquitin moieties from RORγt.
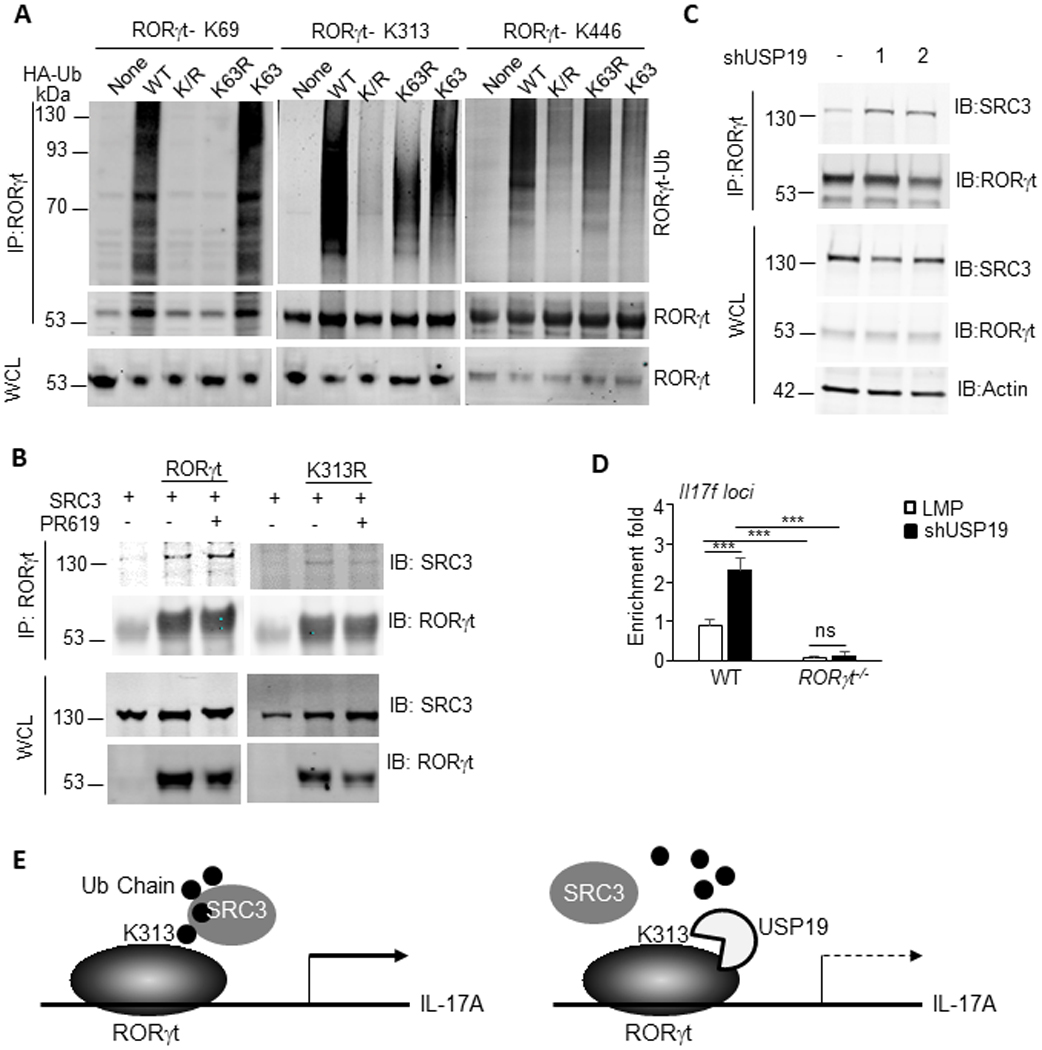
(A) HEK293T cells transfected to express RORγt mutants (Methods) and indicated ubiquitin mutants (Methods), were immunoblotted for ubiquitinated RORγt. IP, immunoprecipitation. IB: Immunoblot. WCL, whole cell lysate. Data are representative of two independent experiments. (B) SRC3 association with immunoprecipitated RORγt in vehicle or PR619-treated pTh17 cells. Data are representative of two independent experiments. (C) SRC3 association with immunoprecipitated RORγt in pTh17 cells transduced to express control or USP19-targeting shRNAs. Data are representative of two independent experiments. (D) ChIP-qPCR measurement of the recruitment of SRC3 to Il17f loci in pTh17 cells. Naïve CD4+ T cells from wild-type C57BL/6 (WT) or RORγt−/− mice were transduced to express control or USP19-targeting shRNAs and polarized under pTh17 conditions. ChIP assay was performed on GFP+ cells with SRC3 specific antibody. The recruitment of SRC3 to Il17f loci was evaluated with RT-PCR using primers flanking RORγt-binding site in Il17f promoter. P values calculated by student’s t-test, ns: not significant, *** P<0.001. (E) Schematic diagram of the function of USP19 in regulating pTh17 differentiation.
K63-linked chain type has many well-studied non-degradative roles and is engaged in inflammatory signaling and NF-κB activation through affiliating protein interactions21–22. We previously reported that replacing RORγt K313 with arginine (RORγt-K313R) interrupted the recruitment of coactivator SRC3, which is indispensable for pTh17 differentiation19. However, the mechanism underlying this phenomenon is yet known. We speculated that K313R mutation prevented RORγt-K313 ubiquitination, which is needed for SRC3 binding. To test this hypothesis, we examined SRC3-RORγt binding in mock and PR-619-treated pTh17 cells via co-IP assay. As shown in Fig. 6B, PR-619 treatment increases the affinity of SRC3 for RORγt, but not its mutant RORγt-K313R, revealing important roles of RORγt-K313 ubiquitination in recruiting SRC3. Next, we investigated SRC3-RORγt interaction in pTh17 transduced with USP19-targeting or control shRNAs (Fig. 6C). The results showed that reduced expression of USP19 facilitated the formation of RORγt-SRC3 complex in pTh17 cells.
As a coactivator, SRC3 lacks intrinsic DNA-binding capability. The amount of SRC3 recruited to RORγt-binding sites within promoters reflected the affinity between these two proteins. We therefore validated the impact of USP19-mediated deubiquitination on SRC3-RORγt interaction through ChIP assay. Il17f promoter, occupied by SRC3, was quantified with RT-qPCR using primers flanking RORγt-binding site. In pTh17 cells with USP19 knocked down, SRC3 exhibits significantly increased affinity to Il17f promoter (Fig. 6D), consistent with elevated expression of Il17f in these cells (Fig. 3E). However, the SRC3-Il17f promoter interaction was hardly detected in RORγt-deficient cells even polarized with pTh17-priming cytokines. These data indicate that silencing USP19 enhances SRC3 recruitment by RORγt in pTh17 cells.
Taken together, USP19 suppresses pTh17 differentiation and pathogenic potential through the removal of K63-linked ubiquitin chain from lysine 313 of RORγt, which is required for recruiting coactivator SRC3 and essential for the expression of proinflammatory effectors (Fig. 6E).
Discussion
Post-translational modifications can rapidly and reversibly regulate transcription factor functions, including subcellular localization, stabilization, interactions with cofactors, and transcriptional activities in order to create layers of transcriptional regulation. Here, we examined the roles of USP19 in the differentiation and functions of Th17 cells. We revealed that Th17-lineage specific transcription factor RORγt directly bound to the promoters of genes engaged in the ubiquitination pathway and thus upregulated their expression in pTh17 cells. The ubiquitination level correlated with Th17-related pathology in the context of autoimmunity. Consistent with these findings, a deubiquitinase USP19 suppressed pTh17 differentiation in vitro as well as Th17-mediated pathogenesis in murine autoimmune models. Mechanistically, USP19 removed the K63-linked ubiquitin chain from RORγt lysine 313, which is essential for recruiting coactivator SRC3. Collectively, our findings indicate that the USP19 selectively suppresses the pathogenic potential of Th17 cells and suggest strategies for treating autoimmune diseases.
RORγt is a transcription factor with pleiotropic functions in thymocytes, innate lymphoid cells, γδT cells and Th17 cells. We reported previously that the ubiquitination at RORγt-K6923 and sumoylation at K3119 are selectively required by Th17 functions but not T cell development. Here we found that ubiquitination at RORγt-K313 correlates with the severity of autoimmunity in murine models, which is consistent with the observations that pTh17 cells had greater expression of ubiquitination-related genes than npTh17 cells. Our data suggest that RORγt adapts to the context within distinct cells and responds to environmental signals by recruiting specific cofactors, most likely through different post-translational modification. Our data strongly argue that ubiquitination, combining with other post-translational modification such as phosphorylation and sumoylation, serves as a physical basis for differential RORγt functions in distinct cells.
The highly dynamic process of protein ubiquitination reflects the continuously changing steady-state of enzymes by adding and removing ubiquitin chains from substrates, serving as a rapid mechanism of adaptation to the surrounding signals. Of note, several other molecules besides RORγt might be ubiquitinated in pTh17 cells in order to regulate Th17-mediated pathogenesis. It is not uncommon that ubiquitination is involved in transcriptional regulation, as many transcription factors, including SRC330, were identified as substrates of this modification. One mechanism is that UIM (Ubiquitin-interacting motif) mediated protein-protein interaction is required to facilitate the formation of transcriptional complexes31. The absence of UIM in SRC3 is likely because of the involvement of a SRC3-binding molecule, which can recognize a K63-linked ubiquitin chain conjugated to Lys313 of RORγt. SRC3 interacts not only with a ubiquitin chain on this residue, but also physically binds to the AF2 domain in the C-terminal of RORγt19. The level of ubiquitination at Lys313 determines the SRC3-binding affinity of RORγt and facilitates the regulation of Th17 pathogenic potential.
Although our results clearly demonstrate that USP19 interacts directly with RORγt and constrains pathogenic Th17 cell programming and response amplification, deubiquitinases also have effects on multiple other substrates. USP19 was recently reported to be involved in regulating innate immunity as a negative regulator of TLR3/4-mediated signaling through the deubiquitination of TRIF. It also deconjugates K27-linked polyubiquitin chains from TAK1 after IL-1β stimulation, leading to impairment of TAK1 activity and the inhibition of IL-1β-triggered inflammatory response32. This impairment may also contribute to the suppression of Th17-mediated pathogenesis. Although we cannot exclude the possibility that USP19 functions through other substrates during in vivo pTh17 cell differentiation, our results strongly support a substantial contribution through direct interaction with and deubiquitinase RORγt.
The general importance of the USP19 as a checkpoint for the pathogenic programming of Th17 cells is underscored by worsened EAE observed when USP19 is diminished by specific shRNAs in T cells. The combined results show that USP19 provides previously unappreciated means for constraining inflammatory Th17 cells and that adoptively transferred antigen-specific T cells confer pathogenicity based, at least in part by, the ubiquitination of RORγt. Together, these findings suggest that a better understanding of differential RORγt functions modulated by USP19 will facilitate therapeutic efforts to ameliorate inflammatory diseases. In addition, the E3 ligase, which catalyzes the ubiquitination of RORγt-Lys313, is to elucidate and may serve as an ideal drug target for Th17-mediated inflammatory disorders. With the insight of increasing inhibitors of E3 ligases will be approved for clinical treatment, identification of this enzyme will lead to novel therapy for autoimmune diseases.
Supplementary Material
KEY POINTS:
USP19 suppresses the pathogenic potential of Th17 cells.
USP19 removes the K63-linked ubiquitin chain from RORγt lysine 313.
Ubiquitination of RORγt at lysine 313 is essential for recruiting SRC3.
Acknowledgements
We appreciate the help of core facilitates and shared resources of Wake Forest School of Medicine including the animal, genomic and flow cytometer cores.
Footnotes: This work was supported by Wake Forest School of Medicine Microbiology and Immunology Start-Up funds and the National Cancer Institute of the National Institutes of Health P30 award under award number P30CA012197. In addition, this work was also supported by National Institutes of Health T32 training grant awarded to Ronald J. Bouch under number 5T32A1007401-28. These funding support the design and conduct of the study, the collection, analysis, and interpretation of the data, and the preparation, review, approval of the manuscript. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- 1.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, and Littman DR 2006. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 126: 1121–1133. [DOI] [PubMed] [Google Scholar]
- 2.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, and Cua DJ 2005. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med 201: 233–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, and Kuchroo VK 2006. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 441: 235–238. [DOI] [PubMed] [Google Scholar]
- 4.Leppkes M, Becker C, Ivanov II, Hirth S, Wirtz S, Neufert C, Pouly S, Murphy AJ, Valenzuela DM, Yancopoulos GD, Becher B, Littman DR and Neurath MF 2009. RORgamma-expressing Th17 cells induce murine chronic intestinal inflammation via redundant effects of IL-17A and IL-17F. Gastroenterology. 136: 257–267. [DOI] [PubMed] [Google Scholar]
- 5.Moser T, Akgün K, Proschmann U, Sellner J, and Ziemssen T. 2020. The role of TH17 ells in multiple sclerosis: Therapeutic implications. Autoimmun. Rev 19:102647. [DOI] [PubMed] [Google Scholar]
- 6.Dendrou CA, Fugger L, and Friese MA 2015. Immunopathology of multiple sclerosis. Nat Rev. Immunol 15: 545–558. [DOI] [PubMed] [Google Scholar]
- 7.Hue S, Ahern P, Buonocore S, Kullberg MC, Cua DJ, McKenzie BS, Powrie F, and Maloy KJ 2006. Interleukin-23 drives innate and T cell-mediated intestinal inflammation. J. Exp. Med 203: 2473–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Geachy MJ, Cua DJ, and Gaffen SL 2019. The IL-17 Family of Cytokines in Health and Disease. Immunity. 50: 892–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patel DD, and Kuchroo VK 2015. Th17 Cell Pathway in Human Immunity: Lessons from Genetics and Therapeutic Interventions. Immunity. 43: 1040–1051. [DOI] [PubMed] [Google Scholar]
- 10.Stockinger B, and Omenetti S. 2017. The dichotomous nature of T helper 17 cells. Nat. Rev. Immunol 17: 535–544. [DOI] [PubMed] [Google Scholar]
- 11.Honda K, and Littman DR 2016. The microbiota in adaptive immune homeostasis and disease. Nature. 535: 75–84. [DOI] [PubMed] [Google Scholar]
- 12.Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, Levy DE, Leonard WJ, and Littman DR 2007. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat. Immunol 8: 967–974. [DOI] [PubMed] [Google Scholar]
- 13.Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, Ramos HL, Wei L, Davidson TS, Bouladoux N, Grainger JR, Chen Q, Kanno Y, Watford WT, Sun HW, Eberl G, Shevach EM, Belkaid Y, Cua DJ, Chen W, and O’Shea JJ 2010. Generation of pathogenic T(H)17 cells in the absence of TGF-β signaling. Nature. 467: 967–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A, Wu C, Kleinewietfeld M, Kunder S, Hafler DA, Sobel RA, Regev A, and Kuchroo VK 2012. Induction and molecular signature of pathogenic TH17 cells. Nat. Immunol 13: 991–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xie H, Sadim MS, and Sun Z. 2005. RORgammat recruits steroid receptor coactivators to ensure thymocyte survival. J. Immunol 175: 3800–3809. [DOI] [PubMed] [Google Scholar]
- 16.He Z, Wang F, Ma J, Sen S, Zhang J, Gwack Y, Zhou Y, and Sun Z. 2016. Ubiquitination of RORγt at Lysine 446 Limits Th17 Differentiation by Controlling Coactivator Recruitment. J. Immunol 197:1148–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sen S, Wang F, Zhang J, He Z, Ma J, Gwack Y, Xu J, and Sun Z. 2018. SRC1 promotes Th17 differentiation by overriding Foxp3 suppression to stimulate RORγt activity in a PKC-θ-dependent manner. Proc. Natl. Acad. Sci. U. S. A 115: E458–E467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.He Z, Zhang J, Huang Z, Du Q, Li N, Zhang Q, Chen Y, and Sun Z. 2018. Sumoylation of RORγt regulates TH17 differentiation and thymocyte development. Nat. Commun 9: 4870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He Z, Zhang J, Du Q, Xu J, Gwack Y, and Sun Z. 2019. SRC3 Is a Cofactor for RORγt in Th17 Differentiation but Not Thymocyte Development. J. Immunol 202: 760–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tanaka K, Martinez GJ, Yan X, Long W, Ichiyama K, Chi X, Kim BS, Reynolds JM, Chung Y, Tanaka S, Liao L, Nakanishi Y, Yoshimura A, Zheng P, Wang X, Tian Q, Xu J, O’Malley BW, and Dong C. 2018. Regulation of Pathogenic T Helper 17 Cell Differentiation by Steroid Receptor Coactivator-3. Cell Rep. 23: 2318–2329. [DOI] [PubMed] [Google Scholar]
- 21.Nathan JA, Kim HT, Ting L, Gygi SP, and Goldberg AL 2013. Why do cellular proteins linked to K63-polyubiquitin chains not associate with proteasomes?. EMBO J. 32: 552–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao S, and Ulrich HD 2010. Distinct consequences of posttranslational modification by linear versus K63-linked polyubiquitin chains. Proc. Natl. Acad. Sci. U. S. A 107: 7704–7709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He Z, Ma J, Wang R, Zhang J, Huang Z, Wang F, Sen S, Rothenberg EV, and Sun Z. 2017. A two-amino-acid substitution in the transcription factor RORγt disrupts its function in TH17 differentiation but not in thymocyte development. Nat. Immunol 18: 1128–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clague MJ, Urbé S, and Komander D. 2019. Breaking the chains: deubiquitylating enzyme specificity begets function. Nat. Rev. Mol. Cell Biol 20: 338–352. [DOI] [PubMed] [Google Scholar]
- 25.Bettelli E, Pagany M, Weiner HL, Linington C, Sobel RA, and Kuchroo VK 2003. Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J. Exp. Med 197:1073–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scheffner M, Nuber U, and Huibregtse JM 1995. Protein ubiquitination involving an E1-E2-E3 enzyme ubiquitin thioester cascade. Nature. 373: 81–83. [DOI] [PubMed] [Google Scholar]
- 27.Zheng N, and Shabek N. 2017. Ubiquitin Ligases: Structure, Function, and Regulation. Annu. Rev. Biochem 86: 129–157. [DOI] [PubMed] [Google Scholar]
- 28.Komander D, and Rape M. 2012. The ubiquitin code. Annu. Rev. Biochem 81:203–229. [DOI] [PubMed] [Google Scholar]
- 29.van Loosdregt J, Fleskens V, Fu J, Brenkman AB, Bekker CP, Pals CE, Meerding J, Berkers CR, Barbi J, Gröne A, Sijts AJ, Maurice MM, Kalkhoven E, Prakken BJ, Ovaa H, Pan F, Zaiss DM, and Coffer PJ 2013. Stabilization of the transcription factor Foxp3 by the deubiquitinase USP7 increases Treg-cell-suppressive capacity. Immunity. 39: 259–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu RC, Feng Q, Lonard DM, and O’Malley BW 2007. SRC-3 coactivator functional lifetime is regulated by a phospho-dependent ubiquitin time clock. Cell. 129: 1125–1140. [DOI] [PubMed] [Google Scholar]
- 31.Fisher RD, Wang B, Alam SL, Higginson DS, Robinson H, Sundquist WI, and Hill CP 2003. Structure and ubiquitin binding of the ubiquitin-interacting motif. J. Biol. Chem 278: 28976–28984. [DOI] [PubMed] [Google Scholar]
- 32.Lei CQ, Wu X, Zhong X, Jiang L, Zhong B, and Shu HB 2019. USP19 Inhibits TNF-α- and IL-1β-Triggered NF-κB Activation by Deubiquitinating TAK1. J. Immunol 203: 259–268. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.