

Abstract
Background:
Complement activation in kidney transplantation is implicated in the pathogenesis of delayed graft function (DGF). This study evaluated the therapeutic efficacy of high-dose recombinant human C1 inhibitor (rhC1INH) to prevent DGF in a nonhuman primate model of kidney transplantation after brain death (BD) and prolonged cold ischemia.
Methods:
BD donors underwent 20-hours of conventional management. Procured kidneys were stored on ice for 44–48 hours, then transplanted into ABO-compatible MHC-mismatched recipients. Recipients were treated with vehicle (n=5) or rhC1INH 500U/kg plus heparin 40U/kg (n=8) before reperfusion, 12-, and 24-hours posttransplant. Recipients were followed 120 days.
Results:
Of vehicle-treated recipients, 80% (4/5) developed DGF versus 12.5% (1/8) rhC1INH-treated recipients (p=0.015). RhC1INH-treated recipients had faster creatinine recovery, superior urinary output, and reduced urinary NGAL and TIMP2·IGFBP7 throughout the first week, indicating reduced allograft injury. Treated recipients presented lower postreperfusion plasma IL-6, IL-8, TNFα, and IL-18, lower day 4 MCP-1, and trended toward lower C5. Treated recipients exhibited less C3b/C5b-9 deposition on day 7 biopsies. RhC1INH-treated animals also trended toward prolonged AMR-free survival.
Conclusions:
Our results recommend high-dose C1INH complement-blockade in transplant recipients as an effective strategy to reduce kidney injury and inflammation, prevent DGF, delay AMR-development, and improve transplant outcomes.
INTRODUCTION
Delayed graft function (DGF), commonly defined as the need for dialysis within the first week posttransplantation, occurs in up to 40% of kidney transplant recipients.1–4 DGF is associated with increased incidence of acute cellular rejection (ACR) and antibody-mediated rejection (AMR), and reduced graft survival rates.2,5–11 DGF imparts in a significant financial burden through increased intensive care unit (ICU) admissions, prolonged hospitalization, posttransplant dialysis treatments, and repeat transplants, increasing the average cost by 10% per patient.12 Despite the high prevalence and costs of DGF, preventative therapeutic regimens remain elusive, and while short-term outcomes in kidney transplantation have improved significantly, long-term outcomes remain suboptimal with over 20% of transplants going to prior allografts recipients.5,13 Additionally, about 20% of deceased donor kidneys are discarded, in part due to prolonged cold ischemia time (CIT) and advanced donor age, both of which are associated with increased risk of developing DGF.14–17 As demand continues to climb in an environment of scarce resources, it is increasingly imperative to develop DGF-prevention strategies to improve long-term graft survival rates, reduce the incidence of AMR, and potentially expand the donor pool to include kidneys that are currently being discarded.
DGF results from acute kidney injury (AKI) suffered during ischemia-reperfusion injury (IRI) in transplantation. Increased inflammatory response, complement activation, and cytokine release are all implicated in the pathogenesis of DGF.18–21 The complement system, consisting of classical (CP), mannose-binding lectin (MBL), and alternative pathways (AP), is an essential component of the innate immune system.18–20 Complement-mediated injury starts in the donor with increased messenger RNA expression of complement components and upregulation of renal tubular anaphylatoxin receptors C3aR and C5aR.22,23 While the liver produces the majority of complement proteins, up to 10–16% of systemically circulating C3, a precursor of C5 convertase, is produced by renal proximal tubular cells.19,22,24–30 These factors foster local complement deposition, leukocyte migration and infiltration, and oxidative damage, exacerbating injury at reperfusion and increasing the risk of DGF and rejection.19,25,31–42
C1 esterase inhibitor (C1INH) is a serine protease inhibitor that blocks components of the complement, contact, fibrinolytic, and coagulation systems.43 FDA-approved for use in hereditary angioedema (HAE), C1INH-products have garnered interest in the transplant community as a mitigator of IRI due to its anti-inflammatory properties.43 In humans, C1INH blocks the classical complement and MBL pathways by inactivation of C1s, C1r, and MASP2.43 This effect is potentiated by heparin, commonly used in organ procurement and transplantation, increasing the activity of C1INH up to 11-fold.44–46
We previously demonstrated that donor-pretreatment with recombinant human C1INH (rhC1INH) plus heparin can prevent DGF in a nonhuman primate (NHP) model of brain death (BD), prolonged CIT, and kidney transplantation; however, ACR and AMR incidences were unaffected.47 In this study, we tested perioperative and postoperative rhC1INH-treatment in recipients to determine the impact on DGF and acute rejection within the first 120 days. We assessed markers of inflammation, complement activity, kidney function, and rejection to provide a comprehensive assessment and potential mechanism of rhC1INH-therapy in kidney transplantation.
MATERIALS AND METHODS
2.1. Animals
Rhesus macaques obtained from the Wisconsin National Primate Research Center (WNPRC) and Alpha Genesis Inc. (Yemassee, SC) were housed in accordance with National Institutes of Health (NIH) and U.S. Department of Agriculture animal welfare guidelines; all protocols were approved by the Institutional Animal Care and Use Committee at the University of Wisconsin-Madison. All animals were prescreened for TB, Herpes B, SRV, SIV, and STLV-1. Each donor-recipient pair was ABO-compatible, nonsensitized, and fully-mismatched for MHC class I and II alleles identified with microsatellite analysis as previously described.48 Recipient animals were divided into treatment groups: vehicle (n=5) and rhC1INH (n=8).
2.2. Experimental Design
Pharming Technologies B.V. (Leiden, The Netherlands) provided the rhC1INH (half-life of 2.5 hours49).
Brain death (BD) was induced and donors were maintained 20-hours in an ICU environment following previously published guidelines (Figure 1A).50 Briefly, animals were anesthetized and placed on a ventilator. A 16F Foley catheter was inserted into the extradural space and inflated until hemodynamic and neurologic signs of brain-stem herniation were observed. Animals were managed for 20 hours with standard donor management, including fluid resuscitation and vasopressor support (Table S1). Donors subsequently underwent aortic cannulation, retrograde infusion of UW preservation solution (Organ Recovery Systems, IL) supplemented with heparin (5 U/mL), and bilateral nephrectomy. Recovered kidneys were stored in UW solution on ice for 44–48 hours. Recipients underwent bilateral native nephrectomy and heterotopic kidney allotransplantation (Figure 1B).
Figure 1:
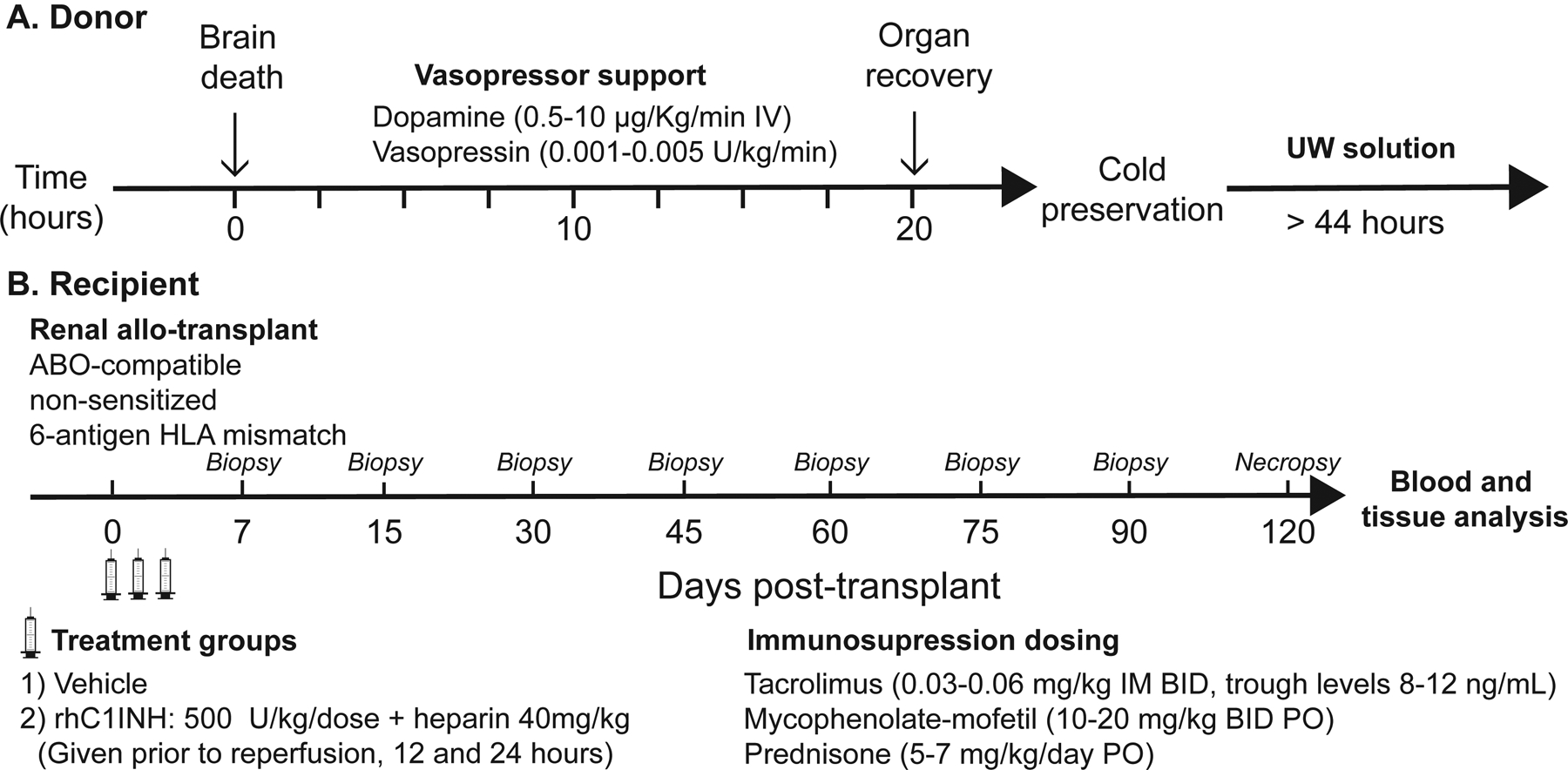
Experimental design
Immediately before arterial reperfusion of the graft, recipients received an IV bolus of vehicle (3.33mL/kg normal saline) in the control group or rhC1INH (500U/kg) plus heparin (40U/kg) for the experimental group. The same dose was repeated at 12- and 24-hours posttransplant. Recipients were followed 120 days. A heparin-alone group was not used due to detrimental outcomes when donors received heparin alone in our previous study.47 Immunosuppressive induction therapy was not administered. Maintenance immunosuppression included prednisone, mycophenolate-mofetil (MMF), and tacrolimus guided by biweekly trough measurements to maintain 8–12 ng/ml. Basic metabolic panel (including creatinine) and blood gasses were measured by i-STAT 1 Analyzer (Abaxis, Union City, CA). Termination criteria included 1) survival for 120 days or 2) severe azotemia, acute renal failure, or progressive antibody-mediated rejection not responsive to medical management.
2.3. Definitions: DGF, AMR, ACR
DGF was defined as a failure of serum creatinine to fall by least 10% on 3 consecutive days in the first-week posttransplant and/or serum creatinine at posttransplant day 7 >2.5 mg/dL.4,51 AMR and ACR were diagnosed using the 2017 Banff classification.52 AMR diagnosis required 2 of 3 criteria: a) clinical acute increase in serum creatinine and/or oliguria/anuria, b) histopathology on H/E-staining with C4d-positive immunohistochemistry, and/or c) serological evidence of increased donor-specific antibody (DSA) median fluorescence intensity shift ≥1.5.
2.4. Resistive Index
Intrarenal resistive indices (RI) of kidney transplants [RI=(peak systolic velocity – end diastolic velocity)/(peak systolic velocity)] were measured using ultrasonography on postoperative day 1. RI >0.7 was considered elevated.
2.5. Urinary Marker Evaluation
Collected urine was stored at −80°C. Urinary neutrophil gelatinase-associated lipocalin (NGAL) levels were quantified using the NHP NGAL ELISA kit (Bioporto, Copenhagen, Denmark) according to manufacturer recommendations. Urinary tissue-inhibitor of metalloproteinases (TIMP2) and insulin-like growth-factor-binding protein (IGFBP7) were measured with LEGENDplex NHP Mix-and-Match Subpanel (Biolegend, San Diego, CA, USA) according to manufacturer recommendations.
2.6. Complement and Cytokine Assessments
Blood was collected into Vacuettes (Greiner Bio-One, Austria). Serum tubes were allowed 15 minutes to clot before centrifugation with EDTA-plasma tubes for 10 minutes at 3000g, aliquots were stored at −80°C. Circulating serum C1INH was measured via Sino Biological human C1 inhibitor/SerpinG1 ELISA kit (KIT10995). Activation of the complement CP, MBL, and AP pathways were tested using the Wieslab Complement Kits (CP310, LP320, and AP330, EuroDiagnostica, Sweden). Circulating sC5b-9 was measured using sC5b-9 ELISA (Quidel, San Diego, CA). Detection of C5 and C5a were measured using C5 ELISA (Abnova, Taipei City, Taiwan) and C5a ELISA (Novus, Abingdon, UK). All assays were performed according to manufacturer recommendations. Circulating IL-6, MCP-1, IL-8, TNFα, and IL-18 were measured with LEGENDplex NHP Mix-and-Match Subpanel (Biolegend, San Diego, CA, USA) according to manufacturer recommendations. Assay results for serum and plasma proteins were normalized to albumin, measured by VetTest Analyzer (Idexx, Westbrook, ME), to account for hemodilution.
2.7. Kidney Biopsy Histopathology and Immunohistochemistry
Recipient native kidney samples, ultrasound-guided core biopsies of the allografts, and necropsy specimens were fixed in 10% formalin or frozen in optimal cutting temperature (OCT) compound. Slides cut at 4μm thickness were stained with Hematoxylin and Eosin (H/E), periodic acid-Schiff (PAS), anti-CD68 (KP1, DAKO-Agilent), anti-MPO (MPO, Abcam), malondialdehyde (MDA, Abcam), C4d, C3b and C5b-9 (Ventana-Roche, AZ). Diagnostic histopathology was blindly interpreted by 2 renal pathologists and graded according to the Banff 2017 criteria.52 Images for quantification were acquired from 6 to 12 random fields as available within each slide at appropriate magnification using an Olympus BX51 microscope (Olympus, Tokyo, Japan) and processed using ImageJ software (NIH, Bethesda, MD). Cell counts or area fraction measurements were collected using color-separation, automatic-threshold, and particle-analysis algorithms.
2.8. DSA
Donor-specific alloantibody production was assessed by flow-cytometric crossmatch of donor splenocytes with serially collected recipient EDTA-plasma samples. Donor cells incubated with recipient plasma were stained with FITC-labeled anti-monkey IgG (NIH NHP Reagent Resource, Boston, MA), PerCP-Cy5.5 CD20 (BD Pharmingen, San Diego, CA), and APC CD3 (BD Pharmingen, San Diego, CA). Data was acquired on a Cytek DxP upgraded BD FACSCalibur and analyzed with FlowJo (Treestar Inc., USA).
2.9. Statistics
Statistical analyses were performed using GraphPad Prism V8.2 (GraphPad, USA) and represented as mean ± standard error of the mean (SEM). Chi-square test was used to analyze DGF incidence and intrarenal RI. Normality and variance were tested using the Shapiro-Wilk test and F-test, whereupon independent comparisons of 2 groups were performed using 2-tailed Student’s T-test or Mann-Whitney U test as appropriate. For single-timepoint samples where normality could not be tested, parametric tests were used if equal variance. For histology comparisons involving native kidneys, differences between the groups were tested by 1-way ANOVA with Tukey’s correction. Kruskal-Wallis test with Dunn’s correction was used for nonnormally distributed data. Data sets were tested for outliers using the Grubbs’ test. Kaplan-Meier death-censored analysis was performed on AMR and ACR outcomes with significance by log-rank test. Differences between groups were considered significant at p<0.05.
RESULTS
3.1. RhC1INH Reduces Renal Injury and Incidence of DGF, Improves Urine Output and Resistive Index
DGF-incidence was significantly reduced among rhC1INH-treated animals (12.5%, 1/8) compared to vehicle-treated animals (80.0%, 4/5) (Table 1, p=0.015). RhC1INH-treated animals exhibited faster serum creatinine recovery (Figure 2A, p=0.046) and urine output (Figure 2B, p=0.011) in the first week posttransplant, including significantly lower creatinine levels at days 6 and 7 (p=0.018, p=0.031) and significantly higher urine output as early as day 2, proceeding through 5 (p=0.020, p=0.002, p=0.026, p=0.026).
Table 1:
Recipient characteristics and incidence of DGF
| Recipient ID | Age (years) | Weight (kg) | Donor ID | CIT (hours) | Meet DGF criteria | Peak creatinine (Days 0–7) | AMR diagnosis | Graft survival (days) |
|---|---|---|---|---|---|---|---|---|
| Vehicle group | ||||||||
| R11051 | 4.3 | 8.3 | Rhas84 | 44.5 | Yes | 14.8 | No | 3 |
| R11107 | 4.2 | 4.7 | 47.9 | Noa,b | 6.7 | Yes | 90 | |
| Rh2612 | 4.0 | 4.0 | Rh2602 | 46.7 | Yes | 10.9 | Yes | 39 |
| Rh2636 | 5.6 | 5.4 | Rh2030 | 44.1 | Yes | 15.7 | Yes | 29 |
| R13020 | 3.6 | 5.9 | 47.2 | Yes | 18.4 | No | 10 | |
| Mean/SEM | 4.34 ± 0.34 | 5.66 ± 0.73 | 46.08 ± 0.75 | 80.00% | 13.30 ± 2.04 | 34.20 ± 15.37 | ||
| RhC1INH group | ||||||||
| Rh2714 | 6.0 | 13.5 | Rh2715 | 48.0 | Yes | 16.9 | Yes | 56 |
| Rh2707 | 4.2 | 9.0 | Rh2638 | 44.5 | Nob | 6.6 | Yes | 84 |
| Rh2723 | 4.2 | 6.7 | Rh2728 | 46.5 | Noa,b | 12.8 | No | 50 |
| Rh2733 | 4.2 | 5.6 | 44.8 | Noa,b | 9.0 | Yes | 119 | |
| Rh2725 | 4.3 | 5.0 | Rh2731 | 44.4 | Noa,b | 12.6 | Yes | 54 |
| Rh2727 | 5.2 | 7.2 | 46.4 | Noa,b | 10.1 | No | 119 | |
| Rh2719 | 4.6 | 6.6 | Rh2626 | 44.0 | Noa,b | 9.0 | Yes | 36 |
| Rh2732 | 4.3 | 6.8 | 46.5 | Noa,b | 7.2 | No | 14 | |
| Mean/SEM | 4.63 ± 0.23 | 7.54 ± 0.94 | 45.62 ± 0.50 | 12.5% | 10.53 ± 1.21 | 66.50 ± 13.39 | ||
| p-value | 0.484 | 0.189 | 0.608 | 0.015 | 0.235 | 0.928 | 0.150 | |
AMR, antibody-mediated rejection; CIT, cold ischemia time; DGF, delayed graft function; POD, postoperative day;
SEM, standard error of the mean
Failure for serum creatinine to drop at least 10% over 3 consecutive days
serum creatinine <2.5 mg/dL by day 7
Figure 2: RhC1INH reduces renal injury and incidence of DGF, improves urine output and resistive index.

(a) Daily serum creatinine levels (mg/dL) and (b) urinary output (mL/day) for vehicle-treated (n=5) versus rhC1INH-treated (n=8) recipients in the first postoperative week with the AUC measured for creatinine (mg/dL2) are presented as mean ± SEM. Rh2714 and Rh2733 had an acute increase in creatinine after renal biopsy at day 6, followed by a significant drop in creatinine on day 8, creatinine values from days 6 and 7 were excluded for analysis. Urine kidney injury marker (c) NGAL (ng/mL) is presented as individual values and AUC. Our historical control data found baseline NGAL levels to be below the detectable assay limit in naïve samples. Calculated (d) urinary TIMP2·IGFBP7 and AUC (ng2/1000 for sample, (ng2/1000)2 for AUC) are presented as mean ± SEM. (e) Representative Doppler tracings of the renal arterial waveforms. Statistics: Individual and AUC data for creatinine, urine output, and urinary NGAL was normally distributed with equal variance and parametric testing was used. Urinary TIMP2·IGFBP7 individual and AUC data were normally distributed, however, variance was not equal and nonparametric testing was used. RI significance was calculated by chi-square test (*p<0.05; **p<0.01).
Urinary kidney injury marker NGAL was significantly lower in rhC1INH-treated animals during the first week (p=0.008) and daily at days 1, 3, and 5 (Figure 2C, p=0.034, p=0.007, p=0.008). TIMP2·IGFBP7 was also lower in rhC1INH recipients (p=0.019), with a significant difference on day 1 (Figure 2D, p=0.030). Day 1 RI measured by Doppler ultrasound revealed a significant elevation in 100% (4/4) vehicle animals versus only 14% (1/7) of rhC1INH animals (Figure 2E–F, p=0.002). Ultrasounds were unavailable for 1 vehicle-treated and 1 rhC1INH-treated animal.
3.2. RhC1INH Reduces Classical and MBL Complement Pathway Activity and C3b/C5b-9 deposition but Does Not Impact Alternative Pathway Activity or C5/C5a Levels
We assessed exogenous and endogenous serum C1INH levels, CP, MBL, and AP activity at baseline and 60 minutes postreperfusion (T60). Serum C1INH was significantly elevated in rhC1INH recipients at T60, demonstrating the presence of the drug, while vehicle recipients remained near baseline (Figure 3A, p=0.016). CP and MBL pathway activities were both significantly higher in vehicle animals at T60 when compared to rhC1INH-treated animals who remained near or below baseline levels (Figure 3B, p=0.036; Figure 3C, p=0.022). While AP activity trended higher at T60 for both groups, no significant differences were observed between groups or time points (Figure 3D).
Figure 3: RhC1INH reduces classical and MBL complement pathway activity and C3b/C5b-9 deposition but does not impact alternative pathway activity.
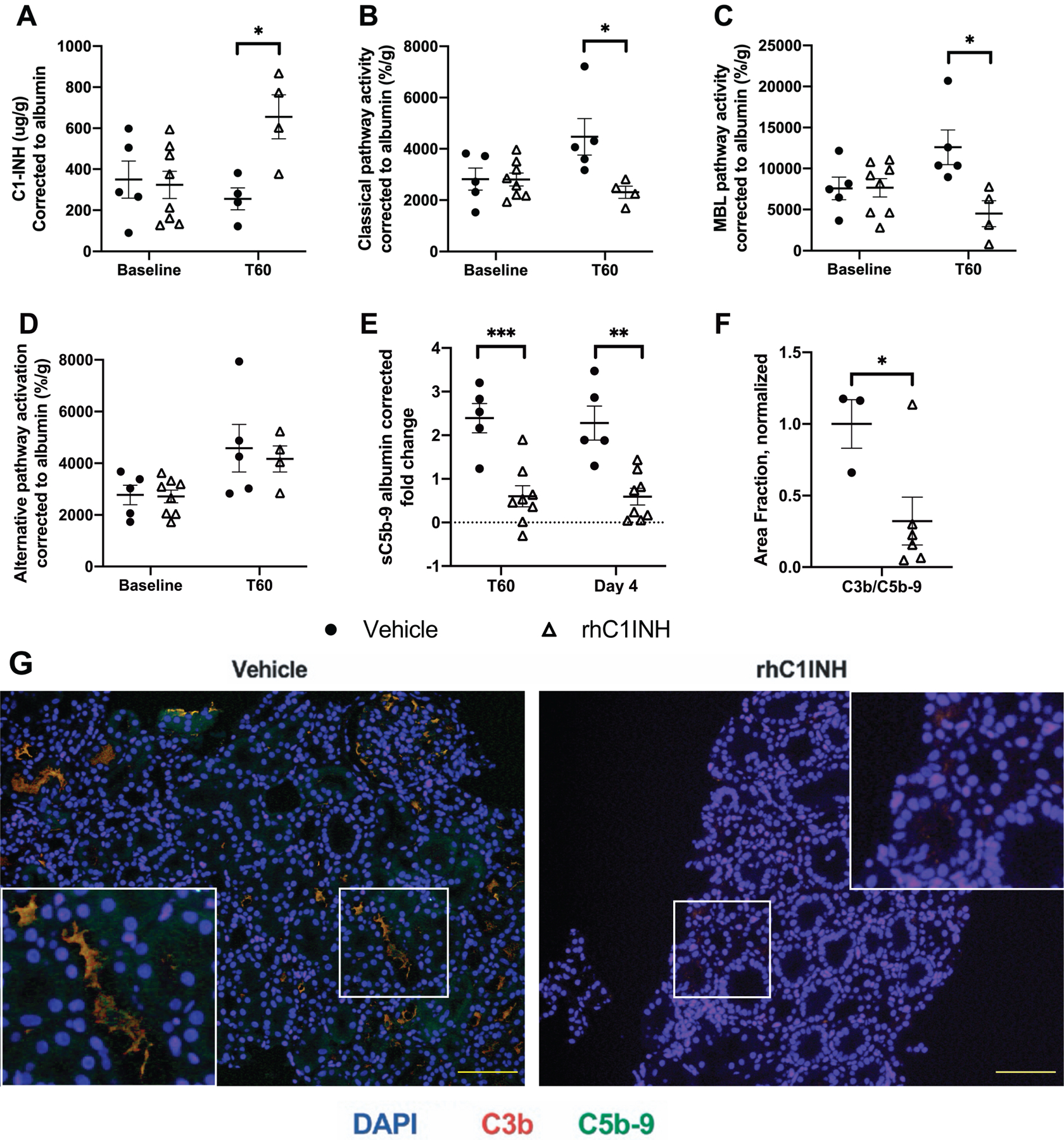
Data in a-d are shown as individual values ± SEM. (a) Serum levels of C1INH for vehicle-treated (n=5) vs rhC1INH-treated (n=4) recipients at baseline and 60-minutes (T60) after reperfusion presented as sample C1INH (μg) normalized to serum albumin to correct for hemodilution. Complement activation was measured by the complement screening assay for (b) CP, (c) MBL, and (d) AP. Four rhC1INH-treated T60 samples were excluded due to hemolysis. (e) Vehicle-treated (n=5) and rhC1INH-treated (n=8) downstream complement products from terminal complement activation sC5b-9 (e) in EDTA-plasma is expressed as sample ng/ml, normalized to albumin and to baseline values due to hemodilution. (f-g) Immunofluorescence staining of biopsies for C3b, C5b-9, and DAPI at 1-week after transplantation are measured by area fraction normalized to vehicle control. Statistics: Data for C1INH, CP, MBL, AP, sC5b-9, and area fraction of C3b/C5b-9 were normally distributed with equal variance, therefore parametric testing was used (*p<0.05; **p<0.01; ***p<0.001).
Plasma sC5b-9 increased 2.39-fold in the vehicle-treated group at T60 and 2.28-fold on day 4, significantly higher than the rhC1INH-treated group (Figure 3F, p<0.001 and p=0.001). There was no significant difference in plasma C5 or C5a between groups or timepoints (Figure S1A–B). Immunofluorescent staining of biopsies at 1-week posttransplant revealed significantly less deposition of C3b/C5b-9 in rhC1INH-treated animals (Figure 3F–G, p=0.048).
3.3. RhC1INH Reduces Inflammatory Cytokines
Plasma cytokines were quantified at baseline, T60 and day 4 (Figure 4A–E). RhC1INH-treated animals presented significantly less IL-6, IL-8, TNFα and IL-18 than vehicle-treated animals at T60 (p=0.030, p=0.030, p=0.005, p=0.030 respectively) and significantly less MCP-1 and IL-18 on day 4 (p=0.030, p=0.011).
Figure 4: RhC1INH reduces inflammatory cytokines.
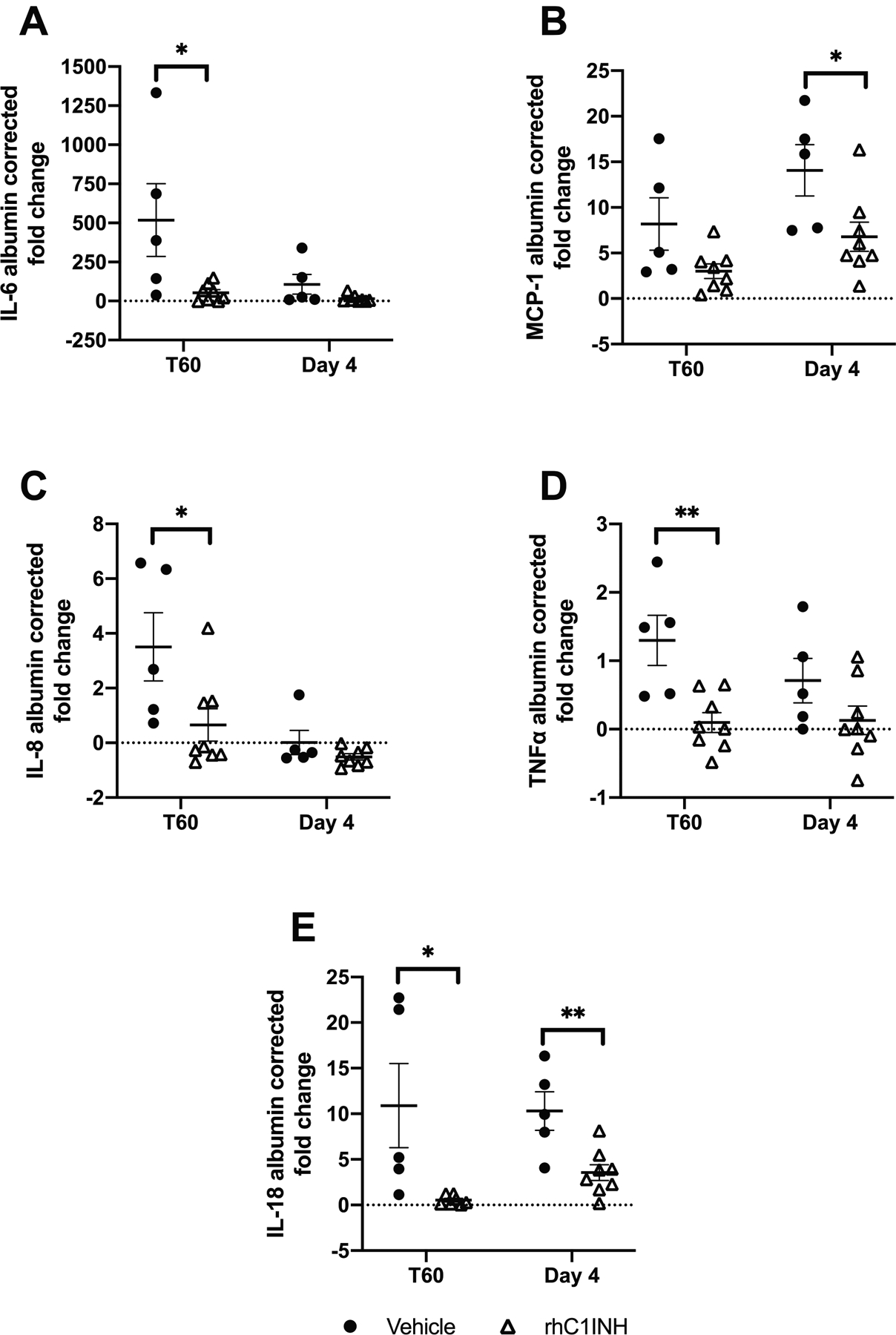
Data are shown as individual values ± SEM. EDTA-plasma levels of (a) IL-6, (b) MCP-1, (c) IL-8, (d) TNF-α, and (e) IL-18 in vehicle-treated (n=5) vs rhC1INH-treated (n=8) was expressed as individual values (ng/mL) normalized to albumin and then to baseline values due to hemodilution. Statistics: IL-6 and MCP-1 were normally distributed with unequal variance, hence nonparametric testing was used. IL-8 and IL-18 were not normally distributed with unequal variance, hence nonparametric testing was used. TNF-α was normally distributed with equal variance, parametric testing was used (*p<0.05; **p<0.01).
3.4. RhC1INH-treatment Trends Toward Reduced Cellular Infiltration and Oxidative Damage in the First Posttransplant Week
Immunohistochemistry in biopsies 1-week posttransplant revealed no significant difference in macrophage (CD68+) or neutrophil (MPO+) infiltration between groups, though there was a trend toward increased macrophage infiltration compared to native biopsies and a significant increase in neutrophil infiltration between vehicle-treated graft-recipients and native kidneys (Figure 5A, p=0.015) – this difference was not observed between rhC1INH-treated and native kidneys. No significant difference was observed in MDA-staining comparing oxidative damage in rhC1INH-treated to native kidneys or rhC1INH-treated to vehicle kidneys, but a significant increase was again observed comparing vehicle to native kidneys (Figure 5B, p=0.038).
Figure 5: RhC1INH-treatment trends toward reduced cellular infiltration and oxidative-damage in the first posttransplant week.
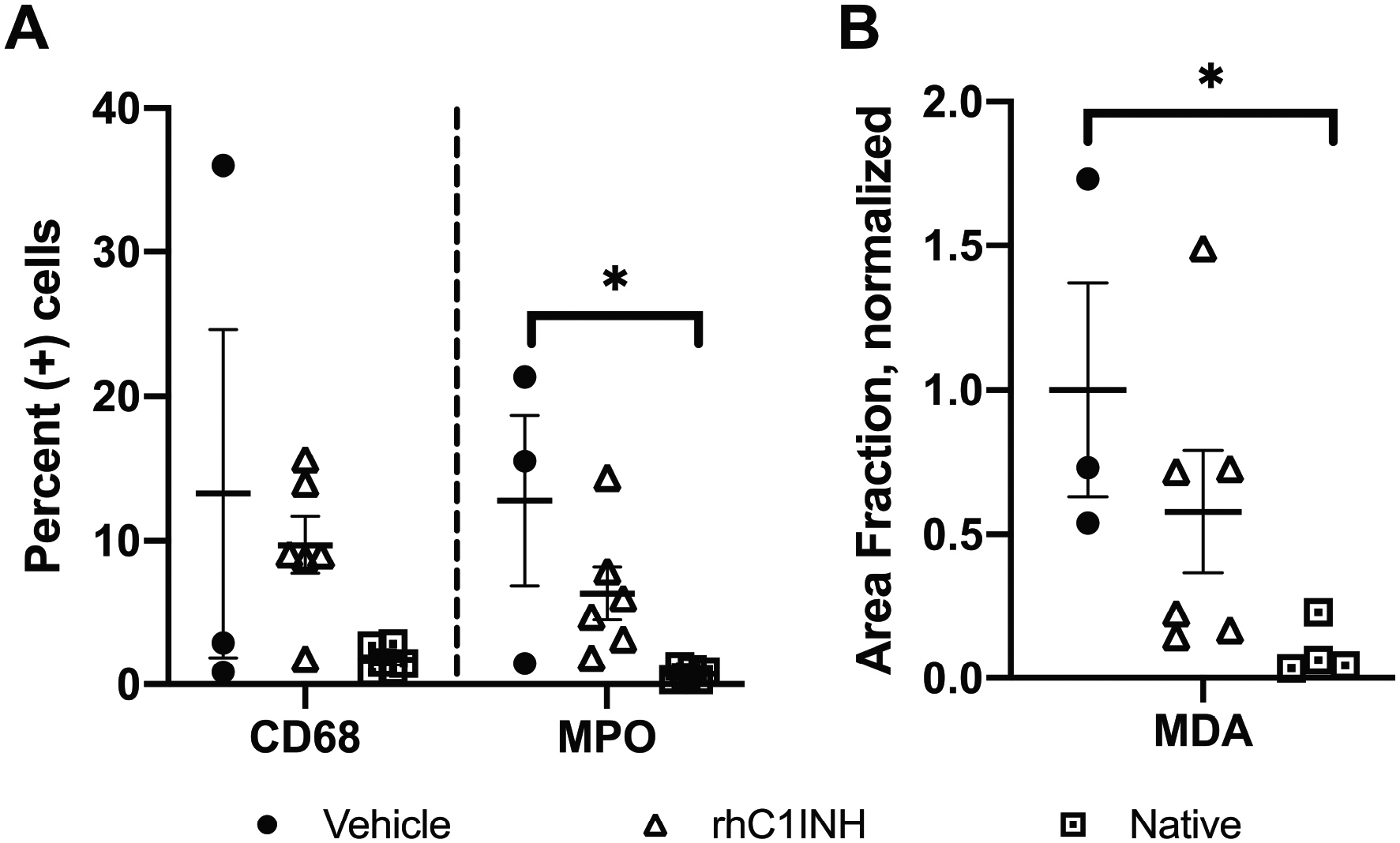
Day 7 biopsies from vehicle (n=3), rhC1INH-treated (n=6), and native (n=5) were stained for (a) macrophages (measured by CD68) and neutrophils (measured by MPO), represented as percent positive cells for individual animals. (b) Oxidative damage (measured by MDA) represented as individual values. Statistics: CD68 and MPO was normally distributed with equal variance and parametric tests were used. MDA exhibited was not normally distributed so nonparametric testing was used (*p<0.05).
3.5. DGF Prevention with RhC1INH Delays Antibody-mediated Rejection
Recipients were followed for up to 120 days with serial biopsies for histopathology and immunohistochemistry to diagnose injury, rejection, and cellular infiltration; DSA and clinical parameters were also used to diagnose AMR (Table S2).
During the 120-day period, vehicle recipients averaged 44.7 days before AMR diagnosis while rhC1INH-treated animals averaged 65.3 days; animals euthanized ≤14 days were excluded from the averages. The trends for AMR presentation were not significantly different between treatment groups (Figure 6A, p=0.226); however, a subanalysis comparing the DGF+ vehicle animals (n=4) and rhC1INH animal (n=1) against the DGF- vehicle animal (n=1) and rhC1INH animals (n=7) demonstrates a significant correlation between DGF-incidence and AMR-free survival (Figure 6B, p=0.008). ACR incidence was similar between the groups (Figure S2).
Figure 6: DGF prevention with rhC1INH delays antibody-mediated rejection.
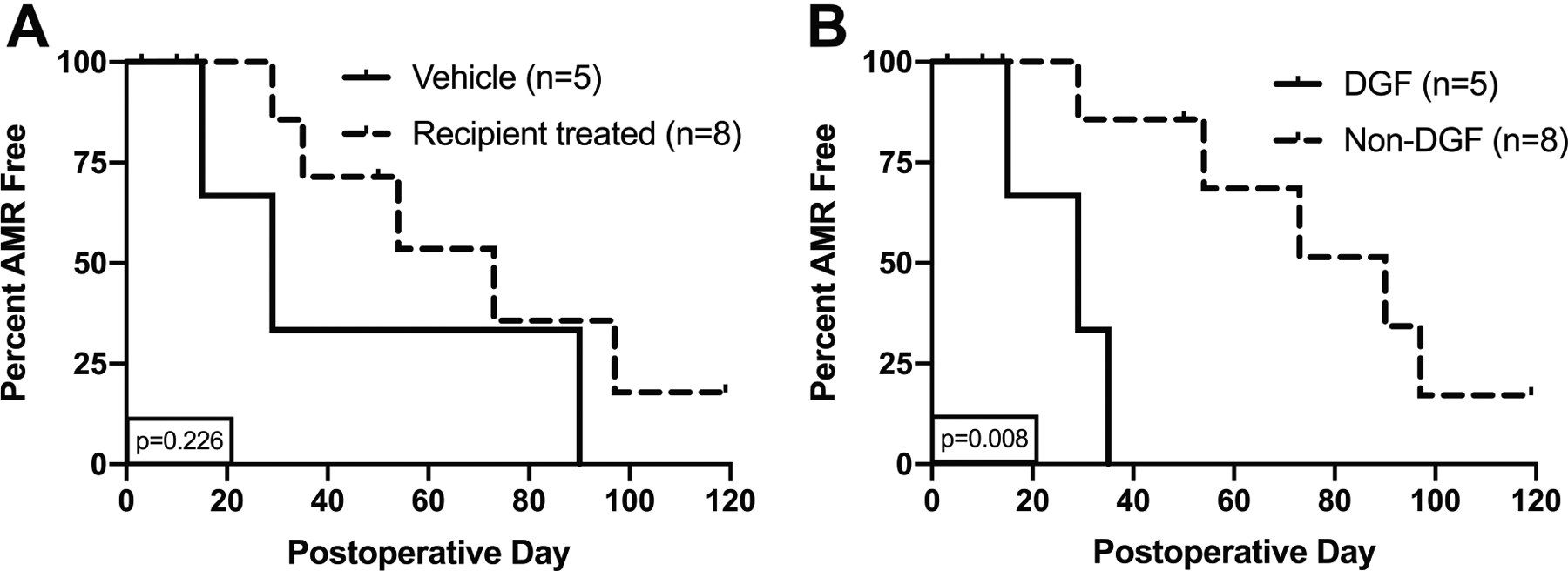
Kaplan-Meier survival analysis of (a) Vehicle-treated recipients (n=5) averaged 44.7 days before AMR diagnosis (A), while rhC1INH-treated recipients (n=8) averaged 65.3 days (p=0.226). Two animals in the vehicle group and 3 in the rhC1INH group were death censored. (b) Recipients who developed DGF (n=5) vs recipients without DGF (n=8). Significance calculated with log-rank test (p=0.008). Statistics: Log-rank test was used to compare groups.
DISCUSSION
Our NHP model of kidney transplantation after donor BD and prolonged CIT (44–48 hours) presents the unique opportunity to evaluate factors contributing to DGF in the transplant environment, and to test therapeutic regimens against those factors in isolation, without confounding factors that are mostly unavoidable in clinical trials. This includes complement and cytokine activation resulting from chronic kidney disease or donation after cardiocirculatory death (DCD), which are known to have a higher incidence of DGF than BD donation in clinical practice.1–3,7,47,50,53–56 Previously, we demonstrated that BD donor-intervention with high-dose rhC1INH prevents DGF – the drug-effect carrying over to untreated recipients.9 In this study of recipient-treatment, 3 doses of rhC1INH at 500 U/kg magnified by combining with heparin, delivered over the first 24 hours posttransplant, only 1 of 8 rhC1INH-treated recipients developed DGF.
Reduced allograft injury was evident both through the enhanced renal recovery seen by superior serum creatinine levels, urine output, and RI; and through reduced urinary injury markers, NGAL and TIMP2·IGFBP7, in the first-week posttransplant. Previous studies of swine kidney autotransplantation showed no significant increase of NGAL in the first week posttransplant after 60 minutes of warm ischemia plus 24 hours CIT.57 While this may be species specific, other studies have indicated that donor urinary NGAL increases during brain death, early evidence of injury that may carry over to recipients of those kidneys if left unmanaged in the donor.58,59 The elevation of urinary TIMP2 and IGFBP7, cell-cycle arrest biomarkers produced by renal tubular cells, has been attributed to increased filtration, reduced absorption by renal tubules, and proximal tubular cell leakage in the setting of AKI.60 On average, TIMP2·IGFBP7 levels observed in our rhC1INH-treated recipients remained under the 0.3 ng2/1000 threshold predictive of AKI.61 In contrast, vehicle animals remained >0.3 ng2/1000 until day 7.
We investigated complement activity and the presence of key complement activation products, including anaphylatoxin C5a, C3b, and terminal complex C5b-9, as the primary agents precipitating AKI leading to DGF. Naesens et al showed that expression of upstream complement proteins significantly increased in renal biopsies from deceased human donors compared to living donors.31 C5a, involved in inflammatory response and priming of alloreactive T-cells due to widespread C5aR expression in immune cells and renal tubules, is upregulated after IRI.18,19,23,33,62–65 In mice, C5aR-blockade reduces inflammatory cytokines, cellular infiltration, and protects against IRI.19 In our study, circulating C5 and C5a levels were not statistically different between groups at T60.
Peak levels of C5a may have been missed due to its rapid breakdown by the serum enzyme carboxypeptidase B.34 In a study evaluating complement activator ISIS 3202, a complement activator, in cynomolgus monkeys, C5a levels peaked near 10 minutes, followed by a reduction almost to baseline within 30 minutes.66 We cannot here confirm the role of C5a in AKI-mediated DGF. Instead, our model presents a significant increase in circulating sC5b-9 and local C3b/C5b-9 deposition in vehicle-treated recipients ameliorated by rhC1INH-treatment. C3b/C5b-9 deposition directly disrupts cellular function as well as increasing the endothelial expression of P-selectin that assists leukocyte adhesion to the renal vasculature.35,36 By blocking complement activity upstream, we are able to abrogate direct complement-mediated AKI which is otherwise sufficient to culminate in DGF.
Our investigation of proinflammatory cytokines in the first week revealed significantly lower levels of IL-6, MCP-1, IL-8, TNFα, and IL-18 in rhC1INH-treated recipients. IL-8 and MCP-1 are especially important in directing innate immune cells to sites of injury, and all play a role in immune cell activation and differentiation. IL-18 has gained interest as a novel biomarker of renal injury in serum and urine due to upregulation in the distal tubular epithelia during injury.67–72 IL-18 can recruit and prime neutrophils as well as stimulate the production of proinflammatory IL-6, IL-1β, IFN-γ, and MCP-1; which in turn, can promote T-cell differentiation into Th1 and Th17 cells, both of which promote later allograft rejection.73–76 The reduced levels of these markers at T60, and especially MCP-1 and IL-18 at day 4, the trend towards reduced macrophage and neutrophil infiltration, and reduced oxidative damage exhibited by MDA-staining, all further illustrate the reduction in kidney injury as a result of recipient treatment.
A number of studies have evaluated the use of complement inhibition in humans, baboons, pigs, and rodents for amelioration of IRI and improved transplant outcomes.6,31,47,55,77,78 In rodent models of IRI, AP activity predominates, while CP and MBL activity predominate in NHP, swine, and humans.6,31,47,55,79 In 2 recent clinical trials by Schröppel et al, eculizumab, a C5-inhibitor that primarily inhibits the AP,80 was evaluated for the prevention of IRI but failed to prevent DGF.81 Harder et al determined through in vitro hemolytic studies that robust activation of the classical complement pathway is sufficient to overcome eculizumab therapy even amidst full abrogation of AP activity, and only through combination with direct upstream inactivation of C3 (significantly produced in the kidney itself) is suppression of CP and presumably MBL pathway-mediated tissue injury accomplished.19,22,24–30,82 Chun et al demonstrated in murine heart transplants that peritransplant C1INH administration or recipient C3 and MBL deficiencies, but not factor B (AP) deficiency, yielded a survival benefit against cold ischemic injury.83 Use of rhC1INH to block complement activity upstream of C3 without impacting AP activity may prove beneficial to protect immunosuppressed recipients against pathogens in the posttransplant period.84 In our study, rhC1INH recipient-treatment blocked peritransplant increases in CP and MBL activity. Sustained AP activity did not prevent reduction of sC5b-9 or local C3b/C5b-9 deposition, nor hamper prevention of DGF.
Notably, Jordan et al evaluated the use of C1INH at 50U/kg (used for the treatment of HAE) to prevent DGF in human kidney transplant recipients – while this reduced the number of dialysis sessions, it did not prevent DGF.7 At their 3.5 years follow-up, the C1INH-treated recipients exhibited superior renal function and graft survival when compared to placebo-group.85 When they evaluated the use of C1INH (20U/kg/dose) in HLA sensitized patients to prevent AMR, they observed lower DGF incidence versus placebo but had similar rates of AMR despite reduced C1q+ HLA antibodies, which increase the risk of AMR.86–90
In our own preclinical studies, we failed to prevent DGF through donor-intervention at doses as high as 200 U/kg (data not shown), which informed the strategy employed in this study. Our efficacious use of 500 U/kg rhC1INH, 10-times the recommended dose used by Jordan et al, supplemented with heparin to further potentiate the activity of the inhibitor to obtain maximal blockade of complement-associated pathways and also protect against thrombus formation, provides a significant enhancement to the clinical regimen under investigation and holds the promise for establishing a best-practice treatment for all future transplant recipients. Further, our model utilizes HLA fully-mismatched donor/recipient pairing to maximize the risk of de novo rejection.
Our recipient rhC1INH-treatment regimen did not prevent the development of ACR or AMR. However, we did observe that recipients with DGF experienced rapid onset of AMR compared to non-DGF recipients, which was limited from attaining significance due to sample size, but confirms previous findings that developing DGF leads to increased risk and acceleration of AMR development.1–5,11,13,91 We are encouraged that a more robust regimen to prevent DGF will lead to a significant difference in AMR-free survival given sufficient sample size. Indeed, even in the absence of transplantation, Cippà et al found that within 12 months of renal IRI in mice, B-cell activation and immunoglobulin production is upregulated, leading to chronic kidney injury.92 They further noted the development of intrarenal ectopic lymphoid organs – we observed a similar phenomenon in some of our recipients (data not shown). By preventing renal injury and subsequent DGF through C1 complement blockade, we may reduce such a response and so delay/prevent rejection.
Our study has several limitations. Statistical power is impeded by small group sizes inherent to NHP models; observed trends may have attained significance with a larger sample size. In the interest of animal well-being, some samples were not collected, resulting in fewer comparable biopsies or blood samples at certain time-points. Four serum samples at T60 in rhC1INH-treated recipients were hemolyzed and could not be used for C1INH or complement pathway evaluation. Individual animals euthanized before AMR development during the study period further limited sample size and histological comparison at 120 days. Rh2714 and Rh2733 had acute increases in creatinine after renal biopsies at day 6, followed by a significant drop in creatinine on day 8 – these were excluded as resulting from procedural artifact and DGF status was already determined at this point.
In this study, we found that perioperative recipient-treatment with rhC1INH at high doses prevents DGF, similar to our donor-intervention study.47 Recipient-treatment with rhC1INH reduced direct complement-mediated injury as well as systemic inflammation through reduced levels of key cytokines and chemokines. Preclinical studies are underway in our laboratory to investigate the potential synergistic effects of dual treatment of donors and recipients with rhC1INH. Further we are developing a model of DCD donation to clarify differences from BD donation and effective treatment regimens.
Supplementary Material
ACKNOWLEDGMENTS
We thank Pharming Technologies B.V. (Leiden, The Netherlands) for their extraordinary support on providing the rhC1INH. We gratefully acknowledge the veterinary and SPI staff at the WNPRC for the extraordinary care for the animals during the observation period. We also thank D. Roenneburg, S. Raglin, W. Zhong, A. Mejia, H. Simmons, S. Larson, C. Boetcher, J. Rose and T. Roehling for expert technical assistance. We thank Kristy Kraemer and Julia Shaw from NIH and Isabella Lussier from Alpha-genesis for their assistance in identifying appropriate nonhuman primate pairs for our experimental design.
Financial Disclosure:
This work was supported by National Institutes of Health NIH #1-R01-AI110617-01A1 (PI: Luis Fernandez), NIH T32 training grants T32DK007665 (Jose Reyes) and T32AI125231 (Juan Danobeitia), and the 2016 American Society of Transplant Surgeons Scientist scholarship (Juan Danobeitia).
ABBREVIATIONS:
- ACR
Acute cellular rejection
- AKI
Acute kidney injury
- AMR
Antibody-mediated rejection
- ANOVA
Analysis of variance
- AP
Alternative (complement) pathway
- AUC
Area under the curve
- BD
Brain death
- BUN
Blood urea nitrogen
- C1INH
C1 esterase inhibitor
- CIT
Cold ischemia time
- CP
Classic (complement) pathway
- DGF
Delayed graft function
- DSA
Donor specific antigen
- ELISA
Enzyme-linked immunosorbent assay
- HAE
Hereditary angioedema
- H/E
Hematoxylin and eosin
- IFNγ
Interferon-gamma
- IGFBP7
Insulin-like growth factor-binding protein 7
- IL-1β
Interleukin-1 beta
- IL-17
Interleukin-17
- IL-6
Interleukin-6
- IL-8
Interleukin-8
- IL-18
Interleukin-18
- IRI
Ischemia-reperfusion injury
- MBL
Mannose-binding lectin (complement) pathway
- MAC
Membrane attack complex
- MAP
Mean arterial pressure
- MCP-1
Monocyte chemoattractant protein 1
- MDA
Malondialdehyde
- MHC
Major histocompatibility complex
- MMF
Mycophenolate-mofetil
- NGAL
Neutrophil gelatinase-associated lipocalin
- NHP
Nonhuman primate
- OCT
optimal cutting temperature
- PBMC
Peripheral blood mononuclear cell
- PTT
Partial thromboplastin time
- RI
Resistive index
- rhC1INH
Recombinant human C1 esterase inhibitor
- ROS
Reactive oxygen species
- SEM
Standard error of the mean
- SRV
Simian type D retrovirus
- SIV
Simian immunodeficiency virus
- STLV-1
Simian T-cell leukemia virus type 1
- TB
Tuberculosis
- TIMP2
Tissue inhibitor of metalloproteinases 2
- TNFα
Tumor necrosis factor-alpha
- WNPRC
Wisconsin National Primate Research Center
Footnotes
Disclaimer:
The authors of this manuscript have no conflicts of interest to disclose as described by Transplantation.
References
- 1.Siedlecki A, Irish W, Brennan DC. Delayed graft function in the kidney transplant. Am J Transplant. 2011;11(11):2279–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yarlagadda SG, Coca SG, Formica RN, et al. Association between delayed graft function and allograft and patient survival: a systematic review and meta-analysis. Nephrol Dial Transplant. 2009;24(3):1039–1047. [DOI] [PubMed] [Google Scholar]
- 3.Ojo AO, Wolfe RA, Held PJ, et al. Delayed graft function: risk factors and implications for renal allograft survival. Transplantation. 1997;63(7):968–974. [DOI] [PubMed] [Google Scholar]
- 4.Mallon DH, Summers DM, Bradley JA, et al. Defining delayed graft function after renal transplantation: Simplest is best. Transplantation. 2013;96(10):885–889. [DOI] [PubMed] [Google Scholar]
- 5.Incerti D, Summers N, Ton TGN, et al. The lifetime health burden of delayed graft function in kidney transplant recipients in the United States. MDM Policy Pract. 2018;3(1):2381468318781811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Castellano G, Melchiorre R, Loverre A, et al. Therapeutic targeting of classical and lectin pathways of complement protects from ischemia-reperfusion-induced renal damage. Am J Pathol. 2010;176(4):1648–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jordan SC, Choi J, Aubert O, et al. A phase I/II, double-blind, placebo-controlled study assessing safety and efficacy of C1 esterase inhibitor for prevention of delayed graft function in deceased donor kidney transplant recipients. Am J Transplant. 2018;18(12):2955–2964. [DOI] [PubMed] [Google Scholar]
- 8.Zeraati AA, Naghibi M, Kianoush S, et al. Impact of slow and delayed graft function on kidney graft survival between various subgroups among renal transplant patients. Transplant Proc. 2009;41(7):2777–2780. [DOI] [PubMed] [Google Scholar]
- 9.Danobeitia JS, Zens TJ, Zitur L, et al. Targeted donor complement blockade after brain-death prevents delayed graft function but not progression to antibody-mediated rejection in a nonhuman primate model of kidney allo-transplantation [abstract]. Am J Transplant. 2017;17 (suppl 3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zens TJ, Danobeitia JS, Leverson G, et al. The impact of kidney donor profile index on delayed graft function and transplant outcomes: A single-center analysis. Clin Transplant. 2018;32(3):e13190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mannon RB. Delayed graft function: The AKI of kidney transplantation. Nephron. 2018;140(2):94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim DW, Tsapepas D, King KL, et al. Financial impact of delayed graft function in kidney transplantation. Clin Transplant. 2020;34(10):e14022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang JH, Skeans MA, Israni AK. Current status of kidney transplant outcomes: Dying to survive. Adv Chronic Kidney Dis. 2016;23(5):281–286. [DOI] [PubMed] [Google Scholar]
- 14.Orlando G, Khan MA, El-Hennawy H, et al. Is prolonged cold ischemia a contraindication to using kidneys from acute kidney injury donors? Clin Transplant. 2018;32(3):e13185. [DOI] [PubMed] [Google Scholar]
- 15.Debout A, Foucher Y, Trébern-Launay K, et al. Each additional hour of cold ischemia time significantly increases the risk of graft failure and mortality following renal transplantation. Kidney Int. 2015;87(2):343–349. [DOI] [PubMed] [Google Scholar]
- 16.Hart A, Smith JM, Skeans MA, et al. OPTN/SRTR 2018 annual data report: Kidney. Am J Transplant. 2020;20 Suppl s1:20–130. [DOI] [PubMed] [Google Scholar]
- 17.Kayler L, Yu X, Cortes C, et al. Impact of cold ischemia time in kidney transplants from donation after circulatory death donors. Transplant Direct. 2017;3(7):e177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schröppel B, Legendre C. Delayed kidney graft function: from mechanism to translation. Kidney Int. 2014;86(2):251–258. [DOI] [PubMed] [Google Scholar]
- 19.Peng Q, Li K, Smyth LA, et al. C3a and C5a promote renal ischemia-reperfusion injury. J Am Soc Nephrol. 2012;23(9):1474–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diepenhorst GM, van Gulik TM, Hack CE. Complement-mediated ischemia-reperfusion injury: lessons learned from animal and clinical studies. Ann Surg. 2009;249(6):889–899. [DOI] [PubMed] [Google Scholar]
- 21.de Vries DK, Lindeman JH, Tsikas D, et al. Early renal ischemia-reperfusion injury in humans is dominated by IL-6 release from the allograft. Am J Transplant. 2009;9(7):1574–1584. [DOI] [PubMed] [Google Scholar]
- 22.Naesens M, Li L, Ying L, et al. Expression of complement components differs between kidney allografts from living and deceased donors. J Am Soc Nephrol. 2009;20(8):1839–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Werkhoven MB, Damman J, van Dijk MCRF, et al. Complement mediated renal inflammation induced by donor brain death: role of renal C5a-C5aR interaction. Am J Transplant. 2013;13(4):875–882. [DOI] [PubMed] [Google Scholar]
- 24.Tang S, Zhou W, Sheerin NS, et al. Contribution of renal secreted complement C3 to the circulating pool in humans. J Immunol. 1999;162(7):4336–4341. [PubMed] [Google Scholar]
- 25.Cravedi P, Leventhal J, Lakhani P, et al. Immune cell-derived C3a and C5a costimulate human T cell alloimmunity. Am J Transplant. 2013;13(10):2530–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou W The new face of anaphylatoxins in immune regulation. Immunobiology. 2012;217(2):225–234. [DOI] [PubMed] [Google Scholar]
- 27.Kelly KJ, Liu Y, Zhang J, et al. Renal C3 complement component: Feed forward to diabetic kidney disease. Am J Nephrol. 2015;41(1):48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Damman J, Nijboer WN, Schuurs TA, et al. Local renal complement C3 induction by donor brain death is associated with reduced renal allograft function after transplantation. Nephrol Dial Transplant. 2011;26(7):2345–2354. [DOI] [PubMed] [Google Scholar]
- 29.Zwarthoff SA, Berends ETM, Mol S, et al. Functional characterization of alternative and classical pathway C3/C5 convertase activity and inhibition using purified models. Front Immunol. 2018;9:1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pratt JR, Basheer SA, Sacks SH. Local synthesis of complement component C3 regulates acute renal transplant rejection. Nat Med. 2002;8(6):582–587. [DOI] [PubMed] [Google Scholar]
- 31.de Vries B, Walter SJ, Peutz-Kootstra CJ, et al. The mannose-binding lectin-pathway is involved in complement activation in the course of renal ischemia-reperfusion injury. Am J Pathol. 2004;165(5):1677–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Vries B, Köhl J, Leclercq WK, et al. Complement factor C5a mediates renal ischemia-reperfusion injury independent from neutrophils. J Immunol. 2003;170(7):3883–3889. [DOI] [PubMed] [Google Scholar]
- 33.Dick J, Gan PY, Ford SL, et al. C5a receptor 1 promotes autoimmunity, neutrophil dysfunction and injury in experimental anti-myeloperoxidase glomerulonephritis. Kidney Int. 2018;93(3):615–625. [DOI] [PubMed] [Google Scholar]
- 34.Rabiet MJ, Huet E, Boulay F. The N-formyl peptide receptors and the anaphylatoxin C5a receptors: An overview. Biochimie. 2007;89(9):1089–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kilgore KS, Ward PA, Warren JS. Neutrophil adhesion to human endothelial cells is induced by the membrane attack complex: the roles of P-selectin and platelet activating factor. Inflammation. 1998;22(6):583–598. [DOI] [PubMed] [Google Scholar]
- 36.Foreman KE, Vaporciyan AA, Bonish BK, et al. C5a-induced expression of P-selectin in endothelial cells. J Clin Invest. 1994;94(3):1147–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zahmatkesh M, Kadkhodaee M, Mahdavi-Mazdeh M, et al. Oxidative stress status in renal transplant recipients. Exp Clin Transplant. 2010;8(1):38–44. [PubMed] [Google Scholar]
- 38.La Manna G, Lanci N, Della Bella E, et al. Reduction of oxidative damage reflects a better kidney transplantation outcome. Am J Nephrol. 2011;34(6):496–504. [DOI] [PubMed] [Google Scholar]
- 39.Biancone L, David S, Della Pietra V, et al. Alternative pathway activation of complement by cultured human proximal tubular epithelial cells. Kidney Int. 1994;45(2):451–460. [DOI] [PubMed] [Google Scholar]
- 40.Fonseca I, Reguengo H, Almeida M, et al. Oxidative stress in kidney transplantation: malondialdehyde is an early predictive marker of graft dysfunction. Transplantation. 2014;97(10):1058–1065. [DOI] [PubMed] [Google Scholar]
- 41.Hariharan N, Zhai P, Sadoshima J. Oxidative stress stimulates autophagic flux during ischemia/reperfusion. Antioxid Redox Signal. 2011;14(11):2179–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Buerke M, Schwertz H, Seitz W, et al. Novel small molecule inhibitor of C1s exerts cardioprotective effects in ischemia-reperfusion injury in rabbits. J Immunol. 2001;167(9):5375–5380. [DOI] [PubMed] [Google Scholar]
- 43.Davis AE, Mejia P, Lu F. Biological activities of C1 inhibitor. Mol Immunol. 2008;45(16):4057–4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rajabi M, Struble E, Zhou Z, et al. Potentiation of C1-esterase inhibitor by heparin and interactions with C1s protease as assessed by surface plasmon resonance. Biochim Biophys Acta. 2012;1820(1):56–63. [DOI] [PubMed] [Google Scholar]
- 45.Caldwell EE, Andreasen AM, Blietz MA, et al. Heparin binding and augmentation of C1 inhibitor activity. Arch Biochem Biophys. 1999;361(2):215–222. [DOI] [PubMed] [Google Scholar]
- 46.Poppelaars F, Damman J, de Vrij EL, et al. New insight into the effects of heparinoids on complement inhibition by C1-inhibitor. Clin Exp Immunol. 2016;184(3):378–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Danobeitia JS, Zens TJ, Chlebeck PJ, et al. Targeted donor complement blockade after brain death prevents delayed graft function in a nonhuman primate model of kidney transplantation. Am J Transplant. 2020;20(6):1513–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Budde ML, Wiseman RW, Karl JA, et al. Characterization of Mauritian cynomolgus macaque major histocompatibility complex class I haplotypes by high-resolution pyrosequencing. Immunogenetics. 2010;62(11–12):773–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cruz MP. Conestat alfa (ruconest): First recombinant c1 esterase inhibitor for the treatment of acute attacks in patients with hereditary angioedema. P T. 2015;40(2):109–114. [PMC free article] [PubMed] [Google Scholar]
- 50.Zens TJ, Danobeitia JS, Chlebeck PJ, et al. Guidelines for the management of a brain death donor in the rhesus macaque: A translational transplant model. PLoS One. 2017;12(9):e0182552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boom H, Mallat MJ, de Fijter JW, et al. Delayed graft function influences renal function, but not survival. Kidney Int. 2000;58(2):859–866. [DOI] [PubMed] [Google Scholar]
- 52.Haas M, Loupy A, Lefaucheur C, et al. The Banff 2017 Kidney Meeting Report: Revised diagnostic criteria for chronic active T cell-mediated rejection, antibody-mediated rejection, and prospects for integrative endpoints for next-generation clinical trials. Am J Transplant. 2018;18(2):293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fearn A, Sheerin NS. Complement activation in progressive renal disease. World J Nephrol. 2015;4(1):31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen SF, Chen M. Complement activation in progression of chronic kidney disease. Adv Exp Med Biol. 2019;1165:423–441. [DOI] [PubMed] [Google Scholar]
- 55.Berger M, Lefaucheur C, Jordan SC. Update on C1 esterase inhibitor in human solid organ transplantation. Transplantation. 2019;103(9):1763–1775. [DOI] [PubMed] [Google Scholar]
- 56.Tsai SF, Chen CH, Shu KH, et al. Current safety of renal allograft biopsy with indication in adult recipients: An observational study. Medicine (Baltimore). 2016;95(6):e2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Delpech PO, Thuillier R, SaintYves T, et al. Inhibition of complement improves graft outcome in a pig model of kidney autotransplantation. J Transl Med. 2016;14(1):277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ganji MR, Alatab S, Naderi GH, et al. Association of brain-dead donor’s urine neutrophil gelatinase-associated lipocalin levels with kidney allograft function. Iran J Kidney Dis. 2015;9(5):394–399. [PubMed] [Google Scholar]
- 59.Hollmen ME, Kyllönen LE, Inkinen KA, et al. Deceased donor neutrophil gelatinase-associated lipocalin and delayed graft function after kidney transplantation: a prospective study. Crit Care. 2011;15(3):R121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Johnson ACM, Zager RA. Mechanisms underlying increased TIMP2 and IGFBP7 urinary excretion in experimental AKI. J Am Soc Nephrol. 2018;29(8):2157–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vijayan A, Faubel S, Askenazi DJ, et al. Clinical use of the urine biomarker [TIMP-2] × [IGFBP7] for acute kidney injury risk assessment. Am J Kidney Dis. 2016;68(1):19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li L, Okusa MD. Macrophages, dendritic cells, and kidney ischemia-reperfusion injury. Semin Nephrol. 2010;30(3):268–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schröppel B, Heeger PS, Thiessen-Philbrook H, et al. Donor urinary C5a levels independently correlate with posttransplant delayed graft function. Transplantation. 2019;103(1):e29–e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Klos A, Tenner AJ, Johswich KO, et al. The role of the anaphylatoxins in health and disease. Mol Immunol. 2009;46(14):2753–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Richard S, Farrell CA, Shaw AS, et al. C5a as a model for chemotactic factor-stimulated tyrosine phosphorylation in the human neutrophil. J Immunol. 1994;152(5):2479–2487. [PubMed] [Google Scholar]
- 66.Henry SP, Beattie G, Yeh G, et al. Complement activation is responsible for acute toxicities in rhesus monkeys treated with a phosphorothioate oligodeoxynucleotide. Int Immunopharmacol. 2002;2(12):1657–1666. [DOI] [PubMed] [Google Scholar]
- 67.Lin CY, Chang CH, Fan PC, et al. Serum interleukin-18 at commencement of renal replacement therapy predicts short-term prognosis in critically ill patients with acute kidney injury. PLoS One. 2013;8(5):e66028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sung WC, Yu HP, Tsai YF, et al. The ratio of plasma interleukin-18 is a sensitive biomarker for acute kidney injury after liver transplantation. Transplant Proc. 2014;46(3):816–817. [DOI] [PubMed] [Google Scholar]
- 69.Liu X, Guan Y, Xu S, et al. Early predictors of acute kidney injury: A narrative review. Kidney Blood Press Res. 2016;41(5):680–700. [DOI] [PubMed] [Google Scholar]
- 70.Liu C, Chen J, Liu B, et al. Role of IL-18 in transplant biology. Eur Cytokine Netw. 2018;29(2):48–51. [DOI] [PubMed] [Google Scholar]
- 71.Hall IE, Yarlagadda SG, Coca SG, et al. IL-18 and urinary NGAL predict dialysis and graft recovery after kidney transplantation. J Am Soc Nephrol. 2010;21(1):189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Striz I, Krasna E, Eliska K, et al. Interleukin 18 (IL-18) upregulation in acute rejection of kidney allograft. Immunol Lett. 2005;99(1):30–35. [DOI] [PubMed] [Google Scholar]
- 73.Sullivan JA, Adams AB, Burlingham WJ. The emerging role of TH17 cells in organ transplantation. Transplantation. 2014;97(5):483–489. [DOI] [PubMed] [Google Scholar]
- 74.Afzali B, Lombardi G, Lechler RI, et al. The role of T helper 17 (Th17) and regulatory T cells (Treg) in human organ transplantation and autoimmune disease. Clin Exp Immunol. 2007;148(1):32–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xie XJ, Ye YF, Zhou L, et al. Th17 promotes acute rejection following liver transplantation in rats. J Zhejiang Univ Sci B. 2010;11(11):819–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ma L, Zhang H, Hu K, et al. The imbalance between Tregs, Th17 cells and inflammatory cytokines among renal transplant recipients. BMC Immunol. 2015;16:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tillou X, Poirier N, Le Bas-Bernardet S, et al. Recombinant human C1-inhibitor prevents acute antibody-mediated rejection in alloimmunized baboons. Kidney Int. 2010;78(2):152–159. [DOI] [PubMed] [Google Scholar]
- 78.Poppelaars F, Jager NM, Kotimaa J, et al. C1-inhibitor treatment decreases renal injury in an established brain-dead rat model. Transplantation. 2018;102(1):79–87. [DOI] [PubMed] [Google Scholar]
- 79.van der Pol P, Schlagwein N, van Gijlswijk DJ, et al. Mannan-binding lectin mediates renal ischemia/reperfusion injury independent of complement activation. Am J Transplant. 2012;12(4):877–887. [DOI] [PubMed] [Google Scholar]
- 80.Willrich MAV, Andreguetto BD, Sridharan M, et al. The impact of eculizumab on routine complement assays. J Immunol Methods. 2018;460:63–71. [DOI] [PubMed] [Google Scholar]
- 81.Schröppel B, Akalin E, Baweja M, et al. Peritransplant eculizumab does not prevent delayed graft function in deceased donor kidney transplant recipients: Results of two randomized controlled pilot trials. Am J Transplant. 2020;20(2):564–572. [DOI] [PubMed] [Google Scholar]
- 82.Harder MJ, Kuhn N, Schrezenmeier H, et al. Incomplete inhibition by eculizumab: mechanistic evidence for residual C5 activity during strong complement activation. Blood. 2017;129(8):970–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chun N, Fairchild RL, Li Y, et al. Complement dependence of murine costimulatory blockade-resistant cellular cardiac allograft rejection. Am J Transplant. 2017;17(11):2810–2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Katschke KJ, Stawicki S, Yin J, et al. Structural and functional analysis of a C3b-specific antibody that selectively inhibits the alternative pathway of complement. J Biol Chem. 2009;284(16):10473–10479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Huang E, Vo A, Choi J, et al. Three-year outcomes of a randomized, double-blind, placebo-controlled study assessing safety and efficacy of c1 esterase inhibitor for prevention of delayed graft function in deceased donor kidney transplant recipients. Clin J Am Soc Nephrol. 2020;15(1):109–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vo AA, Zeevi A, Choi J, et al. A phase I/II placebo-controlled trial of C1-inhibitor for prevention of antibody-mediated rejection in HLA sensitized patients. Transplantation. 2015;99(2):299–308. [DOI] [PubMed] [Google Scholar]
- 87.Yell M, Muth BL, Kaufman DB, et al. C1q binding activity of de novo donor-specific HLA antibodies in renal transplant recipients with and without antibody-mediated rejection. Transplantation. 2015;99(6):1151–1155. [DOI] [PubMed] [Google Scholar]
- 88.Cozzi E, Biancone L. C1q-binding donor-specific antibody assays help define risk and prognosis in antibody-mediated rejection. Kidney Int. 2018;94(4):657–659. [DOI] [PubMed] [Google Scholar]
- 89.Viglietti D, Gosset C, Loupy A, et al. C1 inhibitor in acute antibody-mediated rejection nonresponsive to conventional therapy in kidney transplant recipients: A pilot study. Am J Transplant. 2016;16(5): 1596–1603. [DOI] [PubMed] [Google Scholar]
- 90.Loupy A, Lefaucheur C, Vernerey D, et al. Complement-binding anti-HLA antibodies and kidney-allograft survival. N Engl J Med. 2013;369(13):1215–1226. [DOI] [PubMed] [Google Scholar]
- 91.Weber S, Dienemann T, Jacobi J, et al. Delayed graft function is associated with an increased rate of renal allograft rejection: A retrospective single center analysis. PLoS One. 2018;13(6):e0199445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cippà PE, Liu J, Sun B, et al. A late B lymphocyte action in dysfunctional tissue repair following kidney injury and transplantation. Nat Commun. 2019;10(1):1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.