

Abstract
Data-independent acquisition (DIA) proteomics is a recently-developed global mass spectrometry (MS)-based proteomics strategy. In a DIA method, precursor ions are isolated into pre-defined isolation windows and fragmented; all fragmented ions in each window are then analyzed by a high-resolution mass spectrometer. DIA proteomics analysis is characterized by a broad protein coverage, high reproducibility, and accuracy, and its combination with advances in other techniques such as sample preparation and computational data analysis could lead to further improvements in assay performances. DIA technology has been increasingly utilized in various proteomics studies, including quantifying drug-metabolizing enzymes and transporters. Quantitative proteomics study of drug-metabolizing enzymes and transporters could lead to a better understanding of pharmacokinetics and pharmacodynamics and facilitate drug development. This review summarizes the application of DIA technology in proteomic analysis of drug-metabolizing enzymes and transporters.
Keywords: Proteomics, Data-Independent Acquisition, Data-Dependent Acquisition, Drug-metabolizing Enzyme, Drug Transporter
Introduction
Global protein profiling can provide a vast amount of information with relevance to many research areas where consistent, accurate, and large-scale protein quantification data are required; examples include but are not limited to biomarker discovery, drug screens, and personalized medicine. It has been widely recognized that compared to mRNA expression, protein expression is a better surrogate of protein function due to its better correlation with protein activity [1]. As such, protein quantification is essential for understanding the biological function of proteins. Liquid chromatography-tandem mass spectrometry (LC-MS/MS)-based proteomics technology offers a high-throughput approach for quantifying complex protein mixtures, with the capacity quantifying more proteins than those conventional immunoaffinity-based quantification methods (e.g., Western blotting and enzyme-linked immunosorbent assays [ELISAs]) by orders of magnitude. The past decade has witnessed significant advances in LC-MS/MS-based proteomics technology, with increasingly improved performance in terms of sensitivity, selectivity, reproducibility, protein coverage, and cost-effectiveness.
There are four commonly used LC-MS/MS-based quantitative proteomics strategies: two targeted approaches: selective/multiple-reaction monitoring (SRM/MRM) and parallel-reaction monitoring (PRM), and two untargeted approaches: data-dependent acquisition (DDA), and data-independent acquisition (DIA) [2]. The basic principles and performance comparison of these methods are shown in Figure 1 and 2, respectively. Targeted proteomics utilizes pre-selected surrogate peptides to quantify proteins of interest, and its prototypical method is SRM/MRM that is usually carried out on a triple quadrupole mass spectrometer (QqQ MS) [3]. In SRM/MRM mode, targeted peptide precursors are filtered in the first quadrupole (Q1) for fragmentation, and only selected product ions are monitored for each precursor in the third quadrupole (Q3) [3]. This generates multiple transition (precursor/fragment ion pair) ion chromatograms, allowing for the detection and quantification of targeted peptides [4]. PRM is a more recently developed targeted proteomics technique that adopts a hybrid mass spectrometer system by replacing the third quadrupole of a QqQ MS with a high-resolution accurate-mass (HRAM) mass analyzer, such as an Orbitrap or time of flight (TOF) analyzer [5, 6]. Overall, both SRM/MRM and PRM feature high sensitivity, selectivity, and reproducibility but have limited protein coverage [4, 7, 8]. Unlike targeted proteomics methods, the DDA approach has a much broader coverage, allowing for global proteome-scale profiling. DDA isolates the most intensive precursor ions in Q1, and the precursors are subsequently fragmented in q2 for peptide identification. In DDA mode, all the product ions are monitored, usually by an HRAM mass analyzer (Orbitrap or TOF). The DDA method is effort-saving because there is no need to select precursors and product ions. However, its accuracy and reproducibility are often lower than targeted methods because its precursor selection is stochastic with a risk of under-sampling [9].
Figure 1.
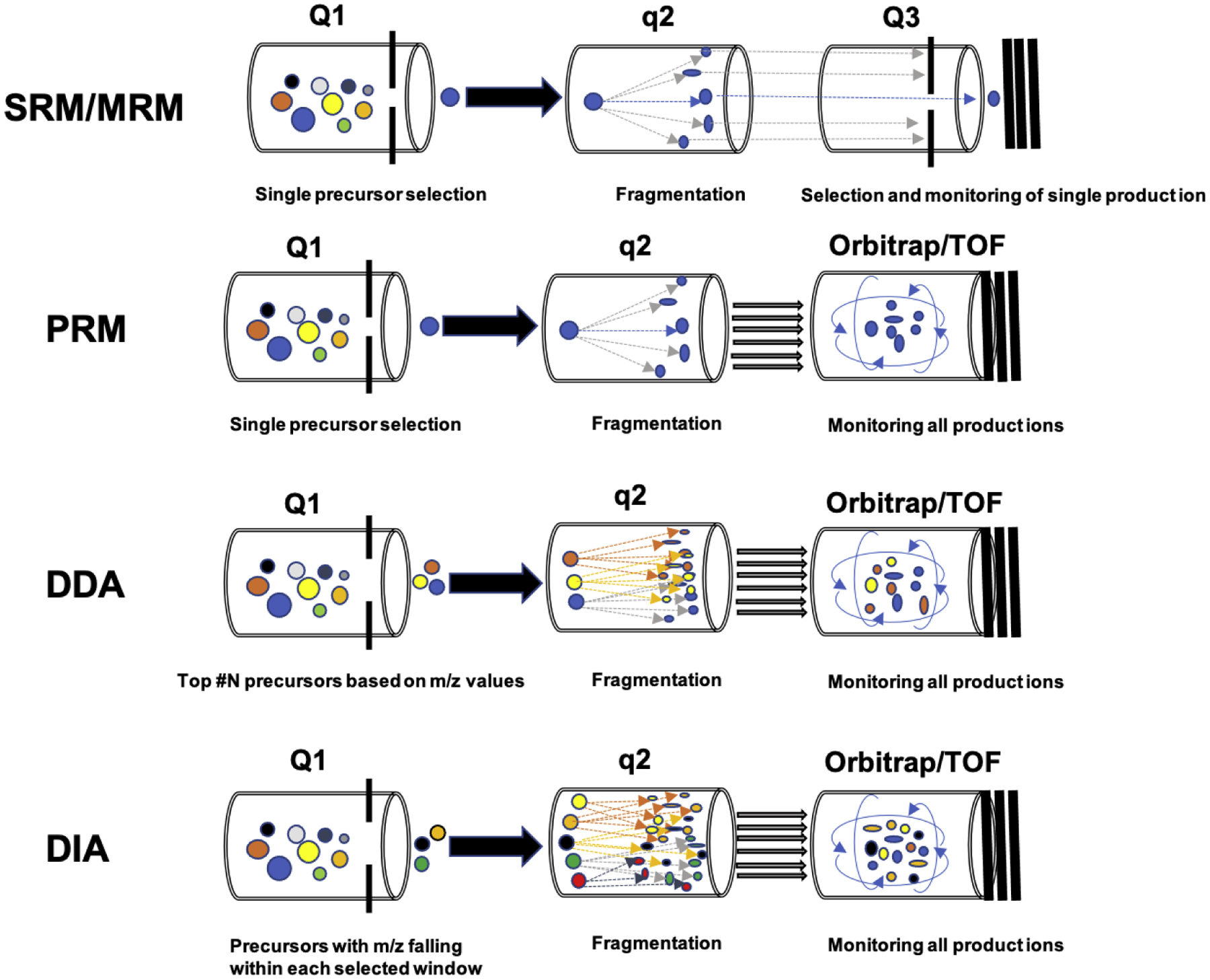
Schematic diagrams of SRM/MRM, PRM, DDA, and DIA proteomics. The SRM/MRM method isolates and fragments pre-defined precursor ions and monitors pre-defined product ions derived from the precursor. PRM isolates and fragments pre-defined precursors in the same way as SRM/MRM but records all product ions of a pre-defined precursor. DDA automatically selects the most abundant precursor ions for isolation and fragmentation and monitors all product ions. DIA isolates precursors into pre-defined isolation mass windows and fragments all precursors in each window to generate product ions being monitored by an HRAM mass analyzer.
Figure 2.
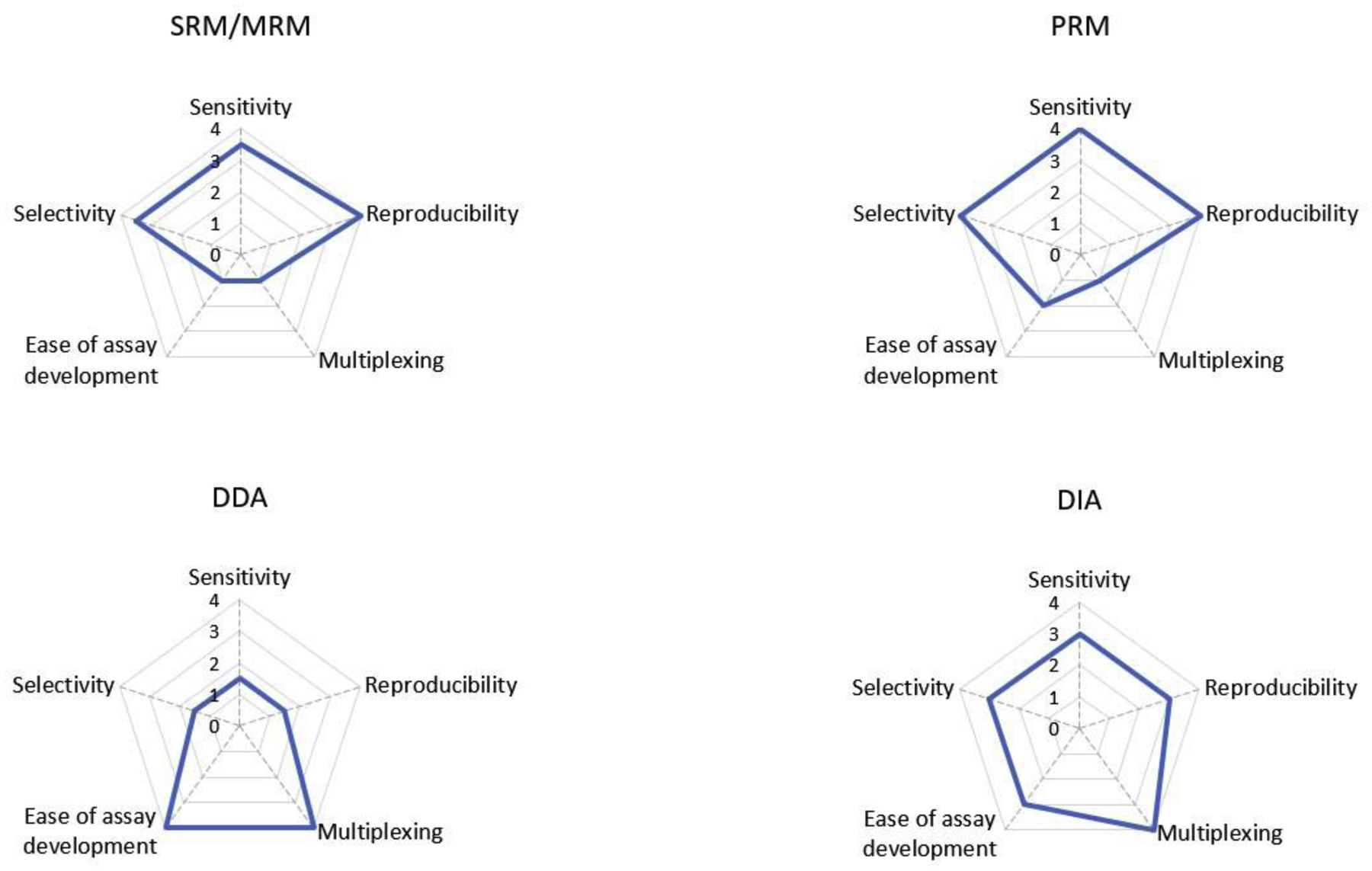
Performance profiles of SRM, PRM, DDA, and DIA. The radar graphs depict the performance of the four methods regarding selectivity, sensitivity, reproducibility, multiplexing, ease of assay development. The highest value “four” indicates top performance, whereas “0” indicates the worst performance.
The DIA approach is a more recently-developed technique that is believed to have the strengths of both PRM (high sensitivity and reproducibility) and DDA (broad protein coverage) [10]. A common DIA method is Sequential Windowed acquisition of All THeoretical fragment ion Mass Spectra (SWATH-MS) [11], and the term “SWATH” was a registered trademark of SCIEX for the DIA technology implemented on quadrupole time-of-flight (Q-TOF) instrumentation [12]. In this review, the generic term “DIA” will be used to refer to all DIA strategies regardless of the underlying instrument type. In DIA, precursors are separated into consecutive small mass-to-charge (m/z) windows (5–25 Da) in Q1, and all precursors within an isolation window are fragmented in q2. All product ions within a particular window are monitored by an HRAM mass analyzer in a systematic and unbiased manner [8, 11]. The co-fragmentation of many peptides co-eluted from the same precursor window usually results in highly complex MS2 spectra, analysis of which usually requires a peptide spectral library established by DDA studies because DIA data lack the information linking a precursor and its fragments [10, 12, 13]. However, recent development in DIA bioinformatics has permitted direct analysis of DIA data without the need for a DDA-based spectral library [14]. DIA proteomics can thus sometimes be described as a large-scale PRM with high protein coverage that is comparable to DDA [2, 10, 12]. Additionally, compared to DDA, DIA proteomics has the advantage of better reproducibility and accuracy for protein quantification as it overcomes the issue of biased sampling inherent in DDA [10]. DIA requires the predefined precursor isolation window width and MS2 scans frequency. As such, its development process is relatively more sophisticated than for DDA but still more effort-saving than the establishment of targeted methods. The selectivity and sensitivity of DIA are slightly inferior to targeted methods (i.e., MRM/SRM and PRM) due to its highly complex MS spectra, but its performance is gradually improving with the development of data processing tools and instrumentations. Multiple studies have shown that the quantification values obtained via DIA for many proteins were generally comparable to those obtained with targeted methods [15–18]. Currently, DIA is still a rapidly-developing technology but has been demonstrated well-suited for numerous applications, especially those that require a large-scale quantification of complex protein samples, such as precision medicine, biomarker discovery, and drug screening. With further advances in instrumentation and software, DIA technology is expected to be established as a powerful tool for proteome quantification because of its versatility, excellent reproducibility, and high throughput.
The performance of a DIA assay regarding sensitivity, selectivity, and proteome coverage is affected by multiple factors, such as instrumentation, MS1 isolation window schemes, and spectral library. Q-TOF and Q-Orbitrap are the two types of mass analyzers currently used for DIA proteomics. Q-Orbitrap can reach higher mass resolution relative to Q-TOF, and higher resolution is associated with better performance on peptide identification and quantification. However, unlike Q-TOF, Q-Orbitrap’s mass resolution decreases as the scan speed increases. In comparison, modern Q-TOF mass analyzers can maintain high mass resolution (e.g., 30,000 FWHM) at high scan speed (e.g., 100 HZ). The higher scan speed results in shorter cycle times, which is essential for optimal chromatographic performance. Various widths of MS1 isolation windows have been adopted for DIA analysis. In general, narrower isolation windows can improve the assay performance by reducing the number of co-fragmented precursors, and thus, enhancing selectivity and sensitivity. A limitation of narrower isolation windows is the increased cycle time, leading to the reduction of the number of data points over a chromatographic peak. Moreover, isolation window width can be adjusted based on the distribution of precursor m/z (i.e., variable isolation window scheme), which could improve the selectivity of DIA proteomics (Zhang et al., 2015). Both organism-specific and tissue/experiment-specific libraries can be utilized in DIA data analysis. Using a publicly available comprehensive organism-level library (e.g., Pan-Human library) eliminates the need to generate project-specific libraries, but this approach is associated with a higher risk of false discovery. Thus, a DIA study was often companied by a DDA analysis to generate a library specific to the samples of interest.
Drug metabolizing enzymes (DMEs) and transporters are the determinants of drug metabolism and disposition. Critically, interindividual variability in DMEs and transporters could lead to variations in pharmacokinetics (PK) and drug response. A wealth of data suggests that the functions of many DMEs and transporters are more closely correlated with their protein expression levels than with mRNA expressions [19, 20]; thus, quantification of DMEs and transporters can provide more insights into the functions of these proteins pertaining to PK and treatment outcomes and facilitate the understanding and prediction of PK and pharmacodynamics (PD) [1, 2]. In this review, we will summarize the applications of DIA in DMEs and transporters proteomics and discuss how DIA technology can aid drug development and application.
DIA-based quantification of drug-metabolizing enzymes and transporters
The studies that applied DIA methods for quantitative analysis of DMEs and drug transporters are summarized in Table 1. Overall, DIA allows for large-scale, multiplexed quantification of proteins in complex biological samples and is featured by the merits of high reproducibility and reasonable accuracy. Coupled with appropriate sample preparation and data processing approaches, DIA-based methods have been well established for different applications. In many cases, DIA proteomics methods were developed for the purpose of simplifying assay development and improving the accuracy and reproducibility of quantification. Label-based quantification usually entails spiking samples with a known amount of stable isotope-labeled peptides, allowing for relative and absolute quantification. Stable isotope labeling methods have a higher quantification accuracy than label-free assays but are more resource demanding, and their applications are often limited to a certain type of sample [21]. For example, the stable isotope labeling using amino acids in cell culture/mammals (SILAC) is widely used in in vitro cell culture systems but rarely adopted for in vivo experiments due to the high cost of stable isotope compounds [22]. In contrast, label-free methods are versatile, efficient, and cost-effective, offering an alternative to labeling proteomics [21].
Table 1.
DIA quantitative proteomics of DMEs and drug transporters.
| Proteins of interest | Sample | Method features | Normalization method | Major findings and implications | Ref. |
|---|---|---|---|---|---|
| CYPs | HLM | Label-free; relative quantification; multiplex | Normalized to β-galactosidase tryptic peptides | The protein abundance correlated better with enzyme activity than mRNA for most CYPs. | [24] |
| CYPs | HLM and microsomes of HepG2, Hep3B, and Huh7 cells | Label-free; relative quantification; multiplex | Normalized to the internal standard bovine serum albumin | The results measured by DIA were highly correlated with those obtained with PRM (r2=0.87–0.90). The expression levels of most DMEs in the hepatic cell lines were lower than those in human liver tissues. | [15] |
| CESs, UGTs, CYPs | HLS9 | Label-free; absolute quantification; multiplex | Total protein in the sample | Broader coverage than DDA-TPA; the results were comparable to those obtained from labeling and targeted methods. | [16] |
| DMEs, transporters | HLM, HLS9 | The differences in the protein concentrations of DMEs and transporters between HLM and HLS9 were profiled. | [26] | ||
| DMEs, transporters | HLM, HIM, HKM | Labeling; absolute quantification; large-scale multiplex | Stable isotope-labeled internal standard peptides | DIA was capable of multiplex quantifying proteins with accuracy comparable to targeted methods. | [17] |
| Uptake transporters | Human hepatic membrane proteins extraction | Total protein in the sample | The quantification results obtained with the DIA method was consistent with the measurements from a targeted method and data from the literature. | [18] | |
| DMEs, transporters | Mice liver and kidney fractions | Label-free and relative quantification for DIA; Labeling and absolute quantification for PRM | Stable isotope-labeled internal standard peptides for PRM | Two rounds of proteomics analysis: a DIA study for identifying proteins that differentially expressed between the control and the model groups; a PRM study for quantifying the expression changes of proteins identified by the DIA analysis. | [23] |
HLM: Human liver microsomes; HIM: Human intestine microsomes; HKM: Human kidney microsomes; HLS9: Human liver S9 fractions; CYP: cytochrome P450; UGT: uridine-diphosphate glucuronosyl transferase; TPA: total protein approach; MRM: multiple reaction monitoring; SRM: selected reaction monitoring; DIA: data independent acquisition; PRM: parallel reaction monitoring.
Label-free relative quantification
In 2016, Kuno et al. applied a DIA-based method to quantify the major DMEs and transporters in mouse liver and kidney tissues [23], with the aim of evaluating the effect of dysbiosis (alteration of intestinal flora) on the protein expression of DMEs and transporters in the liver and kidney. They used antibiotic-treated mice as the dysbiosis model and compared the differences in protein expression of DMEs and transporters between the dysbiosis model and germ-free control mice [23]. DIA proteomics analysis was first performed to screen the DMEs or transporters whose protein expression was significantly changed by dysbiosis. This analysis demonstrated a large proteome coverage with approximately 1,600 proteins being detected in germ-free mice and 825 proteins showed altered expressions in the livers, among which 52 were DMEs and drug transporters. After candidate protein identification, targeted proteomics analysis (PRM) was conducted to validate and quantify the changes in protein expression [23]. This study thus exemplified a workflow combining DIA’s large-scale multiplex quantification capability with the high confidence of targeted proteomics for biomarker discovery and validation.
Rohitash et al. (2017) developed a DIA-based label-free relative quantification method for the study of human liver microsomes [24]. They then used the Skyline software for targeted data extraction from the DIA data to analyze ten major cytochrome P450 (CYP) enzymes. Notably, trypsin-digested β-galactosidase was included as an internal standard during the analysis, and relative quantification was achieved after the ion intensities of peptides of interest were normalized to the average intensity of β-galactosidase peptides. The authors evaluated the correlations between the protein abundance, mRNA expression, and activity of the ten CYPs [24]. The results were consistent with another study that used an MRM method for CYP quantification [19]. The activity of the tested CYPs, except for CYP2B6, CYP 2C8, and CYP 3A4, was demonstrated to better correlate with their protein abundance than with mRNA expression levels, suggesting that protein abundance is a better surrogate than mRNA expression for predicting the activity of these enzymes [19, 24]. Meanwhile, both studies showed that the activity of CYP 2B6 had a relatively better correlation with its mRNA expression than its protein abundance. In the case of CYP 3A4, both protein abundance and mRNA expression strongly correlated with activity, again in both cases [19, 24]. However, there was an inconsistency between the two studies regarding CYP 2C8, for which the DIA study reported low correlations between mRNA expression and activity (Pearson correlation coefficient, r2=0.27) and between protein abundance and activity (r2=0.11) [24], while the MRM study reported strong correlations for both RNA-activity (r2=0.75) and protein-activity (r2=0.80) [19]. The reasons for this inter-study difference needs further investigation.
Similarly, in 2018, Shi et al. used a DIA proteomics method with bovine serum albumin (BSA) as the internal standard to compare the abundance of DMEs in several commonly used hepatic cell lines (i.e., HepG2, Hep3B, and Huh7) with those in human liver tissue.[15] The study also utilized a PRM proteomics method to quantify DMEs in the same samples. The results showed that the selected hepatic cell lines generally had fewer DME proteins, and the DME expression levels were lower than their counterparts in human liver tissues, suggesting that caution needs to be exercised when using hepatic cell lines for drug metabolism study [15]. The Log2-fold change values in DME expression between the cell lines and liver tissues obtained through the DIA and PRM methods were well correlated (r2 = 0.87–0.90). The PRM method detected several more low-abundance DMEs than the DIA method, suggesting a higher sensitivity and selectivity of PRM. The data indicate that DIA is a reliable quantification method for untargeted proteomics study, while PRM is more suitable for quantification of targeted low-abundance proteins.
Label-free “absolute” quantification
He et al. recently established a DIA-based, label-free, “absolute” quantification method, named DIA-TPA, that combined DIA with the total protein approach (TPA) algorithm [16]. The TPA algorithm calculates the amount of protein i by multiplying the ratio of the MS signal of protein i to the total MS signal with the total amount of protein in the sample (equation 1)[16, 25].
Proteini concentration in samplej
| (1) |
This algorithm is based on the assumption that all peptides have similar MS responses at the same concentrations; however, MS signal intensity could vary markedly among different peptides. Therefore, DIA-TPA is expected to perform reasonably well for high-abundance proteins because the variations in MS responses can be canceled out when multiple peptides are detected and utilized for quantification of a protein. However, a bias could be introduced when DIA-TPA is used for quantifying low-abundance proteins based on few or even a single signature peptide. Therefore, appropriate internal standards are warranted for reliable quantification of low-abundance proteins.
In He et al.’s study, 36 individual human liver S9 fractions (HLS9) were analyzed by DIA-TPA and DDA-TPA, with the only difference being the data acquisition mode (i.e., DIA vs. DDA). They found that DIA-TPA was able to quantify more than twice the number of proteins that identified by DDA-TPA, indicating a broader coverage for DIA-TPA. The additional proteins measured by DIA-TPA were mainly those of low abundance. The superior coverage of DIA-TPA can be attributed to its use of MS2 signals, whereas DDA-TPA uses MS1 signals for quantification. In addition, the DIA-TPA algorithm was able to accurately quantify protein isoforms by allocating the MS signals of shared peptides to individual protein isoforms. As such, DIA-TPA is suitable for quantifying proteins with multiple isoforms, such as DMEs and drug transporters. Furthermore, the quantifications of 23 major DMEs obtained from the DIA-TPA analysis were comparable to previously reported data obtained with labeling-based methods; approximately half of the measurements fell within the ranges of previously reported values, and most of the rest were within two-fold of the previously-reported mean values [16].
More recently, Wang et al. applied this DIA-TPA method to a global, absolute, quantitative proteomics analysis of 102 individual HLS9 and corresponding human liver microsomes (HLM) samples with a focus on DMEs and drug transporters [26]. The resulting protein expression profiles between HLM and HLS provide a reference for making sample choices for in vitro studies.
Labeling-based quantification
Labeling-based quantification is characterized with high accuracy and reliability. In 2016, Nakamura et al. evaluated the performance of a DIA labeling-based method in quantifying DMEs and transporters in HLM, human intestine microsomes (HIM), and human kidney microsomes (HKM) [17]. Prior to LC-MS/MS injections, they spiked the trypsin-digested peptides with stable isotope-labeled internal standard peptides and quantified targeted peptides according to the peak area ratios of peptides of interest to the internal standard peptides. This study quantified 27 DMEs and 54 transporters with the DIA method. Most values obtained by the DIA method were comparable to those obtained by targeted proteomics methods (MRM and PRM) with differences less than 50%. This study thus demonstrated the large coverage and high reliability of the DIA method when used with isotope-labeled internal standards. More recently, another study used isobaric tandem mass tags (TMT) to label peptide samples and performed both targeted and DIA proteomics analysis on the same batch of human liver tissue samples [18]. They adopted triple-stage mass spectrometry (MS3) to address the ratio distortions that result from TMT labeling. The quantifications of hepatic uptake transporters obtained with the DIA method were well correlated to those from the targeted method [18].
The reported DIA quantification data for human hepatic DMEs and transporters [16, 17, 26] were summarized and compared to the values obtained from targeted methods (i.e., MRM) [19, 27–32] (Figure 3A and 3B). All the DME proteomics studies were conducted with HLM samples. The DME protein concentrations determined by the DIA methods were generally agreeable to the values obtained from the targeted methods. Absolute DME proteins levels measured in human liver S9 fractions were not included in the comparison [33]. For hepatic drug transporters, the DIA studies [17, 26] used HLM while the MRM studies adopted liver membrane proteins [19, 32]. The concentrations of several transporters reported by a label-free DIA study [26] were lower than those from a labeling DIA study [17] and two MRM studies [19, 32]. The observed differences could be attributed to different sample resources, sample preparation methods, and instrumentations. A study revealed that most transporters’ concentrations in cellular membrane fractions were considerably higher than those in tissue lysates [34]. As such, it is pertinent to use same samples with identical preparation procedures to compare the performance of DIA and targeted proteomics methods. Of note, among the three absolute DIA quantification studies included in Figure 3 [16, 17, 26], one investigation compared the quantitative performance of DIA with MRM and PRM with identical samples [17]. The results showed that the DIA data highly correlated with those from the MRM and PRM studies, with the determination coefficient (r2) of 0.936 and 0.898, respectively [17].
Figure 3.
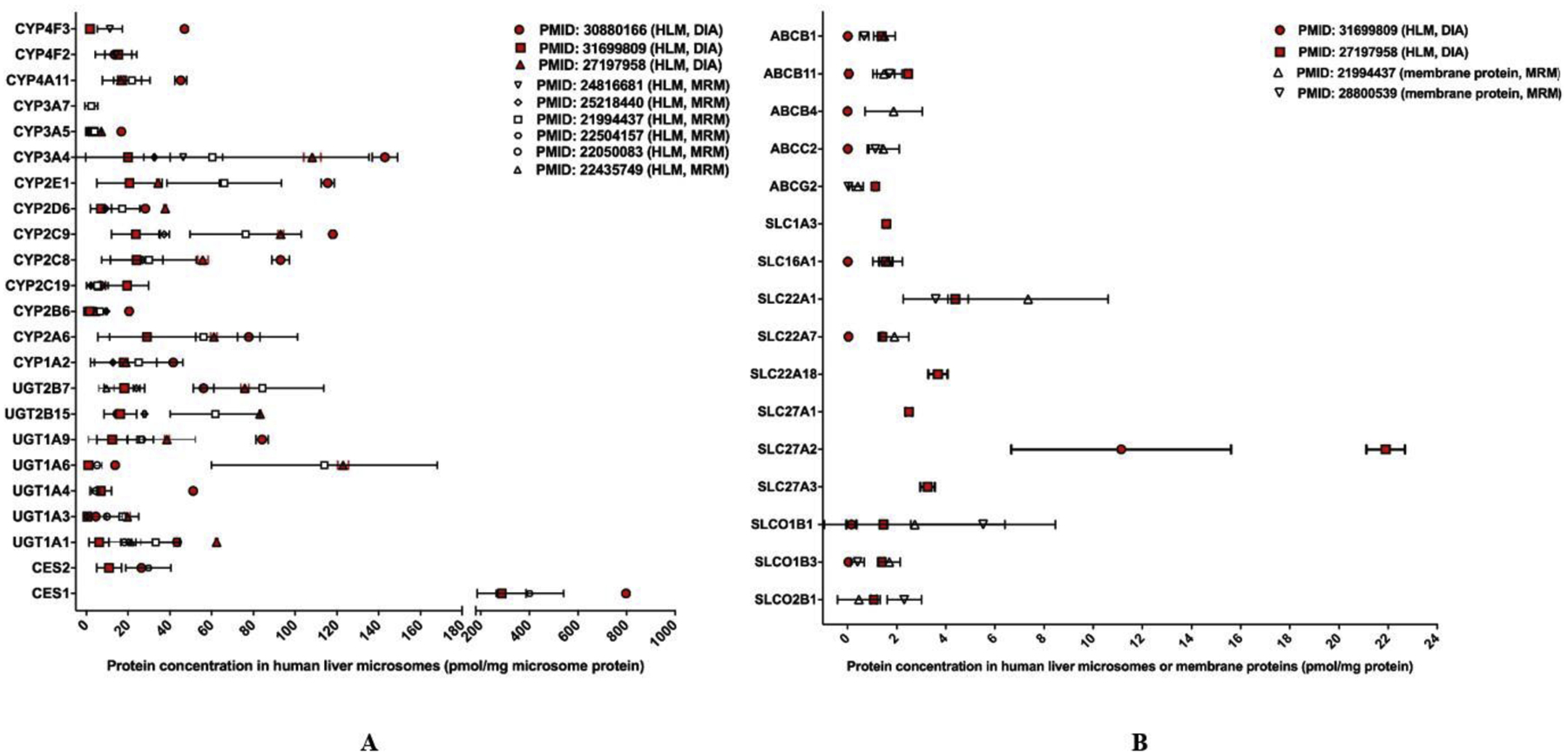
Absolute quantification of major clinically-relevant hepatic DMEs (A) and transporters (B). Protein expression levels are presented as mean with standard deviation (SD). For human hepatic DMEs, all studies used HLM samples. For transporters, the DIA studies used HLM samples while the targeted proteomics studies used membrane protein preparations. Values determined by DIA methods are highlighted in red whereas values determined by targeted assays are in black and white. For transporters, the different sample preparation methods (i.e., HLM vs. membrane proteins) should be taken into consideration when comparing results obtained from DIA and targeted proteomics methods.
Summary
Targeted proteomics is admittedly the most commonly-used approach for protein quantification in the study of DMEs and transporters [2]. Furthermore, targeted proteomics has been recognized as the gold standard for MS-based protein quantification [4]. However, the inherently restricted number of proteins that can be detected in a single assay and the labor intensiveness of assay development limit the application of targeted proteomics in large-scale proteomics studies. DIA methods offer an alternative due to their superior performance in terms of protein coverage, ease-of-development, and reproducibility.
In the past, DIA methods were mainly used for discovery studies; [35] however, recent advances have offered several strategies for greatly improving the quantitative performance of DIA methods, especially in sensitivity and selectivity [35]. Multiple studies exemplify the combination of DIA with optimized sample preparation strategies to improve assay performance for both label-free [15, 16, 24, 26] and labeling studies [17, 18, 23]. The DIA-TPA approach developed in a recent study [16] demonstrated that an innovative computational approach could improve quantitative capability over the typical DIA proteomics method. Recent significant advance in instrumentation also facilitates the improvement of DIA quantifications. One such emerging advance is the trapped ion mobility spectrometer (TIMS), which traps ions at different positions in an ion tunnel by counteracting the gas stream from the source with the force of the electric field [36]. These trapped ions are then consecutively released by lowering the electrical potential, with the release of ions over time being a function of their mobility. The introduction of TIMS into the quadrupole-TOF system allows for adding the ion mobility separation to chromatographic and mass separation in a strategy named parallel accumulation-serial fragmentation (PASEF), which synchronizes the release of precursor ions with the quadrupole selection for fragmentation [37]. The integration of PASEF with DIA (dia-PASEF) brings ion mobility separation into the DIA method and subsequently increases the selectivity for precursor identification and reduces analysis complexity [38]. Furthermore, dia-PASEF displayed high reproducibility and reliable accuracy when dealing with highly complex biological samples.
In sum, DIA methods allow for global/targeted and relative/absolute protein quantification in multiplex biological samples and have demonstrated performance comparable to conventional targeted proteomics methods. For the future, we expect that DIA proteomics will be increasingly utilized as a versatile tool for DME and transporter proteomics study with the continued improvement of both hardware and software.
Acknowledgements
This work was partially supported by the National Institutes of Health National Heart, Lung, and Blood Institute [R01 HL126969, Hao-Jie Zhu], the Eunice Kennedy Shriver National Institute of Child Health and Human Development [R01 HD093612, John S. Markowitz and Hao-Jie Zhu].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interest
The Authors declare that there is no conflict of interest regarding the submission of the manuscript entitled “Application of data-independent acquisition (DIA) to proteomic analysis of drug-metabolizing enzymes and transporters”
References
- [1].Jain KK. Role of proteomics in the development of personalized medicine. Advances in protein chemistry and structural biology. Elsevier; 2016, p. 41–52. [DOI] [PubMed] [Google Scholar]
- [2].Li J, Zhu H-J. Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS)-Based Proteomics of Drug-Metabolizing Enzymes and Transporters. Molecules 2020;25(11):2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Shi T, Su D, Liu T, Tang K, Camp II DG, Qian W-J, et al. Advancing the sensitivity of selected reaction monitoring-based targeted quantitative proteomics. Proteomics 2012;12(8):1074–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Orlando E, Aebersold R. On the contribution of mass spectrometry-based platforms to the field of personalized oncology. TrAC Trends in Analytical Chemistry 2019;110:129–42. [Google Scholar]
- [5].Bourmaud A, Gallien S, Domon B. Parallel reaction monitoring using quadrupole-Orbitrap mass spectrometer: Principle and applications. Proteomics 2016;16(15–16):2146–59. [DOI] [PubMed] [Google Scholar]
- [6].Schilling B, MacLean B, Held JM, Sahu AK, Rardin MJ, Sorensen DJ, et al. Multiplexed, Scheduled, High-Resolution Parallel Reaction Monitoring on a Full Scan QqTOF Instrument with Integrated Data-Dependent and Targeted Mass Spectrometric Workflows. Analytical chemistry 2015;87(20):10222–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Vidova V, Spacil Z. A review on mass spectrometry-based quantitative proteomics: Targeted and data independent acquisition. Analytica Chimica Acta 2017;964:7–23. [DOI] [PubMed] [Google Scholar]
- [8].Shi T, Song E, Nie S, Rodland KD, Liu T, Qian W-J, et al. Advances in targeted proteomics and applications to biomedical research. Proteomics 2016;16(15–16):2160–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gillet LC, Leitner A, Aebersold R. Mass Spectrometry Applied to Bottom-Up Proteomics: Entering the High-Throughput Era for Hypothesis Testing. 2016;9(1):449–72. [DOI] [PubMed] [Google Scholar]
- [10].Gillet LC, Navarro P, Tate S, Röst H, Selevsek N, Reiter L, et al. Targeted Data Extraction of the MS/MS Spectra Generated by Data-independent Acquisition: A New Concept for Consistent and Accurate Proteome Analysis. 2012;11(6):O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Huang Q, Yang L, Luo J, Guo L, Wang Z, Yang X, et al. SWATH enables precise label-free quantification on proteome scale. Proteomics 2015;15(7):1215–23. [DOI] [PubMed] [Google Scholar]
- [12].Ludwig C, Gillet L, Rosenberger G, Amon S, Collins BC, Aebersold R. Data-independent acquisition-based SWATH-MS for quantitative proteomics: a tutorial. Molecular systems biology 2018;14(8):e8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Suomi T, Elo LL. Enhanced differential expression statistics for data-independent acquisition proteomics. Scientific reports 2017;7(1):5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Tsou C-C, Avtonomov D, Larsen B, Tucholska M, Choi H, Gingras A-C, et al. DIA-Umpire: comprehensive computational framework for data-independent acquisition proteomics. Nature Methods 2015;12(3):258–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Shi J, Wang X, Lyu L, Jiang H, Zhu H-J. Comparison of protein expression between human livers and the hepatic cell lines HepG2, Hep3B, and Huh7 using SWATH and MRM-HR proteomics: Focusing on drug-metabolizing enzymes. Drug metabolism and pharmacokinetics 2018;33(2):133–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].He B, Shi J, Wang X, Jiang H, Zhu H-J. Label-free absolute protein quantification with data-independent acquisition. Journal of Proteomics 2019;200:51–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Nakamura K, Hirayama-Kurogi M, Ito S, Kuno T, Yoneyama T, Obuchi W, et al. Large-scale multiplex absolute protein quantification of drug-metabolizing enzymes and transporters in human intestine, liver, and kidney microsomes by SWATH-MS: Comparison with MRM/SRM and HR-MRM/PRM. Proteomics 2016;16(15–16):2106–17. [DOI] [PubMed] [Google Scholar]
- [18].Vildhede A, Nguyen C, Erickson BK, Kunz RC, Jones R, Kimoto E, et al. Comparison of Proteomic Quantification Approaches for Hepatic Drug Transporters: Multiplexed Global Quantitation Correlates with Targeted Proteomic Quantitation. Drug Metabolism and Disposition 2018;46(5):692–6. [DOI] [PubMed] [Google Scholar]
- [19].Ohtsuki S, Schaefer O, Kawakami H, Inoue T, Liehner S, Saito A, et al. Simultaneous Absolute Protein Quantification of Transporters, Cytochromes P450, and UDP-Glucuronosyltransferases as a Novel Approach for the Characterization of Individual Human Liver: Comparison with mRNA Levels and Activities. 2012;40(1):83–92. [DOI] [PubMed] [Google Scholar]
- [20].Deo AK, Prasad B, Balogh L, Lai Y, Unadkat JD. Interindividual Variability in Hepatic Expression of the Multidrug Resistance-Associated Protein 2 (MRP2/ABCC2): Quantification by Liquid Chromatography/Tandem Mass Spectrometry. Drug Metabolism and Disposition 2012;40(5):852–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Neilson KA, Ali NA, Muralidharan S, Mirzaei M, Mariani M, Assadourian G, et al. Less label, more free: Approaches in label-free quantitative mass spectrometry. Proteomics 2011;11(4):535–53. [DOI] [PubMed] [Google Scholar]
- [22].Hanke S, Besir H, Oesterhelt D, Mann M. Absolute SILAC for Accurate Quantitation of Proteins in Complex Mixtures Down to the Attomole Level. Journal of Proteome Research 2008;7(3):1118–30. [DOI] [PubMed] [Google Scholar]
- [23].Kuno T, Hirayama-Kurogi M, Ito S, Ohtsuki S. Effect of Intestinal Flora on Protein Expression of Drug-Metabolizing Enzymes and Transporters in the Liver and Kidney of Germ-Free and Antibiotics-Treated Mice. Molecular pharmaceutics 2016;13(8):2691–701. [DOI] [PubMed] [Google Scholar]
- [24].Jamwal R, Barlock BJ, Adusumalli S, Ogasawara K, Simons BL, Akhlaghi F. Multiplex and Label-Free Relative Quantification Approach for Studying Protein Abundance of Drug Metabolizing Enzymes in Human Liver Microsomes Using SWATH-MS. Journal of Proteome Research 2017;16(11):4134–43. [DOI] [PubMed] [Google Scholar]
- [25].Wiśniewski JR. Chapter Four - Label-Free and Standard-Free Absolute Quantitative Proteomics Using the “Total Protein” and “Proteomic Ruler” Approaches. Methods in Enzymology. Academic Press; 2017, p. 49–60. [DOI] [PubMed] [Google Scholar]
- [26].Wang X, He B, Shi J, Li Q, Zhu H-JJDM, Disposition. Comparative Proteomics Analysis of Human Liver Microsomes and S9 Fractions. 2020;48(1):31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Michaels S, Wang MZJDM, Disposition. The revised human liver cytochrome P450” Pie”: absolute protein quantification of CYP4F and CYP3A enzymes using targeted quantitative proteomics. 2014:dmd. 114.058040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Gröer C, Busch D, Patrzyk M, Beyer K, Busemann A, Heidecke C, et al. Absolute protein quantification of clinically relevant cytochrome P450 enzymes and UDP-glucuronosyltransferases by mass spectrometry-based targeted proteomics. 2014;100:393–401. [DOI] [PubMed] [Google Scholar]
- [29].Sato Y, Miyashita A, Iwatsubo T, Usui TJDM, Disposition. Simultaneous Absolute Protein Quantification of Carboxylesterases 1 and 2 in Human Liver Tissue Fractions using Liquid Chromatography Tandem Mass Spectrometry. 2012:dmd. 112.045054. [DOI] [PubMed] [Google Scholar]
- [30].Harbourt DE, Fallon JK, Ito S, Baba T, Ritter JK, Glish GL, et al. Quantification of Human Uridine-Diphosphate Glucuronosyl Transferase 1A Isoforms in Liver, Intestine, and Kidney Using Nanobore Liquid Chromatography–Tandem Mass Spectrometry. Analytical Chemistry 2012;84(1):98–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Nagata M, Kawamura A, Miyashita A, Usui T. Protein quantification of UDP-glucuronosyltransferases 1A1 and 2B7 in human liver microsomes by LC-MS/MS and correlation with glucuronidation activities AU - Sato, Yuichiro. Xenobiotica 2012;42(9):823–9. [DOI] [PubMed] [Google Scholar]
- [32].Wang L, Rubadue KJ, Alberts J, Bedwell DW, Ruterbories KJ. Development of a rapid and sensitive multiple reaction monitoring proteomic approach for quantification of transporters in human liver tissue. Journal of chromatography B, Analytical technologies in the biomedical and life sciences 2017;1061–1062:356–63. [DOI] [PubMed] [Google Scholar]
- [33].Basit A, Neradugomma NK, Wolford C, Fan PW, Murray B, Takahashi RH, et al. Characterization of Differential Tissue Abundance of Major Non-CYP Enzymes in Human. Molecular pharmaceutics 2020. [DOI] [PubMed] [Google Scholar]
- [34].Wegler C, Gaugaz FZ, Andersson TB, Wiśniewski JR, Busch D, Gröer C, et al. Variability in Mass Spectrometry-based Quantification of Clinically Relevant Drug Transporters and Drug Metabolizing Enzymes. Molecular pharmaceutics 2017;14(9):3142–51. [DOI] [PubMed] [Google Scholar]
- [35].Pino LK, Rose J, O’Broin A, Shah S, Schilling B. Emerging mass spectrometry-based proteomics methodologies for novel biomedical applications. Biochemical Society Transactions 2020;48(5):1953–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ridgeway ME, Lubeck M, Jordens J, Mann M, Park MAJIJoMS. Trapped ion mobility spectrometry: A short review. 2018;425:22–35. [Google Scholar]
- [37].Meier F, Brunner A-D, Koch S, Koch H, Lubeck M, Krause M, et al. Online Parallel Accumulation–Serial Fragmentation (PASEF) with a Novel Trapped Ion Mobility Mass Spectrometer. Molecular & Cellular Proteomics 2018;17(12):2534–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Meier F, Brunner A-D, Frank M, Ha A, Bludau I, Voytik E, et al. diaPASEF: parallel accumulation–serial fragmentation combined with data-independent acquisition. Nature Methods 2020;17(12):1229–36. [DOI] [PubMed] [Google Scholar]