

ABSTRACT
The vitamin D hormone, 1,25dihydroxyvitamin D3 (1,25(OH)2D3), and related compounds derived from vitamin D3 or lumisterol as a result of metabolism via the enzyme CYP11A1, have been shown, when applied 24 hours before or immediately after UV irradiation, to protect human skin cells and skin from DNA damage due to UV exposure, by reducing both cyclobutane pyrimidine dimers (CPD) and oxidative damage in the form of 8‐oxo‐7,8‐dihydro‐2′‐deoxyguanosine (8‐OHdG). We now report that knockdown of either the vitamin D receptor or the endoplasmic reticulum protein ERp57 by small, interfering RNA (siRNA) abolished the reductions in UV‐induced DNA damage with 20‐hydroxyvitamin D3 or 24‐hydroxylumisterol3, as previously shown for 1,25(OH)2D3. Treatment with 1,25(OH)2D3 reduced oxygen consumption rates in UV‐exposed and sham‐exposed human keratinocytes and reduced phosphorylation of cyclic AMP response binding element protein (CREB). Both these actions have been shown to inhibit skin carcinogenesis after chronic UV exposure, consistent with the anticarcinogenic activity of 1,25(OH)2D3. The requirement for a vitamin D receptor for the photoprotective actions of 1,25(OH)2D3 and of naturally occurring CYP11A1‐derived vitamin D–related compounds may explain why mice lacking the vitamin D receptor in skin are more susceptible to UV‐induced skin cancers, whereas mice lacking the 1α‐hydroxylase and thus unable to make 1,25(OH)2D3 are not more susceptible. © 2021 The Authors. JBMR Plus published by Wiley Periodicals LLC on behalf of American Society for Bone and Mineral Research.
Keywords: 1α,25‐DIHYDROXYVITAMIN D3 ; LUMISTEROL; UV‐INDUCED DNA DAMAGE; VITAMIN D RECEPTOR; ERp57; CREB PHOSPHORYLATION; OXIDATIVE PHOSPHORYLATION
Introduction
Vitamin D3 is primarily made in skin through the absorption of UVB photons by 7‐dehydrocholesterol, opening the B‐ring of the sterol to form pre‐vitamin D3. At body temperature, pre‐vitamin D3 isomerizes to vitamin D3.( 1 ) However, the same sunlight exposure that produces vitamin D3 also causes several types of DNA damage in skin cells.( 2 , 3 ) Some DNA bases directly absorb photons. Upon UV absorbance, adjacent pyrimidine bases of the same DNA strand form dimeric photolesions.( 4 , 5 ) The most prevalent UV‐induced lesions are the cis‐syn cyclobutane pyrimidine dimers (CPDs), mostly formed between the 5–6 bonds of adjacent thymine and cytosine pyrimidines.( 6 ) The commonest forms of CPD are thymine dimers, which are present in numbers proportional to total CPD of all types.( 7 ) Although thymine dimers (even if not properly repaired) are not in theory mutagenic, they have been shown to cause mutations in practice.( 8 , 9 ) Thymine‐cytosine dimers and cytosine‐cytosine dimers are highly mutagenic if not correctly repaired by nucleotide excision repair.( 9 )
Indirect DNA damage or oxidative damage to purine bases has been shown to contribute to mutagenesis, cancer, aging, and other pathological conditions.( 10 ) The main endogenous agents that cause this damage are free radicals such as reactive oxygen species (ROS)( 11 ) and reactive nitrogen species (RNS).( 12 ) Oxidative DNA damage, with free radicals targeting guanine, produces the main photolesion, 8‐oxo‐7,8‐dihydro‐2′‐deoxyguanosine (8‐oxoguanine/8‐OHdG).( 13 ) 8‐OHdG is used as a biomarker for DNA damage by oxidative stress. Oxidative stress may cause noncanonical base pairing and incorrect pairing by DNA polymerase, leading to DNA damage,( 8 , 9 ) mutations,( 9 , 14 ) and contributing to photocarcinogenesis.( 15 )
Pre‐vitamin D3 and vitamin D3 are not the only vitamin D compounds made in skin. Vitamin D3 is metabolized in skin cells through 25‐hydroxyvitamin D3 (25(OH)D3) to the active hormone, 1,25‐dihydroxyvitamin D3 (1,25(OH)2D3).( 16 , 17 ) Vitamin D3 in skin can also be metabolized by the enzyme CYP11A1,( 18 , 19 ) which increases in expression after exposure to UV,( 20 ) to produce 20‐hydroxyvitamin D3 (20(OH)D3) and other compounds (Fig. 1).( 21 , 22 , 23 ) Furthermore, continued absorption of UV by pre‐vitamin D3 produces so called, “overirradiation products,” including the major product, lumisterol.( 24 ) Lumisterol can also be metabolized by CYP11A1 to produce several hydroxylated metabolites including 24‐hydroxylumisterol3 (24(OH)L3) (Fig. 1).( 25 , 26 )
Fig. 1.

Production and metabolism of vitamin D3 and related compounds in the skin. Pre‐vitamin D3 is synthesized when the B‐ring of 7‐dehydrocholesterol is broken on the absorption of a photon of UVB. At body temperature, pre‐vitamin D3 is converted to vitamin D3. Continued absorption of UV photons by pre‐vitamin D or vitamin D3 results in conversion to overirradiation products such as lumisterol3, tachysterol3, or suprasterols, or 5,6‐transvitamin D3.Vitamin D3 is converted in the skin to 25‐hydroxyvitamin D3 by CYP2R1 (or possibly CYP27A1) and then to 1,25‐dihydroxyvitamin D3 by CYP27B1. The cholesterol side‐chain cleavage enzyme CYP11A1 is also expressed in the skin and upregulated by UV. It can convert vitamin D3 into 20‐hydroxyvitamin D3 and at least 10 other products. CYP11A1 can also convert lumisterol3 into 24‐hydroxylumisterol3 and several other lumisterol derivatives. (Adapted from Tuckey( 25 )).
Sunlight produces both vitamin D3 and DNA damage. Although DNA damage occurs relatively quickly, from immediately to a few hours,( 27 , 28 ) production of vitamin D3 takes many hours.( 1 ) This gives a window of opportunity to test whether vitamin D compounds added topically, either before UV exposure, or in most cases immediately after UV exposure, could reduce UV‐induced DNA damage. Many studies, including studies carried out with Tony Norman (deceased; Anthony W. Norman, PhD, Department of Biochemistry, University of California, Riverside, Riverside, California, USA) reported that the vitamin D hormone, 1,25(OH)2D3, reduced UV‐induced DNA damage in skin cells, skin explants and in human subjects.( 28 , 29 , 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 ) Vitamin‐D like compounds such as 20(OH)D3 and the synthetic compound 1,25‐dihydroxylumisterol3, synthesized by Bill Okamura in Tony Norman's group,( 39 ) also have been shown to reduce UV‐induced CPD in separate studies.( 33 , 34 , 38 , 40 , 41 , 42 , 43 ) It seems likely then, that vitamin D compounds, synthesized in skin as a result of sunlight exposure, contribute to protection of DNA in skin cells from the next exposure to UV. This skin adaptation is similar in timing to the development of a thickened stratum corneum,( 44 ) the outer layer of skin that attenuates UV penetration,( 45 ) and pigmentation (tan), both of which contribute to reduced UV damage and photocarcinogenesis.( 46 , 47 )
Indeed, in mice, topical application of 1,25(OH)2D3 immediately after UV exposure over 10 weeks, significantly reduced skin tumor development over the subsequent 30 weeks.( 34 ) 1,25(OH)2D3 normally requires the vitamin D receptor (VDR), a member of the steroid hormone receptor superfamily, to effect changes in cellular function.( 48 , 49 ) In support of the hypothesis that 1,25(OH)2D3 contributes to skin adaptation to sunlight exposure, mice with a nonfunctional VDR developed more skin tumors than wild‐type (WT) mice after UV exposure( 50 ) or after oral administration of a chemical carcinogen.( 51 ) Somewhat surprisingly, mice with ablation of CYP27B1, the 1α‐hydroxylase that converts 25‐hydroxyvitamin D to 1,25(OH)2D3, are not more susceptible to UV‐induced skin tumors.( 52 ) This raises the likelihood that other vitamin D compounds made in skin (Fig. 1) may also contribute to protection of DNA from UV exposure. There is some evidence to support this proposal,( 38 , 41 , 42 , 43 , 53 ) but more is needed.
1,25(OH)2D3 binds to the VDR, which acts principally as a modifier of transcription in the nucleus.( 54 ) Tony Norman's group was the first to demonstrate a nonclassical pathway of 1,25(OH)2D3 response: a rapid intestinal calcium uptake response to 1,25(OH)2D3 was shown to be via plasma membrane‐associated VDR and/or endoplasmic reticulum protein, ERp57, a member of the protein disulphide isomerase family (PDIA3).( 55 , 56 , 57 , 58 , 59 ) The receptor has been called 1,25D3–membrane associated rapid response steroid binding (1,25D3‐MARRS).( 60 , 61 ) This nonclassical pathway was also shown to be present in bone cells.( 59 , 62 , 63 )
In collaboration with Tony Norman, we published evidence that, at least in part, the protective effect of 1,25(OH)2D3 to reduce UV‐induced CPD depends on both VDR and ERp57.( 64 ) These earlier studies in skin cells from patients with Hereditary Vitamin D Resistant Rickets type 1, due to anomalies in the VDR,( 65 , 66 , 67 ) showed that 1,25(OH)2D3 did not reduce CPD in fibroblasts from a patient with an early stop codon in the VDR gene, but was effective in reducing CPD in fibroblasts from patients with mutations in either the DNA binding domain or the ligand binding domain.( 64 ) Treatment of skin fibroblasts with a neutralizing antibody (which would not be expected to pass across the cell membrane) to ERp57 or small, interfering RNA (siRNA) to ERp57 also abolished the protective effect of 1,25(OH)2D3 on CPD after UV.( 64 ) Immunoprecipitation experiments on non‐nuclear fractions of these skin cells revealed a VDR‐ERp57 complex. Furthermore, the use of 4,40‐diisothiocyanatostilbene‐2,20‐disulfonic acid (DIDS), a chloride channel blocker shown to prevent 1,25(OH)2D3–induced chloride currents in osteoblasts,( 68 ) also blocked the reduction in post‐UV CPD by 1,25(OH)2D3. ( 37 )
But there are many unknowns. Although 20(OH)D3 and its metabolites derived from vitamin D3 via CYP11A1 reduce both CPD and oxidative DNA damage in the form of 8‐OHdG,( 38 , 42 , 69 ) there is a shortage of similar information on the CYP11A1 derivatives of pre‐vitamin D, such as 24(OH)L3. Recent studies, however, have demonstrated a photoprotective role for CYP11A1‐derived hydroxylumisterol compounds in human keratinocytes.( 69 , 70 ) In addition, although a nonfunctional VDR or knockdown of ERp57 reduced UV‐induced CPD in skin fibroblasts, it is not known whether knockdown of VDR or ERp57 abolishes the protection against UV‐induced CPD by 1,25(OH)2D3 in keratinocytes, the main epidermal cell type. Nor is there data on whether VDR or ERp57 are also required for protection by 1,25(OH)2D3 against oxidative DNA damage or whether any protection against DNA damage by CYP11A1‐derived metabolites of vitamin D3, like 20(OH)D3, or of pre‐vitamin D3, like 24(OH)L3, requires VDR and/or ERp57. Furthermore, evidence has accumulated for vitamin D hydroxyderivatives acting on alternative nuclear receptors including retinoic acid orphan receptor (ROR)α and γ,( 71 ) aryl hydrocarbon receptor (AhR),( 72 ) and liver X receptors (LXR).( 73 ) These questions and the possibility that 1,25(OH)2D3 affects some keratinocyte functions known to play a role in photocarcinogenesis were tested in the current study.
Subjects and Methods
Vitamin D compounds
24(OH)L3 and 20(OH)D3 were enzymatically synthesized using recombinant CYP11A1, procedures as described.( 21 , 26 ) Other compounds were purchased: 1,25(OH)2D3 (Sapphire Bioscience, Sydney, Australia). All compounds were dissolved in spectroscopic grade ethanol (Merck, Darmstadt, Germany) and stored under argon gas at −80°C. The absorbance of these compounds was determined by Nanodrop ND‐1000 Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA), and concentration was calculated using the Beer‐Lambert law. The final concentration of ethanol vehicle used in experiments was 0.1% vol/vol.
Solar simulated irradiation
Cells were exposed to solar simulated UV radiation (ssUV), provided by a Oriel Sol1A™ 94042A 450W solar simulator (Newport Corporation, Irvine, CA, USA) with an inbuilt attenuation filter to eliminate UVC. UVA and UVB output was calibrated using a OL756 spectroradiometer (Optronic Laboratories, Gooch & Housego, Orlando, FL, USA) as described.( 37 ) The UV dose was determined from previous studies to cause reasonable levels of DNA damage in human keratinocytes, but was not high enough to result in a large increases in apoptosis.( 36 , 74 ) Irradiation was carried out as described.( 37 ) All treatments with vitamin D compounds were added immediately after exposure to UV.
Keratinocyte culture
Legal parents or guardians gave written informed consent for collection of skin from circumcision under a protocol approved by the University of Sydney Human Ethics Committee (Reference number 2015/063). Keratinocytes, (passages 1–5) from at least two independent donors were used in all experiments. Keratinocytes were cultured from skin samples and used for studies as described.( 37 ) Keratinocytes at passage 2–5 were used for experiments. All experimental work was carried out in Sydney, NSW, Australia.
siRNA transfection
Opti‐MEM™ Reduced Serum Medium powder (22600134) was from Thermo Fisher Scientific; siRNA transfection reagent, nondirected siRNA sequence (noncomplementary control RNA [siCTRL RNA]), siRNA targeted to VDR (siVDR), and siRNA targeted to ERp57 (siERp57) were all from Santa Cruz Biotechnologies (Dallas, TX, USA). Cell culture media was replaced with 100 μL Opti‐MEM™ serum‐free media (Thermo Fisher Scientific) and incubated at 37°C for 5 to 10 minutes. For VDR, ERp57 knockdown, cells were transfected with 50mM siVDR, siERp57, or siCTRL RNA diluted in siRNA transfection medium/Opti‐MEM™ serum‐free media (Santa Cruz Biotechnologies) in the presence of the recommended concentration of lipid‐based siRNA transfection reagent (Santa Cruz Biotechnologies) according to the manufacturer's instructions, and as described.( 64 , 75 )
Western blots
Western blots used protein lysates from keratinocytes seeded in six‐well plates at a density of 500,000 cells/well as described.( 76 ) Additional antibodies used for protein detection were anti‐ERp57 (mouse monoclonal; Santa Cruz Biotechnologies), anti‐VDR (mouse monoclonal; Santa Cruz Biotechnologies), anti‐phospho–cyclic AMP response binding element protein (CREB)‐Ser133 (mouse monoclonal; Cell Signaling Technology, Danvers, MA, USA), and anti‐tubulin at 1 μg/mL (mouse monoclonal; Santa Cruz Biotechnologies).
Immunochemistry
Cells were incubated for 3 hours at 37°C, based on previous studies,( 34 , 37 ) followed by fixation with ice‐cold 100% methanol at −20°C for 5 minutes and washed three times with MiliQ water (Mili‐Q integral water purification system®; Millipore SAS, Molsheim, France). Cells were left to air‐dry overnight before staining for DNA damage in the form of thymine dimers as an index of CPDs( 7 ) or 8‐OHdG, as described.( 27 ) Briefly, cells were first treated with 1% H2O2 (vol/vol in PBS) for 5 minutes to block endogenous peroxidase activity, while covered in foil, followed by three washes with MiliQ water. Antigen retrieval involved nuclear DNA denaturation with 70mM NaOH diluted in 70% ethanol, followed by aspiration and proteolytic digestion with Proteinase K (Final concentration: 1 μg/mL in 0.1mM CaCl2) at room temperature for 5 minutes for CPD or 10 minutes at 37°C for 8‐OHdG. The wells were gently washed with MiliQ water twice. Nonspecific staining was blocked with 50% horse serum (vol/vol in PBS) (Sigma‐Aldrich, St. Louis, MO, USA). Immunohistochemistry was performed using the anti‐thymine dimer antibody (mouse monoclonal clone H3; Sigma‐Aldrich) at 5 μg/mL or the 8‐OHdG antibody (mouse monoclonal lgG2b, clone 15A3; Santa Cruz Biotechnologies) at 2 μg/mL overnight at 4°C, followed by incubation at room temperature with goat anti‐mouse immunoglobulin G (IgG) F(ab′)2 secondary antibody, biotin conjugate at 1:500 (Thermo Fisher Scientific) for 20 minutes at room temperature, and horseradish peroxidase (HRP)‐Streptavidin conjugate at 1:150 (Invitrogen, Carlsbad, CA, USA) for 15 minutes at room temperature. Isotype control was performed with mouse IgG instead of primary antibody. All steps were separated by washing steps with Tween‐PBS (PBST) for three times. HRP substrate diaminobenzidine (DAB) (Enhanced Liquid substrate System for Immunohistochemistry; Sigma‐Aldrich) was applied for 5 minutes to visualize the staining of DNA damage. Coverslips were rinsed with MiliQ water and mounted with Entellin (Merck) onto glass slides after being air‐dried.
Mouse monoclonal IgG1 ERp57 antibody and mouse monoclonal IgG2a VDR antibody (both from Santa Cruz Biotechnologies) were used for immunohistochemistry of the VDR and ERp57. Cells were fixed with 10% (vol/vol) formaldehyde in PBS for 5 minutes at room temperature and permeabilized with 0.1% (vol/vol) Triton‐X in PBS for 3 minutes at room temperature. Endogenous peroxidase activity was blocked by 6% (vol/vol) H2O2 in PBS for 10 minutes. Nonspecific antibody binding was inhibited by 10% (vol/vol) horse serum (blocker) in PBS for 1 hour. VDR and ERp57 antibodies in 10% horse serum at 2.5 μg/mL concentration were incubated with the cells overnight at 4°C in blocker. Isotype control antibodies were used under similar experimental concentrations and conditions. Cells were rinsed with PBS five times and incubated with anti‐mouse IgG HRP‐linked antibody in blocker for 1 hour at room temperature. Cells were rinsed with PBS, incubated with DAB and rinsed with Mili‐Q water and mounted as described in the previous paragraph for analysis.
Brightfield images were taken on the Stereo Investigator Scope (MBF Bioscience, Williston, VT, USA) or Zeiss‐Axioscan light microscope (Zeiss, Oberkochen, Germany) at the Bosch Advanced Microscopy Facility at The University of Sydney. The image was thresholded in ImageJ imaging software (NIH, Bethesda, MD, USA; https://imagej.nih.gov/ij/) against the isotype control, which showed low staining as reported.( 34 ) Thus, the threshold was set based on the non‐irradiated sample (SHAM) to pick up minimum mean gray value. Mean gray value is the average gray value within the selected area, calculated as the sum of the gray values of all the pixels divided by the number of pixels. The set threshold was applied to all the images. The average of the mean gray value for each coverslip was obtained from 10 fixed‐area regions, where cells were well attached, as reported.( 36 )
These immunohistochemical methods for determining CPD and 8‐OHdG after UV have been validated with COMET assays (otherwise known as single‐cell gel electrophoresis), which use specific endonucleases to detect CPD or 8‐OHdG followed by single cell electrophoresis to detect DNA strand breaks.( 27 , 37 )
Seahorse energetics
The cellular oxygen consumption rates (OCR) were determined in an XF96 Extracellular Flux Analyzer (Seahorse Bioscience, Agilent, Santa Clara, CA, USA) immediately following irradiation or sham irradiation of cells. 1,25(OH)2D3 at 10nM (or vehicle) was injected immediately after irradiation (earliest possible); n = 22–24 per treatment. Data was analyzed using Wave 2.3 (Seahorse Bioscience) and Prism (GraphPad Software, Inc., La Jolla, CA, USA).( 36 )
ROS detection
ROS levels were measured using the ROS‐Glo™ H2O2 assay (Promega, San Luis Obispo, CA, USA) as described.( 36 )
Tunicamycin studies
Tunicamycin (Sigma‐Aldrich, St. Louis, MO, USA), an N‐linked glycosylation inhibitor, was added into the medium at 1 μg/mL, based on previous optimization experiments, and incubated with cells for 48 hours. This was followed by ssUV irradiation and processing for CPD or 8‐OHdG at 3 hours, as described above in Immunohistochemistry.
Statistical analysis
Keratinocyte experiments were performed in triplicate or as otherwise indicated and experiments were repeated at least twice with different tissue donors. Data were graphed and analyzed with use of GraphPad Prism software. Unless stated otherwise, results are presented as mean + standard error of the mean (SEM) and significance between treatment groups was analyzed with one‐way ANOVA with Tukey posttest.( 36 )
Results
Like 1,25(OH)2D3 and 20(OH)D3,( 38 , 41 ) 24(OH)L3 reduced UV‐induced CPD and 8‐OHdG in the nuclei of human primary keratinocytes in a concentration‐dependent manner (Fig. 2A‐D ).
Fig. 2.

Concentration‐dependent reduction in CPD or 8‐OHdG by 24(OH)L3 or 1,25(OH)2D3 in human primary keratinocytes. Representative photomicrographs of immunohistochemical staining of UV‐induced CPD (A) or 8‐OHdG (C), 3 hours after treatment with vehicle [0.1% (vol/vol) ethanol], for five concentrations of 1,25(OH)2D3 or 24(OH)L3. Dark brown staining, as shown by arrows, indicates the presence of CPD‐positive nuclei (scale bar = 100 μm). (B) Image analysis of IHC images presented as CPD‐positive nuclei (B) or 8‐OHdG–positive nuclei (D) as a percentage of total area. Solid gray bars show 1,25(OH)2D3 whereas stippled bars show 24(OH)L3. Results are from a single experiment performed in triplicate, representative of three separate experiments with similar results, mean + SEM; **p < 0.01, ***p < 0.001, ****p < 0.0001, when compared to UV vehicle. IHC = immunohistochemistry; NS = not significant between data sets.
To test whether the VDR or ERp57 was required for the reduction in CPD or oxidative DNA damage by 1,25(OH)2D3 or 24(OH)L3, cells were transfected with siRNA targeted to VDR mRNA (siVDR) or with siRNA to ERp57 (siERp57) or with a nondirected siRNA sequence (siCTRL). VDR or ERp57 knockdown by siRNA was verified by Western blot (Fig. 3A for VDR, or Fig. 3D for ERp57) and, as an additional control, by immunohistochemistry and image analysis for VDR or ERp57 (Fig. 3B, C, E, F ). Significant reductions in VDR protein expression were observed in cells transfected with siVDR compared to siCTRL (***p < 0.001) (Fig. 3B, C ), similar to Western blot results (Fig. 3A ), but there were no significant differences observed in ERp57 expression between either siVDR or siCTRL samples. Likewise, significant reductions in ERp57 protein expression were observed in cells transfected with siERp57 compared to siCTRL (***p < 0.001) (Fig. 3E, F ), similar to Western blot results (Fig. 3D ), but there were no significant differences observed in VDR expression between either siERp57 or siCTRL samples.
Fig. 3.
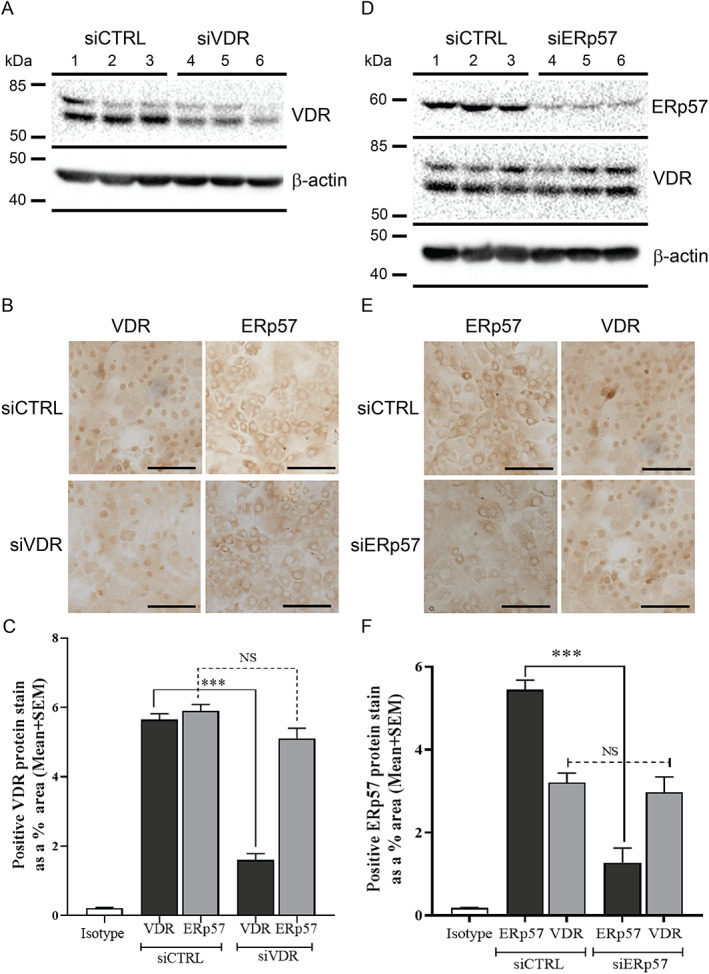
VDR or ERp57 knockdown by siRNA reduced VDR or ERp57 protein expression, respectively, but did not affect the expression of the non‐knocked‐down protein. Representative Western blots showing VDR (A) or ERp57 and VDR (D) in human primary keratinocytes 48 hours following transfection with siRNA targeted to VDR mRNA (siVDR) (A, lanes 4–6), siRNA targeted to ERp57 (siERp57) (D, lanes 4–6), or a nondirected siRNA sequence (siCTRL) (A, D, lanes 1–3). β‐actin is shown as the loading control. Representative photomicrographs of keratinocytes stained for VDR or ERp57 following transfection with siVDR (B) or siERp57 (E) or siCTRL. Image analysis of IHC images presented as positive VDR (C) or ERp57 (F) protein expression as a percentage of total cellular area 48 hours after transfection with siVDR, siERp57, or siCTRL. Results were from a single experiment performed in triplicate, representative of two separate experiments with similar results. Mean + SEM; ***p < 0.001, when compared to siCTRL vehicle. IHC = immunohistochemistry; NS = not significant between data sets.
Keratinocytes were exposed to the solar simulator 48 hours after siRNA transfection and immediately treated with 1,25(OH)2D3, 20(OH)D3, or 24(OH)L3 at 1 × 10−9M. After 3 hours, significant reductions in UV‐induced CPD (Fig. 4A ) or 8‐OHdG (Fig. 4B ) resulted from these treatments in cells treated with siCTRL (***p < 0.001). Reductions of either UV‐induced CPD or 8‐OHdG with any of these compounds were abolished in the presence of siVDR or siERp57 (Fig. 4A,B ).
Fig. 4.

Reductions in UV‐induced CPD or 8‐OHdG by 1,25(OH)2D3, 20(OH)D3, or 24(OH)L3 were abolished by either VDR or ERp57 knockdown in human primary keratinocytes. Image analysis of IHC images of CPD (A) or 8‐OHdG (B). Results are each from a single experiment performed in triplicate, representative of three separate experiments with similar results. Data are presented as positive nuclei as a percentage of total area. Mean + SEM; ****p < 0.0001, ***p < 0.001, when compared to UV vehicle. IHC = immunohistochemistry; NS = not significant between data sets.
In a separate set of experiments, keratinocytes were treated with tunicamycin, an N‐glycosylation inhibitor,( 77 ) at 1 μg/mL for 48 hours. The aim of those experiments was to abolish cell surface expression of the Calcium Sensing Receptor (CaSR),( 78 ) which was achieved, in order to test modulators of CaSR. In the experiments, the vitamin D hormone, 1,25(OH)2D3, was used as a positive control for photoprotection. Rather surprisingly, treatment of keratinocytes with tunicamycin abolished the 1,25(OH)2D3–induced reductions in UV‐induced CPD and 8‐OHdG (Fig. 5A,B ).
Fig. 5.

Tunicamycin pretreatment abolished the protective effect of 1,25(OH)2D3 on UV‐induced CPD and 8‐OHdG. Keratinocytes were treated with 1 μg/mL tunicamycin or vehicle (DMSO) for 48 hours followed by exposure to UV and treatment with vehicle or 1,25(OH)2D3 (1 × 10−9M). (A) CPD or (B) 8‐OHdG (y‐axis) are shown as optical density relative to SHAM. Open white bars show vehicle without UVR; solid black bars show vehicle with UVR; light gray bars show 1,25(OH)2D3 with UVR. Values are means + SEM. Results are from three independent experiments each in triplicate (n = 9). ****p < 0.0001, **p < 0.01, *p < 0.05, and n.s. (not significant compared with UV + Vehicle [non‐tunicamycin treated]) by mixed model analysis and Sidak's multiple comparison posttest. DMSO = dimethylsulfoxide; UVR = ultraviolet radiation.
UV exposure causes mitochondrial damage,( 36 , 79 ) which reduces oxygen consumption rates as measured by Seahorse analysis (Fig. 6A,B ). In contrast, compounds that reduce oxygen consumption rates without causing mitochondrial damage have been characterized as anti‐carcinogenic.( 80 ) Oxygen consumption rates (OCRs), a measure of mitochondrial oxidative phosphorylation, were lower in keratinocytes exposed to 1,25(OH)2D3 compared with vehicle (Fig. 6A ). At 90 minutes after UV, OCRs were significantly lower in keratinocytes treated with 1,25(OH)2D3 (from time shown in arrow), whether SHAM irradiated or exposed to UV (Fig. 6A,B ). Of interest is the observation that treatment with oligomycin, which inhibits oxidative phosphorylation, did not affect the reduction in CPD (Fig. 6C ) or 8‐OHdG (not shown) caused by the addition of 1,25(OH)2D3. Production of ROS was significantly increased by UV exposure and significantly reduced in either SHAM or UV‐exposed keratinocytes, in the presence of 1,25(OH)2D3 (Fig. 6D ).
Fig. 6.

1,25(OH)2D3 reduced oxygen consumption rate and ROS production under basal conditions and after UV exposure but oxidative phosphorylation was not needed for protection from DNA damage after UV. Cells subjected to Seahorse XF after UV irradiation (“UVR”) or non‐irradiation (“SHAM”) ± 1,25(OH)2D3 (“1,25D”) (mean ± SEM, n = 22). (A) OCR following 1,25D/vehicle at injection point (arrow). (B) Graph showing OCR percentage 90 minutes after injection. Bars indicate significant differences between groups: ****p < 0.0001, *p < 0.05. (C) Graph of CPD in keratinocytes measured by image analysis showing the effect of oligomycin treatment (n = 9). (D) ROS in arbitrary luminescence units ± UV and or 1,25D (n = 9); Bars indicate significant differences between groups: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. CPD = cyclobutane pyrimidine dimers; OCR = oxygen consumption rate; ROS = reactive oxygen species.
Increased phosphorylation of CREB is a recently identified marker of (photo)carcinogenic agents.( 81 ) As shown in Fig. 7A,B , CREB phosphorylation is increased by UV exposure in human keratinocytes, but decreased in UV‐exposed keratinocytes by 1,25(OH)2D3.
Fig. 7.

1,25(OH)2D3 reduced UV upregulated p‐CREB. Human keratinocytes cultured in 96‐well plates were irradiated with 400 mJ/cm2 UVB followed by treatment with vehicle or 10nM 1,25(OH)2D3 for 90 minutes. (A) Blot was incubated with anti‐p‐CREB antibody and tubulin, which is shown as a loading control. (B) Densitometry of triplicate blots for p‐CREB expression (mean + SD) was normalized to UV + Vehicle and shown as relative expression. ****p < 0.001 when compared with vehicle‐treated cells after UV. p‐CREB = phospho‐CREB.
Discussion
Several studies reported the photoprotective effect of 1,25(OH)2D3 in reducing UV‐induced DNA damage from in vitro, in vivo, and ex vivo studies (reviewed in McCarthy and colleagues,( 82 ) Mason and colleagues,( 83 ) and De Silva and colleagues.( 84 )) Compounds derived from vitamin D or the “overirradiation” product lumisterol have also been reported to reduce UV‐induced CPD in keratinocytes( 32 , 33 , 69 , 70 ) and in mice.( 34 , 38 ) The data presented here show for the first time that the CYP11A1 derivative of lumisterol, 24(OH)L3, reduces oxidative damage as well as CPD, provide a concentration response curve for 24(OH)L3 in comparison with 1,25(OH)2D3, and also show for the first time that this compound and 20(OH)D3, like 1,25(OH)2D3, reduces DNA damage in human keratinocytes when added immediately after UV exposure. Previous studies in collaboration with Tony Norman reported that both VDR and the endoplasmic reticulum protein ERp57 are required for reduction of UV‐induced CPD by1,25(OH)2D3,( 64 ) but the receptors through which 1,25(OH)2D3 reduces oxidative DNA damage and the receptors required by vitamin‐like compounds to reduce UV‐induced photo lesions were not known. The current study showed for the first time that both VDR and ERp57 are indeed required for reductions in oxidative DNA damage by 1,25(OH)2D3 and for the reduction in both types of UV‐induced DNA damage by the CYP11A1 metabolites, 20(OH)D3 and 24(OH)L3. The absolute requirement for VDR for the reduction in DNA damage, together with the apparent ability of these vitamin D–like compounds derived from vitamin D or lumisterol to reduce UV‐induced DNA damage, go some way toward explaining why knockdown of the VDR leads to increased susceptibility to photocarcinogenesis.( 50 ) In contrast, lack of the 1α‐hydroxylase does not apparently increase susceptibility to UV‐induced skin tumors.( 52 , 85 )
Agents such as 1,25‐dihydroxylumisterol3, 20(OH)D3, and other vitamin D compounds made in skin,( 18 , 19 , 21 , 38 , 86 ) which have been shown to reduce UV‐induced CPD,( 32 , 38 , 41 , 42 ) generally have reduced binding to the G‐pocket of the VDR in comparison to 1,25(OH)2D3. ( 58 , 87 ) They interact with the VDR, however, and the addition of OH at C1α modifies such interaction.( 88 , 89 , 90 ) In this context, it is worth noting that some VDR, but not necessarily a fully functional VDR, seems to be required to enable 1,25(OH)2D3 to reduce CPD after UV, because this protective effect was shown with VDR which had a mutated ligand‐binding domain or a mutation in the DNA binding domain.( 64 )
In that study, we demonstrated the involvement of the endoplasmic reticulum stress protein, ERp57 (also known as PDIA3 or MARRS( 91 )) in the photoprotective response to 1,25(OH)2D3. ( 64 ) As well as siRNA to the protein, a neutralizing antibody to ERp57, which would not enter the cell, abolished the response, supporting the idea of a membrane position for this protein for this function.( 92 ) It is likely that the VDR was also present in the cell membrane as shown by the Norman group.( 57 ) Co‐immunoprecipitation studies of non‐nuclear fractions of human primary fibroblasts showed that ERp57 co‐immunoprecipitated with VDR and VDR with ERp57.( 64 )
The global importance of ERp57 was demonstrated in ERp57−/− embryos where ERp57 deficiency was lethal at embryonic day 13.5, whereas embryos at embryonic day 12.5 were reported to be much smaller than the WT embryos.( 93 ) In the current study, in the presence of siERp57, which significantly reduced ERp57 expression, the reduction of UV‐induced CPD and 8‐OHdG by 1,25(OH)2D3, 20(OH)D3, and 24(OH)L3 was also significantly reduced. Further studies of the role of ERp57 in photoprotection will require the use of animals that have a conditional knockout of this protein in the epidermis.
The current study also showed that when keratinocytes had been incubated with tunicamycin, a known inhibitor of glycosylation,( 77 ) the protection by 1,25(OH)2D3 against UV‐induced CPD was abolished and that against 8‐OHdG was impaired. The explanation for this finding remains unclear and warrants further investigation. In that set of experiments, the tunicamycin was actually used to inhibit glycosylation of the calcium‐sensing receptor in order to prevent its movement to the cell membrane. The 1,25(OH)2D3 was included as a positive control for DNA damage reduction. Glycosylation is not apparently a feature of posttranslational modification of VDR, though slightly higher molecular weight bands have been reported in some tissues,( 94 ) which could be a result of posttranslational modification.( 95 ) These authors reported hyperglycemia‐induced enzyme mediated glycosylation (O‐linked‐N‐acetylglucosaminylation, or OGlcNAcylation) of VDR.( 95 ) Tunicamycin competes with N‐acetylglucosamine phosphotransferase to inhibit N‐glycosylation( 77 ) and would not be expected to inhibit OGlcNAcylation. A search of the literature revealed no evidence of glycosylation of ERp57. Nevertheless, it is possible that tunicamycin interferes with some chaperone protein important for translocating either VDR or ERp57 to the plasma membrane. Testing this proposal was beyond the scope of the current study, but the observations support, at least in part, the hypothesis that membrane associated VDR and/or ERp57 is important for some aspects of photoprotection. Tunicamycin also triggers endoplasmic reticulum stress( 96 ) and is thus is likely have an effect on a typical stress marker, such as ERp57.( 97 )
As noted in the second paragraph of the discussion, 1,25(OH)2D3 reduced UV‐induced CPD in fibroblasts with DNA binding domain mutations of the VDR.( 64 ) 1α,25(OH)2‐lumisterol3 has little ability to modulate gene transcription, but was shown to initiate nongenomic action with a similar potency to 1,25(OH)2D3. ( 58 , 98 ) 1α,25(OH)2‐lumisterol3, similar to 1,25(OH)2D3, reduced UV‐induced CPD in human primary fibroblasts and keratinocytes( 29 , 32 ) and reduced photocarcinogenesis in Skh:hr1 mice, though not to the extent seen with 1,25(OH)2D3. ( 34 ) All these data and the involvement of ERp57 support the proposal that photoprotection by 1,25(OH)2D3 and other vitamin D–like compounds at concentrations similar to 1,25(OH)2D3 signal, at least in part, by nongenomic pathways. The chloride channel inhibitor, DIDS, inhibited UV‐induced CPD reduction by 1,25(OH)2D3 and 20(OH)D3, further supporting involvement of a nonclassical vitamin D pathway in the reduction of UV‐induced DNA lesions by these vitamin‐D like compounds.( 37 , 99 )
Some of the actions of 1,25(OH)2D3 shown here that could contribute to protection from UV damage and subsequent photocarcinogenesis seem unlikely to be mediated by changes in gene transcription. These include suppression by 1,25(OH)2D3 of otherwise upregulated phosphorylation of protein kinase B (AKT) (acutely transforming retrovirus AKT8 in rodent T‐cell lymphoma) and extracellular regulated kinase‐1/2 (ERK1/2), after UV.( 36 ) 1,25(OH)2D3 also increased phosphatase and tensin homolog (PTEN) expression after UV.( 100 ) UVB normally downregulates PTEN in cells and whole skin in an AKT‐dependent and ERK1/2‐dependent manner.( 101 ) In turn, PTEN downregulation impairs global genomic nucleotide excision repair, which is needed to remove UV‐induced DNA lesions such as CPDs.( 102 ) The 1,25(OH)2D3–induced reduction in ROS as soon as 15 minutes after UV probably contributes to reduced oxidative DNA lesions and is likely to result in less damage to repair enzymes and thus to enhance repair.( 36 ) Likewise, after UV, the 1,25(OH)2D3–induced reduction in RNS and damaging nitrosylation of proteins, including DNA repair enzymes, is likely to facilitate DNA repair.( 37 )
In the current study, we observed a reduction in oxygen consumption rate, indicative of a reduction in oxidative phosphorylation, in both sham and UV‐exposed keratinocytes. We had previously reported that energy from glycolysis was required for DNA repair,( 36 ) and it is clear from the results presented here that even treatment with a well‐known inhibitor of oxidative phosphorylation, oligomycin, had no effect of the ability of 1,25(OH)2D3 to reduce DNA lesions. On the other hand, increased activity of the electron transport chain in later stages of oxidative phosphorylation has been shown to be critically important for induction of skin cancers in mice by UV.( 80 ) If this process is blocked, actinic tumors do not develop.( 80 ) Furthermore, agents that interrupt the mitochondrial respiratory chain, such as metformin,( 80 ) and it seems from the current data, 1,25(OH)2D3, reduce tumorigenesis.( 34 , 103 )
As observed in this study, increased phosphorylation of CREB at Serine133 occurs after UV exposure. This increased CREB phosphorylation seems to be important in the early stages of skin tumor development in mice.( 81 ) There is some evidence that the increased CREB phosphorylation after UV is at least, in part, downstream of increased ERK1/2 phosphorylation.( 104 ) So the reduction in ERK1/2 phosphorylation after UV with1,25(OH)2D3, reported earlier,( 36 ) may partly explain the reduced CREB‐phosphorylation after UV noted here. In turn, reduced phosphorylation of CREB‐Ser133 even when the reduction is relatively modest as seen here with 1,25(OH)2D3 and as described for the plant‐derived flavonol, kaempferol, are enough to significantly suppress UV‐induced skin tumor development.( 34 , 105 )
Conclusion
Although Professor Anthony W. Norman did not live to see the extension of his work in photoprotection, the accumulating data further supports the ideas he promulgated for mechanisms of action of the vitamin D hormone, including a role for membrane VDR and nonclassical effects on various cell signaling pathways, as well as the classical steroid hormone modulation of gene transcription.
Conflict of Interest
None.
Acknowledgments
The work was supported by grants from the NHMRC Australia APP1070688 to RSM, KMD, and RCT, and ARC Linkage grant LP100200680. Partial support of NIH grants 1R01AR073004‐01A1, R01AR071189‐01A1, and of a VA merit grant (no. 1I01BX004293‐01A1) to ATS is also acknowledged. Funding support from a Research Incentive Fund grant from Zayed University to MA is acknowledged.
Authors’ roles: RSM, MSR, RCT, ATS, KMD and WGMDS were involvedin the study design. WGMDS, JZRH, CY, WT‐O, BYM, FAI and MSR were involved in data acquisition. AJAH, RCT, MA, KDM and RSM provided resources. MSR, RSM, WGMDS, JZRH, CY, BYM, WT‐O and FAI were involved in data analysis. MSR, RSM, KMD and ATS were involved in interpretation. WGMDS, RSM and ATS drafted the manuscript, which was critically revised by all authors, who approved the final version and agree to be accountable for it. RSM, WGMDS and MSR accept responsibility for the integrity of the data and its analysis.
Contributor Information
Mark Stephen Rybchyn, Email: m.rybchyn@unsw.edu.au.
Rebecca Sara Mason, Email: rebecca.mason@sydney.edu.au.
References
- 1. Holick MF. The cutaneous photosynthesis of previtamin D3: a unique photoendocrine system. J Invest Dermatol. 1981;77(1):51‐58. [DOI] [PubMed] [Google Scholar]
- 2. Gordon‐Thomson C, Tongkao‐on W, Song EJ, Carter SE, Dixon KM, Mason RS. Protection from ultraviolet damage and photocarcinogenesis by vitamin D compounds. Adv Exp Med Biol. 2014;810:303‐328. [DOI] [PubMed] [Google Scholar]
- 3. Thompson JM, Fioletov VE, Marrett LD, Rosen CF, Weinstock MA. Vitamin D at the expense of skin cancer protection: is it worth the risk? J Invest Dermatol. 2016;136(10):2104‐2105. [DOI] [PubMed] [Google Scholar]
- 4. Beukers RT. Isolation and identification of the irradiation product of thymine. Biochim Biophys Acta. 1960;41:550‐551. [DOI] [PubMed] [Google Scholar]
- 5. Setlow RB. Cyclobutane‐type pyrimidine dimers in polynucleotides. Science. 1966;153(734):379‐386. [DOI] [PubMed] [Google Scholar]
- 6. Dumaz N, Drougard C, Sarasin A, Daya‐Grosjean L. Specific UV‐induced mutation spectrum in the p53 gene of skin tumors from DNA‐repair‐deficient xeroderma pigmentosum patients. Proc Natl Acad Sci U S A. 1993;90(22):10529‐10533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Douki T, Court M, Sauvaigo S, Odin F, Cadet J. Formation of the main UV‐induced thymine dimeric lesions within isolated and cellular DNA as measured by high performance liquid chromatography‐tandem mass spectrometry. J Biol Chem. 2000;275(16):11678‐11685. [DOI] [PubMed] [Google Scholar]
- 8. Ichihashi M, Ueda M, Budiyanto A, et al. UV‐induced skin damage. Toxicology. 2003;189(1‐2):21‐39. [DOI] [PubMed] [Google Scholar]
- 9. Lee JW, Ratnakumar K, Hung KF, Rokunohe D, Kawasumi M. Deciphering UV‐induced DNA damage responses to prevent and treat skin cancer. Photochem Photobiol. 2020;96(3):478‐499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ames BN, Gold LS. Endogenous mutagens and the causes of aging and cancer. Mutat Res. 1991;250(1‐2):3‐16. [DOI] [PubMed] [Google Scholar]
- 11. Kvam E, Tyrrell RM. Induction of oxidative DNA base damage in human skin cells by UV and near visible radiation. Carcinogenesis. 1997;18(12):2379‐2384. [DOI] [PubMed] [Google Scholar]
- 12. McAndrew J, Patel RP, Jo H, et al. The interplay of nitric oxide and peroxynitrite with signal transduction pathways: implications for disease. Semin Perinatol. 1997;21(5):351‐366. [DOI] [PubMed] [Google Scholar]
- 13. Douki T, Cadet J. Peroxynitrite mediated oxidation of purine bases of nucleosides and isolated DNA. Free Radic Res. 1996;24(5):369‐380. [DOI] [PubMed] [Google Scholar]
- 14. Robertson AB, Klungland A, Rognes T, Leiros I. DNA repair in mammalian cells: base excision repair: the long and short of it. Cell Mol Life Sci. 2009;66(6):981‐993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Matsumura Y, Ananthaswamy HN. Molecular mechanisms of photocarcinogenesis. Front Biosci. 2002;7:d765‐d783. [DOI] [PubMed] [Google Scholar]
- 16. Bikle DD, Nemanic MK, Whitney JO, Elias PW. Neonatal human foreskin keratinocytes produce 1,25‐dihydroxyvitamin D3. Biochemistry. 1986;25(7):1545‐1548. [DOI] [PubMed] [Google Scholar]
- 17. Lehmann B, Genehr T, Knuschke P, Pietzsch J, Meurer M. UVB‐induced conversion of 7‐dehydrocholesterol to 1alpha,25‐dihydroxyvitamin D3 in an in vitro human skin equivalent model. J Invest Dermatol. 2001;117(5):1179‐1185. [DOI] [PubMed] [Google Scholar]
- 18. Slominski RM, Raman C, Elmets C, Jetten AM, Slominski AT, Tuckey RC. The significance of CYP11A1 expression in skin physiology and pathology. Mol Cell Endocrinol. 2021;530:111238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Slominski AT, Kim TK, Shehabi HZ, et al. In vivo evidence for a novel pathway of vitamin D(3) metabolism initiated by P450scc and modified by CYP27B1. FASEB J. 2012;26(9):3901‐3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Skobowiat C, Dowdy JC, Sayre RM, Tuckey RC, Slominski A. Cutaneous hypothalamic‐pituitary‐adrenal axis homolog: regulation by ultraviolet radiation. Am J Physiol Endocrinol Metab. 2011;301(3):E484‐E493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tuckey RC, Li W, Zjawiony JK, et al. Pathways and products for the metabolism of vitamin D3 by cytochrome P450scc. FEBS J. 2008;275(10):2585‐2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Slominski A, Semak I, Zjawiony J, et al. The cytochrome P450scc system opens an alternate pathway of vitamin D3 metabolism. FEBS J. 2005;272(16):4080‐4090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Slominski AT, Li W, Kim TK, et al. Novel activities of CYP11A1 and their potential physiological significance. J Steroid Biochem Mol Biol. 2015;151:25‐37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. MacLaughlin JA, Anderson RR, Holick MF. Spectral character of sunlight modulates photosynthesis of previtamin D3 and its photoisomers in human skin. Science. 1982;216(4549):1001‐1003. [DOI] [PubMed] [Google Scholar]
- 25. Tuckey RC, Slominski AT, Cheng CY, et al. Lumisterol is metabolized by CYP11A1: discovery of a new pathway. Int J Biochem Cell Biol. 2014;55:24‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Slominski AT, Kim TK, Hobrath JV, et al. Characterization of a new pathway that activates lumisterol in vivo to biologically active hydroxylumisterols. Sci Rep. 2017;7(1):11434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gordon‐Thomson C, Gupta R, Tongkao‐On W, Ryan A, Halliday GM, Mason RS. 1alpha,25 Dihydroxyvitamin D(3) enhances cellular defences against UV‐induced oxidative and other forms of DNA damage in skin. Photochem Photobiol Sci. 2012;11(12):1837‐1847. [DOI] [PubMed] [Google Scholar]
- 28. Song EJ, Gordon‐Thomson C, Cole L, et al. 1α,25‐Dihydroxyvitamin D3 reduces several types of UV‐induced DNA damage and contributes to photoprotection. J Steroid Biochem Mol Biol. 2013;136:131‐138. [DOI] [PubMed] [Google Scholar]
- 29. Wong G, Gupta R, Dixon KM, et al. 1,25‐Dihydroxyvitamin D and three low‐calcemic analogs decrease UV‐induced DNA damage via the rapid response pathway. J Steroid Biochem Mol Biol. 2004;89‐90(1‐5):567‐570. [DOI] [PubMed] [Google Scholar]
- 30. De Haes P, Garmyn M, Verstuyf A, et al. 1,25‐Dihydroxyvitamin D3 and analogues protect primary human keratinocytes against UVB‐induced DNA damage. J Photochem Photobiol B. 2005;78(2):141‐148. [DOI] [PubMed] [Google Scholar]
- 31. Damian DL, Kim YJ, Dixon KM, Halliday GM, Javeri A, Mason RS. Topical calcitriol protects from UV‐induced genetic damage but suppresses cutaneous immunity in humans. Exp Dermatol. 2010;19(8):E23‐E30. [DOI] [PubMed] [Google Scholar]
- 32. Dixon KM, Deo SS, Norman AW, et al. In vivo relevance for photoprotection by the vitamin D rapid response pathway. J Steroid Biochem Mol Biol. 2007;103(3‐5):451‐456. [DOI] [PubMed] [Google Scholar]
- 33. Dixon KM, Deo SS, Wong G, et al. Skin cancer prevention: a possible role of 1,25dihydroxyvitamin D3 and its analogs. J Steroid Biochem Mol Biol. 2005;97(1‐2):137‐143. [DOI] [PubMed] [Google Scholar]
- 34. Dixon KM, Norman AW, Sequeira VB, et al. 1α,25(OH)2‐vitamin D and a nongenomic vitamin D analogue inhibit ultraviolet radiation‐induced skin carcinogenesis. Cancer Prev Res. 2011;4(9):1485‐1494. 10.1186/10.1158/1940-6207.CAPR-11-0165 [DOI] [PubMed] [Google Scholar]
- 35. Mason RS. Photoprotection by 1α,25‐dihydroxyvitamin D and analogs: further studies on mechanisms and implications for UV‐damage. J Steroid Biochem Mol Biol. 2010;121:164‐168. [DOI] [PubMed] [Google Scholar]
- 36. Rybchyn MS, De Silva WGM, Sequeira VB, et al. Enhanced repair of UV‐induced DNA damage by 1,25‐dihydroxyvitamin D3 in skin is linked to pathways that control cellular energy. J Invest Dermatol. 2018;138(5):1146‐1156. [DOI] [PubMed] [Google Scholar]
- 37. Sequeira VB, Rybchyn MS, Gordon‐Thomson C, et al. Opening of chloride channels by 1alpha,25‐dihydroxyvitamin D3 contributes to photoprotection against UVR‐induced thymine dimers in keratinocytes. J Invest Dermatol. 2013;133(3):776‐782. [DOI] [PubMed] [Google Scholar]
- 38. Tongkao‐On W, Carter S, Reeve VE, et al. CYP11A1 in skin: an alternative route to photoprotection by vitamin D compounds. J Steroid Biochem Mol Biol. 2015;148:72‐78. [DOI] [PubMed] [Google Scholar]
- 39. Norman AW, Ishizuka S, Okamura WH. Ligands for the vitamin D endocrine system: different shapes function as agonists and antagonists for genomic and rapid response receptors or as a ligand for the plasma vitamin D binding protein. J Steroid Biochem Mol Biol. 2001;76(1‐5):49‐59. [DOI] [PubMed] [Google Scholar]
- 40. Lee J, Youn JI. The photoprotective effect of 1,25‐dihydroxyvitamin D3 on ultraviolet light B‐induced damage in keratinocyte and its mechanism of action. J Dermatol Sci. 1998;18(1):11‐18. [DOI] [PubMed] [Google Scholar]
- 41. Chaiprasongsuk A, Janjetovic Z, Kim TK, et al. Protective effects of novel derivatives of vitamin D(3) and lumisterol against UVB‐induced damage in human keratinocytes involve activation of Nrf2 and p53 defense mechanisms. Redox Biol. 2019;24:101206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Slominski AT, Janjetovic Z, Kim TK, et al. Novel non‐calcemic secosteroids that are produced by human epidermal keratinocytes protect against solar radiation. J Steroid Biochem Mol Biol. 2015;148:52‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chaiprasongsuk A, Janjetovic Z, Kim TK, et al. CYP11A1‐derived vitamin D3 products protect against UVB‐induced inflammation and promote keratinocytes differentiation. Free Radic Biol Med. 2020;155:87‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lee JH, An HT, Chung JH, Kim KH, Eun HC, Cho KH. Acute effects of UVB radiation on the proliferation and differentiation of keratinocytes. Photodermatol Photoimmunol Photomed. 2002;18(5):253‐261. [DOI] [PubMed] [Google Scholar]
- 45. Gniadecka M, Wulf HC, Mortensen NN, Poulsen T. Photoprotection in vitiligo and normal skin. A quantitative assessment of the role of stratum corneum, viable epidermis and pigmentation. Acta Derm Venereol. 1996;76(6):429‐432. [DOI] [PubMed] [Google Scholar]
- 46. Brenner M, Hearing VJ. The protective role of melanin against UV damage in human skin. Photochem Photobiol. 2008;84(3):539‐549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Scott TL, Christian PA, Kesler MV, et al. Pigment‐independent cAMP‐mediated epidermal thickening protects against cutaneous UV injury by keratinocyte proliferation. Exp Dermatol. 2012;21(10):771‐777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wecksler WR, Mason RS, Norman AW. Specific cytosol receptors for 1,25‐dihydroxyvitamin D3 in human intestine. J Clin Endocrinol Metab. 1979;48(4):715‐717. [DOI] [PubMed] [Google Scholar]
- 49. Mangelsdorf DJ, Koeffler HP, Donaldson CA, Pike JW, Haussler MR. 1,25‐Dihydroxyvitamin D3‐induced differentiation in a human promyelocytic leukemia cell line (HL‐60): receptor‐mediated maturation to macrophage‐like cells. J Cell Biol. 1984;98(2):391‐398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ellison TI, Smith MK, Gilliam AC, MacDonald PN. Inactivation of the vitamin D receptor enhances susceptibility of murine skin to UV‐induced tumorigenesis. J Invest Dermatol. 2008;128(10):2508‐2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zinser GM, Sundberg JP, Welsh J. Vitamin D(3) receptor ablation sensitizes skin to chemically induced tumorigenesis. Carcinogenesis. 2002;23(12):2103‐2109. [DOI] [PubMed] [Google Scholar]
- 52. Teichert AE, Elalieh H, Elias PM, Welsh J, Bikle DD. Overexpression of hedgehog signaling is associated with epidermal tumor formation in vitamin D receptor‐null mice. J Invest Dermatol. 2011;131(11):2289‐2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Slominski AT, Chaiprasongsuk A, Janjetovic Z, et al. Photoprotective properties of vitamin D and lumisterol hydroxyderivatives. Cell Biochem Biophys. 2020;78(2):165‐180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Haussler MR, Norman AW. Chromosomal receptor for a vitamin D metabolite. Proc Natl Acad Sci U S A. 1969;62(1):155‐162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Norman AW, Nemere I, Zhou LX, et al. 1,25(OH)2‐vitamin D3, a steroid hormone that produces biologic effects via both genomic and nongenomic pathways. J Steroid Biochem Mol Biol. 1992;41(3‐8):231‐240. [DOI] [PubMed] [Google Scholar]
- 56. Zhou LX, Nemere I, Norman AW. 1,25‐Dihydroxyvitamin D3 analog structure‐function assessment of the rapid stimulation of intestinal calcium absorption (transcaltachia). J Bone Miner Res. 1992;7(4):457‐463. [DOI] [PubMed] [Google Scholar]
- 57. Huhtakangas JA, Olivera CJ, Bishop JE, Zanello LP, Norman AW. The vitamin D receptor is present in caveolae‐enriched plasma membranes and binds 1 alpha,25(OH)2‐vitamin D3 in vivo and in vitro. Mol Endocrinol. 2004;18(11):2660‐2671. [DOI] [PubMed] [Google Scholar]
- 58. Mizwicki MT, Keidel D, Bula CM, et al. Identification of an alternative ligand‐binding pocket in the nuclear vitamin D receptor and its functional importance in 1alpha,25(OH)2‐vitamin D3 signaling. Proc Natl Acad Sci U S A. 2004;101(35):12876‐12881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Boyan BD, Chen J, Schwartz Z. Mechanism of Pdia3‐dependent 1α,25‐dihydroxy vitamin D3 signaling in musculoskeletal cells. Steroids. 2012;77(10):892‐896. 10.1016/j.steroids.2012.04.018 [DOI] [PubMed] [Google Scholar]
- 60. Nemere I, Farach‐Carson MC, Rohe B, et al. Ribozyme knockdown functionally links a 1,25(OH)2D3 membrane binding protein (1,25D3‐MARRS) and phosphate uptake in intestinal cells. Proc Natl Acad Sci U S A. 2004;101(19):7392‐7397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Nemere I, Ray R, McManus W. Immunochemical studies on the putative plasmalemmal receptor for 1,25(OH)2D3. I. Chick intestine. Am J Physiol Endocrinol Metab. 2000;278(6):E1104‐E1114. [DOI] [PubMed] [Google Scholar]
- 62. Nemere I, Schwartz Z, Pedrozo H, Sylvia VL, Dean DD, Boyan BD. Identification of a membrane receptor for 1,25‐dihydroxyvitamin D3 which mediates rapid activation of protein kinase C. J Bone Miner Res. 1998;13(9):1353‐1359. [DOI] [PubMed] [Google Scholar]
- 63. Pedrozo HA, Schwartz Z, Rimes S, et al. Physiological importance of the 1,25(OH)2D3 membrane receptor and evidence for a membrane receptor specific for 24,25(OH)2D3. J Bone Miner Res. 1999;14(6):856‐867. [DOI] [PubMed] [Google Scholar]
- 64. Sequeira VB, Rybchyn MS, Tongkao‐On W, et al. The role of the vitamin D receptor and ERp57 in photoprotection by 1alpha,25‐dihydroxyvitamin D3. Mol Endocrinol. 2012;26(4):574‐582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Malloy PJ, Pike JW, Feldman D. The vitamin D receptor and the syndrome of hereditary 1,25‐dihydroxyvitamin D‐resistant rickets. Endocr Rev. 1999;20(2):156‐188. [DOI] [PubMed] [Google Scholar]
- 66. Ma NS, Malloy PJ, Pitukcheewanont P, Dreimane D, Geffner ME, Feldman D. Hereditary vitamin D resistant rickets: identification of a novel splice site mutation in the vitamin D receptor gene and successful treatment with oral calcium therapy. Bone. 2009;45(4):743‐746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Feldman D, Malloy JP. Mutations in the vitamin D receptor and hereditary vitamin D‐resistant rickets. Bonekey Rep. 2014;3:510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zanello LP, Norman AW. Rapid modulation of osteoblast ion channel responses by 1a, 25(OH)2‐vitamin D3 requires the presence of a functional vitamin D nuclear receptor. Proc Natl Acad Sci U S A. 2004;101(6):1589‐1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chaiprasongsuk A, Janjetovic Z, Kim TK, et al. Protective effects of novel derivatives of vitamin D3 and lumisterol against UVB‐induced damage in human keratinocytes involve activation of Nrf2 and p53 defense mechanisms. Redox Biol. 2019;24:101206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chaiprasongsuk A, Janjetovic Z, Kim TK, et al. Hydroxylumisterols, photoproducts of pre‐vitamin D3, protect human keratinocytes against UVB‐induced damage. Int J Mol Sci. 2020;21(24):9374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Slominski AT, Kim TK, Takeda Y, et al. RORalpha and ROR gamma are expressed in human skin and serve as receptors for endogenously produced noncalcemic 20‐hydroxy‐ and 20,23‐dihydroxyvitamin D. FASEB J. 2014;28(7):2775‐2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Slominski AT, Kim TK, Janjetovic Z, et al. Differential and overlapping effects of 20,23(OH)2D3 and 1,25(OH)2D3 on gene expression in human epidermal keratinocytes: identification of AhR as an alternative receptor for 20,23(OH)2D3. Int J Mol Sci. 2018;19(10):3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Slominski AT, Kim T‐K, Qayyum S, et al. Vitamin D and lumisterol derivatives can act on liver X receptors (LXRs). Sci Rep. 2021;11(1):8002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Gupta R, Dixon KM, Deo SS, et al. Photoprotection by 1,25 dihydroxyvitamin D3 is associated with an increase in p53 and a decrease in nitric oxide products. J Invest Dermatol. 2007;127(3):707‐715. [DOI] [PubMed] [Google Scholar]
- 75. Rybchyn MS, Islam KS, Brennan‐Speranza TC, et al. Homer1 mediates CaSR‐dependent activation of mTOR complex 2 and initiates a novel pathway for AKT‐dependent β‐catenin stabilization in osteoblasts. J Biol Chem. 2019;294(44):16337‐16350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Rybchyn MS, Slater M, Conigrave AD, Mason RS. An Akt‐dependent increase in canonical Wnt signaling and a decrease in sclerostin protein levels are involved in strontium ranelate‐induced osteogenic effects in human osteoblasts. J Biol Chem. 2011;286(27):23771‐23779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Elbein AD. Inhibitors of the biosynthesis and processing of N‐linked oligosaccharide chains. Annu Rev Biochem. 1987;56:497‐534. [DOI] [PubMed] [Google Scholar]
- 78. Fan G, Goldsmith PK, Collins R, et al. N‐linked glycosylation of the human Ca2+ receptor is essential for its expression at the cell surface. Endocrinology. 1997;138(5):1916‐1922. 10.1210/endo.138.5.5131 [DOI] [PubMed] [Google Scholar]
- 79. Latimer JA, Lloyd JJ, Diffey BL, Matts PJ, Birch‐Machin MA. Determination of the action spectrum of UVR‐induced mitochondrial DNA damage in human skin cells. J Invest Dermatol. 2015;135(10):2512‐2518. [DOI] [PubMed] [Google Scholar]
- 80. Hosseini M, Dousset L, Mahfouf W, et al. Energy metabolism rewiring precedes UVB‐induced primary skin tumor formation. Cell Rep. 2018;23(12):3621‐3634. [DOI] [PubMed] [Google Scholar]
- 81. Rozenberg J, Rishi V, Orosz A, Moitra J, Glick A, Vinson C. Inhibition of CREB function in mouse epidermis reduces papilloma formation. Mol Cancer Res. 2009;7(5):654‐664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. McCarthy BY, Dixon KM, Halliday GM, Reeve VE, Mason RS. The vitamin D saga: breaking dawn. Immunol Endocr Metab Agents Med Chem. 2014;14(3):137‐151. 10.2174/187152221403150521105050 [DOI] [Google Scholar]
- 83. Mason RS, Dixon KM, Sequeira VB, Gordon‐Thomson C. Sunlight protection by vitamin D compounds. In: Hewison M, Bouillon, R , Giovannucci, E & Goltzman, D , eds., Volume 2. Health, Disease and Therapeutics. 4th ed. Academic Press; 2017:1055‐1075. [Google Scholar]
- 84. De Silva WGM, Abboud M, Yang C, Dixon KM, Rybchyn MS, Mason RS. Protection from ultraviolet damage and photocarcinogenesis by vitamin D compounds. Adv Exp Med Biol. 2020;1268:227‐253. [DOI] [PubMed] [Google Scholar]
- 85. Bikle DD. Vitamin D receptor, a tumor suppressor in skin. Can J Physiol Pharmacol. 2015;93(5):349‐354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Slominski AT, Kim TK, Li W, et al. Detection of novel CYP11A1‐derived secosteroids in the human epidermis and serum and pig adrenal gland. Sci Rep. 2015;5:14875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kim TK, Wang J, Janjetovic Z, et al. Correlation between secosteroid‐induced vitamin D receptor activity in melanoma cells and computer‐modeled receptor binding strength. Mol Cell Endocrinol. 2012;361(1‐2):143‐152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Slominski AT, Kim TK, Hobrath JV, et al. Endogenously produced nonclassical vitamin D hydroxy‐metabolites act as "biased" agonists on VDR and inverse agonists on RORalpha and RORgamma. J Steroid Biochem Mol Biol. 2017;173:42‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Lin Z, Marepally SR, Goh ESY, et al. Investigation of 20S‐hydroxyvitamin D3 analogs and their 1alpha‐OH derivatives as potent vitamin D receptor agonists with anti‐inflammatory activities. Sci Rep. 2018;8(1):1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Lin Z, Chen H, Belorusova AY, et al. 1alpha,20S‐Dihydroxyvitamin D3 interacts with vitamin D receptor: crystal structure and route of chemical synthesis. Sci Rep. 2017;7(1):10193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Nemere I, Garbi N, Hammerling GJ, Khanal RC. Intestinal cell calcium uptake and the targeted knockout of the 1,25D3‐MARRS (membrane‐associated, rapid response steroid‐binding) receptor/PDIA3/Erp57. J Biol Chem. 2010;285(41):31859‐31866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Nemere I, Safford SE, Rohe B, DeSouza MM, Farach‐Carson MC. Identification and characterization of 1,25D3‐membrane‐associated rapid response, steroid (1,25D3‐MARRS) binding protein. J Steroid Biochem Mol Biol. 2004;89‐90(1‐5):281‐285. [DOI] [PubMed] [Google Scholar]
- 93. Coe H, Jung J, Groenendyk J, Prins D, Michalak M. ERp57 modulates STAT3 signaling from the lumen of the endoplasmic reticulum. J Biol Chem. 2010;285(9):6725‐6738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Nangia AK, Butcher JL, Konety BR, Vietmeier BN, Getzenberg RH. Association of vitamin D receptors with the nuclear matrix of human and rat genitourinary tissues. J Steroid Biochem Mol Biol. 1998;66(4):241‐246. [DOI] [PubMed] [Google Scholar]
- 95. Hernández‐Sánchez F, Guzmán‐Beltrán S, Herrera MT, et al. High glucose induces O‐GlcNAc glycosylation of the vitamin D receptor (VDR) in THP1 cells and in human macrophages derived from monocytes. Cell Biol Int. 2017;41(9):1065‐1074. [DOI] [PubMed] [Google Scholar]
- 96. Guha P, Kaptan E, Gade P, Kalvakolanu DV, Ahmed H. Tunicamycin induced endoplasmic reticulum stress promotes apoptosis of prostate cancer cells by activating mTORC1. Oncotarget. 2017;8(40):68191‐68207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Xu D, Perez RE, Rezaiekhaligh MH, Bourdi M, Truog WE. Knockdown of ERp57 increases BiP/GRP78 induction and protects against hyperoxia and tunicamycin‐induced apoptosis. Am J Physiol Lung Cell Mol Physiol. 2009;297(1):L44‐L51. [DOI] [PubMed] [Google Scholar]
- 98. Norman AW, Okamura WH, Hammond MW, et al. Comparison of 6‐s‐cis‐ and 6‐s‐trans‐locked analogs of 1alpha,25‐dihydroxyvitamin D3 indicates that the 6‐s‐cis conformation is preferred for rapid nongenomic biological responses and that neither 6‐s‐cis‐ nor 6‐s‐trans‐locked analogs are preferred for genomic biological responses. Mol Endocrinol. 1997;11(10):1518‐1531. [DOI] [PubMed] [Google Scholar]
- 99. Menegaz D, Barrientos‐Duran A, Kline A, et al. 1α,25(OH)2‐Vitamin D3 stimulation of secretion via chloride channel activation in Sertoli cells. J Steroid Biochem Mol Biol. 2010;119(3‐5):127‐134. 10.1016/j.jsbmb.2010.01.011 [DOI] [PubMed] [Google Scholar]
- 100. Shariev A, Mason RS, Dixon KM, editors. NDRG1: a potential target for vitamin D in the inhibition of melanoma metastasis. Australasian Wound and Tissue Repair Society (AWTRS) and the Molecular & Experimental Pathology Society of Australasia (MEPSA); 2016; Melbourne, Australia.
- 101. Ming M, Han W, Maddox J, et al. UVB‐induced ERK/AKT‐dependent PTEN suppression promotes survival of epidermal keratinocytes. Oncogene. 2010;29(4):492‐502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Ming M, Feng L, Shea CR, et al. PTEN positively regulates UVB‐induced DNA damage repair. Cancer Res. 2011;71(15):5287‐5295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Checkley LA, Rho O, Angel JM, et al. Metformin inhibits skin tumor promotion in overweight and obese mice. Cancer Prev Res (Phila). 2014;7(1):54‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Johannessen M, Delghandi MP, Moens U. What turns CREB on? Cell Signal. 2004;16(11):1211‐1227. [DOI] [PubMed] [Google Scholar]
- 105. Yao K, Chen H, Liu K, et al. Kaempferol targets RSK2 and MSK1 to suppress UV radiation‐induced skin cancer. Cancer Prev Res (Phila). 2014;7(9):958‐967. [DOI] [PMC free article] [PubMed] [Google Scholar]