

ABSTRACT
1,25(OH)2D3, the biologically active form of vitamin D3, is a major regulator of mineral and bone homeostasis and exerts its actions through binding to the vitamin D receptor (VDR), a ligand‐activated transcription factor that can directly modulate gene expression in vitamin D‐target tissues such as the intestine, kidney, and bone. Inactivating VDR mutations or vitamin D deficiency during development results in rickets, hypocalcemia, secondary hyperparathyroidism, and hypophosphatemia, pointing to the critical role of 1,25(OH)2D3‐induced signaling in the maintenance of mineral homeostasis and skeletal health. 1,25(OH)2D3 is a potent stimulator of VDR‐mediated intestinal calcium absorption, thus increasing the availability of calcium required for proper bone mineralization. However, when intestinal calcium absorption is impaired, renal calcium reabsorption is increased and calcium is mobilized from the bone to preserve normocalcemia. Multiple cell types within bone express the VDR, thereby allowing 1,25(OH)2D3 to directly affect bone homeostasis. In this review, we will discuss different transgenic mouse models with either Vdr deletion or overexpression in chondrocytes, osteoblasts, osteocytes, or osteoclasts to delineate the direct effects of 1,25(OH)2D3 on bone homeostasis. We will address the bone cell type–specific effects of 1,25(OH)2D3 in conditions of a positive calcium balance, where the amount of (re)absorbed calcium equals or exceeds fecal and renal calcium losses, as well as during a negative calcium balance, due to selective Vdr knockdown in the intestine or triggered by a low calcium diet. © 2021 The Authors. JBMR Plus published by Wiley Periodicals LLC on behalf of American Society for Bone and Mineral Research.
Keywords: GENETIC ANIMAL MODELS, ANIMAL MODELS, PTH/VIT D/FGF23, CELL/TISSUE SIGNALING, ENDOCRINE PATHWAYS, CELLS OF BONE, CHONDROCYTE AND CARTILAGE BIOLOGY, DISORDERS OF CALCIUM/PHOSPHATE METABOLISM
Endocrine Loops Regulate Mineral and Bone Homeostasis
The hormonally active form of vitamin D3, 1,25(OH)2D3, is an important mediator of mineral and bone homeostasis. 1,25(OH)2D3, primarily produced in the kidneys, exerts its effects through the vitamin D receptor (VDR), which is most abundantly expressed in tissues involved in the maintenance of mineral and bone homeostasis, such as the parathyroid glands, intestine, kidney, and bone.( 1 ) Mutations in the VDR gene that result in a defective receptor lead to the development of vitamin D‐dependent rickets type 2, an autosomal recessive disorder characterized by the early onset of rickets, hypocalcemia, secondary hyperparathyroidism, and hypophosphatemia.( 2 )
When serum calcium concentrations drop, they elicit an elevated secretion of parathyroid hormone (PTH) by the parathyroid glands, which in turn stimulates renal synthesis of CYP27B1, the rate‐limiting enzyme that is responsible for the hydroxylation of 25(OH)D3 to the biologically active form 1,25(OH)2D3. In addition, PTH induces calcium mobilization from the bone, which represents a major calcium reservoir, and enhances renal calcium reabsorption. The rise in serum 1,25(OH)2D3 concentrations induces, on its turn, calcium absorption in the intestine and reabsorption in the kidney. Moreover, to preserve normocalcemia during a negative calcium balance, 1,25(OH)2D3 can mobilize calcium from the bone.( 3 ) In a first negative feedback loop regulating calcium homeostasis, elevated levels of 1,25(OH)2D3 inhibit PTH secretion by the parathyroid glands, and in a second negative feedback loop, 1,25(OH)2D3 suppresses CYP27B1 and stimulates CYP24A1 in the kidney thereby limiting its own production and enhancing its catabolism.
During the process of correcting hypocalcemia, elevated levels of PTH and 1,25(OH)2D3 induce phosphate mobilization from the bone, leading to elevated serum phosphate levels. This hyperphosphatemia is partially corrected by PTH itself by mediating internalization of the sodium/phosphate cotransporters NPT2A and NPT2C within the renal tubules, resulting in urinary phosphate wasting. In addition, increased serum phosphate levels are sensed by osteoblasts and osteocytes, leading to an elevated production of fibroblast growth factor (FGF)23. FGF23 binds to FGFR1C/αKlotho in the kidney, which transcriptionally inhibits the expression of Npt2a and Npt2c and thereby promotes phosphaturia. Furthermore, 1,25(OH)2D3 induces FGF23, and as such indirectly stimulates phosphate wasting. On the other hand, 1,25(OH)2D3 is able to transcriptionally induce Npt2a expression in mouse and NPT2A/C in human renal cell lines, suggesting a positive effect of 1,25(OH)2D3 on renal phosphate reabsorption.( 4 , 5 ) The in vivo importance of this regulation is, however, questioned, as short‐term treatment of mice with supraphysiological doses of 1,25(OH)2D3 increases NPT2A protein levels in total renal homogenates, whereas urinary phosphate levels are not altered.( 6 ) Therefore, further research is required to establish the role of 1,25(OH)2D3 in the regulation of renal phosphate reabsorption.( 7 ) Of note, 1,25(OH)2D3 also increases the expression of NPT2B in the intestine, thereby stimulating intestinal transcellular phosphate absorption.( 8 , 9 , 10 )
Elevated FGF23 levels not only induce renal phosphate excretion but also affect renal vitamin D metabolism by decreasing Cyp27b1 expression and stimulating Cyp24a1 expression, which together reduce circulating levels of 1,25(OH)2D3. Thus, normal serum concentrations of calcium and phosphate are guaranteed by a complex interplay between 1,25(OH)2D3, PTH, and FGF23, which is controlled by multiple feedback mechanisms operating in the parathyroid glands, kidney, and bone.
1,25(OH) 2D3 Indirectly Regulates Bone Homeostasis by Enhancing Intestinal Calcium Absorption
One of the major activities of 1,25(OH)2D3 is to stimulate intestinal calcium absorption. Adequate dietary calcium absorption indirectly contributes to bone homeostasis by ensuring sufficient calcium supply to the bone, which is crucial for bone mineralization. Intestinal calcium absorption occurs through a saturable, transcellular pathway that is energy‐dependent as well as through a non‐saturable, paracellular passive pathway. The passive transport takes place throughout the whole intestine and is dependent on the intestinal luminal calcium concentration. Although it was previously assumed that 1,25(OH)2D3 only induces transcellular calcium uptake in the duodenum, recent findings indicate a more complex situation. First, the paracellular calcium transport seems also regulated by the action of 1,25(OH)2D3, as the expression of the paracellular calcium channels Claudin 2 and 12 is induced by 1,25(OH)2D3 and decreased in the intestine of Vdr null mice.( 11 ) Second, it now appears that 1,25(OH)2D3 also stimulates the transcellular process in the large intestine.( 12 )
The essential role of 1,25(OH)2D3‐mediated intestinal calcium absorption for bone and mineral homeostasis is clearly demonstrated by the phenotype of Vdr null mice. Vdr null mice are phenotypically normal at birth, but after weaning, they develop hypocalcemia, secondary hyperparathyroidism, and hypophosphatemia despite very high levels of 1,25(OH)2D3, and they become growth retarded with severe rickets.( 13 , 14 , 15 , 16 ) Mice deficient in Cyp27b1 display a similar phenotype.( 17 , 18 ) This bone and calcium phenotype can be largely corrected by a high dietary calcium intake (especially in combination with a high lactose intake) in both knockout models, indicating the importance of 1,25(OH)2D3‐mediated intestinal calcium absorption.( 14 , 19 , 20 , 21 , 22 ) Treatment with 1,25(OH)2D3 also rescues the phenotype of Cyp27b1 mice.( 23 , 24 , 25 ) In contrast to systemic Vdr null mice, intestine‐specific Vdr null mice (Villin‐Vdr‐cKO mice) are normocalcemic. In response to the reduced intestinal calcium absorption in these mice, circulating levels of PTH and 1,25(OH)2D3 are increased, which not only enhance bone resorption but also suppress bone matrix mineralization. Both processes contribute to the maintenance of normal serum calcium levels, as the increased bone resorption releases calcium from the bone, whereas the suppressed bone mineralization prevents calcium incorporation in the bone (as explained later in more detail). These findings indicate that the maintenance of normocalcemia occurs at the expense of skeletal integrity.( 26 ) Interestingly, mice with Vdr inactivation in the distal intestine reveal that also the large intestine significantly contributes to whole‐body calcium homeostasis.( 27 ) In agreement, transgenic expression of human VDR exclusively in the ileum, caecum, and colon of Vdr null mice prevents abnormalities in calcium and bone homeostasis, confirming the essential role of VDR‐mediated calcium absorption in the distal gastrointestinal tract.( 12 , 28 ) Of note, villin‐driven overexpression of the VDR in the intestine of wild‐type mice does not improve the intestinal response to 1,25(OH)2D3 nor does it prevent the bone loss when mice are fed a low calcium diet, indicating that supraphysiological intestinal VDR levels do not provide additional benefit.( 29 )
During transcellular calcium transport, 1,25(OH)2D3 enhances the apical entry of calcium across the brush border membrane into the enterocyte by upregulating the expression of the epithelial channels transient receptor potential cation channel subfamily V (TRPV) member 6 and TRPV5.( 14 ) The intracellular calcium transfer is mainly dependent on the calbindin‐D9K protein, which is encoded by the S100g gene.( 30 ) The transfer of calcium from the cytoplasm to the extracellular space requires energy expenditure because of an uphill concentration gradient and an unfavorable electrochemical gradient. Both the plasma membrane calcium pump PMCA1, a low‐affinity Ca‐ATPase, and a sodium‐calcium exchanger NCX1 play important roles in this process. Although the expression of Trpv5 and 6 as well as S100g is highly induced by 1,25(OH)2D3, the exact molecular mechanism of 1,25(OH)2D3‐mediated transcellular calcium transport remains elusive. Indeed, the finding that significant 1,25(OH)2D3‐inducible active intestinal calcium absorption occurs in the absence of Trpv6 and S100g suggests that additional calcium transporters or channels are involved.( 31 ) Recent studies show a strong induction of Slc30a10, which encodes a manganese efflux transporter, by 1,25(OH)2D3 both in the proximal and distal intestine, possibly suggesting an interrelationship between vitamin D signaling and manganese efflux.( 12 , 32 , 33 )
Taken together, 1,25(OH)2D3‐regulated intestinal calcium absorption is crucial for mineral and bone homeostasis, although the molecular mechanisms are still incompletely characterized.
Renal Synthesis of 1,25(OH) 2D3 Contributes to Renal Calcium and Phosphate Handling
Calcium reabsorption in the kidney is an important contributor to whole‐body mineral handling and is suggested to become more dominant when intestinal calcium absorption is suboptimal or when bone turnover is low.( 34 , 35 ) Most of the filtered calcium (98% to 99%) is reabsorbed in the kidney via paracellular and transcellular transport pathways. Calcium reabsorption occurs mainly in the proximal convoluted tubules (PCTs) (60% to 70%) and in the thick ascending loop of Henle (TAL) (20%) through a passive, paracellular transport that depends on electrochemical gradients. However, fine‐tuning of calcium reabsorption occurs in the distal convoluted tubules (DCTs). Here, active transcellular calcium transport takes place in a three‐step pathway, comparable to the process in the intestine. Apical entry is mediated by TRPV5, whereas intracellular transport to the basolateral side occurs via the calbindin proteins, S100g and CALB1. Basolateral exit occurs through the calcium‐ATPase PMCA4 and the sodium calcium exchanger NCX1.( 34 , 36 , 37 ) Next to PTH and FGF23, 1,25(OH)2D3 also regulates the active uptake of calcium in the DCT.( 3 ) The role of VDR in 1,25(OH)2D‐induced calcium reabsorption is illustrated by the reduced urinary calcium excretion in mice with intestinal‐specific Vdr deletion( 26 ) and the elevated urinary calcium excretion in Vdr null mice with transgenic expression of low levels of Vdr exclusively in the gut.( 35 ) Moreover, the crucial role of 1,25(OH)2D‐regulated renal calcium reabsorption is demonstrated by persistent hypercalciuria and reduced bone mass in Trpv5‐deficient mice.( 38 )
Vitamin D signaling also affects renal phosphate handling, although mainly indirectly. Upon a normal dietary phosphate intake, approximately 80% of the filtered phosphate is reabsorbed, mostly via sodium‐dependent cotransport in the PCTs. Type I (NPT1) as well as type 2 (NPT2a, NPT2c) and type 3 (PIT1, PIT2) sodium/phosphate cotransporters are present in the apical membrane of the PCTs, whereas the transport mechanism at the basolateral side remains to be identified.( 39 , 40 ) PTH and FGF23 promote renal phosphate loss by directly decreasing the abundance of NPT2A and NPT2C. 1,25(OH)2D3 influences phosphate homeostasis indirectly by stimulating FGF23 synthesis by the osteocytes and by inhibiting PTH production in the parathyroid glands.( 3 ) Direct regulation of NPT2A and NPT2C by 1,25(OH)2D3 is demonstrated in cultured renal epithelial cells, but its in vivo significance is still undefined.( 4 , 5 )
Thus, 1,25(OH)2D3 signaling is important for fine‐tuning renal calcium reabsorption by regulating its active transport.
Direct Effects of 1,25(OH) 2D3 on Bone Homeostasis
As already outlined above, calcium malabsorption in Vdr null mice causes hypocalcemia after weaning and leads to the induction of hyperparathyroidism and hypophosphatemia.( 13 , 14 , 15 ) The deficit in calcium supply hampers bone mineralization and ultimately gives rise to the development of hyperosteoidosis and osteomalacia.( 13 , 14 , 15 ) Moreover, rickets will develop, characterized by an enlargement and disorganization of the zone of hypertrophic chondrocytes, which leads to a progressive widening of the epiphyseal growth plates. When calcium stores in Vdr null mice are replenished by feeding them a diet rich in calcium and lactose, their bone and mineral phenotype is fully restored, which emphasizes the indirect importance of 1,25(OH)2D3 for bone health by ensuring sufficient calcium (re)absorption.( 13 , 14 , 15 ) Nevertheless, 1,25(OH)2D3 may also directly affect bone homeostasis as chondrocytes, osteoblasts, osteocytes, and osteoclasts express the VDR. Because VDR is most abundantly expressed in osteoblasts and osteocytes, these cells are considered to be the main mediators of 1,25(OH)2D3 signaling in bone. By analysis of different transgenic mouse models with either Vdr deletion or overexpression in specific bone cell types, we try to delineate the direct effects of 1,25(OH)2D3 on bone homeostasis (Fig. 1). {FIG1} We will discuss the bone cell type–specific effects of VDR signaling in conditions of a positive calcium balance, where the amount of (re)absorbed calcium equals or exceeds the amount of excreted calcium (fecal or urinary loss) as well as during a negative calcium balance, resulting from insufficient calcium absorption.
Fig 1.
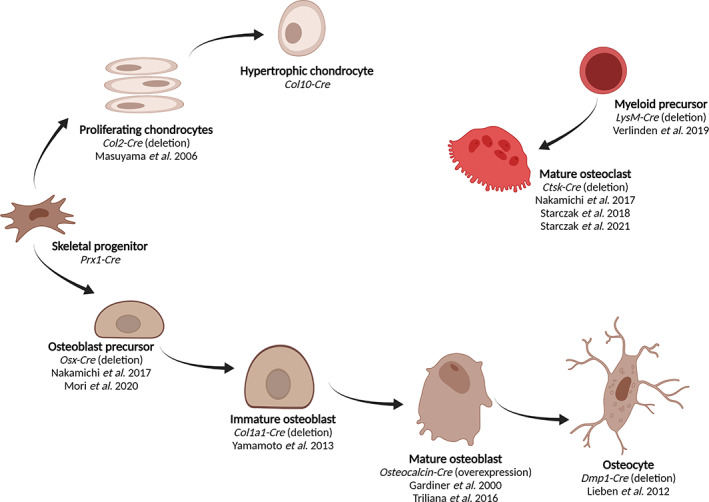
Overview of skeletal cell types in which Vdr expression is targeted. (Figure created with BioRender.com.)
Vdr signaling in chondrocytes is not crucial for growth plate development but transiently regulates bone development and phosphate homeostasis
Chondrocytes develop from mesenchymal progenitor cells that have multilineage potential. These progenitors give rise to hematopoietic‐supporting bone marrow stromal cells, chondrocytes, osteoblasts, and adipocytes through highly controlled processes.( 41 ) Long bones develop through endochondral ossification, and chondrocytes in the growth plate are responsible for their lengthening, not only by proliferating but also by volume expansion during their differentiation to hypertrophic chondrocytes. During this differentiation, chondrocytes express stage‐specific extracellular matrix (ECM) proteins including different types of collagens. Promoters of these genes can therefore be used to enable chondrocyte‐specific expression of transgenes.( 42 ) As such, the Col2a1 promoter targets resting and proliferating chondrocytes, whereas the Col10a1 promoter marks more differentiated hypertrophic chondrocytes.
As outlined above, Vdr null mice develop a rachitic bone phenotype characterized by an expansion of the hypertrophic chondrocyte layers. The finding that this rachitic bone phenotype can be prevented by normalization of calcium and phosphate homeostasis indicates that the VDR is not required for normal growth plate maturation.( 43 ) Growth plate expansion in Vdr null mice originates from a defect in programmed cell death within the hypertrophic chondrocyte layer, as a consequence of the low serum phosphate levels.( 44 ) To investigate whether VDR in chondrocytes has a role in bone development, the consequences of Vdr inactivation by Col2‐Cre‐driven excision was investigated in Col2‐Vdr‐cKO mice.( 45 ) As expected, chondrocyte maturation occurs normally in Col2‐Vdr‐cKO mice. Yet, trabecular bone mass is increased in juvenile Col2‐Vdr‐cKO mice and is accompanied by a reduction in blood vessels and osteoclasts at the border between the growth plate and trabecular bone. This decreased osteoclast number originates from the reduced capacity of Col2‐Vdr‐cKO‐derived chondrocytes to produce receptor activator of NF‐κB ligand (RANKL) and stimulate osteoclastogenesis. In addition, serum FGF23 levels are lower in Col2‐Vdr‐cKO mice, although chondrocytes hardly express FGF23. Coculture experiments show that VDR signaling in chondrocytes indirectly regulates FGF23 secretion by osteoblasts. This reduced FGF23 level in juvenile Col2‐Vdr‐cKO mice has two consequences: first, renal Cyp27b1 expression is increased, resulting in higher circulating 1,25(OH)2D3 levels; and second, renal NPT2A expression is enhanced, which leads to elevated serum phosphate levels. Of note, this phenotype disappears in adult mice when the contribution of VDR signaling in growth plate chondrocytes becomes less important compared with osteoblasts. In summary, these data demonstrate that VDR signaling in chondrocytes affects bone mass, by secreting RANKL, thereby inducing osteoclast formation, and it regulates phosphate homeostasis by participating in the endocrine loop between FGF23 and 1,25(OH)2D3. ( 45 )
Inactivation of Vdr at different stages during osteoblast differentiation points to a minor role of osteoblastic Vdr in bone homeostasis
Osteoblasts derive from the same skeletal progenitors as chondrocytes, and their differentiation is mediated by multiple transcription factors, including Runx2 and Osterix (Osx).( 46 ) Cells in the osteogenic lineage differentiate from osteoblast precursors, to immature osteoblasts, mature osteoblasts, and finally to osteocytes. For each of these maturation stages, stage‐specific promoters can be used to silence target gene expression. The Col1a1 promoter, which encodes the ECM protein type 1 collagen α1, is often used to target immature osteoblasts, whereas the osteocalcin promoter is commonly used to modify gene expression in mature osteoblasts. Terminally differentiated osteocytes, which are embedded in the bone matrix and are involved in the coordination between bone formation and resorption, can be targeted with the dentin matrix protein 1 (Dmp1)‐promoter.( 47 )
Vdr expression has been specifically deleted or overexpressed at different stages of osteoblast maturation( 26 , 48 , 49 , 50 , 51 , 52 ) (Fig. 1). In osteoblast precursors, Vdr expression is successfully (90%) deleted by Osx‐driven recombination (Osx‐Vdr‐cKO mice), which does not alter bone mass, resorption, or formation, nor serum calcium, phosphate, 1,25(OH)2D3, or PTH levels.( 51 ) Only FGF23 levels are slightly but significantly decreased.( 51 ) Interestingly, treatment with the vitamin D analog eldecalcitol has a different effect in Osx‐Vdr‐cKO mice compared with wild‐type mice. First, eldecalcitol induces bone mass in wild‐type mice by inhibiting bone resorption, which is not observed in Osx‐Vdr‐cKO mice, (51 ) and second, it enhances FGF23 levels in wild‐type mice but much less in Osx‐Vdr‐cKO mice, indicating that eldecalcitol‐mediated VDR signaling in osteoblast‐lineage cells suppresses bone resorption and regulates FGF23 expression.( 51 )
Moreover, administration of high doses of 1,25(OH)2D3 to Osx‐Vdr‐cKO mice fails to increase Rankl expression in bone and does not lead to elevated bone resorption( 52 ) (Fig. 2). {FIG2} Together, these data indicate that VDR signaling in osteoprogenitors is not required for bone and mineral homeostasis in basal conditions, but it contributes to bone resorption when 1,25(OH)2D3 levels are increased.
Fig 2.
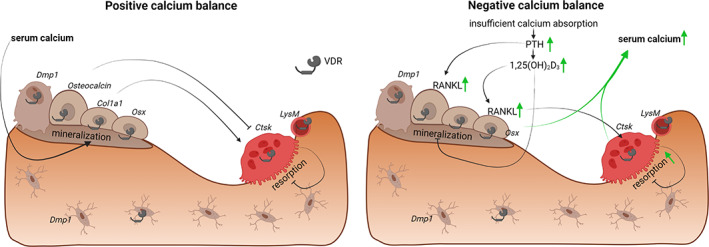
During a positive calcium balance (left panel), normal serum calcium concentrations allow proper bone mineralization. Transgenic models with Vdr inactivation in osteoprogenitors (Osx‐Cre( 51 )) or in mature osteoblasts and osteocytes (Dmp1‐Cre( 26 )) display normal mineral and bone homeostasis. Vdr knockdown in immature osteoblasts (Col1a1‐Cre( 49 )) induces bone mass, whereas Vdr overexpression in mature osteoblasts (Osteocalcin‐Cre( 48 , 50 )) also leads to an elevated bone mass. Vdr inactivation in osteoclast precursors (LysM‐Cre( 57 )) does not affect bone homeostasis, whereas Vdr‐silencing in mature osteoclasts (Ctsk‐Cre) has either no effect( 51 ) or leads to a slight reduction in bone mass.( 58 ) During a negative calcium balance (right panel), insufficient intestinal calcium absorption leads to increased PTH and 1,25(OH)2D3 levels, which stimulate RANKL expression by osteoblasts and enhance bone resorption.( 52 ) Moreover, 1,25(OH)2D3 inhibits bone mineralization by directly inducing the expression of mineralization inhibitors.( 26 ) Elevated resorption and decreased mineralization both contribute to the maintenance of normocalcemia. Mice with selective Vdr knockdown in mature osteoclasts (Ctsk‐Cre( 59 )) experience a greater bone loss during a negative calcium balance. (Figure created with BioRender.com.)
Targeting Vdr expression in immature osteoblasts by Col1a1‐mediated excision (Col1a1‐Vdr‐cKO mice)( 49 ) does not affect mineral homeostasis, as evidenced by normal serum calcium, phosphate, and 1,25(OH)2D3 concentrations. In contrast to Osx‐Vdr‐cKO mice, Col1a1‐Vdr‐cKO mice have normal circulating levels of FGF23, possibly due to incomplete osteoblastic Vdr deletion (only 50%) or pointing to the importance of additional mediators that regulate serum FGF23 levels. On the other hand, adult Col1a1‐Vdr‐cKO mice show increased trabecular bone mass, accompanied by a reduced osteoclast surface due to decreased Rankl expression, whereas no differences in osteoblast numbers or dynamic bone parameters are present. In summary, these findings suggest that VDR signaling in immature osteoblasts has a negative effect on bone homeostasis, possibly by enhancing RANKL‐mediated osteoclastogenesis.
In contrast, Vdr overexpression (threefold) in mature osteoblasts under control of the osteocalcin promoter (osteocalcin‐Vdr‐cOE mice), evaluated on two different genetic mouse backgrounds, points to a positive effect of VDR expression in osteoblasts on bone mass.( 48 , 50 ) Whereas serum calcium, PTH, and 1,25(OH)2D3 levels are comparable between osteocalcin‐Vdr‐cOE and wild‐type mice, cortical bone in adult osteocalcin‐Vdr‐cOE mice is wider and associated with an increased periosteal mineral apposition rate. Also, vertebral trabecular bone content is elevated in osteocalcin‐Vdr‐cOE mice, possibly due to an increased expression of Opg, a decoy receptor of RANKL, and subsequent decreased osteoclastic bone resorption.( 53 )
The role of VDR signaling in mature osteoblasts and osteocytes is studied by targeted Vdr knockdown (90%) under control of the Dmp1 promoter (Dmp‐Vdr‐cKO mice).( 26 ) Mineral and bone homeostasis is not disturbed in Dmp‐Vdr‐cKO mice as evidenced by normal serum levels of calcium, phosphate, and 1,25(OH)2D3 and normal cortical and trabecular bone mass. Interestingly, administration of high doses 1,25(OH)2D3 increases serum calcium levels to a much lower extent in Dmp‐Vdr‐cKO mice compared with wild‐type littermates, whereas bone mass tends to be better preserved. These effects are not due to differences in osteoclastogenesis or Rankl/Opg ratios, suggesting that VDR signaling in early osteoblasts is sufficient to account for the 1,25(OH)2D3‐mediated bone resorption. On the other hand, high doses of 1,25(OH)2D3 lead to decreased mineralization of bone matrix in wild‐type mice, which is accompanied by increased expression of the mineralization inhibitors osteopontin and progressive ankylosis (Ank). These effects were not observed in Dmp‐Vdr‐cKO mice, indicating that VDR‐mediated signaling in the osteocytes impairs bone matrix mineralization in response to high circulating levels of 1,25(OH)2D3. This mechanism also contributes to the decrease in mineralized bone mass in Vdr‐Villin‐cKO mice. The Vdr‐Villin‐cKO mice represent a negative calcium balance model, with impaired intestinal calcium absorption leading to increased circulating levels of PTH and 1,25(OH)2D3. These hormonal changes likely stimulate compensatory mechanisms to preserve normocalcemia in Vdr‐Villin‐cKO mice, including increased renal calcium reabsorption, next to decreased bone calcium content, resulting from enhanced osteoclastogenesis and impaired bone matrix mineralization.( 26 ) Recently, Misof and colleagues showed that, when systemic Vdr null mice are given a rescue diet from weaning until the age of 4 months, bone homeostasis is normal, as previously reported. When these adult Vdr null mice are subsequently switched to a low calcium diet (0.5% calcium, 0.4% phosphate), they develop osteomalacia with a marked decrease in bone calcium content and an increased cortical porosity, indicative of enhanced osteoclastic bone resorption but without signs of osteocytic osteolysis.( 54 )
In summary, VDR signaling in osteogenic cells does not seem to play a major role in the control of bone homeostasis, when calcium balance is normal, although some small but discordant effects are reported. Currently, it is unclear how to reconcile the puzzling findings that both osteoblast‐specific Vdr deletion and Vdr overexpression result in a slight increase in bone mass.( 48 , 49 , 50 ) A possible explanation is that the effects of osteoblastic VDR signaling on bone homeostasis is dependent on the maturation stage of the osteoblasts, as suggested by in vitro data.( 55 ) Moreover, the differences in experimental settings such as gene dosage effects (deletion versus overexpression), genetic background of the mice, experimental diets, and animal age may alter VDR‐mediated signaling in bone. On the other hand, VDR signaling in osteogenic cells contributes to maintaining normocalcemia, when intestinal calcium absorption is reduced, by decreasing mineralized bone mass.
Osteoclast‐specific VDR signaling is not a major determinant of bone homeostasis
The role of VDR signaling in osteoclasts remains controversial. Immunohistochemical analysis reveals no( 56 ) or faint( 51 ) VDR staining in osteoclasts; however, osteoclasts are shown to be responsive to 1,25(OH)2D3, as illustrated by the VDR‐mediated induction of Cyp24a1 expression in 1,25(OH)2D3‐treated primary osteoclast cultures.( 28 , 51 ) To investigate whether osteoclastic Vdr expression affects bone homeostasis, its expression was specifically deleted either in osteoclast precursors under control of the M lysozyme promoter (LysM‐Vdr‐cKO mice)( 57 ) or in mature osteoclasts by Cathepsin K‐driven Cre recombination (Ctsk‐Vdr‐cKO mice).( 51 , 58 , 59 )
Vdr inactivation in myeloid cells leads to an approximately 70% reduction of Vdr transcript levels in primary osteoclast cultures, derived from LysM‐Vdr‐cKO mice. Mineral and bone homeostasis are normal in young adult LysM‐Vdr‐cKO mice, as is in vitro osteoclast differentiation.( 57 ) Two research groups independently investigated the consequences of Vdr knockdown specifically in mature osteoclasts.( 51 , 58 , 59 ) Nakamichi and colleagues( 51 ) confirmed the osteoclast‐specific Vdr knockdown by a reduced induction of Cyp24a1 expression in osteoclasts after stimulation with 1,25(OH)2D3, yet in vivo analysis does not reveal any alterations in bone mass of Ctsk‐Vdr‐cKO mice. However, the studies of Starczak and colleagues( 58 , 59 ) do suggest a role of osteoclastic Vdr expression in the control of bone homeostasis. In their studies, Vdr transcript levels in whole bone homogenates of Ctsk‐Vdr‐cKO mice are reduced by 70%. Serum calcium and phosphate levels are normal in young adult Ctsk‐Vdr‐cKO mice, whereas trabecular bone mass is slightly decreased.( 58 , 59 ) In vitro osteoclastogenesis is enhanced in Ctsk‐Vdr‐cKO‐derived splenocytes, although in vivo bone resorption parameters are normal.
The physiological consequences of osteoclastic Vdr knockdown are also investigated during a negative calcium balance in young adult LysM‐Vdr‐cKO( 57 ) and Ctsk‐Vdr‐cKO mice,( 59 ) by feeding a low‐calcium diet from weaning onward. Bone loss as well as osteoclast activity in LysM‐Vdr‐cKO mice induced by the low‐calcium diet are comparable to that in wild‐type littermates.( 57 ) However, trabecular bone loss is slightly more pronounced in Ctsk‐Vdr‐cKO mice on the low‐calcium diet, despite no effect on bone resorption parameters. Similar to the osteoblast‐specific Vdr knockdown models, differences in experimental settings of the osteoclast‐specific Vdr silencing (gene promoter, genetic background, animal age, experimental diet, site of bone analysis) may underlie the divergent findings in the different studies. Indeed, the Ctsk promoter is also expressed in other skeletal cells such as osteocytes and periosteal stem cells, which may explain the strongly reduced Vdr expression in Ctsk‐Vdr‐cKO bones and may account for the observed differences in bone phenotype.( 60 )
However, overall these data seem to suggest that VDR signaling in osteoclasts is not or minimally involved in bone homeostasis during a positive or a negative calcium balance.
Conclusion
Multiple cell types within the skeleton express the VDR. High VDR expression is present in osteoblasts and osteocytes, whereas its expression is low in chondrocytes and osteoclasts.( 56 ) VDR expression in each of these different bone cell types allows 1,25(OH)2D3 to affect bone development and remodeling. During a positive calcium balance, when the amount of absorbed calcium exceeds fecal and renal calcium losses, 1,25(OH)2D3 regulates bone mass and mineralization mainly indirectly by ensuring sufficient calcium supply through intestinal and renal calcium (re)absorption.( 3 , 61 ) At this moment, it remains unclear whether 1,25(OH)2D3 also directly affects bone homeostasis in this condition, as overexpression or deletion of Vdr specifically at different stages of osteoblastic differentiation yielded contradictory findings, including none, positive, and negative effects on bone resorption. Deletion of Vdr expression in the osteoclast lineage has little or no consequences on bone homeostasis as specific Vdr deletion in osteoclast precursors or in mature osteoclasts hardly affects bone resorption or alters bone mass.
During a negative calcium balance, intestinal calcium absorption does not meet the daily calcium demands required to maintain normocalcemia and to ensure proper calcium incorporation in bone. In case of intestinal calcium malabsorption, as is the case in intestine‐specific Vdr null mice, elevated levels of circulating PTH and 1,25(OH)2D3 stimulate production of RANKL by osteoblasts, which enhances osteoclastic bone resorption. Moreover, high 1,25(OH)2D3 levels induce the expression of mineralization inhibitors in osteoblasts and osteocytes and reduce bone matrix mineralization. The elevated bone resorption as well as the decreased mineralization contribute to the maintenance of normocalcemia by releasing calcium from bone and preventing calcium incorporation into the bone, respectively.
Disclosures
The authors state that they have no conflicts of interest.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1002/jbm4.10577.
Acknowledgments
This article is dedicated to the memory of Dr Anthony W Norman, an outstanding scientist and inspirational to many scientists, a giant in the vitamin D field, and a major promoter of international collaboration in this field. GC acknowledges funding from Fund for Scientific Research‐Flanders (FWO: G.0A24.16); LV acknowledges funding from Fund for Scientific Research‐Flanders (FWO: G0D0120N).
Authors' roles: LV: conceptualization; visualization; writing—original draft; writing—review and editing. GC: conceptualization; formal analysis; investigation; project administration; supervision; writing—original draft; writing—review and editing.
References
- 1. Haussler MR, Livingston S, Sabir ZL, Haussler CA, Jurutka PW. Vitamin D receptor mediates a myriad of biological actions dependent on its 1,25‐dihydroxyvitamin D ligand: distinct regulatory themes revealed by induction of Klotho and fibroblast growth Factor‐23. JBMR Plus. 2021;5(1):e10432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Malloy PJ, Tasic V, Taha D, et al. Vitamin D receptor mutations in patients with hereditary 1,25‐dihydroxyvitamin D‐resistant rickets. Mol Genet Metab. 2014;111:33‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Christakos S, Dhawan P, Verstuyf A, Verlinden L, Carmeliet G. Vitamin D: metabolism, molecular mechanism of action, and pleiotropic effects. Physiol Rev. 2016;96(1):365‐408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yamamoto H, Tani Y, Kobayashi K, et al. Alternative promoters and renal cell‐specific regulation of the mouse type IIa sodium‐dependent phosphate cotransporter gene. Biochim Biophys Acta. 2005;1732(1–3):43‐52. [DOI] [PubMed] [Google Scholar]
- 5. Kido S, Kaneko I, Tatsumi S, Segawa H, Miyamoto K. Vitamin D and type II sodium‐dependent phosphate cotransporters. Contrib Nephrol. 2013;180:86‐97. [DOI] [PubMed] [Google Scholar]
- 6. Kagi L, Bettoni C, Pastor‐Arroyo EM, Schnitzbauer U, Hernando N, Wagner CA. Regulation of vitamin D metabolizing enzymes in murine renal and extrarenal tissues by dietary phosphate, FGF23, and 1,25(OH)2D3. PLoS One. 2018;13(5):e0195427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fukumoto S. Phosphate metabolism and vitamin D. BoneKey Rep. 2014;3:497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Katai K, Miyamoto K, Kishida S, et al. Regulation of intestinal Na+−dependent phosphate co‐transporters by a low‐phosphate diet and 1,25‐dihydroxyvitamin D3. Biochem J. 1999;343(Pt 3):705‐712. [PMC free article] [PubMed] [Google Scholar]
- 9. Marks J, Srai SK, Biber J, Murer H, Unwin RJ, Debnam ES. Intestinal phosphate absorption and the effect of vitamin D: a comparison of rats with mice. Exp Physiol. 2006;91(3):531‐537. [DOI] [PubMed] [Google Scholar]
- 10. Hernando N, Pastor‐Arroyo EM, Marks J, et al. 1,25(OH)2 vitamin D3 stimulates active phosphate transport but not paracellular phosphate absorption in mouse intestine. J Physiol. 2021;599(4):1131‐1150. [DOI] [PubMed] [Google Scholar]
- 11. Fujita H, Sugimoto K, Inatomi S, et al. Tight junction proteins claudin‐2 and ‐12 are critical for vitamin D‐dependent Ca2+ absorption between enterocytes. Mol Biol Cell. 2008;19(5):1912‐1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li S, De La Cruz J, Hutchens S, et al. Analysis of 1,25‐Dihydroxyvitamin D3 genomic action reveals calcium‐regulating and calcium‐independent effects in mouse intestine and human Enteroids. Mol Cell Biol. 2020;41(1):e00372‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yoshizawa T, Handa Y, Uematsu Y, et al. Mice lacking the vitamin D receptor exhibit impaired bone formation, uterine hypoplasia and growth retardation after weaning. Nat Genet. 1997;16(4):391‐396. [DOI] [PubMed] [Google Scholar]
- 14. Van Cromphaut SJ, Dewerchin M, Hoenderop JG, et al. Duodenal calcium absorption in vitamin D receptor‐knockout mice: functional and molecular aspects. Proc Natl Acad Sci U S A. 2001;98(23):13324‐13329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li YC, Pirro AE, Amling M, et al. Targeted ablation of the vitamin D receptor: an animal model of vitamin D‐dependent rickets type II with alopecia. Proc Natl Acad Sci U S A. 1997;94(18):9831‐9835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Erben RG, Soegiarto DW, Weber K, et al. Deletion of deoxyribonucleic acid binding domain of the vitamin D receptor abrogates genomic and nongenomic functions of vitamin D. Mol Endocrinol. 2002;16(7):1524‐1537. [DOI] [PubMed] [Google Scholar]
- 17. Dardenne O, Prud'homme J, Arabian A, Glorieux FH, St‐Arnaud R. Targeted inactivation of the 25‐hydroxyvitamin D(3)‐1(alpha)‐hydroxylase gene (CYP27B1) creates an animal model of pseudovitamin D‐deficiency rickets. Endocrinology. 2001;142(7):3135‐3141. [DOI] [PubMed] [Google Scholar]
- 18. Panda DK, Miao D, Tremblay ML, et al. Targeted ablation of the 25‐hydroxyvitamin D 1alpha ‐hydroxylase enzyme: evidence for skeletal, reproductive, and immune dysfunction. Proc Natl Acad Sci U S A. 2001;98(13):7498‐7503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dardenne O, Prud'homme J, Hacking SA, Glorieux FH, St‐Arnaud R. Correction of the abnormal mineral ion homeostasis with a high‐calcium, high‐phosphorus, high‐lactose diet rescues the PDDR phenotype of mice deficient for the 25‐hydroxyvitamin D‐1alpha‐hydroxylase (CYP27B1). Bone. 2003;32(4):332‐340. [DOI] [PubMed] [Google Scholar]
- 20. Li YC, Amling M, Pirro AE, et al. Normalization of mineral ion homeostasis by dietary means prevents hyperparathyroidism, rickets, and osteomalacia, but not alopecia in vitamin D receptor‐ablated mice. Endocrinology. 1998;139(10):4391‐4396. [DOI] [PubMed] [Google Scholar]
- 21. Amling M, Priemel M, Holzmann T, et al. Rescue of the skeletal phenotype of vitamin D receptor‐ablated mice in the setting of normal mineral ion homeostasis: formal histomorphometric and biomechanical analyses. Endocrinology. 1999;140(11):4982‐4987. [DOI] [PubMed] [Google Scholar]
- 22. Song Y, Kato S, Fleet JC. Vitamin D receptor (VDR) knockout mice reveal VDR‐independent regulation of intestinal calcium absorption and ECaC2 and calbindin D9k mRNA. J Nutr. 2003;133(2):374‐380. [DOI] [PubMed] [Google Scholar]
- 23. Dardenne O, Prudhomme J, Hacking SA, Glorieux FH, St‐Arnaud R. Rescue of the pseudo‐vitamin D deficiency rickets phenotype of CYP27B1‐deficient mice by treatment with 1,25‐dihydroxyvitamin D3: biochemical, histomorphometric, and biomechanical analyses. J Bone Miner Res. 2003;18(4):637‐643. [DOI] [PubMed] [Google Scholar]
- 24. Hoenderop JG, Dardenne O, Van Abel M, et al. Modulation of renal Ca2+ transport protein genes by dietary Ca2+ and 1,25‐dihydroxyvitamin D3 in 25‐hydroxyvitamin D3‐1alpha‐hydroxylase knockout mice. FASEB J. 2002;16(11):1398‐1406. [DOI] [PubMed] [Google Scholar]
- 25. Rowling MJ, Gliniak C, Welsh J, Fleet JC. High dietary vitamin D prevents hypocalcemia and osteomalacia in CYP27B1 knockout mice. J Nutr. 2007;137(12):2608‐2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lieben L, Masuyama R, Torrekens S, et al. Normocalcemia is maintained in mice under conditions of calcium malabsorption by vitamin D‐induced inhibition of bone mineralization. J Clin Invest. 2012;122(5):1803‐1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Reyes‐Fernandez PC, Fleet JC. Compensatory changes in calcium metabolism accompany the loss of vitamin D receptor (VDR) from the distal intestine and kidney of mice. J Bone Miner Res. 2016;31(1):143‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dhawan P, Veldurthy V, Yehia G, et al. Transgenic expression of the vitamin D receptor restricted to the ileum, cecum, and colon of vitamin D receptor knockout mice rescues vitamin D receptor‐dependent rickets. Endocrinology. 2017;158(11):3792‐3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fleet JC, Reyes‐Fernandez P. Intestinal responses to 1,25 dihydroxyvitamin D are not improved by higher intestinal VDR levels resulting from intestine‐specific transgenic expression of VDR in mice. J Steroid Biochem Mol Biol. 2020;200:105670. [DOI] [PubMed] [Google Scholar]
- 30. Feher JJ, Fullmer CS. Facilitated diffusion of calcium by calcium‐binding protein: its role in intestinal calcium absorption. Prog Clin Biol Res. 1988;252:121‐126. [PubMed] [Google Scholar]
- 31. Benn BS, Ajibade D, Porta A, et al. Active intestinal calcium transport in the absence of transient receptor potential vanilloid type 6 and calbindin‐D9k. Endocrinology. 2008;149(6):3196‐3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Huet T, Laverny G, Ciesielski F, et al. A vitamin D receptor selectively activated by gemini analogs reveals ligand dependent and independent effects. Cell Rep. 2015;10(4):516‐526. [DOI] [PubMed] [Google Scholar]
- 33. Lee SM, Riley EM, Meyer MB, et al. 1,25‐Dihydroxyvitamin D3 controls a cohort of vitamin D receptor target genes in the proximal intestine that is enriched for calcium‐regulating components. J Biol Chem. 2015;290(29):18199‐18215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Moor MB, Bonny O. Ways of calcium reabsorption in the kidney. Am J Physiol Renal Physiol. 2016;310(11):F1337‐F1350. [DOI] [PubMed] [Google Scholar]
- 35. Lieben L, Verlinden L, Masuyama R, et al. Extra‐intestinal calcium handling contributes to normal serum calcium levels when intestinal calcium absorption is suboptimal. Bone. 2015;81:502‐512. [DOI] [PubMed] [Google Scholar]
- 36. Friedman PA. Mechanisms of renal calcium transport. Exp Nephrol. 2000;8(6):343‐350. [DOI] [PubMed] [Google Scholar]
- 37. Blaine J, Chonchol M, Levi M. Renal control of calcium, phosphate, and magnesium homeostasis. Clin J Am Soc Nephrol. 2015;10(7):1257‐1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Renkema KY, Nijenhuis T, van der Eerden BC, et al. Hypervitaminosis D mediates compensatory Ca2+ hyperabsorption in TRPV5 knockout mice. J Am Soc Nephrol. 2005;16(11):3188‐3195. [DOI] [PubMed] [Google Scholar]
- 39. Lederer E. Regulation of serum phosphate. J Physiol. 2014;592(18):3985‐3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Khalil R, Kim NR, Jardi F, Vanderschueren D, Claessens F, Decallonne B. Sex steroids and the kidney: role in renal calcium and phosphate handling. Mol Cell Endocrinol. 2018;465:61‐72. [DOI] [PubMed] [Google Scholar]
- 41. Thomas S, Jaganathan BG. Signaling network regulating osteogenesis in mesenchymal stem cells. J Cell Commun Sig. 2021. Jul 8. DOI: 10.1007/s12079‐021‐00635‐1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Blaney Davidson EN, van de Loo FA, van den Berg WB, van der Kraan PM. How to build an inducible cartilage‐specific transgenic mouse. Arthritis Res Ther. 2014;16(3):210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Donohue MM, Demay MB. Rickets in VDR null mice is secondary to decreased apoptosis of hypertrophic chondrocytes. Endocrinology. 2002;143(9):3691‐3694. [DOI] [PubMed] [Google Scholar]
- 44. Sabbagh Y, Carpenter TO, Demay MB. Hypophosphatemia leads to rickets by impairing caspase‐mediated apoptosis of hypertrophic chondrocytes. Proc Natl Acad Sci U S A. 2005;102(27):9637‐9642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Masuyama R, Stockmans I, Torrekens S, et al. Vitamin D receptor in chondrocytes promotes osteoclastogenesis and regulates FGF23 production in osteoblasts. J Clin Invest. 2006;116(12):3150‐3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Huang W, Yang S, Shao J, Li YP. Signaling and transcriptional regulation in osteoblast commitment and differentiation. Front Biosci. 2007;12:3068‐3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Elefteriou F, Yang X. Genetic mouse models for bone studies—strengths and limitations. Bone. 2011;49(6):1242‐1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gardiner EM, Baldock PA, Thomas GP, et al. Increased formation and decreased resorption of bone in mice with elevated vitamin D receptor in mature cells of the osteoblastic lineage. FASEB J. 2000;14(13):1908‐1916. [DOI] [PubMed] [Google Scholar]
- 49. Yamamoto Y, Yoshizawa T, Fukuda T, et al. Vitamin D receptor in osteoblasts is a negative regulator of bone mass control. Endocrinology. 2013;154(3):1008‐1020. [DOI] [PubMed] [Google Scholar]
- 50. Triliana R, Lam NN, Sawyer RK, Atkins GJ, Morris HA, Anderson PH. Skeletal characterization of an osteoblast‐specific vitamin D receptor transgenic (ObVDR‐B6) mouse model. J Steroid Biochem Mol Biol. 2016;164:331‐336. [DOI] [PubMed] [Google Scholar]
- 51. Nakamichi Y, Udagawa N, Horibe K, et al. VDR in osteoblast‐lineage cells primarily mediates vitamin D treatment‐induced increase in bone mass by suppressing bone resorption. J Bone Miner Res. 2017;32(6):1297‐1308. [DOI] [PubMed] [Google Scholar]
- 52. Mori T, Horibe K, Koide M, et al. The vitamin D receptor in osteoblast‐lineage cells is essential for the proresorptive activity of 1alpha,25(OH)2D3 in vivo. Endocrinology. 2020;161(11):bqaa178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Baldock PA, Thomas GP, Hodge JM, et al. Vitamin D action and regulation of bone remodeling: suppression of osteoclastogenesis by the mature osteoblast. J Bone Miner Res. 2006;21(10):1618‐1626. [DOI] [PubMed] [Google Scholar]
- 54. Misof BM, Blouin S, Hofstaetter JG, Roschger P, Zwerina J, Erben RG. No role of osteocytic osteolysis in the development and recovery of the bone phenotype induced by severe secondary hyperparathyroidism in vitamin D receptor deficient mice. Int J Mol Sci. 2020;21(21):7989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. St John HC, Bishop KA, Meyer MB, et al. The osteoblast to osteocyte transition: epigenetic changes and response to the vitamin D3 hormone. Mol Endocrinol. 2014;28(7):1150‐1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang Y, Zhu J, DeLuca HF. Identification of the vitamin D receptor in osteoblasts and chondrocytes but not osteoclasts in mouse bone. J Bone Miner Res. 2014;29(3):685‐692. [DOI] [PubMed] [Google Scholar]
- 57. Verlinden L, Janssens I, Doms S, Vanhevel J, Carmeliet G, Verstuyf A. Vdr expression in osteoclast precursors is not critical in bone homeostasis. J Steroid Biochem Mol Biol. 2019;195:105478. [DOI] [PubMed] [Google Scholar]
- 58. Starczak Y, Reinke DC, Barratt KR, et al. Absence of vitamin D receptor in mature osteoclasts results in altered osteoclastic activity and bone loss. J Steroid Biochem Mol Biol. 2018;177:77‐82. [DOI] [PubMed] [Google Scholar]
- 59. Starczak Y, Reinke DC, Barratt KR, et al. Vitamin D receptor expression in mature osteoclasts reduces bone loss due to low dietary calcium intake in male mice. J Steroid Biochem Mol Biol. 2021;210:105857. [DOI] [PubMed] [Google Scholar]
- 60. Dai R, Wu Z, Chu HY, et al. Cathepsin K: the action in and beyond bone. Front Cell Dev Biol. 2020;8:433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Goltzman D. Functions of vitamin D in bone. Histochem Cell Biol. 2018;149(4):305‐312. [DOI] [PubMed] [Google Scholar]